
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Supervalent doping and its effect on the thermal, structural and electrochemical properties of Li7La3Zr2O12 solid electrolytes†
Janez
Košir
 a,
Seyedabolfazl
Mousavihashemi‡
a,
Milla
Suominen
a,
Anna
Kobets
a,
Benjamin P.
Wilson
b,
Eeva-Leena
Rautama
a and
Tanja
Kallio
*a
a,
Seyedabolfazl
Mousavihashemi‡
a,
Milla
Suominen
a,
Anna
Kobets
a,
Benjamin P.
Wilson
b,
Eeva-Leena
Rautama
a and
Tanja
Kallio
*a
aDepartment of Chemistry and Materials Science, School of Chemical Engineering, Aalto University, 02150 Espoo, Finland. E-mail: tanja.kallio@aalto.fi
bDepartment of Chemical and Metallurgical Engineering, School of Chemical Engineering, Aalto University, 02150 Espoo, Finland
First published on 3rd May 2024
Abstract
Supervalent doping is one of the most common methods used to stabilize the highly conductive cubic phase of Li7La3Zr2O12 (LLZO) solid electrolytes. While several different doping elements have shown that they are capable of stabilizing the cubic LLZO structure, there is still no clear consensus as to an optimal doping strategy. In this study we present one of the most extensive comparative analyses on supervalent doping of LLZO done to date. Herein, we compare the effects of eight different doping elements (Al, Ga, Fe, Ta, Nb, Sb, W and Mo) on the synthesis, crystal structure, morphology, and electrochemical properties of LLZO. We also propose a new guideline that would allow for the quick and easy identification of doping elements in LLZO and the estimation of their concentration using Raman spectroscopy. Our results show how Ga doping leads to exceptionally high ionic conductivities (1.30 × 10−3 S cm−1) and low activation energies (0.26 eV) due to changes in the crystal symmetry of LLZO. On top of that, Ga doping also significantly lowers the required synthesis temperatures and increases the relative density of the LLZO structure, making Ga the most suitable element for LLZO doping. On the other hand, Nb doping shows the lowest ionic conductivity (1.91 × 10−4 S cm−1) and a high activation energy (0.44 eV) of the investigated dopants, due to a poor sintering performance.
1 Introduction
Ever since their widespread introduction in the early 90's, Li-ion batteries have garnered significant attention as systems for electrochemical energy storage due to their high energy density and good cycling stability. However, current Li-ion batteries are still limited in their performance mainly due to the operational limits and safety concerns of the liquid electrolyte.1,2 With an increasing need for even better and safer energy storage solutions, all-solid-state batteries have presented themselves as the next major step in the development of Li-ion energy storage technology.2,3 By replacing traditional liquid electrolytes with novel inorganic solid-state ionic conductors, we can greatly improve the power density, operational voltage, and long-term stability of Li-ion batteries while at the same time making them safer. Additionally, solid electrolytes have shown to be relatively stable against Li metal and are able to effectively suppress Li-dendrite growth, enabling the implementation of a Li–metal anode which would significantly increase the batteries energy density.4–6 Although solid electrolytes possess numerous advantages over their liquid counterparts, they still face several challenges before they can be implemented in a more widespread use, such as lower ionic conductivities than liquid electrolytes and interfacial issues with the electrodes.7,8 Today, numerous different inorganic materials have been developed to be used as solid electrolytes in Li-ion batteries.9 Among them, garnet-type Li7La3Zr2O12 (LLZO) has been shown to be one of the most promising due to its relatively high ionic conductivity, good chemical and high thermal stability, as well as its wide electrochemical stability window.10,11 Although, recent reports indicate that the electrochemical stability and stability against Li–metal is not as high as previously thought of.12LLZO is a polymorph with two different crystal structures, a tetragonal (space group I41/acd) and cubic phase (space group Ia![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) d).10,13,14 Generally, the cubic LLZO (c-LLZO) structure is more desirable as it maintains a higher ionic conductivity (>10−4 S cm−1) and lower activation energy when compared to the tetragonal LLZO (t-LLZO) structure (∼10−6 S cm−1).10,13 However, while the c-LLZO phase is more desirable for practical applications, LLZO is significantly more thermodynamically stable in its tetragonal form at room temperatures.15,16 Thus, various methods have been developed in order to stabilize the highly conductive c-LLZO phase at room temperatures.17,18
d).10,13,14 Generally, the cubic LLZO (c-LLZO) structure is more desirable as it maintains a higher ionic conductivity (>10−4 S cm−1) and lower activation energy when compared to the tetragonal LLZO (t-LLZO) structure (∼10−6 S cm−1).10,13 However, while the c-LLZO phase is more desirable for practical applications, LLZO is significantly more thermodynamically stable in its tetragonal form at room temperatures.15,16 Thus, various methods have been developed in order to stabilize the highly conductive c-LLZO phase at room temperatures.17,18
One of the most common methods of stabilizing the c-LLZO phase is through supervalent elemental doping. By introducing ions with a higher oxidation state into the structure of LLZO, the material retains the neutral charge of the system by removing Li+ ions, thus creating Li-site vacancies and disrupting the Li-ion sublattice ordering. This, in-turn, increases the entropy and lowers the free energy of the system, thereby stabilizing the c-LLZO structure.18–21 Previous studies show that a minimum concentration of approximately 0.4–0.5 Li vacancies per formula unit (pfu) are required in order to fully stabilize the c-LLZO structure.16,19,22–26
To date, several different supervalent dopants have been used in order to stabilize the c-LLZO phase including, Al3+,16,22,27 Ta5+,23,28,29 Nb5+,29,30 Ga3+,25,31,32 Fe3+,33,34 W6+,35,36 Mo6+,24,26 Sb5+,29,37 and Te6+.38 Although all these elements have shown that they can stabilize the c-LLZO structure, the choice of dopant can affect various other properties of the material that can have a wide range of effects on the electrochemical performance of LLZO and its use as a solid electrolyte in practical applications. Some of these effects include:
• Changes in the bulk ionic conductivity based on the specific site of the doping element and its effect on the crystal structure of LLZO.29,39,40
• Formation of secondary phases between the LLZO grains, which can either enhance or hinder Li-ion migration across the grain boundary and increase the relative density of the material during sintering.35,41–46
• Stability against Li–metal and the formation of interfacial layers on the electrolyte surface, that can impede the mobility of Li-ions.36,47–49
• Stability of the bulk electrolyte during cycling and the effect of the applied voltage on the formation of secondary phases.12,36,50
• The critical current density for the formation and growth of Li dendrites.51,52
Often times it is shown that while a dopant may form c-LLZO with exceptionally good properties for some of these characteristics, they can also reduce the performance of others.
Several comparative studies have been conducted in the past on the properties and performance of c-LLZO using different doping elements.29,32,47,53,54 While these studies provide us with a better understanding on the effects of LLZO doping, they typically compare only two or three dopants and rarely include more uncommon elements, such as Sb and Mo. Since the properties of LLZO also heavily depend on the synthesis method and processing parameters, it can be rather difficult to properly compare results between different studies. Therefore, more extensive comparative studies are required in order to better understand the effects of doping and come to a consensus on an optimal doping strategy of LLZO.
In this work we present one of the most comprehensive comparative analyses on the effects of elemental doping on the properties of LLZO done to date. For this we prepared c-LLZO samples through a conventional solid-state synthesis method using eight different supervalent dopants, Al3+, Ga3+, Fe3+, Ta5+, Nb5+, Sb5+, W6+ and Mo6+. The samples were analysed in terms of the dopants effect on the synthesis performance, crystal structure, morphology, and electrochemical properties of LLZO. The results demonstrate that Ga doping not only produces LLZO samples with exceptionally high conductivities and low activation energies, but also lowers the required synthesis temperature and densifies the LLZO structure.
2 Experimental
2.1 Sample preparation
A series of supervalent doped c-LLZO pellets were prepared by means of a conventional solid-state reaction method using eight different dopants. The precursor mixtures were prepared from stoichiometric amounts of Li2CO3 (≥99.0%, Alfa Aesar), La2O3 (99.99%, Alfa Aesar, dried at 900 °C for 12 h), ZrO2 (99.7%, Alfa Aesar) and the doping precursor, in accordance with Table 1. The concentrations of the doping elements were chosen to induce 0.5 Li-site vacancies per formula unit (pfu), which should fully stabilize the c-LLZO phase whilst maintaining a high ionic conductivity. An additional 15 mol% of Li was added to each precursor mixture to compensate for any Li-loss during the high-temperature treatment. The mixtures were ball milled in zirconia milling vessels with a MM 400 mixer mill (Retsch GmbH) for 2 h at a frequency of 20 Hz, using 12 mm zirconia balls and isopropanol as the milling medium. The suspensions were then dried in a UNB 100 convection oven (Memmert GmbH) at 75 °C.| Doping element | Concentration [pfu] | Doping site | Nominal LLZO composition | Doping precursor |
|---|---|---|---|---|
| Al3+ | 0.25 | Li | Li6.25Al0.25La3Zr2O12 | Al2O3-γ (99.997%, Alfa Aesar) |
| Ga3+ | 0.25 | Li | Li6.25Ga0.25La3Zr2O12 | Ga2O3 (99.99%, Alfa Aesar) |
| Fe3+ | 0.25 | Li | Li6.25Fe0.25La3Zr2O12 | Fe2O3 (99.9%, Thermo Scientific) |
| Ta5+ | 0.5 | Zr | Li6.5La3Zr1.5Ta0.5O12 | Ta2O5 (99.85%, Alfa Aesar) |
| Nb5+ | 0.5 | Zr | Li6.5La3Zr1.5Nb0.5O12 | Nb2O5 (99.9%, Thermo Scientific) |
| Sb5+ | 0.5 | Zr | Li6.5La3Zr1.5Sb0.5O12 | Sb2O5 (99.998%, Thermo Scientific) |
| W6+ | 0.25 | Zr | Li6.5La3Zr1.75W0.25O12 | WO3 (99.998%, Alfa Aesar) |
| Mo6+ | 0.25 | Zr | Li6.5La3Zr1.75Mo0.25O12 | MoO3 (99.998%, Alfa Aesar) |
Precursor mixtures were placed in ZrO2 crucibles and synthesized in a CWF 13/5 muffle furnace (Carbolite Ltd) at 950 °C for 12 h, with a heating rate of 200 °C h−1. The synthesized powders were then cold pressed uniaxially into round pellets with a ∼1 mm thickness and 13 mm diameter. The pellets were placed in an MgO crucible and sintered at 1200 °C for 12 h, with the remaining LLZO powder placed in the bottom of the crucible to prevent contamination of the pellets by the crucible. Sintered pellets were dry polished in air with P1200 and P2500 grit SiC grinding papers down to a thickness of ∼0.5 mm and stored in an Ar-filled glovebox.
2.2 Characterization
Phase composition and crystal structure of the LLZO samples were analyzed with X-ray diffraction (XRD) and Raman spectroscopy. XRD patterns were measured of both the LLZO powders after synthesis and of the sintered pellets, with a Panalytical X’Pert Pro MPD Alpha1 XRD (CuKα1; 45 kV, 40 mA). The pellets were ground into a powder and the data was collected in a 2θ range of 10°–110°. XRD data was analyzed by means of Rietveld refinement with the program FullProf.The chemical compositions were determined by inductively coupled plasma-optical emission spectroscopy (ICP-OES; Agilent 5900 SVDV) from dissolved samples. A detailed description of the sample preparation and the measurement procedure is provided in the ESI.†
Raman spectroscopy was carried out using a confocal Renishaw inVia Qontor Raman spectrometer equipped with a Leica microscope, a 532 nm wavelength laser, 2400 l mm−1 grating, and a CCD detector.
The cross-sectional morphology and elemental distribution of the sintered pellets was analyzed with a JEOL JIB-4700F scanning electron microscope (SEM) coupled with an energy dispersive X-ray spectrometer (EDS) by Oxford Instruments. Images were taken using secondary electrons, with an operational voltage of 10 kV while 15 kV was used for the EDS analysis.
Relative densities of the pellets were investigated with a Quantachrome Ultrapyc 1200e He-pycnometer using a 4.25 cm3 microcell. Samples were allowed to equilibrate for at least 30 minutes in the equipment prior to measurement and each sample was measured 50 times.
The ionic conductivities and activation energies of the sintered LLZO pellets were investigated by means of electrochemical impedance spectroscopy (EIS). The polished pellets were sputtered on both sides with a ∼10 nm layer of an Li-ion blocking 80![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :20 Au/Pd electrode and assembled in a HS Flat Cell (Hohsen). The cell was connected to an Autolab PGSTAT302N potentiostat (Nova 2.1.3 software) and the EIS spectra were obtained by applying a 10 mVrms potential amplitude over a frequency range from 1 MHz to 1 Hz at room temperature. The cell was then placed in a G-Cell oven (Fratelli Galli) and the measurements were repeated in a temperature range between 30 and 80 °C. The impedance data was analyzed with ZView (Scribner Associates, ver. 3.3).
:20 Au/Pd electrode and assembled in a HS Flat Cell (Hohsen). The cell was connected to an Autolab PGSTAT302N potentiostat (Nova 2.1.3 software) and the EIS spectra were obtained by applying a 10 mVrms potential amplitude over a frequency range from 1 MHz to 1 Hz at room temperature. The cell was then placed in a G-Cell oven (Fratelli Galli) and the measurements were repeated in a temperature range between 30 and 80 °C. The impedance data was analyzed with ZView (Scribner Associates, ver. 3.3).
The electronic conductivity of the LLZO pellets was measured at room temperature by means of a DC polarization method, where a 0.2 V potential was applied to the sputtered pellets and the resulting current was measured over a period of 30 min.
3 Results and discussion
3.1 Synthesis and phase formation of cubic LLZO
Doping of LLZO not only affect its functionality as a solid electrolyte but also the required synthesis parameters of the material. One of the ways in which the doping element affects the synthesis of LLZO is by changing the thermodynamic and kinetic factors of the reaction mechanism for the formation and stabilization of c-LLZO.27,55–57 This can be observed by analyzing the XRD patterns of doped LLZO powders after synthesis at 950 °C, presented in Fig. 1. By analyzing the powders phase fractions, as well as the c-LLZO:t-LLZO ratios, we can assess a dopant's level of affinity towards integrating into the LLZO structure, where a higher phase fraction of c-LLZO indicates a greater affinity of the doping element towards LLZO. For example, synthesized Ga–LLZO powder shows a fully stabilized c-LLZO phase with minor amounts of unreacted La2O3 and Li2ZrO3. This means that the doping element is fully integrated into the LLZO structure, which would indicate a very high chemical affinity and good diffusion kinetics of Ga3+ towards LLZO. On the other hand, Al–LLZO and Fe–LLZO powders show only a partially stabilized c-LLZO phase, with the majority of the LLZO structure present in its tetragonal form and a significant amount of the doping element located in secondary phases, such as Li0.5La2Al0.5O4 and LaFeO3. The high phase fraction of t-LLZO indicates mass transport limitations of the doping element and a low affinity of the intermediary phases towards integrating into the LLZO structure.34
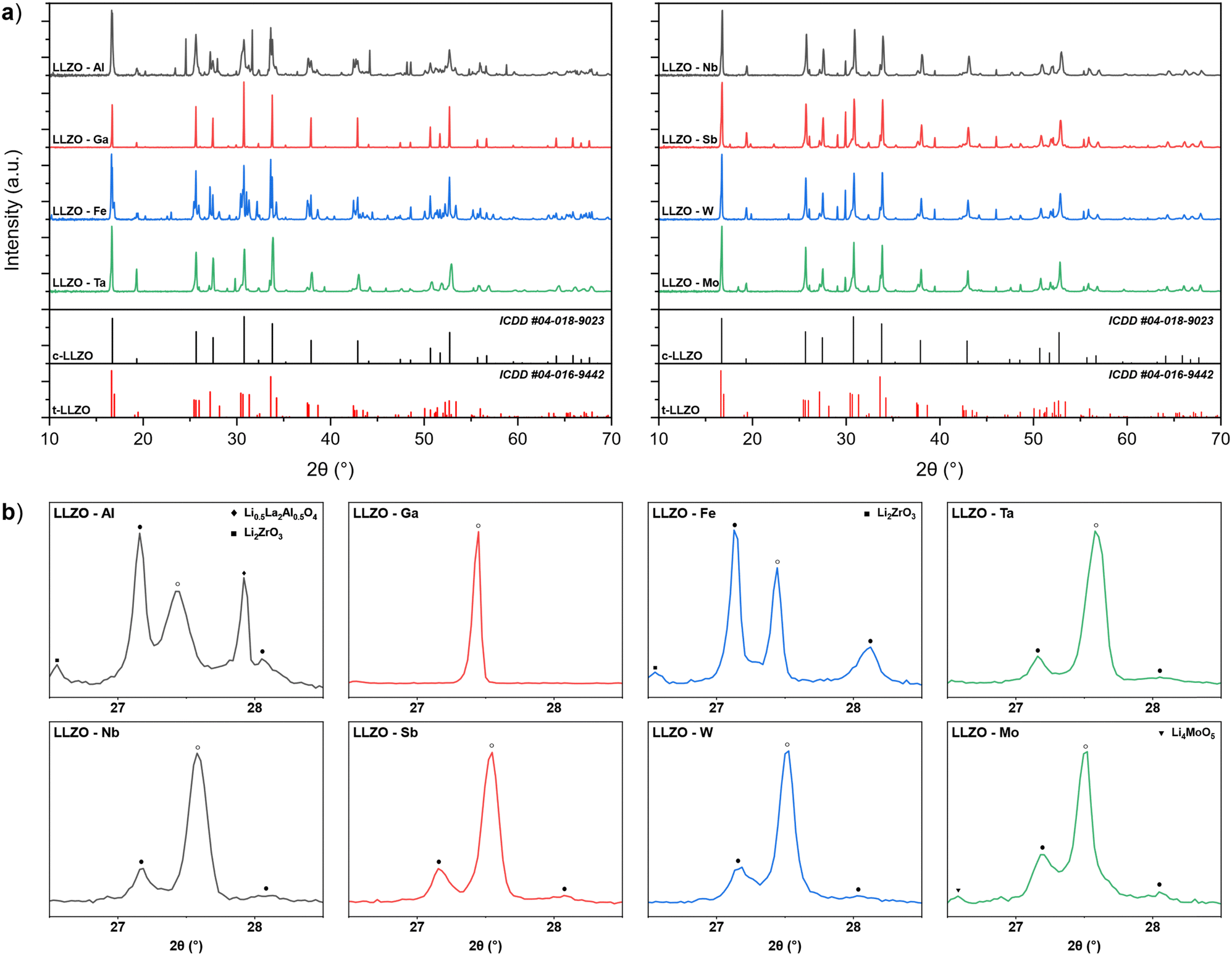 | ||
| Fig. 1 (a) XRD patterns of doped LLZO powders after synthesis at 950 °C. (b) Partially enlarged XRD patterns highlighting the intensity ratio between the t-LLZO and c-LLZO peaks (● – t-LLZO, ○ – c-LLZO). | ||
Zr site dopants also show a partially stabilized c-LLZO structure, with similar c-LLZO:t-LLZO ratios. Nevertheless, the c-LLZO:t-LLZO ratios are notably higher than with Al and Fe doped LLZO, indicating a higher affinity of these dopants towards LLZO. Based on the LLZO phase fraction composition of the synthesized powders shown in Table 2, the dopants level of affinity towards LLZO integration and c-LLZO phase stabilization can be presented as follows: Ga > (Ta, Nb, Sb, W) > Mo > (Al, Fe). These results highlight that Ga doping would be most beneficial for synthesizing c-LLZO as it would allow for lower synthesis temperatures and shorter synthesis times thus allowing for a more optimized synthesis process. In contrast, Al and Fe doping require much higher synthesis temperatures and prolonged synthesis times which can cause additional issues such as Li-loss and decomposition of LLZO.27
| Sample | LLZO phase fraction [wt%] | c-LLZO phase fraction [wt%] | t-LLZO phase fraction [wt%] | c-LLZO:t-LLZO phase ratio |
|---|---|---|---|---|
| LLZO–Al | 83.8 | 23.5 | 60.3 | 28:72 |
| LLZO–Ga | 97.2 | 97.2 | 0 | 100:0 |
| LLZO–Fe | 92.5 | 17.5 | 75 | 19:81 |
| LLZO–Ta | 98 | 75 | 23 | 77:23 |
| LLZO–Nb | 97.5 | 67 | 30.5 | 69:31 |
| LLZO–Sb | 92 | 57 | 35 | 62:38 |
| LLZO–W | 92.5 | 57.5 | 35 | 62:38 |
| LLZO–Mo | 89 | 37.2 | 51.8 | 42:58 |
To confirm that the differences in the degree of dopant integration into the LLZO structure are related to the chemical affinity of the doping elements towards LLZO and not purely due to physical processes, we analysed the morphology of the dopant precursor particles for Al and Ga doped LLZO. Fig. S1 (ESI†) shows SEM images of Al2O3 and Ga2O3 particles within the LLZO precursor mixture. Both Al2O3 and Ga2O3 particles possess a similar size, ranging between approximately 3–5 μm. While Ga2O3 particles appear to have a slightly higher surface area, this difference cannot solely explain the significant variation in the level of dopant integration between Al and Ga–LLZO. This would indicate that the chemical affinity plays a significant role in the reaction mechanism through which the doping elements integrate into the LLZO structure.
The results presented here also correlate well with findings from previous studies, which show that Ga doping can fully stabilize the cubic structure of pre-synthesized t-LLZO at temperatures as low as 150 °C.58 On the other hand, Al and Fe doping provide only a partially stabilized c-LLZO structure after synthesis at 950 °C.34,59 The complete phase fraction composition of the synthesized LLZO powders is presented in Table S1 (ESI†).
3.2 Crystal structure of LLZO
A visual inspection of the sintered LLZO pellets after polishing shows that there is a variation in colours between the samples depending on the doping element used. As presented in Fig. 2, Al and Ga doped pellets are beige in appearance, while Zr-site doped LLZO samples appear white in colour. In contrast, Fe–LLZO shows a distinct dark brown colour. | ||
| Fig. 2 Image of polished LLZO pellets after sintering at 1200 °C. | ||
During sintering, the reactions progress through to completion so the pellets obtained have a pure c-LLZO structure without any signs of Li-loss, as evidenced by ICP-OES analysis presented in Table 3 and XRD patterns of the sintered pellets presented in Fig. 3(a) and (b). For Fe–LLZO, the XRD pattern also indicates minor amounts of LaFeO3 still present in the pellet, further indicating the low affinity of Fe secondary phases towards LLZO integration and giving Fe–LLZO a dark brown color. Although all LLZO pellets crystalize in a cubic structure, the XRD patterns reveal crystal symmetries belonging to two different space groups. Al, Ta, Nb, W and Mo doped LLZO possess a cubic structure with the commonly observed Iad (no. 230) symmetry, whereas Ga and Fe doped LLZO are observed to have a cubic structure with a symmetry that belongs to an acentric space group I![[4 with combining macron]](https://www.rsc.org/images/entities/char_0034_0304.gif) 3d (no. 220). The change in the crystal symmetry for Ga and Fe doped LLZO is identified by the presence of (310), (530) and (730) reflections at approximately 2θ = 21.65°, 40.50° and 53.75°, respectively. Fig. 3(c) shows partially enlarged XRD patterns highlighting the presence of a (310) peak for Ga and Fe doped LLZO.
3d (no. 220). The change in the crystal symmetry for Ga and Fe doped LLZO is identified by the presence of (310), (530) and (730) reflections at approximately 2θ = 21.65°, 40.50° and 53.75°, respectively. Fig. 3(c) shows partially enlarged XRD patterns highlighting the presence of a (310) peak for Ga and Fe doped LLZO.
| Dopant | Nominal composition | Experimental La:Zr ratio |
Dopant concentration (pfu) | x Li | Experimantal composition |
|---|---|---|---|---|---|
| Al | Li6.25Al0.25La3Zr2O12 | 1.49 | 0.17 | No result | LixAl0.17La3Zr2O12−δ |
| Ga | Li6.25Ga0.25La3Zr2O12 | 1.51 | 0.21 | 6.227 | Li6.23Ga0.21La3Zr2O12−δ |
| Fe | Li6.25Fe0.25La3Zr2O12 | 1.51 | 0.27 | 6.246 | Li6.25Fe0.26La3Zr2O12 |
| Ta | Li6.5La3Zr1.5Ta0.5O12 | 2.00 | 0.50 | 6.500 | Li6.5La3Zr1.5Ta0.5O12 |
| Nb | Li6.5La3Zr1.5Nb0.5O12 | 2.00 | 0.50 | 6.500 | Li6.5La3Zr1.5Nb0.5O12 |
| Sb | Li6.5La3Zr1.5Sb0.5O12 | 2.01 | 0.48 | 6.482 | Li6.48La3Zr1.5Sb0.48O12 |
| W | Li6.5La3Zr1.75W0.25O12 | 1.71 | 0.26 | 6.501 | Li6.5La3Zr1.75W0.26O12 |
| Mo | Li6.5La3Zr1.75Mo0.25O12 | 1.71 | 0.25 | 6.503 | Li6.5La3Zr1.75Mo0.25O12 |
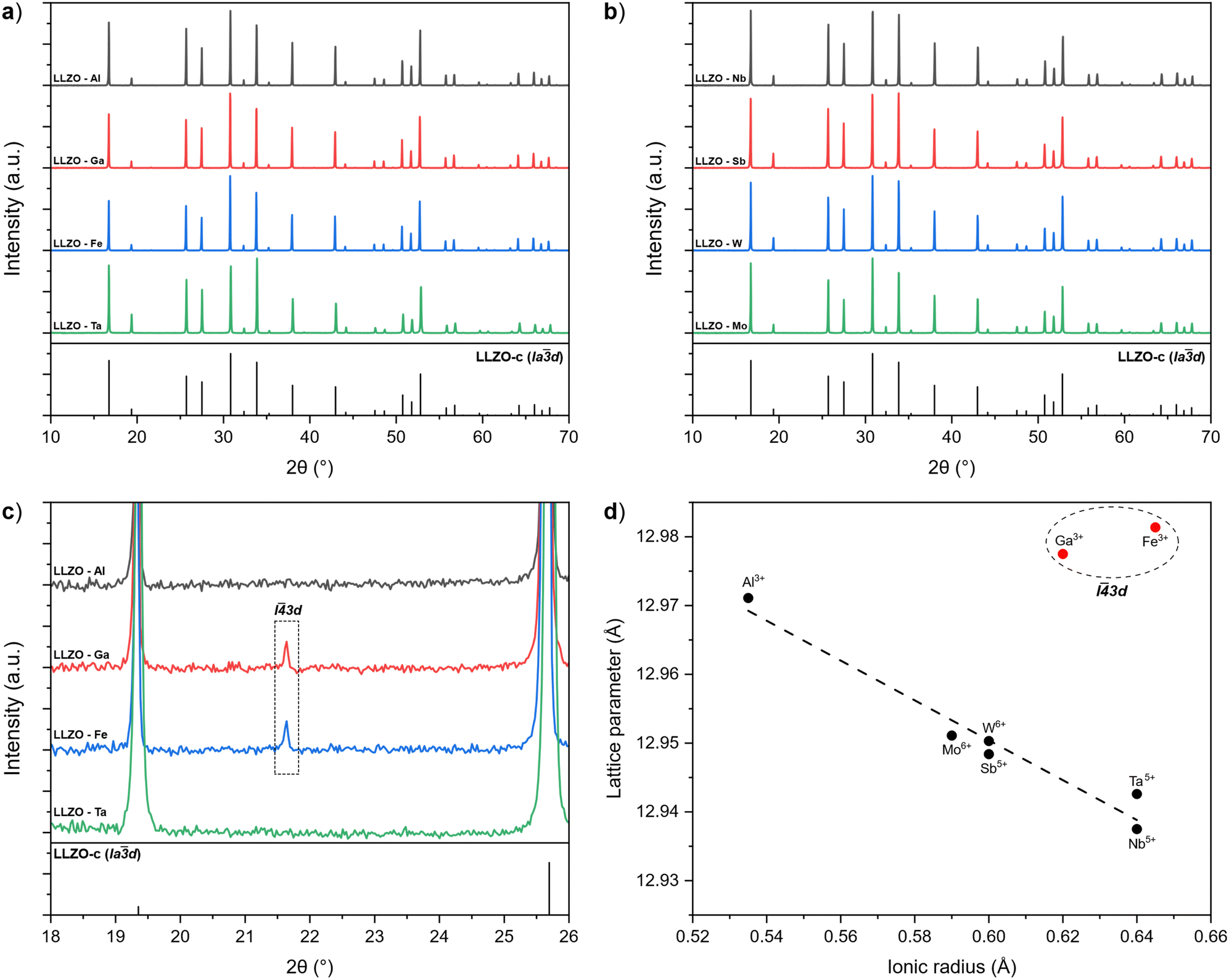 | ||
| Fig. 3 (a) and (b) XRD patterns of supervalent doped LLZO pellets with the reference pattern for c-LLZO (ICDD #04-018-9023). (c) Partially enlarged XRD patterns showcasing the presence of a I3d symmetry peak for Ga and Fe doped LLZO. (d) Relationship between the lattice parameter of LLZO and the ionic radius of the corresponding dopant.60 | ||
Evidence of changes in the crystal symmetry can also be determined from the relationship between the lattice parameters and ionic radius (Shannon,60 CN = 6) of the doping elements, as outlined in Fig. 3(d). For samples that maintain a Iad symmetry, the lattice parameter becomes smaller with a larger ionic radius of the doping element in an almost linear-like fashion due to a stronger Coulombic repulsion between the cations which shortens the metal–oxygen bonds.29 Conversely, Ga and Fe doped LLZO show a disproportionately larger lattice parameter due to a lower degree of symmetry of the I3d space group. A high-spin configuration was assumed for Fe3+.
Fig. 4 shows the crystal structures of c-LLZO belonging to space group Iad and I3d. Both crystal structures are comprised of LaO8 dodecahedrons and ZrO6 octahedrons with sharing edges. The main difference between these configurations is in the ordering of the Li-ion sublattice and migration pathways. In c-LLZO belonging to space group Iad, Li-ions occupy two crystallographic sites. Li1 sites (24d) are comprised of LiO4 tetrahedrons and act as junction points between four adjacent Li2 sites (96h) comprised of partially occupied LiO6 octahedrons. In this case, Li-ions move along a Li1–Li2–Li1 pathway, as illustrated in Fig. 4(a).14 This means that Al3+ ions can effectively block Li+ migration pathways and decrease the bulk ionic conductivity of c-LLZO.32,39,54 In c-LLZO belonging to space group I3d, Li ions occupy three crystallographic positions. Li1 (12a) and Li2 (12b) sites are comprised of LiO4 tetrahedra. These sites are equivalent to the Li1 site in the Iad space group but differ in the bond length from the Li3 site. Li3 (48e) sites consist of heavily distorted LiO6 octahedra. These sites are equivalent to the Li2 sites in the Iad space group but contain only one off-center Li site, with the Li3 site being closer to the Li1 site.31,33 Two Li-ion migration pathways have been proposed for the I3d space group, a Li2–Li3–Li1–Li3–Li2 pathway and a second Li3–Li3 pathway around the Li1 site.31,32 The formation of Li3–Li3 pathways is significant for Li-ion mobility as it opens up additional pathways for Li-ion movement. On top of that, the formation of Li3–Li3 pathways also allows Li-ions to bypass blocked Li1 sites, especially since Ga3+ and Fe3+ ions have a high preference for occupying Li1 sites, thus providing very high bulk ionic conductivities of c-LLZO.31–33
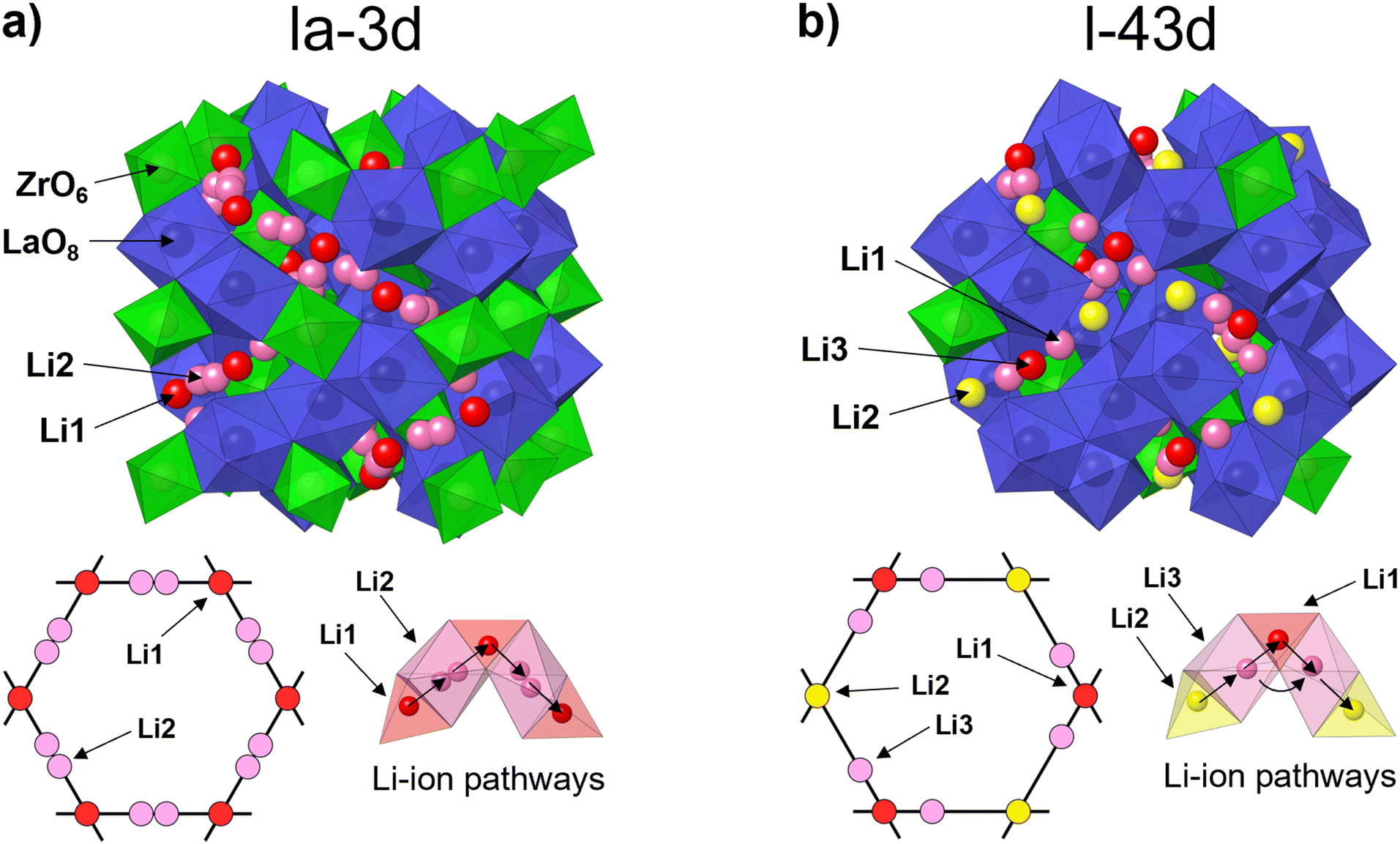 | ||
| Fig. 4 Crystal structure, Li-ion sublattice and Li-ion diffusion pathways of c-LLZO belonging to (a) space group Iad and (b) space group I3d. | ||
Although very few studies have been conducted as to why Ga3+ and Fe3+ doping causes c-LLZO to crystalize with an acentric I3d space group, there are some prevailing theories that suggest that the shift in crystal symmetry is caused by the strong preference of Ga3+ and Fe3+ ions for doping on the Li1 sites, compared to that of Al3+ ions31,33 which is indeed what was discovered also in the present work especially for Fe–LLZO (Table 4). Another possible explanation is that the shift in crystal symmetry is caused by the doping of Ga3+ and Fe3+ ions on La3+ sites.31,33,61 Due to the smaller size of these ions, doping on La3+ sites would cause the 96h oxygen positions to split into two 48e positions, thus lowering the symmetry of the crystal structure and distorting the Li-ion sublattice.61 Compared to Li-site dopants, Zr-site dopants crystalize only in a c-LLZO structure belonging to space group Iad. Although these dopants do not actively block Li+ migration pathways, they can still affect the bulk ionic conductivities of LLZO by influencing the size of the Li-ion pathways as well as the distance between Li1 and Li2 sites.29,40,62
| Dopant | Al | Ga | Fe | Ta | Nb | Sb | W | Mo |
|---|---|---|---|---|---|---|---|---|
| Space group |
Iad |
I3d |
I3d |
Iad |
Iad |
Iad |
Iad |
Iad |
| Lattice parameter [Å] | 12.9711 (1) | 12.9775 (1) | 12.9814 (1) | 12.9426 (1) | 12.9375 (1) | 12.9484 (1) | 12.9503 (1) | 12.9511 (1) |
| Lattice volume [Å3] | 2182.38 (1) | 2185.61 (1) | 2187.56 (1) | 2168.02 (2) | 2165.46 (1) | 2171.02 (1) | 2171.90 (1) | 2172.30 (2) |
| Li1–Li2 [Å] | 1.54 (0) | — | — | 1.50 (4) | 1.55 (0) | 1.55 (4) | 1.54 (4) | 1.58 (6) |
| Li2–Li2 [Å] | 1.02 (0) | — | — | 1.00 (8) | 0.89 (0) | 0.90 (8) | 0.94 (6) | 0.88 (11) |
| Li1–Li3 [Å] | — | 1.82 (8) | 1.74 (8) | — | — | — | — | — |
| Li2–L3 [Å] | — | 2.19 (8) | 2.28 (8) | — | — | — | — | — |
| g (Li1) | 0.50 | 0.90 | 0.82 | 0.43 | 0.40 | 0.31 | 0.37 | 0.45 |
| g (Li2) | 0.40 | 0.93 | 0.99 | 0.44 | 0.44 | 0.47 | 0.45 | 0.43 |
| g (Li3) | — | 0.58 | 0.58 | — | — | — | — | — |
| R wp | 13.8 | 13.2 | 12.6 | 9.78 | 10.1 | 9.65 | 9.09 | 12.4 |
| R Bragg | 4.51 | 4.53 | 5.73 | 2.66 | 3.17 | 4.09 | 3.11 | 8.06 |
| χ 2 | 29.1 | 27.7 | 24.6 | 15.3 | 13.7 | 19.6 | 11.3 | 21.24 |
Basic crystal structure parameters of the LLZO samples, obtained from the refined XRD data, are compiled in Table 4. The occupancy refinements were conducted using two to four linear restraints to improve the reliability of these complex refinements. The fractional crystallographic occupancies and chemical compositions of the refined sites were constrained to precisely match the stoichiometry. The released atoms were allowed to move under these limitations. The Zr site composition was fixed for the heavier dopants once it was found they cannot be found at the Li sites. Under these conditions, the refinements were stable. Yet, receiving meaningful data for the occupancies of Li+ from laboratory XRD data is challenging. Unfortunately, the conventional goodness-of-fit factors are somewhat higher than aimed at due to observed right-side anisotropic broadening of certain reflections (mostly when k = odd in hkl notation), as observed also in our previous work on LLZO materials.27 However, the RBragg, an indicator of the goodness of the used structural model, seems to produce very nice results. It was also found the RBragg decreases along the occupancy refinements. Representative Rietveld refinement fits (Al, Ga and Ta doped LLZOs) with example of peak profile issue are shown in Fig. S1–S3 (ESI†).
Further investigation of the crystal structure was undertaken with Raman spectroscopy. The Raman spectra of the supervalent doped LLZO pellets are presented in Fig. 5, where several bands can be observed between 100 and 900 cm−1. According to Tietz et al.63 and Larraz et al.64 the Raman spectrum of c-LLZO can be divided into three regions. Bands located at the lower end of the Raman spectrum (<150 cm−1) are related to the internal modes of LaO8 dodecahedrons. Bands associated with vibrational modes of Li–O bonds are confined to the intermediary region (200–550 cm−1) and are particularly sensitive to the distribution of Li-ions. Peaks related to the vibrational modes of LiO6 octahedra are in the 200–300 cm−1 range, while vibrational modes of LiO4 tetrahedra are found between 350–550 cm−1. The Raman band at the high end of the spectrum (∼650 cm−1) is assigned to the stretching of ZrO6 octahedra. The specific vibrational modes of individual Raman bands are presented in Fig. S2 (ESI†).
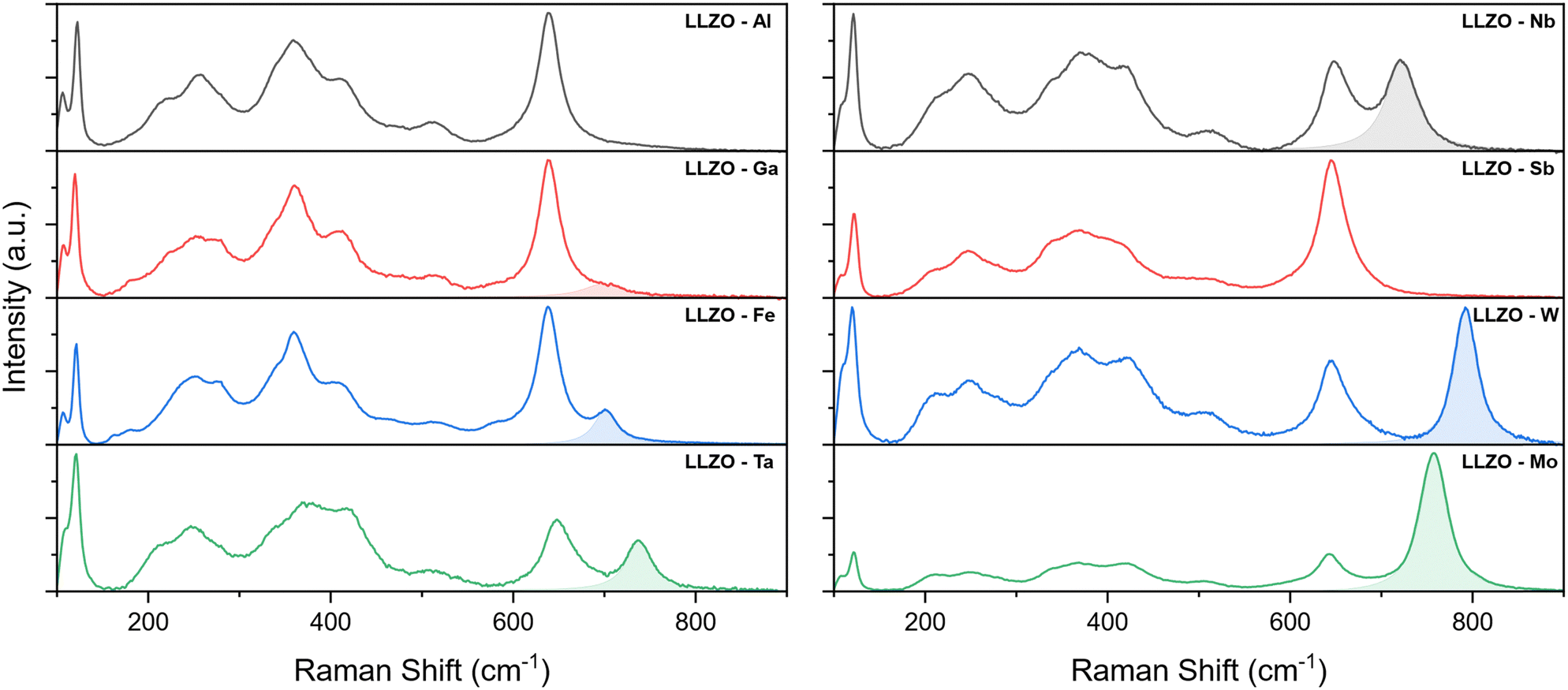 | ||
| Fig. 5 Raman spectra of the supervalent doped LLZO pellets between 100–900 cm−1. The highlighted peaks at the high end of the spectrum are ascribed to the vibrational modes of the dopant–oxygen (M–O) bonds. | ||
For the majority of LLZO samples, additional bands can be observed at wavenumbers above the Zr peak, with varying positions and relative intensities. These bands are highlighted in the Raman spectra presented in Fig. 5. As these peaks maintain the same position and relative intensity between different samples containing the same doping element, we can ascribe them to the vibrational modes of dopant–oxygen (M–O) bonds (the dopant metal here is noted as M). The positions and relative intensities of these dopant-related peaks are given in Table 5.
| Sample | Doping concentration [pfu] | Zr–O peak position [cm−1] | M–O peak position [cm−1] | M/Zr intensity ratio |
|---|---|---|---|---|
| LLZO–Al | 0.17 | 639 | — | — |
| LLZO–Ga | 0.21 | 639 | 698 | 0.08 |
| LLZO–Fe | 0.27 | 639 | 701 | 0.21 |
| LLZO–Ta | 0.50 | 648 | 738 | 0.54 |
| LLZO–Nb | 0.50 | 650 | 721 | 1.12 |
| LLZO–Sb | 048 | 646 | — | — |
| LLZO–W | 0.26 | 643 | 792 | 1.34 |
| LLZO–Mo | 0.25 | 645 | 757 | 4.05 |
For Al and Sb doped LLZO, no dopant related peak could be observed in the Raman spectra. One possible explanation for the absence of the M–O peak is that these peaks overlap with the Zr band. A good indicator of this is that the Zr peak of Sb–LLZO has a much greater intensity, compared to the La and Li peaks, than observed with other dopants. Another possible explanation is that the specific M–O bonds are not Raman active. While no specific dopant peak can be observed for these two dopants, the Raman spectra can still be distinguished from one another by the presence of a more pronounced LiO4 peak at ∼360 cm−1 for Al–LLZO. For Ga–LLZO, the dopant peak is very subtle and manifests itself almost as a shoulder of the Zr peak, which is why it is often overlooked.
Raman spectra of Ga and Fe doped LLZO also show two additional bands at ∼ 180 and 280 cm−1. These bands could potentially be related to changes in the Li-ion sublattice and a reduction in the crystal structure symmetry of the I3d space group, especially as these bands occur in the region associated with vibrational modes of Li–O bonds in LiO6 octahedra.
Further analysis of the Zr peak positions also shows the peaks shift towards higher wavenumbers with Zr-site dopants when compared to Li-site dopants. This would indicate that the Zr–O bonds become shorter with Zr-site doping.65,66 The shift of the peak position is also proportional to changes in the lattice parameter between the different dopants, as presented in Fig. S3 (ESI†), where a smaller lattice size shows a greater shift of the peak towards higher wavenumbers. This means that the Zr–O bonds become shorter with a smaller volume of the crystal cell. The Zr peak positions are listed in Table 5.
Another aspect of the Raman spectrum to consider is the integrated intensity of the dopant peaks. Since the relative intensity of the M–O peak can be correlated to the doping concentration, the intensity ratio of the M/Zr bands can potentially provide a good estimate on the concentration of the doping element in LLZO, where a higher intensity ratio indicates a higher doping concentration.23,67,68 The M/Zr peak intensity ratios obtained in this study are provided in Table 5 along with the related dopant concentrations. These values can be used as a good starting point to determine the concentrations of specific doping elements in future studies. It should however be noted that these intensity ratios cannot be used to accurately determine the doping concentrations, but rather can be used as a guideline to estimate the doping concentrations quickly and easily. It should also be noted that because the relative intensity of the M peak depends on how Raman-active the specific M–O bonds are, the M/Zr intensity ratios cannot be directly compared between different doping elements, but rather can only be used between samples containing different concentrations of the same dopant.
Since almost all doping elements used in this investigation have a rather distinguishable position and intensity ratio of the M–O band, we have shown how Raman spectroscopy can be used as a fast and inexpensive alternative to not only determine the specific doping element used in LLZO, but also approximate their concentrations based on the values provided in Table 5. Due to a considerable overlapping of the LiO4 and LiO6 Raman modes, a full deconvolution of the Li-related bands was not possible.
3.2 Morphology of sintered LLZO pellets
SEM images of the pellet cross-sections are presented in Fig. 6. The images show how the LLZO grains vary in shape, size, and density depending on the doping element used. For example, Fe–LLZO exhibits tightly packed polyhedral grains with an average size between 5–25 μm and a 93% relative density. On the other hand, Nb–LLZO contains loosely packed spherical grains with an average size ranging from 3–15 μm and an 85% relative density, which would indicate a poor sintering performance of the sample. For Al–LLZO and Mo–LLZO, abnormally large grains, with sizes up to 50 μm, can also be observed. In general, larger and more tightly packed grains are desirable as they reduce the grain boundary area, that can act as a bottleneck for Li-ion mobility and a site for Li-dendrite propagation, whilst simultaneously increasing the air stability of LLZO by reducing the surface area exposed to H2O and CO2.69–72 | ||
| Fig. 6 Cross-section SEM images of supervalent doped LLZO pellets. | ||
Grain connectivity can also be determined based on the fracture type they exhibit. For example, Sb–LLZO experiences a very high degree of transgranular fracture implying a very strong connectivity between the grains. In contrast, Nb–LLZO shows a high level of intergranular fracture that is indicative of a poor connection between the grains. Of particular interest is Ga–LLZO, which exhibits a pure transgranular fracture with a 94% relative density and a morphology that resembles c-LLZO obtained through sol–gel methods.27 In this case, the grains are so tightly interconnected and densified that they cannot be readily distinguished from each other. This would infer that the effects of the grain boundary are minimized which gives Ga–LLZO the best morphology among the used dopants for practical applications. The relative densities of the LLZO samples are listed in Table 6 while the grain shape, size and fracture type are listed in Table S2 (ESI†).
| Sample | σ total (S cm−1) | E a (eV) | σ elec (S cm−1) | Relative density (%) |
|---|---|---|---|---|
| LLZO–Al | 3.72 × 10−4 (± 0.44 × 10−4) | 0.31 | 4.78 × 10−9 (± 0.14 × 10−9) | 89 |
| LLZO–Ga | 13.0 × 10−4 (± 0.38 × 10−4) | 0.26 | 1.09 × 10−9 (± 0.04 × 10−9) | 94 |
| LLZO–Fe | 11.2 × 10−4 (± 0.13 × 10−4) | 0.22 | 3.90 × 10−9 (± 0.07 × 10−9) | 93 |
| LLZO–Ta | 2.38 × 10−4 (2) (± 0.12 × 10−4) | 0.37 | 3.32 × 10−9 (± 0.27 × 10−9) | 88 |
| LLZO–Nb | 1.91 × 10−4 (± 0.17 × 10−4) | 0.44 | 2.80 × 10−9 (± 015 × 10−9) | 85 |
| LLZO–Sb | 3.41 × 10−4 (3) (± 0.14 × 10−4) | 0.41 | 4.13 × 10−9 (± 0.24 × 10−9) | 91 |
| LLZO–W | 5.43 × 10−4 (± 0.02 × 10−4) | 0.37 | 5.04 × 10−9 (± 0.19 × 10−9) | 90 |
| LLZO–Mo | 3.03 × 10−4 (± 0.06 × 10−4) | 0.36 | 4.09 × 10−9 (± 0.18 × 10−9) | 90 |
One of the main driving forces for the differences in the grains shape, size and relative density, between the different LLZO dopants, is the presence of secondary phases during sintering.35,44,46 Evidence of this can be seen from the EDS maps presented in Fig. 7, where dopant-rich secondary phases can be observed on the grain boundaries of Al, Mo and Fe doped LLZO. For Al–LLZO, this phase can be identified as a dark layer surrounding the LLZO grains. As these layers contain a substantial amount of Al and O while lacking La and Zr, it can be assumed that the structure is that of LiAlO2 and Li5AlO4 which melt at temperatures over 1050 °C and flow between the grains. This melt acts as a liquid sintering aid that promotes abnormal grain growth, increases the relative density and enhances Li-ion diffusion across the grain boundary.44,73 Since the phase does not appear in the XRD patterns of Al–LLZO it is most likely present in an amorphous-like glassy state.
 | ||
| Fig. 7 Cross-section EDS elemental maps of Al, Mo, Fe and Ga doped LLZO, showcasing the various distribution of doping elements. | ||
A similar observation is also made for Mo–LLZO where a Mo-rich phase is found segregated on the LLZO grain boundaries, albeit to a lesser extent than with Al doping. No previous reports have been made on such a phase and it is most likely that this is an intermediary Li–Mo–O phase, such as Li4MoO5 or Li2MoO4, which melt at temperatures above 950 °C.74 Considering that Mo–LLZO also shows abnormal grain growth, it is very likely that this phase also acts as a sintering aid. A dopant rich secondary phase can also be observed on the grain boundaries of Fe–LLZO. Since this phase contains La it is identified as residual LaFeO3 which gives Fe–LLZO its distinct dark brown color. These results further showcase the low affinity of Al, Fe and Mo dopants towards LLZO integration.
Other LLZO samples do not show any segregation of a dopant-rich phase on the grain boundaries, but rather a homogeneous distribution of the dopant across the grains, as is the case with Ga–LLZO presented in Fig. 7. This, however, does not mean that a secondary phase is not present during sintering as a secondary phase can be present during the initial stages of sintering and then later fully integrates itself into the LLZO structure during the final stages of sintering. The presence of a secondary phase, that acts as a sintering aid, could also explain the exceptionally high density and very strong grain connectivity of Ga–LLZO. Given that the eutectic point of the LiGaO2–Li5GaO4 system is at approximately 950 °C, we can speculate that an amorphous Li–Ga–O phase forms during synthesis, acts as a liquid sintering aid during the initial stages of sintering and eventually fully integrates into the LLZO structure.41 Previous studies have shown the presence of a Li–Ga–O liquid phase during sintering of Ga doped LLZO, which greatly increases the relative density and size of the LLZO grains.46 Similarly, the high density of Fe–LLZO grains could also potentially be explained by the presence of a liquid LiFeO2 phase during sintering. Although no such phase was detected with EDS, it is most likely that the phase was fully integrated into the LLZO structure during the final stages of sintering.
3.3 Electrochemical properties of LLZO
Fig. 8(a) shows the Nyquist plots of the supervalent doped LLZO samples at room temperature. The plots show a single semicircle at higher frequencies, followed by a tail at lower frequencies. The intercept of the semicircle with the real axis Z′ at lower frequencies represents the ionic conductivity of LLZO, while the low frequency tail is ascribed to the polarization of the Au/Pd electrodes.75 As only one semicircle for each sample is evident, the semi-circles have been ascribed to the total conductivity of LLZO (grain and grain boundary) and have been fitted with the equivalent circuit shown in Fig. 8(a).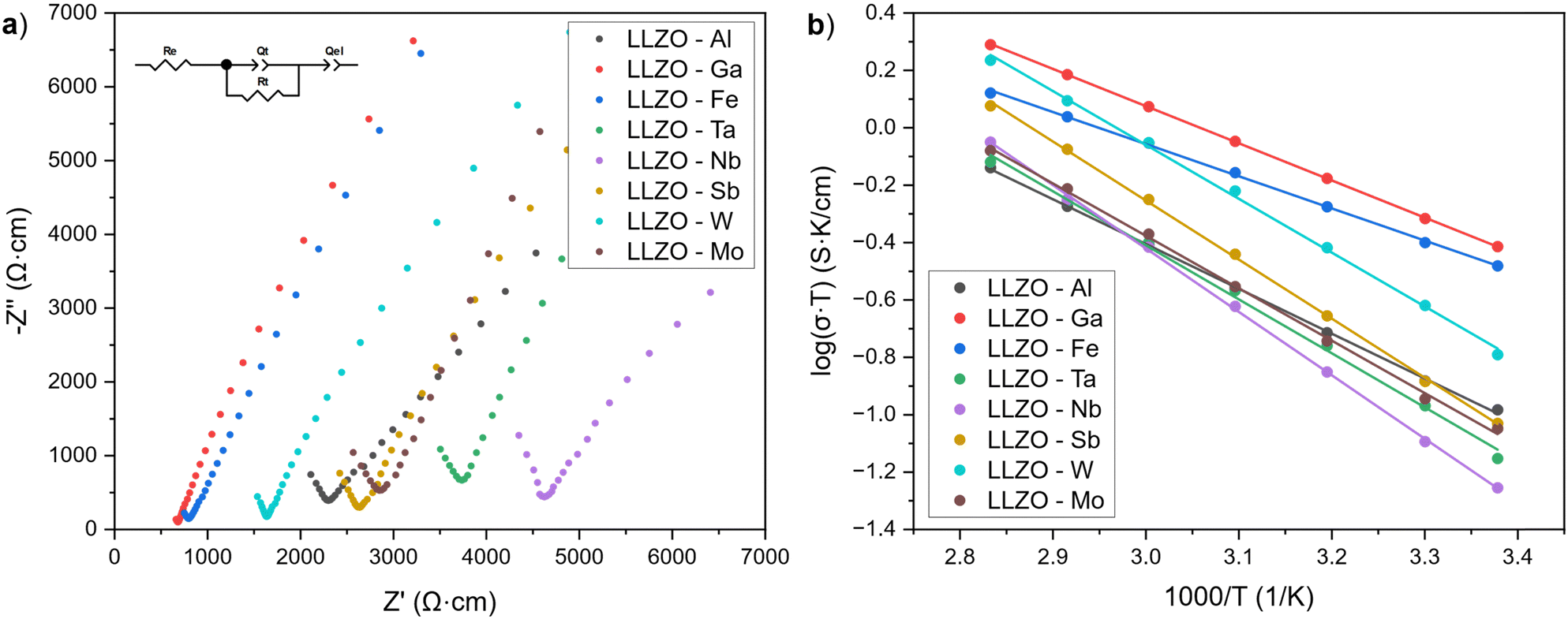 | ||
| Fig. 8 (a) Nyquist plots of the supervalent doped LLZO pellets obtained at room temperature and (b) Arrhenius plots showing the temperature dependence on the total ionic conductivity of the LLZO pellets. | ||
The ionic conductivities of the LLZO samples are presented in Table 6. The listed values represent the average ionic conductivities measured over 8 sample for each dopant and range from 1.91 × 10−4 to 1.30 × 10−3 S cm−1. The highest conductivities were achieved with Ga and Fe doped LLZO at 1.30 × 10−3 S cm−1 and 1.12 × 10−3 S cm−1, respectively. These conductivities are higher than the conductivities achieved with other dopants by a whole order of magnitude. The exceptionally high conductivities can be ascribed to changes in the crystal symmetry of LLZO from a Iad to a I3d space group, which allows for better mobility of Li-ions due to additional Li3–Li3 pathways. Moreover, Ga and Fe doped LLZO samples also have a very high relative density and tightly interconnected grains which would further lower the resistance for Li-ion transport across grain boundaries.
For LLZO samples belonging to space group Iad, the ionic conductivities range between 1.91–5.43 × 10−4. For these samples, it appears that the conductivities are strongly influenced by the relative densities of the LLZO structure and presence of secondary phases. For example, the presence of LiAlO2 on the grain boundaries of Al–LLZO has a double effect on its conductivity as it not only promotes a tighter binding and higher relative density of the LLZO grains, but also promotes a higher ionic conductivity across the grain boundaries.44,76 On the other hand, Nb–LLZO is not known to have any major Nb-rich secondary phases present during sintering, thereby having a lower relative density and higher grain boundary resistance which results in a lower ionic conductivity. Because of this, Al–LLZO has an almost 2-times higher conductivity than Nb–LLZO at 3.72 × 10−4 S cm−1 and 1.91 × 10−4 S cm−1, respectively. Amongst the LLZO samples belonging to space group Iad, W–LLZO shows the highest conductivity (5.43 × 10−4) and relative density (90%). Although no W-rich secondary phases could be observed on the grain boundaries using EDS mapping, previous studies have shown the presence of a W-rich phase during sintering with W doping.35 This could potentially indicate that the W-rich phase densifies the LLZO grains during the initial stages of sintering and then integrates into the LLZO structures the final stages of sintering.
Another explanation for the lower conductivities of Ta and Nb doped LLZO are the smaller cell parameters compared to other Iad dopants which are caused by a stronger Coulombic repulsion between the cations in Ta– and Nb–LLZO. The stronger Coulombic repulsion hinders Li-ion mobility and reduces the ionic conductivity of LLZO.29 This effect can also be seen from the lower ionic conductivity of Nb–LLZO compared to Ta–LLZO, which has a smaller cell parameter and hence the system maintains stronger Coulombic repulsion forces.
Total ionic conductivities of the LLZO pellets at elevated temperatures were used to construct Arrhenius plots shown in Fig. 8(b). The activation energies (Ea) of the LLZO samples were obtained from the slopes of Arrhenius plots according to equation:
| σ = A/Texp(−Ea/kBT) | (1) |
The activation energy indicates the difficulty at which Li-ions migrate within the sample, where a low activation energy indicates more facile Li-ion mobility. The activation energies of LLZO range from 0.22 to 0.44 eV. Here, Ga and Fe again show the lowest activation energies (0.26 and 0.22, respectively), which would indicate that the transport of Li-ions is notably more facile in these samples due to additional migration channels. For the remaining samples, activation energy values lie between 0.31 and 0.44 eV with Nb–LLZO having the lowest Ea due to a low relative density. The ionic conductivities and activation energies of the doped LLZO samples are listed in Table 6.
Another important factor in the development of solid electrolytes is the electronic conductivity (σelec). High electronic conduction have previously been linked with Li dendrite nucleation, decomposition of the material and short circuiting of the system.50,70,77 This is caused by electronic transfer which reduces Li+ ions to metallic Li0 thus causing the formation of Li-dendrites. Unlike conventional dendrite growth, where the Li-dendrites grow from the electrode–electrolyte interface into the bulk of the electrolyte, with electronic dendrite growth the metallic Li deposits itself on the grain boundaries and voids directly within the bulk of the solid electrolyte thus forming Li dendrites independently of the electrode.77 Recent studies have shown that such Li-dendrite formation can occur relatively easily even in high-density structures (relative density >97%), and requires a critical current density of less than 0.9 mA cm−1.69,70 So, in order to prevent the formation of Li-dendrites the electronic conductivity of LLZO would ideally be need to kept at >10−10 S cm−1.70 Therefore, a good solid electrolyte must possess a high ionic conductivity, whilst also maintaining a very low electronic conductivity.
A recent study by Philipp et al.78 also showed that polycrystalline structures experience a higher electronic conductivity then single crystal samples by roughly two orders of magnitude at 10−8 and 10−10 S cm−1, respectively. This would indicate that the majority of electronic charge carriers are located along the grain boundaries which act as a network for electron mobility. Therefore, by reducing the grain boundary area and number of voids one should be able to lower the electronic conductivity.
The DC polarization curves are presented in Fig. 9. The curves show an initial drop of the current, due to the settling of Li-ions, followed by a steady state where the resulting current is only due to electronic conduction. The steady state conductivities for all samples are in the range of 10−9 S cm−1 as listed in Table 6, where the listed values represent the average electronic conductivities measured over 8 sample for each dopant. This means that the electronic conductivities are several orders of magnitude lower than the ionic conductivity of LLZO thus, the transference number of Li-ions (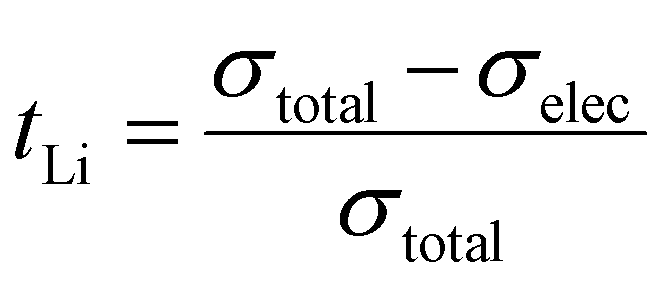 ) is close to unity. This indicates that the influence of the electronic conductivity on the total conductivity of LLZO is practically negligible, making the doped LLZO samples electronic insulators and suitable for practical use as solid electrolytes.
) is close to unity. This indicates that the influence of the electronic conductivity on the total conductivity of LLZO is practically negligible, making the doped LLZO samples electronic insulators and suitable for practical use as solid electrolytes.
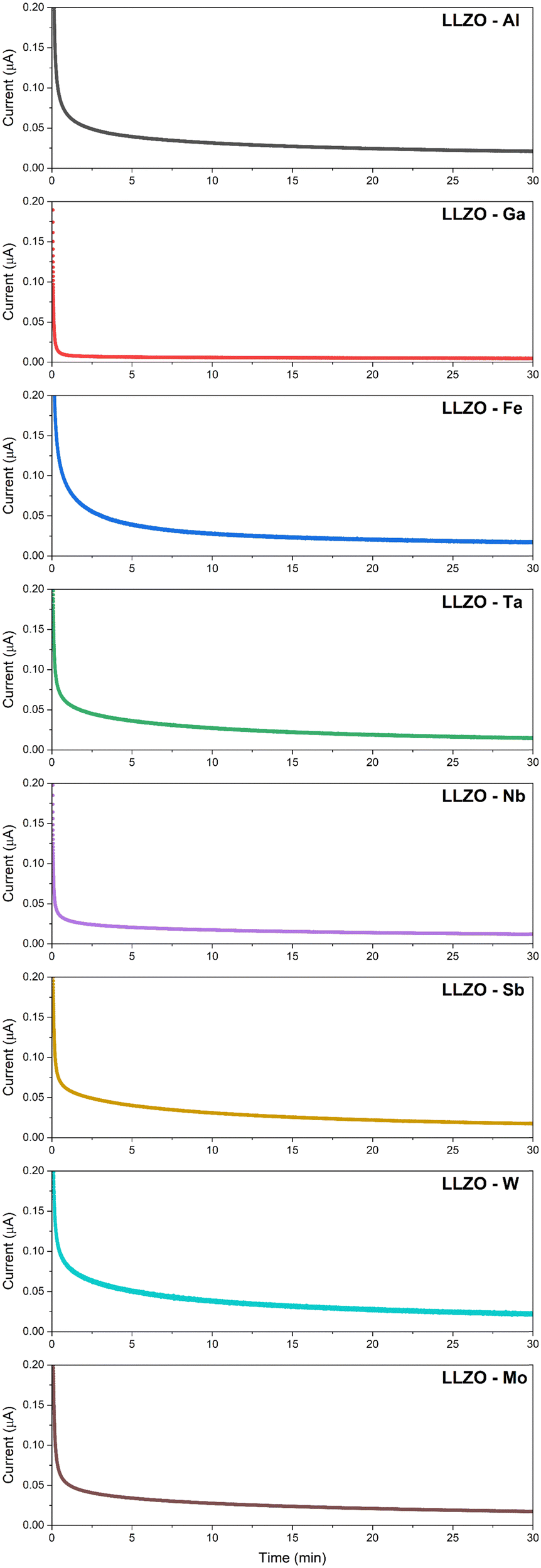 | ||
| Fig. 9 DC polarization curves of doped LLZO samples at room temperature. | ||
Ga–LLZO reaches a steady state especially quickly indicating a very fast settling of Li-ions and retains the lowest electronic conductivity at 1.09 × 10−9. The exceptionally low electronic conductivity is most likely caused due to a significantly reduced grain boundary area and exceptionally large grain size compared to other samples. This would significantly lower the number of electronic charge carriers within the LLZO structure, showcasing how doping can be used to engineer a specific grain structure which would be more favorable to achieve a low electronic conductivity.
It should also be noted that while maintaining a low electronic conductivity is a very important aspect in the development of solid electrolytes, these parameters are rarely reported or discussed. It is therefore highly beneficial for the research community that such values would be reported more commonly in the future.
4 Conclusions
In this study we present the effects of supervalent doping on the properties of LLZO solid electrolytes. Cubic LLZO samples were prepared through a conventional solid state synthesis method using different supervalent dopants (Al3+, Ga3+, Fe3+, Ta5+, Nb5+, Sb5+, W6+ and Mo6+). The effect of the doping element on the synthesis, crystal structure, morphology, and electrochemical performance of LLZO are compared.Ga3+ showed to be the most promising dopant as it produced samples with exceptionally high conductivities (1.30 × 10−3 S cm−1) and low activation energies (0.26 eV) due to a shift in the crystal symmetry from a Iad to a I3d space group. Ga also showed a very low electronic conductivity which is an important factor for effectively reducing Li-dendrite growth. On top of that, Ga doping also showed a very high affinity towards LLZO integration during synthesis, requiring a much lower synthesis temperature, and demonstrated very good sinterability, which resulted in pellets with a very dense morphology. Fe3+ doping also produced samples with very high conductivities (1.12 × 10−3 S cm−1) and low activation energies (0.22 eV) due to a shift in the crystal symmetry. Nonetheless, Fe proved to be less suitable as a dopant due to a very low affinity of Fe towards LLZO, which requires higher synthesis temperatures and prolonged synthesis times that can cause additional problems issues during synthesis, such as Li-loss and decomposition of LLZO. The LLZO samples with a Iad symmetry have conductivities in the range of 1–5 × 10−4 S cm−1 and activation energies between 0.31–0.44 eV, from which it appears that the electrochemical performance is mostly related to the relative density of the LLZO grains and the size of the unit cell. This would indicate that for these samples, the electrochemical performance could be improved by densifying the LLZO structure.
Although we have demonstrated in our study the superiority of Ga doping in virtually every aspect, further studies need to be conducted in order to fully understand the performance of these dopants in practical applications. Recent studies have shown that the electrochemical stability and stability against Li–metal of LLZO is lower than what was previously thought of.12 This means that a more thorough evaluation of these dopants must be conducted in future studies so that we may better understand their behavior in practical application.
Author contributions
Janez Košir: conceptualization, investigation, formal analysis, visualization, writing – original draft; Seyedabolfazl Mousavihashemi: investigation, formal analysis; Milla Suominen: investigation, formal analysis; Anna Kobets: investigation; Benjamin P. Wilson: investigation, formal analysis, writing – review & editing; Eeva-Leena Rautama: formal analysis, writing – review & editing; Tanja Kallio: writing – review & editing, funding acquisition, supervision.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank financial support from Business Finland (the NextGenBat project, grant no. 44811/31/2020). We greatfully acknowledge M.Sc. Ilkka Välimaa for performing the ICP-OES measurements. This work made use of Aalto University RawMatTERS facilities, supported by the Academy of Finland. We acknowledge the provision of facilities by OtaNano – Nanomicroscopy Center (NMC) at Aalto University.References
- J. B. Goodenough and K. S. Park, J. Am. Chem. Soc., 2013, 135, 1167–1176 CrossRef CAS PubMed.
- S. Chen, K. Wen, J. Fan, Y. Bando and D. Golberg, J. Mater. Chem. A, 2018, 6, 11631–11663 RSC.
- J. B. Goodenough, J. Solid State Electrochem., 2012, 16, 2019–2029 CrossRef CAS.
- J. Janek and W. G. Zeier, Nat. Energy, 2016, 1, 1–4 Search PubMed.
- K. Takada, J. Power Sources, 2018, 394, 74–85 CrossRef CAS.
- Y. Shen, Y. Zhang, S. Han, J. Wang, Z. Peng and L. Chen, Joule, 2018, 2, 1674–1689 CrossRef CAS.
- Q. Liu, Z. Geng, C. Han, Y. Fu, S. Li, Y. Bing He, F. Kang and B. Li, J. Power Sources, 2018, 389, 120–134 CrossRef CAS.
- N. Zhao, W. Khokhar, Z. Bi, C. Shi, X. Guo, L. Z. Fan and C. W. Nan, Joule, 2019, 3, 1190–1199 CrossRef CAS.
- J. C. Bachman, S. Muy, A. Grimaud, H. H. Chang, N. Pour, S. F. Lux, O. Paschos, F. Maglia, S. Lupart, P. Lamp, L. Giordano and Y. Shao-Horn, Chem. Rev., 2016, 116, 140–162 CrossRef CAS PubMed.
- R. Murugan, V. Thangadurai and W. Weppner, Angew. Chem., Int. Ed., 2007, 46, 7778–7781 CrossRef CAS PubMed.
- Y. Zhu, X. He and Y. Mo, ACS Appl. Mater. Interfaces, 2015, 7, 23685–23693 CrossRef CAS PubMed.
- Y. Benabed, A. Vanacker, G. Foran, S. Rousselot, G. Hautier and M. Dollé, Mater. Today Energy, 2023, 35, 101320 CrossRef CAS.
- J. Awaka, N. Kijima, H. Hayakawa and J. Akimoto, J. Solid State Chem., 2009, 182, 2046–2052 CrossRef CAS.
- J. Awaka, A. Takashima, K. Kataoka, N. Kijima, Y. Idemoto and J. Akimoto, Chem. Lett., 2011, 40, 60–62 CrossRef CAS.
- M. Matsui, K. Takahashi, K. Sakamoto, A. Hirano, Y. Takeda, O. Yamamoto and N. Imanishi, Dalton Trans., 2014, 43, 1019–1024 RSC.
- Y. Matsuda, K. Sakamoto, M. Matsui, O. Yamamoto, Y. Takeda and N. Imanishi, Solid State Ionics, 2015, 277, 23–29 CrossRef CAS.
- T. Yang, Z. D. Gordon, Y. Li and C. K. Chan, J. Phys. Chem. C, 2015, 119, 14947–14953 CrossRef CAS.
- C. A. Geiger, E. Alekseev, B. Lazic, M. Fisch, T. Armbruster, R. Langner, M. Fechtelkord, N. Kim, T. Pettke and W. Weppner, Inorg. Chem., 2011, 50, 1089–1097 CrossRef CAS PubMed.
- N. Bernstein, M. D. Johannes and K. Hoang, Phys. Rev. Lett., 2012, 109, 1–5 CrossRef PubMed.
- M. Klenk and W. Lai, Phys. Chem. Chem. Phys., 2015, 17, 8758–8768 RSC.
- F. Chen, J. Li, Z. Huang, Y. Yang, Q. Shen and L. Zhang, J. Phys. Chem. C, 2018, 122, 1963–1972 CrossRef CAS.
- E. Rangasamy, J. Wolfenstine and J. Sakamoto, Solid State Ionics, 2012, 206, 28–32 CrossRef CAS.
- T. Thompson, J. Wolfenstine, J. L. Allen, M. Johannes, A. Huq, I. N. David and J. Sakamoto, J. Mater. Chem. A, 2014, 2, 13431–13436 RSC.
- X. Liu, Y. Li, T. Yang, Z. Cao, W. He, Y. Gao, J. Liu, G. Li and Z. Li, J. Am. Ceram. Soc., 2017, 100, 1527–1533 CrossRef CAS.
- J. F. Wu, E. Y. Chen, Y. Yu, L. Liu, Y. Wu, W. K. Pang, V. K. Peterson and X. Guo, ACS Appl. Mater. Interfaces, 2017, 9, 1542–1552 CrossRef CAS PubMed.
- D. Rettenwander, A. Welzl, L. Cheng, J. Fleig, M. Musso, E. Suard, M. M. Doeff, G. J. Redhammer and G. Amthauer, Inorg. Chem., 2015, 54, 10440–10449 CrossRef CAS PubMed.
- J. Košir, S. Mousavihashemi, B. P. Wilson, E. L. Rautama and T. Kallio, Solid State Ionics, 2022, 380, 1–10 CrossRef.
- R. Inada, K. Kusakabe, T. Tanaka, S. Kudo and Y. Sakurai, Solid State Ionics, 2014, 262, 568–572 CrossRef CAS.
- X. Xiang, F. Chen, W. Yang, J. Yang, X. Ma, D. Chen, K. Su, Q. Shen and L. Zhang, J. Am. Ceram. Soc., 2020, 103, 2483–2490 CrossRef CAS.
- K. Ishiguro, Y. Nakata, M. Matsui, I. Uechi, Y. Takeda, O. Yamamoto and N. Imanishi, J. Electrochem. Soc., 2013, 160, A1690–A1693 CrossRef CAS.
- R. Wagner, G. J. Redhammer, D. Rettenwander, A. Senyshyn, W. Schmidt, M. Wilkening and G. Amthauer, Chem. Mater., 2016, 28, 1861–1871 CrossRef CAS PubMed.
- D. Rettenwander, G. Redhammer, F. Preishuber-Pflügl, L. Cheng, L. Miara, R. Wagner, A. Welzl, E. Suard, M. M. Doeff, M. Wilkening, J. Fleig and G. Amthauer, Chem. Mater., 2016, 28, 2384–2392 CrossRef CAS PubMed.
- R. Wagner, G. J. Redhammer, D. Rettenwander, G. Tippelt, A. Welzl, S. Taibl, J. Fleig, A. Franz, W. Lottermoser and G. Amthauer, Chem. Mater., 2016, 28, 5943–5951 CrossRef CAS PubMed.
- A. Paulus, S. Kammler, S. Heuer, M. C. Paulus, P. Jakes, J. Granwehr and R.-A. Eichel, J. Electrochem. Soc., 2019, 166, A5403–A5409 CrossRef CAS.
- Y. Li, Z. Wang, Y. Cao, F. Du, C. Chen, Z. Cui and X. Guo, Electrochim. Acta, 2015, 180, 37–42 CrossRef CAS.
- I. McClelland, H. El-Shinawi, S. G. Booth, A. Regoutz, J. Clough, S. Altus, E. J. Cussen, P. J. Baker and S. A. Cussen, Chem. Mater., 2022, 34, 5054–5064 CrossRef CAS.
- S. Ramakumar, L. Satyanarayana, S. V. Manorama and R. Murugan, Phys. Chem. Chem. Phys., 2013, 15, 11327–11338 RSC.
- C. Deviannapoorani, L. Dhivya, S. Ramakumar and R. Murugan, J. Power Sources, 2013, 240, 18–25 CrossRef CAS.
- D. O. Shin, K. Oh, K. M. Kim, K. Y. Park, B. Lee, Y. G. Lee and K. Kang, Sci. Rep., 2015, 5, 1–9 Search PubMed.
- F. Chen, L. Xu, J. Li, Y. Yang and Q. Shen, Ionics, 2020, 26, 3193–3198 CrossRef CAS.
- Y. Matsuda, A. Sakaida, K. Sugimoto, D. Mori, Y. Takeda, O. Yamamoto and N. Imanishi, Solid State Ionics, 2017, 311, 69–74 CrossRef CAS.
- L. Zhuang, X. Huang, Y. Lu, J. Tang, Y. Zhou, X. Ao, Y. Yang and B. Tian, Ceram. Int., 2021, 47, 22768–22775 CrossRef CAS.
- H. El Shinawi and J. Janek, J. Power Sources, 2013, 225, 13–19 CrossRef CAS.
- K. Zhang, T. Xu, H. Zhao, S. Zhang, Z. Zhang, Y. Zhang, Z. Du and Z. Li, Int. J. Energy Res., 2020, 44, 9177–9184 CrossRef CAS.
- R. H. Brugge, F. M. Pesci, A. Cavallaro, C. Sole, M. A. Isaacs, G. Kerherve, R. S. Weatherup and A. Aguadero, J. Mater. Chem. A, 2020, 8, 14265–14276 RSC.
- W. Jeong, S. S. Park, J. Yun, H. R. Shin, J. Moon and J. W. Lee, Energy Storage Mater., 2023, 54, 543–552 CrossRef.
- Y. Zhu, J. G. Connell, S. Tepavcevic, P. Zapol, R. Garcia-Mendez, N. J. Taylor, J. Sakamoto, B. J. Ingram, L. A. Curtiss, J. W. Freeland, D. D. Fong and N. M. Markovic, Adv. Energy Mater., 2019, 9, 1–11 Search PubMed.
- D. Rettenwander, R. Wagner, A. Reyer, M. Bonta, L. Cheng, M. M. Doeff, A. Limbeck, M. Wilkening and G. Amthauer, J. Phys. Chem. C, 2018, 122, 3780–3785 CrossRef CAS PubMed.
- C. Tsai, N. T. Thuy Tran, R. Schierholz, Z. Liu, A. Windmüller, C. A. Lin, Q. Xu, X. Lu, S. Yu, H. Tempel, H. Kungl, S. K. Lin and R. A. Eichel, J. Mater. Chem. A, 2022, 10998–11009 RSC.
- F. Han, Y. Zhu, X. He, Y. Mo and C. Wang, Adv. Energy Mater., 2016, 6, 1–9 Search PubMed.
- S. Yu, R. D. Schmidt, R. Garcia-Mendez, E. Herbert, N. J. Dudney, J. B. Wolfenstine, J. Sakamoto and D. J. Siegel, Chem. Mater., 2016, 28, 197–206 CrossRef CAS.
- F. M. Pesci, R. H. Brugge, A. K. O. Hekselman, A. Cavallaro, R. J. Chater and A. Aguadero, J. Mater. Chem. A, 2018, 6, 19817–19827 RSC.
- S. Kim, J. S. Kim, L. Miara, Y. Wang, S. K. Jung, S. Y. Park, Z. Song, H. Kim, M. Badding, J. M. Chang, V. Roev, G. Yoon, R. Kim, J. H. Kim, K. Yoon, D. Im and K. Kang, Nat. Commun., 2022, 13, 1883 CrossRef CAS PubMed.
- X. Xiang, Y. Liu, F. Chen, W. Yang, J. Yang, X. Ma, D. Chen, K. Su, Q. Shen and L. Zhang, J. Eur. Ceram. Soc., 2020, 40, 3065–3071 CrossRef CAS.
- R. P. Rao, W. Gu, N. Sharma, V. K. Peterson, M. Avdeev and S. Adams, Chem. Mater., 2015, 27, 2903–2910 CrossRef CAS.
- Y. Chen, E. Rangasamy, C. R. Dela Cruz, C. Liang and K. An, J. Mater. Chem. A, 2015, 3, 22868–22876 RSC.
- J. M. Weller, J. A. Whetten and C. K. Chan, ACS Appl. Energy Mater., 2018, 1, 552–560 CrossRef CAS.
- S. Afyon, F. Krumeich and J. L. M. Rupp, J. Mater. Chem. A, 2015, 3, 18636–18648 RSC.
- G. Liu, T. Li, Y. Xing and W. Pan, IOP Conf. Ser.: Mater. Sci. Eng., 2019, 678, 012150 CAS.
- R. D. Shannon, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1976, 32, 751–767 CrossRef.
- L. Robben, E. Merzlyakova, P. Heitjans and T. M. Gesing, Acta Crystallogr., Sect. E: Crystallogr. Commun., 2016, 72, 287–289 CrossRef CAS PubMed.
- Y. Zhang, F. Chen, J. Li, L. Zhang, J. Gu, D. Zhang, K. Saito, Q. Guo, P. Luo and S. Dong, Electrochim. Acta, 2018, 261, 137–142 CrossRef CAS.
- F. Tietz, T. Wegener, M. T. Gerhards, M. Giarola and G. Mariotto, Solid State Ionics, 2013, 230, 77–82 CrossRef CAS.
- G. Larraz, A. Orera and M. L. Sanjuán, J. Mater. Chem. A, 2013, 1, 11419–11428 RSC.
- D. J. Kim, J. W. Jang and H. L. Lee, J. Am. Ceram. Soc., 1997, 80, 1453–1461 CrossRef CAS.
- X. Zhou, L. Huang, O. Elkedim, Y. Xie, Y. Luo, Q. Chen, Y. Zhang and Y. Chen, J. Alloys Compd., 2022, 891, 161906 CrossRef CAS.
- S. Mukhopadhyay, T. Thompson, J. Sakamoto, A. Huq, J. Wolfenstine, J. L. Allen, N. Bernstein, D. A. Stewart and M. D. Johannes, Chem. Mater., 2015, 27, 3658–3665 CrossRef CAS.
- S. Saran and Y. Ramazan Eker, Curr. Appl. Phys., 2022, 41, 1–6 CrossRef.
- E. J. Cheng, A. Sharafi and J. Sakamoto, Electrochim. Acta, 2017, 223, 85–91 CrossRef CAS.
- F. Han, A. S. Westover, J. Yue, X. Fan, F. Wang, M. Chi, D. N. Leonard, N. J. Dudney, H. Wang and C. Wang, Nat. Energy, 2019, 4, 187–196 CrossRef CAS.
- C. Zhu, T. Fuchs, S. A. L. Weber, F. H. Richter, G. Glasser, F. Weber, H. J. Butt, J. Janek and R. Berger, Nat. Commun., 2023, 14, 1–14 Search PubMed.
- S. Yu and D. J. Siegel, Chem. Mater., 2017, 29, 9639–9647 CrossRef CAS.
- J. H. Ahn, S. Y. Park, J. M. Lee, Y. Park and J. H. Lee, J. Power Sources, 2014, 254, 287–292 CrossRef CAS.
- M. Moser, D. Klimm, S. Ganschow, A. Kwasniewski and K. Jacobs, Cryst. Res. Technol., 2008, 43, 350–354 CrossRef CAS.
- N. C. Rosero-Navarro, T. Yamashita, A. Miura, M. Higuchi and K. Tadanaga, J. Am. Ceram. Soc., 2017, 100, 276–285 CrossRef CAS.
- Y. Li, J. T. Han, C. A. Wang, H. Xie and J. B. Goodenough, J. Mater. Chem., 2012, 22, 15357–15361 RSC.
- F. Aguesse, W. Manalastas, L. Buannic, J. M. L. Del Amo, G. Singh, A. Llordés and J. Kilner, ACS Appl. Mater. Interfaces, 2017, 9, 3808–3816 CrossRef CAS PubMed.
- M. Philipp, B. Gadermaier, P. Posch, I. Hanzu, S. Ganschow, M. Meven, D. Rettenwander, G. J. Redhammer and H. M. R. Wilkening, Adv. Mater. Interfaces, 2020, 7(16), 2000450 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ma00119b |
| ‡ Current affiliation: VTT Technical Research Centre of Finland, 02150 Espoo, Finland. |
| This journal is © The Royal Society of Chemistry 2024 |