
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Photofabrication of fluorescent nanospheres from de novo designed peptides, and their enzyme-responsive dissociation in living cells†
Sijie
He
a,
Xiang
Shu
a,
Zhaoyang
Wang
a,
Xue-Wang
Gao
b,
Ke
Feng
 b,
Shumin
Yang
a,
Jianqun
Shao
a and
Nan
Xie
*a
b,
Shumin
Yang
a,
Jianqun
Shao
a and
Nan
Xie
*a
aSchool of Pharmaceutical Sciences, Capital Medical University, Beijing 100069, P. R. China. E-mail: nanxie@ccmu.edu.cn
bKey Laboratory of Photochemical Conversion and Optoelectronic Materials, Technical Institute of Physics and Chemistry, The Chinese Academy of Sciences, Beijing 100190, P. R. China
First published on 15th February 2024
Abstract
Photochemical synthesis stands out as an exceptional approach for the bottom-up fabrication of functional assemblies using peptide building blocks. Incorporating a blend of noncovalent and covalent interactions, a series of peptide amphiphiles were designed de novo and synthesized to architect fluorescent nanospheres directly by means of photochemistry. Drawing from our established designs and experience, these peptide sequences were structurally encoded with photoactive tyrosine–tyrosine (YY) motifs at both termini, and the intervening spacer was filled with a specific Pro-Leu-Gly-Leu-Ala (PLGLA) segment and functional charge-bearing residues, including anionic D (aspartic acid), cationic R (arginine), and neutral X (ε-aminocaproic acid). The PLGLA segment acts as a stimuli-responsive element, enabling a targeted response to enzymatic matrix metalloproteinases (MMPs) in the tumor microenvironment (TME), while the charge-bearing residues serve as occupational units for drug hosting, balancing molecular amphiphilicity and regulating charge distribution. As anticipated, these functional nanospheres, efficiently constructed from encoded peptide strands upon exposure to light, were capable of hosting cationic RhB, anionic FL, and neutral DPA as fluorescent indicators, and responding to TME for MMP-responsive release of model drugs. This innovative strategy not only underlines the versatility of peptide-based assemblies but also showcases the potential of these nanospheres in targeted drug delivery and bioimaging applications.
1. Introduction
Peptides have been extensively studied as building blocks due to their chemical versatility, strong biocompatibility, and predictable assembly behavior,1,2 which could be encoded with designable sequences to fabricate biofunctional conformations directly from primary structures and allow various applications in imaging,3–7 drug delivery8–12 and tissue engineering.13–16 Peptide assemblies are ordinarily governed by a combination of noncovalent and covalent interactions, encompassing hydrogen bonds,17 π–π stacking,18 hydrophobic interactions,19,20 coulombic forces,21,22 disulfide S–S bonds,23,24L-phenylalanine polymerization, dityrosine crosslinking,25–27 and so on, which could drive peptide self-assembly processes, stabilize secondary or tertiary structures, and establish conjugation patterns even at the protein level. Tremendous efforts have been made to exploit de novo designed peptides and mimic bioactive assemblies, however, the function-oriented synthesis is still limited by many influencing factors of pH environment,28–30 working temperature,31–33 ionic strength,34,35 hydrophilic/hydrophobic properties,36,37 pre-assembly method,38,39 host–guest modeling,40,41 spacer occupation,42,43 and so on.For the construction of peptide assemblies, photochemical synthesis has emerged as a powerful toolkit.24,44 When subjected to light irradiation, functional assemblies could be created directly from designable peptide sequences via a photoactive crosslinking pathway.45,46 Notably, dityrosine is being elucidated as a versatile photocrosslinker, which exists ubiquitously in resilin, silk, alginate and collagen, and could be facilely utilized to architect covalent coupling networks.47–49 Based on our current understanding, the double-ended dityrosine structure has been exemplified as a prototype peptide strand for photofabrication, the designed spacers within adopted peptide sequences integrate both of hydrophilic and hydrophobic units, allow for a delicate balance between covalent and noncovalent interactions, and offer flexibility in tailoring the functional assemblies.26,27
In application scenarios, stimuli-responsive peptide assemblies had been demonstrated to enhance selectivity toward diseased cells and regulate drug release kinetics.50,51 Influenced by the disparities in the biological microenvironment, matrix metalloproteinases (MMPs) were recognized as typical overexpressed biomarkers in most malignant tumors.52 This characteristic makes them useful stimuli for diverse biomedical applications, particularly in specific imaging and drug delivery.53–56 In this research, we intend to employ MMP-responsive peptides as scaffolds and create functional assemblies directly from de novo designed peptide strands by means of photochemistry. Drawing from our experience and guided by a combinatorial concept of noncovalent and covalent interactions, the designed strands of peptide structure incorporate three functional motifs (Fig. 1): (1) the double-ended dityrosine (YY-spacer-YY) acts as a photocrosslinker to establish covalent peptide skeletons; (2) the specific Pro-Leu-Gly-Leu-Ala (PLGLA) segment serves as a stimuli-responsive component to enable smart responses to enzymatic MMP within the tumor microenvironment (TME); (3) anionic D (aspartic acid), cationic R (arginine), and neutral X (ε-aminocaproic acid) residues act as occupational units for drug hosting to balance molecular amphiphilicity and regulate charge distribution. It is expected that functional nanospheres could be fabricated directly from encoded peptide strands upon light exposure, host ionic or nonionic dye as fluorescent indicator, and respond to TME to realize MMP-responsive release of model molecules. Our goal is to gain deeper insights into the photochemical synthesis of peptide assemblies, unravel the structure–function relationship, and pave the way for potential encapsulation and controlled release of therapeutic drugs in future applications.
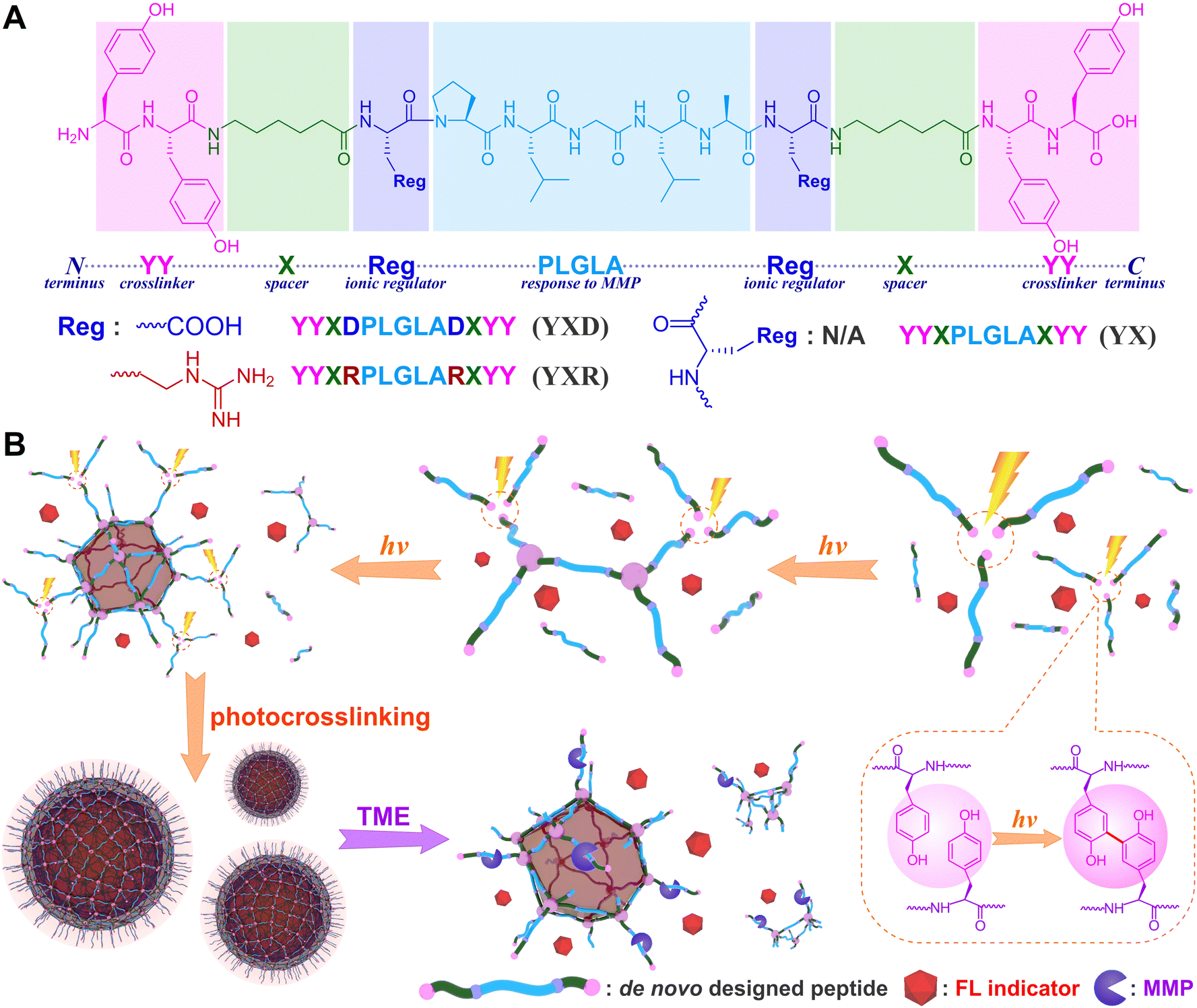 | ||
| Fig. 1 (A) The structure of designed MMP-responsive peptides and (B) schematic photofabrication of peptide nanospheres that respond to protease MMPs action. | ||
2. Experimental section
2.1 Materials
N-α-Fmoc-L-amino acids, 2-chlorotrityl chloride (CTC) resin, o-benzotriazole-N,N,N′,N′-tetramethyluroniumhexafluorophosphate (HBTU), and N-hydroxybenzotriazole (HOBt) were purchased from GL Biochem. Tris(2,2′-bipyridiyl)dichloro ruthenium(II) hexahydrate [Ru(bpy)3Cl2·6H2O], ammonium persulfate (APS), trifluoroacetic acid (TFA), triisopropylsilane (TIPS), 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT), 4-aminophenylmercuric acetate (APMA), and collagenase IV were purchased from Sigma-Aldrich. Rhodamine B (RhB), 9,10-diphenylanthracene (DPA), and fluorescein disodium salt (FL) were purchased from Innochem. MitoTracker Green, LysoTracker Green, LysoTracker Red, Nile Red, and hoechst 33342 were purchased from KeyGEN Biotech. High-glucose Dulbecco's modified Eagle medium (DMEM), trypsin, fetal bovine serum (FBS), penicillin, streptomycin, phosphate-buffered saline (PBS), and trizma hydrochloric acid (Tris–HCl) were purchased from Gibco BRL. N,N-Dimethylformamide (DMF), ethyl ether, and other organic solvents were used as received without further purification.2.2 Synthesis of enzyme-responsive peptides
Enzyme-responsive peptides were synthesized on CTC resin following a standard Fmoc SPPS strategy. The sequence assembly of peptide were performed sequentially using HOBt and HBTU as coupling agents and piperidine for Fmoc deprotection. After removal of the N-terminal Fmoc group, the peptide-resin was cleaved and fully deprotected with TFA/TIPS/H2O (95/2.5/2.5, v/v/v). The peptides were obtained by precipitation in cold ether and could be further purified on HPLC.NH 2-Tyr-Tyr-Acp-Asp-Pro-Leu-Gly-Leu-Ala-Asp-Acp-Tyr-Tyr-COOH (YYXDPLGLADXYY, YXD). HR-ESI MS: m/z = Calcd 1576.7575, found 1576.7601 for [M − H]−, C78H106N13O22−; Calcd 787.8749, found 787.8790 for [M − 2H]2−, C78H105N13O222−. 1H NMR (600 MHz, DMSO-d6, ppm) δ: 12.42 (br, 3H), 9.22 (m, 4H), 8.69 (br, 1H), 7.96 (m, 12H), 7.00 (m, 8H), 6.65 (m, 8H), 4.87 (m, 1H), 4.43 (m, 8H), 3.92 (m, 1H), 3.63 (m, 2H), 3.39 (m, 2H), 2.97 (m, 16H), 1.98 (m, 8H), 1.31 (m, 21H), 0.87 (d, J = 28.2 Hz, 12H).
NH 2-Tyr-Tyr-Acp-Pro-Leu-Gly-Leu-Ala-Acp-Tyr-Tyr-COOH (YYXPLGLAXYY, YX). HR-ESI MS: m/z = Calcd 1348.7193, found 1348.7212 for [M + H]+, C70H98N11O16+; Calcd 1370.7012, found 1370.7031 for [M + Na]+, C70H97N11NaO16+; Calcd 1392.6832, found 1392.6844 for [M + 2Na − H]+, C70H96N11Na2O16+. 1H NMR (600 MHz, DMSO-d6, ppm) δ: 12.71 (br, 1H), 9.22 (m, 4H), 8.70 (br, 1H), 7.99 (m, 10H), 7.00 (m, 8H), 6.65 (m, 8H), 4.33 (m, 7H), 3.92 (m, 3H), 3.42 (m, 2H), 2.96 (m, 12H), 1.98 (m, 8H), 1.32 (m, 21H), 0.85 (m, 12H).
NH 2-Tyr-Tyr-Acp-Arg-Pro-Leu-Gly-Leu-Ala-Arg-Acp-Tyr-Tyr-COOH (YYXRPLGLARXYY, YXR). HR-ESI MS: m/z = Calcd 1660.9215, found 1660.9226 for [M + H]+, C82H122N19O18+. 1H NMR (600 MHz, DMSO-d6, ppm) δ: 12.07 (br, 1H), 9.34 (br, 1H), 9.21 (br, 2H), 9.13 (br, 1H), 8.69 (br, 1H), 8.00 (m, 12H), 7.72 (m, 1H), 7.56 (m, 1H), 7.31 (m, 3H), 7.00 (br, 8H), 6.88 (m, 2H), 6.65 (br, 8H), 6.43 (m, 1H), 4.33 (m, 9H), 3.91 (m, 1H), 3.70 (m, 2H), 3.37 (m, 2H), 2.95 (m, 12H), 2.42 (m, 2H), 2.37 (m, 2H), 2.00 (m, 8H), 1.40 (m, 29H), 0.82 (d, J = 24.6 Hz, 12H).
NH 2-Tyr-Tyr-Acp-Ala-Leu-Gly-Leu-Pro-Acp-Tyr-Tyr-COOH (YYXALGLPXYY, scrambled sequence). HR-ESI MS: m/z = Calcd 1348.7193, found 1348.7199 for [M + H]+, C70H98N11O16+; Calcd 1386.6752, found 1386.6664 for [M + K]+, C70H97KN11O16+. 1H NMR (600 MHz, DMSO-d6, ppm) δ: 12.68 (br, 1H), 9.21 (m, 4H), 8.67 (br, 1H), 7.96 (m, 10H), 7.01 (m, 8H), 6.66 (m, 8H), 4.43 (m, 7H), 3.92 (br, 1H), 3.68 (m, 2H), 3.39 (m, 2H), 2.96 (m, 12H), 1.98 (m, 8H), 1.32 (m, 21H), 0.87 (m, 12H).
2.3 Synthesis of peptide nanospheres (PNS)
Peptide Nanospheres (PNS) were synthesized using a one-step photocrosslinking method with double-ended dityrosine peptides.26 To prepare the photocrosslinking reaction solution, the peptide (1 mM) was dissolved in a 1 mL buffer that comprised 0.1 M NaOH, 4 mM APS, and 0.02 mM Ru(bpy)3Cl2, and subsequently adjusted to the desired pH condition using NaOH/HCl. The solution was transferred to a transparent glass vial, which was then placed within a homemade parallel photoreactor and stirred (700 rpm) at 25 °C, while irradiated upon 405-nm light-emitting diodes (LEDs) (112 mW cm−2, 50 mA DC input) for 10 min at room temperature. After centrifugation, the produced PNS was washed with deionized water, redispersed in aqueous media, and then stored at 4 °C for future utilization.2.4 Synthesis of fluorescent dyes-loaded peptide nanospheres (RhB@YXD-PNS, DPA@YX-PNS, FL@YXR-PNS)
The preassembling of fluorescent dyes was performed by mixing them with enzyme-responsive peptides (1 mM) at a 0.2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 dye-to-peptide weight ratio, followed by stirring the mixture in an aqueous medium at room temperature for 15 min. After addition of the ruthenium photosensitizer and APS, the system was adjusted to optimized pH and immediately irradiated upon 405-nm LEDs for 10 min. As completion of photoencapsulation, the fluorescent dyes-loaded peptide nanospheres were thoroughly washed and collected by centrifugation. Loading efficiency and encapsulation efficiency were determined by a UV-vis spectrometer using calibration curve method.
:1 dye-to-peptide weight ratio, followed by stirring the mixture in an aqueous medium at room temperature for 15 min. After addition of the ruthenium photosensitizer and APS, the system was adjusted to optimized pH and immediately irradiated upon 405-nm LEDs for 10 min. As completion of photoencapsulation, the fluorescent dyes-loaded peptide nanospheres were thoroughly washed and collected by centrifugation. Loading efficiency and encapsulation efficiency were determined by a UV-vis spectrometer using calibration curve method.
2.5 In vitro release of fluorescent dyes from peptide nanospheres
For enzyme-responsive drug release study, 1 mg of fluorescent dyes-loaded peptide nanospheres (RhB@YXD-PNS, DPA@YX-PNS, FL@YXR-PNS) were incubated in 1 mL Tris–HCl buffer (pH 6) at 37 °C in the presence of collagenase IV (MMP-2/9), respectively. At designated time intervals, the supernatant was extracted to quantify the amount of released fluorescent dyes using a UV-vis spectrometer. Collagenase IV (0.5 mg mL−1 in 50 mM Tris–HCl buffer) was pre-activated with the 2.5 mM APMA solution at 37 °C for 1 h prior to the experiment. For comparison, 1 mL of samples was also tested without enzymatic incubation to assess dye release.2.6 Cell viability assay
Human alveolar adenocarcinoma cells (A549) sourced from KeyGEN Biotech were selected for cellular studies. These cells were cultured in DMEM supplemented with 10% FBS and 1% penicillin–streptomycin, and maintained under standard conditions at 37 °C, 5% CO2, and 95% humidity. Cell viability of the peptides was assessed using the tetrazolium reduction (MTT, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) assay. Initially, A549 cells were seeded in 96-well plate at a density of 5 × 104/well and incubated overnight for adhesion. Subsequently, the cells were treated with PBS (used as a control), as well as various concentrations of peptides (50, 100, 400, 800, and 1600 μM) for 24 h, 48 h, and 72 h, respectively. Then, 20 μL of MTT (5 mg mL−1 in PBS) were added to each well, followed by incubation for 4 h. The medium was then replaced with 100 μL of DMSO, and the well-plates were placed on a microplate shaker for over 10 min to ensure complete solubilization of the formazan crystals. Finally, the absorbance was measured at a wavelength of 570 nm using a microplate reader (EnSpire Multimode Plate Reader, PerkinElmer). The viability percentage was calculated by dividing the absorbance of treated cells by that of the control group.2.7 Cellular uptake and intracellular localization
A549 cells were seeded in glass-bottomed confocal dishes at a density of 1 × 105/well and cultured for 24 h. After incubation with RhB@YXD-PNS, DPA@YX-PNS, and FL@YXR-PNS (20 μg mL−1) for 4 h, respectively, the cells were stained with LysoTracker Green, MitoTracker Green, LysoTracker Red, or Nile Red for 30 min, and subsequently imaged on a confocal laser scanning microscope (CLSM, Leica TCS-SP8). For the MMP-responsive release of fluorescent dyes, cells were treated with collagenase IV simultaneously to enzymatically cleave the peptide nanospheres. The control set was conducted for free RhB and DPA. The nuclei of all cells were visualized with Hoechst 33342 except for imaging of DPA.3. Results and discussion
Peptide design and synthesis
For the foundational structure of de novo designed peptide building blocks, MMP-responsive PLGLA segments were adorned with photoactive YY motifs at both ends and interspersed with anionic D, cationic R, or neutral X spacers, leading to the creation of three distinct peptides YYXDPLGLADXYY, YYXRPLGLARXYY, and YYXPLGLAXYY, denoted as YXD, YXR, and YX respectively (Fig. 1A). This design was guided by several considerations: (1) the double-ended dityrosine motifs extended bilaterally from the linear peptide strand, providing multiple sites for photocrosslinking upon light exposure, crucial for constructing the essential nanospheric structures; (2) the hydrophilic D/R, hydrophobic X, and aromatic Y on peptide strands adjusted the molecular amphiphilicity, facilitating the noncovalent assembling of nanospheres; (3) the hydrophobic X, with its flexible pentamethylene structure, effectively regulated the occupancy of spacers in the crosslinkable dityrosine networks; (4) the repetition of negatively charged D or positively charged R residues controlled both the charge distribution in linear peptides and their corresponding nanoassemblies, thus managing the loading capacity of guest indicators like cationic rhodamine B (RhB),57 anionic fluorescein (FL),58 and neutral diphenylalanine (DPA);59 (5) the specific PLGLA segments responded to MMP upregulated tumor microenvironment, enabling the dissociation of peptide encapsules and resulting in a controlled release of model indicators.In the synthesis process, the peptides were prepared step by step on CTC resin utilizing solid-phase peptide synthesis (SPPS) techniques. The MMP-specific peptides were meticulously characterized through high-resolution electrospray ionization mass spectrometry (HR-ESI MS, Fig. S22–S25, ESI†) and proton nuclear magnetic resonance (1H NMR, Fig. S26–S29, ESI†).
Photofabrication of peptide nanospheres
Upon exposure to visible light at 405 nm, all three peptide amphiphiles underwent rapid crosslinking within a mere 10 minutes. It is well-established that tyrosine undergoes photoinduced reactions, generating tyrosyl radicals and solvated electrons, leading to the formation of dityrosine anchors through free radical dimerization processes.60 Various spectral techniques have been employed to investigate the successful crosslinking of tyrosine. We monitored the peptide assembly process within the initial 10 minutes using UV-vis spectra (Fig. 2A and Fig. S1A and S2A, ESI†) and fluorescence emission spectra (Fig. 2B and Fig. S1B and S2B, ESI†). As the irradiation time extended, the shoulder peak at 327 nm gradually intensified in absorbance, and a distinct fluorescence emission peak emerged around 410 nm upon excitation at 330 nm, unequivocally confirming the formation of dityrosine bonds.61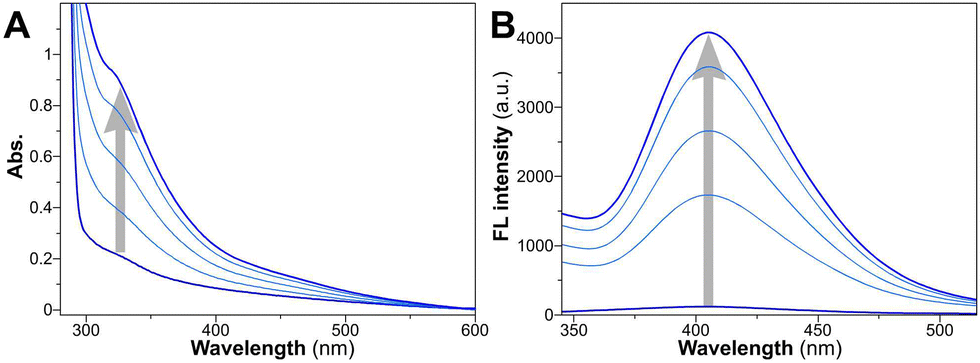 | ||
| Fig. 2 (A) Time-dependent changes of UV-vis absorption and (B) fluorescence emission spectra for YXD peptide upon irradiation of 0, 2, 4, 6, 10 min. | ||
Transmission electron microscopy (TEM) provided a direct morphology of these peptidic assemblies (Fig. 3A and B). The fabrication of peptide nanospheres (PNS) was greatly influenced by pH levels and irradiation parameters. At optimal pH, D and Y residues were partially ionized, striking a balance that prevented aggregation and led to the formation of distinct nanospheres (Fig. S3, ESI†). Meanwhile, extending irradiation to 10 minutes under the ideal conditions (112 mW cm−2 at 405 nm) effectively produced core–shell structured nanospheres through a cascade photocrosslinking pathway (Fig. 1B and Fig. S4, ESI†). The particle size of YXD-PNS, YX-PNS, and YXR-PNS were 152.5 ± 24.3 nm, 156.7 ± 28.1 nm, and 155.6 ± 21.1 nm, respectively (Fig. 3C). Consistently, dynamic light scattering (DLS) measurements revealed corresponding hydrodynamic diameter of 170.7 ± 71.3 nm, 172.5 ± 74.6 nm, 174.3 ± 76.6 nm with unimodal distribution (Fig. 3D).
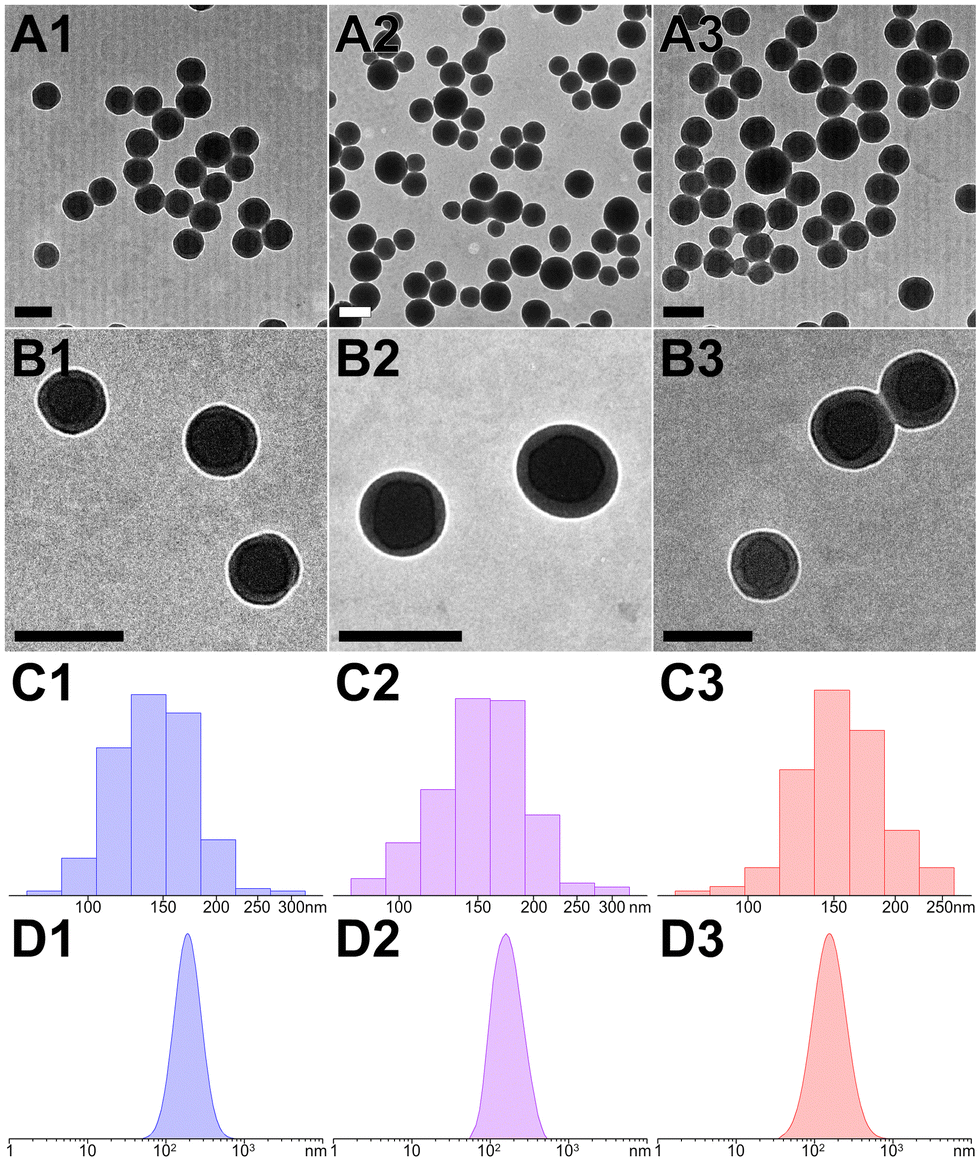 | ||
| Fig. 3 (A) and (B) TEM images and (C) and (D) corresponding particle size distributions ((C), TEM statistics; (D), DLS measurements) for YXD-PNS, YX-PNS, and YXR-PNS, respectively. The scale bar represents 200 nm. | ||
The conformation of the peptides and assemblies was initially assessed using circular dichroism (CD) spectroscopy. Both monomers and nanospheres displayed negative signals at 193 nm, indicating their maintained β-turn conformation (Fig. S5–S7, ESI†).62 Positive signals at 223 nm in monomers signified the presence of tyrosine's phenolic side chains. A slight redshift in the absorption of Tyr residues in peptide nanospheres implied one of the predominant driving forces behind nanosphere formation was the crosslinking of YY motifs. Furthermore, the comparable FT-IR spectra (Fourier transform infrared spectroscopy, Fig. S8–S10, ESI†) of monomers and nanospheres indicated the consistent presence of functional groups, including hydroxyl O–H (stretch, 3290 cm−1), alkyl C–H (methylene stretch, 2930 cm−1), amide (I band, stretch, 1645 cm−1; II band, stretch, 1515 cm−1), C–N (stretch, 1236 cm−1), phenyl–O (stretch, 1170 cm−1), and aromatic ring on tyrosine (bending, 828 cm−1). Additionally, all PNS showed negative zeta potentials (Fig. S11, ESI†), presumably due to the partial ionization of carboxyl groups in aspartate and C-termini, as well as tyrosine residues within the peptide sequences.
Photofabrication of dye-loaded peptide nanospheres
To evaluate the capability of peptide assemblies to host different molecules, we selected three dyes frequently used in biological imaging: cationic rhodamine, anionic fluorescein, and neutral diphenylalanine. These dyes were incorporated into the peptide system, resulting in dye-loaded PNS (RhB@YXD-PNS, DPA@YX-PNS, and FL@YXR-PNS). The integration occurred through electrostatic interactions or π–π stacking, interacting with the peptides’ negatively charged D, positively charged R, or aromatic residues. Remarkably, after the formation of dye-loaded peptide nanospheres through photocrosslinking, there were no significant alterations in their size and shape, as confirmed by DLS and TEM analyses (Fig. 4A–D). The DLS and TEM data showed consistent size distributions for each dye-loaded nanosphere variant (RhB@YXD-PNS, TEM 151.1 ± 26.8 nm, DLS 166.9 ± 73.2 nm; DPA@YX-PNS, TEM 162.0 ± 28.5 nm, DLS 169.5 ± 46.4 nm; FL@YXR-PNS, TEM 153.2 ± 23.9 nm, DLS 173.9 ± 68.9 nm). Additionally, the dye-loaded peptide nanospheres exhibited partially negative zeta potential values, ensuring their suitability for applications within biological systems (Fig. S11, ESI†). The negligible change in surface charge indicated that the dyes were encapsulated within the core, rather than being superficially attached to the assembled structure.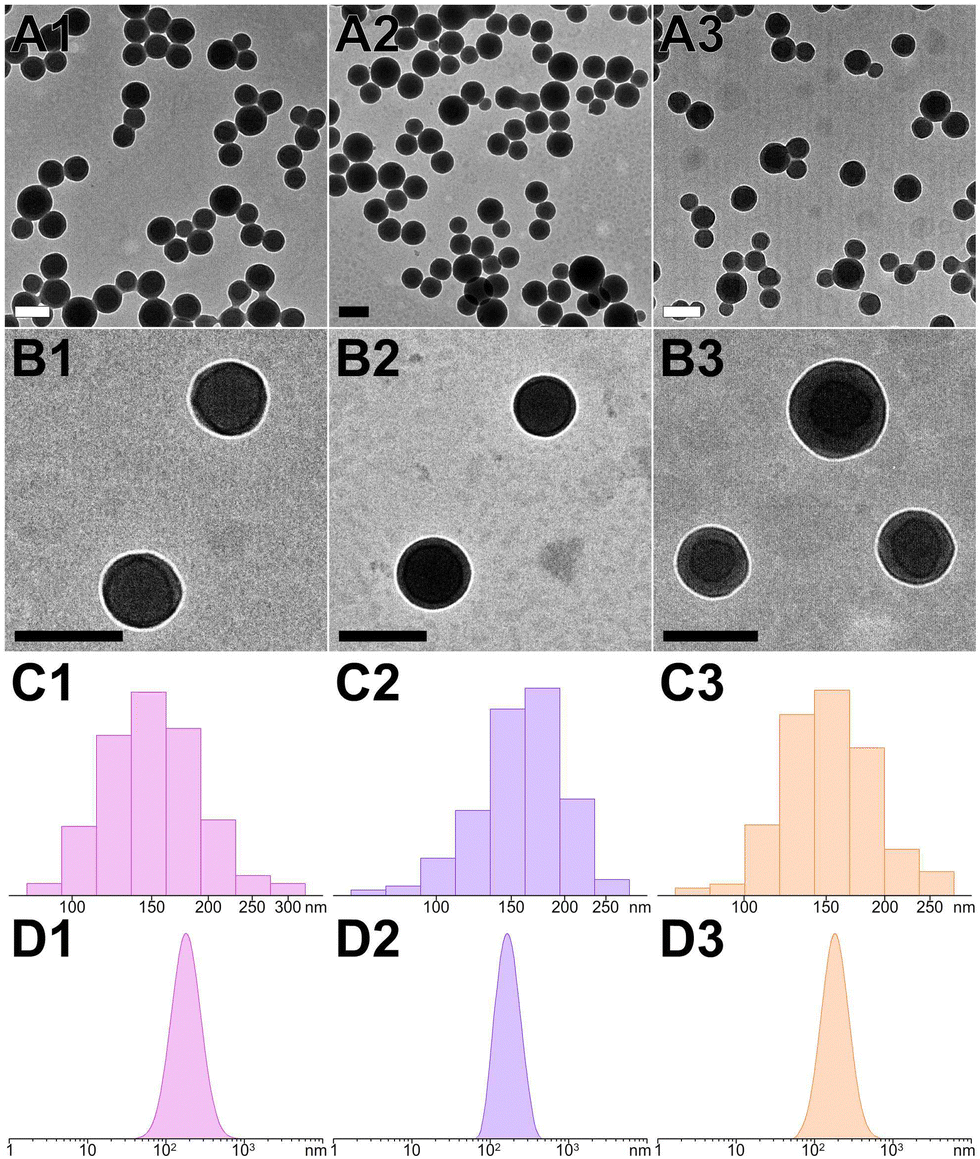 | ||
| Fig. 4 (A) and (B) TEM images and (C) and (D) corresponding particle size distributions ((C), TEM statistics; (D), DLS measurements) for RhB@YXD-PNS, DPA@YX-PNS, and FL@YXR-PNS, respectively. The scale bar represents 200 nm. | ||
Photographic documentation of dye-loaded PNS under both natural and UV light (365 nm) is presented as Fig. S12 (ESI†), the peptide nanospheres infused with different dyes displayed distinct and vivid colors in water: orange for RhB, blue for DPA, and green for FL. The encapsulation of each dye within the PNS system could be quantitatively analyzed using UV-visible absorption spectroscopy. The loading content of RhB, DPA and FL dye in the YXD-PNS, YX-PNS, and YXR-PNS was 17.1%, 12.8%, and 14.0%, respectively, at a dye-to-peptide feeding ratio of 0.2:1, as detailed in Table S1 (ESI†). The corresponding encapsulation efficiencies for RhB, DPA, and FL reached impressive levels of 73.8%, 70.8%, and 72.6%, respectively. Notably, altering the encapsulated peptide monomers led to a variable decrease in encapsulation efficiency, indicating that the charge distribution of the peptide monomers is very crucial to the loading capacity of guest indicators (Table S2, ESI†).
The characteristic absorbances for RhB, FL, and DPA were observed at wavelengths of 550 nm, 497 nm, and 392 nm, respectively (Fig. 5 and Fig. S13 and S14, ESI†). Interestingly, the dye-loaded peptide nanospheres showed emission peaks similar to those of the free dyes, with only slight bathochromic shifts noted. In addition to these observations, FT-IR provided conclusive evidence of the successful encapsulation of these fluorescent dyes within the peptide nanospheres. This encapsulation not only validates the versatility of the PNS system in hosting various molecules but also underscores its potential in diverse bioimaging applications.
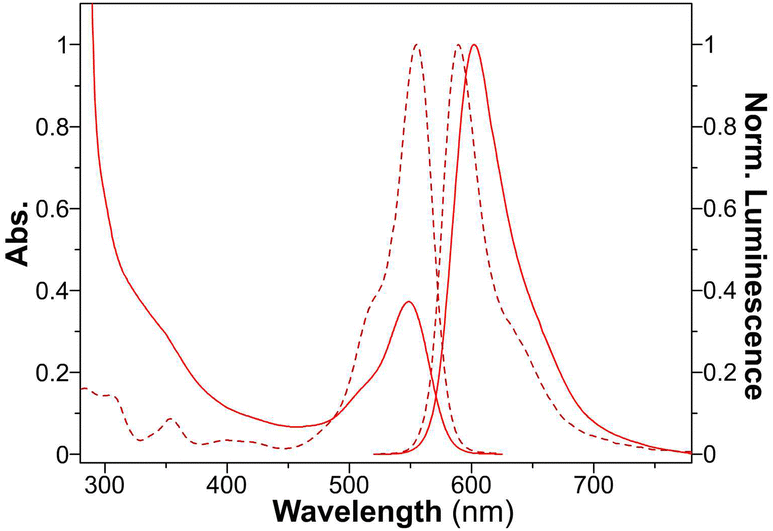 | ||
| Fig. 5 UV-vis absorption and normalized fluorescence spectra of free RhB (dotted line) and RhB@YXD-PNS (solid line). | ||
Stability of PNS and dye-loaded PNS
Utilizing a photochemical synthesis with covalent binding, both the peptide nanospheres and their dye-loaded PNS exhibited exceptional stability in various aqueous environments, including deionized water, PBS, and DMEM (Fig. S15 and S16, ESI†). The covalently linked dityrosine networks within these nanospheres ensured their structural durability and integrity, rendering them highly suitable for diverse biological applications.Enzyme-responsive dissociation and dye release of PNS
To replicate the enzymic response of the peptide nanospheres to elevated levels of matrix metalloproteinases found in tumor environments, MMP protease (collagenase IV) was added to the YXD-NS samples and incubated at 37 °C. As depicted in the TEM images in Fig. 6, the fundamental nanospheres maintained their core structure, albeit becoming less dense, within the initial 12-hour incubation, although they started to loosen and became less compact. From 12 to 24 hours, they evolved into a hierarchical and intricate flower-like morphology. After 48 hours of enzymatic digestion, the peptide nanospheres were completely disintegrated into fragments. As a control, the peptide nanospheres assembled from the scrambled sequence (YYXALGLPXYY), lacking the enzyme-responsive component, exhibited no response to MMP. The morphology of these PNS largely retained, showing no signs of enzymatic cleavage (Fig. S17, ESI†). The MMP-responsive fluorescent dyes release from peptide nanospheres was investigated in Tris–HCl buffer (pH 6) at 37 °C (Fig. 7). Upon addition of activated collagenase IV, the cumulative payload release reached approximately 60% after a 24-hour period. In contrast, in the absence of MMP, the release rate remained as low as 8%. These results demonstrated the substantial impact of MMP-responsive mechanisms on the controlled release of fluorescent dyes from peptide nanospheres, providing valuable insights for potential encapsulation and controlled release of therapeutic drugs in future applications.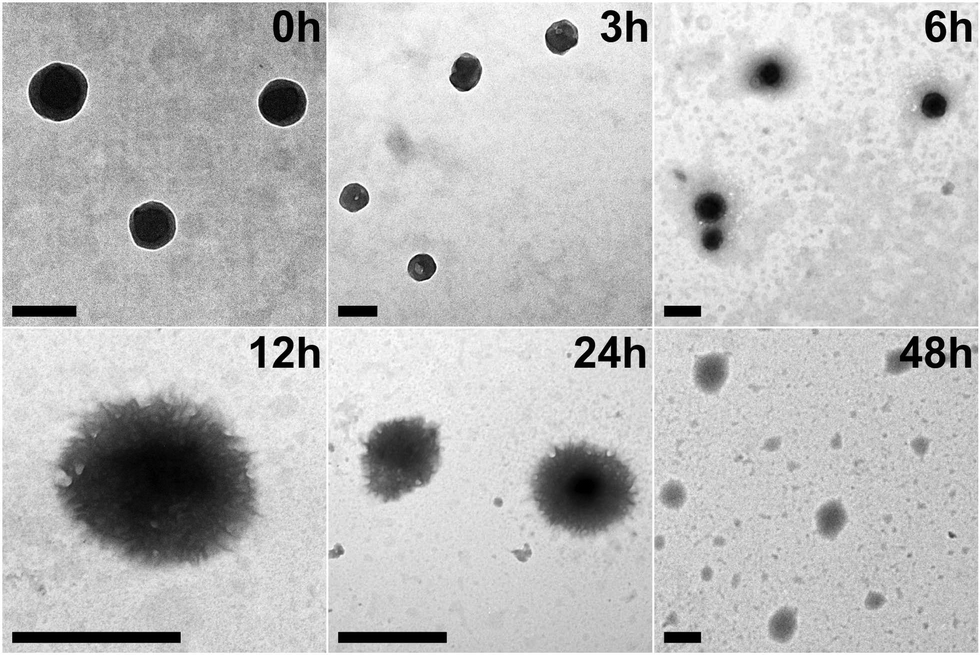 | ||
| Fig. 6 TEM images of YXD-PNS incubated with MMP at 37 °C for different time periods. The scale bar represents 200 nm. | ||
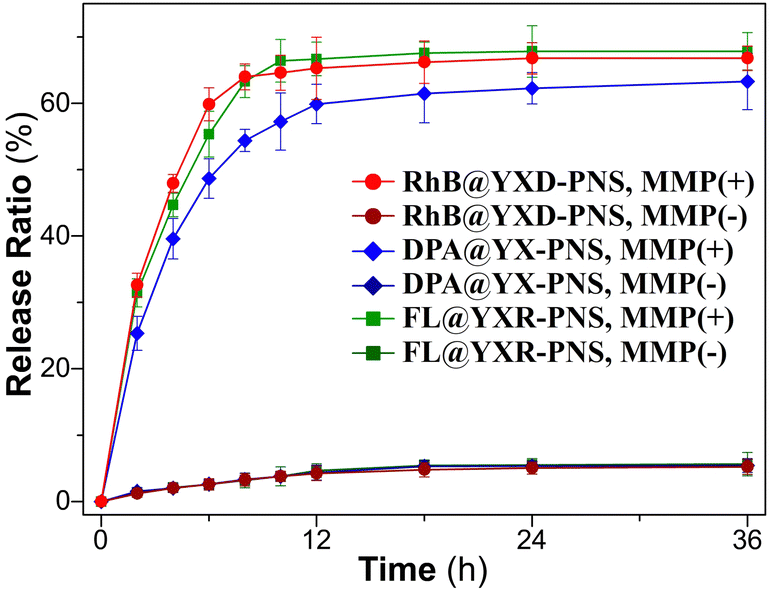 | ||
| Fig. 7 Release profiles of dyes from the PNS in solutions with and without MMP at 37 °C. | ||
Biocompatibility of dye-loaded PNS in vitro
The cytotoxicity of dye-loaded PNS was assessed using the standard MTT assay protocol. Impressively, at various concentrations ranging from 50–1600 μM (Fig. S18–S20, ESI†), all these peptide nanospheres demonstrated no significant cytotoxicity towards A549 cells at various concentrations, even after 3 days of incubation. This minimal cytotoxicity indicates their high biocompatibility, making them suitable for biomedical research and therapeutic applications.Cellular uptake and enzyme-responsive intracellular dissociation behavior of PNS
To explore the cellular uptake and intracellular transport of these peptide nanospheres within A549 cells, a colocalization study was conducted using commercial tracking dyes and a confocal laser scanning microscope (CLSM, Fig. 8). In contrast to the free rhodamine B, which predominantly localizes to mitochondria, RhB-loaded PNS presented a propensity to localize within lysosomes (indicated by overlap with LysoTracker Green, Pearson's r = 0.66 ± 0.12). When activated collagenase IV was introduced, RhB@YXD-PNS responded to the MMP protease, releasing RhB and progressively demonstrating increased colocalization with MitoTracker Green in the confocal profile over time. Extending the incubation time to 6 hours resulted in more pronounced mitochondrial localization of RhB compared to the 1-hour mark, as evidenced by overlap with MitoTracker Green (Pearson's r = 0.64 ± 0.09). | ||
| Fig. 8 (A) and (B) Colocalization of RhB@YXD-PNS with LysoTracker Green and MitoTracker Green in A549 cells. (C) and (D) CLSM images for A549 cells incubated with RhB@YXD-PNS upon stimuli of MMP. (E) and (F) Colocalization of RhB with MitoTracker Green and LysoTracker Green in A549 cells. Cell were counterstained with Hoechst 33342 (blue) for nuclei. | ||
The intracellular behavior of DPA@YX-PNS and FL@YXR-PNS within cells was also examined employing the identical experimental setup in A549 cells, resulting in analogous colocalization outcomes (Fig. 9 and Fig. S21, ESI†). The fluorescence from PNS encapsulating DPA and FL was consistently observed in lysosomes. Upon exposure to MMP, DPA@YX-PNS released free DPA, which then resembled the appearance of classic lipid droplets. This phenomenon could be clearly confirmed through colocalization analysis utilizing the commercial fluorophore Nile Red. In the case of FL@YXR-PNS, owing to the inherent entry of free FL into lysosomes, no significant change in green fluorescence within cells was observed before and after enzymatic digestion. These findings suggest that dye-loaded PNS can effectively respond to the MMP-overexpressed microenvironment in tumor tissues, facilitating a targeted and efficient release of their contents.
 | ||
| Fig. 9 (A) and (B) Colocalization of DPA@YX-PNS with LysoTracker Green and Nile Red in A549 cells. (C) and (D) CLSM images for A549 cells incubated with DPA@YX-PNS upon stimuli of MMP. (E) and (F) Colocalization of DPA with LysoTracker Green and Nile Red in A549 cells. | ||
4. Conclusions
In summary, this research demonstrated the efficacy photochemical synthesis as a powerful toolkit for constructing peptide-based functional assemblies, with significant implications for targeted drug delivery and bioimaging. By integrating both noncovalent and covalent interactions, we have successfully designed de novo and synthesized a series of peptide amphiphiles structurally encoded with photoactive motifs, MMP-responsive segments, and charge/amphiphilicity regulation residues to directly fabricate fluorescent peptide nanospheres by means of photochemistry. The PNS stability across diverse aqueous environments, the ability to host various fluorescent indicators, such as cationic RhB, anionic FL, and neutral DPA, and their minimal cytotoxicity towards A549 cells, further reinforced their potential suitability for biological applications, particularly in the controlled release of therapeutic drugs. Additionally, the controlled release mechanism in response to MMPs and intracellular behavior of these nanospheres in A549 cells revealed their capability for targeted and efficient drug delivery within MMP-upregulated tumor microenvironment. This innovative approach not only highlights the adaptability of peptide-based assemblies but also demonstrates the considerable promise of these peptide nanospheres for use in specific drug delivery systems and bioimaging applications.Author contributions
S. H. contributed to the methodology, data curation, visualization, and writing – original draft. S. X. and Z. W. contributed to methodology, data curation, and validation. X.-W. G. contributed to the peptide characterization. K. F. contributed to the conceptualization. S. Y., and J. S. contributed to the data curation and validation. N. X. contributed to the conceptualization, project administration, and funding acquisition. All authors contributed to writing – review and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by Beijing Natural Science Foundation (2222051). The authors also acknowledge the technical support provided by Core Facility Center at Capital Medical University.References
- A. Levin, T. A. Hakala, L. Schnaider, G. J. L. Bernardes, E. Gazit and T. P. J. Knowles, Nat. Rev. Chem., 2020, 4, 615–634 CrossRef CAS.
- F. Sheehan, D. Sementa, A. Jain, M. Kumar, M. Tayarani-Najjaran, D. Kroiss and R. V. Ulijn, Chem. Rev., 2021, 121, 13869–13914 CrossRef CAS PubMed.
- Q. Jiang, X. Liu, G. Liang and X. Sun, Nanoscale, 2021, 13, 15142–15150 RSC.
- B. Hu, N. Song, Y. Cao, M. Li, X. Liu, Z. Zhou, L. Shi and Z. Yu, J. Am. Chem. Soc., 2021, 143, 13854–13864 CrossRef CAS PubMed.
- Q. Li, J. Zhang, Y. Wang, G. Zhang, W. Qi, S. You, R. Su and Z. He, Nano Lett., 2021, 21, 6406–6415 CrossRef CAS PubMed.
- H. Ren, X. Z. Zeng, X. X. Zhao, D. Y. Hou, H. Yao, M. Yaseen, L. Zhao, W. H. Xu, H. Wang and L. L. Li, Nat. Commun., 2022, 13, 418 CrossRef CAS PubMed.
- Z. Wang, C. Zhao, Y. Li, J. Wang, D. Hou, L. Wang, Y. Wang, X. Wang, X. Liu, H. Wang and W. Xu, Adv. Mater., 2023, 35, e2210732 CrossRef PubMed.
- M. Peng, S. Qin, H. Jia, D. Zheng, L. Rong and X. Zhang, Nano Res., 2015, 9, 663–673 CrossRef.
- X. Wang, Z. Song, S. Wei, G. Ji, X. Zheng, Z. Fu and J. Cheng, Biomaterials, 2021, 275, 120913 CrossRef CAS PubMed.
- J. Yang, H. W. An and H. Wang, ACS Appl. Bio. Mater., 2021, 4, 24–46 CrossRef CAS PubMed.
- L. Wang, C. Li, J. Wang, G. Yang, Y. Lv, B. Fu, L. Jian, J. Ma, J. Yu, Z. Yang, P. Wu, G. Li, X. Liu, Z. Kang, Z. Wang, L. Wang, H. Wang and W. Xu, Adv. Mater., 2022, 34, e2203518 CrossRef PubMed.
- Z. Wang, X. Zhang, M. Han, X. Jiao, J. Zhou, X. Wang, R. Su, Y. Wang and W. Qi, J. Mater. Chem. B, 2023, 11, 8974–8984 RSC.
- P. Chakraborty, H. Oved, D. Bychenko, Y. Yao, Y. Tang, S. Zilberzwige-Tal, G. Wei, T. Dvir and E. Gazit, Adv. Mater., 2021, 33, e2008715 CrossRef PubMed.
- V. P. Gray, C. D. Amelung, I. J. Duti, E. G. Laudermilch, R. A. Letteri and K. J. Lampe, Acta Biomater., 2022, 140, 43–75 CrossRef CAS PubMed.
- Z. Hao, H. Li, Y. Wang, Y. Hu, T. Chen, S. Zhang, X. Guo, L. Cai and J. Li, Adv. Sci., 2022, 9, e2103820 CrossRef PubMed.
- W. Sun, D. A. Gregory and X. Zhao, Adv. Colloid Interface Sci., 2023, 314, 102866 CrossRef CAS PubMed.
- S. Sun, H. W. Liang, H. Wang and Q. Zou, ACS Nano, 2022, 16, 18978–18989 CrossRef CAS PubMed.
- X. Li, H. Zhang, L. Liu, C. Cao, P. Wei, X. Yi, Y. Zhou, Q. Lv, D. Zhou and T. Yi, J. Mater. Chem. B, 2021, 9, 8686–8693 RSC.
- M. H. Sangji, H. Sai, S. M. Chin, S. R. Lee, I. R. Sasselli, L. C. Palmer and S. I. Stupp, Nano Lett., 2021, 21, 6146–6155 CrossRef CAS PubMed.
- A. C. Farsheed, A. J. Thomas, B. H. Pogostin and J. D. Hartgerink, Adv. Mater., 2023, 35, e2210378 CrossRef PubMed.
- B. Wu, S. Zhao, X. Yang, L. Zhou, Y. Ma, H. Zhang, W. Li and H. Wang, ACS Nano, 2022, 16, 4126–4138 CrossRef CAS PubMed.
- R. Liu, X. Dong, D. T. Seroski, B. Soto Morales, K. M. Wong, A. S. Robang, L. Melgar, T. E. Angelini, A. K. Paravastu, C. K. Hall and G. A. Hudalla, Angew. Chem., Int. Ed., 2023, e202314531 CAS.
- H. Dong, M. Wang, S. Fan, C. Wu, C. Zhang, X. Wu, B. Xue, Y. Cao, J. Deng, D. Yuan and J. Shi, Angew. Chem., Int. Ed., 2022, 61, e202212829 CrossRef CAS PubMed.
- F. He, Y. Chai, Z. Zeng, F. Lu, H. Chen, J. Zhu, Y. Fang, K. Cheng, E. Miclet, V. Alezra and Y. Wan, J. Am. Chem. Soc., 2023, 145, 22639–22648 CrossRef CAS PubMed.
- Z. Ma, L. Yang, Y. Wang, M. Wang, W. Qi and Z. He, Chem. Eng. J., 2021, 416, 129149 CrossRef CAS.
- J. Zhao, C. Li, X.-W. Gao, K. Feng, H. Liu, S. He, W. Zhao, S. Yang, J. Shao, L. Ye, B. Chen, N. Xie, C.-H. Tung and L.-Z. Wu, Nano Res., 2023, 16, 4029–4038 CrossRef CAS.
- H. Liu, S. He, L.-Y. Niu, X.-W. Gao, K. Feng, S. Yang, J. Shao, W. Zhao, N. Xie and Q.-Z. Yang, Chem. Commun., 2023, 59, 5059–5062 RSC.
- G. Li, X. Hu, X. Wu and Y. Zhang, Nano Lett., 2021, 21, 3052–3059 CrossRef CAS PubMed.
- P. Tan, C. Wu, Q. Tang, T. Wang, C. Zhou, Y. Ding, H. Fu, S. Xu, Y. Feng, Y. Zhang, Q. Dai and X. Ma, Adv. Mater., 2023, 35, e2210766 CrossRef PubMed.
- Y. Xiang, C. Liu, S. Ma, X. Wang, L. Zhu and C. Bao, Adv. Funct. Mater., 2023, 33, 2300416 CrossRef CAS.
- P. Guo, D. Wang, S. Zhang, D. Cheng, S. Wu, X. Zuo, Y. B. Jiang and T. Jiang, Nano Lett., 2023, 23, 6386–6392 CrossRef CAS PubMed.
- P. Zhou, R. Xing, Q. Li, J. Li, C. Yuan and X. Yan, Matter, 2023, 6, 1945–1963 CrossRef CAS.
- R. D. Chakravarthy, I. Sahroni, C. W. Wang, M. Mohammed and H. C. Lin, ACS Nano, 2023, 17, 11805–11816 CrossRef CAS PubMed.
- S. C. Yuan, J. A. Lewis, H. Sai, S. J. Weigand, L. C. Palmer and S. I. Stupp, J. Am. Chem. Soc., 2022, 144, 16512–16523 CrossRef CAS PubMed.
- Y. Hou, Y. Tian, J. Tian, J. Shi, H. Zhao, J. Hu and Y. Zhang, ACS Appl. Mater. Interfaces, 2023, 15, 29927–29938 CrossRef CAS PubMed.
- G. Gunay, S. Hamsici, G. A. Lang, M. L. Lang, S. Kovats and H. Acar, Adv. Sci., 2022, 9, e2105868 CrossRef PubMed.
- S. H. Hiew, Y. Lu, H. Han, R. A. Goncalves, S. R. Alfarano, R. Mezzenga, A. N. Parikh, Y. Mu and A. Miserez, J. Am. Chem. Soc., 2023, 145, 3382–3393 CrossRef CAS PubMed.
- R. D. Murphy, R. V. Garcia, S. J. Oh, T. J. Wood, K. D. Jo, J. Read de Alaniz, E. Perkins and C. J. Hawker, Adv. Mater., 2023, 35, e2207542 CrossRef PubMed.
- A. Wu, Y. Guo, M. Li, Q. Li, H. Zang and J. Li, Angew. Chem., Int. Ed., 2023, e202314368 CAS.
- X. Yang, B. Wu, J. Zhou, H. Lu, H. Zhang, F. Huang and H. Wang, Nano Lett., 2022, 22, 7588–7596 CrossRef CAS PubMed.
- S. Jia, S. Ji, J. Zhao, Y. Lv, J. Wang, D. Sun and D. Ding, Small Methods, 2023, 7, e2201409 CrossRef PubMed.
- Y. Wang, X. Li, D. Zheng, Y. Chen, Z. Zhang and Z. Yang, Adv. Funct. Mater., 2021, 31, 2102505 CrossRef CAS.
- O. J. Ibukun, M. Gumtya, S. Singh, A. Shit and D. Haldar, Soft Matter, 2023, 19, 3215–3221 RSC.
- Y. Xiang, H. Mao, S. C. Tong, C. Liu, R. Yan, L. Zhao, L. Zhu and C. Bao, ACS Nano, 2023, 17, 5536–5547 CrossRef CAS PubMed.
- K. I. Min, S. W. Lee, E. H. Lee, Y. S. Lee, H. Yi and D. P. Kim, Adv. Funct. Mater., 2018, 28, 1705729 CrossRef.
- M. Vilela-Picos, F. Novelli, A. Pazó, A. Méndez-Ardoy, G. Marafon, M. Amorín, A. Moretto and J. R. Granja, Chem, 2023, 9, 3365–3378 CAS.
- J. Lee, M. Ju, O. H. Cho, Y. Kim and K. T. Nam, Adv. Sci., 2019, 6, 1801255 CrossRef PubMed.
- X. Mu, J. S. K. Yuen, Jr., J. Choi, Y. Zhang, P. Cebe, X. Jiang, Y. S. Zhang and D. L. Kaplan, Proc. Natl. Acad. Sci. U. S. A., 2022, 119, e2115523119 CrossRef CAS PubMed.
- D. Kam, A. Olender, A. Rudich, Y. Kan-Tor, A. Buxboim, O. Shoseyov and S. Magdassi, Adv. Funct. Mater., 2023, 33, 2210993 CrossRef CAS.
- Y. Zhou, Q. Li, Y. Wu, X. Li, Y. Zhou, Z. Wang, H. Liang, F. Ding, S. Hong, N. F. Steinmetz and H. Cai, ACS Nano, 2023, 17, 8004–8025 CrossRef CAS PubMed.
- Y. Li, G. Yang, L. Gerstweiler, S. H. Thang and C.-X. Zhao, Adv. Funct. Mater., 2023, 33, 2210387 CrossRef CAS.
- M. De Palma, D. Biziato and T. V. Petrova, Nat. Rev. Cancer, 2017, 17, 457–474 CrossRef CAS PubMed.
- J. Vandooren, G. Opdenakker, P. M. Loadman and D. R. Edwards, Adv. Drug Delivery Rev., 2016, 97, 144–155 CrossRef CAS PubMed.
- J. Son, S. Parveen, D. MacPherson, Y. Marciano, R. H. Huang and R. V. Ulijn, Biomater. Sci., 2023, 11, 6457–6479 RSC.
- Y. Marciano, V. Del Solar, N. Nayeem, D. Dave, J. Son, M. Contel and R. V. Ulijn, J. Am. Chem. Soc., 2023, 145, 234–246 CrossRef CAS PubMed.
- W. Zhang, J. J. Hu, R. Liu, J. Dai, L. Yuan, Y. Liu, B. Chen, M. Gong, F. Xia and X. Lou, Adv. Sci., 2023, 10, e2207228 CrossRef PubMed.
- S. Zeng, X. Liu, Y. S. Kafuti, H. Kim, J. Wang, X. Peng, H. Li and J. Yoon, Chem. Soc. Rev., 2023, 52, 5607–5651 RSC.
- S. L. Porter, S. M. Coulter, S. Pentlavalli and G. Laverty, Macromol. Biosci., 2020, 20, e2000115 CrossRef PubMed.
- Y.-Y. Huang, Y. Tian, X.-Q. Liu, Z. Niu, Q.-Z. Yang, V. Ramamurthy, C.-H. Tung, Y.-Z. Chen and L.-Z. Wu, Mater. Chem. Front., 2018, 2, 1893–1899 RSC.
- D. A. Fancy and T. Kodadek, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 6020–6024 CrossRef CAS PubMed.
- S. W. P. Greg, S. Harms, J. F. Hedstrom and C. K. Johnson, J. Fluoresc., 1997, 7, 273–282 CrossRef.
- M. L. Chiara Cabrele and A. G. Beck-Sickinger, J. Org. Chem., 1999, 64, 4353–4361 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ma00024b |
| This journal is © The Royal Society of Chemistry 2024 |