
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Tricolour luminescence in an Au(I) complex controlled by polymorphism and mechanical stress†
Andriani
Furoida
 ,
Misato
Daitani
,
Kohsuke
Matsumoto
,
Kyohei
Hisano‡
and
Osamu
Tsutsumi
*
,
Misato
Daitani
,
Kohsuke
Matsumoto
,
Kyohei
Hisano‡
and
Osamu
Tsutsumi
*
Department of Applied Chemistry, Ritsumeikan University, Kusatsu 525-8577, Japan. E-mail: tsutsumi@sk.ritsumei.ac.jp
First published on 14th May 2024
Abstract
The aggregation-induced emission (AIE) behaviour of gold(I) complexes has been extensively reported. In particular, Au complexes that exhibit colour tunability towards changes in aggregated structures are becoming increasingly attractive in the field of materials science. Herein, we report CP, a rod-shaped Au complex with a cyclohexylphenyl moiety and an isocyanide ligand. The photophysical behaviour of CP in various states is discussed based on their primary and aggregated structures. CP emitted room-temperature phosphorescence (RTP) with high-contrast polymorph-dependent colours of blue (CP-B), green (CP-G), and yellow (CP-Y) depending on the recrystallisation conditions. CP-Y, which has the shortest Au–Au distance, exhibits an efficient direct S0–Tn transition, leading to an enhanced RTP quantum yield (ΦRTP) of 38% under S0–Tn excitation. Interestingly, X-ray diffraction analysis revealed that CP-B and CP-G exhibit identical crystal structures despite their distinct emission colours. Photophysical and differential scanning calorimetry analyses revealed that the presence of small crystal domains, such as those in the ground samples of CP, is responsible for the overall green emission observed in bulk CP-G, indicating that the crystal structure and size determine the RTP colour. Moreover, CP also exhibits a liquid-crystalline (LC) nature and mechanochromic luminescence. Our findings demonstrate that the aggregated structure and AIE properties of Au complexes can be effectively controlled through crystalline-to-LC or crystalline-to-crystalline phase transitions of the complexes.
Introduction
Organic materials showing bright luminescence in solid states have aroused increasing attention due to their potential applications in various light-emitting technologies, such as cell imaging, chemo-sensors, biosensors, and organic light-emitting devices.1 However, the luminescence of typical organic chromophores is quenched in an aggregated state, posing a significant limitation for practical light-emitting applications where emission in aggregated states is imperative.2 Over the past two decades, the pursuit of molecules with aggregation-induced emission (AIE) properties, characterized by a rotor-like structure that renders them non-emissive in dilute solution but strongly emissive in aggregated or solid states, has emerged as a vibrant area of research.3The solid-state luminescence of organic materials is not only intricately linked to their molecular structure but also to their molecular packing and intermolecular interactions.4 Thus, deliberate modulation of aggregated structures is crucial for tuning the luminescence properties of AIE materials. Recently, there has been a growing interest in materials exhibiting distinct solid-state luminescence properties from a single chromophore, capitalising on polymorphic behaviour. Polymorphic materials encompass multiple crystalline phases, each exhibiting a unique emission colour.5 Notably, mechanochromic luminescence (MCL) has garnered considerable attention due to its broad applicability in sensors, probes, and memory devices. In such systems, mechanical forces (e.g., grinding and stretching) induce significant changes in photophysical properties, primarily attributable to changes in the aggregated structure.6 Another noteworthy approach to manipulate the aggregated structure is through the introduction of liquid-crystalline (LC) properties. The integration of LC properties into AIE materials imparts tunability to their luminescence properties through the control of molecular alignment via self-assembly and responsiveness to external fields.7
Gold(I) complexes offer an attractive avenue in this pursuit, offering not only efficient luminescence in aggregated states but also stimuli-responsive luminescence behaviour owing to distinctive Au–Au (aurophilic) interactions. Indeed, numerous studies have reported various Au complexes with tuneable luminescent colours in response to external stimuli (e.g., mechanical force, heat, and pressure).8 An Au complex featuring a linear coordination geometry, reported to exhibit LC properties, enables control of molecular alignment via external stimuli and luminescence properties through the alternation of Au–Au distances.9
The combination of multicolour emission in polymorphic materials with controllable molecular alignment in LC molecules provides organic materials with multifunctional applications. However, the development of luminescent materials exhibiting both polymorphism and LC behaviour remains rare.10 We have previously reported rod-like Au complexes bearing biphenylethynyl and isocyanide ligands that exhibit intense room-temperature phosphorescence (RTP) in aggregates. However, the observation of high-contrast luminescence coupled with polymorphism behavior and LC properties has proven elusive, likely due to the rigid structure of the biphenyl moiety.11 In this study, we designed and synthesised a novel Au complex, CP, featuring a cyclohexylphenyl moiety and isocyanide ligands, as shown in Fig. 1. Due to the non-rigid structure of the cyclohexylphenyl moiety, the complex exhibited both LC properties and crystalline polymorphism. Intriguingly, CP yielded three distinct crystal forms with distinctive RTP colours (blue, green, and yellow) under various recrystallisation conditions. Among these polymorphs, the crystal emitting yellow light, CP-Y, displayed a relatively high RTP quantum yield (Φ) of 38%—even in the presence of air. In addition, CP-B (blue-emitting crystal) and CP-G (green-emitting crystal) shared an identical aggregated structure despite their disparate luminescence behaviours. Upon mechanical grinding of all CP crystals, a small crystal domain emitting green light was obtained. Moreover, luminescence was also apparent in the LC phase at high temperature (>100 °C) and the luminescent colour change was induced by a crystalline-to-LC phase transition. Our results strongly suggest that the aggregated structure and luminescence behaviour of CP can be readily controlled via crystal-to-crystal and crystal-to-LC phase transitions.
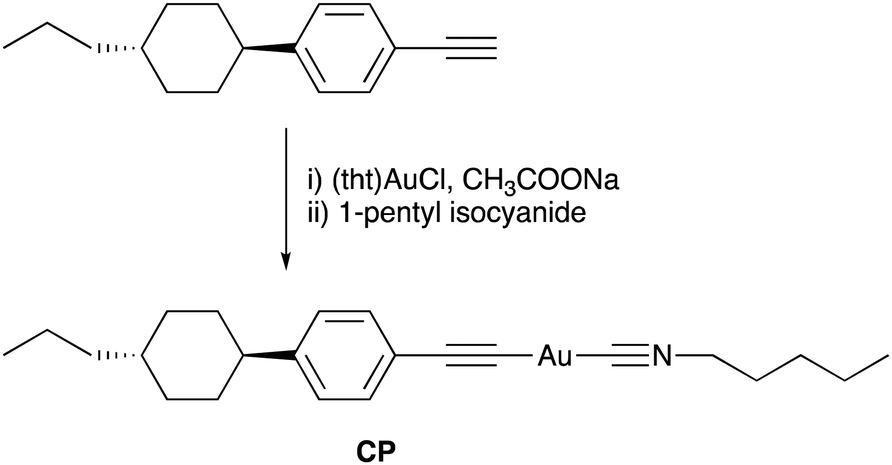 | ||
| Fig. 1 Synthesis and molecular structure of the CP Au complex. | ||
Results and discussion
Synthesis and characterization of the Au complex
The molecular structure of the Au complex used in this study is shown in Fig. 1. CP was prepared from commercially available 1-ethynyl-4-(trans-4-propylcyclohexyl)benzene in two facile steps: complexation with tetrahydrothiophene gold(I) chloride ((tht)AuCl), and subsequent ligand exchange with 1-pentyl isocyanide. The Au complex was successfully purified by silica gel column chromatography, followed by recrystallisation from a mixed solvent system of CH2Cl2 and hexane. The complex was characterised using 1H NMR, 13C NMR, infrared (IR) spectroscopy, high-resolution mass spectroscopy (HRMS), and elemental analysis. All the analytical data (provided in the Experimental section) confirmed that the desired Au complex was obtained in high purity.The thermal stability of the Au complex was evaluated using thermogravimetric/differential thermal analysis (TG–DTA) at a heating rate of 5 °C min−1 in the range of 25–600 °C. The thermal decomposition temperature (Tdec) is defined as the temperature at which 5% weight loss occurs. As shown in Fig. S4 (ESI†), CP decomposed at approximately 186 °C. According to our previous study on the Au isocyanide complex, decomposition is initiated by the disconnection of the isocyanide ligand.11 The calculated weight percentage of isocyanide ligands in the total weight of CP is 19%. Thus, the first weight loss of 18% is attributed to C–Au bond cleavage between the Au atom and the isocyanide ligand. The 37% residual ash corresponded to the calculated percentage of Au (37.9%), suggesting that the residue remaining after heating was in the form of Au ash. This result is also consistent with the ash percentage obtained from the elemental analysis of CP (see the Experimental section).
Photophysical properties in solution
The photophysical properties of CP were investigated in dilute solutions (Fig. 2). An absorption band at 288 nm with a molar extinction coefficient (ε) of 9.4 × 104 L mol−1 cm−1 was observed in CH2Cl2 solution (1 × 10−5 mol L−1), which is attributable to the ligand-base π–π* transition (Fig. 2, black line).12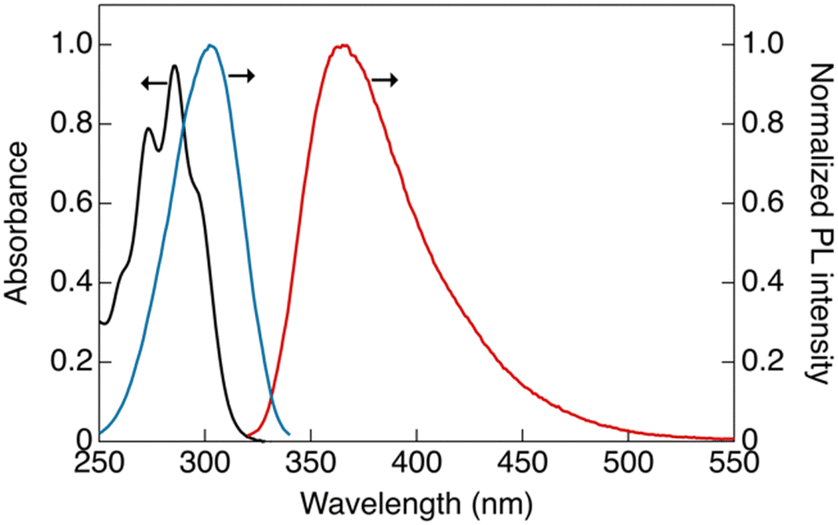 | ||
| Fig. 2 Absorption and corrected photoluminescence spectra of CP in CH2Cl2 solution: black, absorption spectra ([CP] = 1 × 10−5 mol L−1); red, luminescence spectra ([CP] = 1.5 × 10−6 mol L−1, λex = 302 nm); and blue, excitation spectra ([CP] = 1.5 × 10−6 mol L−1, λem = 365 nm). | ||
The dilute solution of CP ([CP] = 1.5 × 10−6 mol L−1) showed a single emission band at 365 nm (band I) under ambient conditions. In the excitation spectrum (Fig. 2, blue line), an excitation band was observed at wavelengths longer than those in the absorption spectrum. This marginal red-shift observed in the excitation band can be attributed to the formation of aggregates in the solution that are likely to be more emissive than the monomeric form of CP.11 The luminescence decay at band I in the dilute solution was within the nanosecond region, indicating that the luminescence at this band is fluorescence (Fig. S9a, ESI†).
Similar luminescence was also obtained in a concentrated THF solution ([CP] = 1 × 10−4 mol L−1). Upon the addition of water as a poor solvent to the THF solution, a gradual reduction in the luminescence intensity at 365 nm was observed until the volume fraction of water in the mixed solution (fw) reached 60% (Fig. 3). Beyond an fw value of 60%, two distinctive emission bands emerged abruptly at 424 (band II) and 500 nm (band III), indicating the formation of aggregates. Consequently, the overall luminescence intensity of CP in the mixed solvent increased significantly (Fig. 3c), confirming the AIE-active nature of the synthesised Au complex. The reduction in the total luminescence intensity in the fw > 80% range is not indicative of the ACQ effect, but rather arises from precipitation due to the formation of relatively large crystals.
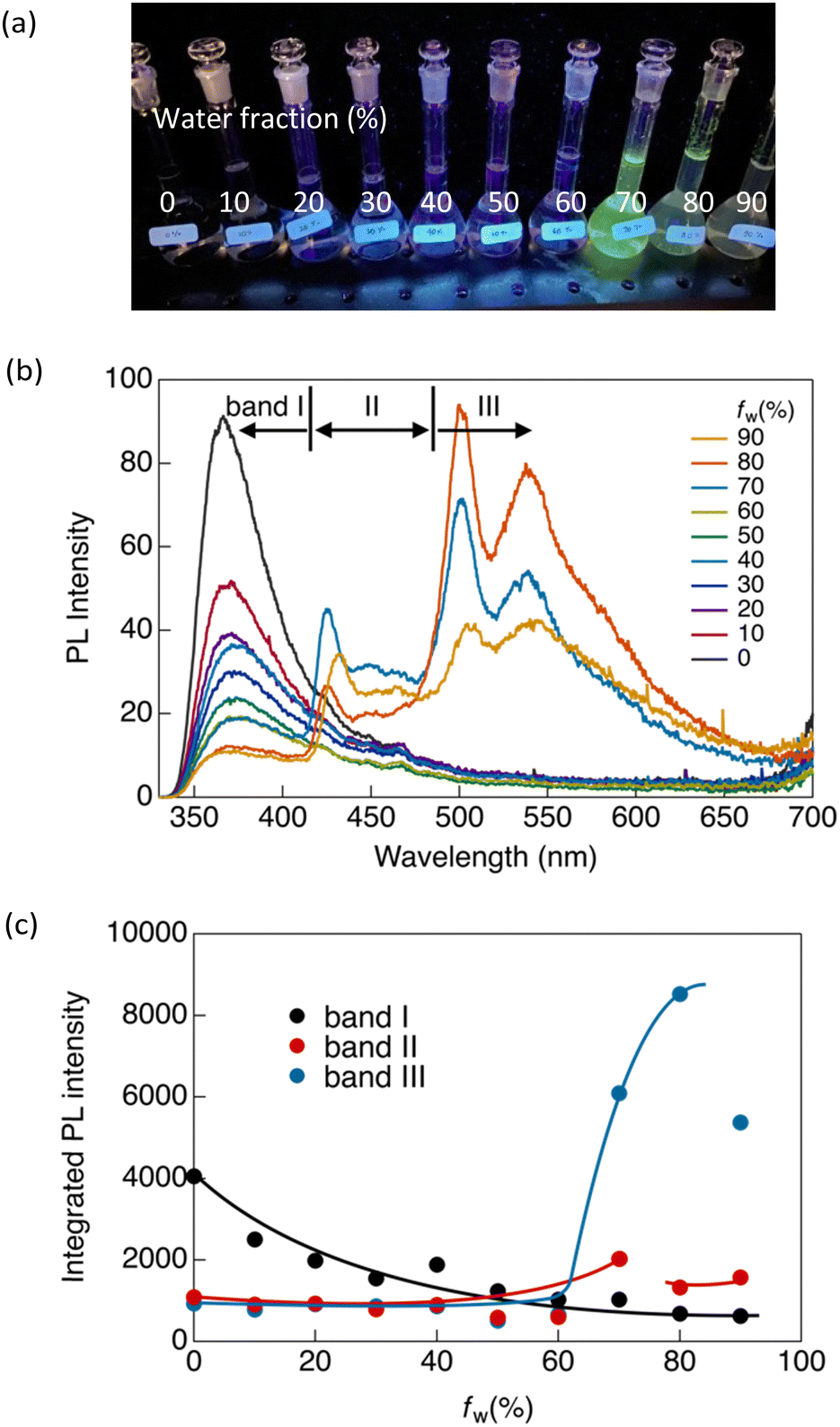 | ||
| Fig. 3 (a) Photograph of CP in a THF/water mixed solvent (fw = 0–90%) under UV irradiation at 365 nm. (b) Emission spectra of CP in fw = 0–90% ([CP] = 1 × 10−4 mol L−1, λex = 300 nm). (c) Integrated photoluminescence intensity in different wavelength ranges: band I, 330–420 nm; band II, 420–480 nm; and band III, 480–700 nm. | ||
To investigate the emission origins of bands II and III, we initially observed the luminescence in a dilute THF/water solution ([CP] = 1.5 × 10−6 mol L−1) under an Ar atmosphere (Fig. S8a, ESI†). At fw = 0%, which initially showed only band I, a pronounced shift towards band II was observed under Ar, indicating that band II corresponded to monomeric phosphorescence. In contrast, a weak band III appeared at fw = 80% under Ar, indicating that the phosphorescence originated from the aggregates.
Luminescence lifetime (τ) measurements in the solution under Ar also supported the emission origin of these three bands. As shown in Fig. S9 and Table S3 (ESI†), bands I and II exhibited nanosecond-scale lifetimes in ambient air. Under Ar, the emission lifetime of band II was significantly increased to the microsecond scale, whereas no significant change was observed in the lifetime of band I. This provides evidence that the observed luminescence at band I can be attributed to fluorescence, and that band II corresponds to phosphorescence. Additionally, as shown in Fig. S9a (ESI†), a rise in the fluorescence intensity of band I was observed during the lifetime measurement. This suggests that the fluorescence of band I originates from an excimer.
In contrast, band III showed biexponential decay with a microsecond-scale lifetime in both air and Ar atmospheres (Fig. S10b and Table S3, ESI†). Because the emission from band III arose from the aggregates, this band was negligible in dilute solutions. We also observed luminescence in a more concentrated solution ([CP] = 1 × 10−5 mol L−1) (Fig. S8b, ESI†). Similar results to those obtained in the dilute solution (10−6 mol L−1) were observed at fw = 0%. However, unlike the dilute solution, at fw = 80%, band III is clearly visible even in the presence of air. This indicated that CP formed more aggregates at high concentrations. This aggregated emission was further amplified and became predominant at fw = 80% under an inert atmosphere, accompanied by an extended lifetime on the microsecond scale (Fig. S10b and Table S3, ESI†). This indicates that band III corresponds to phosphorescence originating from aggregates, and therefore, that CP exhibits aggregation-induced phosphorescence.
In our previous study, we demonstrated that the stability of the crystal structures in certain gold(I) complexes depends on crystal size, with crystal-to-crystal phase transitions occurring at critical sizes during crystal growth.13 The present study on CP reveals phenomena similar to those of the previously reported system, in which a crystal-to-crystal phase transition occurs during the crystal growth process in a mixed solvent. Initially, crystal nuclei, corresponding to band II, are formed upon the addition of water. At a higher water fraction (fw > 60%), crystals grew to the critical size, triggering a crystal-to-crystal phase transition and giving rise to band III. We attribute bands II and III to crystals of different sizes, and their formation strongly depends on water content. As shown in Fig. 3c, the ratio of the emission intensities of bands II and III changes as the water content increases. Bands II and III emerged simultaneously at fw > 60%. The intensity of band II peaked at fw = 70% and gradually decreased above this water fraction, whereas the relative emission intensity of band III decreased significantly after fw > 80%. This implies a decrease in the number of small crystal nuclei (associated with the origin of band II) due to the formation of larger crystals (associated with the origin of band III) during the crystal growth process. Larger crystals (band III) predominate at higher water contents; however, they partially precipitate after fw > 80%. This observation aligns with our previous research, where the crystal structures of Au complexes as well as their luminescence properties were contingent on crystal size.13
Luminescence properties of the complex in bulk crystals
CP exhibit intriguing luminescence properties in bulk crystals. By controlling the recrystallisation conditions, three types of plate-like crystals exhibiting different luminescent colours (blue, green, and yellow) were successfully obtained. Rapid and slow evaporation of a solvent mixture of CH2Cl2 and hexane (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 v/v) afforded blue-emitting CP-B and green-emitting CP-G, respectively. CP-Y, which emits yellow light, was obtained via slow recrystallisation from a mixture of CH2Cl2 and ethyl acetate (1:3 v/v). Recrystallisation from a mixture of CH2Cl2 and hexane (2:1 v/v) afforded a mixture of CP-Y and CP-B crystals. As shown in Fig. 4, CP-B and CP-G exhibited vibronic emission bands, with maxima at 423 and 500 nm, respectively, while CP-Y showed an emission band peaked at 560 nm without a vibronic structure. In comparison to luminescence in solution, the solid-state emissions were red-shifted, suggesting that the luminescence arises from lower-energy triplet excited states.14 Similar emission bands at 423 and ∼500 nm were also observed in the degassed THF/water solution, further substantiating that these emissions correspond to phosphorescence.
:3 v/v) afforded blue-emitting CP-B and green-emitting CP-G, respectively. CP-Y, which emits yellow light, was obtained via slow recrystallisation from a mixture of CH2Cl2 and ethyl acetate (1:3 v/v). Recrystallisation from a mixture of CH2Cl2 and hexane (2:1 v/v) afforded a mixture of CP-Y and CP-B crystals. As shown in Fig. 4, CP-B and CP-G exhibited vibronic emission bands, with maxima at 423 and 500 nm, respectively, while CP-Y showed an emission band peaked at 560 nm without a vibronic structure. In comparison to luminescence in solution, the solid-state emissions were red-shifted, suggesting that the luminescence arises from lower-energy triplet excited states.14 Similar emission bands at 423 and ∼500 nm were also observed in the degassed THF/water solution, further substantiating that these emissions correspond to phosphorescence.
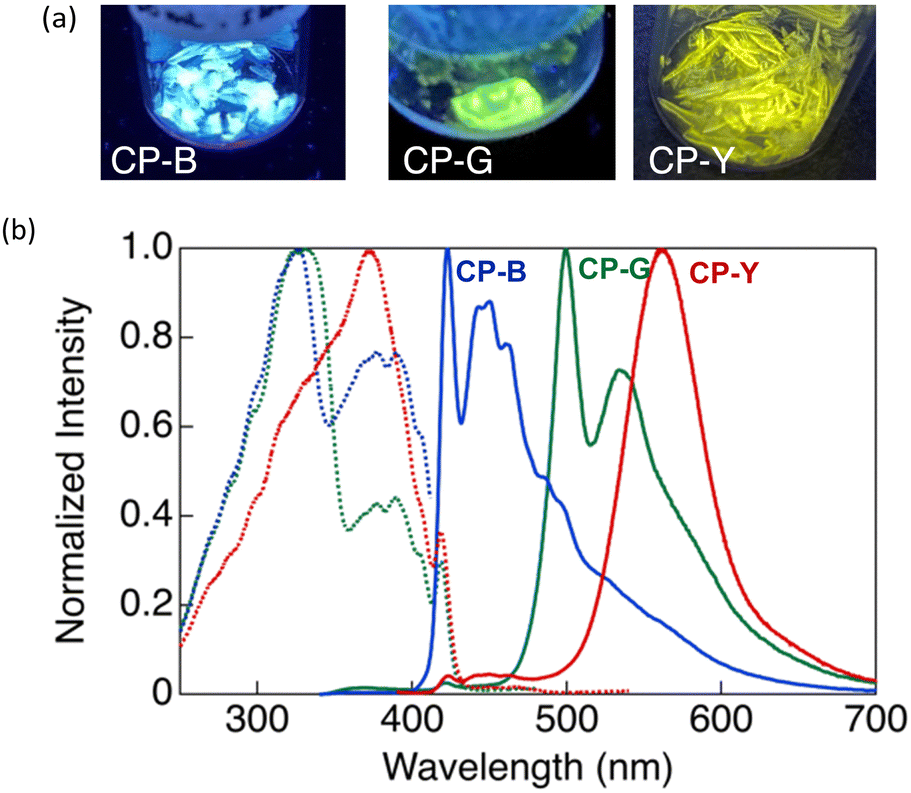 | ||
| Fig. 4 (a) Photographs of crystal polymorphs of CP-B, CP-G, and CP-Y under UV irradiation at 365 nm. (b) Excitation (dashed lines) and emission (solid lines) spectra of the CP crystal polymorphs: CP-B (blue, λex = 320 nm for excitation, λem = 423 nm for emission); CP-G (green, λex = 320 nm, λem = 500 nm); and CP-Y (red, λex = 320 nm, λem = 560 nm). | ||
In our previous study, we investigated the luminescence behaviour of Au complexes with similar molecular structures.9a,15 What distinguishes CP from the previously reported complex is the structure of the acetylide ligand: CP utilises a cyclohexylphenylacetylide ligand instead of a phenylacetylide ligand. Interestingly, the phenylacetylide Au complex did not exhibit any crystalline polymorphism and emitted blue light, similar to CP-B in its crystalline phase. The introduction of a cyclohexyl group in CP played a crucial role in inducing crystal polymorphism and a diverse range of luminescent colours. Our findings suggest the capability of tuning the emission wavelength across a wide range of the visible spectrum, from blue to yellow, in Au complex systems without extending the π-conjugation system.
As shown in Fig. 4, CP-B and CP-G exhibited similar excitation spectra, with a primary excitation band at approximately 320 nm, which was also observed in the absorption spectra of the solutions and is attributable to the S0–Sn transition.15 An additional excitation band was observed at longer wavelengths (370–420 nm). We have previously reported that this excitation band is due to an S0–Tn direct transition that is strictly spin-forbidden.15 However, in this type of rod-like Au complex, an extremely efficient S0–Tn direct transition occurs due not only to the heavy atom effect but also to Au–Au interactions formed by the aggregation of the Au complex.15 The excitation spectra of CP-Y are distinct compared to other polymorphs, with the maxima at 373 nm and a shoulder peak at ∼320 nm. Among the three polymorphs, CP-Y showed an efficient S0–Tn transition, where the luminescence quantum yields (Φ) increased from 33% when excited at 320 nm (S0–Sn transition) to 38% upon excitation at 373 nm (S0–Tn transition) (Fig. 5 and Table 1). This result is in accordance with our previous work wherein direct excitation to the excited triplet state enhanced the Φ value for RTP.15 This RTP enhancement was not observed in CP-B and CP-G (Fig. S11, ESI†), indicating that the S0–Tn transition was less efficient for these polymorphs.
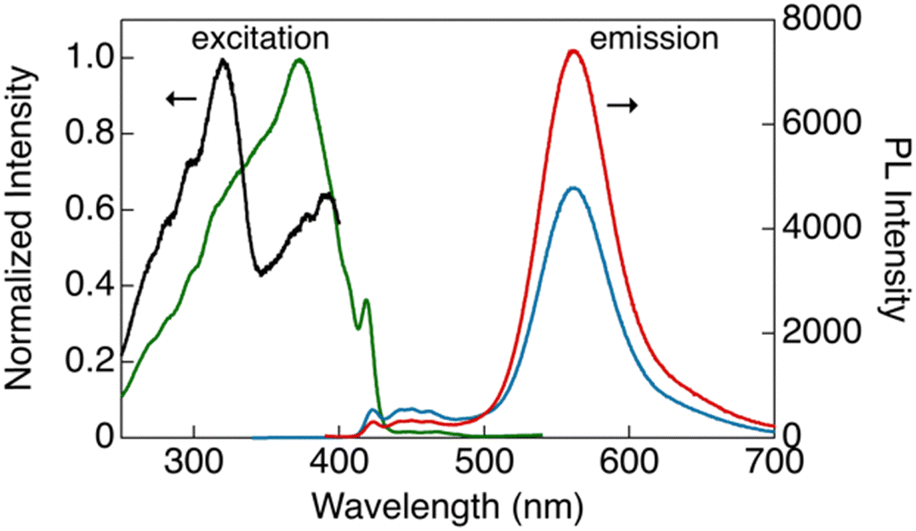 | ||
| Fig. 5 Excitation (black, λem = 423 nm; green, λem = 560 nm) and emission (blue, λex = 320 nm; red: λex = 373 nm) spectra of CP-Y in bulk crystals at room temperature. | ||
| λ lummax (nm) | Φ | τ (μs) | k r (s−1) | k nr (s−1) | |
|---|---|---|---|---|---|
| a Abbreviations: λlummax, luminescence maxima; Φ, luminescence quantum yield; τ, luminescence lifetime; kr, rate constant for radiative transition; knr, rate constant for non-radiative transition. b λ ex = 320 nm for all measurements. c k r was estimated as Φ/τ and knr was estimated as (1 − Φ)/τ; the weighted average of τ was used to estimate kr and knr. d Observed by direct S0–Tn excitation at 373 nm. | |||||
| CP-B | 423 | 0.10 | 11 (50%) | 5.2 × 103 | 4.7 × 104 |
| 27 (50%) | |||||
| CP-G | 500 | 0.23 | 4 (52%) | 1.2 × 103 | 4.0 × 103 |
| 389 (48%) | |||||
| CP-Y | 560 | 0.33 | 13 | 2.9 × 104 | 4.7 × 104 |
| 0.38d | |||||
| Ground | 500 | 5 × 10−3 | 1 (86%) | 1.6 × 103 | 3.5 × 105 |
| 16 (14%) | |||||
To gain a better understanding of the photophysical properties of CP, we investigated the photoluminescence τ and Φ of bulk crystal polymorphs (Table 1 and Fig. S12, ESI†). The three polymorphs exhibit biexponential decay profiles with lifetimes on the order of microseconds, indicating that the luminescence from the crystals is phosphorescence. Despite displaying phosphorescence, the CP polymorphs exhibited considerably good Φ values of up to 38% at room temperature. Generally, it is difficult to demonstrate phosphorescence emission with high Φ under ambient conditions because the triplet excited states tend to be deactivated by molecular oxygen and the thermal motions of the molecules. The strong RTP of the crystal is attributed not only to heavy atom effects but also to the crystallisation-induced phosphorescence due to the densely packed crystal which prevented the penetration of molecular oxygen from quenching the triplet excited states.16 Furthermore, based on the Φ and τ values, the rate constants for radiative transition (kr) and non-radiative deactivation (knr) of the complexes in the crystal were estimated. As summarised in Table 1, the large kr value observed for CP-Y is responsible for its highest Φ value among the studied polymorphs. CP-B and CP-G have similar kr values, and their Φ depends on knr.
Furthermore, we considered whether the luminescence observed in the crystal was thermally activated delayed fluorescence (TADF). We conducted temperature-dependent photoluminescence analyses of the crystal polymorphs (Fig. S13–S15, ESI†). All polymorphs exhibited obvious RTP behaviour, where the photoluminescence intensity gradually increased with decreasing temperature; the emission maxima at 77 K were approximately twice those at 290 K. Thus, the occurrence of TADF can be excluded from the crystal emission. In addition, the emission enhancement upon direct S0–Tn excitation, which was not observed for CP-B or CP-G at room temperature, was observed at 77 K. The emission intensities of CP-B and CP-G under direct S0–Tn excitation (373 nm) at 77 K were twice that under S0–Sn excitation (320 nm). The triplet state was stabilised at low temperatures by a reduction in molecular motion, leading to an efficient S0–Tn transition.17 Notably, the CP-G crystal displayed enhanced emission upon being warmed back to room temperature (298 K) from 77 K under direct S0–Tn excitation at 373 nm, a phenomenon not previously observed during cooling and subsequent heating cycles. This suggests that the cooling to 77 K induced morphological or structural changes in CP-G that persisted upon rewarming to 298 K, thereby enabling efficient direct S0–Tn excitation at room temperature.
The crystal polymorphs were subjected to single-crystal X-ray diffraction (SCXRD) analysis at room temperature to clarify their structures. Typically, polymorphic materials with distinct luminescence properties have different aggregated structures.5 All CP polymorphs crystallized in the triclinic P-1 space group with an anti-parallel orientation, and their Au–Au distances were within the range of 3.7 and 4.0 Å (Fig. 6a). Interestingly, CP-B and CP-G share nearly identical crystal structures, with similar intermolecular interactions, including an Au–Au distance of approximately 4.0 Å. The occurrence of such polymorphic behaviour is rare, and we discuss this intriguing phenomenon further in the following section. CP-Y possess a distinct crystal structure with a shorter Au–Au distance than the other polymorphs (∼3.7 Å), which explains the observed difference in emission colour. As previously reported, an Au isocyanide complex with a short Au–Au distance (∼3.5 Å) exhibited RTP enhancement upon excitation at the S0–Tn region.15 Similarly, the densely packed structure of CP-Y enables the efficient stabilisation of triplet excitations and S0–Tn transition compared to the other polymorphs, leading to enhanced emission upon excitation in the S0–Tn region. In addition, the CH–Au interaction is likely contributed to the crystal packing structure as the distance between the Au and H atom of the phenyl group is short (∼2.9 Å; Fig. S3, ESI†).18
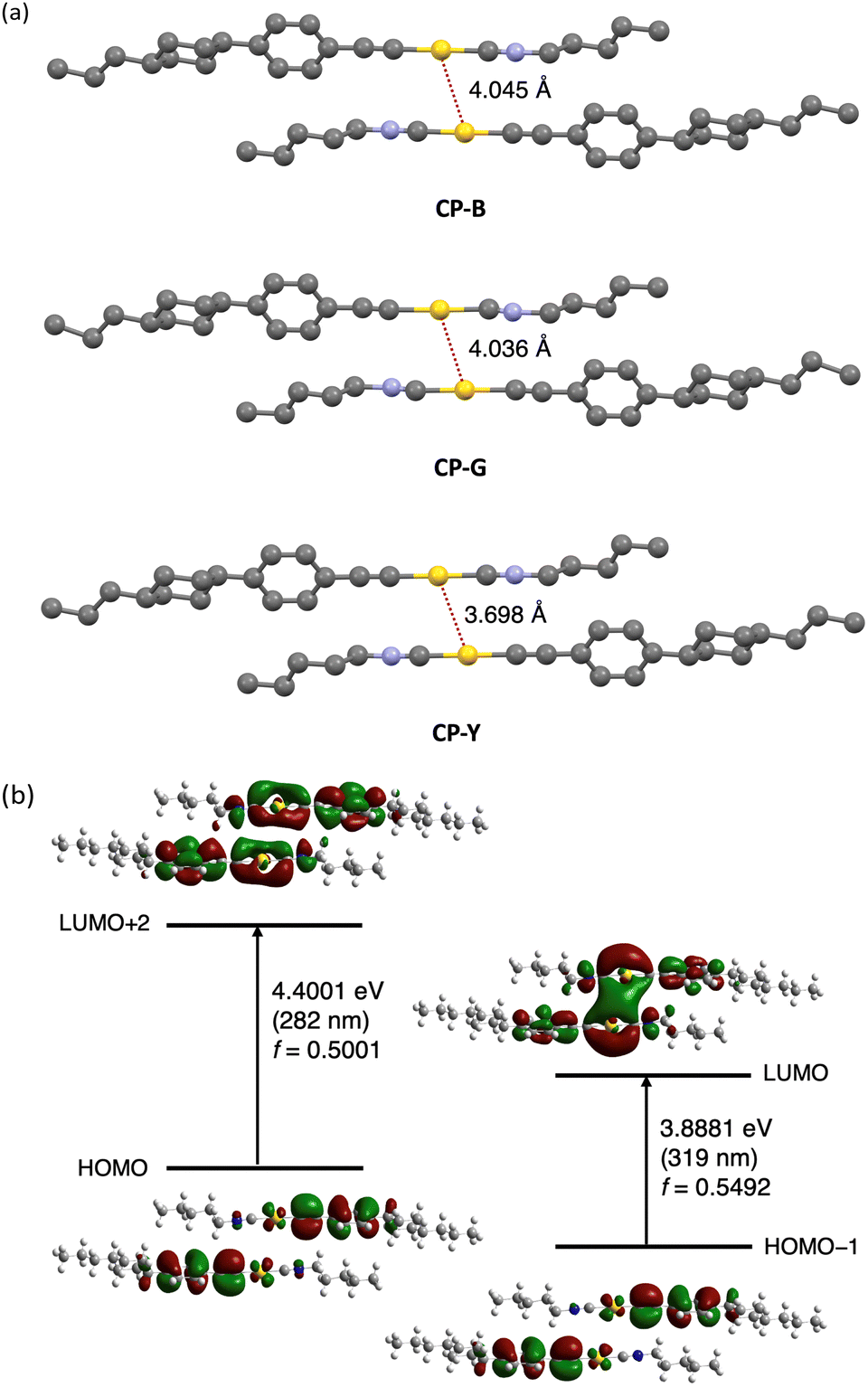 | ||
| Fig. 6 (a) Single-crystal structure of CP polymorphs. Intermolecular Au–Au interactions are indicated with a dashed red line. Hydrogen atoms are omitted for clarity. Atom colour legend: grey, C; blue, N; yellow, Au. (b) Molecular orbitals of the CP-B dimer (extracted from the crystal structure) obtained from DFT calculation using B3LYP and 6-311+G(d,p), except for Au atoms, for which the SDD basis set was employed. | ||
To study the electronic transitions in the present complex, we conducted a computational study using time-dependent density functional theory (TD-DFT) calculations. The B3LYP hybrid functional with the SDD and 6-311+G(d,p) basis sets for the Au and other atoms, respectively, was employed. Calculations were performed for the dimer of the CP polymorphs, whose structures were determined by SCXRD. The oscillator strength (f) distribution was calculated for the transition to the singlet excited state (Table S5, ESI†).
We selected molecular orbitals with large f values corresponding to the allowed electronic transitions from the singlet excited states (Fig. 6b for CP-B and Fig. S17–S18 for CP-G and CP-Y, ESI†). In Fig. 6b, the excitation at 319 nm in CP-B was attributed to the S0–S1 transition with a large f value of 0.5492. The electron density distribution from HOMO−1 to LUMO reveals that this electronic transition arises from the π orbital of the phenylethynyl ligand to the Au–Au atoms in neighbouring molecules. Consequently, it was assigned to the LMMCT transition, indicating the presence of aurophilic interactions within the crystal structure despite the Au–Au distances slightly exceeding the theoretical range, in accordance with our previous finding.11 The excitation at 282 nm, attributed to S0–Sn, exhibited a high f value of 0.5001. According to the electron density distribution from the HOMO to LUMO+2, this transition arises from the π orbital of the phenylethynyl ligand to the Au atom within a single molecule. Thus, this transition was assigned as a ligand-to-metal charge transfer (LMCT) transition within the monomeric unit. Similar outcomes have been observed for other CP polymorphs, and these calculation results are closely aligned with the excitation observed in the crystal. In addition, the excitation energies of the triplet excited states were calculated from the same dimers, indicating that the S0–T1 transitions are located at 409, 372, and 412 nm for CP-B, CP-G, and CP-Y, respectively (Table S6, ESI†). The calculated excitation energy for the S0–Tn transition was consistent with the longer excitation wavelength (370–420 nm) observed in the excitation spectra (Fig. 4b) for the CP crystal polymorphs. In the case of the monomer, the S0–Sn transitions are located at 274 and 347 nm (Table S7, ESI†), which aligns with the absorption spectra of the complex in dilute solution (Fig. 2). Furthermore, the calculations suggested that an intramolecular LMCT transition occurred in the monomer (Fig. S19, ESI†).
Mechanochromic responsive properties
Given the observed correlation between the emission colour of CP and its crystal size during the crystal growth process, we investigated the luminescence behaviour of microsized CP crystals by applying mechanical stimuli (Fig. 7). SEM images of the ground crystals are shown in Fig. S21 (ESI†). Strong grinding of CP-B and CP-Y bulk crystals to microsized crystals induced a substantial emission colour change to green and bluish green, respectively. In the photoluminescence spectra, an obvious red-shift of the emission maxima from 423 to 500 nm was observed for ground CP-B (Fig. S20a, ESI†). In contrast, a blue-shifted emission from 560 to 469 nm was observed in ground CP-Y (Fig. S20c, ESI†). Although the emission maxima were different, ground CP-B and CP-Y exhibited similar broad low-intensity luminescence ranging from 350 to 700 nm. This emission colour change is typical of MCL behaviour and may be attributed to the alteration of the crystal arrangement upon mechanical grinding. Conversely, CP-G did not exhibit prominent MCL, and the emission colour remained green upon grinding. The luminescence intensity of CP-G decreased slightly, whereas the spectral shape was retained relative to that of the bulk crystals (Fig. S20b, ESI†). Despite the absence of emission spectral changes in ground CP-G, its excitation spectrum was identical to those of ground CP-B and CP-Y, suggesting that the luminescence originated from the same excited state (Fig. S20d, ESI†). As summarised in Table 1, the Φ of ground crystals decreased to ∼0.5%, which is significantly lower compared to that in bulk crystals. Moreover, the τ value of the ground sample became shorter. This means that the RTP efficiency of the crystal is significantly quenched upon mechanical grinding; that is, the destruction of the crystal structure upon mechanical grinding promotes nonradiative deactivation, which is in good agreement with the higher knr value of the ground crystals.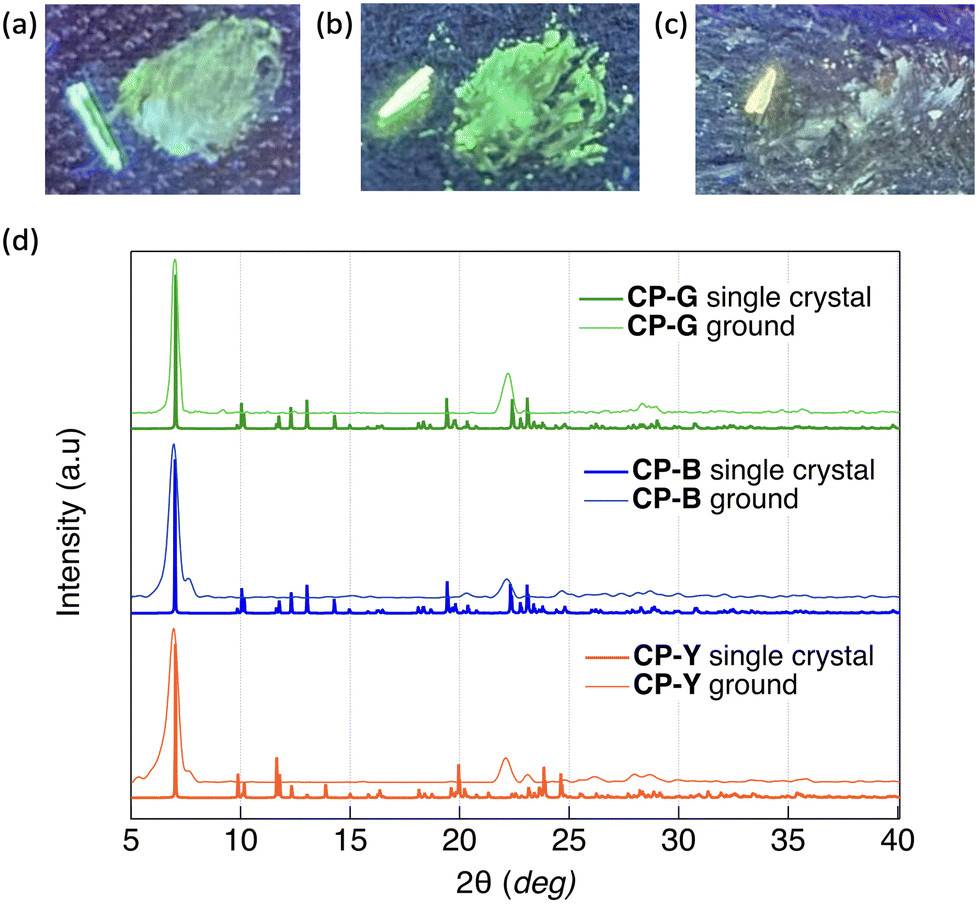 | ||
| Fig. 7 Photographs of (a) CP-B, (b) CP-G, and (c) CP-Y before (left) and after (right) mechanical grinding under 254 nm UV irradiation. (d) Experimental powder XRD patterns of ground crystals (thin lines) and XRD patterns of bulk crystals simulated from the SCXRD structures (bold lines). | ||
The powder X-ray diffraction (PXRD) patterns of the CP polymorphs after mechanical grinding and corresponding patterns simulated from the SCXRD structures are shown in Fig. 7d. The crystal size of the ground samples was determined using the Scherrer equation. Both ground CP-B and CP-Y exhibited a crystal size of 15 nm, while the crystal size of ground CP-G was slightly larger at 26 nm. Upon grinding, CP-B and CP-Y exhibited a change in crystalline structure, which explains the origin of the MCL behaviour. Similarly, the mechanical grinding of CP-G also induced a crystalline structure, even though the emission colour barely changed. Upon comparison of the PXRD patterns of the ground samples, it is evident that both ground CP-B and CP-Y exhibit similar PXRD patterns, which differ from that of ground CP-G. Notably, ground CP-B and CP-Y both display an additional peak close to the main peak at 12.6 Å, along with a wider angle peak relative to ground CP-G. These findings suggest that grinding CP bulk crystals may lead to the formation of new crystal phases with different structures. However, it is noteworthy that despite these structural differences, the comparable Au–Au distances suggest similar luminescence profiles in all ground samples.
We indexed the PXRD patterns of bulk CP-B and CP-G based on the SCXRD data to identify whether there were subtle differences in the crystal structure that could contribute to the differences in emission colours (Fig. S16, ESI†). However, we found similar d(hkl) values for both polymorphs, indicating that the structures were nearly identical and that the difference in emission colour was not caused by the different aggregated structures. Considering the presence of green-emitting CPground with different crystal structures, it is possible that green-emitting CP-G contains small crystal domains of the CPground polymorph, which has a different emission colour than CP-B. Consequently, energy transfer from the main crystal lattice (CP-B) to the small-crystal-domain CPground leads to an overall green emission in CP-G, even though it has the same crystal structure as CP-B. Theoretically, in a Förster resonant energy transfer (FRET) system, the donor moiety absorbs incident light and transfers the energy to a smaller quantity of emitter luminophores.19 This is in accordance with the observed luminescence spectrum in CP-G, where the excitation spectrum mainly consists of the donor characteristics of CP-B as the main crystal lattice at ∼320 nm, whereas the emission spectrum arises from CPground as an emitter at ∼500 nm, resulting in an increased Stokes shift of the overall spectrum. A similar phenomenon in which a small crystal domain caused a change in overall crystal emission has been reported previously in a mechanochromic Au complex20 and in a doped organic crystal.21 The biexponential lifetime observed in CP-G can provide evidence for the presence of a CPground crystal domain within the bulk CP-G crystal. The shorter lifetime of 4 μs is likely attributable to the CPground crystal domain, while the longer lifetime of 389 μs is attributed to FRET from the bulk CP-G crystal. It is generally known that the observed Φ of a FRET system depends largely on the Φ of the emitters.22 The reason is that CP-G exhibits higher Φ (23%) than CP-B (10%) as a donor moiety might be due to the highly emissive CPground crystal domain which acts as emitter in CP-G. The low quantum yield of CPground obtained through mechanical grinding is attributed to poor crystal quality, such as disorder, distortion, and inclination, which promote nonradiative deactivation. In contrast, CPground within the bulk CP-G crystal formed under slow recrystallisation and exhibits more efficient luminescence, likely because the densely packed crystal structure prevented oxygen penetration and emission quenching. The formation of a small crystal domain (CPground) was also observed in THF/water mixed solvents with high water content, identified as band III in Fig. 3. Presently, the crystal structure of CPground remains unclear; however, it is postulated that aurophilic interactions with shorter Au–Au distances, compared to those present in the main CP-B crystal, are involved. These interactions are believed to contribute to the higher RTP quantum yields observed for CP-G.
Thermal phase transition behaviour
The thermal phase transition behaviour of CP was observed by differential scanning calorimetry (DSC) and polarised optical microscopy (POM). The DSC thermograms of all the bulk crystal polymorphs (Fig. S5, ESI†) exhibited small endothermic peaks at approximately 40 °C during the first heating process. However, the optical texture of the crystal was retained at this temperature, as observed using POM. Therefore, it can be concluded that the phase transition at 40 °C corresponds to a crystal-to-crystal phase transition, which also support the polymorphism behaviour observed in the bulk crystal. At high temperatures, CP exhibited an enantiotropic nematic (N) LC phase, as indicated by intense exothermic and endothermic peaks appearing during both heating and cooling. A nematic LC phase was assigned based on the optical texture observed by POM, where a schlieren texture was observed at approximately 122 °C (Fig. 8b). This N phase was retained until the Tdec of the complex. In general, the molecular skeleton of LCs contains a rigid core and flexible alkyl or alkoxy chains. The less rigid structure of the cyclohexyl group is likely to act as a flexible unit to aid the LC phase formation. Upon cooling from the molten states of the bulk crystals, an N-to-crystal phase transition occurs without any observed subsequent crystal-to-crystal phase transitions. This indicates that the original crystal (Cr1) is metastable, and does not recover after its transition to the N phase.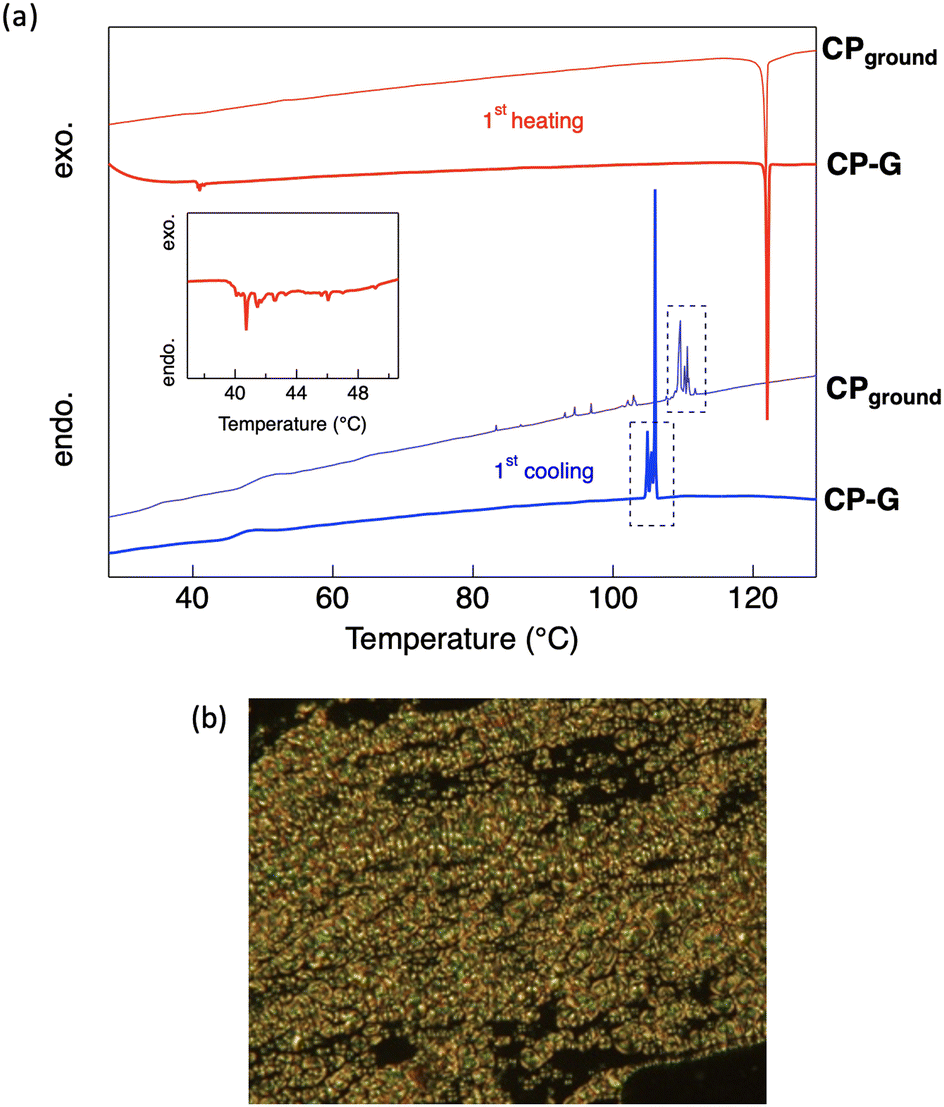 | ||
| Fig. 8 (a) DSC thermograms of CP-G and CPground during the first scan and (b) POM image of CP showing the N phase, observed at 125 °C. The red and blue curves indicate the heating and cooling cycles, respectively. The dashed rectangles represent the region where the peaks are similar between CP-G and CPground. | ||
During the first cooling process in CP-G, multiple exothermic peaks were observed following the N-to-crystal phase transition, which was not seen in other CP polymorphs. Following our previous hypothesis that CP-G bulk crystal contains small crystal domains similar to CPground, we also investigated the phase transition of CPground (Fig. S6, ESI†). Unlike the bulk crystals, the crystal-to-crystal phase transition was not observed in CPground during the first heating, indicating that the original state of CPground is more stable, similar to the Cr2 phase observed in CP crystals. In addition, cooling the molten CPground sample resulted in multiple unidentified crystal-to-crystal phase transitions (Fig. 8a), resembling those observed in bulk CP-G. Similarly, during the third scan, the phase transition of CPground closely matched that of bulk CP-G (Fig. S5b and S6, ESI†), namely, a broad endothermic peak at approximately 120 °C and exothermic peaks at ∼100 °C. These support the presence of CPground within bulk CP-G. The appearance of multiple peaks in CPground after subsequent scans is attributed to crystal defects. These defects or changes in molecular orientation provide nucleation sites for the formation of different crystal phases, resulting in multiple crystal-to-crystal phase transitions during cooling.23
To study the effect of the phase transition on the luminescence behaviour, we investigated the photoluminescence spectra of CP polymorphs in the LC phase (Fig. S22–S29, ESI†). All CP polymorphs showed a decrease in the absolute luminescence intensity upon heating due to the thermal deactivation of excited states. Temperature-dependent luminescence intensity plots for CP-B, CP-G, and CP-Y, are shown in Fig. S24, S27, and S29 (ESI†), respectively. Initially, all polymorphs show a gradual decrease in emission intensity, which was rapidly quenched after reaching the crystal-to-crystal phase transition temperature at 40 °C. Subsequently, the emission intensity continued to decrease until the LC phase temperature was reached. The luminescence spectral shape altered upon the crystal-to-crystal phase transition: for CP-B, the broad shoulder peak at 500–600 nm disappeared above 40 °C (Fig. S22, ESI†); for CP-Y, the emission at 560 nm rapidly quenched while the emission at 424 nm persisted, resulting in a pronounced blue shift in the emission maxima (Fig. S28a, ESI†). These results indicate that the longer wavelength emission band originated from the original Cr1 phase, and the crystal-to-crystal phase transition altered the luminescence. In contrast, CP-G exhibited no significant change in its emission spectral shape upon undergoing the crystal-to-crystal phase transition. Instead, heating to the N phase temperature led to a broadening emission band, where the ratio between shorter-wavelength emission (∼420 nm) and the main emission at 500 nm became nearly equal (Fig. S25, ESI†). It is noteworthy that in CP-Y, the emission at 560 nm remained observable at 50 °C despite this temperature corresponding to the Cr2 phase. Prolonging the holding time at 50 °C to approximately 20 min led to a reduction in the relative emission intensity at 560 nm (Fig. S28c, ESI†). This implies that some crystals did not immediately transition from Cr1 to Cr2 at 50 °C, which is considered to also be applicable to other CP polymorphs. Notably, the difference in the luminescence spectrum of CP-Y at room temperature in Fig. S28 (ESI†) compared to Fig. 5 is attributed to the lower quality of the crystal used in the temperature-dependent luminescence measurement. Even in high-quality crystals, CP-Y exhibited a shorter wavelength peak at 423 nm under excitation at 320 nm (Fig. 5). In high-quality crystals, this shorter wavelength peak is significantly lower compared to the main emission at 560 nm.
After cooling the molten sample to room temperature, all polymorphs retained only 25% of their luminescence intensity, which is attributed to less-ordered crystals. Moreover, in both CP-B and CP-Y the emission maxima of the frozen crystal (Cr2) were red-shifted by 39 nm, and the shoulder peak at longer wavelengths was recovered (Fig. S23b and S28d, ESI†). In contrast, cooling molten CP-G to room temperature recovers its green emission (∼500 nm) (Fig. S26, ESI†). Because the crystal-to-LC phase transition temperature is close to the Tdec of CP, the LC temperature (130 °C) was maintained for less than 5 min before the cooling process. Due to this short holding time, a small number of CP-G crystals might persist and not completely transition to the LC phase, resulting in continued observation of the green emission band at 500 nm even after cooling. These results suggest that the temperature-induced phase transition of CP polymorphs can significantly alter their luminescence behaviour.
To gain deeper insights into the thermal behaviour of the luminescence process, Arrhenius-type plots were utilised (Fig. S30, ESI†). The estimation of activation energy (Ea) for thermal quenching associated with the phase transitions of CP crystals was derived from the slope of these plots. Note that plots close to the phase transition temperature, with a significant margin of error, were excluded from subsequent analyses. For CP-B and CP-G, the initial slope observed during heating the sample up to 40 °C indicated Ea values of 5.5 and 13 kJ mol−1, respectively. Between 40 and 50 °C, discrete changes in the plot were observed for both polymorphs. Beyond 50 °C, the Ea values of CP-B and CP-G increased to 13 and 24 kJ mol−1, respectively, until the phase transition to the LC phase. This rise in Ea between 40 and 50 °C is attributable to the crystal-to-crystal phase transition, aligning closely with the DSC results. These results indicate that alteration in the crystal packing structure affects the rate of thermal deactivation of the excited states. CP-Y exhibited similar behaviour under both S0–Sn and direct S0–Tn excitation (Fig. S30c and d, ESI†). In contrast, during cooling, a decline in the logarithm of luminescence intensity was observed in the N phase for all samples, while it remained constant in the crystal phase until approximately 80 °C (Fig. S30e–g, ESI†). We hypothesise that the transient decrease in the N phase is linked to the instability of the LC phase at temperatures close to their N-to-crystal phase transition. Subsequently, the crystals gradually grew from the N phase below the phase transition temperature. However, immediately after the phase transition temperature, a biphasic region exists until ∼80 °C because the cooling rate (2 °C s−1) was too rapid for crystal growth, resulting in a constant region in log intensity. The crystal domains grow during the cooling process via Ostwald ripening. Below 80 °C, there is a gradual increase in log emission intensity along with a change in spectral shape. These results further support the dependence of luminescence properties on polymorphism.
Conclusions
In summary, the CP Au complex exhibited multicolour RTP in the bulk crystal, which was attributed to its polymorphic behaviour. Three types of crystals with high-contrast luminescent colours were obtained by controlling the recrystallisation conditions. CP exhibited a strong RTP with ΦRTP of up to 38%. The SCXRD results revealed that CP-Y exhibited a shorter Au–Au distance, which can explain the observed distinct luminescent colour. In contrast, CP-B and CP-G have longer Au–Au distances and identical crystal structures, despite exhibiting distinct luminescence properties. This type of polymorphism is rare, and our study revealed that the presence of a small CPground crystal domain was responsible for the overall green emission observed in CP-G. The FRET process occurs from the main crystal domain to CPground as an emitter. A switch in luminescent colour in CP can also be induced by mechanical stimuli due to alternation of the aggregated structure. Moreover, CP exhibited an N phase at higher temperatures, where luminescence was also observed. Our study demonstrates that the luminescence behaviour of CP Au complexes can be effectively controlled through crystal-to-crystal and crystal-to-LC phase transitions. Thus, we expect that this LC Au complex has great potential as a colour-controllable emitting material for applications in various optoelectronic devices.Experimental
Materials
The Au complex CP was synthesised in two facile steps, as shown in Fig. 1. All solvents and reagents were of reagent grade, commercially available, and used without further purification unless otherwise noted. 1H and 13C NMR spectra were recorded using a JEOL ECS-400 spectrometer (1H: 400 MHz and 13C: 100 MHz) in CDCl3. Chemical shifts are reported in parts per million (ppm), with residual protons or carbon atoms in the NMR solvent as an internal reference. IR spectra were obtained using the KBr disk method with a JASCO FT/IR-4100 spectrometer, and all spectra were reported as wavenumbers (cm−1). HRMS was performed on a JEOL JMS-700 spectrometer. Elemental analysis was conducted using a MICRO CORDER JM10 apparatus (J-SCIENCE).Synthesis of complex CP
Ethynyl-4-(trans-4-propylcyclohexyl)benzene (86 mg, 0.38 mmol) and (tht)AuCl (0.12 g, 0.42 mmol) were dissolved in CH2Cl2 (7 mL), and a freshly prepared solution of CH3COONa (0.26 g, 1.9 mmol) in MeOH (4 mL) was added to the solution at room temperature with stirring. After stirring at room temperature for 2 h, the precipitate was collected by filtration and washed with MeOH, H2O, and CH2Cl2. The obtained yellow solid was suspended in CH2Cl2 (15 mL), and 1-pentyl isocyanide (0.047 mL, 0.42 mmol) was added dropwise to the resultant suspension at room temperature. The reaction mixture was stirred at room temperature for 2 h, and then subjected to Celite® filtration, followed by the removal of solvent from the filtrate using a rotary evaporator. The resultant white solid was purified by silica-gel column chromatography (eluent: CH2Cl2), followed by recrystallization from a mixture of CH2Cl2 and n-hexane (1:2 v/v), yielding colourless plate crystals of CP in 39% yield (75 mg, 0.14 mmol) in two steps. mp 122 °C. 1H NMR (CDCl3, δ): 7.39 (dd, J = 6.3, 1.8 Hz; 2H; 2,6-H in phenyl), 7.08 (dd, J = 6.3, 1.6 Hz; 2H; 3,5-H in phenyl), 3.61 (t, J = 5.6 Hz; 2H; C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) NCH2), 2.41 (m, 1H; 1-H in cyclohexyl), 1.83 (m, 6H; CNCH2CH2, and 2,3,5,6-H in cyclohexyl (equatorial)), 1.50–1.16 (m, 11H; 4-H in cyclohexyl, 1-CH2 in propyl, CNCH2CH2(CH2)2, and 2,3,5,6-H in cyclohexyl (axial)), 1.09–0.89 (m, 5H; 2-CH2 in propyl, CN(CH2)4CH3), 0.87 (t, J = 7.2 Hz; 3H; CH3 in propyl). 13C NMR (CDCl3, δ): 146.95, 132.429, 126.561, 121.920, 120.569, 103.741, 44.530, 44.051, 39.797, 37.072, 34.231, 33.604, 28.368, 27.851, 21.805, 20.110, 14.515, 13.844. FTIR (KBr, ν): 3033, 2952, 2923, 2251 cm−1. HRMS (ESI, m/z): calculated for C23H32AuNNa (M + Na)+, 542.2093; found, 542.2090. Anal. calcd for C23H32AuN: C, 53.18; H, 6.21; N, 2.70; Au, 37.92. Found: C, 53.02; H, 5.91; N, 2.69; Ash, 38.1.
NCH2), 2.41 (m, 1H; 1-H in cyclohexyl), 1.83 (m, 6H; CNCH2CH2, and 2,3,5,6-H in cyclohexyl (equatorial)), 1.50–1.16 (m, 11H; 4-H in cyclohexyl, 1-CH2 in propyl, CNCH2CH2(CH2)2, and 2,3,5,6-H in cyclohexyl (axial)), 1.09–0.89 (m, 5H; 2-CH2 in propyl, CN(CH2)4CH3), 0.87 (t, J = 7.2 Hz; 3H; CH3 in propyl). 13C NMR (CDCl3, δ): 146.95, 132.429, 126.561, 121.920, 120.569, 103.741, 44.530, 44.051, 39.797, 37.072, 34.231, 33.604, 28.368, 27.851, 21.805, 20.110, 14.515, 13.844. FTIR (KBr, ν): 3033, 2952, 2923, 2251 cm−1. HRMS (ESI, m/z): calculated for C23H32AuNNa (M + Na)+, 542.2093; found, 542.2090. Anal. calcd for C23H32AuN: C, 53.18; H, 6.21; N, 2.70; Au, 37.92. Found: C, 53.02; H, 5.91; N, 2.69; Ash, 38.1.
X-ray crystallography
Single crystals of the Au complex were obtained by slow evaporation from mixed solvents (1:3 v/v mixture of CH2Cl2 and n-hexane for CP-B and CP-G, and 1:3 v/v mixture of CH2Cl2 and ethyl acetate for CP-Y). The reflection data were collected using the omega scanning technique with a Bruker D8 goniometer. Monochromatic Mo Kα radiation (λ = 0.71075 Å) was used to analyse the complexes and all measurements were performed at ambient temperature (293 K). The initial structure of the unit cell was determined using APEX2 using direct methods. The structural model was refined by a full-matrix least-squares method using SHELXL. The crystal structure data for CP are provided in the ESI.† The indexed data of CP complex were deposited in the Cambridge Crystallographic Data Centre (CCDC) database under reference number 2327717 (CP-B), CCDC 2327718 (CP-G), and CCDC 2327741 (CP-Y). The indexed database contains additional supplementary crystallographic data for this study and may be accessed without charge at https://www.ccdc.cam.ac.uk/conts/retrieving.html (accessed on 2024).
PXRD measurements were operated using the X’ Pert PRO system (PANalytical) at 45 kV and 40 mA with Cu Kα radiation (λ = 1.5405 Å).
Thermal properties
The phase transition properties of the Au complex were observed by POM using an Olympus BX51 microscope equipped with a temperature-controlled stage (Instec HCS302 microscope hot and cold stages, and an mK1000 temperature controller). To assess thermal stability, TG–DTA was carried out using a DTG-60AH analyser (Shimadzu) at a heating rate of 5.0 °C min−1. The thermodynamic parameters were determined using DSC (SII X-DSC7000) at heating and cooling rates of 1.0 °C min−1. At least three scans were conducted to check reproducibility.Photophysical properties
The UV-Vis absorption spectra were recorded using a JASCO V-550 absorption spectrophotometer. Steady-state photoluminescence spectra were recorded using a Hitachi F-7500 fluorescence spectrophotometer. Photoluminescence quantum yields were estimated using a calibrated integration sphere system (Hitachi). Photoluminescence decay profiles were recorded using a Hamamatsu Quantaurus-Tau photoluminescence lifetime measurement system (C1136-21).Computation
All calculations were performed using DFT with the B3LYP hybrid functional and 6-311+G(d,p) (for C, H, and N atoms) and SDD (for Au atoms) basis sets in the Gaussian 16 (revision C.01) program package.24 Single point calculations were conducted for the dimers using the conformation obtained from the crystal structure. The optimised geometry of the monomer was determined using DFT calculations at the same level of theory. The stationary points were characterised by frequency calculations to confirm the correct number of imaginary frequencies; it was confirmed that the minimum energy structures had no imaginary frequencies. The vertical excitation energies and oscillator strengths were estimated for the ten lowest transitions for the ground state equilibrium geometries using TD-DFT at the same level of theory.Author contributions
A.F.: data curation, formal analysis, methodology, investigation, validation, and writing – original draft; M.D.: data curation, formal analysis, methodology, investigation; K.M.: formal analysis, investigation, and writing – original draft; K.H.: formal analysis, investigation, writing – review and editing. O.T.: conceptualisation, formal analysis, supervision, project administration, resources, writing – review and editing, and funding acquisition.Conflicts of interest
The authors declare there are no conflicts of interest.Acknowledgements
This research was supported by the Japan Science and Technology agency (JST) A-STEP program (JPMJTR22T1), the Toshiaki Ogasawara Memorial Foundation, and the Cooperative Research Program of the Network Joint Research Centre for Materials and Devices.References
- (a) C. Murawski and M. C. Gather, Adv. Opt. Mater., 2021, 9, 2100269 CrossRef CAS; (b) S. Negi, P. Mittal, B. Kumar and P. K. Juneja, Microelectron. Eng., 2019, 218, 111154 CrossRef CAS; (c) X. Zhang, X. Zhang, B. Yang, J. Hui, M. Liu, Z. Chi, S. Liu, J. Xu and Y. Wei, Polym. Chem., 2014, 5, 318 RSC.
- K. Zhang, J. Liu, Y. Zhang, J. Fan, C.-K. Wang and L. Lin, J. Phys. Chem. C, 2019, 123, 24705 CrossRef CAS.
- (a) B. Z. Tang, X. Zhan, G. Yu, P. P. Sze Lee, Y. Liu and D. Zhu, J. Mater. Chem., 2001, 11, 2974 RSC; (b) B. Jiang, C.-W. Zhang, X.-L. Shi and H.-B. Yang, Chin. J. Polym. Sci., 2019, 37, 372 CrossRef CAS; (c) P. Alam, C. Climent, P. Alemany and I. R. Laskar, J. Photochem. Photobiol., C, 2019, 41, 100317 CrossRef CAS; (d) S. Suzuki, S. Sasaki, A. S. Sairi, R. Iwai, B. Z. Tang and G. Konishi, Angew. Chem., Int. Ed., 2020, 59, 9856 CrossRef CAS PubMed.
- (a) A. L. Balch, Angew. Chem., Int. Ed., 2009, 48, 2641 CrossRef CAS PubMed; (b) Y. Sagara and T. Kato, Angew. Chem., Int. Ed., 2011, 50, 9128 CrossRef CAS PubMed; (c) M.-J. Teng, X.-R. Jia, X.-F. Chen and Y. Wei, Angew. Chem., Int. Ed., 2012, 51, 6398 CrossRef CAS PubMed.
- (a) S. Kohmoto, R. Tsuyuki, H. Masu, I. Azumaya and K. Kishikawa, Tetrahedron Lett., 2008, 49, 39 CrossRef CAS; (b) W. Lu, N. Zhu and C. M. Che, J. Am. Chem. Soc., 2003, 125, 16081 CrossRef CAS PubMed; (c) G. Zhang, J. Lu, M. Sabat and C. L. Fraser, J. Am. Chem. Soc., 2010, 132, 2160 CrossRef CAS PubMed.
- (a) Y. Sagara and T. Kato, Angew. Chem., Int. Ed., 2008, 47, 5175 CrossRef CAS PubMed; (b) Y. Sagara and T. Kato, Nat. Chem., 2009, 1, 605 CrossRef CAS PubMed; (c) K. Nagura, S. Saito, H. Yusa, H. Yamawaki, H. Fujihisa, H. Sato, Y. Shimoikeda and S. Yamaguchi, J. Am. Chem. Soc., 2013, 135, 10322 CrossRef CAS PubMed.
- K. Hisano, S. Panthai and O. Tsutsumi, in Handbook of Aggregation-Induced Emission, ed. Y. Tang and B. Z. Tang, John Wiley & Sons Ltd., 2022, vol. 9, p. 555 Search PubMed.
- (a) T. Seki, K. Sakurada and H. Ito, Angew. Chem., Int. Ed., 2013, 52, 12828 CrossRef CAS PubMed; (b) A. Sathyanarayana, S. Nakamura, K. Hisano, O. Tsutsumi, K. Srinivas and G. Prabusankar, Sci. China: Chem., 2018, 61, 957 CrossRef CAS; (c) R. Kawano, O. Younis, A. Ando, Y. Rokusha, S. Yamada and O. Tsutsumi, Chem. Lett., 2016, 45, 66 CrossRef CAS; (d) K. Fujisawa, S. Yamada, Y. Yanagi, Y. Yoshioka, A. Kiyohara and O. Tsutsumi, Sci. Rep., 2015, 5, 7934 CrossRef CAS PubMed.
- (a) K. Fujisawa, N. Kawakami, Y. Onishi, Y. Izumi, S. Tamai, N. Sugimoto and O. Tsutsumi, J. Mater. Chem. C, 2013, 1, 5359 RSC; (b) K. Fujisawa, Y. Okuda, Y. Izumi, A. Nagamatsu, Y. Rokusha, Y. Sadaike and O. Tsutsumi, J. Mater. Chem. C, 2014, 2, 3549 RSC; (c) S. Yamada, Y. Rokusha, R. Kawano, K. Fujisawa and O. Tsutsumi, Faraday Discuss., 2017, 196, 269 RSC.
- (a) S. Roy, A. Hazra, A. Bandyopadhyay, D. Raut, P. L. Madhuri, D. S. S. Rao, U. Ramamurty, S. K. Pati, S. Krishna Prasad and T. K. Maji, J. Phys. Chem. Lett., 2016, 7, 4086 CrossRef CAS PubMed; (b) M. Krikorian, S. Liu and T. M. Swager, J. Am. Chem. Soc., 2014, 136, 2952 CrossRef CAS PubMed.
- A. Furoida, M. Daitani, K. Hisano and O. Tsutsumi, Molecules, 2021, 26, 7255 CrossRef CAS PubMed.
- (a) E. R. T. Tiekink and J.-G. Kang, Coord. Chem. Rev., 2009, 253, 1627 CrossRef CAS; (b) I. O. Koshevoy, E. S. Smirnova, M. Haukka, A. Laguna, J. C. Chueca, T. A. Pakkanen, S. P. Tunik, I. Ospino and O. Crespo, Dalton Trans., 2011, 40, 7412 RSC.
- Y. Kuroda, M. Tamaru, H. Nakasato, K. Nakamura, M. Nakata, K. Hisano, K. Fujisawa and O. Tsutsumi, Commun. Chem., 2020, 3, 139 CrossRef CAS PubMed.
- (a) R. C. Evans, P. Douglas and C. J. Winscom, Coord. Chem. Rev., 2006, 250, 2093 CrossRef CAS; (b) I. E. Pomestchenko, C. R. Luman, M. Hissler, R. Ziessel and F. N. Castellano, Inorg. Chem., 2003, 42, 1394 CrossRef CAS PubMed.
- A. Ando, K. Ozaki, U. Shiina, E. Nagao, K. Hisano, K. Kamada and O. Tsutsumi, Aggregate, 2022, 3, e125 CrossRef CAS.
- (a) L. Favereau, C. Quinton, C. Poriel, T. Roisnel, D. Jacquemin and J. Crassous, J. Phys. Chem. Lett., 2020, 11, 6426 CrossRef CAS PubMed; (b) X. Bi, Y. Shi, T. Peng, S. Yue, F. Wang, L. Zheng and Q.-E. Cao, Adv. Funct. Mater., 2021, 31, 2101312 CrossRef CAS; (c) O. Tsutsumi, M. Tamaru, H. Nakasato, S. Shimai, S. Panthai, Y. Kuroda, K. Yamaguchi, K. Fujisawa and K. Hisano, Molecules, 2019, 24, 4606 CrossRef CAS PubMed.
- W. Shao and J. Kim, Acc. Chem. Res., 2022, 55, 1573 CrossRef CAS PubMed.
- M. Vaddamanu, A. Sathyanarayana, Y. Masaya, S. Sugiyama, O. Kazuhisa, K. Velappan, K. Subramaniyam, K. Hisano, O. Tsutsumi and G. Prabusankar, Organometallics, 2020, 39, 2202 CrossRef CAS.
- B. Zhang, P. Zhao, L. J. Wilson, J. Subbiah, H. Yang, P. Mulvaney, D. J. Jones, K. P. Ghiggino and W. W. H. Wong, ACS Energy Lett., 2019, 4, 1839 CrossRef CAS.
- T. Seki, K. Sakurada and H. Ito, Chem. Commun., 2015, 51, 13933 RSC.
- R. Yoshida, T. Tachikawa and S. Ito, Chem. Commun., 2022, 58, 6781 RSC.
- B. Zhang, G. Lyu, E. A. Kelly and R. C. Evans, Adv. Sci., 2022, 9, 2201160 CrossRef PubMed.
- (a) G. Shi, S. Li, P. Shi, J. Gong, M. Zhang and W. Tang, IUCrJ, 2021, 8, 584 CrossRef CAS PubMed; (b) O. Al Rahal, B. M. Kariuki, C. E. Hughes, P. A. Williams, X. Xu, S. Gaisford, D. Iuga and K. D. M. Harris, Cryst. Growth Des., 2023, 23, 3820 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision C.01, Gaussian, Inc., Wallingford CT, 2019 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2327717, 2327718 and 2327741. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4ma00314d |
| ‡ Present address: Laboratory for Chemistry and Life Science, Institute of Innovative Research, Tokyo Institute of Technology, 4259 Nagatsuta, Midori-ku, Yokohama 226-8501, Japan. |
| This journal is © The Royal Society of Chemistry 2024 |