
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Strengthening eco-friendly packaging from pectin by filling with poly(ε-caprolactone) nanoparticles and tailoring the degree of methyl-esterification†
Marcos Vinicius
Lorevice‡
*ab,
Graziela Solferini
Baccarin
ab,
Juliana Reghine
Souza
ab,
Pedro Ivo Cunha
Claro
bc,
Márcia Regina
de Moura
d,
Caio Gomide
Otoni
 *ce and
Luiz Henrique Capparelli
Mattoso
acd
*ce and
Luiz Henrique Capparelli
Mattoso
acd
aPPGQ, Department of Chemistry, Federal University of São Carlos – Rodovia Washington Luís km 235, São Carlos, SP 13566-905, Brazil. E-mail: marcos.lorevice@lnnano.cnpem.br; caio.otoni@ufscar.br
bNational Nanotechnology Laboratory for Agriculture (LNNA), Embrapa Instrumentation – Rua XV de Novembro 1452, São Carlos, SP 13560-970, Brazil
cGraduate Program in Materials Science and Engineering (PPGCEM), Federal University of São Carlos – Rodovia Washington Luís km 235, São Carlos, SP 13566-905, Brazil
dDepartment of Physics and Chemistry, FEIS, São Paulo State University – Av. Brasil 56, Ilha Solteira, SP 15385-000, Brazil
eDepartment of Materials Engineering (DEMa), Federal University of São Carlos – Rodovia Washington Luís km 235, São Carlos, SP 13566-905, Brazil
First published on 24th June 2024
Abstract
In light of the environmental damage caused by conventional polymers and the challenge of making polysaccharides competitive in food packaging, there is a growing investment in the development of nanoparticles to enhance their physicochemical properties. Further advancing this concept, we have produced poly(ε-caprolactone) nanoparticles (PCLNPs) via solvent displacement and incorporated them into low- and high-methoxyl pectin films by solvent casting. The PCLNPs display a quasi-spherical morphology with a uniform size (ca. 300 nm), exhibiting excellent colloidal stability due to their hydrophobic PCL core and a hydrophilic Tween 80 surface. PCLNPs moderately improved the thermal stability (reaching ca. 250 °C) and preserved the water vapor barrier (<2 g mm k−1 Pa−1 h−1 m−2) of pectin-based films, while providing significant flexibility and achieving a 3.5-fold increase in tensile strength (75 MPa) compared to neat pectin films due to favorable matrix/filler compatibility. Therefore, our findings contribute to the avenue paved for nanocomposites based on polysaccharides to serve as alternatives to nonbiodegradable and/or nonrenewable packaging, reducing environmental impact.
Introduction
Plastic pollution has become a major global concern, as its annual production increases 4% per year and over 10 million tonnes will be produced by 2050, and no effective strategy has been used to deal with this environmental issue. Moreover, concerns regarding microplastic particles1 have grown, since they can be potentially harmful when carrying contaminants, and more than 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 additives and hydrophobic organic compounds can accumulate in living organisms through the ingestion of contaminated water.2–4
000 additives and hydrophobic organic compounds can accumulate in living organisms through the ingestion of contaminated water.2–4
Recently, these concerns have been uniting efforts to produce eco-friendly packaging from materials that exhibit biodegradability and renewable characteristics, with physicochemical properties comparable or superior to conventional plastics.5,6 These biopolymers and biodegradable polymers can produce bio-based plastics, which align with sustainable packaging, as they can reduce the environmental carbon footprint and generation of microplastics through complete biodegradation, which is an effective route towards circular economy. In this context, polysaccharides and polypeptides have risen as effective and sustainable alternatives to produce eco-friendly food packaging.2,7 Pectin, specifically, has been widely exploited owing to its great film-forming capability, leading to cohesive and transparent films that may also be edible4,8–10 and it is recognized as safe for human consumption (GRAS) by the US Food and Drug Administration (FDA).11,12
Pectin is an anionic polysaccharide having D-α-1,4-galacturonic acid as 65% of its monomeric units, plus two smooth and hairy regions, where neutral sugars such as galactose and glucose are located.13,14 Pectin isolation from biomass determines the methyl-esterification degree (MD): high (HMD, MD > 50%) and low (LMD, MD < 50%) degree of methoxylation.15 The MD interferes with the physical properties of pectin (e.g., gelling behavior) and of the materials assembled from it, and they can be driven by environmental factors such as temperature, pH, and the presence of sugars or metal ions.16 Low pH induces gelling in HMD, while LMD is only gelled when added by electrolytes in solutions. In this case, divalent cations can interact with the anionic moieties of the side carboxylic groups of LMD pectin, generating the so-called “egg-box” structure.17
Although the characteristics of pectin favor its use as a food packaging material, films based on pectin lack mechanical (rigid and brittle) properties and thermoplasticity.18,19 Furthermore, although pectin-based films present a good oxygen barrier, their bio-based intrinsic hydrophilic characteristic makes them permeable to water vapor, reducing their applicability in food packaging.20,21 On the other hand, traditional polymers such as polyethylene terephthalate (PET) exhibit high mechanical strength (∼60 MPa) with malleable characteristics at room temperature and high thermal stability (>300 °C), and are also an excellent barrier to water vapor in food packaging.22
Filling biopolymer matrices with nanostructures has been widely exploited to pave a way to overcome these drawbacks and produce nanocomposites with new or superior properties.23–26 In this context, poly(ε-caprolactone) (PCL) nanoparticles (PCLNPs) resurface as simple nanostructures that can be used as additives to enhance the properties of polysaccharide matrices.26 The main processes for obtaining PCLNPs involve solvent displacement or precipitation of a pre-synthesized polymer, allowing the encapsulation of various compounds.27 Due to this characteristic, PCLNPs have been used in several applications, encompassing the controlled release of drugs and active ingredients and tissue engineering systems.28–32 It is important to note that PCLNPs have shown greater notoriety due to being based on PCL, which is both biocompatible and biodegradable.33,34 Furthermore, PCL is a semicrystalline polymer with a glass transition temperature of ca. −60 °C and a melting temperature of ca. 60 °C, desirable for several food packaging and biomedical purposes.34
In summary, while PCLNPs offer excellent chemical stability and mechanical strength, making them highly durable in various applications, the durability of pectin depends on factors such as its biodegradability, mechanical properties, and environmental sensitivity. Both materials have unique characteristics and suitability for different purposes (scaffolds, films, and porous materials), with PCLNPs often preferred for long-term applications and pectin chosen for its biodegradability and environmental friendliness.35 Considering this scenario, this study hypothesized that the characteristics of the PCL combined with the PCLNP surface constituted by a non-ionic surfactant could promote physicochemical improvements in pectin nanocomposites. Moreover, the surfactant, as well as pectin MD would be responsible for compatibilizing filler/matrices, promoting nanoparticle–polysaccharide chain interactions, and consequently producing a robust and competitive nanocomposite.
We report on the incorporation of PCLNPs into pectin films, a combination of hydrophobic nanofillers with hydrophilic biopolymers. The effect of the methyl-esterification degree was also investigated, which surprisingly indicated more affinity of the LMD matrix to the nanofiller. PCLNPs had their morphological and surface attributes characterized by SEM and DLS analysis, while PCLNP-added pectin films were characterized in terms of morphology (SEM) as well as physical, thermal (TGA and DSC), and mechanical properties (tensile strength, Young's modulus), revealing exceptional mechanical improvement, without losing an eco-friendly packaging.
Experimental section
Materials
PCL CAPA TM 6500 (Mw = 50000 g mol−1) was provided by Perstop Winnin Formulas (Malmö, Sweden). Acetone and Tween 80 (polyoxyethylene 20 sorbitol monoleate) were obtained from Sigma-Aldrich Ltd (São Paulo, Brazil). High-methyl (HMD; MD = 74%) (GENUSET-Z, Mw = 130000 g mol−1) and low-methoxyl (LMD; MD = 8.4%) (USPB – Mw = 170000 g mol−1) pectins from citrus peel were obtained from CP Kelco (Limeira, Brazil).
Methods
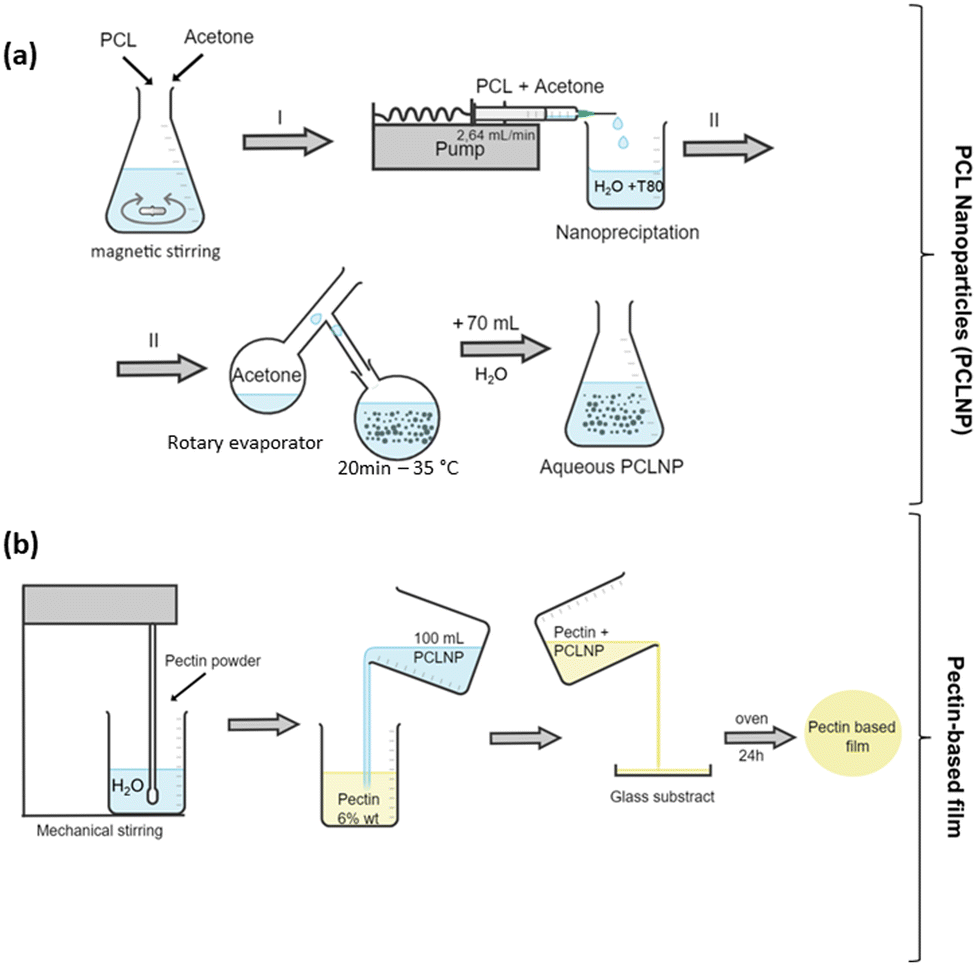 | ||
| Fig. 1 Illustration of the experimental procedure of the PCL NPs’ synthesis (a) by nanoprecipitation and Pectin-based film production (b). | ||
Fourier-transform infrared (FT-IR) spectra of films (1 mg film sample plus 100 mg KBr mashed into pellets) were obtained by 128 scans at a resolution of 2 cm−1 at wavenumbers ranging from 4000 to 600 cm−1, on a Paragon 1000 infrared spectrometer (Perkin-Elmer, Inc., USA). The films were pre-conditioned to 0% humidity before the FT-IR measurements.
X-ray photoelectron spectroscopy (XPS) (Thermo Scientific K-α) was carried out with Mg Kα (hυ = 1253.6 eV) at low pressure and 10 eV. The dried films were cut to 2 mm × 2 mm and examined for C 1s measurements. The films were pre-conditioned to 0% humidity before the measurements, which were performed in three different regions of each sample. The raw data and peak deconvolution were determined using an Avantage Data System (Thermo ScientificTM).
The morphology of the PCLNPs and the films (surface and cross-sections) was analyzed by scanning electron microscopy (SEM) on a Carl Zeiss Supra 35 (VP, Germany). PCLNPs were diluted (1:10 v/v) and poured into silicon surfaces. The internal morphology of the films was imaged after cryogenic fracture in nitrogen liquid. All samples were fixed onto stubs and coated with a gold layer (Denton Vaccum Inc.,USA) for 45 s at 20 mA before being analyzed.
Film thermal stability was evaluated by thermogravimetry on Q-500 TGA equipment (TA Instruments, Inc., USA): 5–7 mg sample was heated from 30 to 700 °C at 10 mL min−1 within a synthetic air atmosphere. Differential scanning calorimetry (DSC) was performed on a Q100 equipment (TA Instruments, Inc., USA) using dried films, which were heated at 10 mL min−1 from −80 °C to 200 °C within a nitrogen atmosphere (50 mL min−1).
Film thickness was measured in five randomly different regions per sample using a micrometer (Mitutoyo Corp., Japan). The mechanical attributes such as tensile strength (TS), Young's modulus (E), and elongation at break (EB) were acquired through uniaxial tensile assay (ASTM D882, 2012): rectangular specimens (100 × 15 mm2) were equilibrated at 50% relative humidity (RH) for 48 h at room temperature and then tested on a tensile testing machine (Instron Corp., USA) at a stretching rate of 10 mm min−1 and with a load cell of 0.01 kN. At least eight replicates were performed for each sample.38 TS was calculated by dividing the maximum tensile force by the original cross-sectional area of the film. The difference in the final sample length (at break) and initial length was normalized by the onset dimension and multiplied by 100 to give percentage values of EB. Young's modulus was calculated from the slope of tensile stress versus strain curves in the initial, linear portion of the curve.
Water vapor permeability (WVP) was determined through the modified gravimetric method.39 Briefly, film samples were cut in circular shapes (65 mm in diameter) and allocated in poly(methyl methacrylate) cups filled with ultrapure water (6 mL), creating a semipermeable barrier between a low- (external) and high-(internal) RH environment. Chamber RH was held by dried silica as low as possible during the WVP tests. For a period of 24 h at 25 °C, the weight of the cups was monitored. Intervals of two hours between the measurements were used to allow chamber RH stabilization. Four replications were used to calculate WVP (g mm kPa−1 h−1 m−2).
The Origin (version 10.1.0.178) software (OriginLab, Inc. USA) was used to perform the data processing. The Minitab (version 14.12.0) software (Minitab, Inc., USA) was used to analyze all obtained quantitative data through analysis of variance (ANOVA) followed by Tukey's test at 5% of significance for mean comparison.
Results and discussion
Poly(ε-caprolactone) nanoparticles
The morphology and effective functionalization of the PCLNP with Tween 80 are shown in Fig. 2. The size and shape homogeneity of the PCLNP (Fig. 2a and b) is consistent with other reports on particles with diameters around 400 nm.36,40–42 The hydrodynamic diameter of the PCLNP obtained in the three different syntheses did not differ (p > 0.05) from each other: ca. 131 nm (Fig. 1c and Table S1, ESI†). The polydispersity index (PdI) below 0.3 (p > 0.05; Table S1, ESI†) indicates a monomodal size dispersion with a great degree of reproducibility (Fig. 1c).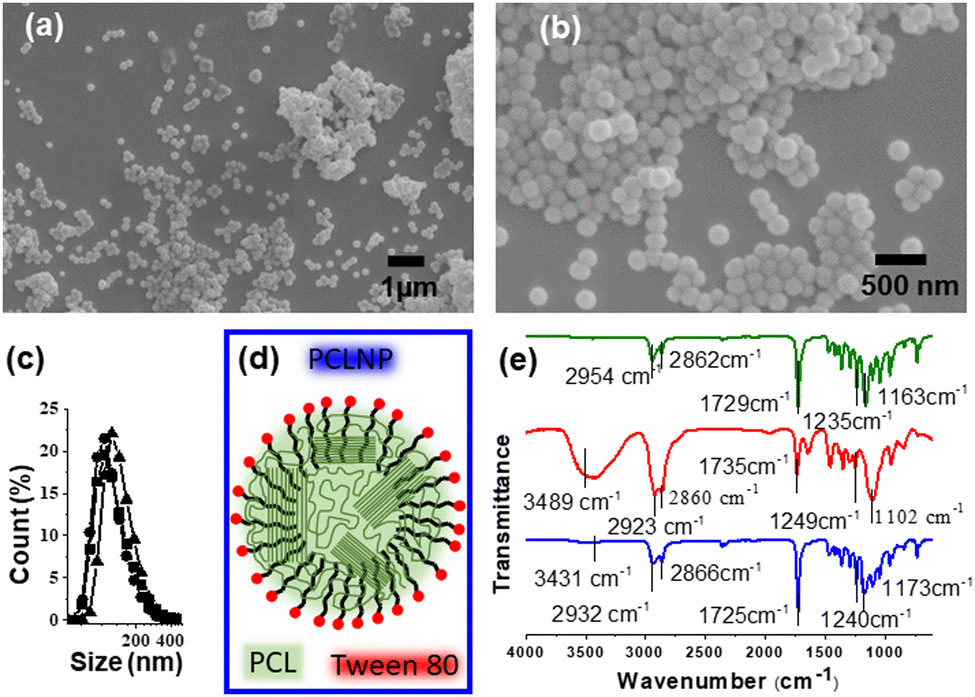 | ||
| Fig. 2 (a and b) Scanning electron microscopy images of poly(ε-caprolactone) nanoparticles (PCLNPs) at different magnifications. (c) Representation of the size distribution of three different syntheses of poly(ε-caprolactone) (PCL) nanoparticles obtained by dynamic light scattering (DLS). (d) Schematic structure of PCLNPs. (e) FT-IR spectra of PCL (green), Tween 80 (red), and PCLNPs (blue). | ||
PCLNPs exhibited negative and constant zeta potential values (ca. −17 mV) for all syntheses (p > 0.05) (see Table S1, ESI†), which might have been enough to ensure electrostatic repulsion in suspension and to lessen aggregation during the drying of the nanocomposite film. The negative values can be related to the particle surface composition of PCL and Tween 80 (Fig. 2d). The surface potential is influenced by changes in the interface of the nanoparticles (inner region, Stern layer) and the dispersing environment (outer region), due to dissociation or ionization of the functional groups present on the surface of the nanoparticles, or due to adsorption of ion species onto the particles’ surface.43 In this case, PCL tends to show a negative character promoted by the carbonyl groups,42 which could orient themselves to the surface of the PCLNP, mainly due to the higher electron density around the oxygen atom; however, the low absolute zeta potential value suggests that this conformation is not predominant for the surface of the PCLNP produced here.
The surfactant adsorption onto the nanoparticle surface can interfere with its surface charge. Tween 80 acts by stabilizing the suspension, avoiding particle agglomeration and precipitation. Thus, once PCL is insoluble, the surfactant is expected to adhere to the surface of the PCLNP, interacting with the nonpolar region of the structure (Fig. 2d). The hydrophobic region of polysorbate interacts with the PCLNP surface, directing the hydrophilic part to the external region, which has ionized hydroxyl groups, releasing protons for the water molecule, and thus leaving the anionic oxygen (O−), leading to the negative zeta potential of PCLNPs. Such interactions could also suggest interaction with the polysaccharide matrix to which it was added.
Fig. 2e shows the infrared spectra of PCL, Tween 80, and PCLNPs. Possible changes in the specific functional groups are identified in Table S2 (ESI†), related to the interactions between PCL and the surfactant in the nanostructure. The characteristic bands of PCL can be identified at 2944 cm−1 (asymmetric stretching of the CH2 group), 2867 cm−1 (symmetric stretching of the CH2 group), 1729 cm−1 (stretching of the carbonyl group), 1238 cm−1 and 1163 cm−1 (asymmetric and symmetric stretching of the –C–O–C– group, respectively).44 Due to the existence of similar functional groups in PCL and Tween 80, the spectrum of Tween 80 showed similar bands: 2923 and 2872 cm−1 related to asymmetric and symmetric stretching (–CH); 1735 cm−1 linked to stretching of carbonyls (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O); 1235 and 1136 cm−1, relating respectively to the asymmetrical and symmetrical stretches of the –C–O–C– group. The spectrum of PCLNPs showed slight deviations in the characteristic PCL bands, referring mainly to the groups that coexist in the hydrophobic region of the surfactant: 2932 and 2866 cm−1 (asymmetrical and symmetrical elongations –CH, respectively); 1725 cm−1 (CO stretching); 1240 and 1173 cm−1 (symmetrical and asymmetrical group elongations of –C–O–C–); which suggests interactions of the surface of PCL with the surfactant, corroborating the hypothesis that it is stabilizing the nanoparticle surface (Fig. 2d). Considering that the PCLNP surface is coated by the surfactant, this implies that the hydrophilic region of the surfactant will interact with the adjacent pectin chains (Fig. 2d). Thus, during this discussion, when a PCLNP is cited, the discussion will rely on the surface of the surfactant-coated PCLNP that makes the interface with the pectin matrix.
O); 1235 and 1136 cm−1, relating respectively to the asymmetrical and symmetrical stretches of the –C–O–C– group. The spectrum of PCLNPs showed slight deviations in the characteristic PCL bands, referring mainly to the groups that coexist in the hydrophobic region of the surfactant: 2932 and 2866 cm−1 (asymmetrical and symmetrical elongations –CH, respectively); 1725 cm−1 (CO stretching); 1240 and 1173 cm−1 (symmetrical and asymmetrical group elongations of –C–O–C–); which suggests interactions of the surface of PCL with the surfactant, corroborating the hypothesis that it is stabilizing the nanoparticle surface (Fig. 2d). Considering that the PCLNP surface is coated by the surfactant, this implies that the hydrophilic region of the surfactant will interact with the adjacent pectin chains (Fig. 2d). Thus, during this discussion, when a PCLNP is cited, the discussion will rely on the surface of the surfactant-coated PCLNP that makes the interface with the pectin matrix.
Pectin/PCLNP nanocomposite films
The control pectin and pectin/PCLNP nanocomposite films were produced using the casting method, and their visual appearance and microstructure are shown in Fig. 3. SEM images of the film cross-sections indicate the morphology, homogeneity, and possible imperfections in the pectin/PCLNP matrix. Fig. 3(b) and (d) compare the cryo-fractured cross-sections of control pectin films (both HMD and LMD), while Fig. 3(d) and (h) compares HMD/PCLNP and LMD/PCLNP films. The HMD film (Fig. 3b) shows some deformations and cracks, although the LMD pectin film resulted in a compact and smooth matrix (Fig. 3d). PCLNP incorporation resulted in no evidence of pores or imperfections, a desirable characteristic that is expected not to impair the mechanical performance of the pectin/PCLNP nanocomposites. | ||
| Fig. 3 Digital photographs of control HMD (a) and LMD (c) pectin films and HMD/PCLNP (e), and LMD/PCLNP (g) nanocomposite films. SEM images of the cryo-fractured cross-sections of HMD (b), LMD (d), HMD/PCLNP (f), and LMD/PCLNP (h) films. FT-IR spectra of PCLNP ((i) and (j); blue line), HMD ((i); black line), LMD ((j); red line), HMD/PCLNP ((i); green), and LMD/PCLNP ((j); orange line) films. | ||
Fig. 3(i) and (j) exhibit the infrared spectra of PCLNPs as well as the control and nanocomposite films. Peaks between 3000 and 2900 cm−1, related to O–CH3 elongations that are characteristic of both pectin and PCLNPs, can be identified with no significant band shifts, as observed for the peak at 1637 cm−1, related to the stretching of the non-esterified carboxylic group characteristic of pectin.45,46 However, a shift in the peak related to the carboxyl elongation for the two nanocomposites can be noted: 1731 cm−1 (HMD/PCLNP) and 1739 cm−1 (LMD/PCLNP). This suggests that somehow the PCLNP surface and pectin chains are interacting by hydrogen bonds.
The XPS analysis was performed to investigate the composition of the samples and the potential interactions between the film components. Fig. S1 (ESI†) illustrates the C 1s deconvolutions, which resulted in four distinct C peaks: C–C/C–H (∼285 eV), C–O (∼287 eV), O–C–O (∼288 eV), and O–CO (∼289 eV), the latter is associated with the contribution of ester groups. Interestingly, shifts in binding energy were observed from pectin-based films (HMD and LMD) to nanocomposites (Table S3, ESI†). For instance, in the LDM-nanocomposite, the O–C–O peak shifted from 287 to 287.1 eV (C–O–H), 285.4 to 285.1 eV (C–C), 288.4 to 286.8 eV (O–C–O), and 289.7 to 289.6 eV, indicating that these functional groups may be involved in the interactions between pectin chains and PCLNP surfaces, as previously mentioned in the infrared data (Fig. 3). Furthermore, the presence of functional groups such as C–O, O–C–O, C–O–H, and O–CO is consistent with the composition of polysaccharide-based nanocomposites.47–49 Additionally, LMD pectin exhibited higher quantities of COH and O–CO due to its lower MD value. In HMD nanocomposites, variations in the C–O and O–C–O peaks occur due to the addition of PCL nanoparticles coated with Tween 80 (Fig. 2) to the composition, and these components contain higher amounts of carbonyl and hydroxyl groups. In contrast, in LMD nanocomposites, the presence of these groups was more pronounced in the aliphatic regions of the compounds. This was a result of the addition of both an aliphatic chain surfactant and a hydrophilic region composed of hydroxyls and carbonyls, as well as PCL, as observed by the increase in the magnitude of the C–C/C–H peak (Table S4, ESI†).
Fig. 4 exhibits the TGA, DTG, and DSC curves related to thermal analyses of the pectin-based films. Table 1 presents the parameters extracted from these curves. No interference of the MD is noticed on TGA and DTG profiles (Fig. 4(a)–(d)). From TGA curves, the event around 150 °C is related to matrix dehydration and water evaporation, representing ca. 12% of the initial mass.50,51 Additionally, two further degradation stages at ca. 250 and ca. 440 °C are clearly identified in TGA and DTG curves (Fig. 4(b) and (d)), where the onset values related to the first stage are described in Table 1. The first degradation stage at ca. 250 °C is related to pectin degradation.52–54 The second stage at ca. 440 °C is related to polysaccharide carbonization.55 The incorporation of PCLNPs into pectin increased in around 10 °C the thermal stability of the films, suggesting that the high thermal stability of PCL (>330 °C) may have influenced the thermal stability of pectin films.33 The DSC curves indicate that the MD did not influence the endothermic peak temperature (EPT) and the endothermic event (ΔH) of pectin films (Fig. 4e). In neat pectin films, the EPT values ranging from 100 to 150 °C, are related to water evaporation from the polysaccharide matrix.56,57 HMD and LMD/PCLNP films displayed two endothermic processes: at 60 °C and around 110 °C. The first event is correlated to the melting of PCL, and the second is probably due to water desorption. The ΔH values involved in this process indicate a larger amount of energy is required for water evaporation.
 | ||
| Fig. 4 Thermogravimetric (a)–(d) and differential scanning calorimetric (e) curves of high (HMD – red line) and low (LMD – black line) pectin films and incorporated with poly (ε-caprolactone) (green and blue line, respectively). | ||
| Samples | DT (°C) | EPT (°C) | ΔH (J g−1) | |
|---|---|---|---|---|
| HMD | 229 | 122 | 367.9 | |
| LMD | 227 | 124 | 426.2 | |
| NPPCL | 356 | 69 | 115 | |
| HMD/PCLNP film | 238 | 53 | 112 | 478.9 |
| LMD/PCLNP film | 237 | 54 | 110 | 462.1 |
The tensile strength, Young's modulus, and elongation at break of the films are summarized in Fig. 5. All attributes were statistically constant (p > 0.05) for LMD and HMD pectin films, in contrast with previous works where LMD promoted a compact network generated by the higher quantity of hydrogen bonds.15
 | ||
| Fig. 5 Mechanical properties of pectin-based films: (a) tensile strength, (b) Young's modulus, and (c) elongation at break. Different letters in which graph (a) or (b) mean values with statistical difference (p < 0.05) in the same graphs. Tensile strength vs. elongation curves for HMD (d) and (e) LMD pectin films. (f) Proposed scheme of PCLNPs dispersed in a pectin matrix: in the blue dot–dot square the illustration of the percolation phenomenon, and in the red dot–dot square the intermolecular interactions of pectin chains and PCLNP surface. (g) Schematics of the tortuous pathway of water molecule diffusion in the pectin matrix (right) and pectin/PCLNP matrix (left). | ||
Interestingly, the addition of PCLNPs significantly increased the tensile strength in comparison with neat pectin films (HMD and LMD) (p < 0.05) (Fig. 5a). HMD/PCLNP and LMD/PCLNP films showed 2.5- (Fig. 5b) and 3.5-fold (Fig. 5c) enhancements in tensile strength compared to neat pectin films, respectively. The tensile strength of the HMD/PCLNP produced in this work exceeded that of pectin films reinforced with nanoparticles reported in previous studies. For example, HMD/AgNPs (25 MPa),58 HMD/CNC/ZnNPs (13 MPa),59 or HMD/garlic essential oil nanoemulsion60 (30 MPa) pectin films reached values lower than 72.5 MPa of HMD/PCLNP produced in this work. Similarly, LMD/PCLNP (74 MPa) exhibited higher tensile strength compared to LMD pectin films incorporated with chitosan (58 MPa)51 or cooper/betanin pigment61 (7.49 MPa). It is worth noting that the tensile strength of LMD/PCLNP surpassed that of polyethylene terephthalate (PET) (∼60 MPa).22
Although the Young's modulus and elongation at break remained statistically constant, the nanocomposites exhibited notable stiffness, reaching values over 5 GPa (Fig. 5(d) and (e), respectively), values comparable or superior to common plastics used in food packaging (see Fig. S2, ESI†).
The remarkable mechanical reinforcement can be attributed to (1) the good dispersion level of PCLNPs within the pectin matrix; (2) the intrinsic mechanical stiffness of PCL (Young's modulus up to 386 MPa),62 as well as (3) the favorable interaction between PCLNPs and pectin, as indicated by SEM, FT-IR, and XPS.
The large specific surface area of the PCLNP with electrochemical charges can result in a greater chemical affinity of PCLNP/pectin by hydrogen bonds, creating a cohesive interface PCL–surfactant–pectin. This hydrogen bond hypothesis is corroborated by the significant increase of tensile strength in LMD pectin films, suggesting that PCLNPs interact more effectively with carboxylic groups (LMD pectin) than with esterified groups (HMD pectin), as evidenced by the FT-IR spectra in Fig. 3(i) and (j), and the peak shifts in binding energy observed in XPS analyses (Table S3, ESI†).
Beyond the interface interactions, understanding terms like the percolation threshold and interparticle layer may clarify the considerable results in mechanical improvement.63,64 The percolation threshold refers to the point at which PCLNPs, at a concentration of 3.2 wt% in relation to the dried film, aggregate into ever-growing clusters, forming an infinite structure above this threshold. This percolated network enhances mechanical properties by increasing the energy required to disrupt polymer/nanoparticle and nanoparticle/nanoparticle interactions, thereby reinforcing the polymeric matrices (Fig. 5f).65–67 Moreover, the interparticle layer, composed of the surfactant, also contributes to the mechanical improvement of the nanocomposites. This layer facilitates the transmission of external forces through the nanoparticle–interlayer–nanoparticle chain.68–70 Additionally, it is proposed that this interlayer may interpenetrate with the LMD polymer chain, further enhancing mechanical properties.
The WVP of neat pectin and nanocomposite films was evaluated, as per Table 2. The WVP values did not show significant changes (p > 0.05), neither by the difference in pectin MD (HMD or LMD) nor by the addition of PCLNPs. The process of water vapor permeation through polymer films, semipermeable membranes, occurs mostly by diffusion and it can be altered by the characteristics of particulate additives incorporated into the matrix, e.g. plasticizer,71 besides effects connected to the material hydrophobicity,72 In parallel, WVP changes may be attributed to the existence of pores in the film matrix, or the tortuous pathway caused by the incorporation of components (or particles) to the film.73–77
| Sample | Thickness (μm) | WVP (g mm k−1 Pa−1 h−1 m−2) |
|---|---|---|
| ab Mean values ± standard deviations in the column followed by the same lowercase superscript letters within a column are not different (p > 0.05). | ||
| HMD film | 29 ± 8ab | 1.5 ± 0.2a |
| LMD film | 19 ± 4a | 1.4 ± 0.3a |
| HMD/PCLNP film | 35 ± 8b | 1.9 ± 0.3a |
| LMD/PCLNP film | 32 ± 6b | 1.8 ± 0.9a |
For the system analyzed here (pectin and PCLNPs), we propose a complementary understanding of the WVP mechanism (Fig. 5g), comprising the electrostatic repulsion of matrixes/fillers to water droplets. The zeta potential of PCLNPs and the carboxylic (in higher quantity in LMD pectin) and carbonyl (in higher quantity in HMD pectin) groups can exhibit negative charges, due to the dielectric bilayer on the particle surface and ionized groups of pectin in polymer dispersion. In the HMD and LMD control pectin films, the carboxylic (–COOH) and carbonyl (CO) groups, respectively, present a higher electronic density in the oxygen atoms due to their electronegativity, resulting in a negative formal charge in this region. In the water vapor permeation process, the diffusion of water molecules was not easy due to repulsion of –COOH (or –CO) groups in the polymer matrix or PCLNP surface (Fig. 5g). The unchanged WVP (p > 0.05) when PCLNPs were added suggests that, owing to the negative zeta potential of PCLNPs, the repulsion of water molecules during the permeation resulted in unchanged WVP values.
This contrasts with nanocomposites from pectin and chitosan nanoparticles, a system already studied by this group, where the positive zeta potential values of chitosan nanoparticles cancelled the repulsion of molecules of water, helping the permeation of water vapor.77 Additionally, the existence of the surfactant on the PCLNP surface or dissolved/micellized throughout the pectin matrix could influence WVP. Surfactant molecules from PCLNPs have a hydrophilic head (red spherical structure, Fig. 1d), which could interact with the carboxylic groups from lateral chains of pectin, and could also interact with water molecules, swelling the polymeric matrix and enabling water diffusion through the films. The surfactant, in this case, would be acting as a plasticizer, decreasing the film barrier properties.
In summary, the findings of this study demonstrate that pectin-based films with PCLNPs are competitive compared to conventional polymers widely used in the packaging industry.78 This is mainly due to their thermal stability, reaching up to 250 °C, which allows for versatile use under various food storage conditions. Furthermore, the enhanced mechanical strength enables the application of these biopolymers in packaging requiring resistance to forces during handling and transportation. Additionally, the lower WVP values observed for pectin-based films can potentially reduce mass loss, color alteration, pH variation, and microbial proliferation in packaged foods.
Finally, when considering the characteristics of PCL and pectin, the environmental impact of PCLNP-enhanced pectin films can be assessed based on the biodegradability features of both pectin and PCL.79 Additionally, the natural sourcing of pectin from fruit peels and the water solvent approach used to produce pectin-based films contribute to their sustainability.80 These combined characteristics make the PCLNP system with pectin a highly sustainable option compared to traditional polymers for food packaging applications.
Conclusions
Poly(ε-caprolactone) nanoparticles (PCLNPs) were successfully produced with monomodal size distribution around 300 nm and spherical morphology. The particle surface composition of PCL and Tween 80 drove PCLNP colloidal stability and negative charge on the nanoparticle surface. PCLNPs were homogeneously dispersed over the pectin matrix, regardless of the degree of methyl-esterification, demonstrating reasonable flexibility and transparency. Interestingly, PCLNPs slightly enhanced the thermal stability of pectin films, and decreased the degree of hydration. In addition, the PCLNPs maintained the high resistance of pectin films against the permeation of water vapor. The remarkable increment in mechanical resistance suggests good pectin–PCLNP compatibility, especially for LMD–pectin films. This increment was related to the higher occurrence of carboxyl groups in the LMD matrix promoting more hydrogen bonds with the PCLNP surface. These findings indicate the efficiency of the nanostructures in improving the physicochemical properties of pectin films, further promoting polysaccharide-based films for use in eco-friendly packaging.Author contributions
MVL: conceptualization, methodology, formal analysis, investigation, writing – original draft, and review & editing. GSB: methodology, writing and editing. CGO: methodology, conceptualization, formal analysis, writing – review & editing. JRS: methodology. PICC: writing – review & editing. MRM: supervision, conceptualization, writing – review & editing. LHCM: supervision, conceptualization, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded by the São Paulo Research Foundation (FAPESP; grant no. 2012/24362-6 and 2021/12071-6) and the Brazilian National Council for Scientific and Technological Develpment (CNPq) through the INCT Circularity in Polymer Materials (grant no. 406925/2022-4). The authors are also grateful to Embrapa Instrumentation for the infrastructure and to MCTI/SISNANO, REDE AGRONANO, CAPES, and CNPq (grant no. 304753/2022-0) for support. The authors also thank the Department of Physics and Chemistry of UNESP/FEIS for the mechanical assays and to LNNano – Brazilian Nanotechnology National Laboratory (CNPEM/MCTI) for access to the Spectroscopy and Scattering Laboratory (proposal no. 20240993).References
- D. Li, Y. Shi, L. Yang, L. Xiao, D. K. Kehoe, Y. K. Gun’ko, J. J. Boland and J. J. Wang, Nat. Food, 2020, 1, 746–754 CrossRef CAS PubMed.
- J. G. Rosenboom, R. Langer and G. Traverso, Nat. Rev. Mater., 2022, 7, 117–137 CrossRef PubMed.
- R. Geyer, J. R. Jambeck and K. L. Law, Sci. Adv., 2017, 3, 25–29 CrossRef PubMed.
- A. Satsum, W. Busayaporn, W. Rungswang, S. Soontaranon, K. Thumanu and C. Wanapu, Polym. J., 2022, 54, 921–930 CrossRef CAS.
- C. G. Otoni, R. J. Avena-Bustillos, H. M. C. Azeredo, M. V. Lorevice, M. R. Moura, L. H. C. Mattoso and T. H. McHugh, Compr. Rev. Food Sci. Food Saf., 2017, 16, 1151–1169 CrossRef PubMed.
- T. F. da S. Saranti, P. T. S. Melo, M. A. Cerqueira, F. A. Aouada and M. R. de Moura, Polymers, 2021, 13(24), 4298 CrossRef CAS PubMed.
- N. Dedhia, S. J. Marathe and R. S. Singhal, Carbohydr. Polym., 2022, 287, 119355 CrossRef CAS PubMed.
- C. Mellinas, M. Ramos, A. Jiménez and M. C. Garrigós, Materials, 2020, 13(3), 673 CrossRef CAS PubMed.
- A. Nesic, S. Meseldzija, G. Cabrera-Barjas and A. Onjia, Foods, 2022, 11, 1–11 CrossRef PubMed.
- V. G. L. Souza, I. P. Mello, O. Khalid, J. R. A. Pires, C. Rodrigues, M. M. Alves, C. Santos, A. L. Fernando and I. Coelhoso, Coatings, 2022, 12(2), 108 CrossRef CAS.
- U. S. F. and D. Administration, Code of Ferederal Regulation.
- P. J. P. Espitia, W. X. Du, R. de J. Avena-Bustillos, N. de F. F. Soares and T. H. McHugh, Food Hydrocolloids, 2014, 35, 287–296 CrossRef CAS.
- H. Würfel, K. Geitel, H. Qi and T. Heinze, BioResources, 2021, 16, 8457–8488 Search PubMed.
- S. Basak and U. S. Annapure, Carbohydr. Polym., 2022, 278, 118967 CrossRef CAS PubMed.
- F. K. V. Moreira, L. A. De Camargo, J. M. Marconcini and L. H. C. Mattoso, J. Agric. Food Chem., 2013, 61, 7110–7119 CrossRef CAS PubMed.
- E. D. Ngouémazong, S. Christiaens, A. Shpigelman, A. Van Loey and M. Hendrickx, Compr. Rev. Food Sci. Food Saf., 2015, 14, 705–718 CrossRef.
- M. Celus, C. Kyomugasho, A. M. Van Loey, T. Grauwet and M. E. Hendrickx, Compr. Rev. Food Sci. Food Saf., 2018, 17, 1576–1594 CrossRef CAS PubMed.
- J. Deng, E. Q. Zhu, G. F. Xu, N. Naik, V. Murugadoss, M. G. Ma, Z. Guo and Z. J. Shi, Green Chem., 2022, 24, 480–492 RSC.
- T. Qiang, W. Ren and L. Chen, Food Hydrocolloids, 2024, 149, 109539 CrossRef CAS.
- V. Souza, I. Mello, O. Khalid, J. Pires, C. Rodrigues, M. Alves, C. Santos, A. Fernando and I. Coelhoso, Coatings, 2022, 12, 108 CrossRef CAS.
- M. R. Sharaby, E. A. Soliman, A. B. Abdel-Rahman, A. Osman and R. Khalil, Sci. Rep., 2022, 12, 20673 CrossRef CAS PubMed.
- M. M. Ben Zair, F. M. Jakarni, R. Muniandy and S. Hassim, Sustainability, 2021, 13, 1303 CrossRef CAS.
- N. Chausali, J. Saxena and R. Prasad, J. Agric. Food Res., 2022, 7, 100257 CAS.
- A. Ashfaq, N. Khursheed, S. Fatima, Z. Anjum and K. Younis, J. Agric. Food Res., 2022, 7, 100270 CAS.
- H. M. Fahmy, R. E. Salah Eldin, E. S. Abu Serea, N. M. Gomaa, G. M. AboElmagd, S. A. Salem, Z. A. Elsayed, A. Edrees, E. Shams-Eldin and A. E. Shalan, RSC Adv., 2020, 10, 20467–20484 RSC.
- J. Sarfraz, T. Gulin-Sarfraz, J. Nilsen-Nygaard and M. K. Pettersen, Nanomaterials, 2020, 11, 10 CrossRef PubMed.
- S. Łukasiewicz, A. Mikołajczyk, E. Błasiak, E. Fic and M. Dziedzicka-Wasylewska, Pharmaceutics, 2021, 13, 191 CrossRef PubMed.
- P. Kotcharat, P. Chuysinuan, T. Thanyacharoen, S. Techasakul and S. Ummartyotin, Sustainable Chem. Pharm., 2021, 20, 100404 CrossRef CAS.
- V. Maingret, C. Chartier, J. L. Six, V. Schmitt and V. Héroguez, Carbohydr. Polym., 2022, 284, 119146 CAS.
- S. Fahimirad, H. Abtahi, P. Satei, E. Ghaznavi-Rad, M. Moslehi and A. Ganji, Carbohydr. Polym., 2021, 259, 117640 CrossRef CAS PubMed.
- F. Asghari, D. Rabiei Faradonbeh, Z. V. Malekshahi, H. Nekounam, B. Ghaemi, Y. Yousefpoor, H. Ghanbari and R. Faridi-Majidi, Carbohydr. Polym., 2022, 278, 118926 CrossRef CAS PubMed.
- F. Zhou, C. Cui, S. Sun, S. Wu, S. Chen, J. Ma and C. M. Li, Carbohydr. Polym., 2022, 282, 119131 CrossRef CAS PubMed.
- M. A. Woodruff and D. W. Hutmacher, Prog. Polym. Sci., 2010, 35, 1217–1256 CrossRef CAS.
- M. Bartnikowski, T. R. Dargaville, S. Ivanovski and D. W. Hutmacher, Prog. Polym. Sci., 2019, 96, 1–20 CrossRef CAS.
- G. Ö. Kayan and A. Kayan, ChemEngineering, 2023, 7, 104 CrossRef CAS.
- I. C. Külkamp, K. Paese, S. S. Guterres and A. R. Pohlmann, Quim. Nova, 2009, 32, 2078–2084 CrossRef.
- W. Badri, K. Miladi, S. Robin, C. Viennet, Q. A. Nazari, G. Agusti, H. Fessi and A. Elaissari, Pharm. Res., 2017, 34, 1773–1783 CrossRef CAS PubMed.
- ASTM D5988-03, ASTM International, 2012, D5988-12, 1–6.
- T. H. McHugh, R. Avena-BustilloS and J. M. Krochta, J. Food Sci., 1993, 58, 899–903 CrossRef CAS.
- M. Ansari, S. Salahshour-Kordestani, M. Habibi-Rezaei and A. A. M. Movahedi, J. Macromol. Sci., Part B: Phys., 2015, 54, 71–80 CrossRef CAS.
- M. A. Woodruff and D. W. Hutmacher, Prog. Polym. Sci., 2010, 35, 1217–1256 CrossRef CAS.
- S. R. Schaffazick, S. S. Guterres, L. D. L. Freitas, A. R. Pohlmann, J. Sarfraz, T. Gulin-Sarfraz, J. Nilsen-Nygaard and M. K. Pettersen, Quim. Nova, 2003, 26, 726–737 CrossRef CAS.
- G. V. Lowry, R. J. Hill, S. Harper, A. F. Rawle, C. O. Hendren, F. Klaessig, U. Nobbmann, P. Sayre and J. Rumble, Environ. Sci.: Nano, 2016, 3, 953–965 RSC.
- T. Elzein, M. Nasser-Eddine, C. Delaite, S. Bistac and P. Dumas, J. Colloid Interface Sci., 2004, 273, 381–387 CrossRef CAS PubMed.
- J. Lim, J. Yoo, S. Ko and S. Lee, Food Hydrocolloids, 2012, 29, 160–165 CrossRef CAS.
- X. Wang, X. Sun, H. Liu, M. Li and Z. Ma, Food Bioprod. Process., 2011, 89, 149–156 CrossRef CAS.
- J. S. Stevens and S. L. M. Schroeder, Surf. Interface Anal., 2009, 41, 453–462 CrossRef CAS.
- F. V. Ferreira, M. Mariano, S. C. Rabelo, R. F. Gouveia and L. M. F. Lona, Appl. Surf. Sci., 2018, 436, 1113–1122 CrossRef CAS.
- Y. Zhao, R. Tian, M. Cui, Y. Zhang, L. Jiang, B. Tian and X. Sui, Food Hydrocolloids, 2023, 145, 109071 CrossRef CAS.
- M. R. Martelli, T. T. Barros, M. R. de Moura, L. H. C. Mattoso and O. B. G. Assis, J. Food Sci., 2013, 78, N98–N104 CrossRef CAS PubMed.
- M. V. Lorevice, C. G. Otoni, M. R. de Moura and L. H. C. Mattoso, Food Hydrocolloids, 2016, 52, 732–740 CrossRef CAS.
- U. Einhorn-Stoll, H. Kunzek and G. Dongowski, Food Hydrocolloids, 2007, 21, 1101–1112 CrossRef CAS.
- U. Einhorn-Stoll and H. Kunzek, Food Hydrocolloids, 2009, 23, 856–866 CrossRef CAS.
- T. Giancone, E. Torrieri, P. Di Pierro, S. Cavella, C. V. L. Giosafatto and P. Masi, Food Bioprocess Technol., 2011, 4, 1228–1236 CrossRef CAS.
- F. López, M. J. Díaz, M. E. Eugenio, J. Ariza, A. Rodríguez and L. Jiménez, Bioresour. Technol., 2003, 87, 255–261 CrossRef PubMed.
- M. Iijima, Carbohydr. Polym., 2000, 41, 101–106 CrossRef CAS.
- U. Einhorn-Stoll, H. Kastner and S. Drusch, oids, 2014, 35, 565–575 CAS.
- S. Shankar, N. Tanomrod, S. Rawdkuen and J.-W. Rhim, Int. J. Biol. Macromol., 2016, 92, 842–849 CrossRef CAS PubMed.
- M. R. Sharaby, E. A. Soliman, A. B. Abdel-Rahman, A. Osman and R. Khalil, Sci. Rep., 2022, 12, 20673 CrossRef CAS PubMed.
- V. S. dos Santos, M. V. Lorevice, G. S. Baccarin, F. M. da Costa, R. da Silva Fernandes, F. A. Aouada and M. R. de Moura, Polymers, 2023, 15, 2244 CrossRef CAS PubMed.
- Z. Shabahang, L. Nouri and A. M. Nafchi, J. Polym. Environ., 2022, 30, 3985–3998 CrossRef CAS.
- L. Bastarrachea, S. Dhawan and S. S. Sablani, Food Eng. Rev., 2011, 3, 79–93 CrossRef.
- V. I. Irzhak, Colloid J., 2021, 83, 64–69 CrossRef CAS.
- A. Redondo, N. Mortensen, K. Djeghdi, D. Jang, R. D. Ortuso, C. Weder, L. T. J. Korley, U. Steiner and I. Gunkel, ACS Appl. Mater. Interfaces, 2022, 14, 7270–7282 CrossRef CAS PubMed.
- M. Mariano, N. El Kissi and A. Dufresne, J. Polym. Sci., Part B: Polym. Phys., 2014, 52, 791–806 CrossRef CAS.
- V. Favier, J. Y. Cavaille, G. R. Canova and S. C. Shrivastava, Polym. Eng. Sci., 1997, 37, 1732–1739 CrossRef CAS.
- S. Boufi, H. Kaddami and A. Dufresne, Macromol. Mater. Eng., 2014, 299, 560–568 CrossRef CAS.
- Y. Zare and K.-Y. Rhee, Phys. Mesomech., 2020, 23, 531–537 CrossRef.
- Y. Zare and S. W. Lee, Colloid Polym. Sci., 2017, 295, 1535–1540 CrossRef CAS.
- Y. Zare and K. Y. Rhee, Phys. Mesomech., 2020, 23, 332–339 CrossRef.
- P. J. P. Espitia, R. J. Avena-Bustillos, W. X. Du, B. Sen Chiou, T. G. Williams, D. Wood, T. H. Mchugh and N. F. F. Soares, J. Food Sci., 2014, 79(5), M903–M910 CrossRef CAS PubMed.
- C. G. Otoni, M. R. de Moura, F. A. Aouada, G. P. Camilloto, R. S. Cruz, M. V. Lorevice, N. de, F. F. Soares and L. H. C. Mattoso, Food Hydrocolloids, 2014, 41, 188–194 CrossRef CAS.
- P. Mangiacapra, G. Gorrasi, A. Sorrentino and V. Vittoria, Carbohydr. Polym., 2006, 64, 516–523 CrossRef CAS.
- M. R. de Moura, F. A. Aouada, R. J. Avena-Bustillos, T. H. McHugh, J. M. Krochta and L. H. C. Mattoso, J. Food Eng., 2009, 92, 448–453 CrossRef CAS.
- J. Vartiainen, T. Tammelin, J. Pere, U. Tapper and A. Harlin, Carbohydr. Polym., 2010, 82, 989–996 CrossRef CAS.
- W. X. Yu, Z. W. Wang, C. Y. Hu and L. Wang, Int. J. Food Sci. Technol., 2014, 49, 2592–2601 CrossRef CAS.
- M. V. Lorevice, C. G. Otoni, M. R. de Moura and L. H. C. Mattoso, Food Hydrocolloids, 2016, 52, 732–740 CrossRef CAS.
- S. Mangaraj, A. Yadav, L. M. Bal, S. K. Dash and N. K. Mahanti, J. Packag. Technol. Res., 2019, 3, 77–96 CrossRef.
- G. Ö. Kayan and A. Kayan, ChemEngineering, 2023, 7, 104 CrossRef CAS.
- S. Kumar, A. R. L. Reddy, I. B. Basumatary, A. Nayak, D. Dutta, J. Konwar, M. Das Purkayastha and A. Mukherjee, Int. J. Biol. Macromol., 2023, 239, 124281 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ma00033a |
| ‡ Present address: Brazilian Nanotechnology National Laboratory (LNNano), Brazilian Center for Research in Energy and Materials (CNPEM), 13083-970 Campinas, São Paulo, Brazil. |
| This journal is © The Royal Society of Chemistry 2024 |