
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSoft-templated, mesoporous Co3O4 thin films for electrocatalysis of the oxygen evolution reaction†
Qingyang
Wu
a,
Maximilian
Mellin
 a,
Stefan
Lauterbach
b,
Chuanmu
Tian
a,
Christian
Dietz
c,
Jan P.
Hofmann
a and
Marcus
Einert
*a
a,
Stefan
Lauterbach
b,
Chuanmu
Tian
a,
Christian
Dietz
c,
Jan P.
Hofmann
a and
Marcus
Einert
*a
aSurface Science Laboratory, Department of Materials and Earth Sciences, Technical University of Darmstadt, Otto-Berndt-Strasse 3, 64287 Darmstadt, Germany. E-mail: meinert@surface.tu-darmstadt.de
bInstitute for Applied Geosciences, Geomaterial Science, Technical University of Darmstadt, Schnittspahnstrasse 9, 64287 Darmstadt, Germany
cInstitute of Material Science, Physics of Surfaces, Technical University of Darmstadt, Peter-Gruenberg-Strasse 2, 64287 Darmstadt, Germany
First published on 29th January 2024
Abstract
In order to decrease the overpotentials required for the electrocatalytic water splitting reaction, high-performance and abundant-element based-electrocatalysts have to be developed. Especially, high-surface-area materials are considered as promising candidates to drive the oxygen evolution reaction (OER) at low overpotentials in alkaline and acidic media owing to a large amount of available active surface sites. In this context, mesoporous spinel cobalt oxide (meso-Co3O4) thin films on conductive substrates were prepared for the first time by the dip-coating and soft-templating approach using cobalte nitrate as precursor and commercially available, low-cost Pluronic® F-127 as structure-directing agent. By making use of the evaporation-induced self-assembly (EISA) process, citric acid was applied as structural stabilizer reported yet to be only compatible with customized polymers for formation of mesoporous thin films. The usage of commercial Pluronic® F-127 was reported to result in breakdown of the mesoporous framework. However, this work shows that both Pluronic® F-127 and citric acid are compatible and can be utilized to produce uniform mesoporous networks after annealing. The meso-Co3O4 thin film was found to be crack-free on the nano- and macroscale, thus offering a highly accessible nanoarchitecture ensuring a large interface to any electrolyte allowing for efficient mass transport. The surface and bulk morphology, crystallographic structure, and surface composition of the mesostructured Co3O4 thin film were correlated to its OER activity. The Co3O4 nanostructure showed, when applied as OER catalyst, a low overpotential of 340 mV vs. RHE at 10 mA cm−2 in 1 M KOH. The electrochemical performance was evaluated by means of impedance spectroscopy and Tafel plot analysis confirming enhanced electron transfer at the electrode/electrolyte interface. The Co3O4 electrocatalyst exhibited a promising stability under alkaline conditions over more than 4 hours upon chronopotentiometry (OER-activity loss of only 2% at 10 mA cm−2). Our preparation procedure reveals the general capability of combining commercial copolymer templates with citric acid and can be transferred to other metal oxides to produce low-cost and high-surface-area materials for electrochemical applications.
Introduction
To slow down CO2 emission and substitute fossil fuels, sustainable energy sources need to be utilized.1,2 Electrocatalytic water splitting is a promising approach to produce hydrogen as renewable energy vector. In principle, in an electrochemical cell, the hydrogen evolution reaction (HER) occurs through reduction of protons at the cathode, and by oxidation of water, the oxygen evolution reaction (OER) is driven at the anode.3–5 However, considerable overpotentials (η) are necessary to drive both reactions, while it is known that the OER is the efficiency-limiting process of water splitting since it requires four electrons to be transferred for the production of one molecule of O2. In contrast, for the HER, only two electrons are involved.6 Therefore, the exploration of high-performance, cheap and durable electrocatalysts for the OER is essential to make electrocatalytic water splitting economically competitive to already existing hydrogen production technologies (such as the steam reforming process). Noble metal oxides such as iridium oxide (IrO2) and ruthenium oxide (RuO2) were thoroughly examined as OER catalysts in acidic and alkaline media,7 respectively, and have been most frequently utilized for the past decades.8–10 However, the high cost in production and low resources of ruthenium and iridium as elements are inevitably drawbacks for industrial up-scaling purposes. As a consequence, the search for non-noble (transition) metal-based electrocatalysts with comparable electrocatalytic performance, which are abundant, and thus low in cost, seems to be vital toward commercialisation. In this context, as early as 1987, it has been revealed that incorporating Fe into nickel oxide can significantly enhance the OER performance.11 The NiFe-based materials offered a comparable OER performance to those of noble metal oxides, meanwhile they are more cost-efficient and abundant as elements on earth. Thus, the alloy of nickel, iron, and chromium is still to date the most utilized catalyst for water electrolysis industrial-wise.12 However, the mechanism of their OER process still remains partially unclear and the NiFe-based nanomaterials still suffer from insufficient stability.13As an alternative, cobalt-based oxides have attracted attention as electrocatalysts since cobalt is also an abundant element on earth, its oxides show good stability in aqueous electrolytes, and they have been proven to lower the OER overpotential substantially comparable to other catalytically active transition metal oxides (TMOs) such as NiOx14,15 or Fe3O4.16 The electrocatalytically active reaction sites in spinel cobalt oxide (Co3O4), which are known to be mainly Co3+ centres, allow Co3O4 nanoparticles with spinel structure to possess an overpotential as low as 328 mV at a current density of 10 mA cm−2 in 1 M KOH.17 In principal, Co3O4 suffers, like TMOs in general, from poor intrinsic electronic conductivity.18 However, this drawback can be overcome by doping19 and/or nanostructuring strategies.20 It is described in literature that the (nano-) morphology of Co3O4 plays a significant role for the overall electrocatalytic performance since it affects the effective surface area and migration paths of the charge carriers.21 In a comparative study between mesoporous and compact/dense high entropy oxide (HEO) thin films applied as OER catalysts, it has been demonstrated that indeed nanostructured materials possess substantially lower charge transfer resistances and an increased number of catalytically active reaction sites resulting in reduced overpotentials.22 In 2018, Liu's group has synthesized Co3O4 structures in form of nanosheets, nanoparticles, and nanospheres and performed electrochemical measurements in 1 M KOH.23 The obtained overpotentials varied from 300 mV to 400 mV, which indicated that the nanomorphology significantly impacted the electrochemical performance mainly via changes in the contact area between the surface of the electrocatalyst and the electrolyte. In another study, the effect of crystal faceting of Co3O4 was investigated by exposing different crystal planes to the electrolyte and it was found that the ratio of Co3+ and Co2+ ions differs with varied crystal planes, which in turn significantly affected the activity of the OER.24 Furthermore, a recent work of Haase et al., who quantitatively investigated a size-dependent catalytic activity of CoOx(OH)y nanoparticles, uncovered a size-dependent oxidation activity leading to a near-surface Co–O bond contraction during OER.21 It has also been revealed that this sort of contraction as well as the accumulation of oxidatively induced charge carriers increases with the decrease in nanoparticle size. Consequently, nano- and mesostructuring can be considered as promising strategies to improve the electrocatalytic activity of cobalt oxides. Mesoporous networks provide additional catalytically active reaction centres due to increase of the internal surface area, which represents the contact area with the electrolyte. Additionally, mesoporous frameworks shorten the transfer paths of migrating charge carriers and endows the material with mechanical flexibility: all parameters allowing for the tailor and control of the material properties.25 Making use of the evaporation-induced self-assembly (EISA) process of metallic precursors and block-copolymers, dip-coating is an easy-to-handle and low-cost technique to create high-quality mesoporous thin films by soft-templating.26 Amphiphilic surfactants, such as the di-block copolymer Kraton LiquidTM- poly(ethylene-co-butylene)-block-poly (ethylene oxide) (KLE-)27 or the tri-block copolymer poly (ethylene oxide)106-poly (propylene oxide)70-poly (ethylene oxide)106 (PEO106-PPO70-PEO106, Pluronic® F-127) contribute to microphase separation by generation of micellular structures owing to their hydrophilic and hydrophobic character.26 Upon dip-coating, the solution, which contains the dissolved block-copolymer and metal precursors, can be readily coated on polar and non-polar substrates, and with the formation and self-assembly of micelles along with the hydrolysis and condensation reactions of the inorganic precursors, an amorphous and mesostructured hybrid network can be formed (Fig. 1). By subsequent thermal treatment of the as-deposited amorphous composite, the decomposition of the organic template and crystallisation of the inorganic nanoarchitecture are initiated (Fig. 1).28 In terms of preparation of mesoporous Co3O4 (further ascribed to meso-Co3O4) by soft-templating, structure directing agents (SDA) from the Pluronic® family have been utilized in a few studies. In Song's work, poly(ethylene glycol)-block-poly(propylene glycol)-block-poly(ethylene glycol) (PEO20–PPO70–PEO20, Pluronic® P-123) was used as surfactant to prepare Ni- and Mn-doped meso-Co3O4 with pore sizes ranging from 7.7 nm to 12.3 nm.29 It was also reported by Feng et al. that mesoporous Co3O4/Fe3O4 nanocomposite materials (pore diameter from 3.4 nm to 6.1 nm) can be synthesized through a ligand-assisted self-assembly method using Pluronic® P-123 in which the synthesized compound exhibited a relatively low overpotential of 322 mV at 10 mA cm−2 in 1 M KOH.30 Utilization of Pluronic® F-127 as SDA has also been reported to produce mesoporous metal oxides,31 however never in the context of direct fabrication of crack-free and structurally intact meso-Co3O4 thin film electrodes for application in OER catalysis. Eckhardt et al. developed a general synthesis concept to prepare various mesoporous metal oxides using citric acid as chelating agent.32 The presented method addresses the general drawback that inorganic precursors with good solubility in polar solvent often decompose (in the case of cobalt nitrate hexahydrate, Tm = 55 °C) during calcination before they can be stabilized e.g. by complexation. This typically leads to failure of production of both the desired metal oxide phase and nanomorphology. They reported that the precursor-citric acid complex was transformed into a metal carbonate phase by applying relatively low temperatures (in the case of cobalt carbonate, the range is between 170 °C and 250 °C) in air. Since carbonates are thermally more stable, they can be directly converted into the corresponding metal oxide structure at elevated temperatures.32,33 Such transition from carbonates to oxides has been proven to be beneficial to the formation of porous structures.34 This modified EISA method has mostly been adaptable for costume polymer materials (such as PIB-PEO or KLE) as they typically possess a tailored hydrophilic–hydrophobic character and higher thermal stability35 compared to commercial polymers, such as from the Pluronic family. Hence, the main challenges which have to be considered by the selection of the SDA in terms of forming mesoporous structures are the hydrophilicity of the formed complex and the volume change upon the conversion from the precursor to the oxide.36 As consequence, it was reported that utilization of common commercial templates (like Pluronic® F-127) did not result in mesoporous networks based on the prior formation of a metal nitrate–citric acid complex.32–34
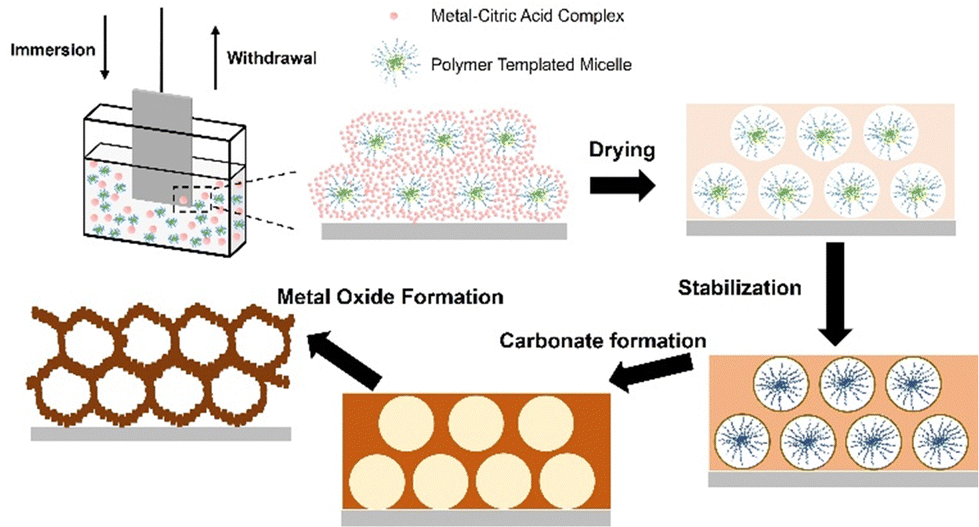 | ||
| Fig. 1 Schematic drawing of the consecutive steps during the applied dip-coating process: the drying process leads to an amorphous hybrid structure, followed by formation of a composite network, which is stabilized through condensation at 200 °C and further transformed into a nanocrystalline mesoporous framework after annealing at 300 °C (template removal and calcination). | ||
However, in this study, the compatibility of both Pluronic® F-127 and citric acid for the direct preparation of meso-Co3O4 thin films on conductive substrates by dip-coating is reported. The successful preparation of mesoporous Co3O4 networks with Pluronic® F-127 is highly indicated since the polymer is comparatively cheap and its usage results in the formation of small mesopores leading to high surface areas.
The underlying study aims to demonstrate a novel synthesis approach for the creation of meso-Co3O4 thin film electrodes readily available on conductive substrates by utilizing both Pluronic® F-127 and citric acid upon the EISA process. By correlating the electrochemical performance to the nanomorphology, crystallographic structure, elemental bulk and surface composition, and atomic configuration in/on meso-Co3O4, it was found that high surface areas, short charge carrier migration paths, phase-purity, and the presence of defective oxides and Co3+ species are synergistically responsible for the enhanced OER activity.
Experimental
Chemicals
All chemicals were of analytical grade and used as received: cobalt(II) nitrate hexahydrate (Co(NO3)2·6H2O, 99.999%, Sigma Aldrich), citric acid (≥99.5%, Carl Roth), Pluronic® F-127 (Mw = 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 600 g mol−1, Sigma Aldrich), ethanol (EtOH, ≥99.8%, Sigma Aldrich), potassium hydroxide (KOH, 1 mol L−1, Carl Roth).
600 g mol−1, Sigma Aldrich), ethanol (EtOH, ≥99.8%, Sigma Aldrich), potassium hydroxide (KOH, 1 mol L−1, Carl Roth).
Sample preparation
For preparation of the dip-coating solution, which was adapted from Eckhardt et al.,33 276 mg Co(NO3)2·6H2O, 91.3 mg citric acid (CA), and 45 mg Pluronic® F-127 were dissolved in a mixture of 1.2 mL EtOH and 1.1 mL of double distilled H2O. The solution was stirred using a magnetic stirrer for 30 min until a transparent and homogeneous solution was obtained. Before dip-coating, all substrates were initially cleaned with ethanol and acetone, and subsequently treated by an UV ozone cleaner (L2002A3-EU, Ossila) to remove organic surface contamination. Dip-coating was conducted with a dip-coater (L2006A1-EU, Ossila) equipped with a self-constructed chamber for control of the relative humidity at 15% RH for a homogenous thin film deposition. Fluorine-doped tin oxide (FTO) glass (from XOP glass) and silicon (100) wafers (from Silchem, n-type, resistivity <3 mΩ) were used as substrates applying a constant withdrawal speed between 2–8 mm s−1 upon dip-coating.After dip-coating, the samples were dried in the humidity-controlled chamber for 5 min, then transferred into a pre-heated muffle furnace (Nabertherm LT3/11) set to 125 °C, und subsequently heated to 200 °C with the rate of 10 °C min−1 and hold for 1 h, which was necessary for the formation of metal carbonates. A following heating step to 300 °C within 10 min was proceeded, and this temperature was kept for additional 30 min. The synthetic procedure of meso-Co3O4 is schematically illustrated in Fig. 1.
Structural characterization
Scanning electron microscopy (SEM) investigations were carried out on a Philips XL30 FEG at acceleration voltages of 30 keV and working distances of 3–4 cm. In transmission electron microscopy (TEM) measurements, post-treatment of samples was conducted by using a razor blade. Flakes of the thin film were scraped off the silicon substrate and collected in a small sample glass. After adding 2 mL of ethanol, they were dispersed with the help of an ultrasonic bath. The dispersion was allowed to settle for 20 s to separate large particles. Two to three droplets of the upper part of the dispersion were applied on a carbon coated copper grid (holey type, Plano GmbH, Wetzlar, Germany). After evaporation of the ethanol, the samples were coated with a thin carbon film (carbon coater MED 010, Bal-Tec AG, Balzers, Liechtenstein) to eliminate charging from the incident electron beam in the TEM. Examination of the samples were performed using the TEM mode in a FEG TEM (JEM2100F, JEOL Ltd, Tokyo, Japan). X-ray diffraction was conducted on a Rigaku Smartlab (Cu Kα radiation) diffractometer in a grazing incidence setup with θ source = 5°. Data were recorded between 10° and 60° (in units of 2θ) using a step size of 0.01°. We acquired topography images of the sample surface using an atomic force microscope (AFM) from Bruker (Santa Barbara, CA, Type: Icon Dimension) in the amplitude modulation mode in combination with a PPP-FMAuD cantilever (NanoAndMore GmbH, Wetzlar, Germany). In this mode, the sample surface is tracked by keeping the oscillation amplitude of the cantilever at a constant value (here: 63% of the free amplitude A0, A0 = 206 nm) varying the tip-sample distance. The cantilever had a nominal spring constant of 1.6 N m−1 as determined by the thermal noise method,37 a resonance frequency of 71.966 kHz with a guaranteed tip radius of <10 nm provided by the manufacturer. The scan speed of the tip was set to 2 μm s−1. The inverse optical lever sensitivity of the cantilever was calibrated using a sapphire sample. By pushing the tip towards the stiff surface, the oscillation amplitude detected by the photodiode (in the unit of volts) could be correlated with the motion (in the unit of nanometres) of the z-piezo and resulted in S = 137 nm V−1. Topography images were first-order flattened to remove sample tilt and to correct for thermal drift which is unavoidable during image acquisition in ambient conditions. The film thicknesses were determined with a Bruker profilometer (Dektak XT) using a scan rate of 10 μm s−1 and a stylus force of 3 mg. For each sample, the thickness was collected on three different positions on the sample surface, and the values were averaged assuming an error of ±10 nm for determination of the film thickness.Spectroscopic characterization
X-Ray photoelectron spectroscopy (XPS) experiments were accomplished in a vacuum-cluster tool on a ULVAC-PHI VersaProbe II instrument with a base pressure in the analysis chamber below 5 × 10−9 mbar. Monochromatized Al Kα radiation (1486.6 eV) was utilized as excitation source. Detailed spectra were measured applying a pass energy of 23.5 eV and a step size of 0.1 eV per step. Survey spectra were determined by applying a pass energy of 187.9 eV and the step size was set to by 0.8 eV. Binding energy calibration was performed by setting the Au 4f7/2 emission of Au foil to 84.0 eV. Backgrounds of the acquired spectra were subtracted by using the Shirley method in CasaXPS,38 version 2.3.25. Raman spectra were acquired with a LabRAM Horiba HR-800 Raman microscope with a 785 nm laser. Using a neutral density filter the power density was 25 mW at a spot size of ≈ 1.5 μm for a 50× LWD objective. As a 600 g mm−1 grating was chosen, the diameter of the pinhole was set to 200 μm and the entrance slit size to 100 μm. Spectra were accumulated 15 times for 10 s acquisition time per scan. For each sample equivalent spectra were collected on two different positions.Optical characterization
UV-visible light spectroscopy was accomplished by a UV/Vis/NIR spectrometer from Agilent (Cary 7000). The absorption spectra were determined by measuring the transmittance between 300 nm and 2500 nm using a scan speed of 600 nm min−1 and 6° angle of incidence. By conversion of the corresponding absorbance spectra into the Tauc plots, the direct and indirect optical band gaps were derived by extrapolation of the slope with the intercept of the x-axis and the baseline of the plot, respectively.Electrochemical measurements
All electrochemical experiments were conducted in a three-electrode setup (Zahner cell PECC-2) utilizing a Hg/HgO reference electrode and a platinum wire ring as counter electrode. The electrolyte was chosen to be 1 M KOH (pH ≈ 13.6). The potential was controlled by a GAMRY Interface 1000E potentiostat. Linear sweep voltammetry (LSV) was acquired between 0.9 V and 1.9 V versus RHE using a step size of 1 mV and a scan rate of 10 mV s−1. Cyclic voltammetry (CV) was conducted between 0.6 V and 1.5 V versus RHE with a scan rate of 100 mV s−1 and a step size of 1 mV. The potentials in the LSV curves were corrected by iR-drop through evaluation of the electrolyte resistance derived from electrochemical impedance spectroscopy (EIS) data. EIS was measured at 1.64 V versus RHE for applied frequencies between 100 kHz and 1 Hz and by using an AC modulation of 10 mV. The double-layer capacitance was derived after the method of McCrory39 by analysing the differential current densities at distinct scan rates of 20–200 mV s−1 in a non-faradaic potential range (found between 0.57 V and 0.67 V vs. RHE). Chronopotentiometry (CP) was carried out at constant current density of 10 mA cm−2 for 14000 s. The reference potential of the Hg/HgO electrode was measured against a reversible hydrogen electrode (RHE, HydroFlex, Gaskatel) in the above-mentioned electrolyte before the experiments and used as conversion value taking the Nernst equation into account.
Results and discussion
Structural properties
The surface structure and morphology were reported to have major impact on the electronic conductivity and OER activity, which was demonstrated, for example for mesoporous HEO thin films also prepared by dip-coating and soft-templating.22 In order to investigate these relevant surface structures and to reveal the impact of utilizing citric acid as complexing agent, scanning electron microscopy (SEM) were performed for Co3O4 samples treated at distinct calcination temperatures as well as in the presence (Fig. 2(A)) and absence of citric acid (Fig. 2(B)). As shown in Fig. 2(A), the surface morphology of the meso-Co3O4 thin films calcined at 300 °C for 30 min displays a homogenous and crack-free mesostructured framework with nanopores of 16 nm in average size (see ESI,† Fig. S1 B1–B2 for mesopore size determination). The pore size distribution in the underlying thin films was found to be slightly different compared to Pluronic® F-127 templated samples from literature, which is attributed to the balance of interaction between the phase separation effects,40,41 the stabilization effect of citric acid, and application of distinct annealing protocols.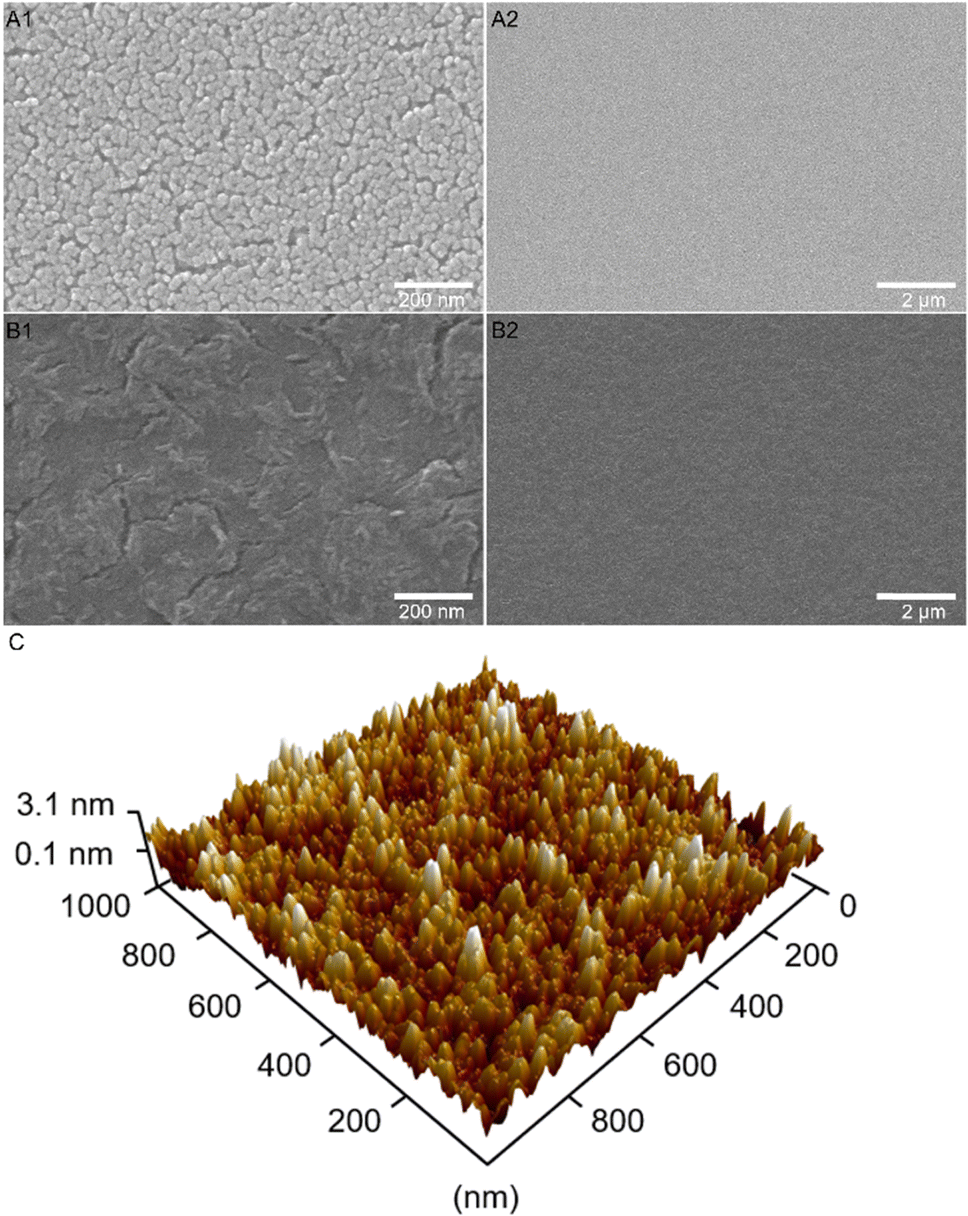 | ||
| Fig. 2 SEM images of Co3O4 thin films recorded at distinct magnifications prepared (A1) and (A2) with citric acid (mesoporous thin films) and (B1) and (B2) without citric acid (dense thin films). Both materials were pre-heated for 10 min at 125 °C, stabilized at 200 °C for 1 h, and calcined at 300 °C for 30 min. (C) Three-dimensional representation of the topography of meso-Co3O4 thin films (using citric acid) measured by atomic force microscopy in the amplitude modulation mode. | ||
For investigating the morphological evolution of the surface upon annealing, meso-Co3O4 samples were treated at 400 °C, 500 °C, and 600 °C and the corresponding top-view SEM pictures are presented in the ESI† (Fig. S2). Fig. S2 (ESI†) illustrates that the size of the pore wall domains and the average pore diameter increases with annealing temperature, which was assigned to the thermally induced growth of nanocrystals.42–44 After delicate optimisation of the intrinsic (e.g., solution and precursor concentration) and extrinsic (e.g., humidity, temperature, and withdrawal speed) parameters, homogeneous and reproducible surface morphologies were formed at each temperature, showing neither macro- nor mesoscopic cracks. In this study, meso-Co3O4 annealed at 300 °C for 30 min was chosen as control sample, since the dependency of the OER activity of nanostructured TMOs on their crystallinity and surface area (which is a function of calcination temperature for soft-templated metal oxides) have already been thoroughly investigated in literature.39–42 The temperature dependent OER activity of mesoporous oxides was, for example, studied for XCo2O4 (X = Mn, Ni, and Zn)45 and IrO246,47 revealing ideal calcination temperatures between 300 °C and 375 °C depending on the used surfactants. It is known that the OER activity benefits from high surface areas and nanocrystalline and/or partially amorphous structures due to the presence of unsaturated reaction sites, such as catalytically active oxygen vacancies.48,49
Fig. 2(B1)–(B3) demonstrates the top-view SEM images of Co3O4 annealed at 300 °C without usage of citric acid. It can be observed that no mesoporous framework has been formed in the absence of citric acid giving empirical evidence that this stabiliser is essential for the complexation of cobalt ions during the stabilisation and annealing process. Citric acid is vital for the formation of a mesoporous framework.
Interestingly, most mesoporous TMO thin films are reported to be prepared without chelating agents such as citric acid, as was shown, for example, for the synthesis of ordered mesoporous CoFe2O4,50 CuFe2O4,51 indium tin oxide,52 and high-entropy oxide.53 In conclusion, the feasibility of forming those mesoporous TMOs thin films strongly depends on the complexation behaviour of the corresponding TM. The formation of TMO mesostructures does not only depends on experimental conditions, but also on the metal-oxide sources, solvent composition, and coordinating agents.36
Atomic force microscopy (AFM) in the amplitude modulation mode was accomplished to visualize and analysis the three-dimensional surface topography of the meso-Co3O4 sample (Fig. 2(C)). It is exhibited that the sample possesses a highly nanoporous surface structure with peak elevations representing the pore walls. The sample exhibited a typical root-mean-square roughness of 0.82 nm measured within an area of 1000 × 1000 nm2 indicating a rather smooth and uniform surface morphology in spite of the fact that mesopores are present, thus further proving the homogeneity of the produced thin films. Additional topography and phase-mode images of the sample can be found in the ESI† (Fig. S3). The thickness of the meso-Co3O4 thin film was measured to be 240 ± 10 nm on silicon substrate by profilometry (Fig. S4, ESI†).
To further study the structural bulk features of meso-Co3O4 on the nanoscale, TEM was carried out as shown in Fig. 3 and Fig. S5 (ESI†). As indicated, the material consists of 3–4 nm sized crystals embedded in an amorphous matrix (Fig. 3(B)). The HR-TEM image (compare inverse fast Fourier transition IFFT images in Fig. 3(B)) displays the lattice fringes associated to (311) and (400) crystal planes. Also, the selected area electron diffraction (SAED) pattern can be indexed applying the lattice constant of the cubic Co3O4 spinel phase, which was determined to be a = 8.05 Å being in good accordance with literature.54 To further assess the crystallographic structure, gracing-incidence wide-angle X-ray diffraction (GIWAXS) analysis was conducted (Fig. 4(A)). Six characteristic diffraction peaks at 19.2°, 31.0°, 36.7°, 44.9°, 60.4°, and 65.4° were identified to correspond to the (111)-, (220)-, (311)-, (400)-, (511)-, and (440)- lattice plane, respectively, which were consistently assigned to the cubic Co3O4 spinel phase (JCPDS no. 42-1467). No diffraction peaks of the as-prepared samples were related to Co, CoO, CoOOH, and/or Co(OH)2 phases. The general low signal-to-noise ratio in the diffraction patterns has to be attributed to the low crystallinity of the thin films, which agrees with TEM observations (Fig. 3) and the fact that the films were treated at low calcination temperatures of 300 °C for a relative short time frame of 30 min. The average crystallite size of the Co3O4 sample was determined by applying the Scherer equation to the (311) peak and was evaluated to be 3.7 nm allowing the description as a quantum-sized nanocrystalline system (nanoconfinement effect). Raman spectroscopy, which is more sensitive to the formation of secondary phases, was carried out to further elucidate and verify the phase-purity, and thus, to exclude the presence of additional crystal phases. Co3O4 typically crystallizes in the spinel structure Co2+(Co3+)2O42− with Co2+ located at tetrahedral sites (A-site) and Co3+ at octahedral sites (B-site), respectively, considering a normal spinel configuration.55 According to Rousseau et al., the vibrational modes at k = 0 can be reduced from a 42-dimensional representation of Γ into:56
| Γ = A1g + Eg + 3F2g + 5F1u + 2A2u + 2Eu + 2F2u |
 | ||
| Fig. 3 (A) TEM and (B) HRTEM images of meso-Co3O4 thin films. Inset in (A) shows SAED patterns from the bulk area indexed in agreement with the cubic spinel phase. (B) IFFT images from the marked areas including the indicated atomic distances for (311) and (400) lattice planes. | ||
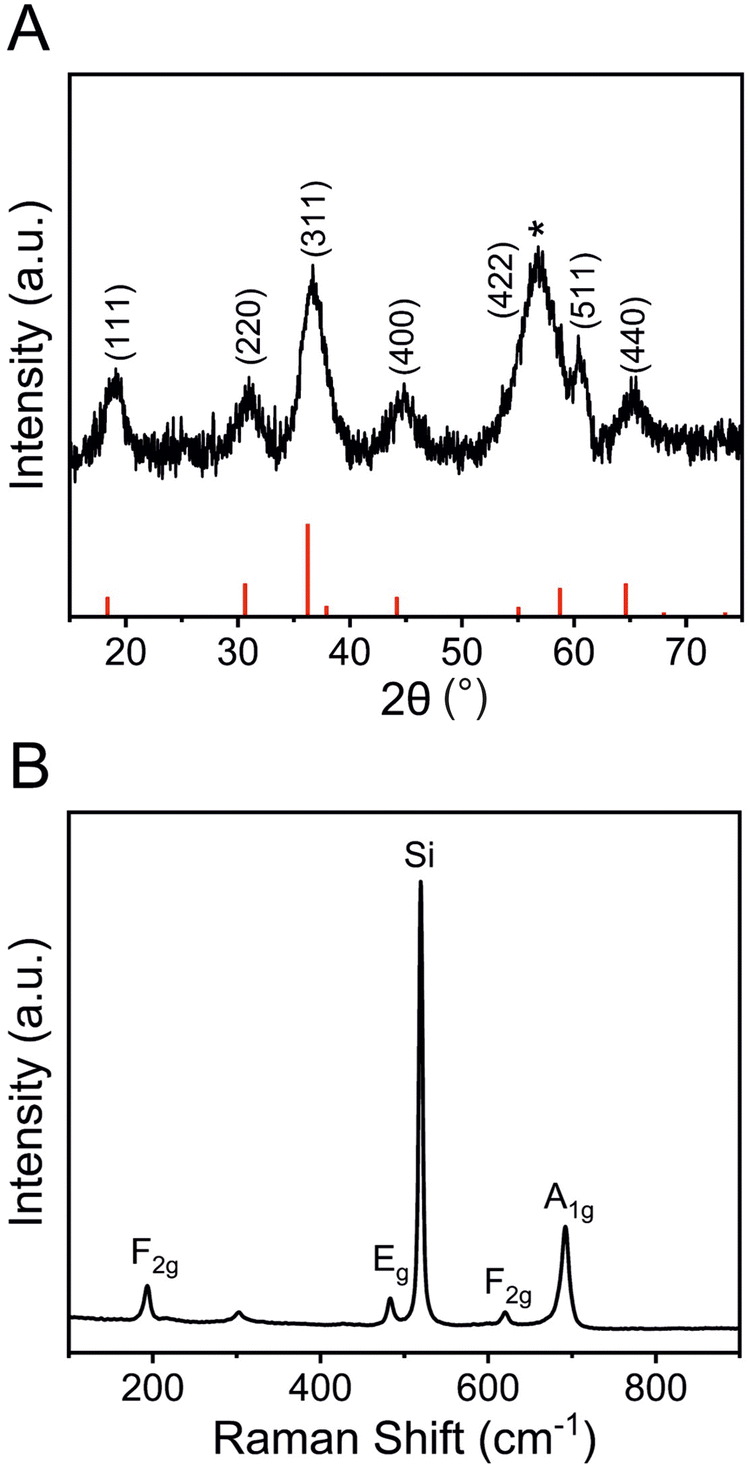 | ||
| Fig. 4 (A) GIWAXS pattern including reference bars (red lines) from JCPDS no. 42-1467 and the asterisk indicating the diffraction peak by the silicon substrate, and (B) Raman spectrum of the cubic spinel meso-Co3O4 thin film calcined at 300 °C for 30 min. | ||
Surface analysis
The chemical oxidation states and binding configurations of the elements on the surface of meso-Co3O4 were analysed through X-ray photoelectron spectroscopy (XPS). The pronounced peaks in the survey spectrum (Fig. S7A, ESI†) correspond to the photoemission of cobalt and oxygen orbitals,58 which can be found along with the carbon C 1s signal (Fig. S7B, ESI†) at 285 eV. For the detailed scan of the O 1s spectrum (Fig. 5(A)) recorded between 538 eV and 524 eV in terms of binding energy, the main peak at 529.8 eV can be attributed to lattice oxide (i.e., metal oxide), and the less intense peak at 531.0 eV presents hydroxide hydrated and/or potential defective oxide groups.59 It has been reported that the electrocatalytic activities of spinel metal oxides largely benefit from the presence of defective oxygen sites, e.g., in the form of oxygen vacancies, which are more likely to be present in nanocrystalline cobalt oxide samples due to the less organized nature on the molecular scale.60,61 It has also been reported that the peak located at 532.3 eV can be ascribed to the signals of organic ligands, such C–O, which most likely originate from the residual carbonized Pluronic® F-127.59 In the Co 2p spectra (Fig. 5(B)), two prominent peaks located at the binding energy of ∼795 eV and ∼780 eV are observed, which were attributed to the Co 2p1/2 and Co 2p3/2 spin-orbital splitting.62 The associated charge-transfer satellites at 805 eV and 790 eV indicate that cobalt ions are present in the oxidation state Co3+ and Co2+ occupying both tetrahedral and octahedral sites in the spinel configuration.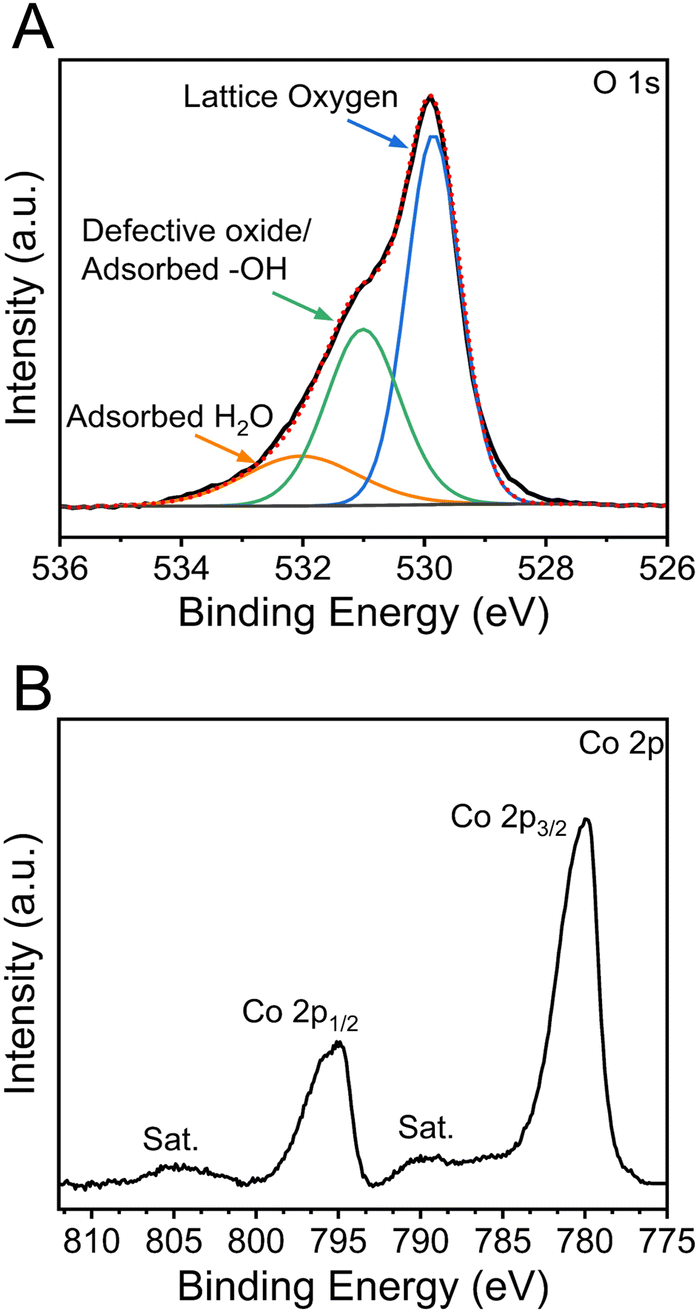 | ||
| Fig. 5 XPS spectra of the (A) O 1s spectrum (black) with the fitting curves (red dots) including fitting component of lattice oxygen (blue), oxygen defects or adsorbed hydroxyl groups (green), adsorbed water (orange). (B) Co 2p photoemission lines of meso-Co3O4 showing spin–orbital splitting and the presence of satellites. | ||
UV-vis spectroscopy was additionally conducted to analyse the optical band gap energy of the material and to determine the energy diagram. Therefore, the absorbance spectrum (Fig. S8A, ESI†) was converted into a Tauc plot (Fig. S8B, ESI†). By extrapolation of the recorded data to the x-axis, the optical band gap energy of meso-Co3O4 was determined to be 1.5 eV and 2.1 eV, when assuming direct optical transitions. It is well known that in the case of spinel Co3O4, the valence band has a strong oxygen 2p character, and the conduction band consists of Co(II) 3d orbitals, while a sub-band inside the band gap is formed due to the presence of Co(III) centres. As the Co(III) d states are closer to the p states of O (2−), the system can undergo an electron transition from p(O2−) to eg(Co3+).63 For the pure Co3O4 sample, the direct allowed optical transitions can be divided into two excitations from p(O2−) to eg(Co3+) and p(O2−) to eg(Co2+) levels, corresponding to optical band gap energies found at 1.5 eV and 2.1 eV (Fig. S8B, ESI†), respectively.64,65 However, according to the study of Qiao et al.,66 in the lower range of photon energies (between 0.5 eV and 1.0 eV), the fundamental band gap of Co3O4 was demonstrated to be located at 0.74 eV, as verified by using synergetic infrared optical and in situ photoemission spectroscopy combined with density functional theory (DFT) calculations. The authors ascribed the energy band gap of Co3O4 to a direct dipole-forbidden d–d transition between tetrahedral-site Co2+ cations.66 Such a related significant absorbance characteristic in a wavelength range between 1000 nm and 2500 nm has also been observed for the underlying meso-Co3O4 thin films (Fig. S8A, ESI†). The main absorbance feature starts at an energy of 0.9 eV (Fig. S8B, ESI†). These data are in accordance with the study by Lohaus et al., also reporting characteristic absorption maxima at 0.8 eV and 1.7 eV.58 Hence, the evaluated band gap energy of 0.9 eV was used to draw the energy diagram as shown in Fig. 6. Due to the nanoconfinement effect, a band gap widening is likely to being present for the mesostructured Co3O4 since the crystallite size was analysed to be 3–4 nm (by GIWAXS and TEM). For example, this phenomenon was also observed for quantum-sized TiO2 particles.67,68 Based on the evaluated direct optical band gap energies and the valence band maximum (VBM) along with the analysed work function (see Fig. S9, ESI†) derived from XPS experiments, the energy diagram of the meso-Co3O4 was constructed (Fig. 6). It can be stated according to the energy diagram that meso-Co3O4 is a p-type semiconductor as its Fermi level, which was determined by measuring the work function, is close to the VBM (0.4 eV distance), which agrees well with literature data.58
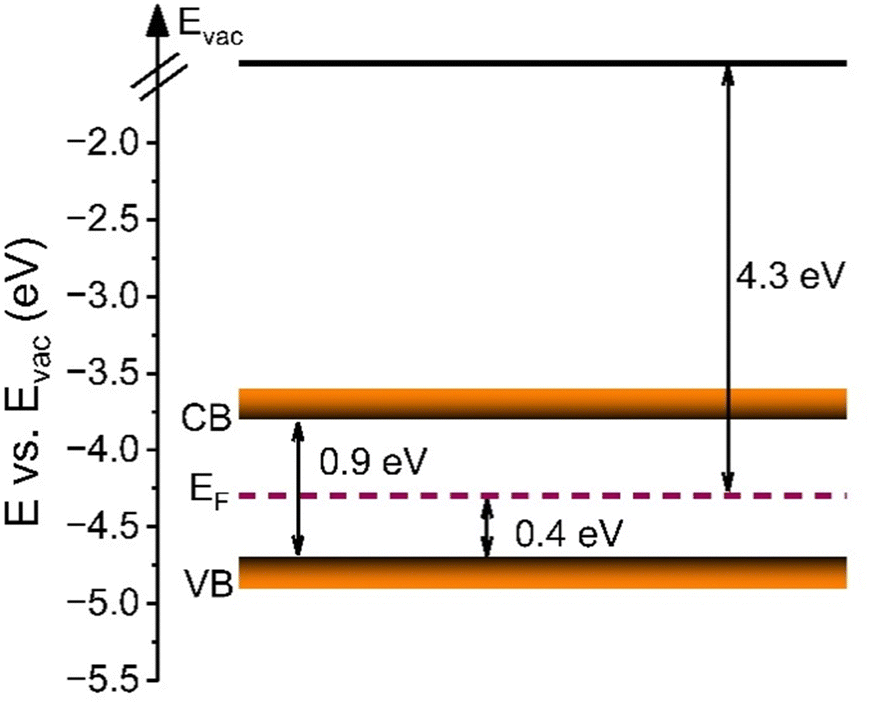 | ||
| Fig. 6 Band energy diagram representing the VB, CB, and EF alignment derived from determination of the valence band maximum by XPS analysis and UV-vis spectroscopy. The energy gap between CB and VB illustrates the direct optical band gap energy derived by UV-vis spectroscopy for the meso-Co3O4 thin film. | ||
Electrochemistry
To show the beneficial effect of mesostructuring on the electrochemical properties and the OER activity of sol–gel-derived Co3O4 thin films, mesoporous (with citric acid) and dense Co3O4 thin films (without citric acid, see Fig. 2) were analysed and compared by linear sweep voltammetry (LSV) in 1 M KOH in a potential range between 1.0 V and 1.7 V vs. RHE (Fig. 7(A)). For comparison, a nickel oxide (NiO) thin film known to form highly reactive NiOOH species under OER conditions in alkaline media,14 was characterized as reference material under very same conditions as illustrated in Fig. 7(A). An anodic peak present at 1.45 V reveals the oxidation of electrocatalytically active surface species, which was attributed to the oxidation of Co(III) to Co(IV) ions, where the Co(IV) in the form of CoO2 is suggested as the active catalytic species.69 Such behaviour has also been reported by Surendranath et al. who describes that the generation of Co(IV) species is a further oxidation channel besides the oxidation of Co(II) to Co(III).70 From the LSV curves, the onset potential of the current density was determined to be 1.50 V vs. RHE, which is a relatively low value compared with the data from literature usually reporting onsets between 1.52 and 1.56 V vs. RHE.71 The reason for that is most likely ascribed to the mesostructure of the Co3O4 sample facilitating the involved charge transfer at the interface, which will be discussed in more detail in the electrochemical impedance spectroscopy (EIS) section. The overpotential of the OER is usually determined at a geometric current density of 10 mA cm−2, thus making a general comparison of distinct OER electrocatalysts possible. The presented meso-Co3O4 thin film exhibits a voltage of 1.56 V vs. RHE identified at current density of 10 mA cm−2 and corresponding to an overpotential of 340 mV. This is a comparatively low value among cobalt-oxide-based catalysts from previous studies reporting overpotentials ranging from 310 mV to 400 mV (at the same current density), and even exceed the OER performance of cobalt containing, mesoporous HEO thin films.22,72 The dense Co3O4 thin films, produced in the absence of citric acid as chelating agent, exhibit an overpotential of 355 mV, which is higher compared to the mesoporous thin film, and exceed the performance of NiO (η10 = 450 mV). The high OER activity can be assigned to the large surface area arising from the ex- and internal mesoporous structure offering a plenty of reaction sites, most likely in the form of defective oxides and Co3+ ions (the presence which was proofed by XPS) preferably occupying octahedral sites in the spinel structure.73 Co3+ is known to be the active species in cobalt oxide-based spinel structures due to the increased charging, appropriate adsorption energetics of the reaction intermediates, and the preferred localisation of octahedrally coordinated units at surface sites of an electrocatalyst.74 As described earlier, the intrinsic properties (e.g., electronic conductivity) have a tremendous impact on the charge carrier transfer processes, and thus the overall OER performance. In order to interpret the kinetics for the oxygen evolution reaction and to compare the meso-Co3O4 with the performance of other electrocatalysts, the Tafel slope (Fig. 7(B)) was determined. Here, the slope was determined to be 57 mV dec−1, which is a rather low value compared to other cobalt oxide-based materials typically ranging between 60 mV dec−1 and 70 mV dec−1.72 The dense Co3O4 thin film electrocatalyst showed a value of (62 mV dec−1). The low Tafel slope values suggest that the rate-determining step for OER is an activation-controlled process involving a reversible one-electron transfer as reported in literature.75 Prospective work will focus on operando analysis of the meso-Co3O4 to analyse the catalytically active surface phase in detail.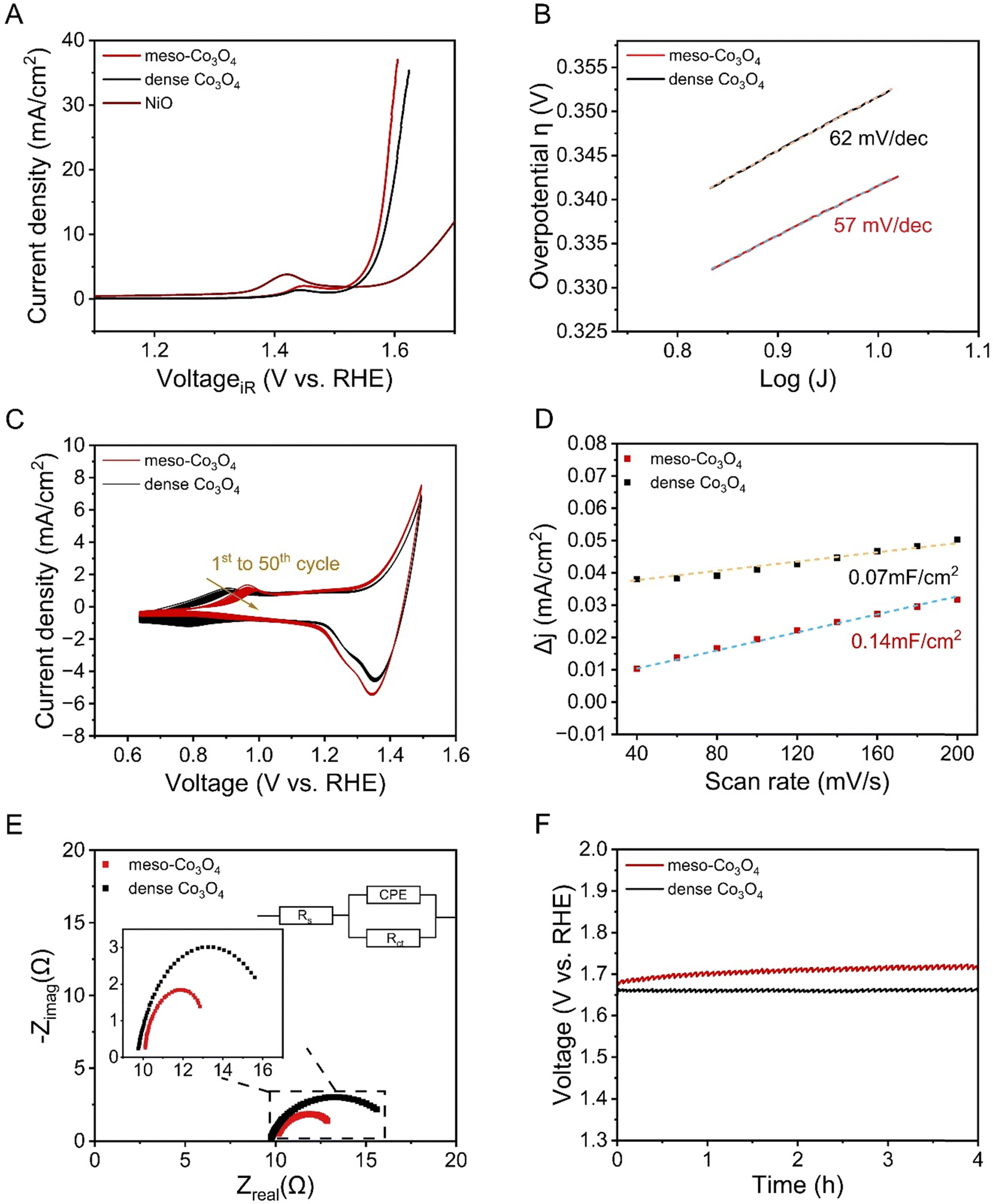 | ||
| Fig. 7 Electrochemical characterization of mesoporous and dense Co3O4 thin films including (A) linear sweep voltammetry curves collected at a scan rate of 10 mV s−1 and the performance of the NiO films taken from reference14 as comparison, (B) Tafel plot analysis, (C) 50 cyclic voltammetry scans measured from 0.6 to 1.5 V and recorded at 100 mV s−1, (D) differential current densities plotted versus the scan rate for the evaluation of the double layer capacitance, (E) the Nyquist plot determined by EIS at 1.6 V, and (F) chronopotentiometry analysis. | ||
Cyclic voltammetry (CV) was measured in 1 M KOH at a scan rate of 100 mV s−1 for 50 cycles. As indicated in Fig. 7(C), pronounced redox peaks were observed in the potential range between 0.6 V and 1.5 V vs. RHE. Note that the broad peak appearing in the range of 0.8 V to 1.1 V, represents the redox couple of Co(II) (most likely located at the tetrahedral sites) and Co(III). It is widely agreed that the surface of Co3O4 experiences a partial oxidation into a CoOOH phase,72,76 which is in accordance with the Tafel plot evaluation also suggesting a possible formation of oxyhydroxide species. As mentioned in the discussion for LSV, Co(III) species – preferentially located at octahedral sites – are catalytically active towards OH-oxidation during OER.70 The main oxidation peaks of mesoporous and dense Co3O4 were observed with a difference in the peak potential of 80 mV (Fig. 7(C)), which can be most likely ascribed to the change in surface morphology, resulting increased surface areas (see Fig. 2 and 7(D)), and thus, enhanced surface reactivity. Upon cyclisation, the oxidation peaks decrease substantially from the 1st to the 50th cycle (Fig. 7(C)) indicating the consumption and thus reduced amount of Co(II) active sites on the (internal) surface.72
The electrochemical active surface area (ECSA) is another important electrochemical parameter for the characterisation of electrocatalysts, as it provides information about the mesoporous architecture and amount of reaction sites at the surface.77 The ECSA is directly related to the ratio of the double-layer capacitance (CDL) and specific capacitance (Cs):
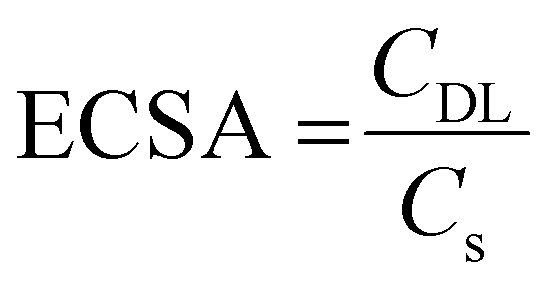
By measuring scan rate-dependent cyclic voltammetry curves (Fig. S10, ESI†), the difference in the anodic current density collected at varied scan rates were plotted in Fig. 7(D). From the slopes of the linear fittings, CDL can be directly derived and were evaluated to be 0.14 mF and 0.07 mF, which correspond to an ESCA of 3.5 cm2 and 1.75 cm2 (when applying 40 μF cm−2 for CS as suggested by McCrory et al.39 for 1 M KOH electrolytes)78 for mesoporous and dense Co3O4 samples, respectively. The comparison of the ECSA values suggests that the mesoporous structure provides an increased surface area, and thus, high amount of catalytically active reaction centres (compared to the dense thin films) contributing to the improved OER performance. Additionally, the mesoporous network still might be partially blocked by non-degraded, residual carbon from the synthesis (see Fig. S7, ESI†).
EIS is a powerful tool to investigate the electrical resistance in an electrochemical device. Therefore, EIS was carried out for the mesoporous and dense Co3O4 samples at a fixed potential of 1.6 V vs. RHE and within a frequency range of 100 kHz to 1 Hz. Fig. 7(E) presents the Nyquist plots of the mesoporous and dense Co3O4 thin film electrodes, and the upper inset shows the applied equivalent circuit including the solution resistance Rs (which was further used for the iR-correction of the current density applied in Fig. 7(A)), charge transfer resistance Rct, and constant phase element (CPE). From EIS analysis the electrolyte resistance, Rs, was determined as 10 Ω, while the resistance of the charge transfer, Rct, was calculated to be 3.5 Ω for mesoporous Co3O4. This value was found to be substantially lower compared to dense Co3O4 (7 Ω) and to most literature values,71 indicating a significantly facilitated charge carrier transfer process from the catalyst to the electrolyte through the mesoporous architecture. Mesostructuring is known to ease the charge transport owing to the presence and accessibility of short charge carrier conduction paths, which are with respect to the surface, distances in the dimension of the pore walls. Graberg et al. reported for mesoporous ITO thin films that the electric conductivity increases for smaller mesopore sizes owing to a higher conduction path cross section.52 Therefore, Pluronic® F-127 based mesoporous frameworks can be generally considered as a suitable nanoarchitecture for electrochemical applications, since this polymer is capable of creating mesopores below 15 nm (depending on the calcination temperature).51 Conclusively, the presented citric acid route in combination with Pluronic® F-127 as SDA and cobalt nitrate as precursor, results in the formation of structurally intact Co3O4 mesostructures with good electronic conductivity, high surface areas, and thus a remarkable OER performance.
Since the stability of an electrocatalyst is an important property with respect to long-term applications,79,80 chronopotentiometry at a constant applied current density of 10 mA cm−2 for 4 hours was performed in order to check the electrochemical stability of the material in alkaline solution. As shown in Fig. 7(F), a stable current response was obtained over four hours of CP analysis. Accordingly, the catalytic activity loss was determined to be only ∼2% with respect to the change in the measured electrode potential. For comparison, purely amorphous CoOx is highly unstable upon proceeding OER owing to dissolution of metal ions into the electrolyte.48 In the ESI† in Fig. S11, a SEM image of meso-Co3O4 after electrochemical performances is exhibited, which presents – despite of a few cracks on the macroscale – that the mesoporous morphology remains mainly structurally intact. However, the mesostructure seems to be less developed and densification has occurred most likely due to reconstruction of the surface upon electrochemical treatment, a phenomenon which was also observed for e.g. metal oxide nanofibers electrochemically treated in alkaline media.44 For comparison, a tabular overview (Table S1, ESI†) of the OER performances of various nanostructured Co3O4 OER-electrocatalysts from literature are presented. The comparison under identical testing conditions (electrolyte, substrate, scan rate etc.) indicates that the underlying meso-Co3O4 thin films are among the best-performing wet-chemically derived electrocatalysts with promising stability for driving the OER reaction in water splitting under alkaline conditions.
Conclusions
In the search for high-performance electrocatalysts possessing high surface areas aiming for high reactivity, the dip-coating and soft-templating method offers an easy-to-handle and scalable technology to produce uniform and mesoporous Co3O4 networks on conductive substrates. Investigations towards the bulk and surface morphology, crystallography, elemental composition, and energetic alignment of the band positions were carried out for the mesoporous Co3O4 thin films in detail by combining several structural X-ray-based characterization techniques. The mesoporous Co3O4 framework was presented to possess a nanocrystalline bulk structure composed of quantum-sized nanocrystals (3–4 nm) hosting small, disordered mesopores averagely 16 nm in size. The mesostructuring resulted in a rich amount of accessible, electrochemically active reaction sites being the reason for the comparatively low overpotentials of 340 mV at a geometric current density of 10 mA cm−2 and when analysed in alkaline solution. Long-term stability tests by chronopotentiometry at 10 mA cm−2 presented stable oxygen evolution for at least four hours. In essence, the presented sol–gel process proves the compatibility of Pluronic® F-127 and citric acid in alcoholic solutions and the successful transformation of the amorphous composite into a nanocrystalline, mesoporous Co3O4 network during calcination. The presented synthesis approach can be understood as conceptual strategy to synthesize earth-abundant element-based high-surface-area TMOs thin film electrodes by utilization of a commercially available, inexpensive block-copolymer serving as organic template. This preparation route might be utilized to fabricate novel mesoporous metal oxide-based electrode systems, which benefit from the presence of large surfaces especially in the steadily developing research field of (electrochemical) energy conversion and storage.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge funding by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation, Walter Benjamin Program to Marcus Einert) under project no. 469377211, and the support from China Scholarship Council (CSC) of no. 202208320036 (Qingyang Wu) and no. 202008420222 (Chuanmu Tian).References
- S. Chu and A. Majumdar, Nature, 2012, 488, 294–303 CrossRef PubMed.
- K. Khan, A. K. Tareen, M. Aslam, Y. Zhang, R. Wang, Z. Ouyang, Z. Gou and H. Zhang, Nanoscale, 2019, 11, 21622–21678 RSC.
- B. You and Y. Sun, Acc. Chem. Res., 2018, 51, 1571–1580 CrossRef.
- J. Hou, Y. Wu, B. Zhang, S. Cao, Z. Li and L. Sun, Adv. Funct. Mater., 2019, 29, 1808367 CrossRef.
- X. Peng, C. Pi, X. Zhang, S. Li, K. Huo and P. K. Chu, Sustainable Energy Fuels, 2019, 3, 366–381 RSC.
- K. Zhang and R. Zou, Small, 2021, 17, 2100129 CrossRef PubMed.
- S. Trasatti, J. Electroanal. Chem. Interfacial Electrochem., 1980, 111(1), 125–131 CrossRef.
- Yu. E. Roginskaya, T. V. Varlamova, M. D. Goldstein, I. D. Belova, B. Sh Galyamov, R. R. Shifrina, V. A. Shepelin and V. N. Fateev, Mater. Chem. Phys., 1991, 30, 101–113 CrossRef.
- Y. Lee, J. Suntivich, K. J. May, E. E. Perry and Y. Shao-Horn, J. Phys. Chem. Lett., 2012, 3, 399–404 CrossRef.
- T. Audichon, T. W. Napporn, C. Canaff, C. Morais, C. Comminges and K. B. Kokoh, J. Phys. Chem. C, 2016, 120, 2562–2573 CrossRef.
- D. A. Corrigan, J. Electrochem. Soc., 1987, 134, 377 CrossRef.
- M. Gong and H. Dai, Nano Res., 2015, 8, 23–39 CrossRef.
- J. Zhao, J.-J. Zhang, Z.-Y. Li and X.-H. Bu, Small, 2020, 16, 2003916 CrossRef.
- J. Gallenberger, H. M. Fernández, A. Alkemper, M. Li, C. Tian, B. Kaiser and J. Philipp Hofmann, Catal. Sci. Technol., 2023, 13, 4693–4700 RSC.
- H. Radinger, P. Connor, S. Tengeler, R. W. Stark, W. Jaegermann and B. Kaiser, Chem. Mater., 2021, 33, 8259–8266 CrossRef.
- R. Elakkiya and G. Maduraiveeran, Langmuir, 2020, 36, 4728–4736 CrossRef.
- A. J. Esswein, M. J. McMurdo, P. N. Ross, A. T. Bell and T. D. Tilley, J. Phys. Chem. C, 2009, 113, 15068–15072 CrossRef.
- J. Yin, P. Zhou, L. An, L. Huang, C. Shao, J. Wang, H. Liu and P. Xi, Nanoscale, 2016, 8, 1390–1400 RSC.
- T. Tran-Phu, M. Chatti, J. Leverett, T. K. A. Nguyen, D. Simondson, D. A. Hoogeveen, A. Kiy, T. Duong, B. Johannessen, J. Meilak, P. Kluth, R. Amal, A. N. Simonov, R. K. Hocking, R. Daiyan and A. Tricoli, Small, 2023, 19, 2208074 CrossRef.
- A. Miquelot, M. Despotopoulou, C. Vahlas, C. Villeneuve, N. Dragoe, N. Prud’homme and O. Debieu, Mater. Chem. Phys., 2020, 240, 122059 CrossRef.
- F. T. Haase, A. Bergmann, T. E. Jones, J. Timoshenko, A. Herzog, H. S. Jeon, C. Rettenmaier and B. R. Cuenya, Nat. Energy, 2022, 7, 765–773 CrossRef.
- M. Einert, M. Mellin, N. Bahadorani, C. Dietz, S. Lauterbach and J. P. Hofmann, ACS Appl. Energy Mater., 2022, 5, 717–730 CrossRef.
- S. Liu, R. Zhang, W. Lv, F. Kong and W. Wang, Int. J. Electrochem. Sci., 2018, 3843–3854 CrossRef.
- Q. Liu, Z. Chen, Z. Yan, Y. Wang, E. Wang, S. Wang, S. Wang and G. Sun, ChemElectroChem, 2018, 5, 1080–1086 CrossRef.
- E. Celik, Y. Ma, T. Brezesinski and M. T. Elm, Phys. Chem. Chem. Phys., 2021, 23, 10706–10735 RSC.
- B. Smarsly and M. Antonietti, Eur. J. Inorg. Chem., 2006, 1111–1119 CrossRef.
- A. Thomas, H. Schlaad, B. Smarsly and M. Antonietti, Langmuir, 2003, 19, 4455–4459 CrossRef.
- C. J. Brinker, Y. Lu, A. Sellinger and H. Fan, Adv. Mater., 1999, 11, 579–585 CrossRef.
- W. Song, Z. Ren, S.-Y. Chen, Y. Meng, S. Biswas, P. Nandi, H. A. Elsen, P.-X. Gao and S. L. Suib, ACS Appl. Mater. Interfaces, 2016, 8, 20802–20813 CrossRef.
- D. Feng, T.-N. Gao, M. Fan, A. Li, K. Li, T. Wang, Q. Huo and Z.-A. Qiao, NPG Asia Mater., 2018, 10, 800–809 CrossRef.
- M. S. Amer, M. A. Ghanem, P. Arunachalam, A. M. Al-Mayouf and T. A. Aljohani, Adv. Mater. Lett., 2019, 10, 136–144 CrossRef.
- B. Eckhardt, E. Ortel, J. Polte, D. Bernsmeier, O. Görke, P. Strasser and R. Kraehnert, Adv. Mater., 2012, 24, 3115–3119 CrossRef.
- B. Eckhardt, E. Ortel, D. Bernsmeier, J. Polte, P. Strasser, U. Vainio, F. Emmerling and R. Kraehnert, Chem. Mater., 2013, 25, 2749–2758 CrossRef.
- H. Du, L. Jiao, Q. Wang, Q. Huan, L. Guo, Y. Si, Y. Wang and H. Yuan, CrystEngComm, 2013, 15, 6101–6109 RSC.
- T. Brezesinski, B. Smarsly, K. Iimura, D. Grosso, C. Boissière, H. Amenitsch, M. Antonietti and C. Sanchez, Small, 2005, 1, 889–898 CrossRef PubMed.
- X. Xia and B. D. Vogt, Langmuir, 2020, 36, 1288–1297 CrossRef PubMed.
- H.-J. Butt and M. Jaschke, Nanotechnology, 1995, 6, 1–7 CrossRef.
- N. Fairley, V. Fernandez, M. Richard-Plouet, C. Guillot-Deudon, J. Walton, E. Smith, D. Flahaut, M. Greiner, M. Biesinger, S. Tougaard, D. Morgan and J. Baltrusaitis, Appl. Surf. Sci. Adv., 2021, 5, 100112 CrossRef.
- C. C. L. McCrory, S. Jung, J. C. Peters and T. F. Jaramillo, J. Am. Chem. Soc., 2013, 135, 16977–16987 CrossRef CAS PubMed.
- H. K. Christenson, J. Fang and J. N. Israelachvili, Phys. Rev. B: Condens. Matter Mater. Phys., 1989, 39, 11750–11754 CrossRef.
- T. Huang, W. Huang, C. Zhou, Y. Situ and H. Huang, Surf. Coat. Technol., 2012, 213, 126–132 CrossRef CAS.
- W. Xing, F. Li, Z. Yan and G. Q. Lu, J. Power Sources, 2004, 134, 324–330 CrossRef.
- M. Zheng, J. Cao, S. Liao, J. Liu, H. Chen, Y. Zhao, W. Dai, G. Ji, J. Cao and J. Tao, J. Phys. Chem. C, 2009, 113, 3887–3894 CrossRef.
- M. Einert, T. Weller, T. Leichtweiß, B. M. Smarsly and R. Marschall, ChemPhotoChem, 2017, 1, 326–340 CrossRef.
- A. Amirzhanova, N. Akmanşen, I. Karakaya and Ö. Dag, ACS Appl. Energy Mater., 2021, 4, 2769–2785 CrossRef.
- E. Ortel, T. Reier, P. Strasser and R. Kraehnert, Chem. Mater., 2011, 23, 3201–3209 CrossRef.
- M. Bernicke, E. Ortel, T. Reier, A. Bergmann, J. Ferreira de Araujo, P. Strasser and R. Kraehnert, ChemSusChem, 2015, 8, 1908–1915 CrossRef PubMed.
- S. Anantharaj and S. Noda, Small, 2020, 16, 1905779 CrossRef CAS.
- W. Liu, H. Liu, L. Dang, H. Zhang, X. Wu, B. Yang, Z. Li, X. Zhang, L. Lei and S. Jin, Adv. Funct. Mater., 2017, 27, 1603904 CrossRef.
- T. E. Quickel, V. H. Le, T. Brezesinski and S. H. Tolbert, Nano Lett., 2010, 10, 2982–2988 CrossRef CAS PubMed.
- M. Einert, A. Waheed, D. C. Moritz, S. Lauterbach, A. Kundmann, S. Daemi, H. Schlaad, F. E. Osterloh and J. P. Hofmann, Chem. – Eur. J., 2023, 29, e202300277 CrossRef.
- T. von Graberg, P. Hartmann, A. Rein, S. Gross, B. Seelandt, C. Röger, R. Zieba, A. Traut, M. Wark, J. Janek and B. M. Smarsly, Sci. Technol. Adv. Mater., 2011, 12, 025005 CrossRef.
- M. Einert, A. Waheed, S. Lauterbach, M. Mellin, M. Rohnke, L. Q. Wagner, J. Gallenberger, C. Tian, B. M. Smarsly, W. Jaegermann, F. Hess, H. Schlaad and J. P. Hofmann, Small, 2023, 19, 2205412 CrossRef.
- J. van Elp, J. L. Wieland, H. Eskes, P. Kuiper, G. A. Sawatzky, F. M. F. de Groot and T. S. Turner, Phys. Rev. B: Condens. Matter Mater. Phys., 1991, 44, 6090–6103 CrossRef.
- W. L. Roth, J. Phys. Chem. Solids, 1964, 25, 1–10 CrossRef.
- D. L. Rousseau, R. P. Bauman and S. P. S. Porto, J. Raman Spectrosc., 1981, 10, 253–290 CrossRef.
- V. G. Hadjiev, M. N. Iliev and I. V. Vergilov, J. Phys. C Solid State Phys., 1988, 21, L199–L201 CrossRef.
- C. Lohaus, J. Morasch, J. Brötz, A. Klein and W. Jaegermann, J. Phys. Appl. Phys., 2016, 49, 155306 CrossRef.
- Z. Xiao, Y.-C. Huang, C.-L. Dong, C. Xie, Z. Liu, S. Du, W. Chen, D. Yan, L. Tao, Z. Shu, G. Zhang, H. Duan, Y. Wang, Y. Zou, R. Chen and S. Wang, J. Am. Chem. Soc., 2020, 142, 12087–12095 CrossRef PubMed.
- J. Bao, X. Zhang, B. Fan, J. Zhang, M. Zhou, W. Yang, X. Hu, H. Wang, B. Pan and Y. Xie, Angew. Chem., 2015, 127, 7507–7512 CrossRef.
- X. Yan, L. Tian, M. He and X. Chen, Nano Lett., 2015, 15, 6015–6021 CrossRef PubMed.
- M. C. Biesinger, B. P. Payne, A. P. Grosvenor, L. W. M. Lau, A. R. Gerson and R. St. C. Smart, Appl. Surf. Sci., 2011, 257, 2717–2730 CrossRef.
- S. A. Makhlouf, Z. H. Bakr, K. I. Aly and M. S. Moustafa, Superlattices Microstruct., 2013, 64, 107–117 CrossRef.
- C.-S. Cheng, M. Serizawa, H. Sakata and T. Hirayama, Mater. Chem. Phys., 1998, 53, 225–230 CrossRef CAS.
- D. Barreca, C. Massignan, S. Daolio, M. Fabrizio, C. Piccirillo, L. Armelao and E. Tondello, Chem. Mater., 2001, 13, 588–593 CrossRef CAS.
- L. Qiao, H. Y. Xiao, H. M. Meyer, J. N. Sun, C. M. Rouleau, A. A. Puretzky, D. B. Geohegan, I. N. Ivanov, M. Yoon, W. J. Weber and M. D. Biegalski, J. Mater. Chem. C, 2013, 1, 4628–4633 RSC.
- M. Einert, P. Hartmann, B. Smarsly and T. Brezesinski, Sci. Rep., 2021, 11, 17687 CrossRef CAS PubMed.
- M. Schröder, S. Sallard, M. Böhm, M. Einert, C. Suchomski, B. M. Smarsly, S. Mutisya and M. F. Bertino, Small, 2014, 10, 1566–1574 CrossRef.
- M. Hamdani, R. N. Singh and P. Chartier, Int. J. Electrochem. Sci., 2010, 5, 556–577 CrossRef CAS.
- Y. Surendranath, M. W. Kanan and D. G. Nocera, J. Am. Chem. Soc., 2010, 132, 16501–16509 CrossRef CAS.
- B. Sidhureddy, J. S. Dondapati and A. Chen, Chem. Commun., 2019, 55, 3626–3629 RSC.
- X. Deng and H. Tüysüz, ACS Catal., 2014, 4, 3701–3714 CrossRef CAS.
- Y. J. Sa, K. Kwon, J. Y. Cheon, F. Kleitz and S. H. Joo, J. Mater. Chem. A, 2013, 1, 9992 RSC.
- Y. Zhou, S. Sun, C. Wei, Y. Sun, P. Xi, Z. Feng and Z. J. Xu, Adv. Mater., 2019, 31, 1902509 CrossRef CAS PubMed.
- R.-N. Singh, M. Hamdani, J.-F. Koenig, G. Poillerat, J. L. Gautier and P. Chartier, J. Appl. Electrochem., 1990, 20, 442–446 CrossRef CAS.
- N. Weidler, S. Paulus, J. Schuch, J. Klett, S. Hoch, P. Stenner, A. Maljusch, J. Brötz, C. Wittich, B. Kaiser and W. Jaegermann, Phys. Chem. Chem. Phys., 2016, 18, 10708–10718 RSC.
- F. P. Lohmann-Richters, B. Abel and Á. Varga, J. Mater. Chem. A, 2018, 6, 2700–2707 RSC.
- P. Connor, J. Schuch, B. Kaiser and W. Jaegermann, Z. Phys. Chem., 2020, 234, 979–994 CrossRef.
- L. Zhang, J. Zhu, X. Li, S. Mu, F. Verpoort, J. Xue, Z. Kou and J. Wang, J. Interdiscip. Math., 2022, 1, 51–87 Search PubMed.
- R. Paul, Q. Zhai, A. K. Roy and L. Dai, J. Interdiscip. Math., 2022, 1, 28–50 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ma01054f |
| This journal is © The Royal Society of Chemistry 2024 |