
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
High-performance BiVO4 photoanodes: elucidating the combined effects of Mo-doping and modification with cobalt polyoxometalate†
Fan
Feng
a,
Dariusz
Mitoraj
b,
Ruihao
Gong
c,
Dandan
Gao
 a,
Mohamed M.
Elnagar
b,
Rongji
Liu
a,
Radim
Beranek
*b and
Carsten
Streb
*a
a,
Mohamed M.
Elnagar
b,
Rongji
Liu
a,
Radim
Beranek
*b and
Carsten
Streb
*a
aDepartment of Chemistry, Johannes Gutenberg University Mainz, Duesbergweg 10-14, 55128 Mainz, Germany. E-mail: carsten.streb@uni-mainz.de
bInstitute of Electrochemistry, Ulm University, Albert-Einstein-Allee 47, 89081 Ulm, Germany. E-mail: radim.beranek@uni-ulm.de
cInstitute of Inorganic Chemistry I, Ulm University, Albert-Einstein-Allee 11, 89081 Ulm, Germany
First published on 26th April 2024
Abstract
Bulk doping of BiVO4 with molybdenum combined with surface modification with a cobalt polyoxometalate water oxidation catalyst (CoPOM = Na10[Co4(H2O)2(PW9O34)2]) is reported. The best-performing Mo–BiVO4/CoPOM photoanode exhibits a photocurrent density of 4.32 mA cm−2 at 1.23 V vs. RHE under AM 1.5G (1 sun) illumination and an applied-bias photoconversion efficiency (ABPE) of ∼0.73%, which is an improvement by a factor of ∼24 with respect to pristine BiVO4. Mechanistic analyses are used to prove the contributions of Mo-doping and CoPOM modification. Mo-doping is shown to result in enhanced electronic conductivity and passivation of surface states, whereby these beneficial effects are operative only at relatively high applied bias potentials (>0.9 V vs. RHE), and at lower bias potentials (<0.7 V vs. RHE) they are counterbalanced by strongly detrimental effects related to increased concentration of electron polaronic states induced by the Mo-doping. CoPOM deposition is related to the enhancement of water oxidation catalysis. The molecular CoPOM acts as a pre-catalyst and undergoes (partial) conversion to cobalt oxide under the PEC operating conditions. The study demonstrates CoPOM-derived catalysts as effective water oxidation catalysts at BiVO4 photoanodes and suggests that further progress in BiVO4 photoanode development depends on alternative strategies for conductivity enhancement to avoid detrimental polaronic effects associated with the conventional bulk doping of BiVO4.
1. Introduction
Hydrogen generation via photoelectrochemical (PEC) water splitting represents one of the promising strategies to secure the future supply of free (low-entropy) energy in an environmentally sustainable manner.1–4 Compared to the hydrogen evolution reaction (HER), which is a proton-coupled two-electron process, the oxygen evolution reaction (OER) is a typical kinetic bottleneck of PEC devices since the mechanism of oxygen generation is much more complex, involving concerted transfer of four electrons and four protons.5,6 Accordingly, the development of efficient, stable and low-cost photoanodes for water oxidation is of paramount importance for the realization of economically viable PEC water splitting devices.7 Some of the most intensely investigated photoanodes are based on passivated high-quality semiconductors (e.g., Si or III–V compounds),8–10 hybrid inorganic–organic architectures,11–14 or on low-cost metal oxides, such as Fe2O3,15,16 CuWO417,18 or BiVO4.3,19–27 In particular, BiVO4 is an attractive material owing to its bandgap energy of ∼2.4 to 2.6 eV, which translates to the maximum theoretically achievable photocurrents of 6.4–8.9 mA cm−2 and the solar-to-hydrogen (STH) efficiencies of 8–11% under AM 1.5G (1 sun) illumination. However, the PEC performance of pristine BiVO4 photoanodes is typically far below the theoretical limit, both in terms of maximum photocurrents and achievable photovoltages, the latter manifested by rather positive photocurrent onset potentials (with respect to the reversible hydrogen electrode, RHE) and generally poor fill-factors of the photocurrent density–potential (J–V) curves. The reasons for this suboptimal performance have been identified as (i) poor bulk electronic conductivity of BiVO4 and the presence of surface intragap states, both of which result in severe electron–hole recombination,21,28,29 and (ii) ineffective hole extraction and slow rate of water oxidation at pristine BiVO4 surfaces.3,20,22 Concerning the former, significant improvements in performance have been demonstrated, for example, by doping BiVO4 with molybdenum,30–34 whereby the beneficial effect of Mo doping has been ascribed to increased electronic conductivity and/or passivation of surface recombination centers.28,35–37 In addition, further improvements have been reported after modification of BiVO4 with various additional hole extractors and/or OER catalysts, such as rhodium oxide,30 vanadium oxide,38 iron oxyhydroxide,31 nickel hydroxide,39 cobalt oxide,33,40,41 cobalt hexacyanoferrate,42 iron/nickel oxide,3,20,37,43,44 or cobalt/nickel sulfide,45 whereby the question of whether the improvement should be ascribed to enhanced water oxidation kinetics or to other factors (e.g., enhanced charge extraction or passivation of surface recombination centers) is still a source of intense debate and seems to depend on specific catalysts and photoanode architectures.42,46–50Well-defined polyoxometalates (POMs) – molecular metal oxide clusters – comprising redox-active transition metals, e.g. Co or Mn, are known to be effective catalysts in various (photo)electrocatalytic systems.51–54 A prototypical example is the complex CoPOM (Na10[Co4(H2O)2(PW9O34)2]) with a Co4O4 core stabilized by oxidatively resistant polytungstate ligands.54 Notably, this CoPOM has been reported to be an efficient homogeneous water oxidation catalyst,54–56 though there is also some evidence that – depending on specific experimental conditions (e.g., pH and composition of the electrolyte, catalyst concentration, applied electrode potential) – this CoPOM can also undergo conversion to cobalt oxide, and thus behave rather as a pre-catalyst for the formation of a heterogeneous water oxidation catalyst (CoOx) which then often accounts for the major part of activity in water oxidation.57,58 Very recently, there have been several reports of successful immobilization of CoPOM onto various photoanodes (e.g., TiO2, Fe2O3, BiVO4,59 or TiO2/carbon nitride hybrids60), typically using diverse cationic polyelectrolytes (e.g., polyethyleneimine) or N-doped carbon61 as an interlayer or linker. However, the scope of CoPOM-modified photoanodes investigated so far is rather limited, and our knowledge of the factors governing the PEC performance of CoPOM-containing photoelectrochemical architectures is still underdeveloped, which makes the rational design of high-performance Co-POM-based photoanodes very challenging.
In the present work, we report significant improvements of the PEC performance of mesoporous BiVO4 photoanodes using a combined strategy of Mo-doping and direct deposition of CoPOM via a simple impregnation protocol without any linkers or binders. The best-performing optimized Mo–BiVO4/CoPOM photoanode exhibits a photocurrent density of 4.32 mA cm−2 at 1.23 V vs. RHE under AM 1.5G (1 sun) irradiation and an applied-bias photoconversion efficiency (ABPE) of ∼0.73%, which is an improvement by a factor of 24 and 2.5 compared to pristine BiVO4 (∼0.03%) and Mo–BiVO4 (∼0.29%), respectively. Furthermore, we disentangle the respective contributions of the Mo-doping and modification with CoPOM to the performance enhancement and show compelling evidence that the beneficial role of CoPOM is related to enhanced kinetics of water oxidation. Finally, we provide several unprecedented insights into the effects of both Mo-doping (e.g., Mo-induced limitation of photovoltage) and CoPOM modification (e.g., compositional changes under operating conditions and significant electrolyte effects), which are of great importance for the further design and development of efficient photoanodes for water splitting.
2. Results and discussion
The detailed descriptions for the fabrication of BiVO4 and Mo–BiVO4/CoPOM films are given in the ESI.† The fabrication protocols (including doping concentration and co-catalyst loading) were optimized by screening for the highest photoelectrocatalytic activity in each case. In short, the BiVO4 and Mo–BiVO4 films were prepared via spin coating onto FTO glass substrates, followed by drying and calcination. The CoPOM complex was synthesized according to a previously reported method,54 and deposited via impregnation to obtain Mo–BiVO4/CoPOM photoelectrodes. The X-ray diffraction (XRD) patterns of all BiVO4-based photoanodes exhibit the typical diffraction peaks of monoclinic scheelite BiVO4 (Fig. S1a, ESI†), indicating that the crystal structure of BiVO4 is not significantly altered upon Mo-doping or deposition of CoPOM. No shifts of diffraction peaks or new reflexes related to molybdenum oxide were detected, which suggests that Mo is present in low amounts as a homogenously distributed dopant without affecting the structure of BiVO4. As expected, as the amount of deposited CoPOM was very low, it could be detected neither in XRD nor by using Fourier-transform infrared (FT-IR) spectroscopy (Fig. S1b, ESI†). However, the presence of CoPOM in Mo–BiVO4/CoPOM was verified by transmission electron microscopy, energy dispersive X-ray spectroscopy (EDX) and X-ray photoelectron spectroscopy (XPS) (see below).The morphology and film thickness of as-prepared BiVO4, Mo–BiVO4 and Mo–BiVO4/CoPOM photoanodes were investigated by field emission scanning electron microscopy (FESEM). As shown in Fig. 1a, all of the films are composed of uniform and homogeneous worm-like nanoparticles with an average diameter of ∼100 to 200 nm, forming a three-dimensional mesoporous structure with pore sizes in the range of tens of nanometers. The introduction of F-108 as a structural agent during the synthesis is responsible for inducing this porous architecture,62 which serves as the foundation for all subsequent surface-modified photoanodes. Notably, both Mo doping shown in Fig. 1b and surface modification with CoPOM shown in Fig. 1c have no appreciable effect on the morphology of BiVO4, retaining the typical porous structure. Fig. S2a–c (ESI†) shows the cross-section SEM images of all three photoanodes, showing that the material layers have a comparable thickness of 600 ± 20 nm. Since the structure of the Mo–BiVO4 material is porous and the CoPOM was deposited via the impregnation method, a homogenous distribution of the co-catalyst throughout the porous layer is expected. To investigate the distribution of CoPOM, EDX elemental mapping was carried out. Both in the cross-section and the surface of Mo–BiVO4/CoPOM, the elements Mo, Bi, V and O (representing Mo–BiVO4) and Co, W (representing CoPOM) have been detected. These EDX mapping results demonstrate a very dense and uniform distribution of CoPOM within the mesoporous Mo–BiVO4 structure (Fig. S2d and e, ESI†). A closer look with the help of high-resolution transmission electron microscopy (HRTEM) revealed that Mo–BiVO4/CoPOM contains uniformly distributed amorphous nanospheres with a diameter of less than 10 nm (Fig. 1f and g). Since these nanospheres are completely absent in the (CoPOM-free) Mo–BiVO4 photoanode (Fig. 1d and e), we assume the nanospheres to be CoPOM nanoparticles. These CoPOM nanospheres are closely spaced and homogeneously distributed on the surface of the Mo–BiVO4 porous support (Fig. 1f and g). Furthermore, Fig. 1h shows the scanning transmission electron microscopy (STEM) image of Mo–BiVO4/CoPOM with EDX mapping images of Bi, V, O, Mo, Co, and W, which again confirms the uniform distribution of the Mo dopant and of the CoPOM co-catalyst.
 | ||
| Fig. 1 FESEM images of (a) BiVO4, (b) Mo–BiVO4 and (c) Mo–BiVO4/CoPOM. High-resolution TEM images of Mo–BiVO4 (d) and (e) and Mo–BiVO4/CoPOM (f) and (g). (h) EDX elemental mappings of Mo–BiVO4/CoPOM. The lattice spacing of 0.47 nm is characteristic of the (110) planes of BiVO4 (JCPDS card number 14-0688).63 | ||
In order to further verify the presence of CoPOM in the Mo–BiVO4/CoPOM and to investigate the surface chemical composition of the photoelectrode material, XPS analysis was carried out at BiVO4, Mo–BiVO4 and Mo–BiVO4/CoPOM electrodes. As shown in Fig. S3a (ESI†), all expected signals of Mo, Bi, V, O, Na, Co and W elements are observed in the XP survey spectrum of Mo–BiVO4/CoPOM, which indicates the successful preparation of the composite material. Two typical spin–orbit coupling peaks of Bi (at 164.3 eV and 159.1 eV for Bi 4f5/2 and Bi 4f7/2, respectively) are assigned to Bi3+ in BiVO4 (Fig. 2a and Fig. S3b, ESI†).64,65 The O 1s XP spectrum (Fig. 2b) can be fitted to two peaks located at 529.8 eV and 530.7 eV, which are assigned to the lattice oxygen in BiVO4 and surface hydroxyl oxygen, respectively.61,66 The vanadium signals (Fig. 2b and Fig. S3c, ESI†) can be fitted into two spin–orbit coupling peaks located at 524.0 eV (V 2p1/2) and 516.6 eV (V 2p3/2), which are characteristic of V5+ species in BiVO4.67–69 As shown in Fig. S3d (ESI†), the binding energies of the Mo 3d3/2 (235.1 eV) and Mo 3d5/2 (232.0 eV) peaks indicate that the molybdenum dopant is in the oxidation state of +VI.61,65,70 The high-resolution XP spectrum of Co 2p (Fig. 2c) shows two core-level spin–orbit coupling peaks located at 797.1 (Co 2p1/2) and 781.2 eV (Co 2p3/2) with typical satellite peaks at 805.4 and 785.7 eV, indicating the presence of Co2+.70–72 In Fig. 2d, the two spin–orbit splitting peaks at 37.6 eV and 35.4 eV attributed to W 4f5/2 and W 4f7/2 confirm the presence of W6+.73 Importantly, the Co 2p and W 4f XPS signals of authentic CoPOM powder (Fig. S4, ESI†) are nearly identical to the corresponding XPS signals of the CoPOM in the electrode, which further corroborates the successful loading of CoPOM clusters into the porous structure of the Mo–BiVO4/CoPOM photoelectrode.
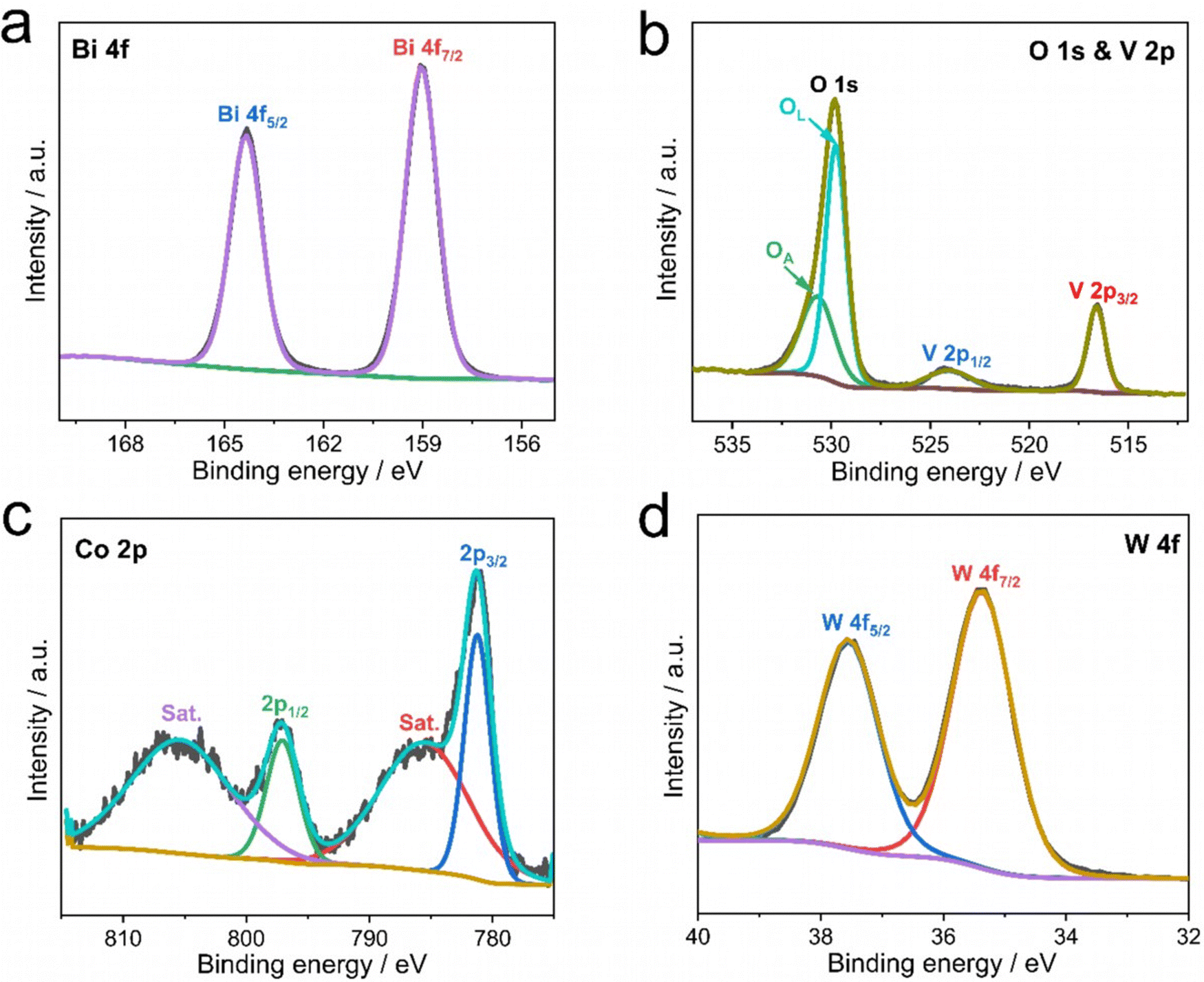 | ||
| Fig. 2 XP spectra of the Mo–BiVO4/CoPOM photoanode: (a) Bi 4f, (b) O 1s and V 2p, (c) Co 2p and (d) W 4f. | ||
The UV-vis electronic absorption spectra of BiVO4, Mo–BiVO4 and Mo–BiVO4/CoPOM samples displayed in the ESI,† Fig. S5a and b (ESI†) show that all samples exhibit comparable light absorption onsets at ∼515 nm. In this context, we note that in the literature there are conflicting reports about whether BiVO4 is a direct74 or indirect75 bandgap semiconductor. The bandgap energies of all photoelectrodes were estimated to be ∼2.56 eV and ∼2.50 eV for direct and indirect optical transition, respectively, using the Tauc formalism (Fig. S5c and d, ESI†). Importantly, these results reveal that the effects of Mo-doping and CoPOM deposition on the electronic absorption properties of BiVO4 photoanodes are negligible, without any changes of the fundamental absorption edge, nor any significant parasitic light absorption by the CoPOM catalyst.
To investigate the combined effects of Mo-doping and modification by CoPOM on PEC performance of BiVO4 photoanodes, the photocurrent density–potential (J–V) curves of BiVO4, Mo–BiVO4 and Mo–BiVO4/CoPOM photoanodes were measured under intermittent AM 1.5G irradiation (1 sun) using a three-electrode setup in a 0.5 M sodium borate buffer electrolyte at pH 9.0 (Fig. 3a). The PEC performance of the photoanodes was optimized for (i) Mo doping concentration of BiVO4, (ii) calcination temperature of Mo–BiVO4, (iii) loading of the CoPOM in Mo–BiVO4 and (iv) various pH values of borate electrolyte (the results of optimization protocols are summarized in the ESI,† Fig. S6). The content of Mo, Co and W elements in the Mo–BiVO4/CoPOM photoanode optimized for the highest PEC efficiency was 0.79 at%, 0.95 at% and 1.92 at%, respectively, as estimated by the SEM-EDX analysis (Table S1, ESI†), which is also in line with the elemental analysis based on XPS results (Table S2, ESI†). The optimum calcination temperature was 450 °C and the optimal PEC performance was achieved in a borate buffer at pH 9.0.
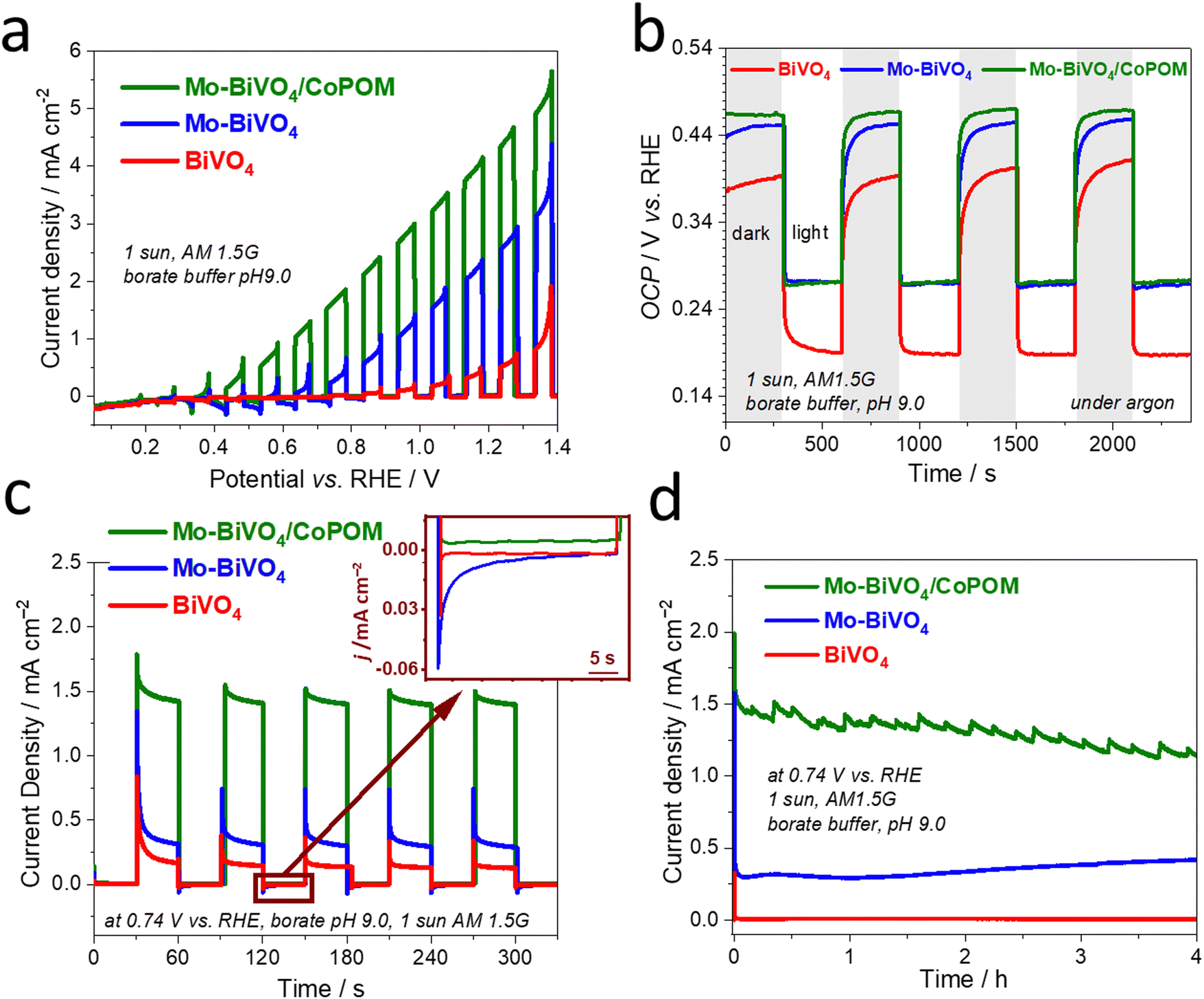 | ||
| Fig. 3 (a) Linear-sweep voltammetry (LSV) curves recorded under intermittent AM 1.5G illumination in borate buffer (0.5 M, pH 9.0) at a cathodic sweep of 10 mV s−1, (b) open-circuit potential (OCP) transients recorded under interrupted AM 1.5G (1 sun) illumination in 0.5 M borate buffer under an argon atmosphere (to prevent electron transfer to O2), (c) photocurrent transients under AM 1.5G illumination, (d) stability chronoamperometric tests measured in borate buffer (0.5 M, pH 9.0) under AM 1.5G illumination (1 sun) at 0.74 V vs. RHE for all three photoanodes. All photocurrent measurements were carried out at least in triplicate, and representative average data are shown. | ||
Fig. 3a shows the J–V curves of various optimized photoanode architectures under back-side illumination (from the FTO-glass side). The BiVO4 and Mo-doped BiVO4 photoanodes reach a photocurrent density of 0.65 mA cm−2 and 2.53 mA cm−2 at 1.23 V vs. RHE, respectively. After the modification by CoPOM, the highest photocurrent density of Mo–BiVO4/CoPOM increases up to 4.32 mA cm−2 at 1.23 V vs. RHE, which is an enhancement by the factor of 6.6, and 1.7 with respect to BiVO4, and Mo–BiVO4, respectively. Importantly, both Mo–BiVO4 and Mo–BiVO4/CoPOM photoanodes exhibit a remarkable cathodic shift of the photocurrent onset potential by ∼0.5 V as compared to the pristine BiVO4 photoanode, whereby the modification with CoPOM significantly increases photocurrents even at very low bias potentials, improving thus the fill-factor of the J–V curve. In other words, the combination of Mo-doping and CoPOM modifications results in a substantial increase of the photovoltage available for driving the water splitting reaction. In order to further disentangle the effects of Mo-doping and Co-POM modification, we also directly compared the J–V curves of Mo-free (undoped) BiVO4/CoPOM with Mo–BiVO4/CoPOM (Fig. S7, ESI†). Interestingly, the Mo–BiVO4/CoPOM shows much higher photocurrents at relatively higher applied potentials (>0.9 V vs. RHE), but at very low bias potentials (<0.7 V vs. RHE) the situation is reversed and the Mo-free BiVO4/CoPOM electrode yields even slightly higher photocurrents than the Mo–BiVO4/CoPOM photoanode. These observations imply that (i) the combination of Mo-doping and Co-POM deposition significantly improves the photocurrent response in the whole potential range with respect to pristine BiVO4, (ii) the enhancement due to the deposition of CoPOM is particularly pronounced at low bias potentials, i.e., the beneficial negative shift of the photocurrent onset potential and improved fill-factor is mainly due to the CoPOM deposition, and (iii) at very low bias potentials the Mo-doping might even partially counteract the positive effects induced by the CoPOM deposition. The observation that the Mo-doping might be detrimental at relatively low bias potentials prompted us to measure the open-circuit photopotential (OCP) of all photoanodes (Fig. 3b). Under illumination, the observed OCP should correspond to the quasi-Fermi level of electrons *EFn (i.e., the electrochemical potential of electrons) in the illuminated semiconductor, which in turn is directly related to the photocurrent onset potential (and the fill-factor at low bias potentials), and thus to the maximum achievable photovoltage of the water-splitting system. Indeed, the OCP results clearly show that for both Mo–BiVO4 and Mo–BiVO4/CoPOM the Mo-doping leads to a positive shift (by ∼90 mV) of the maximum achievable quasi-Fermi level of electrons as compared to pristine BiVO4. It should be noted that this observation is in stark contrast to the data for Mo-doped BiVO4 (with similar doping concentration) reported by Ye et al.34 who observed a slight negative shift of the OCP after Mo-doping. However, our observation of a positive shift of the OCP after the Mo-doping is completely in line with the observed decrease of photocurrent at low bias potentials induced by the Mo-doping (Fig. S7, ESI†), and suggests that the Mo-doping does not have only beneficial effects, such as the enhancement of photocurrents at relatively high bias potentials, but also induces effects that are detrimental to the PEC performance.
Our experimental proof that the maximum achievable quasi-Fermi level of electrons is less negative after Mo-doping is significant and deserves a more detailed discussion. In this context, we point out that it is well-established that the maximum achievable photovoltage in metal oxide photoanodes is typically limited by the presence of intragap electron polaronic states formed upon self-trapping of excess electrons at reduced ionic sites (e.g., Fe2+ in hematite Fe2O3, or V4+ in BiVO4) in the lattice, accompanied by the displacement of surrounding atoms via electron–phonon interactions, forming thus a quasi-particle called polaron.76,77 As the electron polaron level is typically significantly below the conduction band edge, it effectively traps electrons, limits their mobility, acts as a recombination center, and therefore establishes an inherent limitation of the maximum achievable quasi-Fermi level of electrons and photovoltage. For both pristine and doped BiVO4, it is reported that the excess electrons are localized in hybridized vanadium 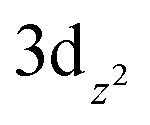 orbitals,78,79 which is accompanied by elongation of the V–O bond by ∼0.1 Å and formation of small polarons located ca. 0.88 eV below the conduction band edge.80 Doping by molybdenum substitutes V5+ with ionized Mo6+ dopants, whereby the excess electron is readily localized at V4+ polaronic states.78 The detrimental polaronic effects can be then (at least partially) avoided by applying a sufficient external electric bias, which, however, compromises the overall photoconversion efficiency. To summarize, the conductivity enhancement upon Mo-doping, which is apparent from enhanced photocurrents at higher bias potentials, is mainly due to the increased electron concentration, whereby the electron mobility is apparently not improved.81 However, at low bias potentials the beneficial effect of the Mo-doping is counterbalanced by enhanced recombination due to the presence of deep electron polaron states,78 which makes itself apparent by decreased photocurrents at low bias potentials and by a less negative OCP (i.e., less negative quasi-Fermi level of electrons) under irradiation.
orbitals,78,79 which is accompanied by elongation of the V–O bond by ∼0.1 Å and formation of small polarons located ca. 0.88 eV below the conduction band edge.80 Doping by molybdenum substitutes V5+ with ionized Mo6+ dopants, whereby the excess electron is readily localized at V4+ polaronic states.78 The detrimental polaronic effects can be then (at least partially) avoided by applying a sufficient external electric bias, which, however, compromises the overall photoconversion efficiency. To summarize, the conductivity enhancement upon Mo-doping, which is apparent from enhanced photocurrents at higher bias potentials, is mainly due to the increased electron concentration, whereby the electron mobility is apparently not improved.81 However, at low bias potentials the beneficial effect of the Mo-doping is counterbalanced by enhanced recombination due to the presence of deep electron polaron states,78 which makes itself apparent by decreased photocurrents at low bias potentials and by a less negative OCP (i.e., less negative quasi-Fermi level of electrons) under irradiation.
In order to assess the dynamics of photogenerated charge carriers, transient photocurrent measurements of pristine BiVO4, Mo–BiVO4 and Mo–BiVO4/CoPOM photoanodes were carried out at a moderate bias potential of +0.74 V vs. RHE under chopped AM 1.5G (1 sun) illumination (Fig. 3c). On the timescale of this experiment (∼minutes), the photocurrent response indicates an excellent short-term stability. Evidently, for both CoPOM-free photoanodes (red and blue line), the photocurrent spikes after switching on the light and negative current overshoots after switching off the light (see inset in Fig. 3c) are significantly more pronounced, demonstrating that in the absence of a water oxidation catalyst the charge carriers are prone to intense surface recombination.82 In other words, in the absence of the CoPOM co-catalyst, the photogenerated holes do not react fast enough with water but accumulate in the surface layer of BiVO4 or Mo–BiVO4, thus enhancing the probability of recombination with electrons. The intense recombination of surface-accumulated holes and electrons leads to a rapid decrease of the photocurrent immediately after switching on the light, and continues even after the light is switched off, as is apparent from the negative current overshoot after switching off the light. In contrast, the current spikes are less pronounced, and the overshoots are nearly absent at the Mo–BiVO4/CoPOM photoanode, which clearly demonstrates that the CoPOM co-catalyst efficiently extracts the holes photogenerated in the Mo–BiVO4 layer and channels them to water molecules in the electrolyte solution. This explains the significant photocurrent enhancement upon the deposition of CoPOM shown in Fig. 3a and c. In this context, we also note that the addition of polyethyleneimine (PEI) as a cationic linker has no effect on the performance of the photoanodes (Fig. S8, ESI†), which highlights the intrinsically good electronic communication between the CoPOM and BiVO4.
The long-term stability of the photoanode and its PEC performance is an important issue not only with respect to possible applications, but in our case also with respect to fundamental questions regarding the stability and/or compositional changes of the used CoPOM polyoxometalate catalyst under PEC operational conditions. As illustrated in Fig. 3d, the photocurrent generated by the investigated photoanodes was measured at a constant potential of +0.74 V vs. RHE for four hours under simulated AM 1.5G (1 sun) illumination, and only a very slight (by ∼15%), yet continuous, decline of photocurrent density was observed for the optimized Mo–BiVO4/CoPOM electrode. The small perturbations of the photocurrent are likely related to the release of oxygen bubbles from the electrode surface, which can be observed by the naked eye (see Video in the ESI†). The XRD patterns of all samples recorded after the PEC experiment show the typical diffraction peaks of monoclinic scheelite BiVO4, indicating that the BiVO4-based light absorber did not undergo any structural changes under the operating conditions (Fig. S9, ESI†), compared with Fig. S1a, ESI†). However, a question arises regarding the stability of the CoPOM co-catalyst under PEC operation since it has been reported that this polyoxometalate can – depending on experimental conditions – undergo conversion to cobalt oxide (CoOx) which then often plays a chief role in water oxidation catalysis.57,58 In order to shed light on the fate of CoPOM in our photoanodes during operation, we analyzed in detail both the composition of the Mo–BiVO4/CoPOM photoanode after the PEC operation and the elemental composition of the electrolyte before and after the PEC experiments. First of all, we note that the small nanospheres attributed to CoPOM that could be seen before the PEC operation (Fig. 1f and g), were not observed anymore by HRTEM after the PEC operation (Fig. S10, ESI†), which indicates that the CoPOM cocatalyst has undergone (at least partially) morphological changes. Notably, the XPS analysis revealed that the signals related to the presence of W and Co are significantly weaker after the PEC operation (Fig. S11a, ESI†), whereby the XPS signature of Co is still well-detectable in the high-resolution XP spectrum and is very similar to the situation before the PEC operation (Fig. S11b, ESI†), compared with Fig. 2c). Notably, the surface content of both Co and W derived from the XPS analysis is significantly decreased and the W/Co atomic ratio increased after the PEC operation (Table S3, ESI†). However, EDX analyses performed both from the top-view (Table S4, ESI†) and from the cross-sectional view (Table S5, ESI†) clearly show that both W and Co are still present in the photoelectrode in significant amounts, albeit with a significantly higher W/Co ratio than before the PEC operation. In this context, it should be noted that quantitative EDX analysis is rather challenging in this case due to the overlap between the Si–K (from the underlying glass) and W–M lines and since the content of Co and W is very low. Yet, the above differences in the composition before and after the PEC operation, in particular the changing ratio of W/Co, indicate that at least a partial dissolution of CoPOM and/or conversion of CoPOM to cobalt and tungsten oxides can occur during the PEC operation. Interestingly, an analysis of the composition of the electrolyte solution before and after the PEC operation using inductively coupled plasma atomic emission spectrometry (ICP-AES) did not detect any Co, and the concentration of W also did not increase (Table S6, ESI†). While we cannot completely rule out the dissolution of CoPOM as the dissolved amount of Co and W might be below the detection limit of ICP-OES, our results indicate that a complete dissolution of CoPOM is not an issue. All the above observations suggest that, though the CoPOM catalyst does not dissolve in the electrolyte solution, it does undergo, at least partially, conversion to cobalt oxide, as previously reported for borate electrolytes by Finke et al.57 In other words, under our PEC operating conditions, the CoPOM co-catalyst should be rather conceived as a pre-catalyst, whereby a substantial part of water oxidation catalysis might be performed by cobalt oxide-based catalyst derived from CoPOM under operating conditions, as suggested by Finke et al. for CoPOM behavior in electrocatalysis.57 However, in this context, it should be also noted that the activity and stability of various cobalt oxide-based water oxidation catalysts can strongly depend on their precursor and operating conditions, as minute changes in composition can often exert strong effects on catalysis. For comparison, we have therefore also tested the performance of our Mo–BiVO4 photoanode after modification with a CoPi co-catalyst (i.e., cobalt oxy-hydroxide formed upon photodeposition in Co2+ containing phosphate buffer) and with a CoO(OH)x co-catalyst (i.e., cobalt oxy-hydroxide deposited via a two-step impregnation process).11 These two photoanodes reached slightly lower (by 9% and 33%, respectively) photocurrents than the Mo–BiVO4/CoPOM photoanode (Fig. S12, ESI†), which confirms that the “pre-history” of the catalyst can make a difference to its activity in photoanodes, and that the CoPOM-derived water oxidation catalyst exhibits optimal performance.
Furthermore, it is well established that the activity and stability of Co-based electrocatalysts can depend strongly on the electrolyte composition,83 and our group previously observed strong electrolyte effects on the stability of CoO(OH)x water oxidation co-catalysts in hybrid photoanodes.11 Therefore, we carried out a comparative analysis of the PEC operational stability of our photoanodes in borate and phosphate electrolytes. Specifically, we measured the linear sweep voltammetry under chopped illumination in the borate and the phosphate electrolyte at pH 9.0, and then repeated the measurement six times. In the borate buffer electrolyte at pH 9.0 (Fig. S13a–c, ESI†), the photocurrents for BiVO4, Mo–BiVO4 and Mo–BiVO4/CoPOM photoanodes remained completely stable over all six cycles. In contrast, in the phosphate electrolyte at pH 9.0 (Fig. S13d–f, ESI†), Mo–BiVO4/CoPOM exhibits a gradual decrease of photocurrents. The much higher operational stability of Mo–BiVO4/CoPOM photoanodes in a borate electrolyte is also clearly apparent from the long-term (4 hours) chronoamperometric measurements at +0.74 V vs. RHE under 1 sun irradiation (Fig. S14, ESI†). In this context, two points are noteworthy. First, the clearly detrimental effect of phosphate anions on the PEC performance stability of Mo–BiVO4/CoPOM is not simply related to the fact that the phosphate electrolyte is operated at pH 9.0 (i.e., beyond its ideal buffering pH range around pH ∼7), whereas the borate electrolyte operates at its optimal buffering capacity (pH ∼9), since comparative experiments performed at pH 7.0 also revealed that the photocurrents at Mo–BiVO4/CoPOM were more stable in the borate electrolyte than in the phosphate electrolyte (Fig. S15, ESI†). Second, similar to the case of the borate electrolyte, no cobalt could be detected in the phosphate electrolyte after PEC operation (Table S6, ESI†), suggesting that the dissolution of the co-catalyst is not the major reason for lower photoanode stability in phosphate electrolytes. We, therefore, conclude that the PEC operational stability of our photoanodes is related to the presence of specific electrolyte anions, whereby borate electrolytes are more beneficial as compared to phosphate electrolytes. Drawing on our previous discussion,11 we assume that the Co(III/IV) ions, which are formed in situ during the water oxidation catalytic cycle and which represent, in terms of the HSAB theory, hard acid species, can interact more strongly with phosphate ions, which are much harder bases than the relatively large borate anions (present mainly as tetraborates). We speculate that the relatively stronger interaction of cobalt ions with phosphate anions can induce, during the PEC operation, changes in the interaction between the BiVO4 light absorber and the co-catalyst, which eventually results in a lower observed stability of PEC performance in phosphate-containing electrolytes.
From a practical point of view, the applied bias photoconversion efficiency (ABPE) is the most important and informative performance metric for water splitting photoanodes. As presented in Fig. 4a, the maximum ABPE of 0.73% was determined for the Mo–BiVO4/CoPOM photoanode at a potential of +0.86 V vs. RHE, which is an enhancement by a factor of 24 and 2.5 compared to pristine BiVO4 (∼0.03% at +1.07 V vs. RHE) and Mo–BiVO4 (∼0.29% at +0.98 V vs. RHE), respectively. (For a comparison of the PEC performance of our Mo–BiVO4/CoPOM photoanodes with other BiVO4-based photoanodes reported in the literature, see Table S7 (ESI†).) Apart from the significant increase of the photoconversion efficiency upon the combined effect of doping with Mo and modification with CoPOM, the fact that the maximum power point is shifted to more negative bias potentials is particularly important in view of more feasible integration with photocathodes for hydrogen evolution in tandem solar-driven water splitting systems.
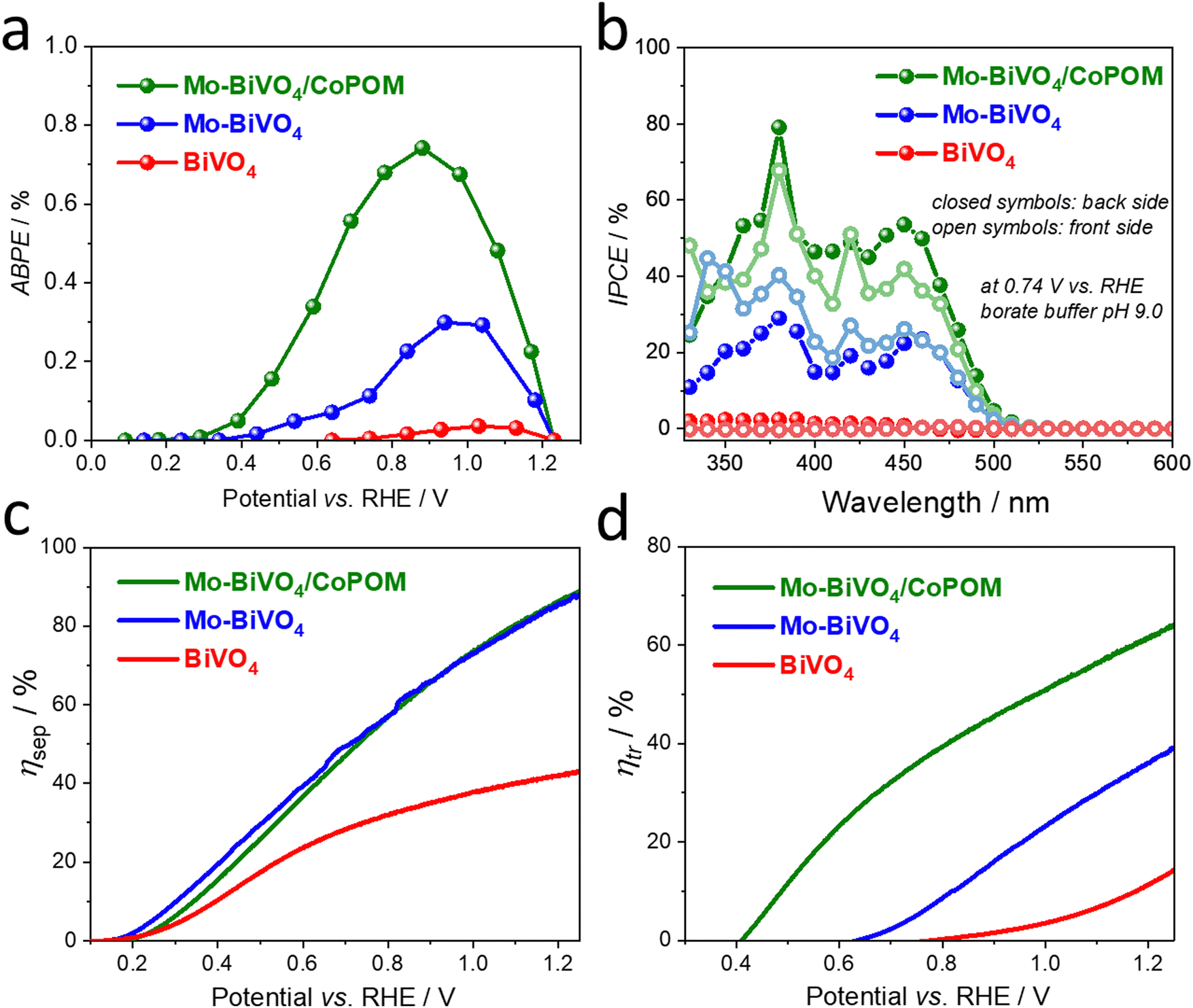 | ||
| Fig. 4 (a) Applied bias photoconversion efficiency (ABPE). (b) IPCE spectra measured under intermittent monochromatic irradiation at 0.74 V vs. RHE. (c) Charge separation efficiency (ηsep) and (d) charge transfer efficiency (ηtr) for BiVO4, Mo–BiVO4 and Mo–BiVO4/CoPOM photoanodes as a function of applied bias. | ||
In order to obtain further insight into the PEC operation of our photoanodes, the wavelength-resolved incident photon-to-current conversion efficiency (IPCE) was measured at +0.74 V vs. RHE under illumination from the backside (BS, i.e. from the substrate side) and frontside (FS, i.e. from the electrolyte side) (Fig. 4b). The photoaction spectra show an onset at ca. 510 nm, which is in line with the electronic absorption properties of the photoanodes (compare with Fig. S5, ESI†). The IPCE values are, independently of the illumination side, the highest for the Mo–BiVO4/CoPOM photoanode, followed by Mo–BiVO4 and pure BiVO4 over the whole wavelength range, which is in line with the photocurrent performance under polychromatic light at the same bias, as shown in Fig. S16 in the ESI† (see also Fig. 3c). From the mechanistic point of view, it is often worth looking at the difference in IPCE values under the FS and BS illumination for a given photoelectrode. The most notable difference is in the behavior of the Mo-free pristine BiVO4 photoanode as compared to Mo–BiVO4 and Mo–BiVO4/CoPOM photoanodes. While the latter two photoanodes show a very small difference in IPCE values recorded under the FS and BS illumination, the Mo-free pristine BiVO4 photoanode exhibits much larger (by the factor of ∼10) IPCE values under the BS illumination than under the FS illumination (for a detailed view see the ESI,† Fig. S17). This is exactly what one expects for a porous photoanode in which the photocurrent is mainly limited by the transport of electrons through the porous network, i.e. by the electronic conductivity of BiVO4. Under the FS illumination, most of the photogenerated electron–hole pairs are generated close to the film/electrolyte interface, and while the holes can readily react with water within the pores of the electrode, the electrons have to travel through the porous electrode to the underlying FTO back contact. Since under the BS illumination the electron–hole pairs are generated much closer to the FTO/film interface, the photogenerated electrons have a much shorter pathway to reach the FTO, resulting in significantly higher IPCE values under the BS illumination. Interestingly, the pristine BiVO4 photoanode even exhibits a wavelength-dependent switching (at ∼460 nm) of the sign of the photocurrent from anodic to cathodic under the BS illumination and from cathodic to anodic under the FS illumination. This behavior can be again rationalized by the dominant effect of the poor electron transport properties of pristine BiVO4 on the photocurrent response, which, in combination with the different penetration depths of short (below 460 nm) and long (above 460 nm) wavelengths of light and with the generally higher intensity of the visible light as compared to UV light in our setup, can even result in the reversal of the photocurrent sign. In this context, we note that while the potential-dependent photocurrent switching has been reported for BiVO4 photoelectrodes,84 the fact that at a specific bias potential (in our case a rather moderate bias potential of +0.74 V vs. RHE) the photocurrent switching at BiVO4 can be controlled even by the light wavelength and by the illumination side has not been reported before, and would deserve further investigation given the importance of such phenomena for the development of photoelectrochemical optoelectronic switches.85–89 In contrast to the behavior of the pristine BiVO4 photoanode that is dominated by its low electronic conductivity, the very small difference in IPCE values under the FS and BS illumination observed for both Mo-doped samples Mo–BiVO4 and Mo–BiVO4/CoPOM (Fig. 4b) nicely illustrate that the beneficial effect of Mo-doping consists mainly in improving the electronic conductivity of Mo–BiVO4 by increasing the electron concentration, as discussed in detail above.
Further valuable insights into the effects of Mo-doping and the role of the CoPOM co-catalyst on the PEC performance can be gained by determining the charge separation efficiency (ηsep) and the hole transfer efficiency (ηtr) as a function of applied potential (Fig. 4c and d), employing the methodology developed by Dotan et al. and Hamann et al. (for details see the ESI†).17,90 Firstly, by integrating the electronic absorption spectra (Fig. S5b, ESI†) over the reference AM1.5G photon flux spectra, the maximum theoretically achievable photocurrents Jmax were calculated to be 7.37 mA cm−2, 7.36 mA cm−2 and 7.34 mA cm−2 for BiVO4, Mo–BiVO4 and Mo–BiVO4/CoPOM, respectively. The photocurrent density of both Mo–BiVO4 and Mo–BiVO4/CoPOM photoanodes in the presence of Na2SO3 acting as a readily oxidizable reducing agent (i.e., sacrificial hole scavenger) is significantly higher than that of pristine BiVO4 and reaches 6.5 mA cm−2 as compared to 3.2 mA cm−2 at 1.23 V vs. RHE, respectively (Fig. S18, ESI†). Based on the above results, the charge separation efficiency ηsep is calculated and plotted in Fig. 4c. Notably, ηsep for Mo–BiVO4 and Mo–BiVO4/CoPOM is significantly higher than for pristine BiVO4 in a wide potential range, whereby the enhancement is increasingly pronounced at more positive bias potentials. This behavior is in line with our finding that the beneficial effects of the improvement of electronic conductivity upon Mo-doping are more pronounced at positive bias potentials, whereby at very low bias potentials the positive effects are traded off against the detrimental effects of Mo-doping associated with increased concentration of intragap electron polaronic states. Interestingly, at higher bias potentials (>0.7 V vs. RHE), the Mo-doped Mo–BiVO4 exhibits also an improved interfacial hole transfer efficiency ηtr with respect to pristine BiVO4 (Fig. 4d). This positive effect of Mo-doping on ηtr can be interpreted as partial mitigation of excessive hole accumulation in the surface states, as reported recently by Selli et al.28 After the deposition of Co-POM, the hole transfer efficiency ηtr (Fig. 4d) is greatly enhanced over the entire potential range and shifted to more cathodic potentials. For example, at 1.23 V vs. RHE, the ηtr increases from 13.1% for BiVO4 to 37.8% after Mo-doping (Mo–BiVO4) up to 63.0% for Mo–BiVO4/CoPOM photoanode. Importantly, the charge separation efficiencies (ηsep) for Mo–BiVO4/CoPOM and Mo–BiVO4 are nearly identical in the whole potential range (Fig. 4c), which clearly indicates that the beneficial effect of CoPOM deposition consists chiefly in enhancing the catalysis of water oxidation. This is a significant result since the effect of co-catalysts on photocurrent enhancement at BiVO4 photoanodes is a much-debated issue and, apart from enhancing the catalysis of water oxidation, other effects of various co-catalysts (e.g., improved charge extraction or passivation of surface states) are often reported to be responsible for photocurrent enhancement in the literature reports.42,46–50 Our results unambiguously demonstrate that in our photoanodes the beneficial effect of CoPOM deposition consists mainly in enhancing the rate of water oxidation by photogenerated holes.
Furthermore, the combined beneficial effects of Mo-doping and modification with CoPOM make themselves also clearly apparent in the results of the electrochemical impedance spectroscopy (EIS) analysis performed under irradiation (for details see the ESI,† Fig. S19). For the sake of simplicity, we used a Randles-type equivalent circuit model, where Rs represents the uncompensated series resistance, and Rct in our case represents the combined charge transport and interfacial charge transfer resistance.29 The fitted values of Rs and Rct are shown in the ESI,† Table S8. The similar Rs values of three films indicate that the effect of doping and modification on the series resistance is negligible,29 while the values of Rct of Mo–BiVO4 (∼385 Ω) and Mo–BiVO4/CoPOM (∼113 Ω) decrease significantly as compared to pristine BiVO4 (∼2620 Ω), indicating that Mo-doping and CoPOM modification greatly enhance both charge transport (i.e., conductivity) and the charge transfer, resulting in enhanced photocurrent density.62,63
Finally, in order to provide direct evidence for dioxygen evolution at our photoanodes, we performed photoelectrocatalytic OER measurements (Fig. 5a) in a borate solution (pH 9.0) at 0.74 V vs. RHE under AM 1.5G (1 sun) illumination. As expected, the Mo–BiVO4/CoPOM photoanode exhibits an excellent oxygen evolution rate with an average faradaic efficiency of oxygen evolution of 102.1% ± 6.2% (calculated from three measurements; the error is taken as ±1σ, σ = standard deviation), which demonstrates that the conversion of H2O to O2 by photogenerated holes is practically quantitative and no side reactions (e.g., photocorrosion or H2O2 evolution) occur. Similar results were obtained for Mo–BiVO4 (100.0% ± 12.0%), whereby at pristine BiVO4 the faradaic efficiency was slightly lower (90.3% ± 8.3%), suggesting a minor role of side reactions, possibly related to the photocorrosion.91 These results suggest that Mo-doping has also a beneficial effect on the stability of the photoanodes under the PEC operation, which again might be related to the decreased charge accumulation in the surface states,28 since excessive hole accumulation in the surface states can be expected to render the dissolution of BiVO4 more facile.
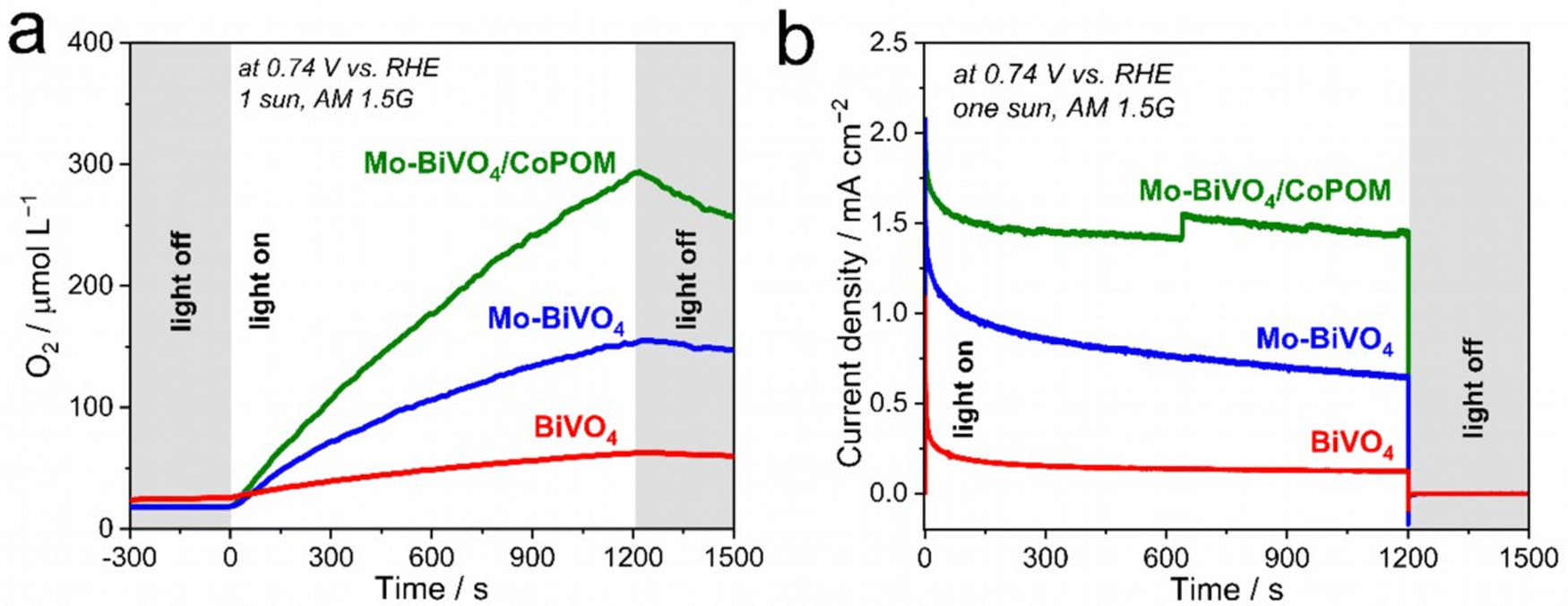 | ||
| Fig. 5 (a) Dioxygen evolution and (b) corresponding photocurrent transients recorded under AM 1.5G one sun illumination at 0.74 V vs. RHE in a borate electrolyte (0.5 M, pH 9.0). All measurements were carried out at least in triplicate, and representative average data are shown. | ||
The key factors influencing the PEC performance of all electrodes at a moderate electrode bias of 0.74 V vs. RHE are summarized in Fig. 6. In pristine BiVO4 (Fig. 6a) the photocurrents are negligible (cf.Fig. 3) due to intense surface recombination in the surface states (SS) and the very low conductivity of the BiVO4 film. The Mo-doping in Mo–BiVO4 (Fig. 6b) enhances the conductivity and passivates partially the surface states, resulting in a moderate photocurrent response (Fig. 3) due to the slow kinetics of water oxidation and the detrimental effect of electron polaronic states on the quasi-Fermi level of electrons (*EFn). The deposition of the CoPOM catalyst (Fig. 6c) results in highly enhanced photocurrents (Fig. 3) due to significantly increased kinetics of water oxidation; however, the negative effects (shift of *EFn) associated with the presence of Mo-induced polaronic states still persist, and represent an inherent limitation for the maximum achievable photovoltage in Mo-doped BiVO4.
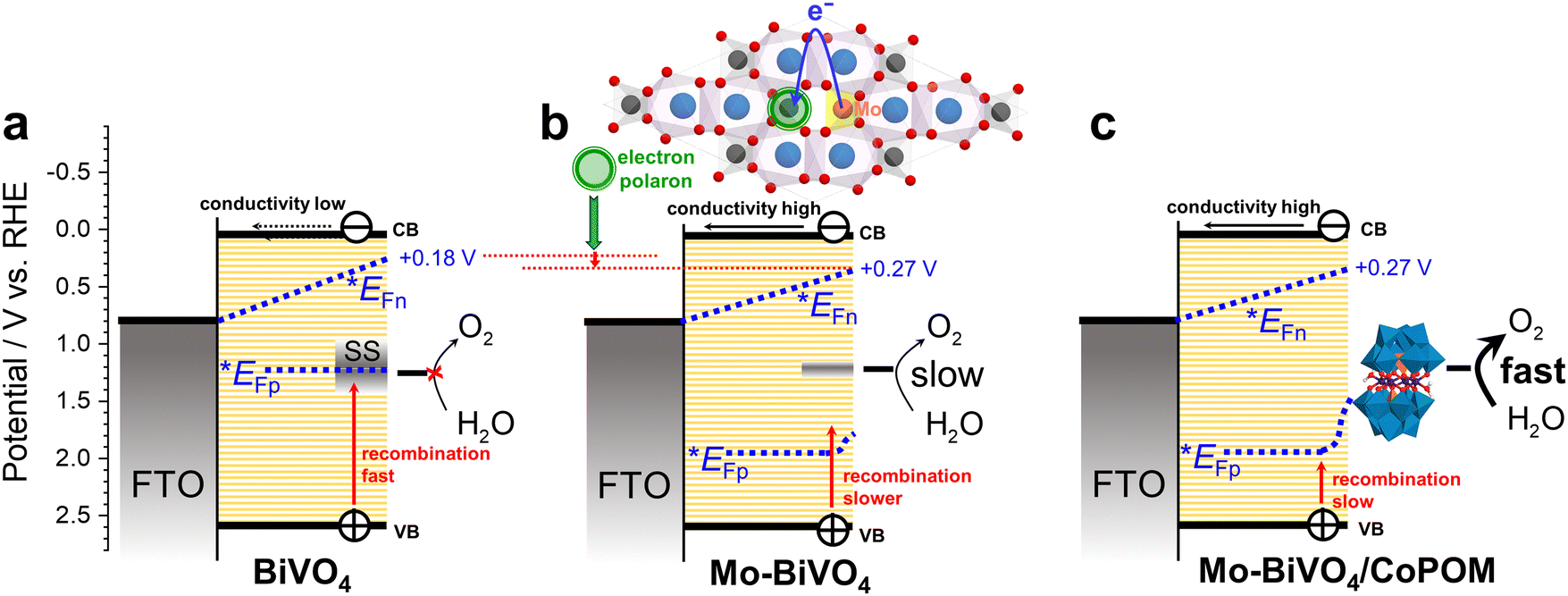 | ||
| Fig. 6 A simplified scheme for (a) BiVO4, (b) Mo–BiVO4 and (c) Mo–BiVO4/CoPOM at a moderate electrode bias of 0.74 V vs. RHE. Note that the band bending is assumed to be negligible due to the mesoporous nature of the BiVO4 film. CB and VB stand for the conduction band edge and valence band edge, respectively; *EFn and *EFp stand for the quasi-Fermi level of electrons and holes in the mesoporous film, respectively. | ||
3. Conclusions
This study reports a facile two-in-one strategy to enhance the performance of BiVO4 photoanodes for water oxidation by combining the bulk doping of BiVO4 with molybdenum and its surface modification with a well-defined molecular cobalt polyoxometalate (CoPOM = Na10[Co4(H2O)2(PW9O34)2]). The resulting modified photoanodes show significantly improved photocurrent densities compared to non-modified references. Mechanistic investigations elucidate the contributions of the Mo-doping and modification with CoPOM to the performance enhancement for water oxidation. Mo-doping leads to enhanced electronic conductivity and passivation of surface states. The deposition of CoPOM enhances photocurrents across the whole potential range, which results in enhanced water oxidation catalysis. Experimental evidence shows that under the PEC operating conditions the molecular CoPOM is, at least partially, disintegrated and converted to cobalt oxide, and is therefore considered as a pre-catalyst. In summary, this work establishes CoPOM-derived catalysts as effective water oxidation catalysts at BiVO4 photoanodes, provides new insights into the combined effects of Mo-doping and modification with molecular cobalt polyoxometalates on the PEC performance of BiVO4, and suggests that further progress in the development of BiVO4 photoanodes depends critically on devising alternative doping strategies to overcome the negative polaronic effects associated with bulk doping of BiVO4, since these detrimental effects set fundamental limits for the maximum achievable photovoltage and thus compromise the overall photoconversion efficiency.Data availability
All datasets related to this work are available from the repository: 10.5281/zenodo.10401296.Author contributions
C. S. and R. B. conceptualized and supervised the study and acquired funding. F. F., D. M., R. G., D. G., M. M. E. and R. L. performed investigations, acquired and analyzed data. All co-authors wrote and reviewed the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge financial support from the Deutsche Forschungsgemeinschaft DFG (TRR 234 “CataLight”, projects A4/B3/B6/C4, project no. 364549901, and project no. BE 5102/5-1). F. F. gratefully acknowledges the China Scholarship Council CSC for a PhD fellowship. R. B. and D. M. acknowledge funding from the European Union's Horizon Europe programme for research and innovation under grant agreement No. 101122061 (SUNGATE). D. G. acknowledges Deutsche Forschungsgemeinschaft (DFG) for a Walter Benjamin Fellowship (project no. 510966757). C. S. and D. G. acknowledge funding from the Top Level Research Area SusInnoScience of the federal state of Rheinland-Pfalz. The authors thank Joachim Bansmann (XPS analysis), Manuel Mundszinger (EDX analysis), and Changbin Im (TOC image) for their help.References
- S. Chu, Y. Cui and N. Liu, Nat. Mater., 2017, 16, 16–22 CrossRef PubMed
.
- I. Roger, M. A. Shipman and M. D. Symes, Nat. Rev. Chem., 2017, 1, 0003 CrossRef CAS
- Z. Zhang, X. Huang, B. Zhang and Y. Bi, Energy Environ. Sci., 2022, 15, 2867–2873 RSC
- M. E. Ivanova, R. Peters, M. Müller, S. Haas, M. F. Seidler, G. Mutschke, K. Eckert, P. Röse, S. Calnan, R. Bagacki, R. Schlatmann, C. Grosselindemann, L.-A. Schäfer, N. H. Menzler, A. Weber, R. van de Krol, F. Liang, F. F. Abdi, S. Brendelberger, N. Neumann, J. Grobbel, M. Roeb, C. Sattler, I. Duran, B. Dietrich, M. E. C. Hofberger, L. Stoppel, N. Uhlenbruck, T. Wetzel, D. Rauner, A. Hecimovic, U. Fantz, N. Kulyk, J. Harting and O. Guillon, Angew. Chem., Int. Ed., 2023, 62, e202218850 CrossRef CAS PubMed
- H. Dau, C. Limberg, T. Reier, M. Risch, S. Roggan and P. Strasser, ChemCatChem, 2010, 2, 724–761 CrossRef CAS
- J. Song, C. Wei, Z.-F. Huang, C. Liu, L. Zeng, X. Wang and Z. J. Xu, Chem. Soc. Rev., 2020, 49, 2196–2214 RSC
- J. Seo, H. Nishiyama, T. Yamada and K. Domen, Angew. Chem., Int. Ed., 2018, 57, 8396–8415 CrossRef CAS PubMed
- A. G. Scheuermann, J. D. Prange, M. Gunji, C. E. D. Chidsey and P. C. McIntyre, Energy Environ. Sci., 2013, 6, 2487–2496 RSC
- S. Hu, M. R. Shaner, J. A. Beardslee, M. Lichterman, B. S. Brunschwig and N. S. Lewis, Science, 2014, 344, 1005–1009 CrossRef CAS PubMed
- I. A. Digdaya, G. W. P. Adhyaksa, B. J. Trześniewski, E. C. Garnett and W. A. Smith, Nat. Commun., 2017, 8, 15968 CrossRef CAS PubMed
- L. Wang, D. Mitoraj, S. Turner, O. V. Khavryuchenko, T. Jacob, R. K. Hocking and R. Beranek, ACS Catal., 2017, 7, 4759–4767 CrossRef CAS
- P. Longchin, D. Mitoraj, O. M. Reyes, C. Adler, N. Wetchakun and R. Beranek, J. Phys. Energy, 2020, 2, 044001 CrossRef CAS
- R. Gong, D. Mitoraj, R. Leiter, M. Mundszinger, A. K. Mengele, I. Krivtsov, J. Biskupek, U. Kaiser, R. Beranek and S. Rau, Front. Chem., 2021, 9, 709903 CrossRef CAS PubMed
- J. T. Kirner and R. G. Finke, J. Mater. Chem. A, 2017, 5, 19560–19592 RSC
- L. M. Peter, K. G. U. Wijayantha and A. A. Tahir, Faraday Discuss., 2012, 155, 309–322 RSC
- J. Park, J. Kang, S. Chaule and J.-H. Jang, J. Mater. Chem. A, 2023, 11, 24551–24565 RSC
- Y. Gao and T. W. Hamann, Chem. Commun., 2017, 53, 1285–1288 RSC
- D. Peeters, O. M. Reyes, L. Mai, A. Sadlo, S. Cwik, D. Rogalla, H.-W. Becker, H. M. Schütz, J. Hirst, S. Müller, D. Friedrich, D. Mitoraj, M. Nagli, M. C. Toroker, R. Eichberger, R. Beranek and A. Devi, J. Mater. Chem. A, 2018, 6, 10206–10216 RSC
- F. F. Abdi, L. Han, A. H. M. Smets, M. Zeman, B. Dam and R. van de Krol, Nat. Commun., 2013, 4, 2195 CrossRef PubMed
- T. W. Kim and K.-S. Choi, Science, 2014, 343, 990–994 CrossRef CAS PubMed
- F. F. Abdi, T. J. Savenije, M. M. May, B. Dam and R. van de Krol, J. Phys. Chem. Lett., 2013, 4, 2752–2757 CrossRef CAS
- J. H. Kim and J. S. Lee, Adv. Mater., 2019, 31, 1806938 CrossRef PubMed
- M. B. Vishlaghi, A. Kahraman, N. Österbacka, E. Usman, E. Erdem, A. Sennaroğlu, J. Wiktor and S. Kaya, J. Mater. Chem. A, 2023, 11, 16648–16658 RSC
- R.-T. Gao, N. T. Nguyen, T. Nakajima, J. He, X. Liu, X. Zhang, L. Wang and L. Wu, Sci. Adv., 2023, 9, eade4589 CrossRef PubMed
- R.-T. Gao and L. Wang, Angew. Chem., Int. Ed., 2020, 59, 23094–23099 CrossRef CAS PubMed
- T. Zhou, S. Chen, J. Wang, Y. Zhang, J. Li, J. Bai and B. Zhou, Chem. Eng. J., 2021, 403, 126350 CrossRef CAS
- T. Zhou, J. Wang, Y. Zhang, C. Zhou, J. Bai, J. Li and B. Zhou, Chem. Eng. J., 2022, 431, 133414 CrossRef CAS
- A. Polo, M. V. Dozzi, I. Grigioni, C. Lhermitte, N. Plainpan, L. Moretti, G. Cerullo, K. Sivula and E. Selli, Sol. RRL, 2022, 6, 2200349 CrossRef CAS
- S. Wang, P. Chen, J.-H. Yun, Y. Hu and L. Wang, Angew. Chem., Int. Ed., 2017, 56, 8500–8504 CrossRef CAS PubMed
- W. Luo, Z. Yang, Z. Li, J. Zhang, J. Liu, Z. Zhao, Z. Wang, S. Yan, T. Yu and Z. Zou, Energy Environ. Sci., 2011, 4, 4046–4051 RSC
- Y. Park, D. Kang and K.-S. Choi, Phys. Chem. Chem. Phys., 2014, 16, 1238–1246 RSC
- H. W. Jeong, T. H. Jeon, J. S. Jang, W. Choi and H. Park, J. Phys. Chem. C, 2013, 117, 9104–9112 CrossRef CAS
- M. Rohloff, B. Anke, S. Zhang, U. Gernert, C. Scheu, M. Lerch and A. Fischer, Sustainable Energy Fuels, 2017, 1, 1830–1846 RSC
- K.-H. Ye, H. Li, D. Huang, S. Xiao, W. Qiu, M. Li, Y. Hu, W. Mai, H. Ji and S. Yang, Nat. Commun., 2019, 10, 3687 CrossRef PubMed
- A. J. E. Rettie, H. C. Lee, L. G. Marshall, J.-F. Lin, C. Capan, J. Lindemuth, J. S. McCloy, J. Zhou, A. J. Bard and C. B. Mullins, J. Am. Chem. Soc., 2013, 135, 11389–11396 CrossRef CAS PubMed
- B. Pattengale and J. Huang, Phys. Chem. Chem. Phys., 2016, 18, 32820–32825 RSC
- A. Polo, I. Grigioni, M. Magni, A. Facibeni, M. V. Dozzi and E. Selli, Appl. Surf. Sci., 2021, 556, 149759 CrossRef CAS
- B. Liu, X. Wang, Y. Zhang, L. Xu, T. Wang, X. Xiao, S. Wang, L. Wang and W. Huang, Angew. Chem., Int. Ed., 2023, 62, e202217346 CrossRef CAS PubMed
- R.-T. Gao, D. He, L. Wu, K. Hu, X. Liu, Y. Su and L. Wang, Angew. Chem., Int. Ed., 2020, 59, 6213–6218 CrossRef CAS PubMed
- S. K. Pilli, T. E. Furtak, L. D. Brown, T. G. Deutsch, J. A. Turner and A. M. Herring, Energy Environ. Sci., 2011, 4, 5028–5034 RSC
- F. F. Abdi and R. van de Krol, J. Phys. Chem. C, 2012, 116, 9398–9404 CrossRef CAS
- B. Moss, F. S. Hegner, S. Corby, S. Selim, L. Francàs, N. López, S. Giménez, J.-R. Galán-Mascarós and J. R. Durrant, ACS Energy Lett., 2019, 4, 337–342 CrossRef CAS
- Y. Yang, S. Wan, S. Li, R. Wang, M. Ou, B. Liu and Q. Zhong, J. Mater. Chem. A, 2023, 11, 1756–1765 RSC
- H. Yin, Y. Guo, N. Zhang, Y. Wang, S. Zhang and R. Jiang, J. Mater. Chem. A, 2023, 11, 24239–24247 RSC
- Y. Lu, J. Su, J. Shi and D. Zhou, ACS Appl. Energy Mater., 2020, 3, 9089–9097 CrossRef CAS
- Y. Ma, A. Kafizas, S. R. Pendlebury, F. L. Formal and J. R. Durrant, Adv. Funct. Mater., 2016, 26, 4951–4960 CrossRef CAS
- Y. Ma, F. L. Formal, A. Kafizas, S. R. Pendlebury and J. R. Durrant, J. Mater. Chem. A, 2015, 3, 20649–20657 RSC
- C. Zachäus, F. F. Abdi, L. M. Peter and R. van de Krol, Chem. Sci., 2017, 8, 3712–3719 RSC
- F. S. Hegner, I. Herraiz-Cardona, D. Cardenas-Morcoso, N. López, J.-R. Galán-Mascarós and S. Gimenez, ACS Appl. Mater. Interfaces, 2017, 9, 37671–37681 CrossRef CAS PubMed
- L. Francàs, S. Selim, S. Corby, D. Lee, C. A. Mesa, E. Pastor, K.-S. Choi and J. R. Durrant, Chem. Sci., 2021, 12, 7442–7452 RSC
- C. Streb, K. Kastner and J. Tucher, Phys. Sci. Rev., 2019, 4, 20170177 Search PubMed
- B. Schwarz, J. Förster, M. K. Goetz, D. Yücel, C. Berger, T. Jacob and C. Streb, Angew. Chem., Int. Ed., 2016, 55, 6329–6333 CrossRef CAS PubMed
- H. Park and W. Choi, J. Phys. Chem. B, 2003, 107, 3885–3890 CrossRef CAS
- Q. Yin, J. M. Tan, C. Besson, Y. V. Geletii, D. G. Musaev, A. E. Kuznetsov, Z. Luo, K. I. Hardcastle and C. L. Hill, Science, 2010, 328, 342–345 CrossRef CAS PubMed
- J. J. Stracke and R. G. Finke, ACS Catal., 2013, 3, 1209–1219 CrossRef CAS
- J. W. Vickers, H. Lv, J. M. Sumliner, G. Zhu, Z. Luo, D. G. Musaev, Y. V. Geletii and C. L. Hill, J. Am. Chem. Soc., 2013, 135, 14110–14118 CrossRef CAS PubMed
- S. J. Folkman, J. Soriano-Lopez, J. R. Galán-Mascarós and R. G. Finke, J. Am. Chem. Soc., 2018, 140, 12040–12055 CrossRef CAS PubMed
- J. J. Stracke and R. G. Finke, J. Am. Chem. Soc., 2011, 133, 14872–14875 CrossRef CAS PubMed
- D. Jeon, H. Kim, C. Lee, Y. Han, M. Gu, B.-S. Kim and J. Ryu, ACS Appl. Mater. Interfaces, 2017, 9, 40151–40161 CrossRef CAS PubMed
- R. Gong, D. Mitoraj, D. Gao, M. Mundszinger, D. Sorsche, U. Kaiser, C. Streb, R. Beranek and S. Rau, Adv. Sustainable Syst., 2022, 6, 2100473 CrossRef CAS
- K. Fan, H. Chen, B. He and J. Yu, Chem. Eng. J., 2020, 392, 123744 CrossRef CAS
- J. Jian, Y. Xu, X. Yang, W. Liu, M. Fu, H. Yu, F. Xu, F. Feng, L. Jia, D. Friedrich, R. van de Krol and H. Wang, Nat. Commun., 2019, 10, 2609 CrossRef PubMed
- C. Li, F. Feng, J. Jian, Y. Xu, F. Li, H. Wang and L. Jia, J. Mater. Sci. Tech., 2021, 79, 21–28 CrossRef CAS
- R. P. Antony, P. S. Bassi, F. F. Abdi, S. Y. Chiam, Y. Ren, J. Barber, J. S. C. Loo and L. H. Wong, Electro. Acta, 2016, 211, 173–182 CrossRef CAS
- G. Talasila, S. Sachdev, U. Srivastva, D. Saxena and S. S. V. Ramakumar, Energy Rep., 2020, 6, 1963–1972 CrossRef
- Y. Liu, P. Deng, R. Wu, R. A. Geioushy, Y. Li, Y. Liu, F. Zhou, H. Li and C. Sun, Front. Phys., 2021, 16, 53503 CrossRef
- G. Silversmit, D. Depla, H. Poelman, G. B. Marin and R. D. Gryse, J. Electron Spectrosc. Relat. Phenom., 2004, 135, 167–175 CrossRef CAS
- S. S. Mali, G. Ryeol Park, H. Kim, H. H. Kim, J. V. Patil and C. K. Hong, Nanoscale Adv., 2019, 1, 799–806 RSC
- L. Chen, E. Alarcón-Lladó, M. Hettick, I. D. Sharp, Y. Lin, A. Javey and J. W. Ager, J. Phys. Chem. C, 2013, 117, 21635–21642 CrossRef CAS
- M. C. Biesinger, B. P. Payne, A. P. Grosvenor, L. W. M. Lau, A. R. Gerson and R. S. Smart, Appl. Surf. Sci., 2011, 257, 2717–2730 CrossRef CAS
- R. Chong, Y. Du, Z. Chang, Y. Jia, Y. qiao, S. Liu, Y. Liu, Y. Zhou and D. Li, Appl. Catal., B, 2019, 250, 224–233 CrossRef CAS
- S. Yi, B. Wulan, J. Yan and Q. Jiang, Adv. Funct. Mater., 2019, 29, 1801902 CrossRef
- U. Prasad, J. Prakash, S. K. Gupta, J. Zuniga, Y. Mao, B. Azeredo and A. N. M. Kannan, ACS Appl. Mater. Interfaces, 2019, 11, 19029–19039 CrossRef CAS PubMed
- A. Walsh, Y. Yan, M. N. Huda, M. M. Al-Jassim and S.-H. Wei, Chem. Mater., 2009, 21, 547–551 CrossRef CAS
- J. K. Cooper, S. Gul, F. M. Toma, L. Chen, Y.-S. Liu, J. Guo, J. W. Ager, J. Yano and I. D. Sharp, J. Phys. Chem. C, 2015, 119, 2969–2974 CrossRef CAS
- C. Lohaus, A. Klein and W. Jaegermann, Nat. Commun., 2018, 9, 4309 CrossRef PubMed
- E. Pastor, M. Sachs, S. Selim, J. R. Durrant, A. A. Bakulin and A. Walsh, Nat. Rev. Mater., 2022, 7, 503–521 CrossRef CAS
- V. Jovic, J. Laverock, A. J. E. Rettie, J.-S. Zhou, C. B. Mullins, V. R. Singh, B. Lamoureux, D. Wilson, T.-Y. Su, B. Jovic, H. Bluhm, T. Söhnel and K. E. Smith, J. Mater. Chem. A, 2015, 3, 23743–23753 RSC
- K. E. Kweon, G. S. Hwang, J. Kim, S. Kim and S. Kim, Phys. Chem. Chem. Phys., 2015, 17, 256–260 RSC
- J. Wiktor, F. Ambrosio and A. Pasquarello, ACS Energy Lett., 2018, 3, 1693–1697 CrossRef CAS
- F. Wu and Y. Ping, J. Mater. Chem. A, 2018, 6, 20025–20036 RSC
- L. M. Peter, Chem. Rev., 1990, 90, 753–769 CrossRef CAS
- Y. Surendranath, M. Dincă and D. G. Nocera, J. Am. Chem. Soc., 2009, 131, 2615–2620 CrossRef CAS PubMed
- M. Antuch, P. Millet, A. Iwase and A. Kudo, Appl. Catal., B, 2018, 237, 401–408 CrossRef CAS
- K. Szaciłowski and W. Macyk, C. R. Chim, 2006, 9, 315–324 CrossRef
- R. Beranek and H. Kisch, Angew. Chem., Int. Ed., 2008, 47, 1320–1322 CrossRef CAS PubMed
- M. Long, R. Beranek, W. Cai and H. Kisch, Electro. Acta, 2008, 53, 4621–4626 CrossRef CAS
- S. Gawęda, A. Podborska, W. Macyk and K. Szaciłowski, Nanoscale, 2009, 1, 299–316 RSC
- A. Podborska, M. Suchecki, K. Mech, M. Marzec, K. Pilarczyk and K. Szaciłowski, Nat. Commun., 2020, 11, 854 CrossRef CAS PubMed
- H. Dotan, K. Sivula, M. Grätzel, A. Rothschild and S. C. Warren, Energy Environ. Sci., 2011, 4, 958–964 RSC
- S. Zhang, I. Ahmet, S.-H. Kim, O. Kasian, A. M. Mingers, P. Schnell, M. Kölbach, J. Lim, A. Fischer, K. J. J. Mayrhofer, S. Cherevko, B. Gault, R. van de Krol and C. Scheu, ACS Appl. Energy Mater., 2020, 3, 9523–9527 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ma00089g |
| This journal is © The Royal Society of Chemistry 2024 |