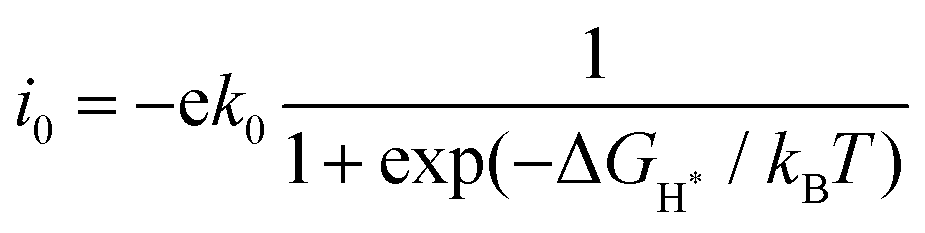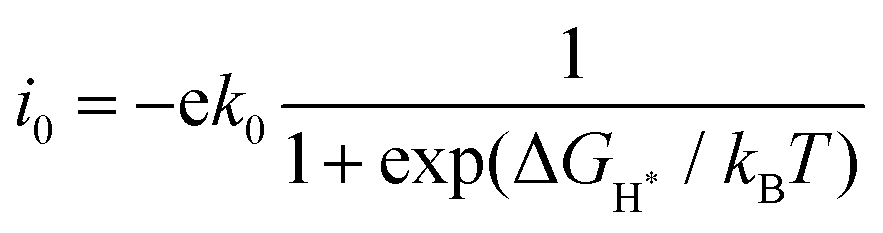DOI:
10.1039/D4MA00161C
(Paper)
Mater. Adv., 2024,
5, 6586-6595
Single-atom transition metals doping two-dimensional BXN materials (X = 2, 3, 5) with promising electrocatalytic activity for efficient hydrogen production in the entire pH range†
Received
17th February 2024
, Accepted 8th July 2024
First published on 24th July 2024
Abstract
Two-dimensional (2D) boron nitrides (BN) have been eagerly and widely used and have tremendous potential in several advanced fields, such as energy harvesting and storage. Due to their significant electronic properties, they are also commonly used as substrates for single-atom catalysts (SACs). Therefore, with the aid of a computer, we designed SACs by doping isolated single atoms of 3d, 4d, and 5d transition metals (TM) on 2D BXN materials (X = 2, 3, 5). In addition, pH regulation is considered to improve the electrocatalytic hydrogen evolution reaction (HER) activity, and the materials’ effectiveness was investigated theoretically based on density functional theory (DFT) calculations. Our results indicate that the low-cost TM SACs can effectively enhance the HER catalytic performance over a wide range of pH. Among all the SACs studied, Ti-, V-, Y- and Zr@B5N show excellent catalytic activity at pH = 0, with the Gibbs free energy change (ΔGH*) of hydrogen adsorption of −0.027, −0.094, 0.073 and −0.040 eV, respectively. We find that Sc- and Y-embedded B2N, Co-, Fe-, and Mo-embedded B3N and Co-, Cr-, V-, Ti-, Y-, Zr-, Nb-, Ru-, and Tc-embedded B5N SACs have excellent catalytic activity in acidic conditions, while Ir-embedded B5N shows high catalytic activity in alkaline conditions. Interestingly, Ti@B2N, Mn@B3N, Fe@B5N and Mo@B5N SACs are highly active in either acidic or alkaline environments. Our work opens new avenues for designing cost-effective SACs with wide-pH-range HER performance.
1. Introduction
Hydrogen is considered to become the cleanest energy source in the world due to its advantages such as abundant raw materials, environmental protection, high energy and recyclability.1–3 Therefore, the development of a safe, efficient and low-cost hydrogen production process is crucial to solve the serious environmental pollution and energy shortage problems. Among them, electrochemical hydrogen evolution reaction (HER) is considered to be the most promising and sustainable industrial hydrogen production method, and the role of catalysts is indispensable.4–8 At present, the precious metal platinum is considered to be the best electrocatalyst for HER because of its excellent kinetic and thermodynamic efficiency.9–13 However, the scarcity and high cost of precious metals are important factors hindering their large-scale and sustainable application, which cannot be ignored.13–15 Therefore, it is very important to find both efficient and low-cost HER electrocatalysts, which have extremely important significance.
In order to develop a practical HER catalyst—in addition to considering some basic requirements such as the cost, stability and conductivity of the catalyst—it must also have the ability to trigger proton reduction with the minimum overpotential, as well as fast kinetic performance. In recent years, the unique surface structure and electronic properties of two-dimensional (2D) materials have provided new opportunities and challenges in the development of HER catalysts.16–23 Two-dimensional materials have the advantages of high durability, low cost and excellent environmental protection, and they have unique physical and chemical properties such as stronger surface activity, faster charge transfer and adjustable band gap, which give them broad application potential in the fields of catalysts, sensors and photodetectors.24–33 So far, 2D HER catalysts based on non-metallic nanomaterials have received a lot of attention. The intrinsic catalytic activity of 2D materials and the formation of single atom catalysts (SACs) by deposition of transition metal (TM) atoms on the surface of 2D materials are two new areas of cutting-edge catalytic research, which have revealed new catalytic processes at the atomic scale.34–36
In recent studies, various synthesized SACs showed excellent catalytic properties in different reactions.37–40 In order to meet the basic requirements of efficient SAC substrate materials, it is necessary to select a low-cost and highly stable substrate material, which should have abundant metal and good electrical conductivity, as well as a large specific surface area and multiple active centers. Among the many 2D materials, materials with metallic properties are a good choice. However, due to the wide variety of 2D materials, sifting through thousands of candidates remains a major challenge. The process of screening hydrogen evolution electrocatalysts can be greatly accelerated by using descriptors based on catalytic performance and density functional theory (DFT) calculations.41–43 To address the current challenges in the screening of 2D materials as SAC substrate materials, we designed a study to focus on the possibility of anchoring transition metal atoms in novel 2D substrate materials, and here we explore and report on the importance of such SAC descriptors. Recently, through first-principles swarm intelligence structure calculations, several 2D BXN materials (X = 2, 3, 5) have been successfully discovered. These BXN materials have high cohesion energy and good kinetic, thermodynamic and mechanical stability, making them highly feasible for experimental synthesis.44 BXN materials also exhibit excellent recyclability and improved metal conductivity after TM loading, showing great potential for hydrogen evolution SAC applications.
In this work, based on first principles, we studied the catalytic activity in HER of TM single atoms loaded on BXN materials’ surface, and discuss the mechanism of the high catalytic activity of the SCAs. Zhou et al. have verified the structural stability of 2D BXN materials through phonon spectrum calculation and ab initio molecular dynamics simulation (AIMD).44 We studied a variety of transition metal elements, including the 3d, 4d, and 5d groups. Here, the catalytic activity of SACs in HER is described by the Gibbs free energy change (GH*) of hydrogen adsorption. The descriptors proposed in this paper only include some inherent properties of substrate materials and provide a simple and rapid method for designing and screening HER electrocatalysts more effectively. Additionally, the optimal HER activity of 2D BXN catalysts in all pH ranges was investigated. Ti-, V-, Y- and Zr-embedded B5N SACs have high HER activity with moderate and good electrical conductivity before and after H binding at pH = 0. In addition, the 3d-B2N, 3d-B3N, 3d-B5N and 4d-B5N SACs studied have high activity over a wide pH range. Specifically, 3d-B2N and 3d-B3N SACs are suitable for acidic environments, and 4d-B5N SACs are suitable for alkaline environments, because the electron state near the Fermi level increases and the charge density on the surface increases. The high concentration of free charges is a key factor in HER progression.
2. Computational methods
All spin-polarized calculations in this section were performed by using the vienna ab initio simulation package (VASP)45,46 based on density functional theory. The generalized gradient approximation (GGA)47 of the Perdew–Burke–Ernzerhof (PEB)48 functional was used to describe the exchange correlation energy, and the projection plane-wave (PAW) method49 was used to describe the pseudopotential of all atoms. In order to eliminate the influence of van der Waals (vdW) forces, Grimme's empirical correction method, namely, DFT+D3,50,51 was used in structural optimizations and property calculations of the 2D BXN materials. A cutoff energy of 500 eV for the plane-wave basis was adopted. The energy and force convergence criteria for self-consistent optimizations were set to 10−5 eV and 10−3 eV Å−1, respectively. In the process of structural optimization, the inverted vector k-point grids of 2D BXN materials were divided into 2π × 0.03 Å−1. It is worth mentioning that when calculating electronic properties, the inverted lattice vector k-point grid for all structures in the self-consistent iteration process was set to a larger value. A 2 × 2 × 1 monolayer of 2D BXN materials was adapted for all calculations, and a vacuum space of 20 Å along the z direction was included to prevent interactions between periodic images. In ab initio molecular dynamics (AIMD) simulations, integration of the equations of motion proceeded with time steps of 1.0 fs for different temperature ranges.52 Typical simulations ran for 3000 steps with the time scale of about 3 ps. The isokinetic ensemble (NVT) was employed for the ions, where the ion temperature Ti was fixed using velocity scaling. As is well known, the influence of the solvation effect on the process of hydrogen adsorption is so slight that the solvation effect can be omitted, so we will not consider the calculations of solvation effect in this work.52–55 For more computational details, please see the ESI.†
3. Results and discussion
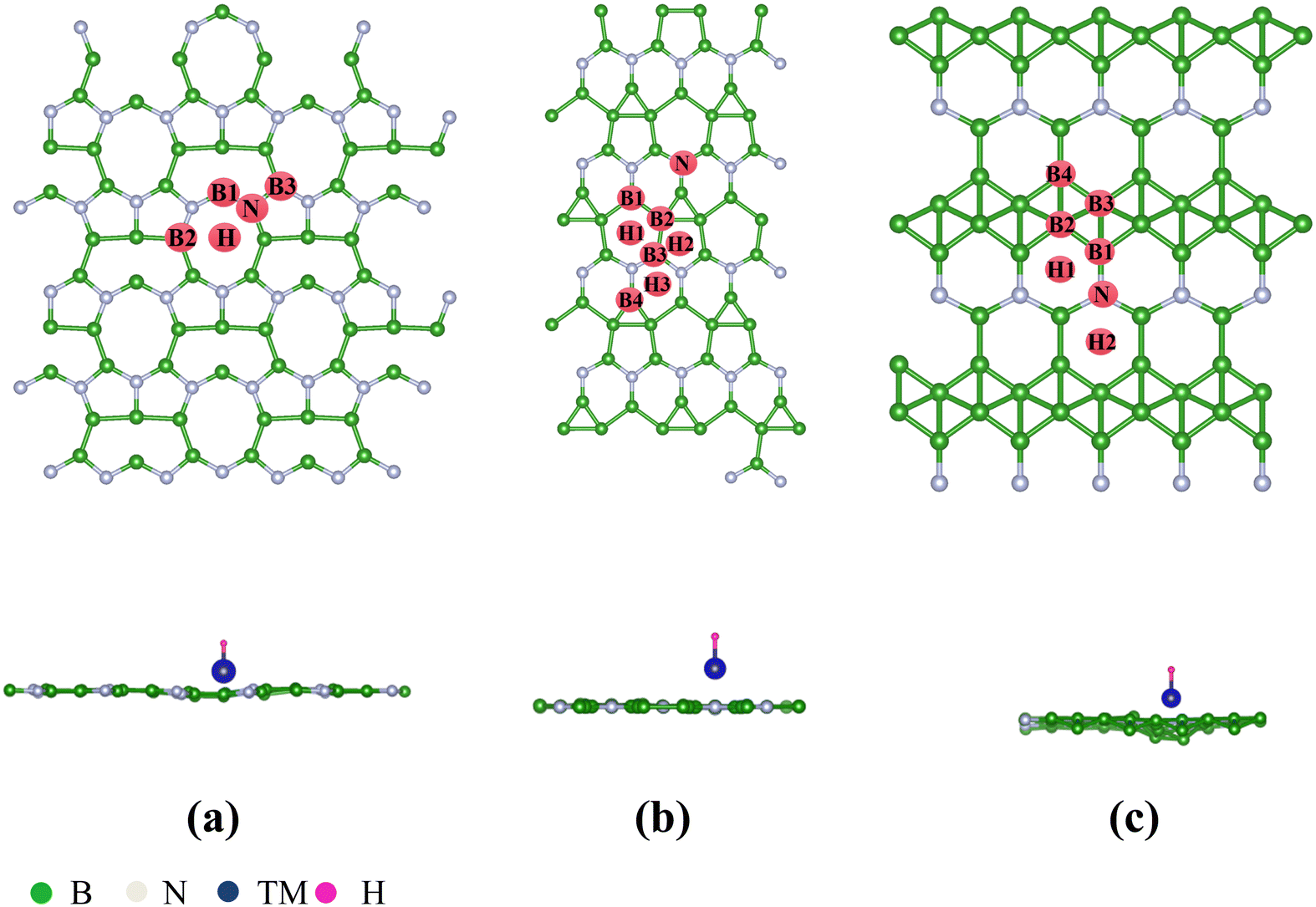
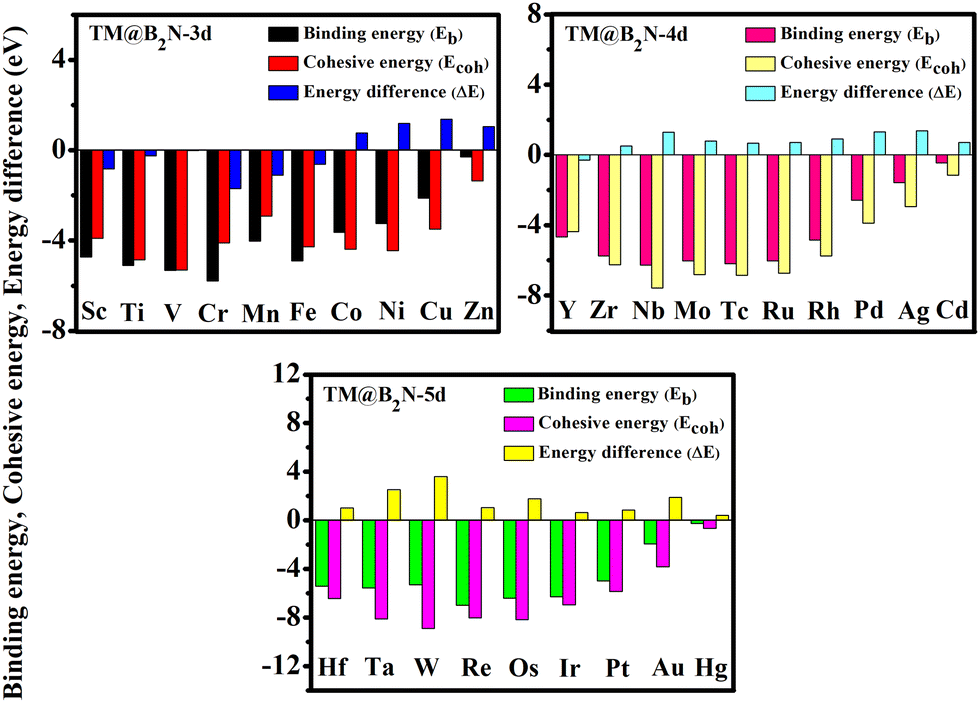
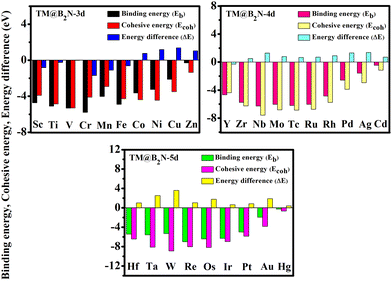
According to previous reports, the B2N, B3N and B5N structures were constructed in diverse orthorhombic configuration with the space group of AMM2, AMM2 and PMC21, respectively.44Fig. 1(a)–(c) illustrate the schematic diagrams of the 2D BXN materials (X = 2, 3, 5), which are composed of three different unit cells, respectively. The common feature is the presence of a five-member ring or even a six-member ring formed by boron atoms crossing with the nitrogen atoms in all the 2D BXN materials’ unit cells. The optimized lattice constant of 2D BXN materials is calculated to be a = 10.29 Å, b = 3.91 Å (X = 2); a = 10.13 Å, b = 4.80 Å (X = 3); and a = 6.41 Å, b = 2.69 Å (X = 5), respectively, in good accordance with previous studies.44 In order to explore the TM adsorption behavior on the surface of 2D BXN materials, a 2 × 2 supercell containing 72 atoms, 2 × 2 supercell including 64 atoms and 2 × 2 supercell involving 48 atoms as the substrate for the adsorption were constructed, correspondingly. Dynamic stability is an inevitable index in material design. So, in order to evaluate the dynamic stability of the BXN materials, we calculated their phonon spectra. As shown in Fig. S1 (ESI†), no negative frequency is observed in the entire Brillouin region, indicating that the B2N, B3N and B5N monolayers have good dynamic stability. Three different sites are considered for TM adsorption on the BXN materials, including the hollow of the surface B–N ring, the top site of B atom and the top site of surface N atom. Then, binding energy (Eb) calculations were implemented to evaluate the stability and feasibility of experimental synthesis of SACs for each configuration based on eqn (S1) (ESI†). Isolated Sc, as the test atom, adsorbs on nine different configurations, and the Eb was calculated between the Sc atom and substrates. Finally, it can be found from Table S1 (ESI†) that among the adsorption configurations, Sc adsorption on the hollow of 2D BXN material (X = 2, 3, 5) surface is the most energetically favorable, with the Eb of −4.75 eV, −3.91 eV and −5.18 eV, respectively. Then, the use of 2D BXN materials as substrates to anchor single atoms of 3d, 4d, and 5d TM was further investigated, and the Eb between TM atom and BXN materials was calculated. The corresponding Eb, cohesive energies (Ecoh) and the energy difference (ΔE) between Eb and Ecoh of all TM@BXN materials studied in the present work are shown in Fig. 2 and Fig. S2, S3 (ESI†). The Ecoh can be calculated by the following equation:where EM(bulk) is the energy of M crystals and n is the number of M atoms in the unit cell of crystals, and these values were compared with the adsorption energies. Because the energy advantage of TM clusters is essentially lower than that of metal crystals, to avoid the formation of clusters among TM atoms, we set a standard for judging system stability, that is, ΔE < 0 eV. As indicated in Fig. 2, the ΔE values are positive for all 5d-TM@B2N systems, and only six TM atoms of 3d state (Fe, Mn, Ti, V, Cr and Sc) and one TM atom of 4d state (Y) can immobilize on the B2N materials with ΔE < 0 eV. Similarly, the calculated ΔE values are positive for all 5d-TM@B3N systems, and only Co, Fe, Cr, Mn and Mo atoms can adsorb stably on B3N materials. For the B5N system, it is obvious that the adsorption of TM atoms by B5N materials is stronger than that by B2N and B3N materials. Most TM atoms, such as Co, Fe, Mn, Ti, V, Cr, Sc, Y, Zr, Nb, Mo, Tc, Ru and Ir, including the 3d, 4d and 5d groups, are effectively adsorbed on the B5N materials. For example, the Eb values of Mn@B5N and Cr@B5N SACs are even as low as −6.37 eV and −6.55 eV, respectively. The reason why the three 2D BXN materials have different adsorption effects on TM atoms is that they have different defect configurations. It is well known that for most 2D materials, the existence of surface atom vacancy is unavoidable to some extent. The main reason is that defect-induced structure disorder always leads to an entropy increase, which is thus in favour of balancing the defects caused a decrease of thermodynamic stability of the material.56–58 This indicates that among 2D materials composed of the same chemical constituents with different configurations, a compound with larger defect concentration always has higher energy and thus shows lower thermodynamic stability than that with lower defect concentration.59–61 This is because the larger structure of the defect configuration has more non-metallic atoms that combine with TM to form strong chemical bonds. Hence, for 2D BXN materials, B5N materials with relatively low defect concentration can accommodate more surface TM atoms, while B2N and B3N materials with relatively small defects have poor ability to adsorb surface TM atoms. The thermal stability of TM@BXN monolayers is simulated by AIMD. Fig. S4 (ESI†) shows that the mean value of the total potential energy during the AIMD simulation oscillates within a narrow range, and the overall configuration of Ti@B2N, Mn@B3N, Fe@B5N and Mo@B5N monolayers remains good after 3 ps AIMD simulation, which confirms their thermal stability.
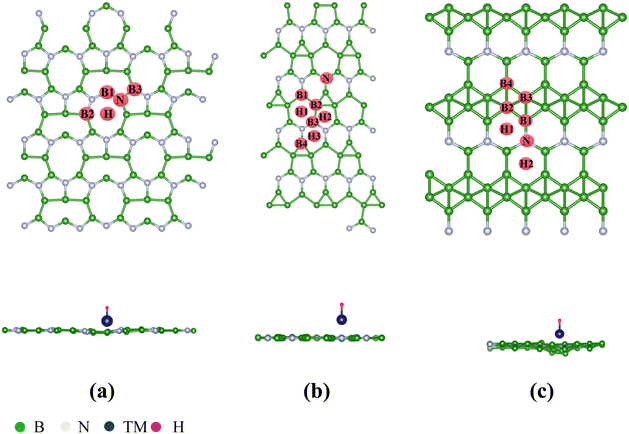 |
| | Fig. 1 Top and side views of the optimized structures of the 2D BXN materials: (a) B2N monolayer, (b) B3N monolayer, and (c) B5N monolayer with the TM and H being adsorbed at the optimal HER sites. | |
 |
| | Fig. 2 . Adsorption of different TM atoms on the B2N monolayer and the corresponding binding energy (Eb), cohesive energy (Ecoh) and their energy difference (ΔE). | |
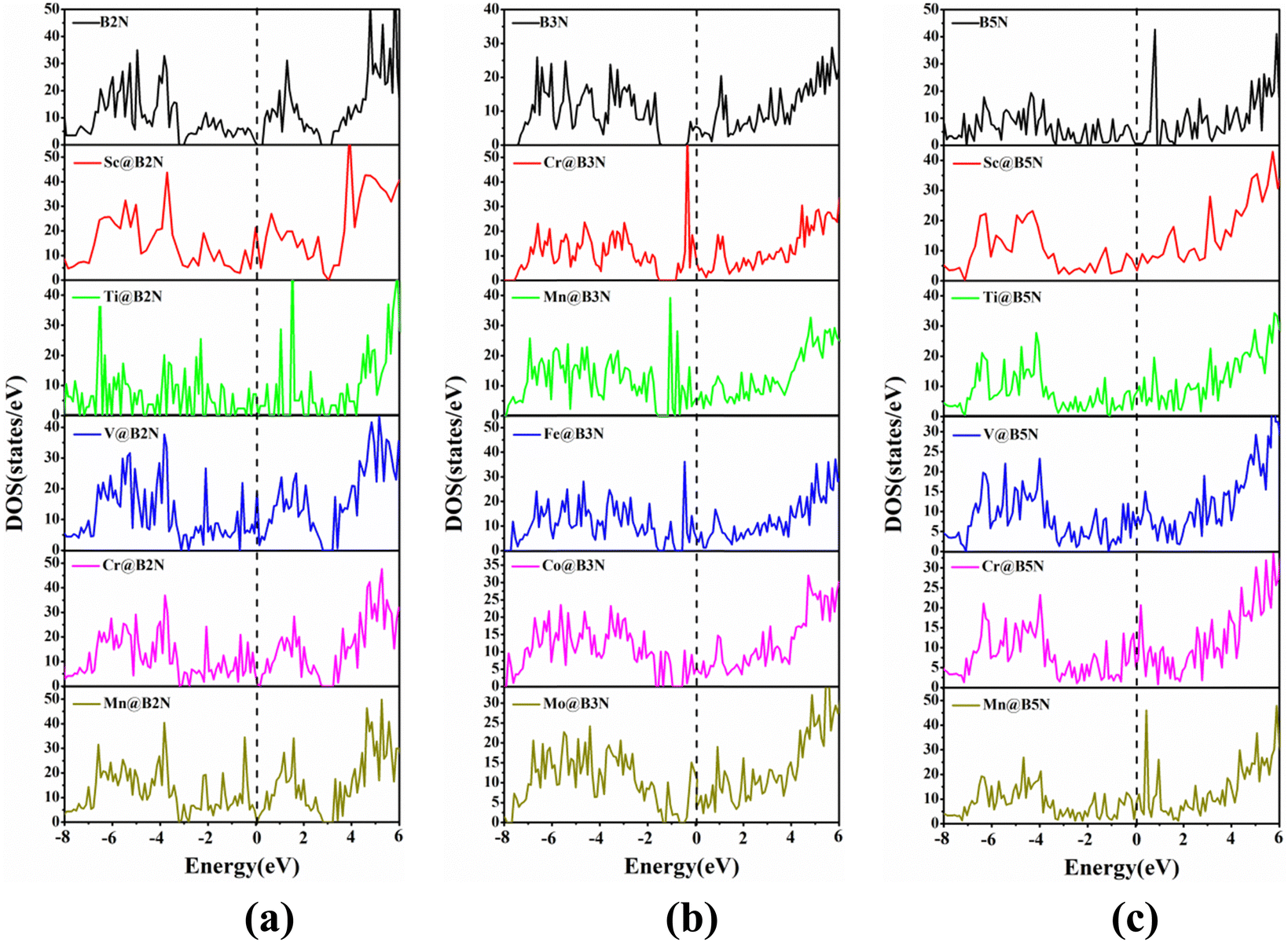
To study the electronic characteristics of the TM@BXN systems, the density of states (DOS) spectra of all pre-screened SACs are given in Fig. 3 and Fig. S5 (ESI†). It is obvious that there are more pronounced electronic states near the Fermi level (EF) of the orbital resolved DOS spectra of TM@BXN materials compared with the DOS spectra of pure 2D BXN materials. This is because the symmetry of 2D BXN materials is broken after the addition of TM atoms, thereby generating more unsaturated electrons. Thus, it can be seen that TM atoms are the major contributors of electron densities near EF, which is offered principally by the d orbitals of TM atoms, rendering the 2D BXN materials metallic. This is conducive to the improvement of surface electrical conductivity of SACs, thus enhancing their catalytic activity.
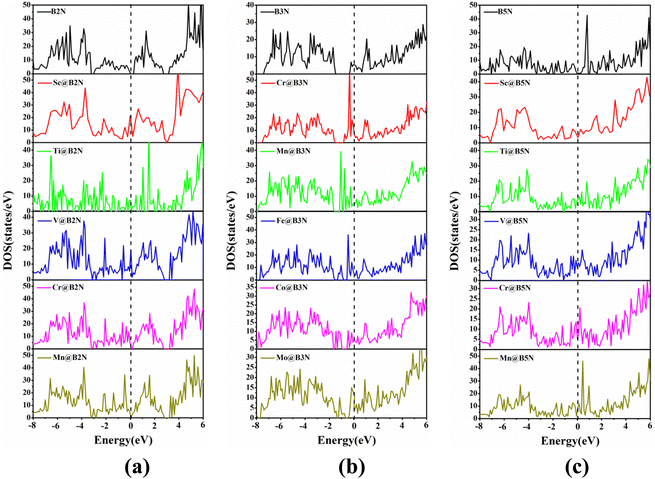 |
| | Fig. 3 The density of states (DOS) spectra for (a) TM@B2N, (b) TM@B3N and (c) TM@B5N. The Fermi levels are set to zero and indicated by black dashed lines. | |
In order to more intuitively demonstrate the metallic nature of SACs, the electronic conductivity of TM@BXN systems was further calculated. According to Fig. S6 (ESI†), it can be clearly found that the studied SACs have good surface conductivity, which is conducive to improving the catalytic activity of SACs. Hence, the HER activity of the pre-screened TM@BXN SACs is achieved by setting TM atoms as the active sites. These stable TM@BXN systems as HER catalysts were next investigated. It is well known that the ΔGH* of atomic hydrogen adsorption is a key descriptor for evaluating HER catalytic activity.62 Thus, we systematically studied the TM@BXN systems in accordance with the calculation of ΔGH* in acidic and alkaline conditions, respectively. In the acid conditions, the HER process can be expressed by Volmer–Tafel and Volmer–Heyrovsky reaction pathways. The two reaction equations are divided into three key steps:
| | | Volmer step: H+ + e− + * → H* | (2) |
| | | Heyrovsky step: H* + H+ + e− → H2+* | (3) |
| | | Tafel step: H* + H* → H2 + 2* | (4) |
where * and H* represent the active site on the catalyst surface and the intermediate hydrogen adsorbed on the surface, respectively. For the Heyrovsky reaction, H
2 is generated when adsorbed hydrogen (H*) interacts with H
3O
+ clusters. As shown in Fig. S7 (ESI
†), the calculated energy barrier for the Heyrovsky pathway of the Ti@B
5N monolayer is 1.78 eV, while the activation energy barrier of the Tafel reaction of the Ti@B
5N monolayer is 0.52 eV. Thus, the HER mechanism of the Ti@B
5N monolayer may follow the Tafel-dominated Volmer–Tafel reaction. Similarly, the HER mechanism of the V@B
5N monolayer also follows the Volmer–Tafel reaction. Nevertheless, under alkaline conditions, water dissociation produces hydrogen ions (H
+), and the two important reaction steps are given as follows:
| | | Alkaline Volmer step: H2O + e− + * → H* + OH− | (5) |
| | | Alkaline Heyrovsky step: H* + H2O + e− → H2 + OH− + * | (6) |
Under alkaline conditions, the apparent HER activity is limited by hydrolytic dissociation, which produces H* and is sensitive to pH, so the energy barrier of hydrolytic dissociation needs to be considered. At the same time, more and more computational and experimental studies have proven that the hydrolytic separation energy barrier plays an important role in the alkaline hydrogen evolution process. As shown in Fig. S8 (ESI†), it is found through calculation that Ti- and V-modified B5N have rapid hydrolysis dissociation ability, with the energy barrier of about 0.23 and 0.09 eV, which can realize rapid proton transfer. Therefore, Ti, V and BS-B1P1 can cooperate well to accelerate alkaline HER.
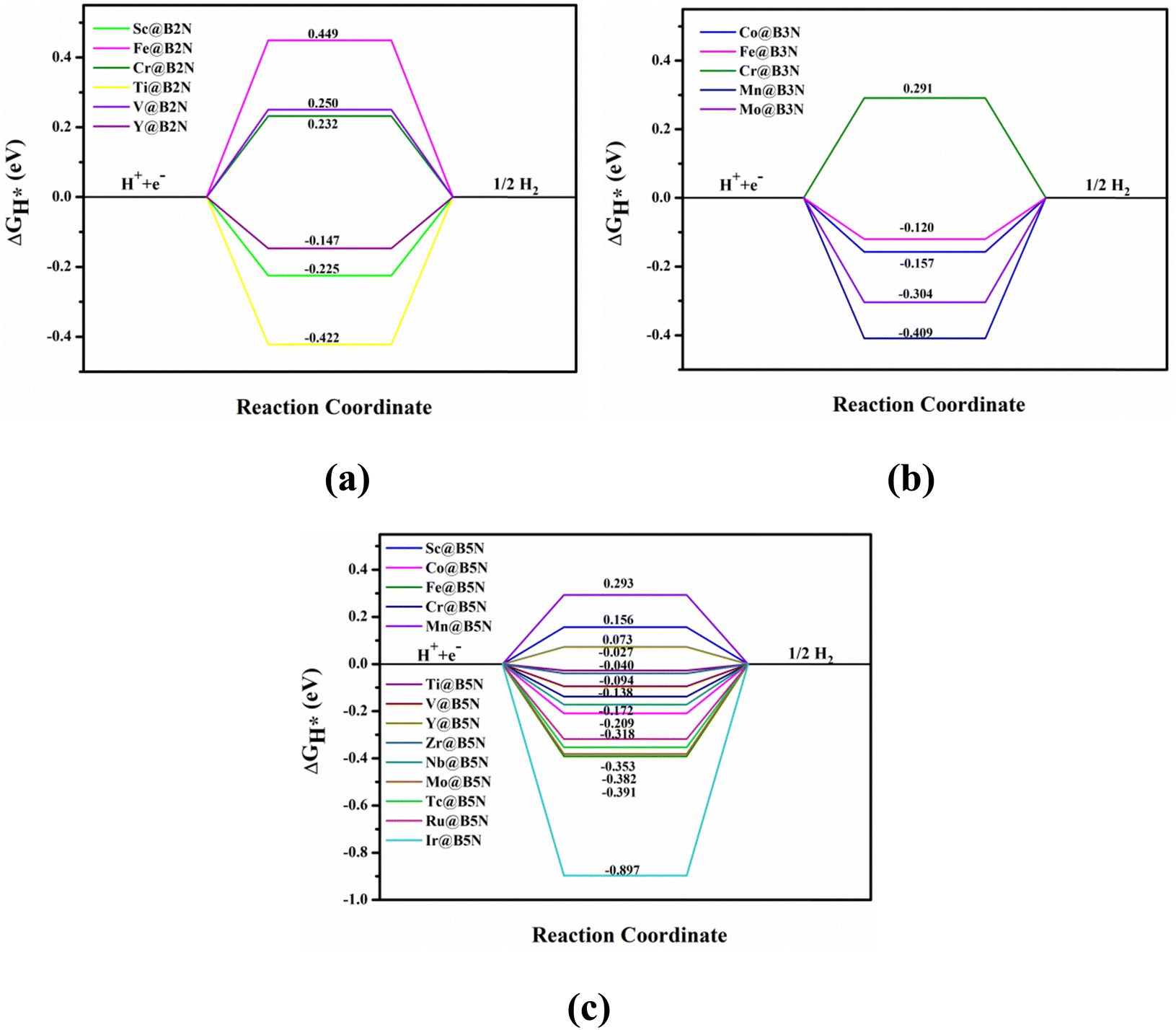
Fig. 4 shows the ΔGH* for single H atom adsorption on TM@BXN systems at pH = 0. Ti@B5N, V@B5N, Y@B5N and Zr@B5N SACs show excellent catalytic activity with ΔGH* values of −0.027, −0.094, 0.073 and −0.04 eV, respectively. It can be seen that the ΔGH* of these SACs are even low as ∼ −0.027 eV, superior to precious metals such as Pt. In reality, electrocatalysts need to undergo charge adjustment to match their Fermi level with the applied electrode potential, and therefore, a grand-canonical ensemble of electrons under the constant potential model (CPM) is better at reflecting practical electrocatalytic conditions.63,64 We supplemented the calculation of ΔGH* corresponding to different potentials under the CPM model. According to Fig. S9 (ESI†), it can be clearly found that TM@B5N SACs with good catalytic performance show little change in ΔGH* at different potentials, and still have excellent catalytic performance. In addition, TM@B5N SACs have the best HER catalytic active sites on the hollow of the 2D B5N surface. Nevertheless, the decoration of TM atoms can reinforce well the binding of H atoms to the B2N and B3N materials, but it performs poorly in enhancing the HER catalytic activity of the TM@B2N and TM@B3N SACs, showing inferior ΔGH* values. This means that at pH = 0, low-cost TM@B5N SACs may become substitutes for precious metal catalysts to improve the performance of HER. To more intuitively compare the catalytic activity of TM@BXN SACs at pH = 0, the corresponding volcano curves are plotted in Fig. 5. According to Norskov's hypothesis, where a simple kinetic model is established to understand the origin of the volcano, the exchange current density (i0) based on ΔGH* was studied and calculated.65 When ΔGH* < 0, the expression of exchange current is expressed as:
| | 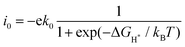 | (7) |
when Δ
GH* > 0, the exchange current is:
| | 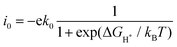 | (8) |
where
k0 is the rate constant and
kB is the Boltzmann constant. Because there are no experimental data available,
k0 is set to 1.
66 First, the volcano map is drawn with Δ
GH* as the horizontal coordinate and
i0 as the vertical coordinate. The HER catalytic activity of these SACs can be better observed based on the position of Δ
GH* and
i0 relative to the volcanic peak, where the closer the location is to the volcanic peak, the higher the HER activity of the SACs. As shown in
Fig. 5(c), Ti@B
5N, V@B
5N, Y@B
5N and Zr@B
5N systems achieve the maximum exchange current rates on account of the appropriate interactions between TM (Ti, V, Y, Zr) and H atoms, implying a promising catalytic activity for HER. However, most TM@B
2N SACs, such as Co@B
5N, Cr@B
5N, Fe@B
5N, Nb@B
5N, Ru@B
5N, Tc@B
5N, Mo@B
5N and Ir@B
5N, exhibit poor HER performance due to the strong bonding strength between H atoms and TM atoms, and are mainly distributed on the left side of the volcano map. Sc@B
5N and Mn@B
5N SACs are on the right side of the volcano map because of the weak interaction between H and TM atoms, which results in positive Δ
GH* values. However, among the pre-screened TM@B
2N (TM = Sc, Cr, V, Ti, Fe and Y) and TM@B
3N (TM = Co, Cr, Fe, Mn and Mo) SACs (shown in
Fig. 5(a) and (b)), Ti-, Mn-, and Y@B
2N; and Co-, Fe-, Mn-, Mo@B
3N are mainly distributed on the left side of the volcano diagram, showing strong hydrogen adsorption, to the disadvantage of the release of atomic hydrogen. V-, Fe-, Cr@B
2N and Cr@B
3N SACs are on the right side of the volcano curve with highly positive Δ
GH* values, which is not conducive to the adsorption of atomic hydrogen on the TM atom, thus exhibiting low
i0 values. It is obvious that most SACs have negative Δ
GH* values because of the strong adsorption between hydrogen atoms and TM atoms. However, pH modification can effectively improve the catalytic activity of these SACs, so as to have a suitable Δ
GH* value. In neutral or alkaline environment, water molecules (H
2O) are first adsorbed and decomposed on the SAC surface (Volmer process: H
2O + e
− + * → H* + OH
−). Then, two adjacent adsorbed hydrogen atoms on the SAC surface will combine to form a molecule of hydrogen (Heyrovsky process: H* + H
2O + e
− → H
2 + OH
− + *). SACs in this process promote the formation of hydrogen molecules.
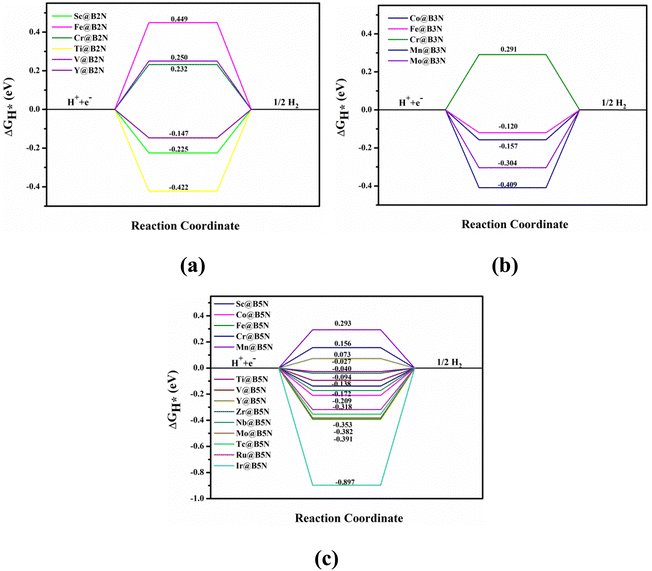 |
| | Fig. 4 Free energy diagram for HER of different TM atoms on the (a) B2N, (b) B3N, and (c) B5N monolayer compounds under standard conditions. | |
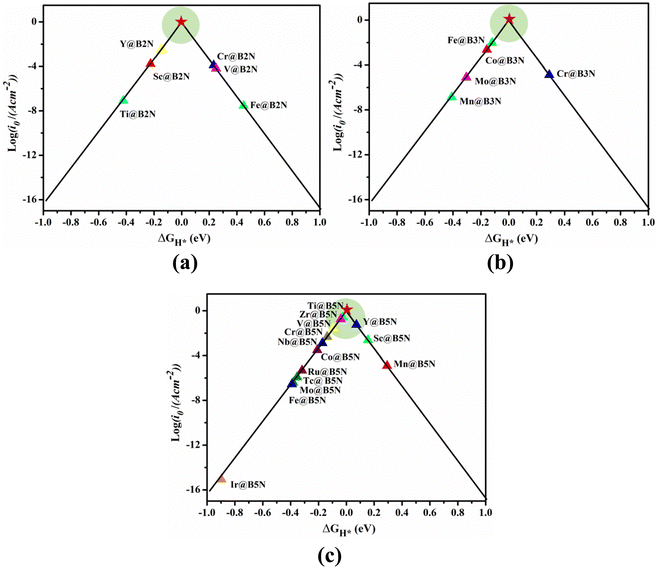 |
| | Fig. 5 HER volcano curve of exchange current (i0) as a function of the Gibbs free energy change (ΔGH*) of hydrogen adsorption on (a) TM@B2N, (b) TM@B3N, and (c) TM@B5N systems. | |
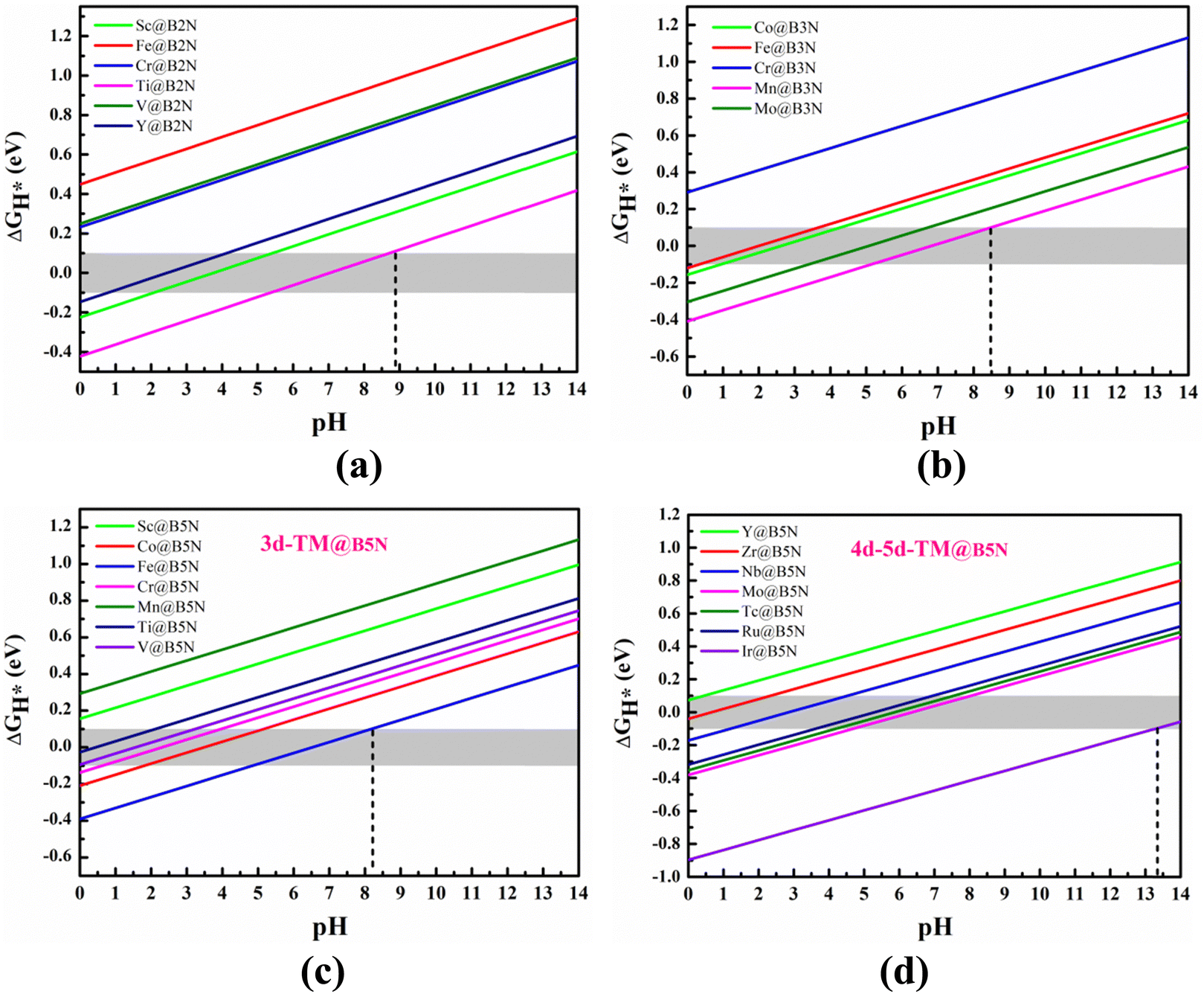
We can regard the effect of pH on ΔGH* as an additional entropic potential. Then the pH-dependent ΔG(pH) can be obtained by eqn (S7) (ESI†): ΔG(pH) = kBTln(10) × pH, which is conducive to understanding the relationship between HER performance and pH. Fig. 6(a)–(d) show the effect of different pH on the HER performance of all pre-screened SACs, including 3d-, 4d and 5d-TM@BXN systems. It is obvious that as the pH value is increased from 1 to 14, ΔGH* value gradually increases, meaning that the adsorption inclination between hydrogen atom and SACs significantly decreases. By exploring the dependence of H adsorption on different SACs with pH, the optimal SACs for HER can be found. Fig. 6(c and d) show the linear relationship between the pH and ΔGH* of TM@B5N SACs. Fig. 6(c) indicates that for 3d-TM@B5N SACs, only V@B5N and Ti@B5N systems lie in the optimal HER activity range at pH = 0. With increasing pH from 0 to 3, HER activity of the V@B5N and Ti@B5N SACs decreases, ΔGH* increases from −0.027 to 0.100 eV, indicating that V@B5N and Ti@B5N SACs can maintain ultra-high HER activity in a strong acid environment. Cr@B5N and Co@B5N SACs can only have excellent catalytic activity in strong acid or weak acid environment, with pH regulation from 2 to 6. It is worth mentioning that Fe@B5N SAC can effectively catalyze HER under weak acid, neutral and alkaline conditions, which means that Fe@B5N SAC has a wide range of choices in different pH environments. Fig. 6(d) gives the ΔGH*vs. pH for the 4d and 5d-TM@B5N SACs. Compared with 3d-TM@B5N SACs, the effect of pH on 4d and 5d-TM@B5N SACs is more obvious, which is reflected in their obvious increase of catalyst activity in the pH range from 0 to 14. As can be seen from the figure at pH = 0, the adsorption between 4d, 5d TM atoms and H atom is too strong, leading to a more negative ΔGH*. As pH increases, the bonding strength between 4d, 5d TM atoms and the H atom is further weakened, bringing ΔGH* down to a suitable value. In the pH range of 0 to 2.5, that is, under strong acid conditions, Y@B5N and Zr@B5N SACs are highly active in HER. In contrast, Ir@B5N SAC has good catalytic activity in a strong alkali environment. Nb@B5N SAC can maintain excellent HER activity under a stronger acid environment with pH range of 2 to 4.5, while only under the condition of weak acid with pH range of 4 to 7 can Ru@B5N and Tc@B5N SACs effectively catalyze HER. Similar to Fe@B5N SAC, Mo@B5N SAC can also play a catalytic role under weak acid, neutral and alkaline conditions. The variation of ΔGH* with pH of TM@B2N SACs is shown in Fig. 6(a). It is obvious that TM@B2N SACs mainly promote HER under acidic conditions. For example, Sc@B2N and Y@B2N SACs are active under the weak-acid or strong-acid condition with pH range of 1 to 5.5, and the HER performance of Ti@B2N SAC can be greatly improved in either acidic, neutral or alkaline conditions with pH range of 5.5 to 9. As seen in Fig. 6(b), TM@B3N SACs also catalyze the conversion process from atomic H to H2 mainly under acidic conditions. Fe@B3N and Co@B3N SACs have excellent catalytic activity in strong acid environment with pH regulation from 1 to 4. Mo@B3N SAC shows stronger HER catalysis in the weak acid environment, with the pH range of 4 to 7. Interestingly, Mn@B3N SAC can be effectively adjusted by pH and gives weak acid, neutral and alkaline HER activity within the pH range of 5 to 8.5. In short, our results suggest that the HER activity of TM@BXN SACs is closely associated with pH. The TM@BXN SACs are not only catalytically active in acidic conditions but also exhibit high HER catalytic activity in alkaline environments. Remarkably, Ti@B2N, Mn@B3N, Fe@B5N and Mo@B5N SACs have potential to be a multifunctional catalyst with a wide pH threshold.
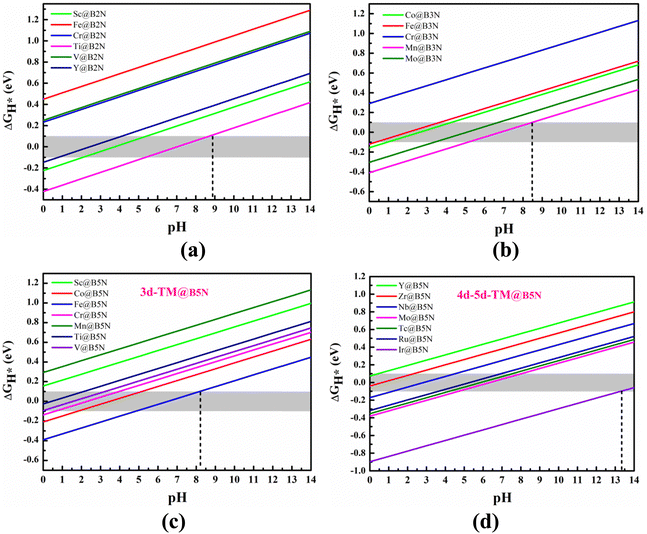 |
| | Fig. 6 The pH dependence of H-adsorption free energy of TM atom adsorption on the (a) B2N, (b) B3N and (c) and (d) B5N monolayer compounds. The pink shaded area in the figures indicates the ideal ΔGH* value range. | |
4. Conclusions
In summary, we designed promising SACs composed of 3d-, 4d-, and 5d-TM atoms and 2D BXN materials toward efficient HER using first-principles calculations. Some stable TM@BXN SACs were screened by theoretical calculations, and DOS calculations show that these SACs have good electrical conductivity, providing favorable conditions for rapid transfer among electrons. In addition, free energy calculations showed that the TM@BXN SACs exhibit different catalytic performance in different pH ranges. Ti-, V-, Y- and Zr-embedded B5N SACs have high HER activity with moderate and good electrical conductivity before and after H binding at pH = 0. TM@B2N and TM@B3N SACs have excellent HER performance in acid environment. Remarkably, three different TM@BXN systems have appropriate SACs which can maintain high HER catalytic activity in acidic, neutral and alkaline environments to promote the conversion of atomic H to H2. Therefore, our works demonstrate that the cost-effective TM@BXN can serve as SACs to facilitate HER, which provides a theoretical reference for future application in practice.
Data availability
All data generated or analyzed during this study are included in this published article and its ESI† files.
Conflicts of interest
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
Acknowledgements
This work is supported by the Zhejiang Engineering Research Center for Edge Intelligence Technology and Equipment, Academy of Edge Intelligence Hangzhou City University, Special Project for Improving Level of Local Universities in Zhejiang Province (Grant No. 202000-584102), Scientific Research Foundation of Hangzhou City University (Grant No. X-202204), Zhejiang Provincial Natural Science Foundation of China (Grant No. LHZY24B030001), and the Scientific Research Project of Zhejiang Provincial Education Department (Grant No. Y202351383)
References
- K. Mazloomi and C. Gomes, Hydrogen as an energy carrier: Prospects and challenges, Renewable Sustainable Energy Rev., 2012, 16, 3024–3033 CrossRef CAS.
- J. Luo, J.-H. Im, M. T. Mayer, M. Schreier, M. K. Nazeeruddin and N.-G. Park,
et al., Water photolysis at 12.3% efficiency via perovskite photovoltaics and Earthabundant catalysts, Science, 2014, 345, 1593–1596 CrossRef CAS PubMed.
- S. Z. Baykara, Hydrogen: a brief overview on its sources, production and environmental impact, Int. J. Hydrogen Energy, 2018, 43, 10605–10614 CrossRef CAS.
- M. S. Dresselhaus and I. L. Thomas, Alternative energy technologies, Nature, 2001, 414, 332–337 CrossRef CAS PubMed.
- S. Chu and A. Majumdar, Opportunities and challenges for a sustainable energy future, Nature, 2012, 488, 294–303 CrossRef CAS PubMed.
- Y. Zheng, Y. Jiao, Y. Zhu, L. Li, Y. Han, Y. Chen, A. Du, M. Jaroniec and S. Qiao, Hydrogen evolution by a metal-free electrocatalyst, Nat. Commun., 2014, 5, 3783 CrossRef PubMed.
- J. Mahmood, F. Li, S.-M. Jung, M. S. Okyay, I. Ahmad, S.-J. Kim, N. Park, H. Y. Jeong and J.-B. Baek, An efficient and pH-universal ruthenium-based catalyst for the hydrogen evolution reaction, Nat. Nanotechnol., 2017, 12, 441–446 CrossRef CAS PubMed.
- G. Zhao, K. Rui, S. X. Dou and W. Sun, Heterostructures for electrochemical hydrogen evolution reaction: a review, Adv. Funct. Mater., 2018, 28, 1803291 CrossRef.
- D. V. Esposito, S. T. Hunt, A. L. Stottlemyer, K. D. Dobson, B. E. McCandless, R. W. Birkmire and J. G. Chen, Low-cost hydrogen-evolution catalysts based on monolayer platinum on tungsten monocarbide substrates, Angew. Chem., Int. Ed., 2010, 49, 9859–9862 CrossRef CAS PubMed.
- L. A. Kibler, Hydrogen Electrocatalysis, ChemPhysChem, 2006, 7, 985–991 CrossRef CAS PubMed.
- Y.-J. Wang, N. Zhao, B. Fang, H. Li, X. T. Bi and H. Wang, Carbon-supported Pt-based alloy electrocatalysts for the oxygen reduction reaction in polymer electrolyte membrane fuel cells: particle size, shape, and composition manipulation and their impact to activity, Chem. Rev., 2015, 115, 3433–3467 CrossRef CAS PubMed.
- R. Subbaraman, D. Tripkovic, D. Strmcnik, K. C. Chang, M. Uchimura, A. P. Paulikas, V. Stamenkovic and N. M. Markovic, Enhancing Hydrogen Evolution Activity in Water Splitting by Tailoring Li+-Ni(OH)2-Pt Interfaces, Science, 2011, 334, 1256–1260 CrossRef CAS PubMed.
- Y. Zheng, Y. Jiao, M. Jaroniec and S. Z. Qiao, Advancing the Electrochemistry of the Hydrogen-Evolution Reaction through Combining Experiment and Theory, Angew. Chem., Int. Ed., 2015, 54, 52–65 CrossRef CAS PubMed.
- H. Xu, D. Cheng, D. Cao and X. C. Zeng, A universal principle for a rational design of single-atom electrocatalysts, Nat. Catal., 2018, 1, 339–348 CrossRef CAS.
- X. Liu and L. Dai, Erratum: Carbon-based metal-free catalysts, Nat. Rev. Mater., 2016, 1, 16064 CrossRef CAS.
- X. Lv, W. Wei, Q. Sun, F. Li, B. Huang and Y. Dai, Two-dimensional germanium monochalcogenides for photocatalytic water splitting with high carrier mobility, Appl. Catal., B, 2017, 217, 275–284 CrossRef CAS.
- T. Liao, L. Kou, A. Du, Y. Gu and Z. Sun, Simplest MOF units for effective photodriven hydrogen evolution reaction, J. Am. Chem. Soc., 2018, 140, 9159–9166 CrossRef CAS.
- C. Liu, Q. Li, C. Wu, J. Zhang, Y. Jin, D. R. MacFarlane and C. Sun, Single-boron catalysts for nitrogen
reduction reaction, J. Am. Chem. Soc., 2019, 141, 2884–2888 CrossRef CAS PubMed.
- L. X. Chen, Z. W. Chen, Y. Wang, C. C. Yang and Q. Jiang, Design of dual-modified MoS2 with nanoporous Ni and graphene as efficient catalysts for the hydrogen evolution reaction, ACS Catal., 2018, 8, 8107–8114 CrossRef CAS.
- G. Gao, Y. Jiao, E. R. Waclawik and A. Du, Single atom (Pd/Pt) supported on graphitic carbon nitride as an efficient photocatalyst for visible-light reduction of carbon dioxide, J. Am. Chem. Soc., 2016, 138, 6292–6297 CrossRef CAS.
- F. Gao, Y. H. Wei, J. G. Du and G. Jiang, Theoretical screening of 2D materials supported transition-metal single atoms as efficient electrocatalysts for hydrogen evolution reaction, Materialia, 2021, 18, 101168 CrossRef CAS.
- Y. H. Wei, F. Gao, J. G. Du and G. Jiang, Modulation of the B4N monolayer as an efficient electrocatalyst for hydrogen evolution reaction, Int. J. Hydrogen Energy, 2022, 47, 11511–11519 CrossRef CAS.
- Y. H. Wei, F. Gao, H. C. Huang and G. Jiang, Two-dimensional B7P2: Dual-purpose functional material for hydrogen evolution reaction/hydrogen storage, Int. J. Hydrogen Energy, 2022, 47, 8338–8347 CrossRef CAS.
- Z. Zhang, Y. Yang, G. Gao and B. I. Yakobson, Two-Dimensional Boron Monolayers Mediated by Metal Substrates, Angew. Chem., Int. Ed., 2015, 54, 13022–13026 CrossRef CAS.
- G. R. Bhimanapati, Z. Lin, V. Meunier, Y. Jung and J. Cha, Recent Advances in Two-Dimensional Materials beyond Graphene, ACS Nano, 2015, 9, 11509–11539 CrossRef CAS PubMed.
- S. Zhang, W. Zhou, Y. Ma, J. Ji, B. Cai, S. A. Yang, Z. Zhu, Z. Chen and H. Zeng, Antimonene Oxides: Emerging Tunable Direct Bandgap Semiconductor and Novel Topological Insulator, Nano Lett., 2017, 17, 3434–3440 CrossRef CAS PubMed.
- S. Milana, The Lab-to-Fab Journey of 2D Materials, Nat. Nanotechnol., 2019, 14, 919–921 CrossRef CAS PubMed.
- H. Shu, D. Zhou, F. Li, D. Cao and X. Chen, Defect Engineering in MoSe2 for the Hydrogen Evolution Reaction: From Point Defects to Edges, ACS Appl. Mater. Interfaces, 2017, 9, 42688–42698 CrossRef CAS.
- M. Zeng, Y. Chen, J. Li, H. Xue, R. G. Mendes, J. Liu, T. Zhang, M. H. Rümmeli and L. Fu, 2D WC Single Crystal Embedded in Graphene for Enhancing Hydrogen Evolution Reaction, Nano Energy, 2017, 33, 356–362 CrossRef CAS.
- P. Manchanda, A. Enders, D. J. Sellmyer and R. Skomski, Hydrogen-Induced Ferromagnetism in Two-Dimensional Pt Dichalcogenides, Phys. Rev. B, 2016, 94, 104426 CrossRef.
- S. Lei, X. Wang, B. Li, J. Kang, Y. He, A. George, L. Ge and Y. Gong, Surface Functionalization of Two Dimensional Metal Chalcogenides by Lewis Acid-Base Chemistry, Nat. Nanotechnol., 2016, 11, 465–471 CrossRef CAS PubMed.
- D. Akinwande, C. J. Brennan, J. S. Bunch, P. Egberts, J. R. Felts and H. Gao, A Review on Mechanics and Mechanical Properties of 2D Materials Graphene and Beyond, Extreme Mech. Lett., 2017, 13, 42–77 CrossRef.
- C. Tan, X. Cao, X.-J. Wu, Q. He, J. Yang, X. Zhang, J. Chen, W. Zhao and S. Han, Recent Advances in Ultrathin Two-Dimensional Nanomaterials, Chem. Rev., 2017, 117, 6225–6331 CrossRef CAS PubMed.
- X.-F. Yang, A. Wang, B. Qiao, J. Li, J. Liu and T. Zhang, Single-atom catalysts: a new frontier in heterogeneous catalysis, Acc. Chem. Res., 2013, 46, 1740–1748 CrossRef CAS PubMed.
- H. Fei, J. Dong, Y. Feng, C. S. Allen, C. Wan and B. Volosskiy, General synthesis and definitive structural identification of MN4C4 single-atom catalysts with tunable electrocatalytic activities, Nat. Catal., 2018, 1, 63–72 CrossRef CAS.
- C. Ling, X. Bai, Y. Ouyang, A. Du and J. Wang, Single molybdenum atom anchored on N-doped carbon as a promising electrocatalyst for nitrogen reduction into ammonia at ambient conditions, J. Phys. Chem. C, 2018, 122, 16842–16847 CrossRef CAS.
- J. Su, R. Ge, Y. Dong, F. Hao and L. Chen, Recent progress in single-atom electrocatalysts: concept, synthesis, and applications in clean energy conversion, J. Mater. Chem. A, 2018, 6, 14025–14042 RSC.
- X. Su, X. F. Yang, Y. Huang, B. Liu and T. Zhang, Single-Atom Catalysis toward Efficient CO2 Conversion to CO and Formate Products, Acc. Chem. Res., 2019, 52, 656–664 CrossRef CAS.
- R. Zhang, L. Jiao, W. Yang, G. Wan and H. L. Jiang, Single-atom catalysts templated by metal–organic frameworks for electrochemical nitrogen reduction, J. Mater. Chem. A, 2019, 7, 26371–26377 RSC.
- X. Zhang, Z. Zhang, D. Wu, X. Zhang, X. Zhao and Z. Zhou, Computational screening of 2D materials and rational design of heterojunctions for water splitting photocatalysts, Small Methods, 2018, 2, 1700359 CrossRef.
- Z. H. Zhang, X. Zhang, X. D. Zhao, S. Yao, A. Chen and Z. Zhou, Algorithm screening to accelerate discovery of 2D metal-free electrocatalysts for hydrogen evolution reaction, ACS Omega, 2019, 4, 7822–7828 CrossRef CAS PubMed.
- S. Yao, X. Zhang and A. Chen, Algorithm screening to accelerate discovery of 2D metal-free electrocatalysts for hydrogen evolution reaction, J. Mater. Chem. A, 2019, 7, 19290 RSC.
- S. Y. Lin, Y. Guo, M. L. Xu, J. J. Zhao and Y. W. Liang, A B2N monolayer: a direct band gap semiconductor with high and highly anisotropic carrier mobility, Nanoscale, 2022, 14, 930–938 RSC.
- X. Y. Zhou, X. F. Chen, C. Z. Shu, Y. Huang, B. B. Xiao, W. T. Zhang and L. L. Wang, Two-Dimensional Boron-Rich Monolayer BxN as High Capacity for Lithium-Ion Batteries: A First-Principles Study, ACS Appl. Mater. Interfaces, 2021, 13, 41169–41181 CrossRef CAS PubMed.
- G. Kresse and J. Furthmuller, Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed.
- G. Kresse and J. Furthmuller, Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- J. P. Perdew and Y. Wang, Accurate and simple analytic representation of the electron-gas correlation energy, Phys. Rev. B: Condens. Matter Mater. Phys., 1992, 45, 13244–13249 CrossRef PubMed.
- P. E. Blöchl, Projector augmented-wave method, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Generalized gradient approximation made simple, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS.
- S. Grimme, S. Ehrlich and L. Goerigk, Effect of the damping function in dispersion corrected density functional theory, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- S. Grimme, Accurate description of van der Waals complexes by density functional theory including empirical corrections, J. Comput. Chem., 2004, 25, 1463–1473 CrossRef CAS PubMed.
- A. Roudgar and A. Groß, Water bilayer on the Pd/Au(111) overlayer system: Coadsorption and electric field effects, Chem. Phys. Lett., 2005, 409, 157–162 CrossRef CAS.
- Y. Gohda, S. Schnur and A. Groß, Influence of water on elementary reaction steps in electrocatalysis, Faraday Discuss., 2009, 140, 233–244 RSC.
- S. Yao, X. Zhang, A. Chen, Z. Zhang, M. Jiao and Z. Zhou, Algorithm screening to accelerate discovery of 2D metal-free electrocatalysts for hydrogen evolution reaction, J. Mater. Chem. A, 2019, 7, 19290–19296 RSC.
- X. Yang, N. Gao, S. Zhou and J. Zhao, MXene nanoribbons as electrocatalysts for the hydrogen evolution reaction with fast kinetics, Phys. Chem. Chem. Phys., 2018, 20, 19390–19397 RSC.
- W. W. Qian, Z. Chen, J. F. Zhang and L. C. Yin, Monolayer MoSi2N4-x as promising electrocatalyst for hydrogen evolution reaction: A DFT prediction, J. Mater. Sci. Technol., 2022, 99, 215–222 CrossRef CAS.
- X. Q. Wan, C. L. Yang, X. H. Li and Y. L. Zhao, Two-dimensional trilayer heterostructures with cascade dual Z-schemes to achieve efficient hydrogen evolution reaction, J. Mater. Chem. A, 2024, 12, 2359–2372 RSC.
- R. Sun, C. L. Yang, M. S. Wang and X. G. Ma, High solar-to-hydrogen efficiency photocatalytic hydrogen evolution reaction with the HfSe2/InSe heterostructure, J. Power Sources, 2022, 547, 232008 CrossRef CAS.
- J. Greeley, T. F. Jaramillo, J. Bonde, I. Chorkendorff and J. K. Nørskov, Computational high-throughput screening of electrocatalytic materials for hydrogen evolution, Nat. Mater., 2006, 5, 909–913 CrossRef CAS PubMed.
- Q. K. Yin, C. L. Yang, M. S. Wang and X. G. Ma, Two-dimensional heterostructures of AuSe/SnS for the photocatalytic hydrogen evolution reaction with a Z-scheme, J. Mater. Chem. C, 2021, 9, 12231 RSC.
- X. Q. Wan, C. L. Yang, X. H. Li, M. S. Wang and X. G. Ma, Insights into Photogenerated Carrier Dynamics and Overall Water Splitting of the CrS3/GeSe Heterostructure, J. Phys. Chem. Lett., 2023, 14, 9126–9135 CrossRef CAS PubMed.
- J. K. Nørskov, T. Bligaard, A. Logadottir, J. R. Kitchin, J. G. Chen and S. Pandelov, Trends in the Exchange Current for Hydrogen Evolution, J. Electrochem. Soc, 2005, 152, J23–J26 CrossRef.
- G. Gao and L.-W. Wang, Substantial potential effects on single atom catalysts for the oxygen evolution reaction simulated via a fixed potential method, J. Catal., 2020, 391, 530–538 CrossRef CAS.
- Y. Ji, Y. Li, H. Dong, L. Ding and Y. Li, Ruthenium single-atom catalysis for electrocatalytic nitrogen reduction unveiled by grand canonical density functional theory, J. Mater. Chem. A, 2020, 8, 20402–20407 RSC.
- G. Gao, A. P. O'Mullane and A. Du, 2D MXenes: a new family of promising catalysts for the hydrogen evolution reaction, ACS. Catal., 2017, 7, 494–500 CrossRef CAS.
- Y. Huang, R. J. Nielsen, W. A. Goddard and M. P. Soriaga, The Reaction Mechanism with Free Energy Barriers for Electrochemical Dihydrogen Evolution on MoS2, J. Am. Chem. Soc., 2015, 137, 6692–6698 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *abc,
Hong
Liu
abc,
Wei
Qi
bc,
Sichao
Du
bc,
Hao
Xie
abc and
Duo
Xiao
abc
*abc,
Hong
Liu
abc,
Wei
Qi
bc,
Sichao
Du
bc,
Hao
Xie
abc and
Duo
Xiao
abc