
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Moisture-cured solvent free silylated poly(ether-urea) pressure-sensitive adhesives (PSAs) for use as skin adhesives for application in transdermal drug delivery (TDD)†
Spyridon
Efstathiou
 a,
Gabit
Nurumbetov
a,
Andrew
Ross
a,
Yongguang
Li
a and
David M.
Haddleton
*ab
a,
Gabit
Nurumbetov
a,
Andrew
Ross
a,
Yongguang
Li
a and
David M.
Haddleton
*ab
aMedherant Ltd., The Venture Centre University of Warwick Science Park, Coventry, CV4 7EZ, UK. E-mail: d.m.haddleton@warwick.ac.uk
bDepartment of Chemistry, University of Warwick, Gibbet Hill Road, Coventry, CV4 7AL, UK
First published on 5th March 2024
Abstract
Improving the adhesion and tack of pressure-sensitive adhesives (PSAs) remains an ongoing challenge. Polyureas often have stronger hydrogen bonding relative to, the more commonly used, polyurethanes which are quite common in adhesive applications. An increased adhesion can reduce the requirements for additives such as tackifier resins and fillers to achieve high levels of adhesion required in certain applications. This approach not only necessitates tedious optimisations but also introduces compatibility issues especially in drug-in-adhesive formulation development. In this work, novel silylated polyurea moisture-curable PSAs are introduced with moisture curing of silyl terminated polyether–urea prepolymers. Prepolymer variants were synthesised using a solvent and catalyst-free step-growth polymerisation by reacting the commercially available polyetheramine Jeffamine® D-4000 with isophorone diisocyanate (IPDI) to yield diprimaryamino-terminated polymers. The polymers’ average molecular weight was controlled by adjusting the –NCO/–NH2 molar ratio. Subsequently, the amino terminal ends were post-functionalised with 3-isocyanatopropyltrimethoxysilane (IPTMS) to attain the silylated moisture-curable prepolymer variant crosslinkers (PUXL). The variants were able to cure to form (–Si–O–Si–) crosslinked PSA gels in the presence of a titanium catalyst and moisture. These materials were characterised by a range of techniques including rheology and thermal characterisation. Although their thermal properties remained unaltered, their adhesion and tack increased by increasing the –NCO/–NH2 molar ratio exceeding the borders of the Chang's classification windows for removable PSAs. This was achieved without the requirement for any additional tackifyer resins. Peel test and rolling ball tack tests agreed with variants demonstrating superior adhesion compared to commercially available transdermal adhesive products. Finally, no cold flow effects were noticed a result of the crosslinked network/gel.
Introduction
Adhesives are used to join materials together either permanently or temporarily by forming bonds with a balance between adhesion and cohesion forces.1 These forces are critical on determining the quality of the adhesive bond without compromising its internal structure. Among the many classes of adhesives, pressure-sensitive adhesives (PSAs) can quickly adhere to a wide variety of surfaces following the application of light pressure for a short time and preferably leaving no residues upon removal which should be easy to achieve.2 The term PSA arises from their unique characteristics to exhibit high tackiness at ambient temperature forming adhesive bonds with substrates through processes that involve wetting for good contact and physical interactions such as van der Waals forces. PSA materials are used in many applications including labels,3 bandages,4 and transdermal drug delivery devices.5Most commercial PSAs are polymers which fall in the category of natural and synthetic rubbers, acrylics, polyurethanes and silicones.6,7 Among these, rubber-based PSAs are used extensively but generally lack the appropriate tack and adhesion which can only be achieved with tackifier resins and fillers. Their poor resistance towards chemical and environmental factors is characteristic.8 Acrylic PSAs are a widely used class of PSAs found in commercial tapes and labels.9,10 They are typically composed of acrylic statistical copolymers combining monomers with low and high glass transition temperatures (Tgs) to impart viscoelastic characteristics. They exhibit average to high adhesion which can be further adjusted with tackifier resins. Despite their high resistance to ultraviolet (UV) radiation, chemical and environmental factors, residual monomers introduce toxicity limiting certain medical applications such as in transdermal patches where skin irritation is critical and being thermoplastics suffer from cold flow.11 On the contrary, silicon PSAs have found application in electronic and transdermal medical devices due to good biocompatibility, flexibility and high-temperature stability.12,13 Their low wetting and tack capabilities are their characteristic with a high cost of production.14 Finally, polyurethane adhesives are considered versatile polymers in manufacturing due to their lower cost as compared to silicone PSAs. Polyurethanes are products of the reaction of diols/polyols (–OH) with diisocyanates/polyisocyanates (–NCO) in a relatively slow chemical reaction which usually requires addition of catalysts (e.g., tin(II) octanoate).15 The soft segments are usually low Tg polymers such as polyethers or polyesters while the hard segments have a high Tg regulated by the chosen isocyanate and chain extender. Despite their application in many fields, they typically exhibit low tack and peel adhesion requiring tackifiers and/or plasticisers to achieve satisfactory adhesion.16 Their use in medical devices is not common due to low moisture permeability and drug loading challenges.17 Nonetheless, recent developments have explored ways to introduce polyurethane adhesives in transdermal drug delivery formulations.18
Recently a new class of PSAs has emerged based on polyureas synthesised from the reaction of diamines/polyamines (–NH2) with diisocyanates/polyisocyanates (–NCO) forming ureido groups (–NH–CO–NH–).19 These reactions are fast relative to urethane formation, due to the increased nucleophilicity of the amine. Polyurea formation is often easy to perform in the absence of solvents and usually with no requirement for a catalyst. Polyureas can demonstrate enhanced mechanical properties and stability relative to polyurethanes as a consequence of the stronger intermolecular interactions between the chains and the higher stability of the urea bonds.20
In 2020, Liu et al.21 used a prepolymer method to synthesise lignin-based reusable polyurea adhesives which had high adhesion strength. Isocyanate-terminated prepolymers were synthesised by reaction of polyetheramines with isophorone diisocyanate (IPDI) which was subsequently combined with modified lignin and a disulfide chain extender achieving high performance dynamic polyurea adhesives. The same year, waterborne dispersions of polyurethane–urea prepolymers coming from the combination of an ether–carbonate polyol copolymer with IPDI and amino–alcohol chain extenders were developed. Polyurethane–urea PSAs were prepared by casting the dispersions in poly(ethylene terephthalate) (PET) films and their properties were assessed demonstrating high tack along with good cohesion and adhesion characteristics.22 More recently, Yang et al.19 investigated the effect of the branching crosslinking-degree on the bonding performance of adhesives. This was achieved by altering the degree of branching in hyperbranched polyureas synthesized by the polycondensation of various branched polyamines with urea. These hyperbranched polyureas demonstrated the ability to adhere to a wide range of substrates such as wood, aluminium, rubber and skin leaving no residues or irritation. In addition, their results showed that the higher the degree of branching the higher the bonding strength.
The development of transdermal drug delivery (TDD) devices is a meticulous process that usually involves extensive feasibility studies to choose the appropriate drug, excipients, permeation enhancers, backing liner, release liner, and, most importantly, a suitable adhesive system.23 Adhesives play a crucial role in TDD devices, where it is essential for the drug to be present in a solubilized state for effective diffusion across the skin with some exceptions to this rule. Apart from the usual requirements of functional PSA properties, like good tack, skin adhesion, and cohesive strength, other parameters must also be considered. These include biocompatibility, ensuring that the adhesive is non-irritant, non-sensitizing to different skin types, and non-toxic.24 In addition, it should provide compatibility with drugs without inducing degradation whilst offering sufficient diffusivity. Furthermore, it should be easily removable without leaving a residue or “black ring” on the skin usually caused by cold flow.
In our previous work, a novel silylated poly(ether-urethane) moisture-curable PSA was introduced as the matrix for a drug-in-adhesive TDD patch designed to deliver ibuprofen.25 The patch was prepared using a solvent-free polyurethane that required up to 50 wt% commercial tackifier resins to give the required levels of adhesion. The delivery of ibuprofen was successful and facilitated penetration into human skin in a controlled manner. Furthermore, the adhesion of the PSA was found to be significantly higher compared to other commercial products with painless removal from the skin and left no residues on the skin following removal and no cold flow.
Herein, inspired by the favourable mechanical and adhesive properties of poly(ether-ureas), we aimed to broaden the scope of these new PSAs and now report novel silylated poly(ether-ureas) moisture-curable PSAs using a prepolymer method. Prepolymer crosslinkers with varying average molecular weight are produced using a two-step procedure. Firstly, amine-terminated poly(ether-urea) prepolymers are synthesised through the step-growth polymerisation of a relatively high molecular weight (4000 g mol−1) commercial polyetheramine with IPDI. Subsequently, the terminal ends are silylated to introduce moisture-curable properties. These prepolymer crosslinkers were able to form crosslinked networks in the presence of a titanium catalyst and moisture/high humidity resulting in a family of transparent and solvent-free PSAs. We hypothesised that the number of urea moieties between crosslinking points along with the change in the polymer chain-length would allow for PSAs with adjustable and tuneable adhesion and tack. The presence of urea groups is intended to fine-tune tackiness and adhesion, eliminating the need for additional tackifier resins, which can lead to compatibility issues and tedious formulation optimisations. In addition, intermolecular attraction between the polyurea segments were hypothesised to enhance cohesion forming bidentate hydrogen bond motifs.20 Acquiring crosslinked PSAs would also mitigate cold flow effects that often contribute to loss of adhesion reducing the long-term performance of materials especially critical for commercial use.26
Materials and methods
Materials
Polyetheramine with Mn = 4000 g mol−1 (Jeffamine® D-4000, total amine: 0.49 meq. g−1) was purchased from Huntsman. Polyetheramine with Mn = 2000 g mol−1 (Baxxodur® EC 303, total amine: 1.01 meq g−1) was purchased from BASF. Isophorone diisocyanate (IPDI, >98% mixed isomers) and (3-aminopropyl)trimethoxysilane (APTMS, 97%) were purchased from ThermoFisher Scientific. 3-Isocyanatopropyltrimethoxysilane (IPTMS, 98.6%) was purchased from Tokyo Chemical Industry Co Ltd. Titanium(IV) butoxide (Ti(OBu)4, 97%) was purchased from Sigma Aldrich. Nurofen® patch (Lot: JJ578), Voltarol® patch (Lot: 8ML36P), Salonpas® (Lot: W604T) and Lidoderm® (Lot: YA138). PRIMELINER™ 75 μm 1S release liners were purchased from Loparex BV. All materials were used as received unless otherwise stated.Characterisation
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500 g mol−1. The instrument had 2× PolarGel Mixed D columns (300 × 7.5 mm) and a PLgel 5 μm guard column. Polymer samples were dissolved in DMF and filtered through GVHP nylon membranes (0.22 μm pore size) prior analysis. Experimental average molecular weights (Mn, Mw) and dispersity (Đ) were calculated using Agilent GPC/SEC software.
500 g mol−1. The instrument had 2× PolarGel Mixed D columns (300 × 7.5 mm) and a PLgel 5 μm guard column. Polymer samples were dissolved in DMF and filtered through GVHP nylon membranes (0.22 μm pore size) prior analysis. Experimental average molecular weights (Mn, Mw) and dispersity (Đ) were calculated using Agilent GPC/SEC software.
Matrix-assisted laser desorption ionization time-of-flight mass spectrometry (MALDI-ToF-MS)
MALDI-ToF-MS was performed on a Bruker Autoflex Speed MALDI-ToF mass spectrometer, equipped with a nitrogen laser delivering 2 ns laser pulses at 337 nm with positive ion ToF detection at an accelerating voltage of 25 kV. Samples were prepared as follows: trans-2-[3-(4-tert-butylphenyl)-2-methyl-2-propenylidene] malononitrile as the matrix (40 mg mL−1) and the polymer (10 mg mL−1) were each dissolved in THF containing 1 mg mL−1 sodium iodide as a cationising agent. Then, 20 μL of matrix and sample were mixed and 0.5 μL of the final solution was applied on the target plate. Spectra recording was made in reflective mode calibrated with PEG 4000 Da.Nuclear magnetic resonance (1H-NMR, 13C-NMR, and HSQC)
NMR spectra were recorded on a Bruker DPX-400 instrument using deuterated chloroform (CDCl3) as the solvent. Chemical shifts are given as δ in parts per million (ppm). All spectra were analysed using ACD/NMR Processor software.Fourier transform infrared spectroscopy (FT-IR)
FT-IR was performed on a PerkinElmer UATR Two spectrometer fitted with an ATR crystal and a pressure tower running at 65 scans per sample with a speed of 0.5 cm s−1 in the range of 4000 to 400 cm−1.Differential scanning calorimetry (DSC)
DSC was carried on a TA DSC 2500 instrument under nitrogen flow (50 mL min−1) with a heating rate of 10 °C min−1 for two heating and cooling cycles. Cured samples were loaded into 40 μL aluminium pans and heated for two cycles in total between −100 and 200 °C. For every measurement, the glass transition temperature (Tg) was determined from the midpoint of the thermogram of the second cycle to erase any thermal history.Thermogravimetric analysis (TGA)
TGA was conducted on a TA SDT 650 instrument which operated under a nitrogen flow of 50 mL min−1. Samples were heated from ambient temperature to 600 °C with a heating rate of 10 °C min−1. The degradation temperature (Tdeg) was defined as the temperature of the 50% mass loss of each sample.Viscometry
The viscosity of uncured prepolymer crosslinkers was measured on a Brookfield AMETEK DV2T equipped with a Thermosel® programmable temperature cell using a number 27 spindle. The polymer sample was heated at 80 °C and added into a crucible (∼10.5 g per measurement). Data were recorded every 1 min for 10 min in total. The test was repeated until concordant results were obtained. The average of ten concordant results were reported as the viscosity value.2790° Peel test
Peel tests were performed on a Mecmensin MultiTest 2.5-dV- peel tester equipped with an Advanced Force Gauge with a 50 N capacity. Test specimens were cured between two release liners and cut into rectangular strips (25 mm × 150 mm). Samples were stored for 24 h at ambient temperature and in an atmosphere with a 50% relative humidity. The strips were fastened over 2/3 of their length (25 mm × 90 mm) to a stainless-steel substrate and the obtained assembly was left for 20 min at ambient temperature. The free end of the rectangular strip (25 mm × 60 mm) is folded in half so that a 25 mm × 30 mm tab is attached to the gauge and the sample is peeled at a 90° angle with a separation rate of 100 mm min−1. The instrument measured the required force to debond the strip from the stainless-steel surface.28Rolling Ball Tack Test
The tackiness of the cured adhesive was evaluated using a ChemInstruments RBT-100 ramp (Fig. S1, ESI†). A ball bearing was used as the test substrate while the cured adhesive samples had a test area of 48 mm × 380 mm. The distance travelled by the ball along the adhesive strip was recorded in triplicate to give a statistically acceptable robust value.29Rheological analysis
Rheology was conducted on an Anton Parr MCR 302 rheometer equipped with a plate-to-plate configuration of 25 mm diameter (PP25). All oscillatory sweep experiments were performed at 25 °C using cured adhesive discs of a 25 mm diameter. Samples were tested under a constant normal force of FN = 1 N throughout each measurement. For sample preparation, adhesives were initially cured between two release liners and cut into the desired discs (Fig. S2, ESI†). Then, the one side of the release liner was removed, and the adhesive was carefully applied on the bottom plate of the rheometer following careful removal of the second release liner. Amplitude sweep experiments were performed at a strain % range of γ = 0.01–710% with a constant angular frequency of ω = 10 rad s−1. Frequency sweep experiments were conducted at an angular frequency range of ω = 0.5–100 rad s−1 with a constant strain % of γ = 1% as dictated by their linear viscoelastic region (LVER). An average of at least three measurements (n = 3) was accepted as a statistically robust run. Measurements at ω = 0.01 rad s−1 were also performed to graph Chang's windows.Cold flow measurement
Small patch discs of 8 mm diameter were cut. The release liner was removed, and each patch was sticked to a glass slide in triplicates for each PUXL variant. The diameter of each disc was measured (D1). Then, the glass slides were left at ambient temperature or placed in ovens at 32 and 40 °C. After 72 h, the diameter of each was remeasured (D2) and the percentage difference was calculated as (eqn (1)):| %CF = (D2 − D1)/D1 × 100 | (1) |
Dynamic mechanical analysis (DMA)
The analysis was performed in a PerkinElmer DMA 8000-Start Pyris system equipped with a 1 L liquid N2 cylinder. Samples were analysed in stainless steel pockets using a single cantilever-rectangle geometry in a temperature sweep mode with a 5 °C min−1 heat rate. A test frequency of 1 Hz and a static force of 2 N were applied. The glass transition temperature (Tg,DMA) was calculated from the peaks of the loss factor (tanδ) curves and the step inflection point of the storage modulus curve.
Scanning electron microscopy (SEM)
Microscopy analysis was conducted on a Zeiss Gemini500 microscope with a field emission electron gun. The accelerating voltage was 10 kV while an InLens detector was used at a working distance of 5.1 mm. For the sample preparation, a patch specimen was initially frozen with liquid nitrogen and further freeze dried to remove water. Then, the sample was loaded onto carbon tabs (9 mm) attached to aluminium specimen stubs. Finally, the sample was sputter coated with gold (Au) for 15 seconds using a plasma coater.Experimental procedures
Synthesis of PUXL1 prepolymer crosslinker
Jeffamine® D-4000 (4700 g, 1.18 mol, 1 eq., amine content: 0.49) was charged in a gas tight oil jacketed reactor vessel equipped with a stainless still impeller and heated to 85 °C under nitrogen with a stirring speed of 120 rpm. The temperature was monitored online by a temperature probe. After the required temperature was reached, the stirring speed was increased and IPDI (121.58 g, 0.55 mol, 0.47 eq.) was added. The mixture was allowed to react for 15 min. Then, IPTMS (235.79 g, 1.19 mol, 0.99 eq.) was added and the reaction was allowed to proceed for 20 min. The final product was collected and analysed by FT-IR to confirm the absence of any residual isocyanate groups then stored under a nitrogen blanket. Yield >96% with high reproducibility per batch.Synthesis of PUXL2 prepolymer crosslinker
Jeffamine® D-4000 (4700 g, 1.18 mol, 1 eq., amine content: 0.49) was charged in a gas tight oil jacketed reactor vessel equipped with a stainless still impeller and heated to 85 °C under nitrogen with a stirring speed of 120 rpm. The temperature was monitored online by a temperature probe. After the required temperature was reached, the stirring speed was increased and IPDI (179.33 g, 0.81 mol, 0.69 eq.) was added. The mixture was allowed to react for 30 min. Then, IPTMS (134.45 g, 0.70 mol, 0.59 eq.) was added and the reaction was allowed to proceed for 20 min. The final product was collected and analysed by FT-IR to confirm the absence of residual isocyanate groups then stored under a nitrogen blanket. Yield >96% with high reproducibility per batch.Synthesis of PUXL3 prepolymer crosslinker
Jeffamine® D-4000 (4700 g, 1.18 mol, 1 eq., amine content: 0.49) was charged in a gas tight oil jacketed reactor vessel equipped with a stainless still impeller and heated to 85 °C under nitrogen with a stirring speed of 120 rpm. The temperature was monitored in real time by a temperature probe. After the required temperature was reached, the stirring speed was increased and IPDI (206.76 g, 0.93 mol, 0.79 eq.) added. The mixture was allowed to react for 45 min. Then, IPTMS (86.32 g, 0.47 mol, 0.40 eq.) was added and the reaction was allowed to proceed for 20 min. The final product was collected and analysed by FT-IR to confirm the absence of residual isocyanate groups then stored under a nitrogen blanket. Yield >96% with high reproducibility per batch.Synthesis of PUXL4 prepolymer crosslinker
Jeffamine® D-4000 (4700 g, 1.18 mol, 1 eq., amine content: 0.49) was charged in a gas tight oil jacketed reactor vessel equipped with a stainless still impeller and heated to 85 °C under nitrogen with a stirring speed of 120 rpm. The temperature was monitored online by a temperature probe. After the required temperature was reached, the stirring speed was increased and IPDI (219.79 g, 0.99 mol, 0.84 eq.) was added. The mixture was allowed to react for 60 min. Then, IPTMS (63.46 g, 0.35 mol, 0.30 eq.) and the reaction was allowed to proceed for 20 min from the addition of IPTMS. The final product was collected and analysed by FT-IR to confirm the absence of residual isocyanate groups then stored under a nitrogen blanket. Yield >96% with high reproducibility per batch.Synthesis of PUXL5 prepolymer crosslinker
Jeffamine® D-4000 (4700 g, 1.18 mol, 1 eq., amine content: 0.49) was charged in a gas tight oil jacketed reactor vessel equipped with a stainless still impeller and heated to 85 °C under nitrogen with a stirring speed of 120 rpm. The temperature was monitored by a temperature probe. After the required temperature was reached, the stirring speed was increased and IPDI (225.98 g, 0.58 mol, 0.86 eq.) was added. The mixture was allowed to react for 75 min. Then, IPTMS (52.60 g, 0.30 mol, 0.25 eq.) was added and the reaction was allowed to proceed for 20 min. The final product was collected and analysed by FT-IR to confirm the absence of residual isocyanate groups then stored under a nitrogen blanket. Yield >96% with high reproducibility per batch.Synthesis of PUXL6 prepolymer crosslinker
Jeffamine® D-4000 (4700 g, 1.18 mol, 1 eq., amine content: 0.49) was charged in a gas tight oil jacketed reactor vessel equipped with a stainless still impeller and heated to 85 °C under nitrogen with a stirring speed of 120 rpm. The temperature was monitored online by a temperature probe. After the required temperature was reached, the stirring speed was increased and IPDI (228.92 g, 0.56 mol, 0.87 eq.) was added. The mixture was allowed to react for 60 min. Then, IPTMS (47.44 g, 0.28 mol, 0.254 eq.) was added and the reaction was allowed to proceed for 20 min. The final product was collected and analysed by FT-IR to confirm the absence of residual isocyanate groups then stored under a nitrogen blanket. Yield >96% with high reproducibility per batch.Synthesis of PUXL7 prepolymer crosslinker
A mixture of 90:10 molar ratio of Jeffamine® D-4000 (4463.1 g, 1.12 mol, amine content: 0.49) and Baxxodur® EC 303 (240.6 g, 0.12 mol, amine content: 1.01) was charged in a reactor vessel and heated to 85 °C under nitrogen stirred at 120 rpm. After the required temperature was reached, the mixing speed was increased and IPDI (238.43 g, 1.34 mol, 1.07 eq. with respect to the total moles of polyetheramines) was added. IPTMS (55.50 g, 0.27 mol, 0.22 eq. with respect to the total moles of polyetheramines) was then added and the reaction was allowed to proceed for 20 min. The final product was analysed by FT-IR to confirm the absence of residual isocyanate groups and stored under a nitrogen blanket. Yield >96% with high reproducibility per batch.
Preparation of cured adhesives
An RK K-Control coater was used with a coating area of up to 325 × 250 mm. As gap applicators, K wedge bars were used at 300 μm. The required humidity for the curing was provided by a modified TaoTronics TT-AH002 humidifier with the capability to control the rate of water vapour generation. The coating setup used is presented in Fig. S3 (ESI†). A representative amount of PUXL prepolymer was pre-weighed in a hot beaker and kept in an oil bath at 80 °C under a continuous nitrogen stream. The polymer was stirred for approximately 5 min using an overhead stirrer with a propeller blade at a stirring speed that gradually increased from 60 to ∼100 rpm. The position and height of the propeller was tuned during mixing to avoid air bubbles. Then, titanium(IV) butoxide catalyst (1%) was added and the resultant mixture was mixed for a further 5 min. In parallel, a release liner sheet was placed on the heated bed (set at 80 °C) of the RK K-Control coater with the siliconized surface facing up. Approximately 10 mL of the hot adhesive were poured onto the heated bed of the coater in front of a K-bar with the desired thickness. The adhesive was spread forming an uncured polymer layer on the release liner. The resultant polymer layer was then subjected to 1.5 min of steam (≥60% humidity) and a further 3.5 minutes of heat to induce the formation of a cured polymer network. The curing of each patch was assessed after 5 min total time using the tack test. After confirmation of curing, the casted formulation was placed on a flat surface and covered with a second layer of release liner with the siliconized surface touching the cured formulation.Results and discussion
Synthesis and characterisation of moisture-curable silylated polyurea prepolymer crosslinkers (PUXL)
Selecting a suitable primary diamine as the soft segment of the PSA was a crucial requirement.30 Polyetheramines were considered ideal as amine terminated polymers that are known for their low viscosity, transparency and low Tg depending on their molecular weight.31,32 A difunctional primary polyetheramine with a molecular weight of MWt = 4000 g mol−1 (Jeffamine® D-4000) was used in this study. This polyetheramine is commercially available providing higher flexibility when compared to lower molecular weight analogues as well as high peel strength.33,34 Selecting an appropriate diisocyanate was equally important as these components typically regulate the viscoelastic and mechanical properties of PSAs.35 IPDI was chosen as the hard segment of these adhesives due to its asymmetric cycloaliphatic structure which reduces conformational changes offering higher cohesion strength and creep resistance.36Commercial Jeffamine® D-4000 was characterised by NMR, s (SEC) and MALDI-ToF-MS. Assignment of the 1H-NMR and 13C-NMR (Fig. S6–S8, ESI†) confirmed the desired structure attributing some low intensity peaks on remaining starting materials. The Mn,NMR was determined by comparing the proton integrals from the initiator –CH3 peaks close to the –NH2 groups (δ = 1.00 ppm, Fig. S6, ESI†) against the –CH3 protons of the polymer backbone (δ = 1.30 ppm, Fig. S6, ESI†). The Mn,NMR = 3600 g mol−1 is in good agreement with the certificate of analysis. Analysis by SEC shows a bimodal distribution trace with an overall dispersity of Đ ∼1.26, a low molecular weight shoulder at Mn,SEC ∼ 2000 g mol−1 and a predominant molecular weight distribution peak at Mn,SEC = 4200 g mol−1 (Fig. S4, ESI†). Further insight about the low molecular weight shoulder was obtained by MALDI-ToF-MS indicating the presence of a mixture of two species (Fig. S9, ESI†). The lower mass distribution was attributed to monofunctional amine terminated polyetheramines seemingly resulting from chain cleavage presumable during the amination process which is assumed to take place at high pressures and temperatures in the presence of hydrogen and ammonia, while the higher mass distribution corresponded to the desired difunctional material. A general schematic describing the synthesis of the prepolymer crosslinkers is shown in Scheme 1. All steps were solvent-free requiring no purification at any stage or addition of catalysts making the process versatile for larger scale manufacturing (up to 5 kg using a 10 kg overhead stirred jacketed glass reactor). Both synthetic steps were performed in a one-pot step at a set temperature of 85 °C insuring effective mixing, even at high viscosities. Firstly, the step-growth polymerisation of Jeffamine® D-4000 with IPDI occurred to afford poly(ether-ureas). The terminal amine groups in Jeffamine® D-4000 react with the isocyanates in IPDI forming urea bonds. Due to the inherent toxicity of isocyanates, complete consumption of IPDI was assured by always keeping an excess of primary amines relative to isocyanates thus giving amine terminated products. Subsequently, the amine terminated poly(ether-ureas) were end-capped with alkoxysilyl groups to serve as moisture-curable crosslinking agents. Incorporation of alkoxysilyl groups through silylation aimed at enhancing the adhesion, cohesion as well as elasticity of the final crosslinked compositions.37,38 Silylation was achieved by end-caping the amine terminated prepolymers with 3-isocyanatopropyltrimethoxysilane (IPTMS) forming the final silylated poly(ether-urea) crosslinker (PUXL) at high yields (typically >95%). Different prepolymer variants were obtained by adjusting the IPDI content. By increasing the IPDI content polymers with theoretically higher average molecular weights were afforded which in turn led to a higher concentration of polyetheramine, IPDI and urea groups per polymer chain. Details concerning the polyurea formation along with corresponding viscosity data after the modification step are presented in Table 1. All variants were synthesised using Jeffamine® D-4000 except for PUXL7 which contained a mixture of Jeffamine® D-4000 and a lower MWt difunctional polyetheramine (Baxxodur® EC 303) with MW = 2000 g mol−1 at a ratio of 90:10. Commercial Baxxodur® EC 303 was also characterised prior use showing a monomodal distribution in both SEC and MALDI-ToF MS (Fig. S10 and S11, ESI†). Viscometry was performed at 80 °C to simulate the process temperature with the results indicating higher viscosity values with increasing –NCO/–NH2 molar ratio. Specifically, PUXL1 had a viscosity of ∼1540 cP which increased to ∼29000 cP for PUXL6 suggesting that a higher content of IPDI potentially formed larger polymer chains. Incorporation of a small percentage of lower MW polyetheramine did not have a significant effect on the viscosity with PUXL7 demonstrating close viscosity values (∼27430 cP) with PUXL5 (∼26000 cP).
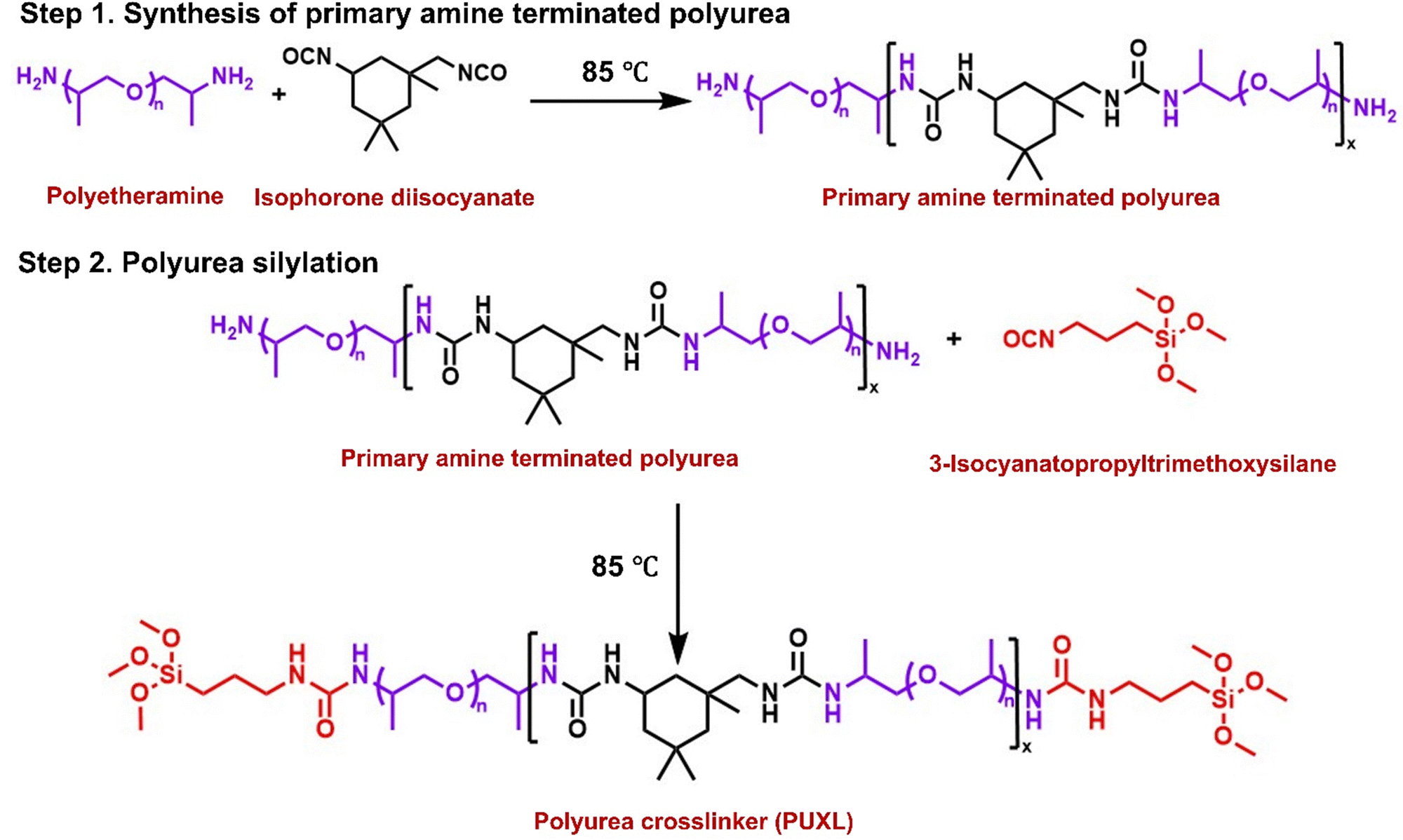 | ||
| Scheme 1 Synthesis of poly(ether-urea) PSA prepolymer crosslinkers (PUXL). | ||
| Entry | IPDI: D-4000 molar ratio | IPTMS (eq.) | Viscosity at 80 °Cb (cP) |
|---|---|---|---|
|
a A 90:10 mixture of Jeffamine® D-4000 and Baxxodur® EC 303 was used.
b Refers to the viscosity of the prepolymer crosslinkers after modification.
|
|||
| PUXL1 | 0.47 | 0.99 | ∼1540 |
| PUXL2 | 0.68 | 0.59 | ∼5820 |
| PUXL3 | 0.79 | 0.40 | ∼14600 |
| PUXL4 | 0.84 | 0.30 | ∼20000 |
| PUXL5 | 0.86 | 0.25 | ∼26000 |
| PUXL6 | 0.87 | 0.25 | ∼29000 |
| PUXL7 | 1.07a | 0.22 | ∼27430 |
1H-NMR assignment confirmed the existence of urea protons appearing in the range of δ = 4.5–5.5 ppm (e + e′, Fig. 1(a)). The noticeable downfield shift of the –CHCH3NH2 proton from δ = 3.06 (e, Fig. S6, ESI†) in Jeffamine® D-4000 to δ = 3.88 ppm (o, Fig. 1(a)) in PUXL5 was a result of resonance effects from the urea further confirming successful reaction of the terminal amines with isocyanates. In the FT-IR spectra, the peaks at 925 and 1096 cm−1 were characteristic of the ether groups found in both the starting material and the finished product suggesting that the backbone of the polymer remained unaltered during manufacturing (Fig. 1(b) and Fig. S12, ESI†). The disappearance of the –NCO peak at 2250 cm−1 suggested no residual isocyanates following IPDI and IPTMS addition. This data coupled with the characteristic proton peaks of IPTMS (c, b, d, Fig. 1(a)) suggest successful modification. Finally, three new peaks appeared in the FT-IR spectrum of the final prepolymer crosslinker compared to the starting material at 1637, 1556 and 3350 cm−1 (Fig. 1(c)). These corresponded to the urea amide with the broad peak at 3350 cm−1 indicating hydrogen bonding between the –NH groups of the formed urea moieties.
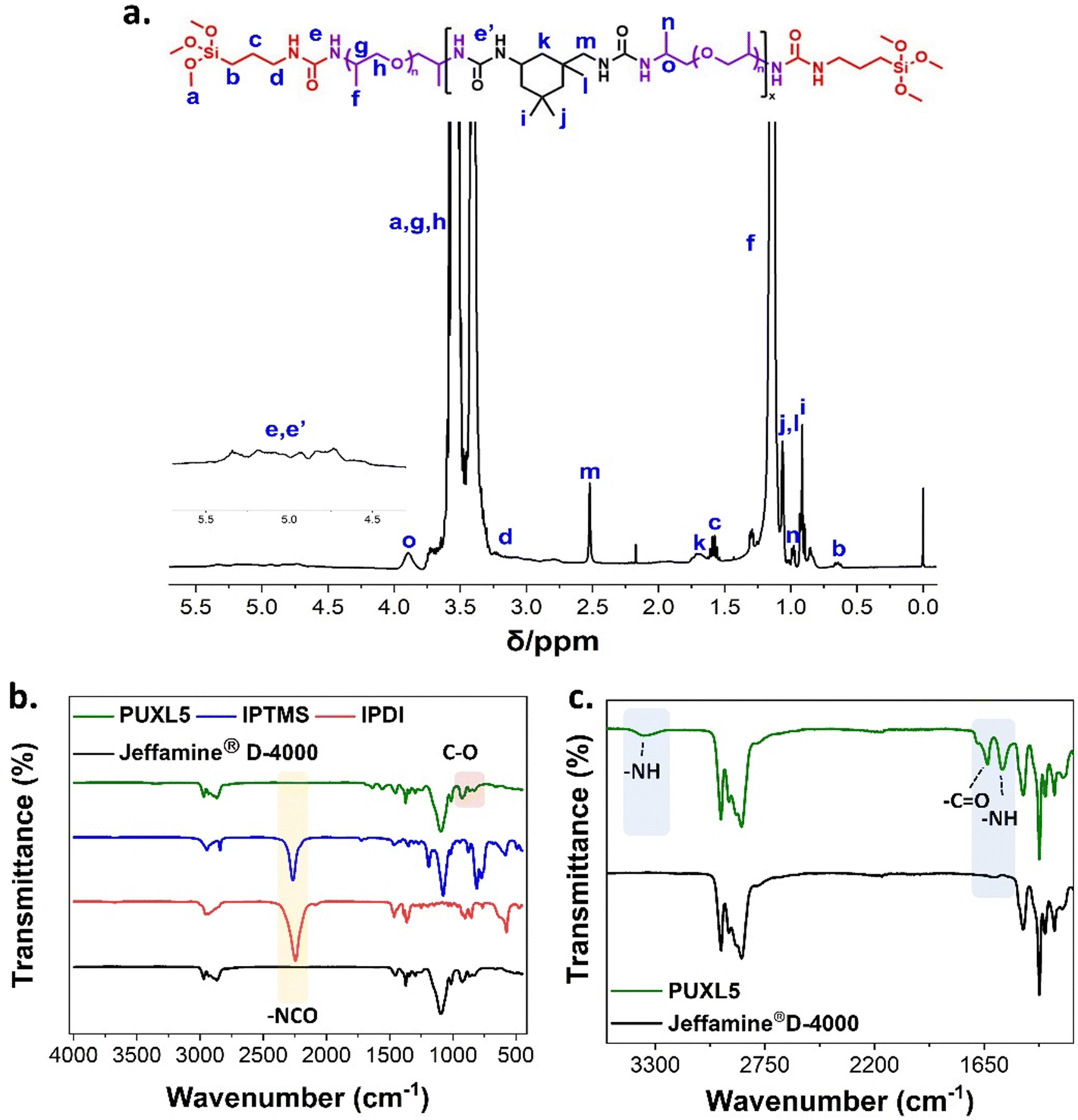 | ||
| Fig. 1 (a) Assigned 1H-NMR (400 MHz, CDCl3) spectrum of PUXL5 prepolymer crosslinker, (b) FT-IR spectra comparison of PUXL5 prepolymer crosslinker with the starting materials Jeffamine® D-4000, IPDI and IPTMS along with (c) expanded region highlighting the appearance of urea peaks. | ||
The unmodified S-PURE variants were analysed by SEC in DMF as eluent revealing shifts to higher molecular weights by increasing the IPDI content (Fig. 2(a)). This indicated successful chain-extension when compared to free Jeffamine® D-4000. The obtained SEC traces were multimodal with a dispersity, ĐRI = 1.64–1.86, typical of step-growth polymerisation processes. The Mn and Mw values obtained from both conventional (differential refractive index, DRI) and universal (viscometer, VS) calibration, showcased that the higher-grade variants were indistinguishable considering a 10% instrumental error (Table 2). Lower average molecular weight shoulders were also found which shifted to higher molecular weights moving from PUXL1 to PUXL6 variants. With the addition of more IPDI these lower average molecular weight species were chain-extended forming larger polymer chains.
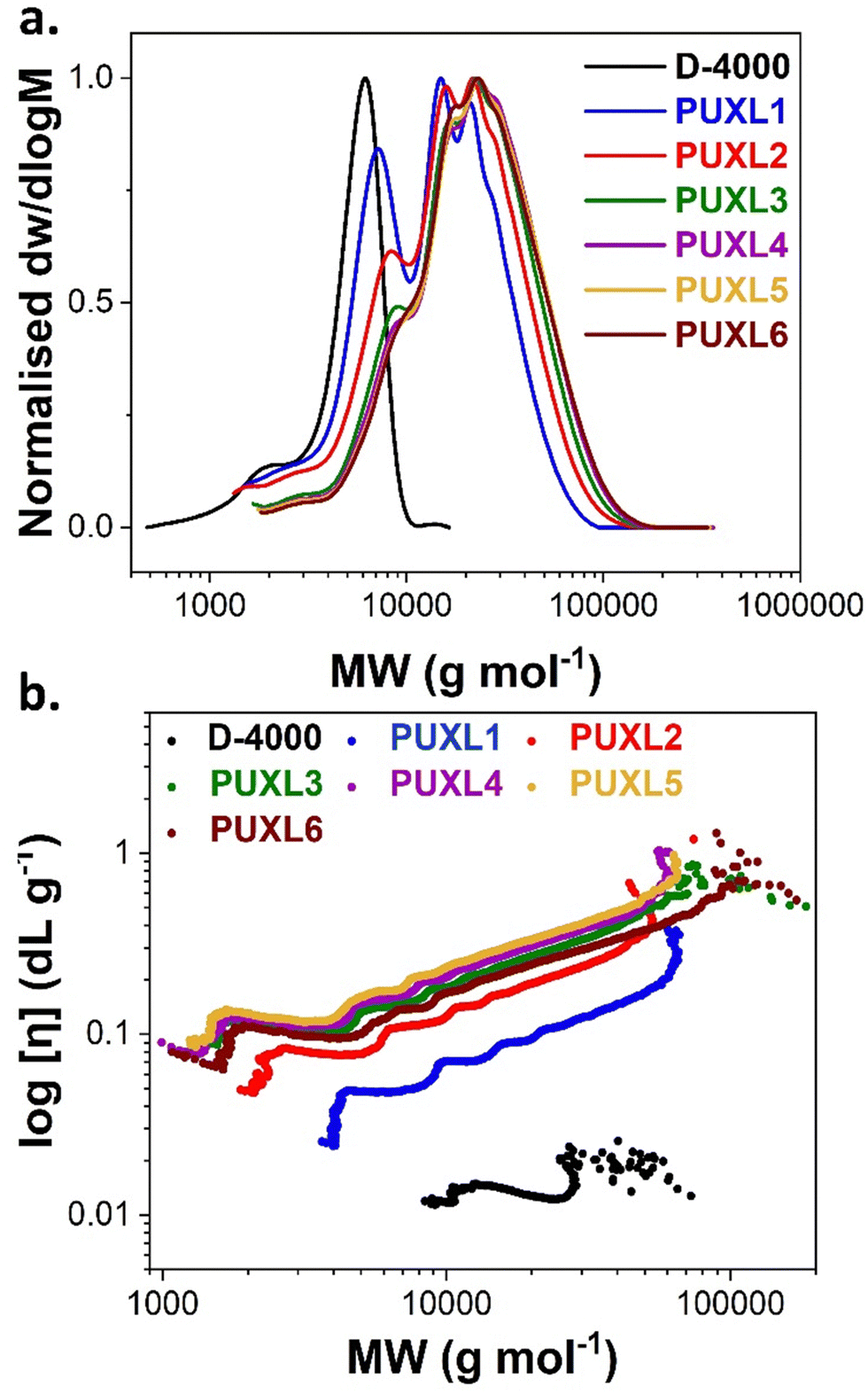 | ||
| Fig. 2 (a) SEC traces and (b) Mark–Houwink–Sakurada (MHS) plots for PUXL synthesised prepolymers prior modification as measured in DMF. | ||
| Entry | M n,RI (g mol−1) | M w,RI (g mol−1) | Đ RI | M n,VS (g mol−1) | M w,VS (g mol−1) | Đ VS | α | k × 10−2 (dL g−1) |
|---|---|---|---|---|---|---|---|---|
| D-4000 | 4200 | 5200 | 1.26 | 8700 | 12500 |
1.45 | 0.11 | 1.037 |
| PUXL1 | 9600 | 17300 |
1.64 | 9500 | 15600 |
1.70 | 0.51 | 0.018 |
| PUXL2 | 11800 |
21700 |
1.86 | 7700 | 13000 |
1.72 | 0.56 | 0.086 |
| PUXL3 | 14500 |
25300 |
1.76 | 7300 | 12900 |
1.78 | 0.49 | 0.158 |
| PUXL4 | 14900 |
27300 |
1.77 | 7400 | 13100 |
1.77 | 0.50 | 0.164 |
| PUXL5 | 15800 |
27900 |
1.77 | 7100 | 12750 |
1.80 | 0.50 | 0.185 |
| PUXL6 | 16100 |
28000 |
1.75 | 8000 | 14400 |
1.81 | 0.49 | 0.129 |
Information concerning the structure of the polymers in the solvent (at T = 50 °C) were obtained by determining the Mark–Houwink–Sakurada (MHS) parameters “α” and “k” from the log–log graphs between the intrinsic viscosity [η] and molecular weight (Fig. 2(b) and Table 2). The [η] values raised by introducing more IPDI, however, there was no clear difference between the PUXL4, PUXL5 and PUXL6 variants in contrast to regular viscometry explained by the absence of solvent in the latter case. The “α” values were determined by the slope of the MHS plots providing information about the polymer size and shape in the examined solvent and temperature (linear fitting of the MHS plots shown in Fig. S13, ESI†). Jeffamine® D-4000 demonstrated an “α” value = 0.11 reflecting a rigid sphere like structure, however, it is noted that this data at low MWt was not convincing, while chain-extensions with IPDI led to polymers with higher “α” values standing at 0.49–0.56. This indicated that the unmodified PUXL variants had a random coil like structure under these conditions.39
Preparation of crosslinked PUXL adhesives
Crosslinking was performed to form the crosslinked PSA which minimises the potential for cold flow, Scheme 2. Curing was via the condensation of the terminal trimethoxy silyl groups upon contact with water (≥60% relative humidity) and in the presence of titanium(IV) butoxide (Ti(OBu)4) catalyst at 80 °C. It is noted that titanium(IV) isopropoxide (Ti(OiPr)4) led curing that was too fast for the process conditions.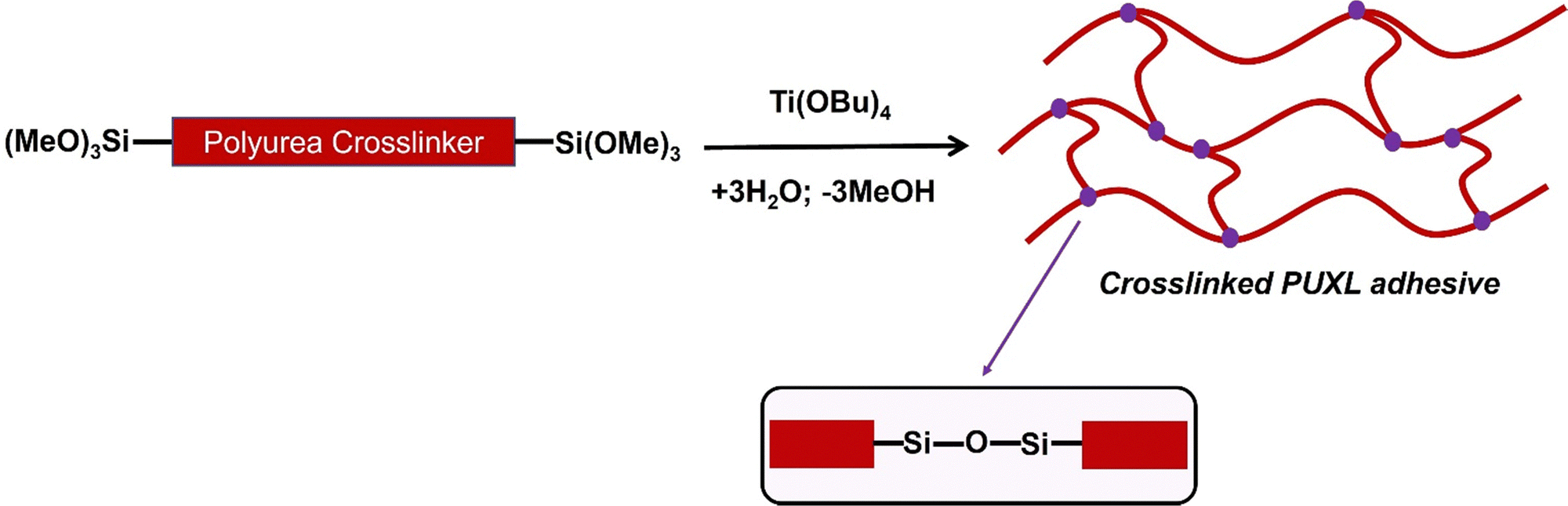 | ||
| Scheme 2 Curing reaction leading to the formation of the crosslinked PUXL adhesives. | ||
An immiscible polymer network was formed via crosslinked siloxane groups (Si–O–Si).40 The biproducts methanol and butanol subsequently evaporated due to the high temperature of the process conditions.
While at ambient conditions the prepolymers were colourless to pale yellow viscous liquids, once crosslinked they turned into transparent tacky viscoelastic solids. Adhesives for rheology characterisation were cured between two release liners. A catalyst content of 1.5 wt% was found sufficient to achieve full crosslinking while a thickness of ∼300 μm was selected to investigate the properties of the cured materials. The curing speed was generally adjusted by the amount of catalyst with more catalyst typically reducing the curing time required. In the case of these variants, all of them, except for PUXL6, cured within 5 min which was confirmed after performing finger tack testing (Fig. S14, ESI†). PUXL6 required 10 min owing to its higher molecular weight which led to the formation of networks with a lower crosslink density.41 Images of the procedure followed can be found in Fig. 3. Specifically, Fig. 3(f) emphasizes the easy peel release of the adhesive from the release liner.
 | ||
| Fig. 3 Curing procedure of PUXL adhesives where: (a) pouring of uncured formulation containing catalyst, (b) casting of a polymer layer onto the heated bed in front of the wire wound bar, (c) placing the humidity chamber over the polymer layer, (d) the cured adhesive, (e) adhesive between two release liners and (f) transparent fully cured adhesive removed from the release liners. | ||
To demonstrate the potential of these adhesives for medical applications, PUXL5 was cured on the siliconized side of a “plastic” release liner and then covered with a fabric backing liner to produce an adhesive patch. The release liner was successfully removed while the adhesive remained intact on the surface of the fabric without leaving residues on the surface of the plastic liner (see Fig. S15, ESI†). The morphology of the cured adhesive was additionally analysed by scanning electron microscopy (SEM) which revealed a smooth polymer layer with no specific structural characteristics (Fig. S16, ESI†).
Thermal characterisation of cured PUXL variants
The thermal properties of the cured PUXL variants were evaluated by thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC), Table S1 (ESI†). The degradation profiles were monitored within the temperature range of 25–600 °C, Fig. 4(a) and (b). Notably, all variants demonstrated identical degradation profiles, with an observed thermal decomposition temperature (Td) ∼380 °C. This loss was attributed to scission reactions of the poly(propylene glycol) backbone, aligning with the degradation temperature of free Jeffamine® D-4000 as reported by Hamciuc et al.42 Due to the small IPDI hard domains, only the decomposition of the longer soft segments was noticed.43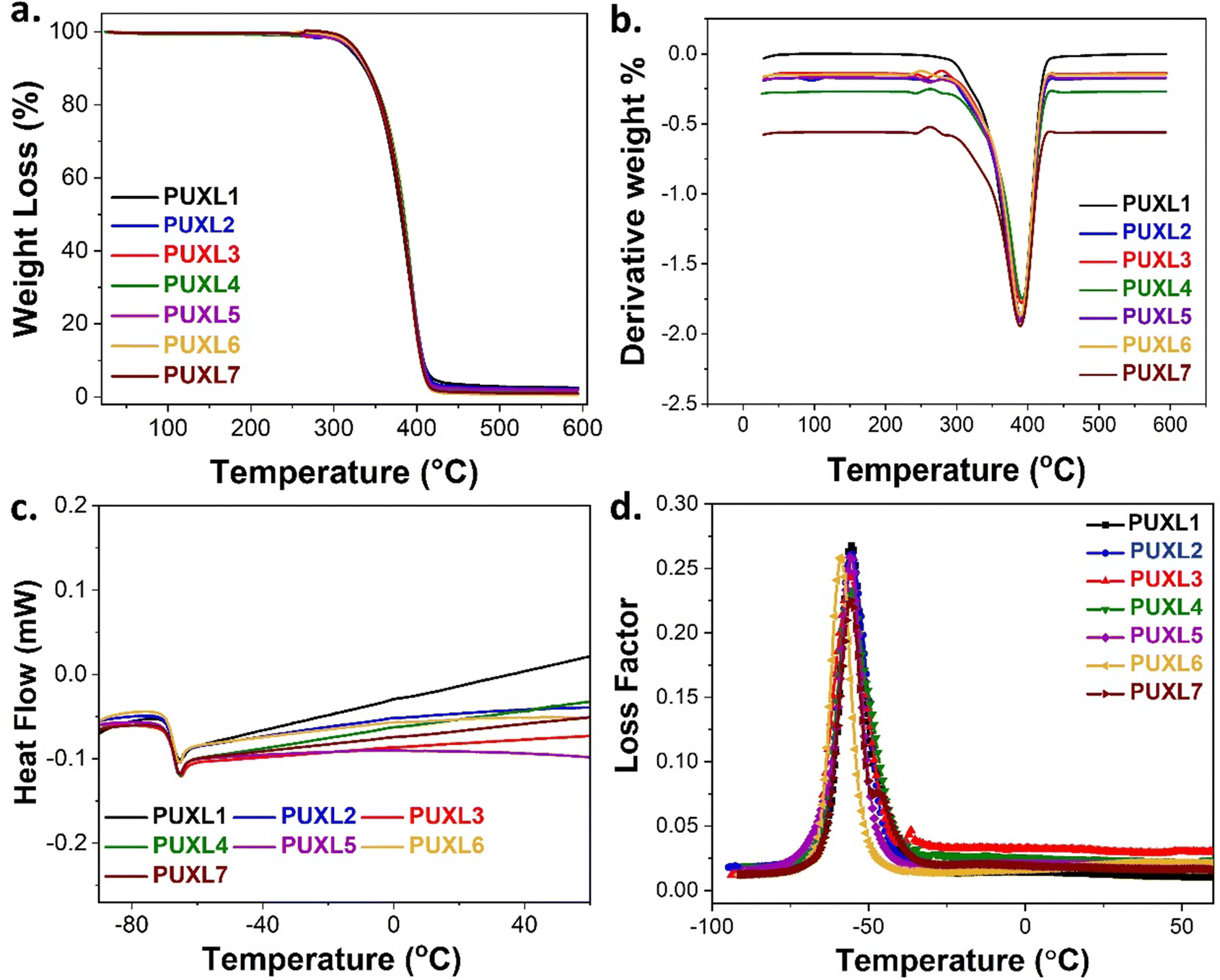 | ||
| Fig. 4 (a) TGA curves along with their (b) first derivatives, (c) DSC thermograms and (d) loss factors as obtained from the DMA analysis of the cured PUXL adhesives. | ||
DSC analysis was performed to explore thermal differences among the variants. For comparison, Jeffamine® D-4000 was analysed revealing a negative Tg,mid = −70 °C (Fig. S5, ESI†). Surprisingly, according to the DSC findings (Fig. 4(c) and Table S1, ESI†), all PUXL variants exhibited a single negative glass transition temperature at Tg,mid ∼ −70 °C which was equivalent to the Tg,mid of free Jeffamine® D-4000. This was unexpected since crosslinking typically raises the Tg of materials, rendering polymer chains rigidity.44 The unaltered negative Tg suggested the possibility of a relatively low crosslink density which allowed for chain flexibility at ambient temperature critical for the performance of these materials as PSAs.18
Additional thermal data were acquired through dynamic mechanical analysis (DMA) with the graphs of the loss factor (tanδ) as a function of temperature in Fig. 4(d). All tanδ were nearly the same exhibiting a main Tg,DMA peak at approximately −56 °C with no significant differences in the peak widths. Despite the variants sharing the same Tg,DMA, a general decrease in their elastic properties was evident as one moved from PUXL1 to PUXL6, depicted by the curves of their elastic modulus (E′) as a function of temperature (Fig. S17, ESI†).
Evaluation of viscoelastic properties
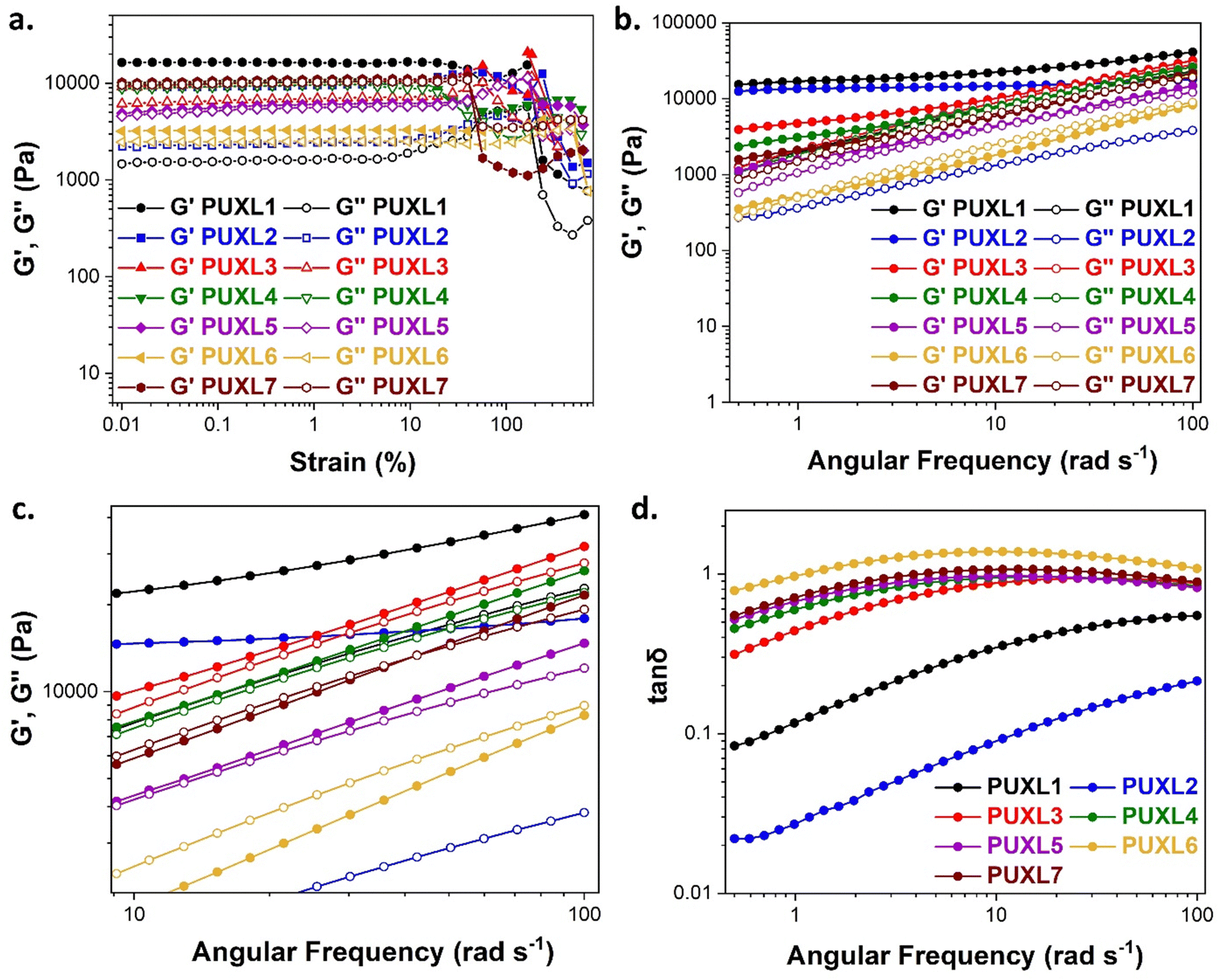
As the performance of PSAs is strongly related to their viscoelastic properties, the rheological characteristics of the cured adhesives were investigated at 25 °C by plate-plate oscillatory rheology monitoring the progress of the storage (G′) and loss (G′′) moduli. The values of G′ and G′′ typically represent the ability of an adhesive to adhere onto a substrate with good performing adhesives exhibiting low G′ at low frequencies (bonding) and equal or higher G′ than G′′ at high frequencies (debonding).45Initially, amplitude sweep experiments were performed at a constant angular frequency of ω = 10 rad s−1 to determine the linear viscoelastic regions (LVR) where G′ and G′′ are independent of strain deformations. The amplitude sweeps graphs, Fig. 5(a), exhibited a large LVR from γ = 0.01 to 20%, though the strain at break (crossover of G′ with G′′) could not be accurately determined due to extensive slippage of the samples at high deformations. For the frequency sweep experiments, a constant strain of γ = 1.0% and the frequency range of ω = 0.5–100 rad s−1 was examined since application of a PSA on skin has been reported to occur in the range of ω = 0.1–10 rad s−1.45 Adhesion and peeling were assessed at ω = 0.5 and 100 rad s−1 respectively with the data presented in Table 3. As depicted by Fig. 5(b), variants with a higher IPDI content demonstrated lower G′ and G′′ values over the entire frequency range agreeing with the modulus trends of DMA. This was indicative of a gradual increase in adhesion from PUXL1 to PUXL6 with the G′ values always being higher than G′′ at low frequencies indicating successful crosslinking. More specifically, PUXL1 and PUXL2 demonstrated the highest modulus values at ω = 0.5 rad s−1 (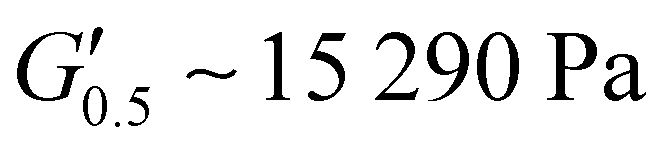 and
and 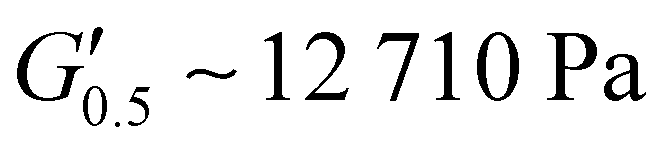 respectively) suggesting poorer adhesion and higher elastic character. From PUXL3 to PUXL6, the
respectively) suggesting poorer adhesion and higher elastic character. From PUXL3 to PUXL6, the  values dropped with PUXL6 showcasing the best adhesion with the lowest value of
values dropped with PUXL6 showcasing the best adhesion with the lowest value of 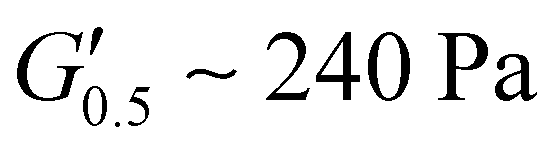 . PUXL7 had a slightly increased
. PUXL7 had a slightly increased 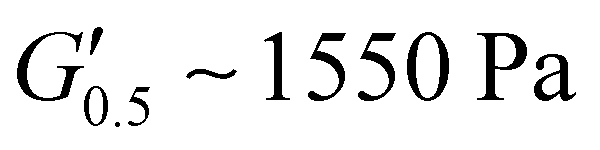 owing to the mix of low and high molecular weight polyetheramines in the network thus forming low and high crosslink density regions. The debonding strength was close to that of PUXL3 and PUXL4 as evidenced from the values of
owing to the mix of low and high molecular weight polyetheramines in the network thus forming low and high crosslink density regions. The debonding strength was close to that of PUXL3 and PUXL4 as evidenced from the values of 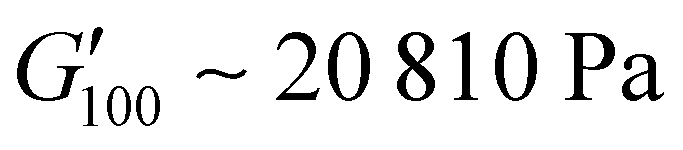 and
and 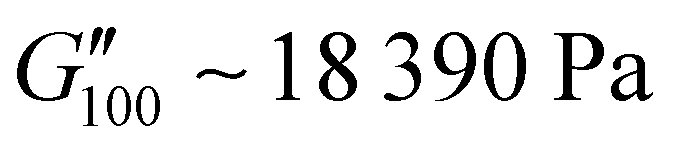 .
.
 | ||
| Fig. 5 (a) Amplitude sweeps of cured PUXL variants at a constant angular frequency of ω = 10 rad s−1. (b) Frequency sweep experiments of cured PUXL variants at a constant strain of γ = 1.0 rad s−1 and (c) zoomed in regions at high angular frequencies. (d) Evolution of the loss factor (tanδ) during frequency sweep measurements. Rheology was conducted at 25 °C using an oscillatory mode where storage modulus (G′, filled symbols) and loss modulus (G′′, empty symbols). | ||
δ) during frequency sweep measurements. Rheology was conducted at 25 °C using an oscillatory mode where storage modulus (G′, filled symbols) and loss modulus (G′′, empty symbols)
| Entry |
|
|
|
|
|---|---|---|---|---|
| PUXL1 | 15290 ± 6000 |
1270± 330 | 40950 ± 16850 |
22750 ± 8200 |
| PUXL2 | 12710 ± 246 |
250 ± 25 | 17100 ± 950 |
3260 ± 400 |
| PUXL3 | 2890 ± 890 |
865 ± 230 | 22300 ± 8300 |
18900 ± 5150 |
| PUXL4 | 2440 ± 981 | 1018 ± 31 | 24720 ± 13020 |
19500 ± 2040 |
| PUXL5 | 1140 ± 70 | 590 ± 18 | 14030 ± 350 |
11670 ± 380 |
| PUXL6 | 240 ± 30 | 207 ± 40 | 4750 ± 370 | 5900 ± 890 |
| PUXL7 | 1550 ± 170 | 840 ± 110 | 20810 ± 3250 |
18390 ± 3180 |
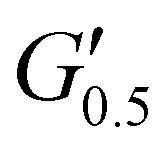
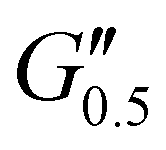
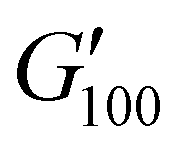
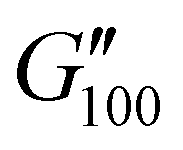
Most of the variants apart from PUXL1 and PUXL2 demonstrated a relative quick increase in their G′ values with increasing oscillation frequency attributing it to physical entanglements between polymer chains.46 With the frequency increase, polymer chains were restrained due to entanglements thus storing more elastic energy. The G′ slopes met Chu's criteria for PSAs with an optimum combination of tack, shear and peel properties.11 Interestingly, in variants PUXL3, PUXL4 and PUXL5, the G′ and G′′ values did not cross over but instead appeared close or parallel at mid frequencies with their loss factors approaching unity, Fig. 5(c) and (d). This indicated no transition from elastic to viscous materials with good cohesion strength at the debonding frequency as 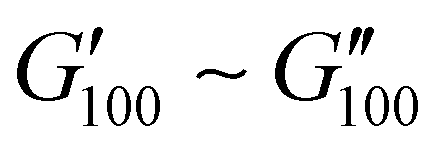 . In contrast, PUXL6 demonstrated an early crossover point at ∼1.0 rad s−1 indicating early cohesion failure at high frequencies.
. In contrast, PUXL6 demonstrated an early crossover point at ∼1.0 rad s−1 indicating early cohesion failure at high frequencies.
The descending rheological trends observed possibly arise from the synergistic effect of higher molecular weight polymer chains (formed by increasing IPDI content) and the subsequent formation of urea groups. Longer polymer chains lead to higher molecular weight between the crosslinks lowering the crosslink density which translates to lower G′, optimum wetting and higher adhesion.42 In parallel, the increase in the number of urea groups between crosslinking points might also be a contributing factor to the increased adhesion due to increased hydrogen bonding. In parallel, the increase in the number of urea groups between crosslinking points might also be a contributing factor to the increased adhesion due to increased hydrogen bonding.
The PSAs behaviour was further investigated using the Chang's classification windows for PSA's.43 The viscoelastic windows of Chang use the rheological properties (G′, G′′) of adhesives at ω = 0.01 and 100 rad s−1 as coordinates to categorise them based on their operating regions. Thus, further rheological characterisation was conducted to attain the values of G′ and G′′ at ω = 0.01 rad s−1, Table S2 (ESI†) and the Chang's viscoelastic windows for the PUXL adhesives given in Fig. 6. All variants exhibited 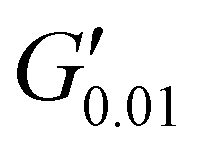 values lower than the requirements of the Dahlquist criterion (
values lower than the requirements of the Dahlquist criterion (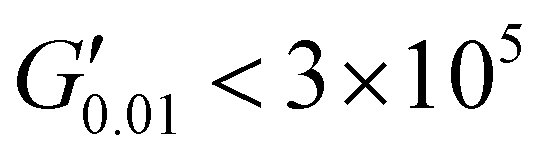 ) for an efficient contact thus anticipating a good tack.44 Generally, a significant shift towards the lower left part quadrant was observed as the IPDI content increased associated with better contact efficiency and low dissipation characteristics of removable PSAs. PUXL1 was in the top part of this window linked to lower tack, but higher shear strength as indicated by the small size and high base of the window.40,45 Similarly, PUXL2 demonstrated high cohesion strength with a slightly higher tack than PUXL1. Based on Chang's work, the range of G′ and G′′ for most commercial PSAs falls between 103 to 106 Pa. In our case, the PUXL2-7 variants exceeded the limits of the bottom left quadrant demonstrating superior bonding properties. PUXL3-5 showed an increasing trend in adhesion with similar cohesion strengths while PUXL6 had the highest adhesion but lowest cohesion strength. Finally, PUXL7 exhibited properties close to that of PUXL4 as evidenced by their overlapping windows.
) for an efficient contact thus anticipating a good tack.44 Generally, a significant shift towards the lower left part quadrant was observed as the IPDI content increased associated with better contact efficiency and low dissipation characteristics of removable PSAs. PUXL1 was in the top part of this window linked to lower tack, but higher shear strength as indicated by the small size and high base of the window.40,45 Similarly, PUXL2 demonstrated high cohesion strength with a slightly higher tack than PUXL1. Based on Chang's work, the range of G′ and G′′ for most commercial PSAs falls between 103 to 106 Pa. In our case, the PUXL2-7 variants exceeded the limits of the bottom left quadrant demonstrating superior bonding properties. PUXL3-5 showed an increasing trend in adhesion with similar cohesion strengths while PUXL6 had the highest adhesion but lowest cohesion strength. Finally, PUXL7 exhibited properties close to that of PUXL4 as evidenced by their overlapping windows.
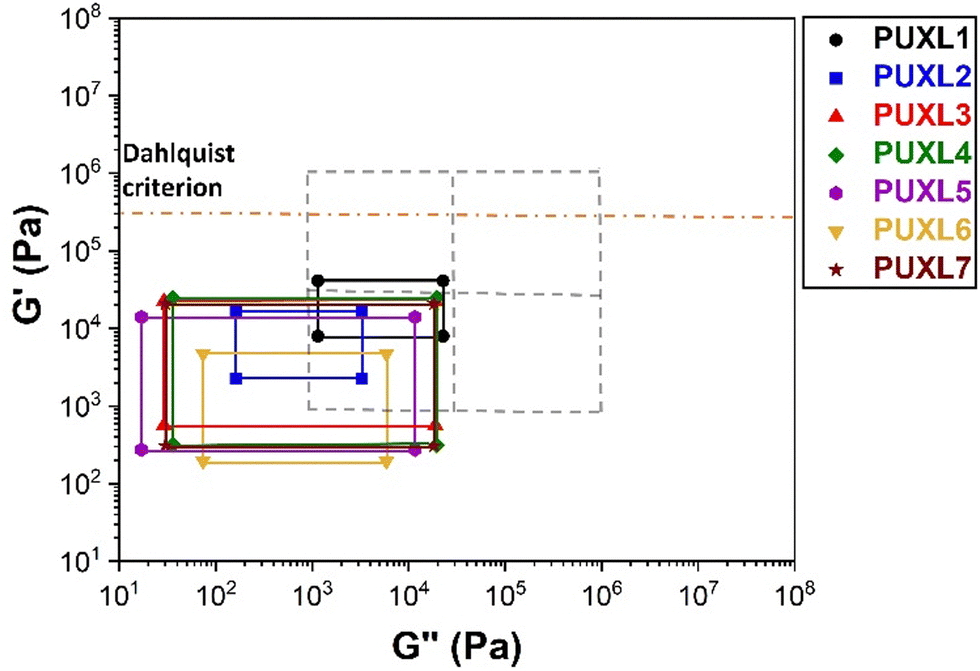 | ||
| Fig. 6 Chang's viscoelastic windows of all PUXL variants at 25 °C with the dotted lines showing Chang's four quadrants found for commercial adhesives and the yellow dashed line the Dahlquist criterion. | ||
Adhesion, tack and cold flow properties
The peel strength and tack properties were determined by performing 90° peel and rolling ball tack tests. The peel strength was evaluated using stainless steel as the substrate, where notable trends were observed (Fig. 7(a)). More specifically, as the molar ratio of IPDI increased, the peel strength values exhibited an upward trend with PUXL1 demonstrating a low value at 0.70 ± 016 N/25 mm and PUXL6 a ∼9 times higher value standing at 6.25 ± 0.22 N/25 mm. Maximum force results are also provided in Fig. S18, ESI.† The outcome aligned with the rheological data, underscoring the increase in adhesion while also justifying the peel strength for PUXL7 (5.19 ± 0.30 N/25 mm) which was in between the properties of PUXL4 and PUXL5. The adhesive strength of four different commercial transdermal patches were also evaluated in comparison to the PUXL adhesives, revealing lower bonding capabilities to the substrate. Among them, Salonpas® exhibited the weakest adhesion (0.56 ± 0.06 N/25 mm) while the other products showcased adhesive properties same to those of the PUXL2 variant. Results suggest that the newly synthesised PSAs had superior bonding strength compared to some commercial products.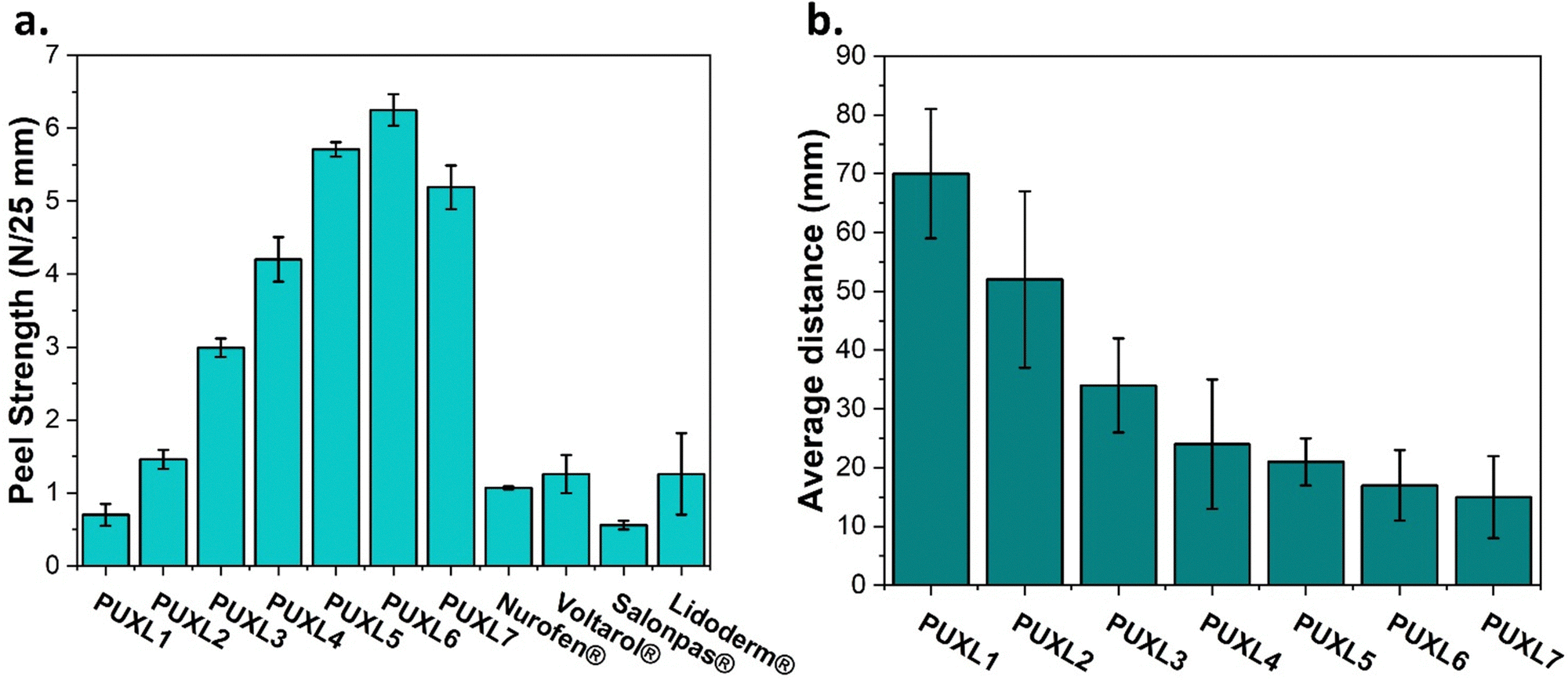 | ||
| Fig. 7 (a) 90° peel strength results and (b) rolling ball tack test results expressed as the average distance the stainless-steel ball travelled in the surface of the adhesives. Data given as mean ± SD of the average of at least n = 3 samples. | ||
A rolling ball tack test demonstrated the ability of the adhesives to quickly adhere onto a surface which was expressed as the distance travelled by the ball after leaving the ramp.47 A reduction in the distance travelled indicated an increase in the materials tackiness. Based on the bar chart of Fig. 7(b), adhesives with a greater IPDI content demonstrated higher tackiness following same trends as the adhesion.
Finally, more insight on the cold flow resistance of the adhesives was attained from cold flow experiments on the cured materials at three different temperatures: ambient temperature, 32 and 40 °C. Each adhesive disc was applied on a glass slide and its diameter was measured at t = 0 and after 72 hr to quantitate the phenomenon as percentage difference cold flow (%CF). As shown in Table S3, none of the PUXL adhesives exhibited strong cold flow phenomena at any of the examined temperatures which may be a result of the formation of a crosslinked network which restricted flow. Interestingly, although the PUXL6 variant showed low G′ values with high peel strength and tack, crosslinking was enough to inhibit cold flow thus ensuring its performance.
Conclusion
In summary, a new series of moisture-curable silylated poly(ether-urea) PSA crosslinkers (PUXL) were successfully synthesised. This was achieved through a two-step process involving the step-growth polymerisation of Jeffamine® D-4000 with IPDI and the subsequent modification with an alkoxysilane to introduce moisture-sensitive properties. The proposed synthetic approach for the PUXL crosslinkers is versatile without requiring the addition of catalysts or involving tedious purifications making it appealing for commercial scale-up. The formed crosslinkers were able to form crosslinked PSAs after addition of a titanium catalyst and exposure to humidity at elevated temperature.The adhesion and viscoelastic properties of the attained adhesives could be fine-tuned by adjusting the IPDI content. More specifically, an increase in the –NCO/–NH2 ratio led to variants with a higher adhesion and a lower modulus as analysed by rheology, thus allowing the selection of a range of variants depending on a particular application. The adhesion and tackiness appeared to be unrelated to their thermal properties which remained constant and equivalent with those of the free polyetheramine. It was likely that the differences in adhesion were influenced by factors such as the average molecular weight of the polymer chains, the crosslink density of the final cured materials and the existence of urea bonds.
According to Chang's viscoelastic windows, all the synthesised PUXL adhesive variants fell within the category of removable PSAs suggesting their potential as adhesives for use in drug-in-adhesive type of transdermal patches without requiring addition of any fillers or tackifier resins. This was further reinforced from the absence of cold flow effects which is critical in achieving drug dosage requirements while preventing medication errors. It should be highlighted that the 90° peel test results demonstrated superior adhesion properties compared to some commercial transdermal patches suggesting the potential to address the adhesion failure of some existing products. This could be a significant benefit to extend the wearability times on skin offering a prolonged drug release delivery.
In another study, the PUXL variant patches were analysed using Terahertz (THz) spectroscopy to assess their impact on skin hydration levels.48–50 Patches with nonpermeable backing materials seemed to hydrate the skin more compared to permeable woven ones in a hydration event that starts the first 30 min of patch application and lasts for 24 h. Finally, preliminary wearability events using PUXL placebo formulations on human skin have also been conducted with most of the variants demonstrating acceptable overall performance.
Author contributions
DMH and GN contributed to the investigation, supervision conceptualization and project administration. SE, GN, AR and YL contributed to the investigation and methodology. SE was responsible for writing the manuscript.Conflicts of interest
Spyridon Efstathiou, Gabit Nurumbetov, Andrew Ross, Yongguang Li are employees of Medherant and David M. Haddleton is CSO Founder and shareholder of Medherant Ltd, the University of Warwick is a shareholder of Medherant Ltd.Acknowledgements
We would like to thank the Warwick Polymer Characterisation Research and Technology Platform (RTP) for the provided characterisation equipment funded in part by EPSRC EP/V036211/1 and EP/V007688/1 and more specifically Dr Daniel Lester, Dr James S. Town and Dr Arkadios Marathianos for their help and support. We would also like to thank Dr Despina Coursari for her help with thermal characterisation, Dr Lucas Al-Shok for conducting the MALDI experiments and Mr Yanpu Yao for rheology. We are also grateful to the University of Warwick, the EPSRC Centre of Doctoral Training in Molecular Analytical Science and the Electron Microscopy RTP.Notes and references
-
Adhesives Technology Handbook, ed. S. Ebnesajjad, William Andrew Publishing, Norwich, NY, 2009, vol. 2, ch. 1, pp.1–19 Search PubMed
.
- S. Mapari, S. Mestry and S. T. Mhaske, Polym. Bull., 2021, 78, 4075–4108 CrossRef CAS
- B.-J. Kim, S. Kim, S.-E. Kim, H.-J. Kim and S.-D. Kim, J. Adhes. Sci. Technol., 2007, 21, 109–123 CrossRef CAS
- J. L. Daristotle, M. Erdi, L. W. Lau, S. T. Zaki, P. Srinivasan, M. Balabhadrapatruni, O. B. Ayyub, A. D. Sandler and P. Kofinas, ACS Biomater. Sci. Eng., 2021, 7, 3908–3916 CrossRef CAS PubMed
- K. Arunprasert, C. Pornpitchanarong, T. Rojanarata, T. Ngawhirunpat, P. Opanasopit, P. Aumklad and P. Patrojanasophon, Pharmaceutics, 2021, 13, 789 CrossRef CAS PubMed
- I. Márquez, N. Paredes, F. Alarcia and J. Velasco, Polymer, 2021, 13, 2627 Search PubMed
- S. V. Pradeep, B. Kandasubramanian and S. Sidharth, J. Adhes., 2023, 99, 2145–2166 CrossRef CAS
- I. Khan, B. T. Poh and K. E. Lee, J. Polym. Environ., 2013, 21, 833–849 CrossRef CAS
- E. Sato, K. Yamanishi, T. Inui, H. Horibe and A. Matsumoto, Polymer, 2015, 64, 260–267 CrossRef CAS
- G. Stîngă, A. Băran, A. Iovescu, L. Aricov and D.-F. Anghel, J. Mol. Liq., 2019, 273, 125–133 CrossRef
- H.-M. Wolff, I. Irsan and K. Dodou, Pharm. Res., 2014, 31, 2186–2202 CrossRef CAS PubMed
- J. Cheng, J. Shang, S. Yang, J. Dou, X. Shi and X. Jiang, Adv. Funct. Mater., 2022, 32, 2200444 CrossRef CAS
- M. Paranjape, J. Garra, S. Brida, T. Schneider, R. White and J. F. Currie, Sens. Actuators, A, 2003, 104, 195–204 CrossRef CAS
- S. Lin, L. Durfee, R. Ekeland, J. McVie and G. Schalau, J. Adhes. Sci. Technol., 2007, 21, 605–623 CrossRef CAS
- R. Gogoi, M. Alam and R. Khandal, Int. J. Basic Appl. Sci., 2014, 3, 118–123 Search PubMed
- M. Fuensanta, M. Vallino and M. Martín, Polymer, 2019, 11, 1608 CAS
- X. Chen, W. Liu, Y. Zhao, L. Jiang, H. Xu and X. Yang, Drug Dev. Commun., 2009, 35, 704–711 CAS
- M. Fuensanta and J. Martin-Martinez, Front. Mech. Eng., 2020, 6, 1–10 CrossRef
- H. Yang, G. Du, Z. Li, X. Ran, X. Zhou, T. Li, W. Gao, J. Li, H. Lei and L. Yang, ACS Appl. Polym. Mater., 2021, 3, 1638–1651 CrossRef CAS
- A. Sánchez-Ferrer, D. Rogez and P. Martinoty, Macromol. Chem. Phys., 2010, 211, 1712–1721 CrossRef
- W. Liu, C. Fang, F. Chen and X. Qiu, ChemSusChem, 2020, 13, 4691–4701 CrossRef CAS PubMed
- M. Fuensanta, A. Khoshnood, L. Rodríguez and M. Martín, Mater., 2020, 13, 627 CrossRef CAS PubMed
- S. Lobo, S. Sachdeva and T. Goswami, Ther. Delivery, 2016, 7, 33–48 CrossRef CAS PubMed
- H. S. Tan and W. R. Pfister, Pharm. Sci. Technol. Today, 1999, 2, 60–69 CrossRef CAS PubMed
- E. L. Tombs, V. Nikolaou, G. Nurumbetov and D. M. Haddleton, J. Pharm. Innov., 2018, 13, 48–57 CrossRef PubMed
- Y. S. R. Krishnaiah, Y. Yang, R. L. Hunt and M. A. Khan, Int. J. Pharm., 2014, 477, 73–80 CrossRef CAS PubMed
- ASTM International, Standard Test Methods for Viscosity of Adhesives, ASTM D1084-97, ASTM International, 1997.
- ASTM International, Standard Test Method For Peel Adhesion Of Pressure-Sensitive Tape, ASTM D3330/D3330M-04, ASTM International, 2018.
- ASTM International, Standard Test Method for Tack of Pressure-Sensitive Adhesives by Rolling Ball, ASTM D3121-17, ASTM International, 2017.
-
K. L. Mittal, in Tribology Series, ed. J. M. Georges, Elsevier, 1981, vol. 7, pp.153–165 Search PubMed
-
S. Alhassan, D. Schiraldi, S. Qutubuddin, T. Agag and H. Ishida, in Handbook of Benzoxazine Resins, ed. H. Ishida and T. Agag, Elsevier, Amsterdam, 2011, ch. 15, pp.309–318 Search PubMed
- T. Agag, S. Geiger, S. M. Alhassan, S. Qutubuddin and H. Ishida, Macromol., 2010, 43, 7122–7127 CrossRef CAS
- HUNTSMAN Jeffamine Polyetheramine, https://www.huntsman.com/docs/Documents/JEFFAMINE%C2%AE%20Polyetheramines%20Formulation%20Guide.pdf, (accessed October 2023).
- A. Sánchez-Ferrer, V. Soprunyuk, M. Engelhardt, R. Stehle, H. A. Gilg, W. Schranz and K. Richter, ACS Appl. Polym. Mater., 2021, 3, 4070–4078 CrossRef
-
H. Fałtynowicz, H. Janik, J. Kucinska-Lipka and M. Sienkiewicz, in Handbook of Thermoset Plastics, ed. H. Dodiuk, William Andrew Publishing, Boston, 4th edn, 2022, pp.231–262 Search PubMed
- N. Akram, K. M. Zia, M. Saeed, M. Usman and W. G. Khan, J. Appl. Polym. Sci., 2019, 136, 47124 CrossRef
- N. Riehle, S. Thude, A. Kandelbauer, G. E. M. Tovar and G. Lorenz, JoVE, 2019, 58590 Search PubMed
-
L. Zander, C. Kunze and J. Klein, US Pat., 8501903B2, 2013 Search PubMed
- M. Masuelli, J. Polym. Biopolym. Phys. Chem., 2014, 2, 37–43 CAS
- F. Weij, Die Makr. Chem., 2003, 181, 2541–2548 CrossRef
- C. M. Kirschner and K. S. Anseth, Acta Mater., 2013, 61, 931–944 CrossRef CAS PubMed
- C. Hamciuc, G. Lisa, D. Serbezeanu, L. M. Grădinaru, M. Asăndulesa, N. Tudorachi and T. Vlad-Bubulac, Polymer, 2022, 14, 4715 CAS
- K. H. Park, D. Y. Lee, S. H. Yoon, S. H. Kim, M. S. Han, S. Jeon, Y. Kim, Y. K. Lim, D. H. Hwang, S. H. Jung and B. Lim, Polymer, 2022, 14, 3963 CAS
- H. Stutz, K. H. Illers and J. Mertes, J. Polym. Sci., Part B: Polym. Phys., 1990, 28, 1483–1498 CrossRef CAS
-
K. S. Gerald II, B. Alexis, O. H. Robert, S. N. Linda and T. Xavier, in Applied Adhesive Bonding in Science and Technology, ed. Ö. Halil, IntechOpen, Rijeka, 2017, ch. 6, pp.94–111 Search PubMed
-
M. G. Anne, B. W. Nicholas and M. G. Lindsey, in Rheology, ed. V. Juan De, IntechOpen, Rijeka, 2012, ch. 3, pp.59–80 Search PubMed
- M. F. Tse, J. Adhes., 1999, 70, 95–118 CrossRef CAS
- X. E. R. Barker, G. Costa, R. I. Stantchev, A. I. Hernandez-Serrano, G. Nurumbetov, D. M. Haddleton and E. Pickwell-MacPherson, IEEE Trans. Terahertz Sci. Technol., 2023, 13, 503–510 CAS
- X. Ding, G. Costa, A. I. Hernandez-Serrano, R. I. Stantchev, G. Nurumbetov, D. M. Haddleton and E. Pickwell-MacPherson, Biomed. Opt. Express, 2023, 14, 1146–1158 CrossRef CAS PubMed
- H. Lindley-Hatcher, J. Wang, A. I. Hernandez-Serrano, J. Hardwicke, G. Nurumbetov, D. M. Haddleton and E. Pickwell-MacPherson, Pharmaceutics, 2021, 13, 2052 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available: Includes supporting data and characterisation. See DOI: https://doi.org/10.1039/d3ma01104f |
| This journal is © The Royal Society of Chemistry 2024 |