
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Zinc oxide–copper model nanocatalysts for CO2 hydrogenation: morphology and interface effects†
Sonia
Hadaoui
ab,
Hang
Liu
a,
Zhang
Lei
a,
Sébastien
Lebègue
 c,
Rabah
Benbalagh
a,
Alexa
Courty
*b and
Ahmed
Naitabdi
*a
c,
Rabah
Benbalagh
a,
Alexa
Courty
*b and
Ahmed
Naitabdi
*a
aLaboratoire de Chimie Physique – Matière et Rayonnement, UMR 7614, Sorbonne Université, 4 place Jussieu, 75005 Paris, France. E-mail: ahmed.nait_abdi@sorbonne-universite.fr
bLaboratoire MONARIS, UMR 8233, Sorbonne Université, 4 place Jussieu, 75005 Paris, France. E-mail: alexa.courty@sorbonne-universite.fr
cLaboratoire de Physique et Chimie Théoriques, UMR 7019, Université de Lorraine, 54000 Nancy, France
First published on 20th December 2023
Abstract
Understanding the mechanism of carbon dioxide (CO2) hydrogenation reaction (HR) to methanol is a major goal as it is an attractive approach to mitigate CO2 emissions by converting them into high-value-added chemicals. Here, we conducted a comparative study on two different ZnO thicknesses grown on Cu(111) as model catalysts. Based on X-ray Photoelectron Spectroscopy (XPS) investigations, we show that the CO2 HR proceeds via two primary paths depending on ZnO thickness: (i) it takes place through a slow path involving bicarbonates, carbonates, and formates, on a thick ZnO film (6.8 monolayers (ML)); (ii) a different and a rapid path was seen for a thin ZnO film (0.9 ML) where the carboxyls were formed readily at room temperature without the formate intermediate. The key effect in the thin ZnO film (ii) (0.9 ML ZnO coverage) involves the Zn–Cu interface which provides activated H atoms. However, both paths exhibit common intermediates as they merge into H2CO which is hydrogenated to CH3O. Additionally, better thermal stability was evidenced for the ZnO thin film (0.9 ML) owing to the presence of a Zn–Cu interface metallic alloy. We demonstrate from DFT computations that a Zn–Cu interface alloy is energetically favorable even in the presence of adsorbed oxygen atoms on Cu(111). The ZnO dewetting phenomenon observed above 550 K was mediated by the desorption of OH species, while CO2 adsorption was found to stabilize ZnO film even at high temperatures (above 550 K). From the morphology point of view, the ZnO films exhibit two distinct structures as a function of the thickness. At low coverage (0.9 ML), ZnO grows into well-ordered Moiré-like patterns with a periodicity of ∼10 Å corresponding to a ZnO-(3 × 3)/Cu(111)-(4 × 4) structure. At high coverage (6.8 ML), the transition towards the ZnO wurtzite structure occurs with the formation of equilateral triangular features on the surface. This study provides insights into the role of the morphology, especially metal–oxide thickness, and the interface in the CO2 HR mechanism.
Introduction
There is now a rising urgency to deal with carbon dioxide emissions, which have continued to increase steadily since the industrial revolution to reach concerning levels as the global average atmospheric concentration in 2019 was 409.8 ppm. Promising strategies are, therefore, being implemented to establish sustainable processes that use CO2 as raw material,1 instead of letting it end up as terminal waste of the utilization of carbon resources. Among these, the catalytic CO2 hydrogenation reaction (HR) to methanol (CH3OH) is the most attractive process and the most studied reaction in heterogeneous catalysis.2–5 The synthesis of methanol occurs through the following hydrogenation process:CO2 + 3H2 → CH3OH + H2, ΔH298![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) K = −49.5 kJ mol−1 K = −49.5 kJ mol−1 | (1) |
The synthesis of methanol has grown tremendously within the last few years.6–8 It is an attractive approach to mitigate CO2 greenhouse gas by converting it into high-value-added chemicals.9,10 Indeed, methanol is not only a key reactant in the synthesis of various important chemicals, such as olefins,11 aromatics,12 and dimethyl ether,13,14 but it is also a highly demanded source of liquid fuel.15,16 CH3OH presents the best compromise as a fuel, since it has a high volumetric energy density (15.9 MJ L−1) and low carbon content.
Cu-based catalysts are the most used catalysts in the CO2 conversion to methanol. In this context, the state-of-the-art industrial catalyst for CH3OH synthesis from the conversion of syngas (CO, H2) consists of Cu/ZnO/Al2O3 composites, where Cu nanoparticles (NPs) are dispersed on a ZnO support and stabilized by an Al2O3 matrix; the reaction takes place at elevated pressures (50–100 bar) and temperatures (200–300 °C).2,17 However, the harsh thermodynamic conditions required for achieving a good CH3OH production yield make this reaction energy-demanding and a costly process.2,17 Besides, the precise role of each component is still widely debated despite the widespread use of these catalysts. Nonetheless, the role of Cu, as claimed and has long been known for high activity toward H2 dissociation,18,19 is essential to provide activated H atoms. These are crucial to the hydrogenation of intermediate species along the overall reaction to form methanol, such as bicarbonates (HCO3−), carbonates (CO32−), formate (HCOO−), carboxyl (OCOH), formyl (HCO), formaldehyde (H2CO), methoxy (H3CO), which eventually transforms into CH3OH. Regarding CO2, previous computational studies have revealed that Cu(111) is particularly inactive toward the dissociation of CO2 due to a high adsorption barrier, while Cu(110) and Cu(100) are capable of activating this molecule.19 On the other hand, ZnO is not a support material only; instead, it ensures the dispersion of Cu nanoparticles (NPs) and contributes to their stabilization against sintering during the reaction. ZnO allows the adsorption of CO2 and its activation in the form of carbonates (CO32−) or HCOO−.3,8 ZnO plays also a major role in the stabilization of key intermediates.2
Concerning the reaction mechanism, which remains under debate, several paths have been proposed for the (CO2 HR) to CH3OH reaction. The first follows the “formate path” in which adsorbed CO2 is hydrogenated to HCOO−,20–25 one of the most important and regularly reported intermediate species in Cu-based catalysts. Subsequently, among the elementary steps proposed in previous studies, depending on experimental conditions or the nature of the metal–oxide, the mechanism then proceeds via the following sequence *CO2 → *HCOO− → *H3CO− → CH3OH.8,23 Wang et al. demonstrated recently that the last step of this sequence, i.e., the conversion of H3CO− to CH3OH, is done by hydrolysis emphasizing the role of H2O in CO2HR conversion,23 while the other ones proceed via hydrogenation processes by capturing adsorbed H atoms. The second proposed path proceeds via “reverse water gas shift” (rWGS) followed by CO hydrogenation.3,8,20
| CO2 + H2 ⇆ CO + H2O, ΔH298K = 41.2 kJ mol−1 | (2) |
Here, the interaction of CO2 with a surface OH leads to the formation of OCOH, another key intermediate in the CO2HR, which is subsequently transformed into CO. One of the sequences proposed herein is *CO2 → *OCOH → *CO → *HCO → *H2CO → *H3CO → CH3OH.3 Despite these advances, the correlation between the structural morphology of the catalysts and the reaction pathway is not yet well established. In particular, the effect of ZnO thickness, the role of the copper–zinc interface interactions, and the synergistic effects at the boundaries of ZnO–Cu in the formation of key intermediates in the CO2HR are still debated. Here, one of the main limitations lies in the difficult access to the surface composition and Cu–ZnO interfaces at the nanoscale.
Consequently, model catalysts consisting of ultrathin films of ZnO grown on crystalline metallic surfaces, including Cu(111), are suitable simple systems to investigate the surface of the catalyst and monitor its evolution as a function of the reaction conditions. Indeed, they constitute ideal systems to identify molecular species occurring during the CO2HR and their coordination as a function of morphology. The question to be answered here is: do the two reaction paths discussed above depend on the thickness of the metal oxide? In this regard, the use of surface characterization tools, such as scanning tunneling microscopy (STM)26 and X-ray photoelectron spectroscopy (XPS),26–28 even if they are operated far from the realistic thermodynamic conditions of the CO2HR, provides valuable insights on the morphology and chemical surface composition of the catalysts.
In the present paper, we have conducted a systematic study on the effect of pure CO2 and H2 reactants and their mixture (CO2 + H2) on ZnO films grown on Cu(111) for two different coverages (0.9 ML and 6.8 ML) as a function of annealing at room temperature (RT) up to 650 K. XPS measurements were conducted before and after exposures at 1 mbar to identify various intermediate species occurring during different CO2HR stages. In addition, we have conducted STM investigations to examine the morphology of the ZnO film as a function of the thickness and its evolution under (CO2 + H2) reaction conditions at 36 mbar. Multiple morphologies are seen as a function of the thickness and annealing conditions throughout the process. This distinctive approach has enabled us to specify the conditions under which the two reaction paths take place with varying ZnO thicknesses and to identify the driving force for the ZnO dewetting phenomenon. In particular, we show that the species formed at the start of the reaction, depending on the ZnO thickness, are those that determine the reaction path that follows. For the thick (6.8 ML) and the thin (0.9 ML) ZnO films, the initial intermediate species formed, HCO3− and OCOH, are the ones responsible for the two proposed pathways, the formate pathway and the carboxyl pathway, respectively. Besides, we provide evidence for the formation of a Zn–Cu interface alloy which plays two roles, (i) it promotes the formation of OCOH as it supplies activated H atoms in the case of 0.9 ML and (ii) it stabilizes the ZnO film. Based on DFT computations, we show that the formation of this interface alloy was energetically favorable even in the presence of adsorbed O atoms on the Cu(111) surface.
Experimental and computational methods
Material
The copper single crystal (111) (purity of 99.9999%, diameter of 8 mm, and thickness of 2 mm) was purchased from MaTeck GmbH. It had one side polished with roughness of less than 0.01 μm and orientation accuracy of <1°. Zinc was used in the form of pellets (purity 99.99%) as supplied by Goodfellow for the growth of Zn.Growth of ZnO on the Cu(111) surface
The Cu(111) surface was prepared by several cycles of argon ion sputtering (15 μA) and annealing at 723 K in UHV. To prepare well-ordered ZnO thin films on Cu(111), we used the metal vapor deposition method. Zinc was then deposited onto the clean Cu(111) surface by evaporation using an effusion cell evaporator; this was performed under ambient O2 atmosphere at a pressure of 10−7 mbar of O2 at room temperature. The zinc oxide film was then further annealed under 10−6 mbar of O2 at 423 K for one hour. The method of ZnO growth based on Zn deposition in O2 ambient offers the advantage of preventing Zn intermixing with Cu during the deposition step and stops subsequent Zn diffusion toward the Cu bulk during oxidation and annealing treatments.29Scanning tunneling microscopy
The STM setup consists of two connected chambers: one is the analysis chamber containing a microscope (a variable temperature XA microscope from Omicron NanoTechnology), and the other is the preparation chamber equipped with a LEED and standard sample preparation tools (for annealing and sputtering). For the exposure to reactive gases, a mixture of CO2 and H2 was introduced into the analysis chamber at room temperature and left to thermalize for 2 hours before scanning. STM images were acquired before gas exposure in the UHV conditions at a base pressure of 10−11 mbar and under in situ conditions of (CO2 + H2) at a base pressure of 30 to 40 mbar at room temperature. The tungsten tip was cleaned by flash heating at UHV before use.X-ray photoelectron spectroscopy
The XPS spectra were recorded on a SPECS spectrometer using an Al kα monochromatized X-ray source (1486.71 eV) and equipped with a PHOIBOS 150 electron analyzer. XPS ex situ analysis of ZnO/Cu(111) samples is performed under UHV conditions after exposition to reactive gases (CO2, H2). Several ZnO/Cu(111) samples were investigated as a function of ZnO thickness. For each film's thickness, the samples were independently exposed to pure CO2 and H2 gases, as well as to a CO2:H2 (1:3) mixture. The nominal ambient pressure for each gas was 5 mbar at room temperature for an hour. Stepwise annealing treatment was conducted from room temperature (RT = 300 K) up to 650 K under each gas exposure. The convolution of C 1s and O 1s was conducted using a home-built procedure in the Igor software of Wavemetrics®. Gaussian functions with a linear background were used. Similar procedures and tools were used for Cu 3p and Zn 3p but with Doniach Sunjic line shapes.
Computational details
We performed our simulations using density functional theory (DFT) and the projector augmented wave method as described by the VASP (Vienna ab initio simulation package) code [1,2].30,31 The generalized gradient approximation (GGA) was used as proposed by Perdew, Burke, and Ernzerhof (PBE)32 as the exchange–correlation potential. The plane wave cutoff of the corresponding basis expansion was set to 500 eV; owing to the large size of the simulated systems (see below), the Γ point was used to sample the Brillouin zone for the relaxation of the structures and was completed by single point calculations on the relaxed structures with a 4 × 4 × 1 k-points mesh to obtain precise total energies. Convergence was reached when the energy difference between the two electronic steps became smaller than a threshold of 10−6 eV and the residual forces on atoms were smaller than 1.0 × 10−2 eV Å−1.Results and discussion
A. The XPS study of the (CO2 + H2) hydrogenation reaction on ZnO/Cu(111)
We have investigated the surface chemical composition as a function of the ZnO ultrathin film thickness grown on Cu(111) and of the annealing treatment from room temperature (RT) up to 650 K. Two ZnO film thicknesses were investigated: 6.8 ML and 0.9 ML (Fig. 1). First, exposures to pure CO2 or H2 were conducted to probe the chemical effects of each reactive gas on the surface. Afterward, we examined the (CO2 + H2) gas mixture exposure to probe the CO2 hydrogenation reaction on these surfaces.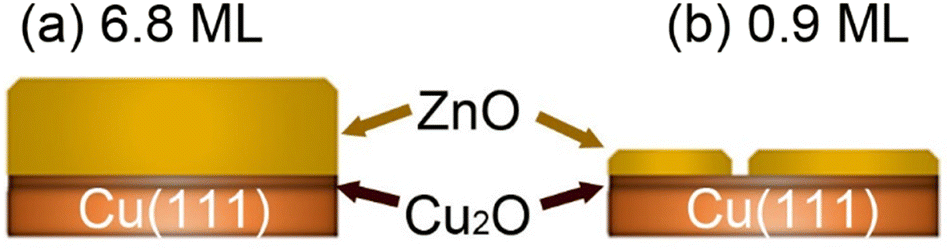 | ||
| Fig. 1 Schematic illustration of the two ZnO/Cu(111) surfaces investigated herein as a function of the ZnO thickness. (a) 6.8 ML, (b) 0.9 ML. | ||
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, noted HxCO*, at BE = 285.6 eV (Fig. 2).34 The HxCO* species include H2CO species, as key intermediates in the CO2 HR to methanol. The specific methoxy species, which arises exactly at 285.2 eV, was only seen for (CO2 + H2) exposure after annealing at 550 K. The methoxy groups are proposed as active and ultimate intermediate species in the CO2 HR pathway towards CH3OH.21,40 However, the hydrogenation of CH3OH is hindered by an energy barrier, Ea = 1.49 eV, as calculated by DFT.40 Concerning the formation of CH3O*, based on DFT calculations, Kattel et al.40 and Wang et al.21 proposed similar reaction pathways, where CH3OH was produced from the hydrogenation of *H2CO across two transient states via CH3O* species on a ZnO/Cu(111) catalyst. H2CO was also proposed as a direct dehydrogenation product in the methanol oxidation on the Pd(111) surface.41 Below 550 K and for other exposures (pure CO2 and H2), HxCO* species dominated this second peak. In the case of pure CO2, this component exhibited even an additional positive chemical shift of 0.2 eV (285.8 eV) from RT to 450 K.
O, noted HxCO*, at BE = 285.6 eV (Fig. 2).34 The HxCO* species include H2CO species, as key intermediates in the CO2 HR to methanol. The specific methoxy species, which arises exactly at 285.2 eV, was only seen for (CO2 + H2) exposure after annealing at 550 K. The methoxy groups are proposed as active and ultimate intermediate species in the CO2 HR pathway towards CH3OH.21,40 However, the hydrogenation of CH3OH is hindered by an energy barrier, Ea = 1.49 eV, as calculated by DFT.40 Concerning the formation of CH3O*, based on DFT calculations, Kattel et al.40 and Wang et al.21 proposed similar reaction pathways, where CH3OH was produced from the hydrogenation of *H2CO across two transient states via CH3O* species on a ZnO/Cu(111) catalyst. H2CO was also proposed as a direct dehydrogenation product in the methanol oxidation on the Pd(111) surface.41 Below 550 K and for other exposures (pure CO2 and H2), HxCO* species dominated this second peak. In the case of pure CO2, this component exhibited even an additional positive chemical shift of 0.2 eV (285.8 eV) from RT to 450 K.
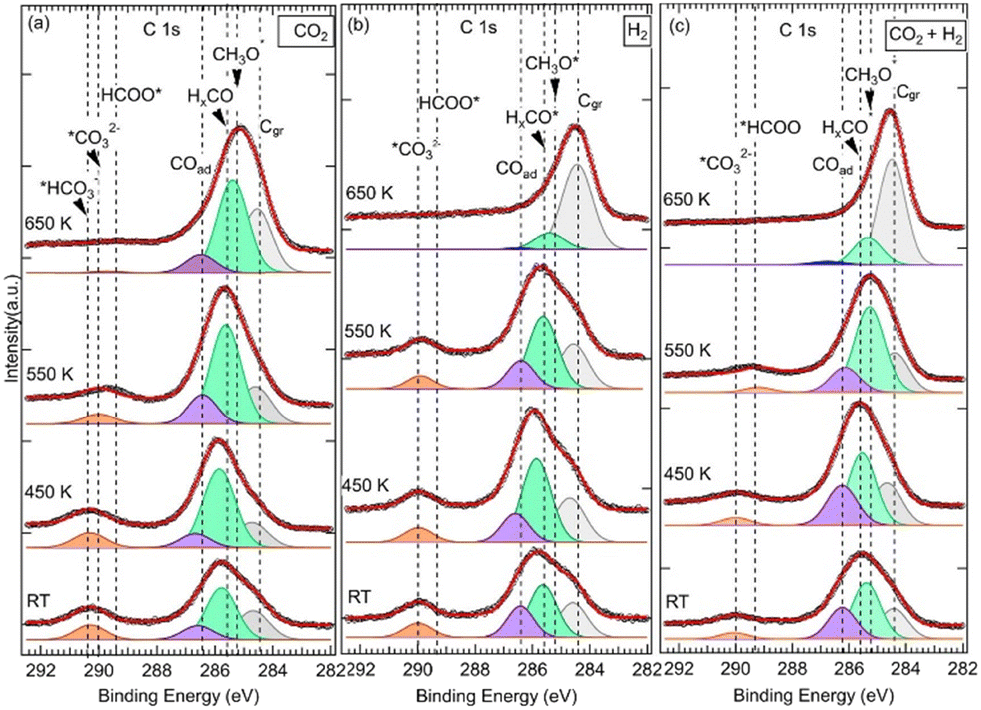 | ||
| Fig. 2 The XPS C 1s core level spectra of three similar ZnO ultrathin film samples with a thickness of 6.8 ML grown on the Cu(111) surface as a function of temperature and gas exposures. (a) Exposure to pure CO2 at 5 mbar nominal pressure. (b) Exposure to pure H2 at 5 mbar nominal pressure. (c) Exposure to the gas mixture (CO2:H2) at 5 mbar nominal pressure at a (1:3) ratio. Cgr stands for graphitic carbon. | ||
The third contribution, which is related to the C 1s spectra, appeared at BE = 286.4 eV for all three exposures from RT up to 550 K (Fig. 2). For the sample exposed to pure CO2, it was detected at RT at slightly higher BE (EB = 286.6 eV), and it was present even after annealing at 650 K. It is attributed to adsorbed CO (CO double-bond) or CO(H) (C–O single-bond) species, (noted COad), resulting from CO2 dissociation.27,28,36,42,43 CO is also considered an important intermediate of the CO2 HR to CH3OH on ZnO/Cu catalysts as it is expected to be the main product along the rWGS + CO hydrogenation pathway.2,3,40 From DFT calculations,3,21,40 it has been proposed that CO acted as an intermediate since it evolved across multiple hydrogenation steps towards H2CO by reacting with surface *H or *OH species. In both CO2HR mechanistic paths, the formate and rWGS, H2CO is a common intermediate that eventually transforms into methoxy.
Interestingly, our XPS data show the presence of both H2CO and CH3O− at BE = 285.6 eV and BE = 285.2 eV, respectively (Fig. 2). In this mechanism, the formation of adsorbed CO occurs due to an indirect process in which CO2 is first activated on ZnO or at the ZnO/Cu interface and converted to hydrocarboxyl intermediate species (OCOH*). Subsequently, CO is formed via the following activation process: OCOH* → *CO + OH* CO can be also generated readily from the dissociation of CO2 without passing through the OCOH* intermediate. In this case, CO2 is first stabilized on the surface as slightly charged COδ−2 before it dissociates into CO* and O* surfaces species via the following reaction:27,43,44
| CO2(g) ⇒ COδ−2(ad) → CO* + O* | (3) |
In both processes, however, as CO was produced at RT, CO2 adsorption sites and the CO bond activation are probably assisted by chemical composition,45–47 morphology,28,47–49 and electronic effects44,50 of the ZnO/Cu(111) surface. Regarding the surface composition (Zn or O termination) and morphology, it was proposed based on experimental investigations that the adsorption of CO2 on ZnO surfaces can occur through a bidentate geometry involving the formation of two bonds with one C and O atom of CO2 bonding to one O and one Zn atoms of the substrate, respectively. However, from theoretical calculations, a more stable configuration corresponding to a tridentate geometry was proposed, where the second O atom of CO2 was also covalently bonded to the ZnO surface.51 In both configurations, the adsorbed CO2 adopted a rather bent geometry. Besides, the ZnO structure considered therein was uniform and similar to the bulk structure, where O and Zn atoms were not located on the same surface plane. The CO2 dissociation is energetically unfavorable for these configurations, especially on reduced ZnO, which is the case in ultrathin films. Indeed, from the perspective of computational modeling, a high energy barrier, E = 1.07 eV, was reported for the activation of CO2 on a ZnO/Cu system.47
Here, however, the formation of CO at RT is facilitated by electronic effects associated with oxygen vacancies (Ov) on the ZnO film, ZnO defects, and ZnO/Cu(111) interface sites. Given the ultrathin film nature of our ZnO and strong ZnO–Cu interactions at the interface, these effects are more severe. Indeed, from the oxidation state point of view, as it will be discussed hereafter from the Auger results (see also Fig. S1, ESI†), the ZnO chemical state at the interface corresponds to Znδ+Ox, which provides interesting oxophilic Znδ+ cations and Ov sites. Indeed, Znδ+ cations are considered important adsorption sites for CO2 activation.44,52 Moreover, regarding the morphology of ZnO here, as it will be discussed below in the STM section, Zn and O are coplanar. This geometry brings CO2 molecules close enough to the surface, facilitating the electron transfer process. Therefore, the transfer of electron density from confined electrons on oxygen vacancies of ZnO film to adsorbed CO2 leads to the occurrence of active COδ−2(ad), via charge transfer to the lowest unoccupied antibonding π*, 2bu (2b2u and 2b3u), and molecular orbitals of CO2. This process causes the weakening of the CO bond and the activated COδ−2(ad) moieties would evolve into intermediate species that act as precursors to CO species.
Indeed, a fourth contribution in the C 1s spectra can be seen at high binding energies of around 290 eV (Fig. 2). These are highly oxidized carbon species corresponding to first intermediates and resulting from COδ−2(ad) moieties. Depending on the temperature and the nature of the reaction treatment, we identified three contributions at BEs of 289.4 eV, 290.0 eV, and 290.4 eV that we attribute to *HCOO and *OCOH,20,28,35,49,53 *CO32−,28,34,49 and bicarbonates (HCO3−),54,55 respectively. The presence of the *HCOO contribution, which is considered as the rate-limiting step at the ZnO–Cu interface during the CO2 hydrogenation to methanol,2,3,40 is relevant here regarding the CO2HR on our ZnO/Cu(111) sample. It can be generated directly on the ZnO/Cu(111) surface owing to the presence of activated adsorbed CO2 and H atoms via the following hydrogenation process:
| COδ−2(ad) + *H + (1 − δ)e− → HCOO− | (4) |
Direct formation of HCOO, considering a Langmuir–Hinshelwood mechanism, resulting from adsorbed CO2 and *H surface interactions, has been proposed by Pathak et al. based on DFT calculations.56 While the values of BEs reported in the literature for COad, CO32−, CH3O−*, were in general slightly or not sensitive to the chemical surface composition (ZnO,27,28,49 ZrO2,34 CeO2,55 Cu2O,34,38 Cu,36,38,42,43 Pt35,57), and the BE value of *HCOO− were clearly sensitive. For example, the BEs of *HCOO− reported on ZnO surfaces are found around ∼289 eV,28,35,49,53,58 which are consistent with our BE at 289.4 eV; they are ∼2 eV higher than that reported on ZrO234 or Cu and Cu2O/Cu(111) surfaces (at ∼287 eV).36–38,43,59 From the XPS spectra in Fig. 2, we have found the *HCOO− contribution mainly happens after annealing at 450K, which points to the existence of an activation barrier for this hydrogenation process and another reaction path. Effectively, from DFT calculation by Kattel,40 the HCOO− formation was produced from adsorbed CO2 and H via a transient state and an activation barrier of Ea ∼1.1 eV. Besides, HCOO− can be produced from the hydrogenation of CO32− as we observed a clear loss of this contribution above 450 K, which was in favor of the formate in the case of (CO2 + H2) exposure (Fig. 2c). In the case of exposure to pure CO2, an additional barrier may apply as this transition was seen partially at 550 K, and the proportion of CO32− remained significant at this temperature (Fig. 2a). Here, the transformation of CO32− into HCOO− is also a hydrogenation reaction that can take place via the following process:
| CO32− + *H + *H → HCOO− + OH− | (5) |
This process reflects the role of CO32− as a key intermediate in the CO2HR on the ZnO/Cu(111) surface. The presence of OH− on the surface was indeed evidenced from the O 1s core level spectra where its contribution appeared at EB = 531.8 eV (not shown here). Interestingly, in the exposure to pure H2 (Fig. 2b), the CO32− did not transform into HCOO− and remained the main contribution even after annealing at 550 K. Thus, the occurrence of a competitive and more favorable reaction for H2 transformation must be considered. In this case, the envisioned process is the reaction leading to OH− formation (H2 + *O–Zn + *O–Zn → 2OH− + 2Zn2+ + 2e−). This sequence is the favored mechanism under exposure to pure H2. As a result, it generates OH− moieties accumulation on the ZnO surface, blocking the active sites for HCOO− formation through the reaction (5). This process will be discussed in relation to the morphology evolution as a function of temperature, exposure, and ZnO thickness.
Regarding the carbonates themselves, here COδ−2(ad) on ZnO can lead to the formation of CO32− readily at RT viareaction (6):
| COδ−2(ad) + CO2(g) + (2 − δ)e− → CO32− + COad | (6) |
This contribution was indeed found starting at RT after exposure to various gases at EB = 290.0 eV (Fig. 2). Carbonates are known to form readily at RT even at UHV from CO2 adsorption on metal–oxide surfaces, such as ZnO.28,51 The formation of CO32− according to the reaction (6) relies on the availability of COδ−2(ad) on the surface. For example, Deng et al found a substantial growth of CO32− concomitant with the depletion of adsorbed CO2 when Zn was deposited on the Cu surface.37
The HCO3− contribution appears readily at RT and EB = 290.4 eV. It is only seen for the ZnO thick film and only for the exposure to pure CO2 (Fig. 2a). Here, the fourth contribution in the C 1s spectra is dominated by HCO3−, for pure CO2 exposure up to 450 K (Fig. 2a), and CO32−, for both pure H2 and (CO2 + H2) mixture exposures, from RT up to 450 K (Fig. 2b and 2c). HCO3− are ubiquitous species that form at RT on hydroxylated metal–oxide surfaces in the presence of adsorbed CO2.60–62 As the reaction to form HCO3− involves surface O–H groups, it is the H2O dissociation on ZnO that provides hydroxyl species in the absence of H2. Indeed, the presence of OH species on the ZnO surface (OH–ZnO) can be confirmed from the O 1s XPS spectra where a component at BE = 531.8 eV can be seen (not shown here). This is consistent with a value reported on the ZnO/Pt(111) system.35 Thus, a mechanism involving adsorbed and activated CO2 in interaction with surface O–H groups can be envisioned in the formation of bicarbonates (eqn (7)).
| COδ−2(ad) + H–O−(surf) → HCO−3(ad) + δe− | (7) |
This mechanism is in line with the model proposed by Baltrusaitis et al. based on quantum chemical calculations, where an intermolecular proton-transfer reaction process was examined.61 They found that bicarbonate species occur through a nucleophilic attack of adsorbed CO2 by surface O–H groups followed by rearrangement on the surface.
A clear evolution of HCO3− toward CO32− was seen with the annealing treatment under CO2 (Fig. 2a). The formation of CO32− species from HCO3− may take place through this process, such that the released H atom bonds to ZnO to form Zn–OH:
| HCO3− → CO32− + H+ | (8) |
Studies clearly show the formation of bicarbonates at low temperatures on various metal–oxides, which evolve toward CO32− or other intermediates upon annealing, such as at 373 K.63 Our results are in line with those reported by Köck et al. that HCO3− species formed upon CO2 adsorption on ZrO2 and Y2O3 vanish upon annealing at 473 K.63 Indeed, in our study, we observe a sharp decrease of the HCO3− component above 450 K in favor of CO32− species that dominate the C 1s spectra at 550 K (Fig. 2a).
 | ||
| Fig. 3 XPS C 1s core level spectra of ZnO/(Cu(111) as a function of the ZnO thickness [(a) 6.8 ML; (b) 0.9 ML] and the temperature from RT up to 650 K under gas mixture (CO2:H2) at 1 mbar nominal pressure at a (1:3) ratio. Cgr stands for graphitic carbon. | ||
It can be seen in the C 1s spectra in Fig. 3, another major difference between the two samples is the appearance of a new component at RT at BE = 288.7 eV in the 0.9 ML ZnO thin film (Fig. 3b). This component, which was not seen in the 6.8 ML sample at RT (Fig. 3a), is attributed to the C 1s in carboxyl species (OCOH). Some studies have suggested that this component may overlap with HCOO−, such as in ZrO2-based samples at 287.7 eV.34 However, on ZnO surfaces there is a clear chemical core-level-shift of ∼0.6 eV between the two species (OCOH and HCOO−), with the formate being at higher binding energy.35 Similarly, carboxylate species, formed on CeOx nanoparticles supported on Cu(111), were reported at 288.5 eV, while HCOO− species were assigned to the C 1s peak at 289.2 eV.59 The formation of OCOH, which was proposed as a key intermediate in CO2HR via the rWGS path,3,8,20 only on the 0.9 ML sample is an indication that it is dependent on the ZnO thickness. More specifically, the interfaces, ZnO–Cu and Zn–Cu, which are exposed for this thickness, must be considered. Hence, we propose a Zn–Cu interface-related mechanism for the formation of OCOH involving adsorbed H atoms (eqn (9)):
| CO2 + *H → OCOH | (9) |
Here, activated H atoms were supplied by Cu atoms at the interface edge forming a Cu–Zn–O–H* group, following an rWGS process.47 Hence, the stabilization of OCOH always takes place through the adsorption of OCO via *H–O–Zn–Cu according to the rWGS mechanism.53 OCOH species would evolve eventually toward CO and OH species OCOH* → *CO + OH* in the 0.9 ML sample. The CO will then be hydrogenated to other reaction intermediates as will be discussed hereafter. Above 550 K, the BE of the OCOH shifts by +0.5 eV to 289.2 eV. The shift can be attributed to a change in the chemical bonding environment and electronic evolution due to a change in the morphology of the ZnO film at this temperature. It is also possible to envision that part of the OCOH may transform to formate at higher temperatures as this binding energy is close to that of the HCOO−.
Species that are common to both samples (6.8 ML and 0.9 ML) are seen in the C 1s spectra in Fig. 3 at 285.2 eV, 285.6 eV, and 286.6 eV. As discussed in detail above, they are attributed to H2CO, CH3O−, and CO, respectively. The presence of these components on both surfaces indicates that, although two different paths for the CO2 HR can be envisaged here as a function of the two ZnO thicknesses, they represent intermediate species that are common to both reaction paths. CO was also seen in both samples, however, its role as an intermediate varies in these two paths depending on the ZnO thickness. In the thin film (0.9 ML), CO adsorbed molecules were produced following the conversion of OCOH as discussed above, as true intermediate species in the second reaction path. Regarding the thick film (6.8 ML), COad was produced as a secondary product following the formation of CO32− at RT from CO2 (see eqn (6) above).
We monitored the evolution of all the intermediates seen in both ZnO thicknesses (6.8 ML and 0.9 ML) as a function of the temperature and gas exposures as shown in Fig. 4. In Fig. 4a, each plot shows the evolution of the sum of all contributions appearing at high binding energy (288.7–290.4 eV) of the C 1s for various exposures and as a function of the thickness. This sum, where ΣCHigh-ox, corresponds to the contributions of highly oxidized carbon species, includes OCOH, HCOO−, CO32−, and HCO3−. The sum does not necessarily contain all the contributions at each temperature since some components may appear or disappear to the detriment or in favor of another as a function of the temperature. In Fig. 4b, each plot shows the evolution as a function of the temperature of adsorbed CO, COad, for various exposures and the 6.8 ML and 0.9 ML ZnO thicknesses. For the exposure to the gas mixture (CO2 + H2), we compared the two ZnO thicknesses, 6.8 ML and 0.9 ML, with respect to the evolution of ΣCHigh-ox and COad. For 6.8 ML, we also examined the effect of exposures to individual pure gases, CO2 and H2. This procedure allowed us to identify intermediates that form owing to a specific gas (CO2 or H2) and highlight the effect of hydrogenation when the mixture (CO2 + H2) is present.
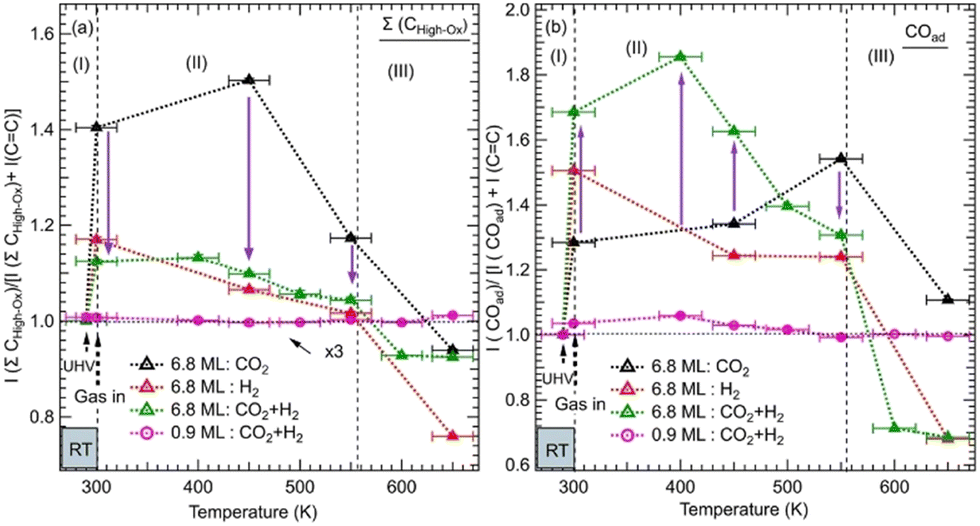 | ||
| Fig. 4 Evolution of the normalized integrated XPS intensity ratios as a function of gas exposure, temperature, and ZnO thickness on Cu(111) of: (a) the XPS peak corresponding to the Σ(HCOO−, OCOH, CO32−) and (b) the peak of COad. The integrated intensities of the XPS peaks were obtained by fitting the C 1s spectra as shown in Fig. 2 and 3. The ratios were calculated with respect to the reference sample of the graphitic carbon (CC). The ratios were normalized to 1 at RT before gas exposure to offset the initial adsorbed species due to residual carbon species. | ||
The variations in the intermediates in Fig. 4 can be decomposed into three regions: (I) corresponds to the initial stage at RT before the annealing. It illustrates the formation of various intermediates readily at RT just after the gas exposure. (II) shows the variations within the [RT–550 K] temperature range. This region corresponds to the evolution of intermediates on a rather stable morphology of ZnO ultrathin film. (III) covers the temperature range of >550 K where ZnO dewetting occurs. In Fig. 4a, the exposure to CO2 alone produced a strong increase in the ΣCHigh-ox contribution at RT in the case of the 6.8 ML thickness. This increase is due to the formation of HCO3− species. The formation of this intermediate indicates that the initial adsorption and activation of CO2 occurs readily at RT in the form of HCO3−. For the same thickness and in the presence of H2, under (CO2 + H2), the ΣCHigh-ox, which contains only the CO32− component, decreased strongly with respect to that for exposure to CO2 alone. Indeed, the ΣCHigh-ox contribution under (CO2 + H2) amounts to only 30% of that under CO2. This evolution underlines a dehydrogenation process that generates CO32− species, from HCO3−via the reaction (8). However, we observe an opposite trend regarding the evolution of the COad component as shown in Fig. 4b. At RT, the COad contribution is 59% higher under (CO2 + H2) than under CO2 alone. Thus, the dehydrogenation under (CO2 + H2) has generated not only CO32− but also CO. Interestingly, while the COad decreased rapidly under (CO2 + H2), when increasing the temperature to >375 K, it continued increasing under CO2 alone for the thick ZnO films (6.8 ML). Hence, CO can be considered as an intermediate that continued to evolve toward other intermediates, especially HCOO− and methoxy species, while it can be considered as a byproduct that accumulates on the surface when H2 is not present (Fig. 4b). In the region (II), the ΣCHigh-ox, which was dominated by CO32−, initiated its transition towards HCOO− intermediates, which was concomitant with the decrease in COad for the 6.8 ML sample. The transformation of CO32− to HCOO− was completed at 550 K. The stabilization of CO32− can be related to oxygen vacancy defects on the ZnO film.37 At this location, oxophilic Znδ+ sites are present, which belong to oxygen-deficient Znδ+Ox areas.44 The presence of surface defects generates an open surface structure with fewer repulsions for CO32− adsorption/stabilization.51
In region (III), both ΣCHigh-ox and COad contributions gradually decreased above 450 K up to 550 K, a temperature at which they rapidly dropped. This decrease may be attributed to further hydrogenation of the ΣCHigh-ox to other species that did not stay on the surface and reached the gas phase.
However, on the 0.9 ML ZnO surface, only a small increase in the ΣCHigh-ox was seen, which corresponded to the formation of OCOH species. The formation of OCOH species instead of HCO3−, CO32−, or HCOO− can be explained by the chemical composition of the 0.9 ML of the ZnO that differs from that of the 6.8 ML. In particular, the 0.9 ML ZnO film exhibits a highly defective structure with a lower concentration of O atoms due to the presence of oxygen vacancies; above all, there is the formation of the Zn–Cu alloy interface at the expense of the ZnO. Thus, the formation of HCO3− or CO32− species that require at least one O atom per molecule from the surface is hindered. Indeed, Auger Zn L3M45M45 spectra acquired on both surfaces exhibit features that are substantially different (see Fig. S1, ESI†). The Zn LMM spectrum acquired on the 0.9 ML exhibits two contributions at kinetic energies (EK), EK = 992.0 eV and EK = 988.3 eV. The former corresponds to a metallic oxidation state Zn0 and the latter to Zn2+ (ZnO) contribution. The Zn0 component, having a metallic character, is attributed to Zn atoms at the Zn–Cu alloy phase at the interface. At 6.8 ML ZnO thickness, the Zn LMM spectrum is dominated by the Zn2+ component at EK = 988.3 eV which reflects the formation of a ZnO structure. As this component coexists with the Zn0 state, it is indicative of the stability of the Zn–Cu metallic interface (Fig. S1, ESI†). Regarding the lack of HCOO− on the thin ZnO film (0.9 ML) and their strong presence on the thick ZnO film, Nie et al. have also recently shown that large thicknesses exhibit superior activities toward the formation of HCOO− owing to the presence of active centers for CO2 activation.25 In Fig. 4b, we noticed a gradual increase of the COad component up 450 K, which is concomitant with the decrease in the OCOH species. This is attributed to the transformation of OCOH to CO. A possible mechanism for this evolution to take place is via this hydrogenation process, where an H atom adsorbed on the Cu-Zn interface is abstracted: OCOH + *H → CO + H2O. The specific occurrence of this reaction only on the thin film (0.9 ML), can be related to favorable stability of the alloyed Cu–Zn interface in the presence of CO. Recently, Amann et al. showed that CO tends to promote the formation of metallic Zn that was alloyed with the Cu near the surface, providing high-density alloyed Cu–Zn sites.39
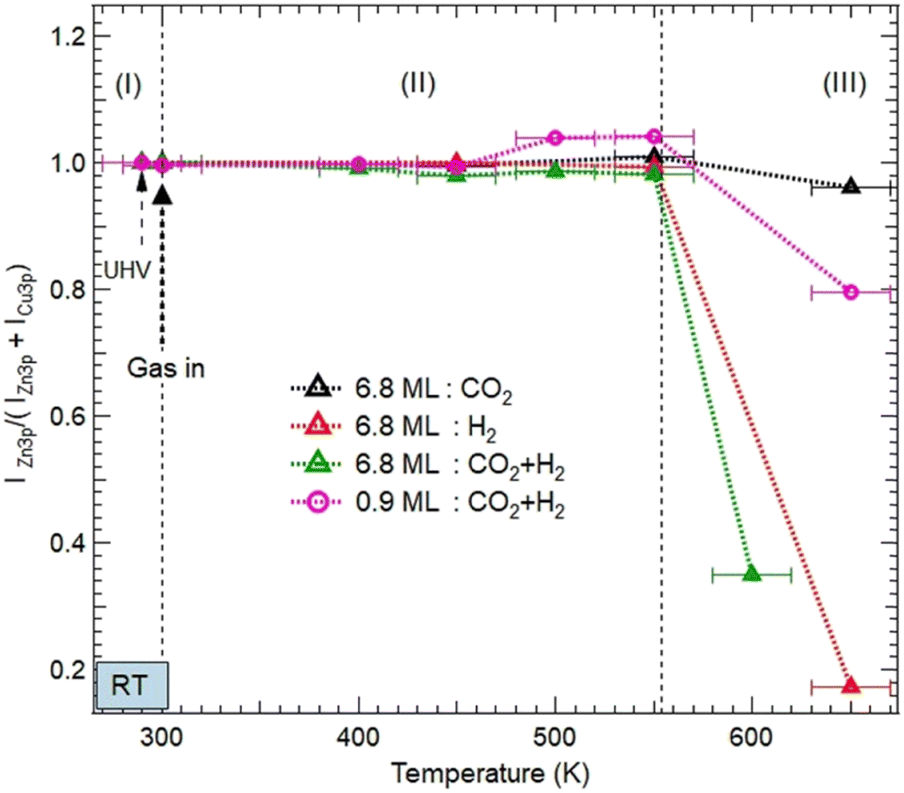 | ||
| Fig. 5 Evolution of the normalized XPS intensity ratios IZn/(IZn + ICu) as a function of gas exposures, temperature, and ZnO thickness on Cu(111). The XPS intensities were obtained from the integrated peak of Zn 3p and Cu 3p after fitting as shown in Fig. S2 and S3 (ESI†). The ratio is normalized to the intensity value obtained in the UHV just before gas dosing at RT. Three areas, (I), (II), and (III) are indicated to illustrate different evolutions of the ZnO morphologies as a function of the temperature. (I) corresponds to just before and after exposure to various reactive gases at room temperature; (II) refers to the temperature interval [RT–550 K] where the ZnO films remain on the surface before dewetting; and (III) (above 550 K) corresponds to the severe dewetting of the ZnO films. | ||
Above 550 K, a decrease in the Zn XPS signal was detected as a result of the ZnO film dewetting. This effect was more severe for the 6.8 ML coverage and in the presence of H2 (Fig. 5). Here, the adsorption of H2 and its subsequent dissociation led to the formation of OH moieties by abstracting O atoms from the ZnO surface. Therefore, the ZnO dewetting takes place through a dehydroxylation process, leading to lattice break-up.
However, under pure CO2 exposure, the ZnO film is still present on the surface even at 650 K. This relative stability is induced by the adsorption of CO2 on the ZnO surface and its subsequent transformation into various chemical intermediates as discussed in the previous section. Unlike H2, the adsorption of CO2 does not disrupt the ZnO lattice. This ZnO stability under pure CO2 exposure is consistent with the recent findings by Amann et al. who showed that under CO2-rich conditions, ZnO stability is energetically favored.39
Similar stability was observed for the ZnO film at 0.9 ML coverage, even in the presence of H2 (Fig. 5). Indeed, ZnO is still present at 550 K. This stability can be readily explained, first by the particular electronic structure of the oxygen-deficient Znδ+Ox occurring on this ultrathin layer of ZnO. Hence, O reaction sites are less available, making the formation of OH groups, a driving mechanism for the ZnO dewetting, less favorable. Second, another effect that promotes the thermal stability of ZnO film originates from the formation of the Zn–Cu interface alloy.
We have examined the possibility of forming an interface alloy using DFT computations. The Cu(111) surface was simulated by a slab composed of 5 layers of Cu with a cell of 8 Å × 8 Å × 28 Å to avoid interactions between periodically repeated images, where a Zn atom was introduced at different initial positions: in the middle of the Cu slab (replacing a Cu atom), at the surface of the Cu slab (replacing a Cu atom), or as an adatom over the slab. The oxygen atom was put over the Cu slab as an adatom. The different possible geometries were subsequently relaxed, and their total energies were compared. It was found that the Zn atom energetically prefers the third configuration (adatom position). Considering different positions of the oxygen atom on the surface, it was found that the situation where Zn and O form a bond (see Fig. 6a) or the situation where Zn and O were separated on the surface (see Fig. 6b) compete energetically, which confirms the possible formation of the Zn–Cu interface alloy in agreement with the Auger experiments (Fig. S1, ESI†). Therefore, the favorable formation of this interface alloy promotes the stability of the ultrathin ZnO film in direct coordination with the surface via Zn–Cu bonds.
 | ||
| Fig. 6 (a) and (b) two possible geometries of Zn and O adsorptions and coordination as obtained from DFT simulations (one ZnO). (c) Adsorption geometry and coordination of a ZnO dimer as obtained from DFT simulations. Atom colors: Cu (brown), Zn (purple), and O (red). | ||
A recent study by Zabilskiy et al.64 reported that in copper-zinc industrial catalysts supported on zeolite-faujasite, a Zn–Cu alloy was not observed and was not a prerequisite for these catalysts to be active for CO2 hydrogenation, despite high pressure of the reactants and temperature conditions used for this reaction. Thus, the preparation method of the ZnO/Cu catalysts must be considered for the possible formation of a Zn–Cu interface alloy, such as in the present system where an epitaxial growth process was conducted at UHV.
B. STM study of the morphology of the as-prepared ZnO/Cu(111) and under (CO2 + H2) hydrogenation reaction
We examined the structure of ZnO films grown on the as-prepared Cu(111) surface in the UHV and under in situ conditions and (CO2 + H2) reactive ambient atmosphere. Before the growth of ZnO, we used STM to examine the pristine Cu(111) surface. The STM images showed the presence of large terraces with an atomic step height of ∼1.9 Å (see Fig. S4a, ESI†). The high-resolution STM images confirmed the expected inter-atomic distance of dCu–Cu = 2.52 Å (Fig. S4b, ESI†). This study provides complementary information with respect to the above XPS results and hence will help better understand the morphology-reactivity relationship of the ZnO/Cu(111) model catalysts.The STM image in Fig. 7 shows a typical morphology of the ZnO film (∼0.9 ML) formed on the Cu(111) surface. It consists of a long-range ordering into hexagonal superlattices. Given the large periodic distance (∼10 Å) between the protrusions along the line profile (the dashed line, “i” in Fig. 7b), this structure suggests a Moiré-like pattern with a periodicity of ∼10 Å. Considering a lattice constant in the surface of the ZnO films, aZnOsurf = 3.3 Å that is strained by ∼2% with respect to the in-plane lattice parameter of ZnO(0001) bulk (aZnOsurf = 3.25 Å)65 and given the lattice constant of Cu(111) (aCu = 3.61 Å), the Moiré pattern can be readily attributed to a structure coincidence, where 3 unit cells of ZnO(0001) coincide with 4 unit cells of Cu(111). Indeed, the periodicity of 10 Å corresponds well to the following values: 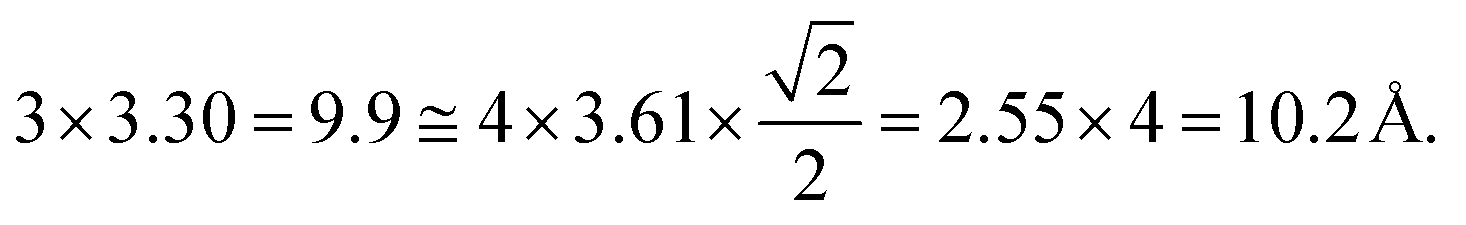 Therefore, the observed ZnO film here can be described as a ZnO-(3 × 3)/Cu(111)-(4 × 4) structure. While several values ranging from 4.8 to 13.5 Å have been reported for the Moiré pattern periodicities observed for ZnO films on Cu(111),26,29,66 our periodicity fits better with ∼10 Å value as reported previously by Zhao et al. for their ZnO structures on Cu(111).66 The variation in values reported in the literature may be attributed to different oxidation and annealing conditions. Various Moiré periodicities were also measured for ZnO grown on other surfaces, ∼17 for ZnO-(5 × 5)/Pt(111)-(6 × 6),35,67 16 Å for ZnO-(5 × 5)/Pd(111)-(6 × 6),68 ∼23 Å for ZnO-(7 × 7)/Au(111)-(8 × 8),66 and ZnO-(7 × 7)/Ag(111)-(8 × 8),65 and ∼15 Å for ZnO-(5 × 5)/Ag(111)-(3√3 × 3√3)R30°.65 These, however, may be readily explained by the lattice mismatch between ZnO and the different monocrystalline metallic surface supports. In these well-ordered ZnO structures (Fig. 7), Zn cations and O anions are coplanar and arranged in 2D planar sheets, similar to the hexagonal BN structure.
Therefore, the observed ZnO film here can be described as a ZnO-(3 × 3)/Cu(111)-(4 × 4) structure. While several values ranging from 4.8 to 13.5 Å have been reported for the Moiré pattern periodicities observed for ZnO films on Cu(111),26,29,66 our periodicity fits better with ∼10 Å value as reported previously by Zhao et al. for their ZnO structures on Cu(111).66 The variation in values reported in the literature may be attributed to different oxidation and annealing conditions. Various Moiré periodicities were also measured for ZnO grown on other surfaces, ∼17 for ZnO-(5 × 5)/Pt(111)-(6 × 6),35,67 16 Å for ZnO-(5 × 5)/Pd(111)-(6 × 6),68 ∼23 Å for ZnO-(7 × 7)/Au(111)-(8 × 8),66 and ZnO-(7 × 7)/Ag(111)-(8 × 8),65 and ∼15 Å for ZnO-(5 × 5)/Ag(111)-(3√3 × 3√3)R30°.65 These, however, may be readily explained by the lattice mismatch between ZnO and the different monocrystalline metallic surface supports. In these well-ordered ZnO structures (Fig. 7), Zn cations and O anions are coplanar and arranged in 2D planar sheets, similar to the hexagonal BN structure.
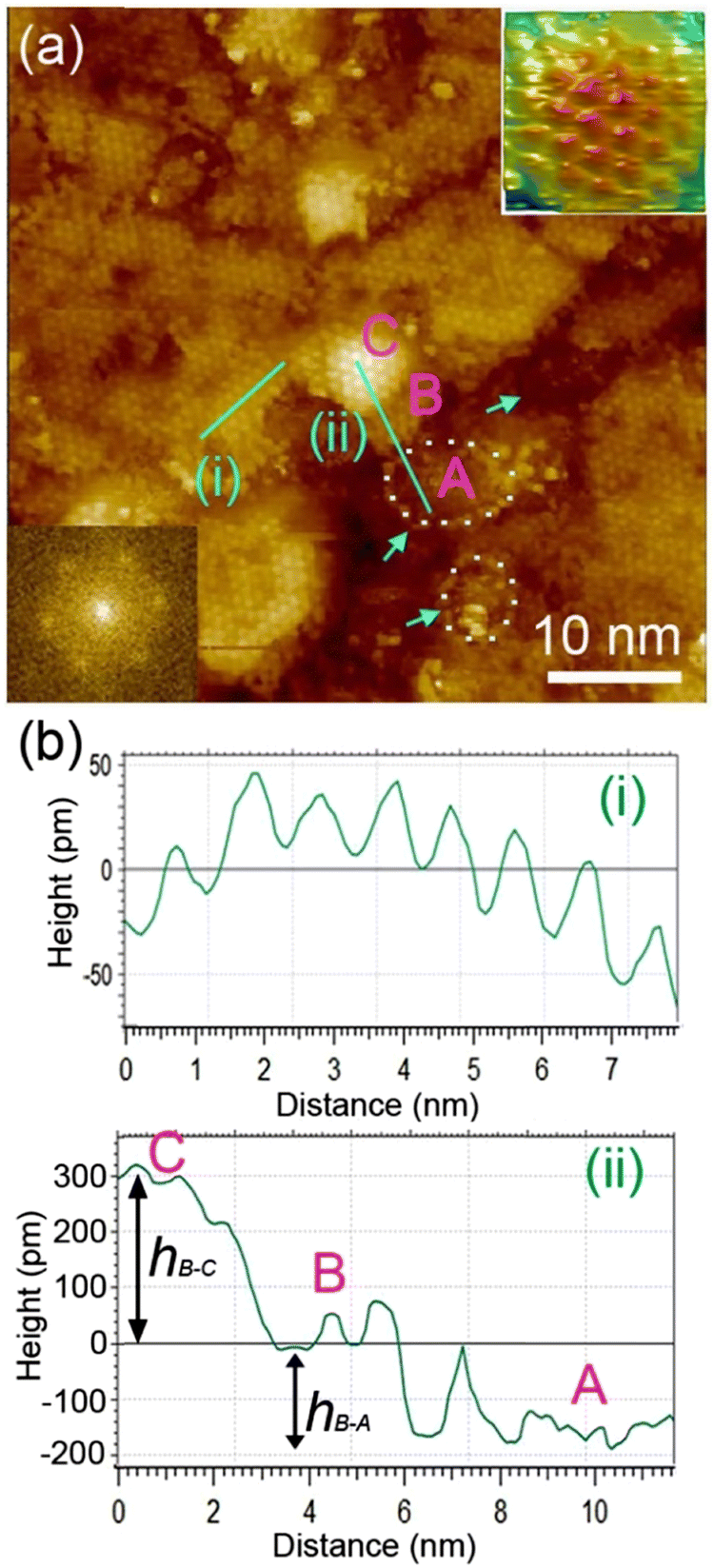 | ||
| Fig. 7 (a) STM image of the as-prepared ZnO ultrathin film on Cu(111) at 0.9 ML coverage. (b) Profiles of two areas as indicated. The profiles (i) and (ii) are acquired on top of the Moiré periodic protrusions and across the (A)-(B)-(C) areas, respectively. The dashed areas, also indicated by arrows, are uncovered surface areas (A). The top right insert image is a 3D display of the Moiré pattern noted C. The bottom left insert displays a Fast Fourier Transform (FFT) that confirms the hexagonal Moiré structure of the ZnO film at this coverage. | ||
While the surface is dominantly covered by the Moiré structure, exhibiting uniform flat domains noted B, some other higher protrusion domains that also display a Moiré pattern, noted C, can be seen in Fig. 7. In addition, featureless areas, noted A, are also present and are assigned to a bare Cu(111) surface. The difference between the apparent heights of B and C domains amounts to hB–C = ∼3 Å; it is larger than that of B and A (hB–A = ∼1.8 Å) and the interlayer height of Cu(111) (hCu = 2.09 Å) (see dashed line profile ‘’ii’’ in Fig. 7). Therefore, B and C domains correspond to the 1st and 2nd layer of the ZnO film, respectively. These values are in line with those reported by others on ZnO films regardless of the support used.29,66,68 It is noteworthy that the bilayer ZnO areas (C) exhibit an apparent height (∼3 Å) that is much lower than that of the wurtzite ZnO (c = 5.309 Å).69,70 Similarly, the 1st ZnO layer (B) exhibits an apparent height that is significantly lower than the value of c/2 in the wurtzite ZnO. These small values are indications of strong interactions between the thin ZnO film and Cu(111) surface. The electronic effect, associated for example with O-vacancies, may impact the tunneling transmission matrix, and hence, the apparent heights can be excluded in these conditions as these different values were confirmed by AFM measurements.65 However, the particular electronic structure of this thin ZnO film is in line with the above XPS discussion. These values are expected to relax towards those expected for the wurtzite ZnO when the film thickness is increased to >5 ML.
The step edges between areas B and A areas are decorated by disordered protrusions as indicated by arrows in Fig. 7 with an apparent height of 2.0 Å. These interface boundaries are reactive sites and, therefore, partially explain the differences seen for the (CO2, H2) interaction with the surface, between the thick and thin ZnO films discussed in the XPS part. In particular, the presence of bare Cu atoms alongside these interfaces is consistent with the formation of OCOH intermediates as discussed in for XPS analyses of the 0.9 ML.
With increasing ZnO film thickness, we observed a smoothening of the ZnO surface with the formation of equilateral triangular shape-like arrangements (Fig. 8). The evolution of the morphology corresponds to the transition into the wurtzite structure of ZnO. With an increase in the ZnO thickness, the size of the triangles increased from 20 nm up to 100 nm in side values. These structures are typical of a ZnO(0001)–Zn terminated surface and result from a charge compensation mechanism.35,69 As this electrostatically driven process occurs in a way that the surface carries the same charge as the bulk atoms, the step-edges of the triangles are two-fold coordinated O anions (Zn are three-fold coordinated inside the triangles). Therefore, the presence of these low-coordinated O anions can provide active sites for H stabilization, which may promote the formation of O–H groups. This reactivity is amplified by the electronic effect occurring in the ZnO ultrathin film. This reactivity is in line with the above XPS discussion regarding the instability of the ZnO thick film (6.8 ML) in the presence of H2. Indeed, considering the geometric model reported by Dulub et al.,69 we can consider the equation for the surface Zn/O atoms ratio to be:
| RZn/O = (n − 1)/(n + 1) | (10) |
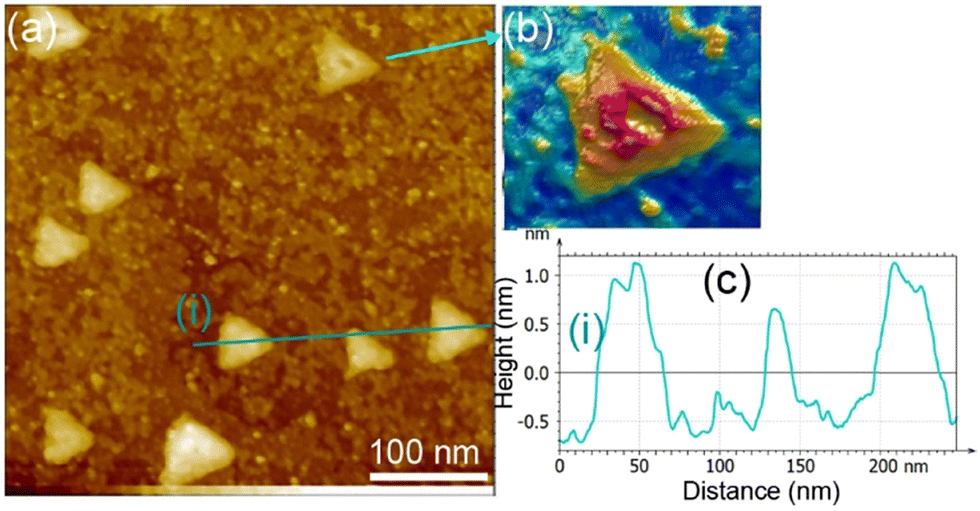 | ||
| Fig. 8 (a) STM image of the as-prepared ZnO ultrathin film on Cu(111) at 6.8 ML coverage. (b) Is a 3D display of a ZnO single equilateral triangle. (c) Gives a line profile including three separate triangles noted (i). | ||
We have investigated the effect of (CO2 + H2) exposure on the structural morphology of ZnO films. We conducted in situ STM investigations under 36 mbar of (CO2 + H2) mixture at room temperature on sub-monolayer ZnO domains on Cu(111) [Fig. 9]. First, before exposure, the surface exhibited isolated ZnO domains covering 25.4% of the surface (Fig. 9a). The uncovered surface areas exhibited well-ordered rows extending along the step edges (as indicated by dashed lines in Fig. 9a. See also Fig. S5a, ESI†). These features are characteristics of a copper surface oxide (Cu2O) having the “29”-structure adlayer (√13R46.1° × 7R21.8°).71–73 The formation of this structure resulted from the adsorption of O2 on Cu(111) and annealing at 423 K performed during the growth of ZnO films. Another copper oxide structure could also be present in the uncovered areas, that is, the “44-structure” which corresponds to (√13R46.1° × 7R21.8°) (Fig. S5b, ESI†). It is noteworthy that under the O2 and annealing conditions used to grow ZnO here, no areas corresponding to the clean Cu(111) were observed.
 | ||
| Fig. 9 STM images of sub-monolayer ZnO domains grown on Cu(111). (a) As-prepared in the UHV conditions. (b) In situ under (CO2 + O2) ambient atmosphere at 36 mbar at room temperature. The dashed lines in (a) indicate well-ordered rows of the “29” adlayer structure of the Cu2O as discussed in the text. | ||
The in situ scanning of the surface under 30 mbar of (CO2 + H2) at room temperature allowed us to monitor the morphology evolution of ZnO domains (Fig. 9b). Indeed, we observed drastic modifications in the morphology of ZnO domains and the bare Cu surface. This is displayed by the strong splintering of the initial ZnO domains in small islands scattered on the surface. This phenomenon can be beneficial for the CO2HR reaction because it generates a significant number of interfacial ZnO–Cu sites. In addition, while the uncovered areas were smooth before the exposure, they had become severely rough. This strong evolution is only due to the reactive gases (CO2 + H2) because no heating was provided yet. The observed behavior reflects the highly reactive character of the ZnO ultrathin films. This structural evolution is consistent with XPS analysis where intermediate species were detected readily upon exposure to CO2 and H2 at room temperatures.
Conclusions
We have investigated the effect of exposure of pure CO2 and H2 exposure and their mixture (CO2 + H2) on a ZnO ultrathin film supported on a Cu(111) surface as a function of annealing at room temperature up to 650 K. Herein, we examined two different thicknesses, thin (0.9 ML) and thick (6.8 ML) ZnO coverages on Cu(111) to establish a correlation between the ZnO thickness and the preferred CO2 hydrogenation reaction pathway. We have also determined the effect of the ZnO–Cu interface.Irrespective of the ZnO thickness, we found that both surfaces were active toward CO2HR. However, the major difference between the two surfaces was related to the nature of the chemical intermediates formed at the first stage of the reaction after exposure to (CO2 + H2) at room temperature. Indeed, while HCO3− were the first reaction intermediate species formed on the thick ZnO film (6.8 ML), only OCOH species were formed readily on the thin ZnO film (0.9 ML) at room temperature. Therefore, we show that the CO2 hydrogenation proceeds via two main paths depending on the film thickness. (i) It takes place through a slow path involving HCO3− → CO32− → HCOO− on a thick ZnO film (6.8 ML). (ii) A different and rapid path was observed for the thin ZnO film (0.9 ML), where the OCOH intermediates were formed readily at room temperature without passing through the HCOO intermediate. We propose that for this thin film (0.9 ML), it is the Zn–Cu interface that promotes the OCOH formation as it provides activated H atoms via Cu–Zn–O–H*. Therefore, the synergistic effect at the Zn–Cu interface is limited to the thin ZnO film. However, both paths exhibit common intermediates as they merge into H2CO which was eventually hydrogenated to CH3O. CO is also another common element, which is possibly a secondary intermediate as it decreased sharply above 400 K, possibly in favor of HCOO− in the case of the thick film.
Concerning the stability of the ZnO films, dewetting phenomena were seen above 550 K, especially in the presence of H2. The exposure to pure CO2 was found to limit this effect and have a stabilizing effect. The mechanism of driving dewetting was linked to OH desorption, a process that removes O atoms from the ZnO lattice. The relative stability was, however, seen for the thin ZnO film owing to the formation of a Zn–Cu alloy at the interface. From DFT computations, we found that this interface alloy was energetically favorable even in the presence of adsorbed O atoms on the Cu(111) surface. These were consistent with the Auger experiments that showed the presence of a metal-like Zn component.
Regarding the morphology of ZnO films, we identified two structures as a function of the thickness, using STM investigations. At low thickness (0.9 ML), the ZnO film, which does not cover the entire Cu surface, exhibits a long-range ordering into hexagonal superlattices corresponding to a Moiré structure with a periodicity of ∼10 Å. This ZnO configuration corresponds to a ZnO-(3 × 3)/Cu(111)-(4 × 4) structure. The uncovered Cu(111) areas exhibits well-ordered rows that are a characteristic of copper surface oxide (Cu2O) having the “29”-structure adlayer (√13R46.1° × 7R21.8°). At high coverage (6.8 ML), a smoothening of the ZnO surface together with the formation of equilateral triangular shape-like features were observed. These structures are typical of a ZnO(0001)–Zn surface and result from a charge compensation mechanism.
From in situ STM imaging of a sub-monolayer ZnO film, the exposure to 36 mbar of (CO2 + H2) mixture at room temperature induced a strong break-up of the initial ZnO domains into small islands scattered on the surface.
This study will thus contribute to reaching a precise understanding of the CO2 hydrogenation reaction mechanism and hence help to design better nanocatalysts by taking into consideration the important role of ZnO thickness and morphology.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We appreciate the valuable contributions from Léa Ghrenassia during her Master internship in our groups. Technical support from Régis Vacheresse (Design Bureau) is highly appreciated. Funding from the Doctoral School (ED 388) (S. H. doctoral thesis fellowship) of the Sorbonne Université and from the China Scholarship Council (CSC) (H. L. doctoral thesis and Z. L. Postdoctoral fellowships) are acknowledged.Notes and references
- M. Perez-Fortes, J. C. Schoneberger, A. Boulamanti and E. Tzimas, Appl. Energy, 2016, 161, 718–732 CrossRef CAS.
- X. Jiang, X. W. Nie, X. W. Guo, C. S. Song and J. G. G. Chen, Chem. Rev., 2020, 120, 7984–8034 CrossRef CAS PubMed.
- J. Zhong, X. Yang, Z. Wu, B. Liang, Y. Huang and T. Zhang, Chem. Soc. Rev., 2020, 49, 1385–1413 RSC.
- L. S. Guo, J. Sun, Q. J. Ge and N. Tsubaki, J. Mater. Chem. A, 2018, 6, 23244–23262 RSC.
- S. Navarro-Jaén, M. Virginie, J. Bonin, M. Robert, R. Wojcieszak and A. Y. Khodakov, Nat. Rev. Chem., 2021, 5, 564–579 CrossRef PubMed.
- N. Podrojkova, V. Sans, A. Orinak and R. Orinakova, ChemCatChem, 2020, 12, 1802–1825 CrossRef CAS.
- C. Hepburn, E. Adlen, J. Beddington, E. A. Carter, S. Fuss, N. Mac Dowell, J. C. Minx, P. Smith and C. K. Williams, Nature, 2019, 575, 87–97 CrossRef CAS PubMed.
- K. Z. Li and J. G. G. Chen, ACS Catal., 2019, 9, 7840–7861 CrossRef CAS.
- R.-P. Ye, J. Ding, W. Gong, M. D. Argyle, Q. Zhong, Y. Wang, C. K. Russell, Z. Xu, A. G. Russell, Q. Li, M. Fan and Y.-G. Yao, Nat. Commun., 2019, 10, 5698 CrossRef CAS PubMed.
- S. C. Mandal, A. Das, D. Roy, S. Das, A. S. Nair and B. Pathak, Coordin. Chem. Rev., 2022, 471, 214737 CrossRef CAS.
- P. Tian, Y. X. Wei, M. Ye and Z. M. Liu, ACS Catal., 2015, 5, 1922–1938 CrossRef CAS.
- T. Shoinkhorova, T. Cordero-Lanzac, A. Ramirez, S.-H. Chung, A. Dokania, J. Ruiz-Martinez and J. Gascon, ACS Catal., 2021, 11, 3602–3613 CrossRef CAS.
- J. Palomo, M. A. Rodriguez-Cano, J. Rodriguez-Mirasol and T. Cordero, Appl. Catal., B, 2020, 270, 118893 CrossRef CAS.
- B. Pathak, A. Das, S. C. Mandal and S. Das, J. Phys. Chem. C, 2022, 126, 21628–21637 CrossRef.
- S. S. Araya, V. Liso, X. T. Cui, N. Li, J. M. Zhu, S. L. Sahlin, S. H. Jensen, M. P. Nielsen and S. K. Kær, Energies, 2020, 13, 596 CrossRef.
- T. J. Deka, A. I. Osman, D. C. Baruah and D. W. Rooney, Environ. Chem. Lett., 2022, 20, 3525–3554 CrossRef CAS.
- J. Sehested, J. Catal., 2019, 371, 368–375 CrossRef CAS.
- L. Alvarez-Falcon, F. Vines, A. Notario-Estevez and F. Illas, Surf. Sci., 2016, 646, 221–229 CrossRef CAS.
- M. D. Higham, M. G. Quesne and C. R. A. Catlow, Dalton Trans., 2020, 49, 8478–8497 RSC.
- X. W. Nie, X. Jiang, H. Z. Wang, W. J. Luo, M. J. Janik, Y. G. Chen, X. W. Guo and C. S. Song, ACS Catal., 2018, 8, 4873–4892 CrossRef CAS.
- Y. H. Wang, S. Kattel, W. G. Gao, K. Z. Li, P. Liu, J. G. G. Chen and H. Wang, Nat. Commun., 2019, 10, 1166 CrossRef PubMed.
- L. C. Grabow and M. Mavrikakis, ACS Catal., 2011, 1, 365–384 CrossRef CAS.
- Y. Wang, W. Gao, K. Li, Y. Zheng, Z. Xie, W. Na, J. G. Chen and H. Wang, Chem, 2020, 6, 419–430 CAS.
- W. F. Yang, S. Chen, W. H. Ren, Y. Zhao, X. J. Chen, C. Jia, J. N. Liu and C. Zhao, J. Mater. Chem. A, 2019, 7, 15907–15912 RSC.
- Z. Nie, L. Yu, L. Jiang, M. Li, S. Ding, B. Xia, C. Cheng, J. Duan and S. Chen, Carbon Neutralization, 2023, 2, 458–466 CrossRef.
- R. Wang, H. W. Wang, X. F. Weng, J. X. Dai, Z. M. Gong, C. B. Zhao, J. L. Lu, Y. Cui and X. H. Bao, J. Energy Chem., 2021, 60, 150–155 CrossRef CAS.
- R. M. Palomino, P. J. Ramirez, Z. Y. Liu, R. Hamlyn, I. Waluyo, M. Mahapatra, I. Orozco, A. Hunt, J. P. Simonovis, S. D. Senanayake and J. A. Rodriguez, J. Phys. Chem. B, 2018, 122, 794–800 CrossRef CAS PubMed.
- T. Koitaya, S. Yamamoto, Y. Shiozawa, Y. Yoshikura, M. Hasegawa, J. Y. Tang, K. Takeuchi, K. Mukai, S. Yoshimoto, I. Matsuda and J. Yoshinobu, ACS Catal., 2019, 9, 4539–4550 CrossRef CAS.
- B. H. Liu, I. M. N. Groot, Q. S. Pan, S. Shailchutdinov and H. J. Freund, Appl. Catal., A, 2017, 548, 16–23 CrossRef CAS.
- G. Kresse and J. Furthmuller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- R. Blume, D. Rosenthal, J. P. Tessonnier, H. N. Li, A. Knop-Gericke and R. Schlogl, ChemCatChem, 2015, 2871–2881 CrossRef CAS.
- Y. Ma, J. Wang, K. R. Goodman, A. R. Head, X. Tong, D. Stacchiola and M. G. White, J. Phys. Chem. C, 2020, 124, 22158–22172 CrossRef CAS.
- H. Liu, A. Zakhtser, A. Naitabdi and F. Rochet, ACS Catal., 2019, 9, 10212–10225 CrossRef CAS.
- Y. J. Ren, K. D. Yuan, X. Zhou, H. C. Sun, K. Wu, S. L. Bernasek, W. Chen and G. Q. Xu, Chem. – Eur. J., 2018, 24, 16097–16103 CrossRef CAS PubMed.
- X. Y. Deng, A. Verdaguer, T. Herranz, C. Weis, H. Bluhm and M. Salmeron, Langmuir, 2008, 24, 9474–9478 CrossRef CAS PubMed.
- I. Orozco, E. W. Huang, M. Mahapatra, R. Shi, J. D. Kang, S. Nemsak, S. D. Senanayake, P. Liu and J. A. Rodriguez, J. Phys. Chem. C, 2021, 125, 558–571 CrossRef CAS.
- P. Amann, B. Klötzer, D. Degerman, N. Köpfle, T. Götsch, P. Lömker, C. Rameshan, K. Ploner, D. Bikaljevic, H. Y. Wang, M. Soldemo, M. Shipilin, C. M. Goodwin, J. Gladh, J. H. Stenlid, M. Börner, C. Schlueter and A. Nilsson, Science, 2022, 376, 603–608 CrossRef CAS PubMed.
- S. Kattel, P. J. Ramirez, J. G. Chen, J. A. Rodriguez and P. Liu, Science, 2017, 355, 1296–1299 CrossRef CAS PubMed.
- M. Borasio, O. R. de la Fuente, G. Rupprechter and H. J. Freund, J. Phys. Chem. B, 2005, 109, 17791–17794 CrossRef CAS PubMed.
- B. Eren, C. Heine, H. Bluhm, G. A. Somorjai and M. Salmeron, J. Am. Chem. Soc., 2015, 137, 11186–11190 CrossRef CAS PubMed.
- M. Favaro, H. Xiao, T. Cheng, W. A. Goddard, J. Yano and E. J. Crumlin, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 6706–6711 CrossRef CAS PubMed.
- M. Heenemann, M. M. Millet, F. Girgsdies, M. Eichelbaum, T. Risse, R. Schlogl, T. Jones and E. Frei, ACS Catal., 2020, 10, 5672–5680 CrossRef CAS.
- Q. L. Tang and Q. H. Luo, J. Phys. Chem. C, 2013, 117, 22954–22966 CrossRef CAS.
- C. Woll, Prog. Surf. Sci., 2007, 82, 55–120 CrossRef.
- T. Reichenbach, K. Mondal, M. Jager, T. Vent-Schmidt, D. Himmel, V. Dybbert, A. Bruix, I. Krossing, M. Walter and M. Moseler, J. Catal., 2018, 360, 168–174 CrossRef CAS.
- X. Y. Deng, D. C. Sorescu and J. Lee, Phys. Chem. Chem. Phys., 2017, 19, 5296–5303 RSC.
- R. Lindsay, E. Michelangeli, B. G. Daniels, T. V. Ashworth, A. J. Limb, G. Thornton, A. Gutierrez-Sosa, A. Baraldi, R. Larciprete and S. Lizzit, J. Am. Chem. Soc., 2002, 124, 7117–7122 CrossRef CAS PubMed.
- G. Dutta, A. A. Sokol, C. R. A. Catlow, T. W. Keal and P. Sherwood, Chem. Phys. Chem., 2012, 13, 3453–3456 CrossRef CAS PubMed.
- Y. Wang, R. Kovacik, B. Meyer, K. Kotsis, D. Stodt, V. Staemmler, H. Qiu, F. Traeger, D. Langenberg, M. Muhler and C. Woll, Angew. Chem., Int. Ed., 2007, 46, 5624–5627 CrossRef CAS PubMed.
- M. B. Fichtl, J. Schumann, I. Kasatkin, N. Jacobsen, M. Behrens, R. Schlogl, M. Muhler and O. Hinrichsen, Angew. Chem., Int. Ed., 2014, 53, 7043–7047 CrossRef CAS PubMed.
- T. Fujitani, I. Nakamura, T. Uchijima and J. Nakamura, Surf. Sci., 1997, 383, 285–298 CrossRef CAS.
- A. H. England, A. M. Duffin, C. P. Schwartz, J. S. Uejio, D. Prendergast and R. J. Saykally, Chem. Phys. Lett., 2011, 514, 187–195 CrossRef CAS.
- C. W. Yang, F. Bebensee, J. Chen, X. J. Yu, A. Nefedov and C. Woll, Chem. Phys. Chem., 2017, 18, 1874–1880 CrossRef CAS PubMed.
- S. C. Mandal and B. Pathak, Mater. Adv., 2020, 1, 2300–2309 RSC.
- A. V. Miller, V. V. Kaichev, I. P. Prosvirin and V. I. Bukhtiyarov, J. Phys. Chem. C, 2013, 117, 8189–8197 CrossRef CAS.
- I. Orozco, E. W. Huang, M. Mahapatra, J. D. Kang, R. Shi, S. Nemsak, X. Tong, S. D. Senanayake, P. Liu and J. A. Rodriguez, J. Phys. Chem. C, 2021, 125, 6673–6683 CrossRef CAS.
- K. Mudiyanselage, S. D. Senanayake, L. Feria, S. Kundu, A. E. Baber, J. Graciani, A. B. Vidal, S. Agnoli, J. Evans, R. Chang, S. Axnanda, Z. Liu, J. F. Sanz, P. Liu, J. A. Rodriguez and D. J. Stacchiola, Angew. Chem., Int. Ed., 2013, 52, 5101–5105 CrossRef CAS PubMed.
- W. Taifan, J. F. Boily and J. Baltrusaitis, Surf. Sci. Rep., 2016, 71, 595–671 CrossRef CAS.
- J. Baltrusaitis, J. H. Jensen and V. H. Grassian, J. Phys. Chem. B, 2006, 110, 12005–12016 CrossRef CAS PubMed.
- S. Furukawa, K. Ehara and T. Komatsu, Catal. Sci. Technol., 2016, 6, 1642–1650 RSC.
- E. M. Kock, M. Kogler, T. Bielz, B. Klotzer and S. Penner, J. Phys. Chem. C, 2013, 117, 17666–17673 CrossRef PubMed.
- M. Zabilskiy, V. L. Sushkevich, M. A. Newton and J. A. van Bokhoven, ACS Catal., 2020, 10, 14240–14244 CrossRef CAS.
- A. Shiotari, B. H. Liu, S. Jaekel, L. Grill, S. Shaikhutdinov, H. J. Freund, M. Wolf and T. Kumagai, J. Phys. Chem. C, 2014, 118, 27428–27435 CrossRef CAS.
- X. F. Zhao, H. Chen, H. Wu, R. Wang, Y. Cui, Q. Fu, F. Yang and X. H. Bao, Acta Phys.-Chim. Sin., 2018, 34, 1373–1380 CAS.
- Y. Martynova, B. H. Liu, M. E. McBriarty, I. M. N. Groot, M. J. Bedzyk, S. Shaikhutdinov and H. J. Freund, J. Catal., 2013, 301, 227–232 CrossRef CAS.
- G. Weirum, G. Barcaro, A. Fortunelli, F. Weber, R. Schennach, S. Surnev and F. P. Netzer, J. Phys. Chem. C, 2010, 114, 15432–15439 CrossRef CAS.
- O. Dulub, U. Diebold and G. Kresse, Phys. Rev. Lett., 2003, 90, 016102 CrossRef PubMed.
- O. Dulub, L. A. Boatner and U. Diebold, Surf. Sci., 2002, 519, 201–217 CrossRef CAS.
- T. Matsumoto, R. A. Bennett, P. Stone, T. Yamada, K. Domen and M. Bowker, Surf. Sci., 2001, 471, 225–245 CrossRef CAS.
- F. Jensen, F. Besenbacher, E. Laegsgaard and I. Stensgaard, Surf. Sci., 1991, 259, L774–L780 CrossRef CAS.
- A. J. Therrien, R. Q. Zhang, F. R. Lucci, M. D. Marcinkowski, A. Hensley, J. S. McEwen and E. C. H. Sykes, J. Phys. Chem. C, 2016, 120, 10879–10886 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ma00872j |
| This journal is © The Royal Society of Chemistry 2024 |