DOI:
10.1039/D3MA01179H
(Paper)
Mater. Adv., 2024,
5, 4848-4856
Synthesis and characterization of N,N′-bis(2-thienylmethylene)-1,X-diaminobenzene isomers (X = 2, 3, 4) and their metal complexes†
Received
28th December 2023
, Accepted 22nd April 2024
First published on 23rd April 2024
Abstract
The synthesis of N,N′-bis(2-thienylmethylene)-1,2-diaminobenzene, N,N′-bis(2-thienylmethylene)-1,3-diaminobenzene, and N,N′-bis(2-thienylmethylene)-1,4-diaminobenzene, three isomers with the molecular formula of (C16H12N2S2) and their metal complexation with Co(II), Fe(III), and Ni(II) is reported in this study. The formation of the ligands and their metal complexes, along with their physicochemical behaviors, are confirmed and characterized using electrochemical and spectroscopic techniques. The coordination chemistry and stoichiometry of the ligand–metal complexes are investigated using UV-vis absorption spectral titration, which reveals that the ligand-to-metal ratio varies from 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 to 4:1 in different cases. The calculated binding constants based on these spectroscopic data, ranging between 103 M−1 and 108 M−1, indicate the isomers' high affinity for the metal ions. Experimental data show that the nitrogen atoms from the azomethine linkage play an important role in coordinating with the metal ions. This observation supports findings reported in the literature for some studied Schiff base ligands.
:1 to 4:1 in different cases. The calculated binding constants based on these spectroscopic data, ranging between 103 M−1 and 108 M−1, indicate the isomers' high affinity for the metal ions. Experimental data show that the nitrogen atoms from the azomethine linkage play an important role in coordinating with the metal ions. This observation supports findings reported in the literature for some studied Schiff base ligands.
1. Introduction
Schiff base reactions are defined by the condensation of an aldehyde or ketone with a primary amine, discovered first by Hugo Schiff in 1864.1 In these reactions, the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O group of the carbonyl compound is typically replaced by a CN–R group through a nucleophilic addition reaction, where R can be aryl or alkyl groups. In this mechanism, the elimination of water during the acid-catalyzed reversible reaction determines the reaction rate, and Schiff bases typically form under neutral conditions,2 but the process is often accelerated by heating or adding an acid catalyst.3 The reaction yields an imine (azomethine) and water as a by-product. In comparison, Schiff bases with aryl substituents exhibit significantly greater stability and ease of synthesis than those with alkyl substituents. Furthermore, Schiff base compounds from aliphatic aldehydes are usually less stable and can easily polymerize, while aromatic aldehydes lead to more stable compounds because of conjugation.3 Functionally substituted Schiff bases, containing additional donor groups, are among the most important heteropolydentate ligands, capable of forming complexes ranging from mononuclear to polynuclear with metal ions.4 Most resulting imines use nitrogen lone pair electrons to bind to metal ions.5 These ligands interact with almost all metal ions to form stable complexes, where ligands are Lewis bases and donate electrons to metal ions, which are Lewis acids. These complexes play a significant role in various fields, including organic and inorganic chemistry, biochemistry,6 and supramolecular chemistry.7 At high temperatures and in the presence of moisture, many Schiff base complexes exhibit catalytic activity which is entirely dependent on metal ions, the type of ligands, and the coordination sites. For example, Schiff base complexes of Co(II)8 showed a profound catalytic effect in the ring-opening of large cycloalkanes, which is a usually complicated process. Cobalt Schiff base complexes have drug properties like antiviral drugs, and chloro(pyridine)cobaloxime is also a well-known model of coenzyme vitamin B12.9 The Ni(II) and Cu(II) complexes in the alkylation reaction of enolates exhibited high activity and stereoselectivity.10–12 For cyclic ester polymerization, aluminum complexes have a high catalytic conversion.13,14 Also, as an example, the Fe(III) of pyridine bis(imine) ligands had significant activity and a high yield in the polymerization of ethylene.15 Schiff bases ligands and their metal complexes have been discovered to possess biological activities,16 such as anticonvulsant, antibacterial,17,18 antimicrobial,19 anti-inflammatory,20 antimalarial,16 anti-tumor,21 anti-HIV,22 anticancer,23 antifungal properties,24 and enzyme immobilization ability.25 The presence of donor atoms, azomethine linkages, and conjugated double bonds in the structure of these organic compounds contributes to their biological functions and industrial applications. Additionally, these compounds are well-known for their role as metal corrosion inhibitors.26 π-conjugated Schiff bases are interesting materials because of their straightforward synthesis, environmental stability, and electrical and optical properties, which have made them useful in electrochemical and non-linear optical devices,27 actuators,28 supercapacitors,29 solar cells,30 anti-corrosion coatings,31 sensors,32 electrochromic devices,33 and batteries.34 They can even be used in Dielse–Alder reactions.35 Last but not least are applications of Schiff base complexes with transition metals as sensing material for selective or non-selective (e-tongue) chemical or electrochemical sensors.36–41 In this study, we have reported Schiff base condensation of o-pheneylediamne, m-pheneylediamne, and p-phenylenediamne with 2-thiophenecarboxaldehyde. These reactions have been characterized using electrochemical techniques such as cyclic voltammetry and spectroscopic techniques like, Fourier-transform infrared spectroscopy (FT-IR), Raman spectroscopy, proton nuclear magnetic resonance (1H-NMR) spectroscopy, gas chromatography–mass spectrometry (GC-MS), and UV-visible (UV-vis) spectrophotometry. Afterward, we studied the complexation of each isomer with transition metals, including Co2+, Fe3+, and Ni2+, and calculated the binding constants.
O group of the carbonyl compound is typically replaced by a CN–R group through a nucleophilic addition reaction, where R can be aryl or alkyl groups. In this mechanism, the elimination of water during the acid-catalyzed reversible reaction determines the reaction rate, and Schiff bases typically form under neutral conditions,2 but the process is often accelerated by heating or adding an acid catalyst.3 The reaction yields an imine (azomethine) and water as a by-product. In comparison, Schiff bases with aryl substituents exhibit significantly greater stability and ease of synthesis than those with alkyl substituents. Furthermore, Schiff base compounds from aliphatic aldehydes are usually less stable and can easily polymerize, while aromatic aldehydes lead to more stable compounds because of conjugation.3 Functionally substituted Schiff bases, containing additional donor groups, are among the most important heteropolydentate ligands, capable of forming complexes ranging from mononuclear to polynuclear with metal ions.4 Most resulting imines use nitrogen lone pair electrons to bind to metal ions.5 These ligands interact with almost all metal ions to form stable complexes, where ligands are Lewis bases and donate electrons to metal ions, which are Lewis acids. These complexes play a significant role in various fields, including organic and inorganic chemistry, biochemistry,6 and supramolecular chemistry.7 At high temperatures and in the presence of moisture, many Schiff base complexes exhibit catalytic activity which is entirely dependent on metal ions, the type of ligands, and the coordination sites. For example, Schiff base complexes of Co(II)8 showed a profound catalytic effect in the ring-opening of large cycloalkanes, which is a usually complicated process. Cobalt Schiff base complexes have drug properties like antiviral drugs, and chloro(pyridine)cobaloxime is also a well-known model of coenzyme vitamin B12.9 The Ni(II) and Cu(II) complexes in the alkylation reaction of enolates exhibited high activity and stereoselectivity.10–12 For cyclic ester polymerization, aluminum complexes have a high catalytic conversion.13,14 Also, as an example, the Fe(III) of pyridine bis(imine) ligands had significant activity and a high yield in the polymerization of ethylene.15 Schiff bases ligands and their metal complexes have been discovered to possess biological activities,16 such as anticonvulsant, antibacterial,17,18 antimicrobial,19 anti-inflammatory,20 antimalarial,16 anti-tumor,21 anti-HIV,22 anticancer,23 antifungal properties,24 and enzyme immobilization ability.25 The presence of donor atoms, azomethine linkages, and conjugated double bonds in the structure of these organic compounds contributes to their biological functions and industrial applications. Additionally, these compounds are well-known for their role as metal corrosion inhibitors.26 π-conjugated Schiff bases are interesting materials because of their straightforward synthesis, environmental stability, and electrical and optical properties, which have made them useful in electrochemical and non-linear optical devices,27 actuators,28 supercapacitors,29 solar cells,30 anti-corrosion coatings,31 sensors,32 electrochromic devices,33 and batteries.34 They can even be used in Dielse–Alder reactions.35 Last but not least are applications of Schiff base complexes with transition metals as sensing material for selective or non-selective (e-tongue) chemical or electrochemical sensors.36–41 In this study, we have reported Schiff base condensation of o-pheneylediamne, m-pheneylediamne, and p-phenylenediamne with 2-thiophenecarboxaldehyde. These reactions have been characterized using electrochemical techniques such as cyclic voltammetry and spectroscopic techniques like, Fourier-transform infrared spectroscopy (FT-IR), Raman spectroscopy, proton nuclear magnetic resonance (1H-NMR) spectroscopy, gas chromatography–mass spectrometry (GC-MS), and UV-visible (UV-vis) spectrophotometry. Afterward, we studied the complexation of each isomer with transition metals, including Co2+, Fe3+, and Ni2+, and calculated the binding constants.
2. Experimental
2.1. Materials and apparatus
In order to synthesize N,N′-bis(2-thienylmethylene)-1,X-diaminobenzene isomers (X = 2, 3, 4), henceforth referred to as BTMD in this paper, 2-thiophenecarboxaldehyde (C5H4OS ≥ 98%), o-phenylenediamine (C6H8N2 ≥ 99.5%), m-phenylenediamine (C6H8N2 ≥ 99%), p-phenylenediamine (C6H8N2 ≥ 99%), and ethanol absolute (C2H6O ≥ 99%) as solvent were obtained from Sigma-Aldrich and Alfa Aesar and used as supplied without further purification. The molar mass of the ligands was measured using GC-MS (Agilent 7890A gas chromatograph coupled with Agilent 5975C MS detector). FT-IR and Raman spectra were recorded using Thermo Fisher Scientific Nicolet iS50 spectrophotometer at room temperature. 1HNMR spectra of the ligands and the metal complexes were recorded in DMSO-d6 using a Bruker 500 MHz spectrometer. UV-vis spectra of all prepared materials were collected on a Shimadzu TCC-240A spectrometer at room temperature in the range of 210–700 nm. PHI 5000 VersaProbe (ULVAC-PHI Inc., Hagisono, Chigasaki, Kanagawa, Japan) spectrometer was used to conduct XPS measurements under the following conditions: monochromatic Al Kα radiation (hν = 1486.6 eV), an X-ray source operating at 25 W, 15 kV, 100 μm spot, pass energy of 23.5 eV, energy step of 0.1 eV. CasaXPS software was used to analyze the obtained XPS spectra using the set of the sensitivity factors native for the hardware. Shirley background and Gaussian–Lorentzian peak shape were used for deconvolution of all spectra. Cyclic voltammetry (CV) was performed using a three-electrode system consisting of a bare platinum (Pt) electrode (2 mm diameter, ItalSens), a silver/silver chloride (Ag/AgCl), and a Pt wire used as working, reference, and counter electrodes, respectively, using an Ivium stat electrochemical workstation (Ivium Technologies, Ivium Stat, Netherlands). The working electrode was polished with alumina, 1 μm, 0.3 μm, and 0.05 μm and rinsed thoroughly with distilled water before each experiment. The electrolytic medium consisted of 0.1 M tetrabutylammonium perchlorate in acetonitrile. The cyclic voltammograms were measured with a scan rate of 5 mV s−1.
2.2. Synthesis of Schiff-base ligands
Three Schiff-base compounds, N,N′-bis(2-thienylmethylene)-1,2-diaminobenzene (o-BTMD), N,N′-bis(2-thienylmethylene)-1,3-diaminobenzene (m-BTMD), and N,N′-bis(2-thienylmethylene)-1,4-diaminobenzene (p-BTMD) with the formula of (C16H12N2S2) were synthesized by reaction of 2-thiophenecarboxaldehyde with ortho-, meta- and para-phenylenediamine respectively with the molar ratio of 2:1. 2-Thiophenecarboxaldehyde (0.02 mol, 1.869 ml) was added to a stirred ethanol solution of phenylenediamine isomers (0.01 mol, 1.0814 g). The reaction mixture was stirred vigorously (∼700 rpm) with a magnetic stirrer for about 3 h at room temperature (RT∼25 °C).42 Two or three drops of acetic acid may serve as a catalyst to enhance the reaction rate and precipitate formation. The completion of the reaction was determined using thin-layer chromatography (TLC). The resulting light-yellow colored precipitate was collected by filtration through a Buchner funnel, washed with cold ethanol, and dried in a vacuum desiccator. Then, the o-BTMD and m-BTMD products were recrystallized from ethanol and dried at room temperature, resulting in yields of 65% and 50%, respectively. Unlike the other isomers, p-BTMD was obtained without recrystallization process with a yield of 70%. The chemical structures of BTMDs (Fig. S1, ESI†), with a molecular mass of approximately 296 g mol−1, were confirmed by mass spectrometry and NMR and further characterized by XPS, Raman, FT-IR, and UV-vis spectroscopy, and CV. The information related to the characterization of BTMD isomers is summarized here, with figures provided in the ESI.†
2.3. Metal complexes of BTMDs
UV-vis absorption spectroscopy technique and molar ratio method were utilized to monitor the complexation. Nine different complexes were prepared by using three isomers (o-BTMD, m-BTMD, and p-BTMD) synthesized already as ligands (Section 2.2), and three metal ions Co(II) chloride hexahydrate, Fe(III) chloride hexahydrate, and Ni(II) nitrate hexahydrate. For all complexes, stock solutions of the BTMDs in acetonitrile with a concentration of 10−2 M were used. Then, the required concentration for UV-vis titration (10−5 M) was prepared by dilution.
Then, the metal salt solution (0.25 × 10−2 M) was used as a titrant. In the molar ratio method,43 the BTMD concentration was maintained constant while the concentration of the metal ion (with Co(II), Fe(III), and Ni(II)) was varied to cover the range of molar ratios [M]:[ligand] from 0.16 to 0.52. The spectra of all prepared solutions of BTMDs and complexes were recorded within the wavelength range of 200–700 nm. The UV-vis spectrum was recorded after each addition to monitor the progress of the complexation reaction during the titration.
3. Result and discussion
3.1. Spectroscopic studies
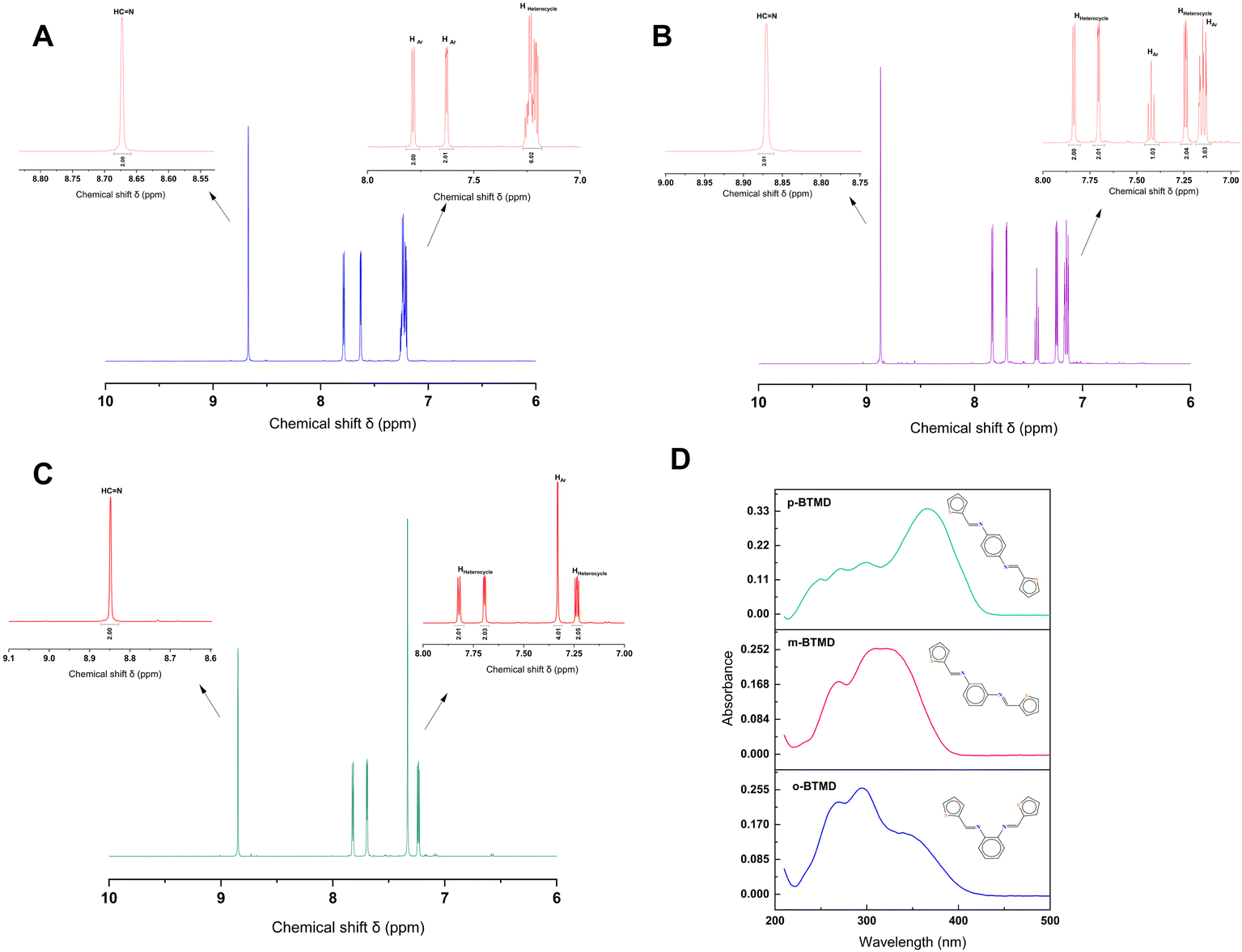
NMR spectroscopy gives valuable information in Schiff base synthesis to confirm the chemical structure of BTMDs, especially about the formation of azomethine linkage, which plays a vital role in the coordination behavior of Schiff base ligands. The 1H-NMR spectra of Schiff base isomers in Fig. 1 show one singlet at 8.5–8.8 ppm attributed to azomethine (–CHN–) protons, downfield doublets, triplets or multiplets attributed to the resonance of aromatic (HAr) and heterocyclic protons in the range of 7–7.8 ppm.42,44–46 According to mass spectra of isomers, ESI-MS (m/z) was calc.: 296.41: found: ∼296 with different retention times of 18.9, 19.9, and 20.1 minutes for o-, m- and p-BTMD, respectively (Fig. S2–S4, ESI†). Further characterizations of BTMDs using FT-IR and Raman spectroscopy (Fig. 2) show a good agreement with proposed chemical structures. Details of bonding analysis are shown in Table S1 (ESI†). In NMR spectra of metal complexes (Fig. S5, ESI†), the azomethine proton peaks of BTMDs with Co(II), Ni(II) slightly shifted, but in complexes with Fe(III), especially in the case of o-BTMD, where changes are more pronounced, proton peaks of azomethine noticeably shifted downfield. In the literature, peak shifting is often attributed to several phenomena, including the formation of metal complexes, configurational changes within complexes that differ from the free ligands, the deshielding of protons due to coordination with metal ions, and the deprotonation of ligands. These interactions can significantly alter the electronic environment around the absorbing species, leading to observable shifts in NMR peaks.47 Significant variations are observed in the behavior of Fe(III) in the ortho- and meta-positions compared to other metal ion complexes. The NMR peaks of heterocyclic protons stayed partially unchanged. This can be attributed to their distance from the metal–ligand binding sites. A trend of line broadening is also seen from the spectrum of the free ligands to the metal complexes. In NMR spectroscopy, when comparing the spectra of a ligand to its metal complexes, line broadening in the metal complexes can often be attributed to the presence of unpaired electrons associated with a paramagnetic metal ion.
 |
| | Fig. 1 (a)–(c) 1H-NMR of (A) o-BTMD, (B) m-BTMD, and (C) p-BTMD in DMSO-d6 and (D) UV-vis spectra of these isomers with the concentration of 10−5 M in acetonitrile. | |
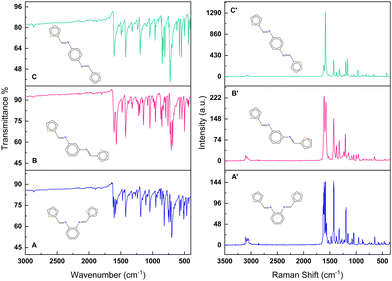 |
| | Fig. 2 FT-IR and Raman spectra of (A) and (A′) o-BTMD, (B) and (B′) m-BTMD, (C) and (C′) p-BTMD. | |
UV-vis absorption spectroscopy was used to study optical properties of BTMDs and their complexes (Fig. 1D and Table 1). According to UV-vis spectra of ligands, o-BTMD showed absorption at 265, 293(λmax), 339 nm,44m-BTMD absorptions happened at 233, 270, 309(λmax), 323 nm and p-BTMD had absorption at 249, 272, 300, and 365 nm (λmax), where λmax is the maximum absorption wavelength.48 In the structure of BTMDs, the presence of aromatic rings and imine groups primarily results in π → π* electronic transitions, which are typically observed in the UV region, around 260–290 nm. Additionally, the ligand possesses lone pairs on nitrogen and sulfur atoms, introducing n → π* transitions that appear at longer wavelengths, approximately in the range of 300–470 nm. When comparing isomers, it is notable that variations in λmax can be observed, which can be attributed to changes in the conjugation length of the isomers. This trend can also be seen in other molecules with ortho, meta, and para substituents.49,50 Regarding the metal complexes of BTMDs, most displayed new peaks with lower absorbance in their UV-vis spectra at wavelengths greater than 360 nm. These peaks can be associated with ligand-to-metal charge transfer (LMCT) transitions51,52 and d–d transition.53,54 Almost all the metal complexes displayed shifts in their UV-vis spectra upon complexation, with some peaks showing a redshift (bathochromic shift) and others a blueshift (hypsochromic shift). An increase in peak intensity, especially for these shifted peaks, can be linked to ligand–metal coordination.55–57
Table 1 Electronic spectral data of Schiff base ligands and their metal complexes
| Compounds |
Absorption bands(nm) |
| Blue/red shift |
New bands |
|
o-BTMD (free ligand) |
265, 293(λmax), 339 |
— |
| [Co(o-BTMD)3]2+ |
261(λmax), 293, 344 |
570, 610, 680 |
| [Fe(o-BTMD)3]3+ |
242(λmax), 315, 360 |
420 |
| [Ni(o-BTMD)3]2+ |
260(λmax), 292, 363 |
— |
|
m-BTMD (free ligand) |
232, 270, 309(λmax), 323 |
— |
| [Fe(m-BTMD)3]3+ |
241(λmax), 310 |
460 |
| [Ni(m-BTMD)4]2+ |
262, 309, 323(λmax) |
— |
|
p-BTMD (free ligand) |
249, 272, 300, 365(λmax) |
— |
| [Co(p-BTMD)3]2+ |
230, 260, 300, 365(λmax) |
570, 616, 680 |
| [Fe(p-BTMD)3]3+ |
241(λmax), 295, 306, 364 |
422 |
| [Ni(p-BTMD)3]2+ |
249, 267, 297, 366(λmax) |
— |
The energy gap between the HOMO and LUMO indicates the molecular chemical stability of compounds, reflecting their chemical hardness or softness.58 The HOMO and LUMO levels, often referred to as the bandgap, can be determined using the Tauc plot method.59 Tauc plot method (eqn (S1) and (S2), ESI†) is frequently employed to calculate both direct and indirect energy gaps of molecules based on their UV-vis spectra. The determination of bandgap values is essential in understanding a compound's intrinsic properties, including the optical absorption characteristics and the electrical conductivity inherent to organic semiconductors, such as Schiff base compounds.56 The bandgap value (Eg) was determined by identifying the point of intersection between the extended linear portion of the (αhυ)n plot and the hυ (photon energy) axis on the abscissa. Fig. S6 and S7 (ESI†) illustrate the plots of (αhυ)2 and (αhυ)0.5versus hυ, respectively, demonstrating the determination of bandgap for all isomers. The (αhυ)2vs. hυ plot reveals that BTMDs possess a direct optical bandgap, with values of 3.14 eV, 3.35 eV, and 3.05 eV for o-BTMD, m-BTMD, and p-BTMD, respectively, as seen in Fig. S6 (ESI†). Additionally, the (αhυ)0.5vs. hυ plot displays a more pronounced linear section for BTMDs, verifying the presence of an indirect optical transition in the isomers. Linear extrapolation to the x-axis yields indirect bandgaps of 2.85 eV, 3.06 eV, and 2.78 eV for o-BTMD, m-BTMD, and p-BTMD, respectively, as shown in Fig. S7 (ESI†). Based on the calculated bandgap values, BTMDs can be classified as organic semiconductors.60,61 The bandgap values of Schiff base complexes, as determined by the Tauc plot method (Fig. S8–S13, ESI†), are presented in Table 2. The data suggests that metal complexation reduces the bandgap in several complexes.62 This reduction in bandgap might be associated with changes in the stability of the Schiff base ligands.
Table 2 Bandgap values for BTMD and their complexes
| Compounds |
Bandgap values |
| Direct (eV) |
Indirect (eV) |
|
o-BTMD (free ligand) |
3.14 |
2.85 |
| [Co(o-BTMD)3]2+ |
3.81 |
2.92 |
| [Fe(o-BTMD)3]3+ |
3.50 |
2.49 |
| [Ni(o-BTMD)3]2+ |
3.81 |
2.64 |
|
m-BTMD (free ligand) |
3.35 |
3.06 |
| [Co(m-BTMD)3]2+ |
3.29 |
3.02 |
| [Fe(m-BTMD)3]3+ |
3.13 |
2.75 |
| [Ni(m-BTMD)4]2+ |
3.34 |
3.05 |
|
p-BTMD (free ligand) |
3.05 |
2.78 |
| [Co(p-BTMD)3]2+ |
2.91 |
2.38 |
| [Fe(p-BTMD)3]3+ |
2.70 |
2.34 |
| [Ni(p-BTMD)3]2+ |
3.01 |
2.80 |
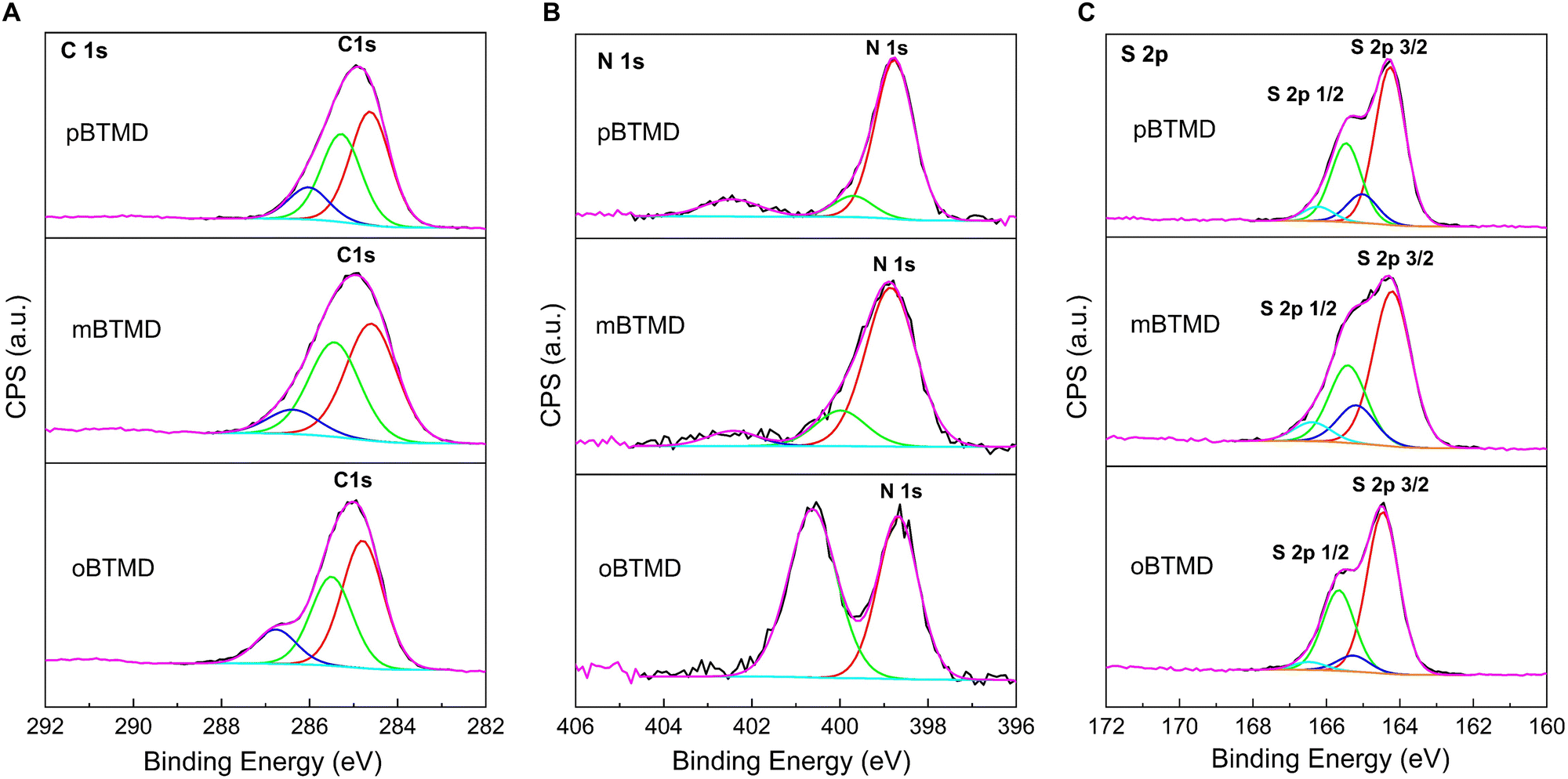
XPS deconvoluted spectra of the BTMD isomers are presented in Fig. 3. Presence of the carbon, nitrogen, and sulfur is discovered. Analysis of the high-resolution C 1s core level spectra shows three component peaks, for instance, at around 284.7, 285.4, and 286.4 eV for m-BTMD molecule. Those could be attributed to CC, C–C/C–H, C–N\C–S carbon bounds in BTMDs;48,63,64 particularly, the peak at 286.8 eV is prominent for o-BTMD. The N 1s core level spectra of the isomers can be deconvoluted into three environments: imine N (399 and 400 eV) and protonated imine N+ (402.4 eW).48,65,66 Protonated imine peak could have been formed in the polymers due to surface oxidation products.67 Due to spin–orbit splitting, the S 2p spectrum appears as a doublet. High-resolution spectra were fitted with 2 S 2p doublets with the S 2p3/2 positions of 164.4 and 165.1 eV with a ratio of 2:1 (S 2p3/2 to S 2p1/2), spin–orbit splitting of 1.2 eV and the same full width at half maximum. Bigger intensity doublet at lower binding energy is ascribed to thiophene-type sulfur.48,68 Second doublet at higher energy could be attributed to an oxidized sulfur species, which means that some sulfur atoms carry a partial positive charge.63
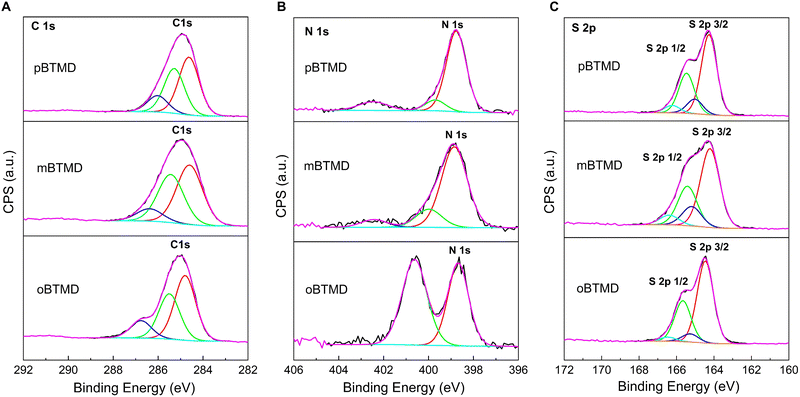 |
| | Fig. 3 High-resolution XPS scan spectra over (A) C 1s, (B) N 1s, and (C) S 2p peaks of the o-, m- and p-BTMD samples. | |
3.1.1. Determination of molar absorption coefficient and binding constant.
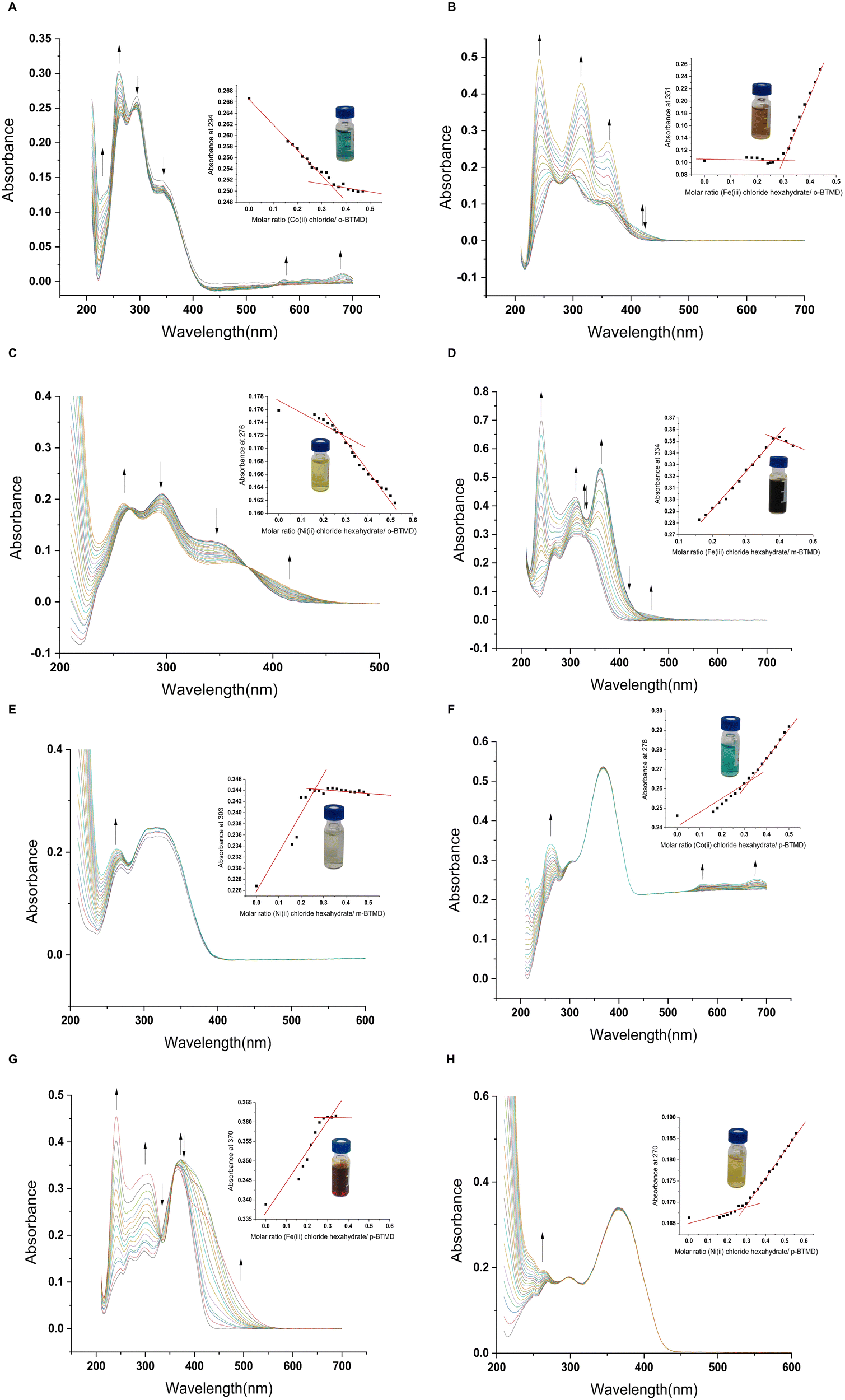
Some methods are commonly used to determine the stoichiometry of metal complexes, such as conductometric titration and spectrophotometric methods, including continuous variation57 and the molar ratio method.69 A UV-vis spectrometer is employed to measure the molar absorption coefficient of compounds at specific wavelengths (Fig. 4 and Table S2, ESI†). The Beer–Lambert law (eqn (1)) allows for the calculation of the molar absorption coefficient based on three key elements: (i) A, which represents the amount of light a compound absorbs; (ii) C, the molarity or concentration of the compound; and (iii) b, the path length of the light in centimeters. A Beer's Law plot is generated by plotting the absorbance values as a function of the differing concentrations. The slope of this line corresponds to the molar absorptivity coefficient multiplied by the light path length.
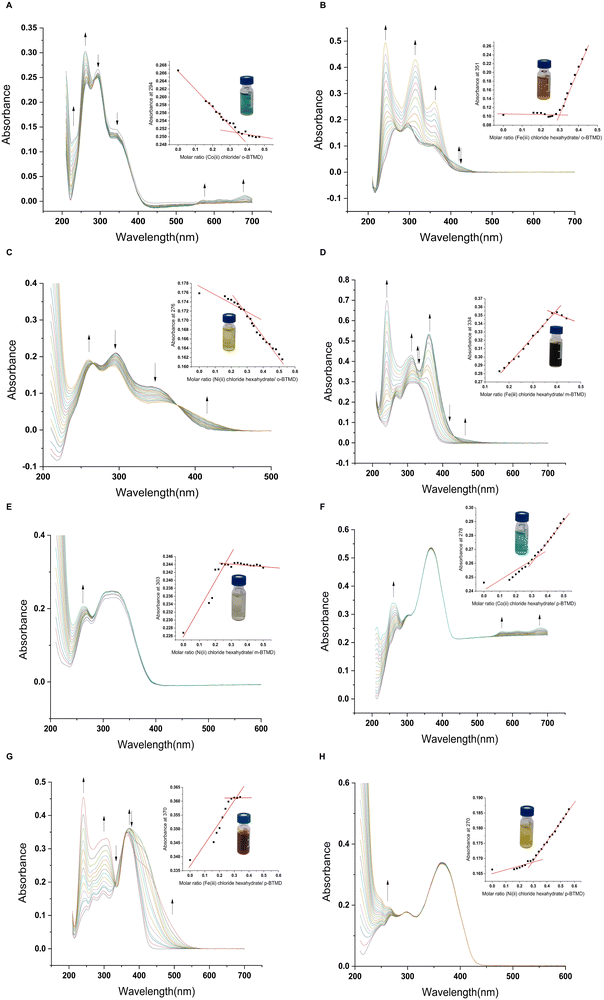 |
| | Fig. 4 UV-vis titration spectra of BTMDs complexes; (A) o-BTMD/Co(II), (B) o-BTMD/Fe(III), (C) o-BTMD/Ni(II), (D) m-BTMD/Fe(III), (E) m-BTMD/Ni(II), (F) p-BTMD/Co(II), (G) p-BTMD/Fe(III), (H) p-BTMD/Ni(II). | |
The Beer–Lambert plots for BTMD isomers are presented in Fig. S14 (ESI†). Subsequently, the concentration of the complex and its binding constant were calculated using the provided formulas (eqn (2) and (3)), respectively.
| | 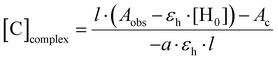 | (2) |
where [C] is the concentration of the complex;
l is the optical path length;
Aobs =
Ac +
Ah where
Ac is the absorbance of the complex while
Ah is the absorbance of the left host;
εh is the host molar absorbance coefficient; [H
0] is the initial (total) concentration of the host, and
a is the stoichiometry of the host. Here, [H
0] = 10
−5, [G
0] = 0.5 × 10
−5| | 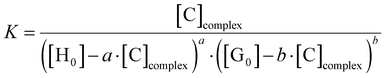 | (3) |
The binding constant values were calculated for ligands, Co(II), Fe(III) and Ni(II) complexes using the equations above. The results revealed that the binding constants of the metal complexes with Schiff base ligands have the following order Co(II) < Ni(II) < Fe(III) in o-BTMD complexes, Fe(III) < Ni(II) for m-BTMD complexes and Fe(III) < Co(II) < Ni(II) in p-BTMD complexes.57 Greater K values indicate stronger binding of a complex.70
3.2. Electrochemical studies
Cyclic voltammetry was conducted over a potential range of 0 to +1.5 V (vs. Ag/AgCl, Fig. S15, ESI†). The observed oxidation potentials for the various BTMD isomers are listed in Table 3. The order of oxidation for BTMD isomers is as follows, from the most resistant to oxidation to the least: m-BTMD, o-BTMD, then p-BTMD. The trend in oxidation for the BTMD isomers corresponds with the UV-vis findings and the bandgap values detailed in Tables 1 and 2. The behavior of m-BTMD may be attributed to the shortest conjugation length of the meta-positioned isomer.71
Table 3 Voltametric data of ligands and their complexes
| Compounds |
E
pa (V) |
E
pc (V) |
|
o-BTMD |
0.9, 1.27, 1.46 |
1.3 |
| [Co(o-BTMD)3]2+ |
1.29 |
— |
| [Fe(o-BTMD)3]3+ |
1.31 |
— |
| [Ni(o-BTMD)3]2+ |
1.27, 1.47 |
— |
|
m-BTMD |
0.96, 1.48, 1.67 |
— |
| [Co(m-BTMD)3]2+ |
0.91, 1.43 |
— |
| [Fe(m-BTMD)3]3+ |
0.68, 0.9, 1.2 |
1.1 |
| [Ni(m-BTMD)4]2+ |
0.9, 1.45 |
— |
|
p-BTMD |
0.78, 1.28, 1.4 |
— |
| [Co(p-BTMD)3]2+ |
0.68, 1.22 |
— |
| [Fe(p-BTMD)3]3+ |
0.73, 1.3 |
1.2, 0.32 |
| [Ni(p-BTMD)3]2+ |
0.66, 1.18 |
1.1, 0.32 |
Several factors influence the electrochemical properties of metal complexes, including substitution pattern,72 degree and distribution, degree and unsaturation,73,74 axial ligation,75 chelate ring size,76 coordination number, and charge type.77 Most BTMD metal complexes exhibit electroactivity in both the ligands and the central metal ion. All relevant electrochemical data of BTMDs and their metal complexes are provided in Table 3. No significant changes were observed for complexes of all BTMDs with Co(II) and Ni(II), this is likely due to their low binding constants, especially in the complexes involving o- and p-BTMDs. Additionally, the electrochemical oxidation of Co2+ to Co3+ occurs at a much higher potential (>2.0 V) than what was used in our study. Meanwhile, Ni2+ is electrochemically stable and is rarely oxidized to a higher oxidation state. In Fe(III) complexes, the oxidation peaks shifting to more positive potentials indicate that the free ligands undergo oxidation more readily than their corresponding complexes. In the [Fe(p-BTMD)3]3+ complex, the quasi-reversible redox couple characterized by an anodic peak at Ea = 0.73 V, and a cathodic peak at Ec = 0.32 V can be attributed to the redox process. Conversely, this redox process exhibits irreversible behavior in the [Fe(m-BTMD)3]3+ complex with an anodic peak at Ea = 0.68 V, and no corresponding peaks are observed for the [Fe(o-BTMD)3]3+ complex. The variations in redox potentials for Fe(III) when complexed with o-, m-, and p-BTMD align well with their respective binding constants. Notably, the [Fe(o-BTMD)3]3+ complex exhibits the highest binding constant.
4. Conclusions
In this study, syntheses of N,N′-bis(2-thienylmethylene)-1,X-diaminobenzene isomers (X = 2, 3, 4) with the molecular formula of C16H12N2S2 and their complexation with Co(II), Fe(III), and Ni(II) were studied. Characterization by 1H NMR, mass spectrometry, FT-IR, Raman, UV-vis, XPS, and cyclic voltammetry demonstrated distinct chemical, electrochemical, and optical properties for each isomer. The NMR spectra confirmed the chemical structures and the presence of azomethine linkages, vital in the coordination chemistry of Schiff base ligands. Notably, shifts in the azomethine proton peaks were observed in the NMR spectra of metal complexes, indicating metal complex formation with significant downfield shifts in Fe(III) complexes, particularly for o-BTMD. UV-vis spectroscopy further revealed conjugation length-related absorption wavelength variations among o-, m- and p-BTMDs, and bandgap reductions upon metal complexation, which indicate altered ligand stability during complexation. Binding constant measurements indicated the relative affinities of the metal complexes, with the [Fe(o-BTMD)3]3+ complex demonstrating the strongest binding. The oxidation trends observed in BTMD isomers indicate that m-BTMD is the most resistant to oxidation, followed by o-BTMD, and p-BTMD as the least. This sequence corresponds with the UV-vis spectroscopy data and the bandgap calculations, suggesting that the oxidation resistance may be influenced by the structural differences among the isomers, particularly the shorter conjugation length in m-BTMD. In metal complexes, the redox potential variations for Fe(III) with o-, m-, and p-BTMDs are consistent with their binding constants.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work is supported by the Academy of Finland (Grant No. 323240) and the FICORE (Indian Finnish Consortia for Research and Education). PVY acknowledges the support from the Magnus Ehrnrooth Foundation.
Notes and references
- H. Schiff, Justus Liebigs Ann. Chem., 1864, 131, 118–119 CrossRef.
- D. Özkır, J. Electrochem. Sci. Technol., 2019, 10, 37–54 Search PubMed.
- W. Qin, S. Long, M. Panunzio and S. Biondi, Molecules, 2013, 18, 12264–12289 CrossRef CAS.
- N. E. Borisova, M. D. Reshetova and Y. A. Ustynyuk, Chem. Rev., 2007, 107, 46–79 CrossRef CAS.
- K. C. Gupta and A. K. Sutar, Coord. Chem. Rev., 2008, 252, 1420–1450 CrossRef CAS.
- A. Z. El-Sonbati, W. H. Mahmoud, G. G. Mohamed, M. A. Diab, S. M. Morgan and S. Y. Abbas, Appl. Organomet. Chem., 2019, 33, e5048 CrossRef.
- E. L. Gavey and M. Pilkington, Coord. Chem. Rev., 2015, 296, 125–152 CrossRef CAS.
- E. N. Jacobsen, F. Kakiuchi, R. G. Konsler, J. F. Larrow and M. Tokunaga, Tetrahedron Lett., 1997, 38, 773–776 CrossRef CAS.
- T. Takeuchi, A. Böttcher, C. M. Quezada, T. J. Meade and H. B. Gray, Bioorg. Med. Chem., 1999, 7, 815–819 CrossRef CAS PubMed.
- Y. N. Belokon, A. A. Petrosyan, V. I. Maleev, T. F. Saveleva, A. V. Grachev, N. S. Ikonnikov and A. S. Sagiyan, Russ. Chem. Bull., 2002, 51, 2086–2089 CrossRef CAS.
- G. Chen, Y. Deng, L. Gong, A. Mi, X. Cui and Y. Jiang,
et al.
, Tetrahedron: Asymmetry, 2001, 12, 1567–1571 CrossRef CAS.
- M. Nakoji, T. Kanayama, T. Okino and Y. Takemoto, Org. Lett., 2001, 3, 3329–3331 CrossRef CAS PubMed.
- N. Nomura, R. Ishii, M. Akakura and K. Aoi, J. Am. Chem. Soc., 2002, 124, 5938–5939 CrossRef CAS.
- P. Hormnirun, E. L. Marshall, V. C. Gibson, A. J. White and D. J. Williams, J. Am. Chem. Soc., 2004, 126, 2688–2689 CrossRef CAS.
- R. Souane, F. Isel, F. Peruch and P. J. Lutz, C. R. Chim, 2002, 5, 43–48 CrossRef CAS.
- C. M. Da Silva, D. L. da Silva, L. V. Modolo, R. B. Alves, M. A. de Resende, C. V. Martins and Â. de Fátima, J. Adv. Res., 2011, 2, 1–8 CrossRef.
- F. K. Ommenya, E. A. Nyawade, D. M. Andala and J. Kinyua, J. Chem., 2020, 1–8 Search PubMed.
- S. Sajal and D. Barnali, Pharma Chem., 2011, 3, 103–111 Search PubMed.
- H. Bayrak, A. Demirbas, S. A. Karaoglu and N. Demirbas, Eur. J. Med. Chem., 2009, 44, 1057–1066 CrossRef CAS PubMed.
- M. S. Alam, J. H. Choi and D. U. Lee, Bioorg. Med. Chem., 2012, 20, 4103–4108 CrossRef CAS PubMed.
- F. Shabani, L. A. Saghatforoush and S. Ghammamy, Bull. Chem. Soc. Ethiop., 2010, 24 Search PubMed.
- S. N. Pandeya, D. Sriram, G. Nath and E. DeClercq, Eur. J. Pharm. Sci., 1999, 9, 25–31 CrossRef CAS PubMed.
- M. A. Malik, O. A. Dar, P. Gull, M. Y. Wani and A. A. Hashmi, MedChemComm, 2018, 9, 409–436 RSC.
- M. Manjunath, A. D. Kulkarni, G. B. Bagihalli, S. Malladi and S. A. Patil, J. Mol. Struct., 2017, 1127, 314–321 CrossRef CAS.
- L. Tahmasbi, T. Sedaghat, H. Motamedi and M. Kooti, J. Solid State Chem., 2018, 258, 517–525 CrossRef CAS.
- A. Y. S. E. L. Yurt, A. Y. L. A. Balaban, S. U. Kandemir, G. Bereket and B. Erk, Mater. Chem. Phys., 2004, 85, 420–426 CrossRef CAS.
- A. Pron and P. Rannou, Prog. Polym. Sci., 2002, 27, 135–190 CrossRef CAS.
- R. Shioiri, H. Kokubo, T. Horii, Y. Kobayashi, K. Hashimoto, K. Ueno and M. Watanabe, Electrochim. Acta, 2019, 298, 866–873 CrossRef CAS.
- H. Zhang, J. Wang, X. Gao, Z. Wang and S. Wang, Synth. Met., 2014, 187, 46–51 CrossRef CAS.
- A. M. Al-Baradi, W. A. Al-Shehri, A. Badawi, A. S. Almalki and A. Merazga, Heliyon, 2019, 5, e01472 CrossRef CAS PubMed.
- D. W. DeBerry, J. Electrochem. Soc., 1985, 132, 1022 CrossRef CAS.
- B. S. Dakshayini, K. R. Reddy, A. Mishra, N. P. Shetti, S. J. Malode and S. Basu,
et al.
, Microchem. J., 2019, 147, 7–24 CrossRef CAS.
- K. Sheng, H. Bai, Y. Sun, C. Li and G. Shi, Polymer, 2011, 52, 5567–5572 CrossRef CAS.
- P. Sengodu and A. D. Deshmukh, RSC Adv., 2015, 5, 42109–42130 RSC.
- A. M. Abu-Dief and I. M. Mohamed, Beni-Suef Univ. J. Basic Appl. Sci., 2015, 4, 119–133 Search PubMed.
- A. S. Castro, C. H. P. Rodrigues, M. M. T. de Menezes, A. B. D. da Silva, A. T. Bruni and M. F. de Oliveira, Forensic Chem., 2021, 25, 100347 CrossRef.
- T. H. Sanatkar, A. Khorshidi, E. Sohouli and J. Janczak, Inorg. Chim. Acta, 2020, 506, 119537 CrossRef.
- Z. Akbari, M. Montazerozohori, G. Bruno, K. Moulaee and G. Neri, Appl. Organomet. Chem., 2022, 36, e6610 CrossRef CAS.
- A. K. Singh and S. Mehtab, Talanta, 2008, 74, 806–814 CrossRef CAS PubMed.
- L. P. Singh and J. M. Bhatnagar, Talanta, 2004, 64, 313–319 CrossRef CAS.
- P. Vahdatiyekta, M. Zniber, J. Bobacka and T. P. Huynh, Anal. Chim. Acta, 2022, 1221, 340114 CrossRef CAS PubMed.
- G. Qin, L. D. Fan, R. Zhang and S. K. Cao, Adv. Mater. Res., 2011, 287, 1761–1764 Search PubMed.
- P. Thordarson, Chem. Soc. Rev., 2011, 40, 1305–1323 RSC.
- Y. B. Zemede and A. Kumar, Int. J. Chem. Tech. Res, 2015, 7, 279–286 Search PubMed.
- V. K. Rao, S. S. Reddy, B. S. Krishna, K. R. M. Naidu, C. N. Raju and S. K. Ghosh, Green Chem. Lett. Rev., 2010, 3, 217–223 CrossRef CAS.
- H. R. Zare, M. Salavati-Niassary, F. Memarzadeh, M. Mazloum and N. Nasirizadeh, Anal. Sci., 2004, 20, 815–819 CrossRef CAS.
- D. Jin, X. Ma, Y. Liu, J. Peng and Z. Yang, Appl. Organomet. Chem., 2019, 33, e4637 CrossRef.
- S. C. Ng, H. S. O. Chan, P. M. L. Wong, K. L. Tan and B. T. G. Tan, Polymer, 1998, 39, 4963–4968 CrossRef CAS.
- A. Golcu, M. Tumer, H. Demirelli and R. A. Wheatley, Inorg. Chim. Acta, 2005, 358, 1785–1797 CrossRef CAS.
- E. L. Spitler, L. D. Shirtcliff and M. M. Haley, J. Org. Chem., 2007, 72, 86–96 CrossRef CAS PubMed.
- W. Benabid, K. Ouari, S. Bendia, R. Bourzami and M. A. Ali, J. Mol. Struct., 2020, 1203, 127313 CrossRef CAS.
- A. Jegan, M. Pannipara, A. G. Al-Sehemi, S. M. Phang, G. Kumar and J. Annaraj, Sens. Actuators, B, 2020, 316, 128082 CrossRef.
- S. Shaygan, H. Pasdar, N. Foroughifar, M. Davallo and F. Motiee, Appl. Sci., 2018, 8, 385 CrossRef.
- C. Capan, S. Uruş and M. Sönmez, J. Saudi Chem. Soc., 2018, 22, 757–766 CrossRef.
- D. Dey, G. Kaur, A. Ranjani, L. Gayathri, P. Chakraborty and J. Adhikary,
et al.
, Eur. J. Inorg. Chem., 2014, 3350–3358 CrossRef CAS.
- A. Gencer Imer, R. H. B. Syan, M. Gülcan, Y. S. Ocak and A. Tombak, J. Mater. Sci.: Mater. Electron., 2018, 29, 898–905 CrossRef CAS.
- U. M. Rabie, A. S. A. Assran and M. H. M. Abou-El-Wafa, J. Mol. Struct., 2008, 872, 113–122 CrossRef CAS.
- C. Alaşalvar, M. S. Soylu, H. Ünver, N. O. Iskeleli, M. Yildiz, M. Çiftçi and E. Banoğlu, Spectrochim. Acta, Part A, 2014, 132, 555–562 CrossRef.
- J. Tauc, Mater. Res. Bull., 1968, 3, 37–46 CrossRef CAS.
- R. Scholz, Encycl. Cond. Matter Phys., 2005, 206–221 Search PubMed.
- J. C. Costa, R. J. Taveira, C. F. Lima, A. Mendes and L. M. Santos, Opt. Mater., 2016, 58, 51–60 CrossRef CAS.
- N. K. Gondia and S. K. Sharma, J. Mol. Struct., 2018, 1171, 619–625 CrossRef CAS.
- M. Bouabdallaoui, Z. Aouzal, S. Ben Jadi, A. El Jaouhari, M. Bazzaoui, G. Lévi and E. A. Bazzaoui, J. Solid State Electrochem., 2017, 21, 3519–3532 CrossRef CAS.
- T. Teslaru, I. Topala, M. Dobromir, V. Pohoata, L. Curecheriu and N. Dumitrascu, Mater. Chem. Phys., 2016, 169, 120–127 CrossRef CAS.
- K. Artyushkova, J. Vacuum Sci. Technol. A, 2020, 38 Search PubMed.
- H. S. O. Chan, S. G. Ang, P. K. H. Ho and D. Johnson, Synth. Met., 1990, 36, 103–110 CrossRef CAS.
- H. S. O. Chan, S. C. Ng, L. S. Leong and K. L. Tan, Synth. Met., 1995, 68, 199–205 CrossRef CAS.
- H. Peng, V. Montes-García, J. Raya, H. Wang, H. Guo and F. Richard,
et al.
, J. Mater. Chem. A, 2023, 11, 2718–2725 RSC.
- J. H. Yoe and A. L. Jones, Ind. Eng. Chem., Anal. Ed., 1944, 16, 111–115 CrossRef CAS.
- M. Bheemarasetti, K. Palakuri, S. Raj, P. Saudagar, D. Gandamalla, N. R. Yellu and L. R. Kotha, J. Iran. Chem. Soc., 2018, 15, 1377–1388 CrossRef CAS.
- S. Mosbah, A. K. Beghdouche, L. Benmekhbi, J. R. Berthelot and L. Bencharif, Int. J. Electrochem. Sci., 2014, 9, 7382–7393 CrossRef CAS.
- J. A. Streeky, D. G. Pillsbury and D. H. Busch, Inorg. Chem., 1980, 19, 3148–3159 CrossRef CAS.
- A. M. Tait, F. V. Lovecchio and D. H. Busch, Inorg. Chem., 1977, 16, 2206–2212 CrossRef CAS.
- J. Telser, Y. C. Fann, M. W. Renner, J. Fajer, S. Wang and H. Zhang,
et al.
, J. Am. Chem. Soc., 1997, 119, 733–743 CrossRef CAS.
- P. A. Connick and K. A. Macor, Inorg. Chem., 1991, 30, 4654–4663 CrossRef CAS.
- E. K. Barefield, G. M. Freeman and D. G. Van Derveer, Inorg. Chem., 1986, 25, 552–558 CrossRef CAS.
- J. Manonmani, R. Thirumuruhan, M. Kandaswamy, V. Narayanan, S. S. S. Raj and M. N. Ponnuswamy,
et al.
, Polyhedron, 2001, 20, 3039–3048 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  b and
Tan-Phat
Huynh
b and
Tan-Phat
Huynh