
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence3D printed porous silicone polymer composites using table salt as a sacrificial template†
Santosh
Adhikari
 *a,
Xavier M.
Torres
a,
John R.
Stockdale
a,
Shelbie A.
Legett
a,
Lindsey B.
Bezek
a,
Jesus A.
Guajardo
a,
Adam
Pacheco
a,
Karthik
Ramasamy
b,
Bart
Benedikt
c,
Matthew
Lewis
c and
Andrea
Labouriau
*a
*a,
Xavier M.
Torres
a,
John R.
Stockdale
a,
Shelbie A.
Legett
a,
Lindsey B.
Bezek
a,
Jesus A.
Guajardo
a,
Adam
Pacheco
a,
Karthik
Ramasamy
b,
Bart
Benedikt
c,
Matthew
Lewis
c and
Andrea
Labouriau
*a
aC-CDE: Chemical Diagnostics and Engineering, Los Alamos National Laboratory, Los Alamos, NM 87545, USA. E-mail: san_adh@lanl.gov; andrea@lanl.gov
bSIGMA-1: Fabrication Manufacturing Science, Los Alamos National Laboratory, Los Alamos, NM 87545, USA
cW-13: Advanced Engineering Analysis, Los Alamos National Laboratory, Los Alamos, NM 87545, USA
First published on 19th September 2024
Abstract
Porous silicone polymer composites (elastomeric foams) with tunable properties and multifunctionalities are of great interest for several applications. However, the difficulties in balancing functionality and printability of silicone polymer based composite resins hinder the development of 3D printed multifunctional porous silicone materials. Here, the direct ink write (DIW) technique and NaCl filler as a sacrificial template were utilized to develop 3D printed porous silicone composites. Three different fillers (hydrophilic and hydrophobic fumed silica, and carbon nanofibers (CNF)) were used to impart additional functionality and to explore their effects on the rheology of the DIW resin, and the mechanical properties of the 3D printed elastomeric foams. While hydrophilic silica was effective in modulating the rheology of the resin, CNFs were effective in improving the tensile strength of the elastomeric foam. Unlike tensile strength, which was found to be dependent on filler type, the uniaxial compressive behavior was found to be more dependent on the porosity of the elastomeric foams. A hyperelastic constitutive model (the Compressive, Hyperelastic, Isotropic, Porosity-based Foam model) was used to simulate the uniaxial compressive behavior of the elastomeric foams, and the model accurately reproduced the experimental stress–strain profiles. The expanded design flexibility of tunable porosity in DIW parts enables the foams to be utilized in a wider variety of applications. For example, the foam with CNF filler demonstrated excellent oil/water separation capacity, with absorbing efficiencies of 450% and 330% respectively for chloroform and toluene. Similarly, a foam with hydrogen getter capacity was developed using the CNF filled foam with hydrogen getter as an additional functional filler, and high performance of the 3D printed hydrogen getter composite was demonstrated.
1. Introduction
Porous polymers have received significant attention due to having both the benefits of porous structures and polymeric properties, such as large surface area, high flexibility and compressibility, and ease of fabrication and functionalization.1,2 Thus, in the past decades, porous polymeric materials have witnessed applications in gas storage/separation,3 water treatment/purification,4 electrochemical energy storage and conversion,5 passive daytime radiative cooling,6 catalysis,7 biomedical devices,8 and wearable electronics.9 Among different porous polymers, silicone polymer (or polydimethylsiloxanes, PDMS) based porous materials (elastomeric foams) have been widely explored.10,11 This is mainly attributed to the outstanding properties of silicone polymers, such as chemical and thermal stability, mechanical robustness and excellent elasticity, and biocompatibility.12,13 In addition, PDMS exhibits high transmittance, high gas permeability, non-flammability, hydrophobicity, and thermal and electrical resistivity, and low bulk density.12 These silicone polymer based porous materials have been used as flexible sensors,14,15 oil/water separators,16 energy generation and storage,17 biomedical devices,18 and wearable electronics.14Different techniques, such as template leaching,16,19 phase separation,20,21 emulsion templating,22 gas foaming,23 3D printing,24,25 and breath figure26 have been implemented to induce porosity and develop silicone polymer based elastomeric foams, which result in distinct porous properties for different applications. Among these techniques, 3D printing provides an opportunity to create unique porous polymeric materials with complex geometries and tailored properties, which would otherwise be difficult to be achievable with conventional techniques. The 3D printing of silicone polymers to create flexible porous materials has been successfully achieved by utilizing direct ink write (DIW). DIW resins require shear thinning, yield stress behavior or non-Newtonian behavior. Because of the low elastic modulus of the liquid silicone pre-polymers, their rheological behavior needs to be modified to meet the printability requirements for DIW. This is achieved by adding fillers, which serve as thixotropic agents to regulate the rheological properties of silicone pre-polymers and impart additional functionality. For example, Zheng et al.27 have used polytetrafluoroethylene micropowder to both serve as a thixotropic agent as well as to endow the PDMS composite materials with high electron affinity for its application in a triboelectric nanogenerator. Similarly, a hydrogen getter active material (the mixture of 1,4-bis(phenylethynyl)benzene with Pd/C catalyst) has been added to PDMS based ink to develop mechanically-flexible, and high-performing porous PDMS based hydrogen getter polymers.28 Recently, Stockdale et al.29 have developed soft, flexible 3D printed PDMS composite materials with radiation-shielding functionality by adding boron as a thixotropic agent as well as functional fillers to PDMS pre-polymers.
In all of the above reported 3D printed PDMS materials, the porosity was imparted based on the printing geometry or lattice structure. However, the printed strands do not bear any porosity within themselves, and there are a very few reports on 3D printed PDMS materials with porosity within the printed strands. Chen et al.30 reported highly porous and hyper elastic 3D printed PDMS material by combining DIW and the salt and solvent leaching method. The researchers used salt and silicone oil as a template to create porosity within the printed strands and to control the rheology of the DIW ink. Similarly, Leob et al.31 fabricated oil-filled silicone microballons and utilized them to impart porosity within the DIW strands. The porosity was created by solvent extraction of oil from the microballons in acetone followed by supercritical drying. Though 3D printed PDMS materials with interconnected hierarchical pores have been reported, there are opportunities to expand the functionality of these porous silicone polymer composite materials and improve the mechanical strength by incorporating additional functional filler into the DIW resin. For example, the performance of hydrogen getter polymer composites could be further improved by introducing porosity within the printed strands, as the performance of these composites is greatly affected by surface area. However, there have not been any reports on DIW of hierarchically porous structures with functional fillers, which could be due to the difficulties in balancing functionality and printability properties of the DIW resin with additional filler.
Here we report formulation of silicone polymer based DIW resins by grinding NaCl with silicone pre-polymers which enabled successful DIW printing with different functional filler to expand functionality of 3D printed porous materials for different applications. The novelty of the work lies in demonstrating the incorporation of different fillers in the DIW resin to impart additional functionality and successful printing of the resin to develop hierarchically porous 3D printed silicone polymer composite material with improved mechanical properties for different application. We study the effect of these fillers on the rheology of the DIW resin and the mechanical properties of the porous materials (elastomeric foams). The experimental results obtained from uniaxial compression studies were compared to the simulation results developed using a hyperelastic constitutive model, namely the Compressive, Hyperelastic, Isotropic Porosity-based Foam (CHIP-Foam) model. Additionally, these materials were explored for oil/water separation and to develop hydrogen getter polymer composites where high surface area is essential for high performance of the materials.
2. Materials and methods
2.1. Materials
Vinyl-terminated (4–6% diphenylsiloxane) dimethylsiloxane copolymer (Gelest PDV-541), and trimethylsiloxyterminated methylhydrosiloxane–dimethylsiloxane copolymer (Gelest HMS-301), used as base polymers in the formulation of the composite resin, were purchased from Gelest Inc., USA. A high-temperature platinum catalyst (Gelest SIP 6829.2; platinum carbonyl cyclovinylmethylsiloxane complex; 1.85–2.1% Pt in cyclomethyl vinyl siloxanes) was used as for curing of all the formulation unless otherwise noted. To prevent premature curing of the suspensions, 1-ethynyl-1-cyclohexanol (ETCH; 99%, Sigma Aldrich, USA) was utilized. Fillers used for the optimization of rheology included OH-functionalized fumed silica (A300; Evonik Aerosil 300; Evonik Industries AG, Germany), a PDMS-functionalized fumed silica (A812; Evonik Aerosil 812; Evonik Industries AG, Germany Cabot Corporation, USA), and carbon nanofibers (PR-25-XT-LHT, Conical carbon nanofibers, Sigma-Aldrich, USA). 1,4-Bis(phenylethynyl)benzene (DEB) was purchased from Ambeed Inc. and palladium on carbon, 5 wt% loading (dry basis), matrix activated carbon support and toluene were purchased from Sigma Aldrich. All chemicals were used as received. DEB–Pd/C catalyst blend (the active material for the hydrogen getter) was prepared by following the same procedure reported in previous literature.28 A mid-temperature platinum catalyst (Gelest SIP 6832.2; platinum–cyclovinylmethylsiloxane complex; 2% Pt in cylcomehtylvinylsiloxanes), also known as Ashby-Karstedt (AK) catalyst, was used to induce cross-linking in the preparation of the hydrogen getter composite.2.2. Resin formulation and preparation of 3D printed and cast samples
The base of all resins consisted of a 9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio of PDV-541 to HMS-301, 50 wt% of NaCl, 5 wt% of filler (A300, A812 or CNFs), and approximately 0.1 wt% of ETCH unless otherwise mentioned. The general procedure for the preparation of DIW resin for cast sheets and 3D printing is explained below in detail. Note that 50 wt% of NaCl (which was calculated to ∼30 vol% in our system) was chosen because below 50 wt% of NaCl, we were not able to quantitative leach out NaCl from the composite material after curing. This could be related to the percolation threshold for NaCl used in our system which we speculated to be ∼50 wt% (∼30 vol%).
:1 ratio of PDV-541 to HMS-301, 50 wt% of NaCl, 5 wt% of filler (A300, A812 or CNFs), and approximately 0.1 wt% of ETCH unless otherwise mentioned. The general procedure for the preparation of DIW resin for cast sheets and 3D printing is explained below in detail. Note that 50 wt% of NaCl (which was calculated to ∼30 vol% in our system) was chosen because below 50 wt% of NaCl, we were not able to quantitative leach out NaCl from the composite material after curing. This could be related to the percolation threshold for NaCl used in our system which we speculated to be ∼50 wt% (∼30 vol%).
PDV-541 and HMS-301 were taken with NaCl and ground using Retsch Mortar Grinder RM 200 in order to reduce the particle size of NaCl. The grinding was conducted for 15 min followed by a 15 min rest to mitigate the increase in temperature and unwanted reaction and degradation of the silicone pre-polymers. This grind-rest cycle was then repeated seven more times (for a total grinding time of 2 h). The fully ground silicone pre-polymer and NaCl mixture was then taken with 5 wt% of each filler (A300, A812 or CNFs) and mixed for 2 min in a Thinky planetary mixer at 2000 rpm for 2 times with hand mixing in between. Then, approximately 0.1 wt% ETCH was added and mixed in the planetary mixer at 2000 rpm for 2 min followed by the addition of 0.1 wt% of high temperature Pt-catalyst and mixing in the planetary mixer for 2 min in a cold fixture to avoid premature curing. The resin having 5 wt% of A300, A812, and CNF fillers and their respective 3D printed samples are named as P1, P2, and P3 respectively, while the resin with no filler is named as P0. The naming and composition of the different DIW resins prepared and studied in this work are listed in Table 1.
| Sample | PDV-541:HMS-301 = 9:1 (wt%) |
NaCl (wt%) | Fillers (wt%) | |||
|---|---|---|---|---|---|---|
| A300 | A812 | CNF | DEB–Pd/C | |||
| P0 | 50 | 50 | — | — | — | — |
| P1 | 45 | 50 | 5 | — | — | — |
| P2 | 45 | 50 | — | 5 | — | — |
| P3 | 45 | 50 | — | — | 5 | — |
| Polymer getter composite | 39 | 50 | — | — | 1 | 10 |
The chemical analysis of the silicone pre-polymer during grinding was carried by FT-IR spectroscopy using a Nicolet iS50 FTIR instrument, while the particle size analysis of ground NaCl was done by laser diffraction using an Anton Parr PSA 1090 Model D instrument. For particle size analysis, 2 g of the sample was taken every 30 min, and the prepolymer was dissolved in toluene. The NaCl suspension in toluene was then filtered and dried to isolate the NaCl powder and determine the particle size.
For 3D printed samples, the blended resin was loaded into a 25 mL stainless steel syringe and centrifuged at 2000 rpm for 1 min to remove any air bubbles. The filled syringes were attached to an EMO-XT print head (Hyrel 3D, USA) and then connected to a Hydra 21 3D printer (Hyrel 3D, USA). Inks were extruded onto the build plate at room temperature using 600 μm plastic luer-lock nozzles (Nordson EFD Precision Tips; Nordson Corporation, USA). The printer was controlled using Repetrel software (Hyrel 3D) and custom G-code which specified the face-centered tetragonal (FCT) lattice geometry and center-to-center spacing between struts. Printed samples with 50 wt% NaCl and 5 wt% filler had center-to-center spacing between struts of 1000 μm, a travel rate of 1600 mm min−1 for all filler types, a flow rate of 175 pulses per μL for P1 and P2 samples, and a flow rate of 200 mm min−1 on the 1st layer and 160 mm min−1 for all other layers for the P3 samples. These settings were used to ensure adequate adhesion of the first layer to the print bed, consistent extrusion of ink, and slight overlap between consecutive layers for each printed sample. For cast samples, the blended resin was cast onto a Teflon sheet using a metal mold to get a rectangular cast sheet of approximately 1.36 ± 0.23 mm in thickness for tensile tests and a circular molded sample of approximately 6.0 ± 0.1 mm for bulk density samples.
Both the cast sheet and printed samples were cured for 2 h at 150 °C in a preheated oven and were then cut into a circular shape (10 mm diameter) to determine density. The cast samples were also cut into dogbones for tensile tests.
The 3D printed hydrogen getter sample consisted of 39 wt% silicone pre-polymers (9:1 ratio of PDV-541 to HMS-301), 50 wt% NaCl, 10 wt% active hydrogen getter (DEB–Pd/C mixture in 3:1 ratio by weight) and 1 wt% CNF. The resin preparation for the hydrogen getter sample is described below. The calculated amount CNFs were added to the 2 h ground pre-polymers and NaCl resin and mixed in a planetary mixer for 2 min. at 2000 rpm under ambient conditions. Then DEB–Pd/C was added and again mixed for additional 2 min. Finally, 0.1 wt% of ETCH followed by 0.1 wt% AK-catalyst were added and mixed for 2 min in a cold fixture. AK catalyst was used so that the resin can be cured effectively at comparatively lower temperature avoiding unwanted reaction between DEB and silane groups present in the prepolymer at high temperature.28 The face centered tetragonal (FCT) lattice geometry, having 1000 μm center-to-center spacing between struts and 325 μm layer height with eight layers, were printed and cured for 5 h at 75 °C in a preheated oven. The particular curing condition was adopted for the polymer getter resin based on our previous work28 to avoid any unwanted reaction between the polymer resin and DEB.
All 3D printed samples were immersed in deionized (DI) water for 24 h at 50 °C with periodical changing of DI water for efficient extraction of NaCl to impart porosity in the samples. The efficiency of NaCl extraction was monitored by measuring the dry mass of the samples before and after the extraction of NaCl.
2.3. Material characterization
Rheological properties of the resins created for this study were determined using a TA Discovery Series Hybrid Rheometer DHR-3. All experiments were conducted using a 25 mm cross-hatched parallel plate fixture geometry with a working gap of 1000 μm. Strain sweeps were conducted from 0.001% to 10% strain at an angular frequency of 10 rad s−1, and stress sweeps were conducted from 10 to 10000 Pa (or until the yield stress was reached). The yield stress (σy) and equilibrium storage modulus (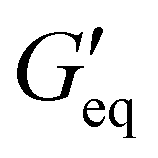 ) were determined as the onset point where G′ starts to decrease and obtained using TA Instruments’ Trios software.
) were determined as the onset point where G′ starts to decrease and obtained using TA Instruments’ Trios software.
FT-IR spectra were recorded in attenuated total reflectance (ATR) using a Nicolet iS50 FTIR spectrophotometer in the range of 500 to 4000 cm−1 with an accumulation of 32 scans in the absorbance mode.
Bulk density was calculated for cast samples using a densimeter, and the densities of the printed porous samples were calculated by measuring the volume and mass of the sample. Porosity of each sample was calculated by measuring the density of each sample and using eqn (1);
| Porosity = (1 – bulk density/density of the porous sample) × 100 | (1) |
An Instron 3343 Low-Force Testing System (Instron; Norwood, MA, USA) with a 1 kN load cell and BlueHill Universal software was used to perform uniaxial compression testing on all printed samples and tensile testing on cast samples. Each compression sample (diameter of 22.23 mm) was subjected to 4 cycles of compression to a maximum stress of 0.6 MPa at a rate of 0.05 mm s−1. The stress–strain curve for each printed sample was determined by the final cycle. Tensile experiments were conducted using a ASTM D638 Type V die on cast samples. Cast samples (thickness of 1.36 ± 0.23) were extended at a rate of 1 mm s−1. At least three samples were tested for each formulation, and ultimate tensile strength and strain at break results were reported.
A confocal microscope (Keyence VHX-6000; Keyence Corporation, Osaka, Japan) was used to obtain optical images as well as to determine the thicknesses of each sample. A Thermo Scientific™ Apreo™ 2 SEM was used to examine the cross sections of the DIW prints. Varying magnification zooms were used (100–5000) with imaging voltage and current operating at 2 kV and 25 pA, respectively.
The evaluation of the surface wettability of the porous 3D printed samples was performed via static contact angle measurements using a Drop Shape Analyzer DSA30E (Krüss GmbH, Hamburg, Germany). DI water (10μL) was dropped at the center of the porous 3D printed samples, which were placed approximately 9 mm from the syringe. The contact angle of the water drop on the surface was measured by the instrument.
For the oil/water separation experiment, organic solvent (chloroform, 2 mL) dyed with indigo was mixed with water (40 mL). The P3 sample was used to demonstrate oil/water separation. For calculating absorption efficiency, the sample was immersed in toluene or chloroform for 10 h until the equilibrium was reached. Then, the sample was removed from the container and held in air until the organic solvent stopped dripping (approximately 20 s), and the mass of the swelled sample was taken immediately. The absorption efficiency (k) is calculated by using eqn (2);
| k = (meq − mo)/mo | (2) |
The hydrogen absorption experiment was performed at low hydrogen partial pressures (750 mtorr of pure hydrogen) for 21 days, which is representative of the working environments using home-built reactor systems and following the procedure reported in our previous work.28
2.4. Modeling
In the modelling part of this study, we calibrated the foam material model using the performed cyclic compression test data. The calibrated model is used to numerically simulate the mechanical response of the fabricated foams. The hyperelastic constitutive model developed in Los Alamos National Laboratory, namely the CHIP-Foam model, was used to simulate the mechanical behavior of the elastomeric foams. The model was coded into an implicit user subroutine UHYPER in Abaqus finite element software, so the model can be used in engineering simulations.The CHIP-Foam model is based on the assumption that the strain energy density function of foam consists of additive terms, where each term models different physical phenomenon taking place during the deformation of a foam. A detailed description of the CHIP-Foam model can be found in literature.32 In this section, only the most fundamental discussion is provided to help the reader to understand the basic principles behind the CHIP-Foam model.
In the theory of hyperelasticity, stress σ is calculated from a potential function, per eqn (3);
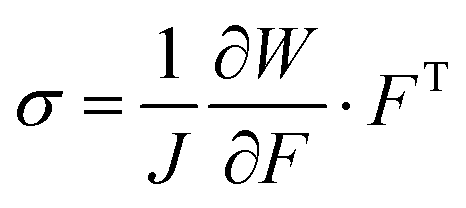 | (3) |
The CHIP-Foam model uses the following formula to define the strain energy density function for a foam following eqn (4);
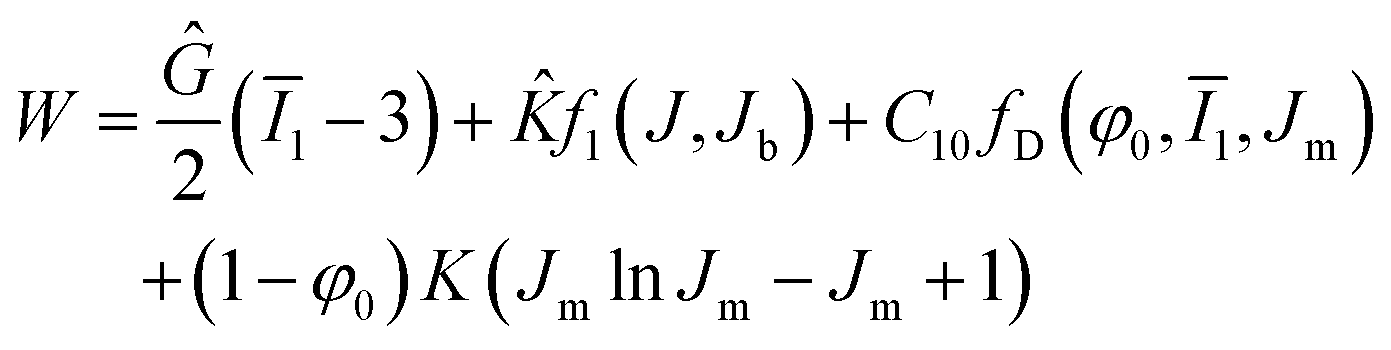 | (4) |
![[K with combining circumflex]](https://www.rsc.org/images/entities/i_char_004b_0302.gif) is small strain bulk modulus of foam, J is the volume change of foam due to deformation, Jb is volume at the onset of buckling, C10 is the Mooney–Rivlin parameter for matrix material, Jm is the volume change of the matrix material, φ0 is the initial porosity, fD is the Danielsson's function,33 and K is the bulk modulus of the matrix material. We note that the strain energy density function for the CHIP-Foam model is the superposition of three different compressive deformation modes:
is small strain bulk modulus of foam, J is the volume change of foam due to deformation, Jb is volume at the onset of buckling, C10 is the Mooney–Rivlin parameter for matrix material, Jm is the volume change of the matrix material, φ0 is the initial porosity, fD is the Danielsson's function,33 and K is the bulk modulus of the matrix material. We note that the strain energy density function for the CHIP-Foam model is the superposition of three different compressive deformation modes:
• Initial compressible deformation of the foam caused mostly by the foam's walls deformation (Ĝ).
• Soft deformation caused by buckling of the foam's walls. This mode takes place in foams with high porosity (>0.6) because the buckling will not take place in thick walls ( and Jb).
• Deformation of a material that progressively becomes almost incompressible due to pores closing with increasing compression loads (K and C10).
To use the CHIP-Foam model in engineering computations, material parameters Ĝ, , Jb, C10, and K have to be determined for a given foam. The numerical work used to determine these parameters is presented in Section 3.4.
3. Results and discussion
3.1. Formulation of the printing resin
For DIW, it is desirable to have the particle size of the filler be significantly small compared to the nozzle size so that the resin with high concentration of filler could be used without any clogging of the nozzle. In general, fillers with small particle size are added during the formulation of DIW resin, but within our experimentation, NaCl was ground with PDV and HMS (silicone pre-polymers) to reduce the particle size and aggregation of NaCl in our DIW suspensions. The particle size of the ground NaCl was analyzed after every 30 min of grinding by laser diffraction. As listed in Table 2, the particle size of NaCl decreases with grinding time. After 2 h of grinding, the volume-weighted particle size distribution D10, D50, D90, and mean size were found to be 0.1 μm, 2.2 μm, 26.6 μm, and 8.8 μm respectively (Table 2) which was small enough to print using the 600 μm nozzle without clogging.| Sample | D10 (μm) | D50 (μm) | D90 (μm) | Mean (μm) |
|---|---|---|---|---|
| Unground NaCl | 20.8 | 265.4 | 445.3 | 263.3 |
| Ground NaCl_0.5 h | 0.1 | 5.9 | 46.3 | 15.0 |
| Ground NaCl_1 h | 0.1 | 4.9 | 38.8 | 13.6 |
| Ground NaCl_1.5 h | 0.1 | 4.1 | 32.8 | 11.5 |
| Ground NaCl_2 h | 0.1 | 2.2 | 26.6 | 8.8 |
While this grinding method was effective in reducing the particle size and enabling DIW printing, it is possible that the grinding procedure may degrade the HMS and PDV pre-polymers. This may lead to inefficient curing of the pre-polymers and poor mechanical properties of final material. FT-IR spectroscopy was conducted on the ground resins at different time intervals with the aim of detecting any spectral changes associated with degradation due to grinding. The FT-IR spectra of the ground resins at different time intervals is shown in Fig. 1. The characteristic peaks of silicone pre-polymers such as C–H stretching (∼2900 cm−1), CH3 symmetric bending in Si–CH3 (∼1250 cm−1), Si–O–Si stretching (900–1150 cm−1) and CH3 rocking in Si–CH3 (∼800 cm−1) remained unchanged and no any noticeable changes in the FT-IR spectra was observed in the spectra of the ground resins even after 2 h of grinding suggesting no significant degradation of the pre-polymers.
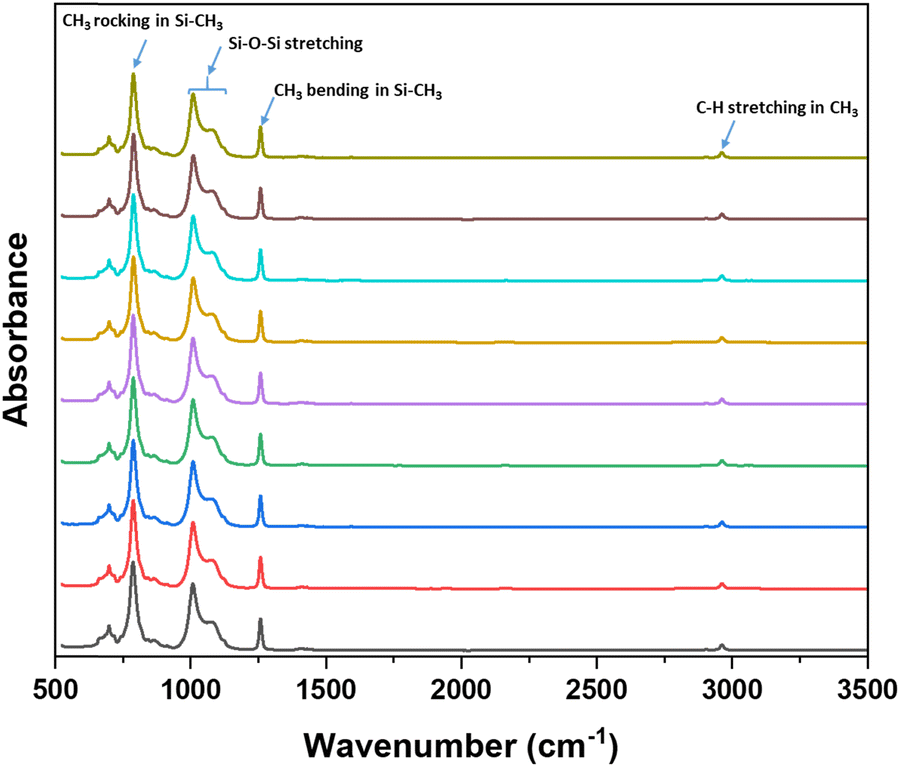 | ||
| Fig. 1 FT-IR spectra of the ground resins taken at different time intervals. | ||
Next, a puck was prepared by curing the 2 h-ground resin at 150 °C for 2 h after addition of Pt-catalyst. The puck was then immersed in toluene for 24 h to determine amount of material extracted (or mass loss). Any unreacted pre-polymers or low molecular weight polymer that did not undergo effective crosslinking will be soluble in toluene and can be extracted. So, we speculate that the mass loss from the puck is due to the extraction of low molecular weight polymer or unreacted pre-polymers that did not undergo effective crosslinking. The mass loss from the puck prepared from the 2 h-ground resin was less than 1% confirming the effective crosslinking and curing of the ground pre-polymers. Thus, 2 h ground resin was used for rest of the study in this work.
3.2. Rheology of different resin composites
For DIW, the rheological property of the printing resin is critical as it determines the printability of the resin. The printing resin must exhibit shear-thinning yield stress behavior (decrease in viscosity with increase in shear rate). Additionally, the rheological parameters such as equilibrium storage modulus,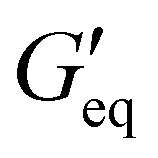 (the stress at which a material behaves like a viscoelastic solid) and yield stress, σy (the stress at which a material flows) need to be in a certain range for the resin to flow through the nozzle and retain its printed shapes after deposition.34 The ground resin containing 50 wt% of NaCl without any filler (P0 resin) showed liquid like behavior (Fig. S1, ESI†) and hence could not be used for DIW. Thus, the addition of thixotropic agents was required to modulate the rheology of the resin. The ability of thixotropic agents to modulate the rheology of the resin depends on their interaction with the resin to make network which is mainly governed by the chemical nature and surface area of the filler. Thus, fillers with high surface area and functionalities that can interact with the resin are good at making network within the material and modulate the rheology of the resin. Equal amounts of different fillers (5 wt%) were added to the ground resin to study the effect of different fillers on the rheological behavior of DIW resins.
(the stress at which a material behaves like a viscoelastic solid) and yield stress, σy (the stress at which a material flows) need to be in a certain range for the resin to flow through the nozzle and retain its printed shapes after deposition.34 The ground resin containing 50 wt% of NaCl without any filler (P0 resin) showed liquid like behavior (Fig. S1, ESI†) and hence could not be used for DIW. Thus, the addition of thixotropic agents was required to modulate the rheology of the resin. The ability of thixotropic agents to modulate the rheology of the resin depends on their interaction with the resin to make network which is mainly governed by the chemical nature and surface area of the filler. Thus, fillers with high surface area and functionalities that can interact with the resin are good at making network within the material and modulate the rheology of the resin. Equal amounts of different fillers (5 wt%) were added to the ground resin to study the effect of different fillers on the rheological behavior of DIW resins.
The rheological results of the ground resins after the addition of 5 wt% of different fillers are shown in Fig. 2 and Table S1 (ESI†). All resins formulated in this study showed shear-thinning behavior and a rubbery plateau region. Among different resins, the P3 resin exhibited the highest 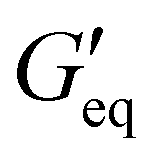 and complex viscosity followed by the P1 and P2 resins (Fig. 2 and Table S1, ESI†). This could be due to the high reinforcing effect of CNF because of the high surface-to-volume ratio.35 However, the P1 resin showed the highest σy, suggesting that a high shear stress is needed to deform and flow the resin. This could be attributed to the stronger interaction of A300 silica filler with the silicone polymer via hydrogen bonding due to the presence of a large number of silanol groups (free, vicinal and germinal silanols) on A300 silica filler.36 The low
and complex viscosity followed by the P1 and P2 resins (Fig. 2 and Table S1, ESI†). This could be due to the high reinforcing effect of CNF because of the high surface-to-volume ratio.35 However, the P1 resin showed the highest σy, suggesting that a high shear stress is needed to deform and flow the resin. This could be attributed to the stronger interaction of A300 silica filler with the silicone polymer via hydrogen bonding due to the presence of a large number of silanol groups (free, vicinal and germinal silanols) on A300 silica filler.36 The low 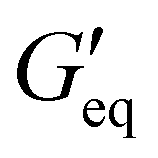 and σy of the P2 resin is attributed to a weaker interaction of A812 silica with the silicone polymer due to its hydrophobic nature and lower surface area.36
and σy of the P2 resin is attributed to a weaker interaction of A812 silica with the silicone polymer due to its hydrophobic nature and lower surface area.36
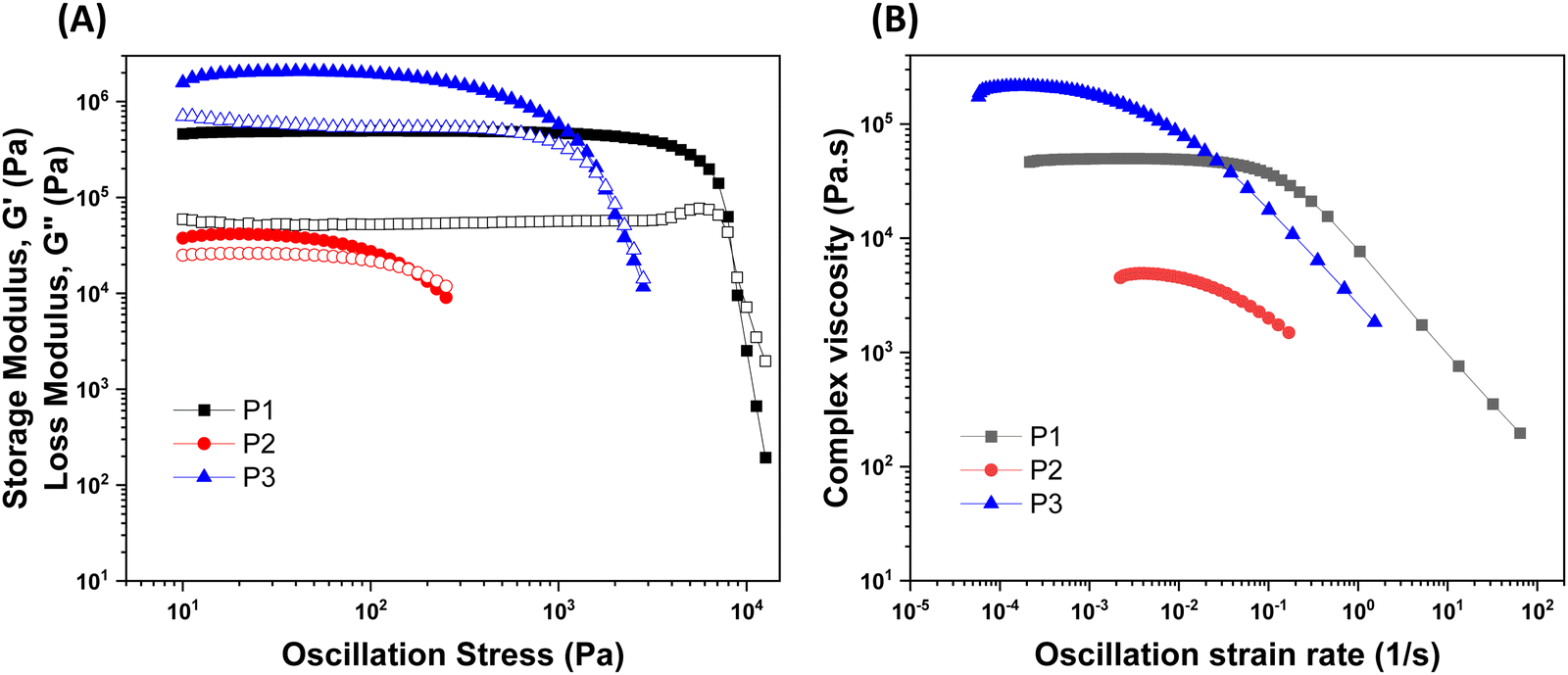 | ||
| Fig. 2 (A) Storage (G′, filled symbols) and loss (G′′, unfilled symbols) moduli as a function of oscillation stress and (B) complex viscosity as a function of oscillation strain rate for the P1, P2, and P3 resins. | ||
The three resins were successfully used as DIW feedstocks to print FCT pads using a 600 μm nozzle, 1000 μm center-to-center spacing between strands, and a layer height of 325 μm. Fig. 3 shows the cross-section view and top view of FCT pads before and after the extraction of NaCl.
 | ||
| Fig. 3 Cross section (left) and top view (right) optical microscope images of P1 (A), P2 (B), and P3 (C) samples before and after extraction of NaCl. | ||
3.3. Salt leaching and porosity calculation
The facile, conventional, and organic solvent free porogen leaching technique was utilized to impart porosity within the printed strands. The complete leaching of the salt was monitored by taking the dry mass of the printed samples before and after the extraction of NaCl in water. As shown in Table 3, the salt was quantitatively removed from the printed sample by extracting in water. Fig. 3 confirms that the lattice geometry and the printed strands were not significantly affected by the extraction process. The porosity of the samples (before and after extraction of NaCl) are reported in Table 2. The samples printed with the same nozzle size, strand distance, and layer height are expected to have similar porosity; however, the porosity of the samples prepared from three different resins using the 600 μm nozzle, 1000 μm strand distance, and 325 μm layer height have different porosity. The porosity of the printed pads (before extraction of NaCl) for the P1, P2, and P3 samples were 36%, 24%, and 30%, respectively. The difference in porosity of the printed pads with different resins could be attributed to the differences in their rheological properties resulting from the chemical structure and reinforcing nature of the different fillers. The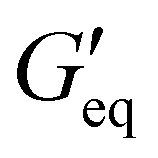 and σy values for the P2 resin containing A812 filler are comparatively low, which may lead to the slight sagging of the printed strands, resulting in an increase in density and a decrease in porosity. The P1 resin with A300 filler has a high σy value, suggesting a high tendency to retain its shape after deposition, which can be seen in Fig. 3(A). This leads to comparatively low density and high porosity in P1 among the other printed resins. The sagging of the printed strand is also evidenced by the difference in the thickness of the printed pads, which were 2.67 mm, 2.40 mm, and 2.64 for P1, P2, and P3, respectively. As expected, after the extraction of NaCl, the porosity of each printed sample increased and the increase in porosity corresponds to the vol% of NaCl i.e. by ∼30% (Table 3). The SEM images after extraction of NaCl (Fig. 4(A)–(F)) show that all samples have a silicone polymer matrix and hierarchical porous frameworks where irregular shaped microscale pores were introduced after extraction of NaCl in addition to the macroscale porosity present within the FCT lattice structures. These demonstrate the successful printing of the resin containing NaCl and functional fillers and development of hierarchically porous 3D printed silicone polymer composite material with functional fillers after leaching out of NaCl in water. Additionally, unlike other porous polymeric materials37 obtained by salt leaching technique where the porosity is determined by the amount of salt used, the combination of DIW and salt leaching techniques could enable tunable porosity based on lattice structure and printing parameters while keeping the salt content same. The pore size within the printed strands has been evaluated using image analysis, in which the pore diameter of the pores present within a 0.126 mm2 area of each sample were measured, and by plotting the relative frequency of the pores against the pore size (Fig. 4(G)–(I)). The average pore size within the printed strands for all samples looks similar and is influenced by, but not completely determined by, the particle size distribution of the ground NaCl. The pore size ranges between 1–50 μm, with the average pore size of 7–10 μm.
and σy values for the P2 resin containing A812 filler are comparatively low, which may lead to the slight sagging of the printed strands, resulting in an increase in density and a decrease in porosity. The P1 resin with A300 filler has a high σy value, suggesting a high tendency to retain its shape after deposition, which can be seen in Fig. 3(A). This leads to comparatively low density and high porosity in P1 among the other printed resins. The sagging of the printed strand is also evidenced by the difference in the thickness of the printed pads, which were 2.67 mm, 2.40 mm, and 2.64 for P1, P2, and P3, respectively. As expected, after the extraction of NaCl, the porosity of each printed sample increased and the increase in porosity corresponds to the vol% of NaCl i.e. by ∼30% (Table 3). The SEM images after extraction of NaCl (Fig. 4(A)–(F)) show that all samples have a silicone polymer matrix and hierarchical porous frameworks where irregular shaped microscale pores were introduced after extraction of NaCl in addition to the macroscale porosity present within the FCT lattice structures. These demonstrate the successful printing of the resin containing NaCl and functional fillers and development of hierarchically porous 3D printed silicone polymer composite material with functional fillers after leaching out of NaCl in water. Additionally, unlike other porous polymeric materials37 obtained by salt leaching technique where the porosity is determined by the amount of salt used, the combination of DIW and salt leaching techniques could enable tunable porosity based on lattice structure and printing parameters while keeping the salt content same. The pore size within the printed strands has been evaluated using image analysis, in which the pore diameter of the pores present within a 0.126 mm2 area of each sample were measured, and by plotting the relative frequency of the pores against the pore size (Fig. 4(G)–(I)). The average pore size within the printed strands for all samples looks similar and is influenced by, but not completely determined by, the particle size distribution of the ground NaCl. The pore size ranges between 1–50 μm, with the average pore size of 7–10 μm.
| Sample | Mass (g) | Change in mass (%) | Density (g cm−3) | Porosity of 3D printed pads (%) | ||||
|---|---|---|---|---|---|---|---|---|
| Before | After | Bulk | Before | After | Before | After | ||
| P1 | 0.849 | 0.422 | 49.7 | 1.29 | 0.820 | 0.515 | 36 | 60 |
| P2 | 0.829 | 0.410 | 49.4 | 1.17 | 0.890 | 0.520 | 24 | 55 |
| P3 | 0.898 | 0.446 | 49.6 | 1.25 | 0.874 | 0.534 | 30 | 57 |
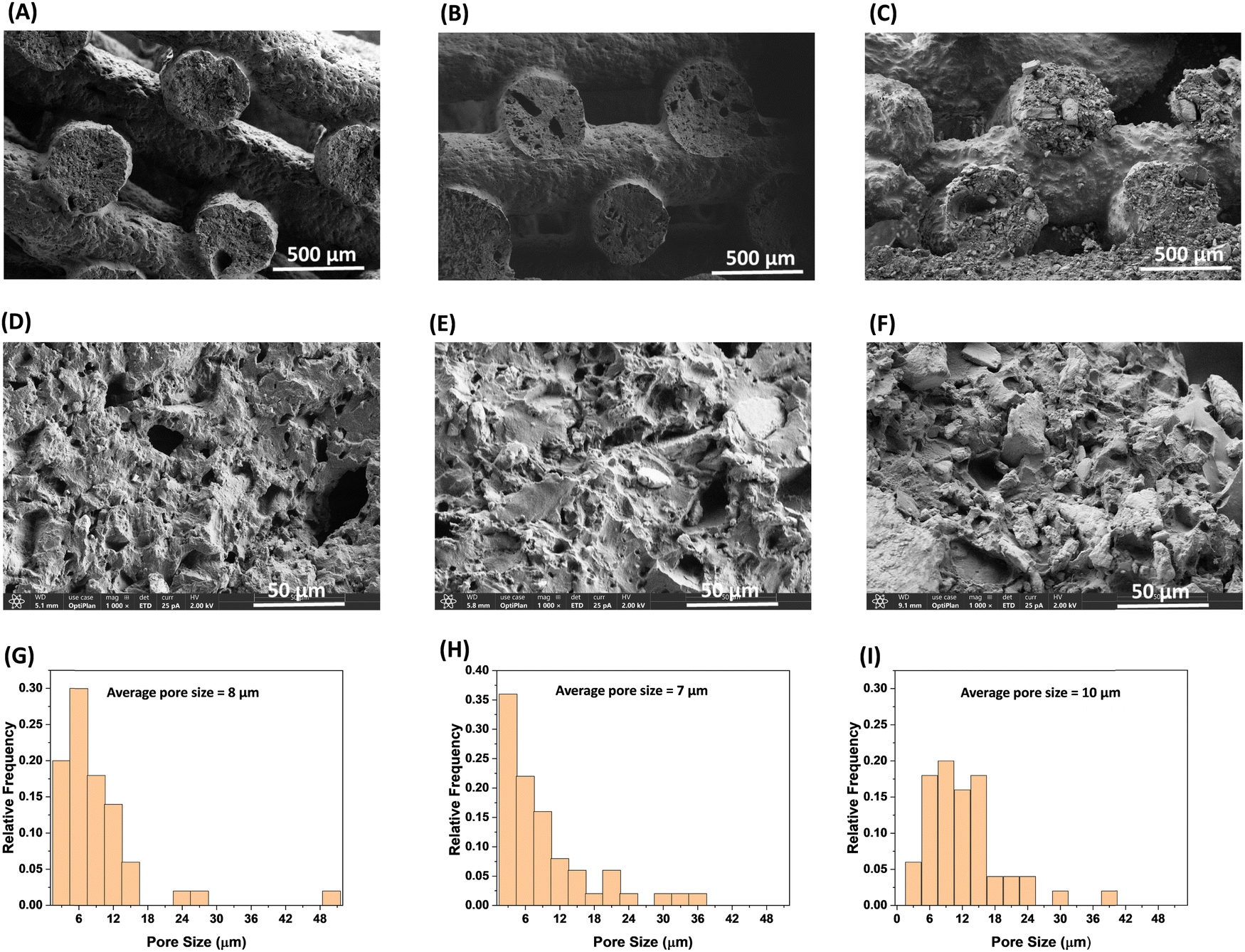 | ||
| Fig. 4 SEM images (A)–(F) at different magnification of cross-section and pore size distribution (D)–(F) of printed samples after extraction of NaCl. ((A), (D) and (G) for sample P1, (B), (E), and (H) for sample P2, and (C), (F), and (I) for sample P3). | ||
3.4. Mechanical characteristics (experimental and modelling)
The effect of these fillers on the mechanical properties of the porous polymer were studied by running tensile and compressive tests. The tensile and compressive stress–strain curves of the porous polymers with different fillers are shown in Fig. 5(A) and (B), respectively, and the data are listed in Table S2 (ESI†). As shown in Fig. 5(A) and Table S2 (ESI†), tensile strength of all three porous polymer samples with fillers (P1, P2 and P3) increased while the strain at break decreased after addition of the fillers compared to the porous sample without any fillers (P0). Among the samples with fillers, the P3 sample showed the highest strength (0.97 MPa) because of the reinforcing effect of the CNF due to having a high aspect ratio. These results indicate that mechanical properties of these porous 3D printed materials could be improved and modulate by selection of different functional fillers without changing their amount in the composite mixture. As expected, all porous samples after extraction of the NaCl reached higher compressive strains than their counterpart (without extraction of NaCl). The samples with high porosity showed high compressive strain. When compared to the previous materials30 obtained from the similar methodology, these material showed higher tensile strength and less compressive strain which could be attributed to the presence of functional fillers and lower porosity in these materials.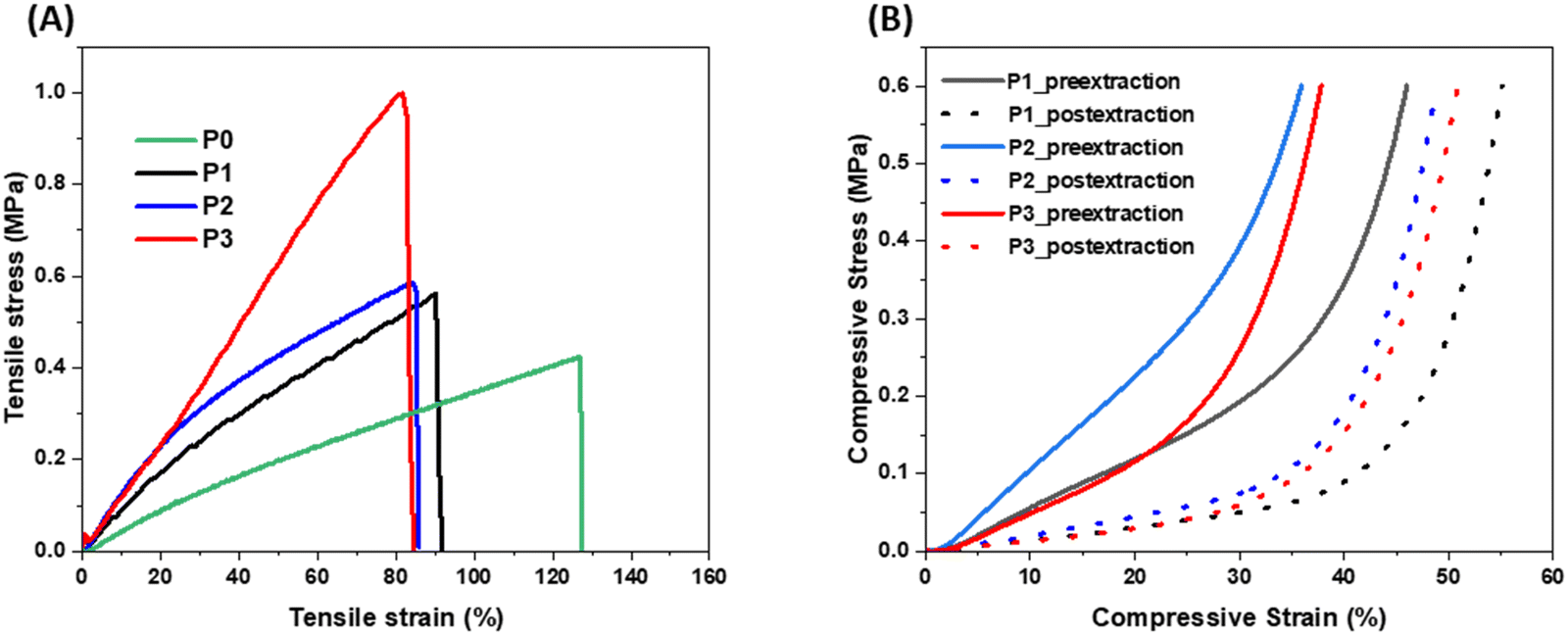 | ||
| Fig. 5 (A) Tensile stress–strain curves of the cast samples after extraction of NaCl, and (B) compressive stress–strain curve of the 3D printed samples before and after extraction of NaCl. Note that P0 sample could not be 3D printed into the FCT lattice structure thus not included for compressive tests. | ||
Uniaxial compression data obtained from the foam samples P1, P2, and P3 was used in the CHIP-Foam material model calibration work. We assumed that the state of deformation in the compressed foams can be approximated by uniaxial strain because the aspect ratio (diameter vs. thickness) of the samples was approximately 15:1. Therefore, the friction between the specimen surfaces and the loading platens minimized the lateral deformation resulting in uniaxial strain deformation. The measured porosity of the samples after NaCl extraction varied between 0.55 and 0.60. Therefore, the stress vs. strain curves for these samples were not expected to show significant plateau type behavior due to wall buckling. Consequently, we have not calibrated and Jb parameters that are used to simulate the buckling behavior. Material parameters and Jb were kept constant at 0.1 MPa and 0.999, respectively. In addition, it was reported that the uniaxial strain compression tests cannot be used to reliably calibrate the porous sample (foam) bulk modulus K. In this work, we used the constant value of K to be 4000 MPa. It has been demonstrated that for moderate compression loads the exact value of K does not affect the stress vs. strain behavior as long as K is large compared to C10. Finally, to calibrate the mechanical model for the post-extraction foams, we used the measured density of the bulk material (parent material) in the model. However, for the calibration of the pre-extraction specimens, we concluded that the measured bulk density would underestimate the actual density of the parent material. This is because the parent material contained embedded NaCl particles that have higher density than the PDMS parent material. Therefore, we allowed the optimization algorithm to find the parent material density for each run for the pre-extraction foam calibrations. Consequently, for pre-extraction specimens, we calibrated Ĝ, C10, and ρbulk material parameters. For post-extraction specimens, we calibrated Ĝ and C10 parameters.
The material model calibration is an optimization procedure. We start with approximating the values for optimized parameters Ĝ, C10, and ρbulk. Next, the stresses at test strains are computed using the model and compared with the experimental stresses. During the optimization procedure, the difference between the computed and measured stresses is minimized by changing the values of parameters Ĝ, C10 and ρbulk within a pre-defined interval (bounded optimization problem). In this work, the L-BFGS-B algorithm was used to perform the optimization of Ĝ, C10, and ρbulk parameters. This algorithm was chosen because it allows the user to specify the bounds on calibrated parameters.
The comparison between the experimental and simulated stress–strain curves for pre- and post-extraction samples P1, P2, and P3 is shown in Fig. 6(A)–(C), respectively. The corresponding material parameters obtained from the calibration of pre- and post-extraction stress vs. strain tests for P1, P2, and P3 samples are shown in Table 4.
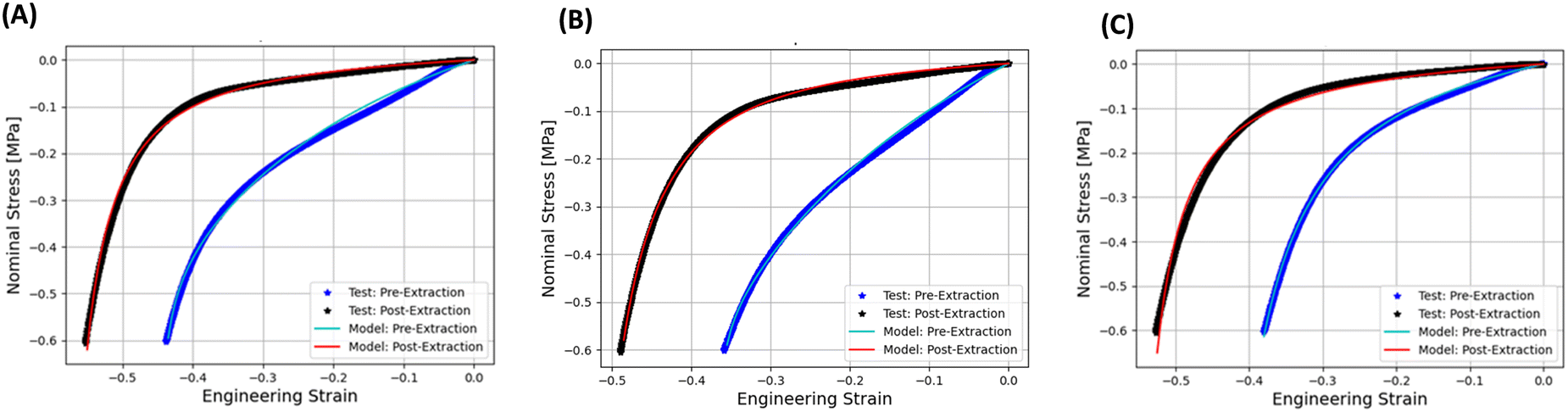 | ||
| Fig. 6 Comparison between physical experiments and simulated stress–strain curves for pre- and post-extraction compressed foam specimens. Data for fourth compression loading cycle is shown for (A) P1, (B) P2, and (C) P3 samples. | ||
| Sample | Material parameter | Pre-extraction | Post-extraction |
|---|---|---|---|
| P1 | Ĝ (MPa) | 0.382 | 0.0331 |
|
(MPa) |
0.1 | 0.1 | |
| J b | 0.999 | 0.999 | |
| C 10 (MPa) | 0.002 | 0.0139 | |
| K (MPa) | 4000 | 4000 | |
| ρ foam (g cm−3) | 0.820 | 0.516 | |
| ρ bulk (g cm−3) | 1.51 | 1.29 | |
| P2 | Ĝ (MPa) | 0.636 | 0.0170 |
|
(MPa) |
0.1 | 0.1 | |
| J b | 0.999 | 0.999 | |
| C 10 (MPa) | 0.0011 | 0.0257 | |
| K (MPa) | 4000 | 4000 | |
| ρ foam (g cm−3) | 0.891 | 0.520 | |
| ρ bulk (g cm−3) | 1.42 | 1.17 | |
| P3 | Ĝ (MPa) | 0.1379 | 0.048 |
|
(MPa) |
0.1 | 0.1 | |
| J b | 0.999 | 0.999 | |
| C 10 (MPa) | 0.042 | 0.015 | |
| K (MPa) | 4000 | 4000 | |
| ρ foam (g cm−3) | 0.874 | 0.534 | |
| ρ bulk (g cm−3) | 1.65 | 1.25 | |
It is seen in Fig. 6(A)–(C) that the calibrated CHIP-Foam model was able to accurately reproduce the measured stress–strain curves for pre- and post-extraction compressed foams. The model was able to reproduce the initial compliant phase of the deformation (deformation of the walls and gradual closing of the pores) and the subsequent stiffening due to the transition to the incompressible deformation (pores mostly closed).
It is seen in the data presented in Table 4 that the calibrated ρbulk for pre-extraction specimens was about 10 to 30% higher than measured ρbulk for the corresponding post-extraction foams. This observation is a consequence of the fact that the parent material has higher density than the post-extraction foams due to the presence of embedded NaCl. We also see that calibrated Ĝ parameter for pre-extraction specimens is higher than Ĝ for post-extraction foams. This can also be explained by the fact that the parent material should have higher stiffness for pre-extraction specimens due to the reinforcing effect of NaCl particles. Finally, we have not observed any clear trends for C10 parameter for pre- and post-extraction specimens. This is probably caused by the fact that C10 parameter has very low magnitude and the numerical optimization procedure is not able to resolve the value of C10 based on the existing test data.
3.5. Applications
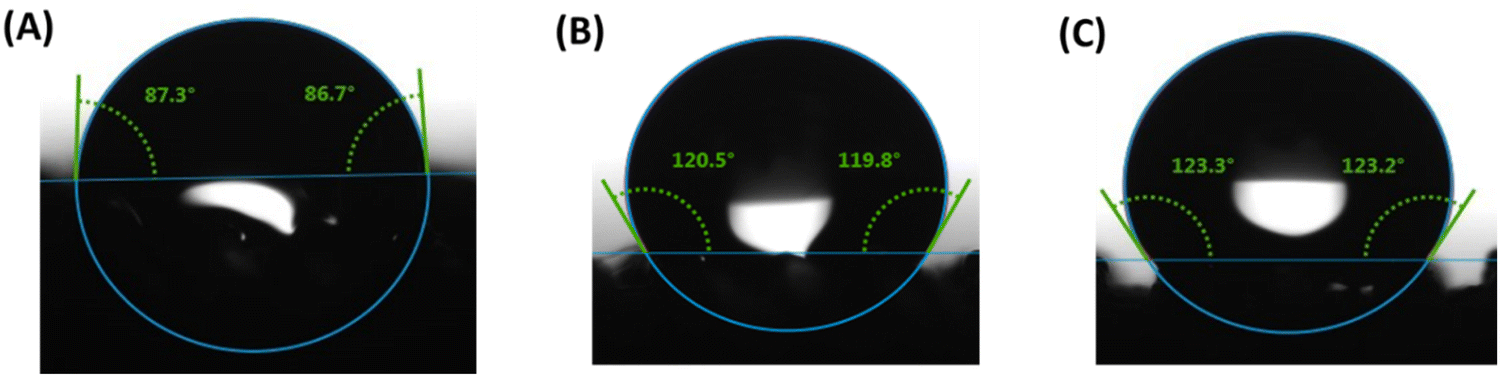 | ||
| Fig. 7 Contact angles from water droplets on the surface of 3D printed pads (A) P1, (B) P2, and (C) P3 after extraction of NaCl. | ||
Since the P3 sample showed higher hydrophobicity (higher water contact angle of ∼123), had comparatively higher porosity (57%), and high tensile strength (0.97 MPa) compared to Pl and P2 samples, the P3 sample was chosen to demonstrate oil/water separation. Fig. 8(A) demonstrates the separation of oil (chloroform, blue color) from water using the P3 sample. When the P3 sample was immersed in the water that contained chloroform, it absorbed chloroform immediately (within a few seconds) as shown in the Video S1 (ESI†). Fig. 8(B) shows the change in dimension of the P3 sample during the absorption of chloroform. We measured the absorption efficiency of the P3 sample for two organic solvents having different density (chloroform and toluene). The absorbing efficiencies of the P3 sample for chloroform and toluene were found to be 440% and 330%, respectively, demonstrating the potential of using this porous structure for functional applications. The difference in absorption efficiencies for two different organic solvents is attributed to the difference in their density (the density of chloroform and toluene is 1.49 g cm−3 and 0.867 g cm−3) respectively.
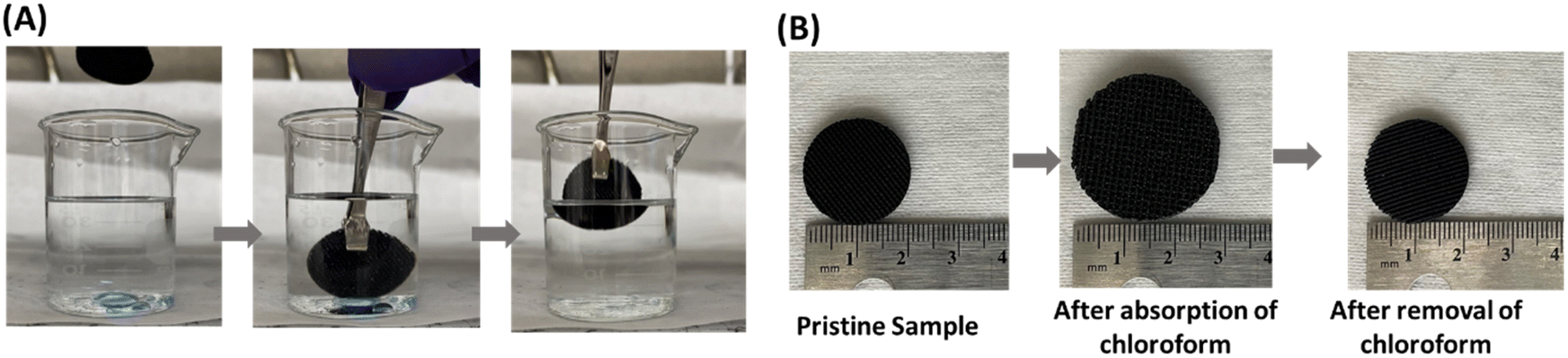 | ||
| Fig. 8 Optical images demonstrating (A) selective chloroform (blue color) absorption in water by the P3 sample and (B) swelling of the P3 sample after absorbing chloroform. | ||
The DIW resin for this polymer getter composite was comprised of 39 wt% of PDMS prepolymers (9:1 ratio of PDV-541 to HMS-301), 50 wt% of NaCl, 10 wt% of active hydrogen getter (DEB–Pd/C mixture in 3:1 ratio by weight) and 1 wt% of CNF. The rheology of this resin is given in the Fig. S2A (ESI†) and the optical microscope picture of 3D printed pads before and after extraction of NaCl are given in Fig. S3 (ESI†). The difference in mass of the printed pads before and after extraction of NaCl confirmed the quantitative extraction of NaCl (Table S3, ESI†). After extraction of NaCl, porosity of the composite increased from 46% to 70% and the compressive strain at 0.6 MPa changed from 46% to 68% (Fig. S2B and Table S3, ESI†). Note that although we have started with 10 wt% of an active hydrogen getter in the resin, the final 3D printed getter composite after extraction of NaCl contained 20 wt% of an active hydrogen getter. Thus, the hydrogen absorption performance of it was compared with our previously reported 3D printed polymer getter composite comprising 20 wt% of an active getter. To minimize the effects of other factors such as geometry on the hydrogen absorption capacity, both of the 3D printed samples had the same FCT lattice geometry having 1000 μm center-to-center spacing between strands and 325 μm layer height with eight layers and were cured for 5 h at 75 °C in a preheated oven.
The hydrogen absorption study was conducted for 21 days under the same setting as reported in our previous work.28 As shown in Fig. 9 and Table 5 the initial specific reaction rate of the 3D printed porous polymer getter composite with porosity within the printed strands is nearly three times that of the 3D printed polymer getter composite without porosity in the printed strands. This suggests that higher surface area resulted in faster consumption of hydrogen gas and DEB conversion in the polymer getter composite. We speculate that higher surface area favors the easy access of hydrogen gas to more active sites (catalyst) in the polymer getter composite resulting in a higher reaction rate. After 21 days, the specific reaction rate of the 3D printed sample with porosity within the strands decreased (35 scc H2 per g sample per year) with 67% of DEB conversion, whereas the specific reaction rate of the 3D printed sample without porosity within the printed strands became 139 scc H2 per g sample per year with 63% of DEB conversion. Although the initial reaction rate was enhanced by an increase in surface area of the polymer getter composite, the final DEB conversion remained almost the same, suggesting a further increase in porosity may not influence the fraction of DEB conversion. Dong et al.38 also reported a similar observation, where the fractional DEB conversion remained almost same above 30 wt% of polyethylene glycol (porogen to create the porosity) in the polymer getter composite.
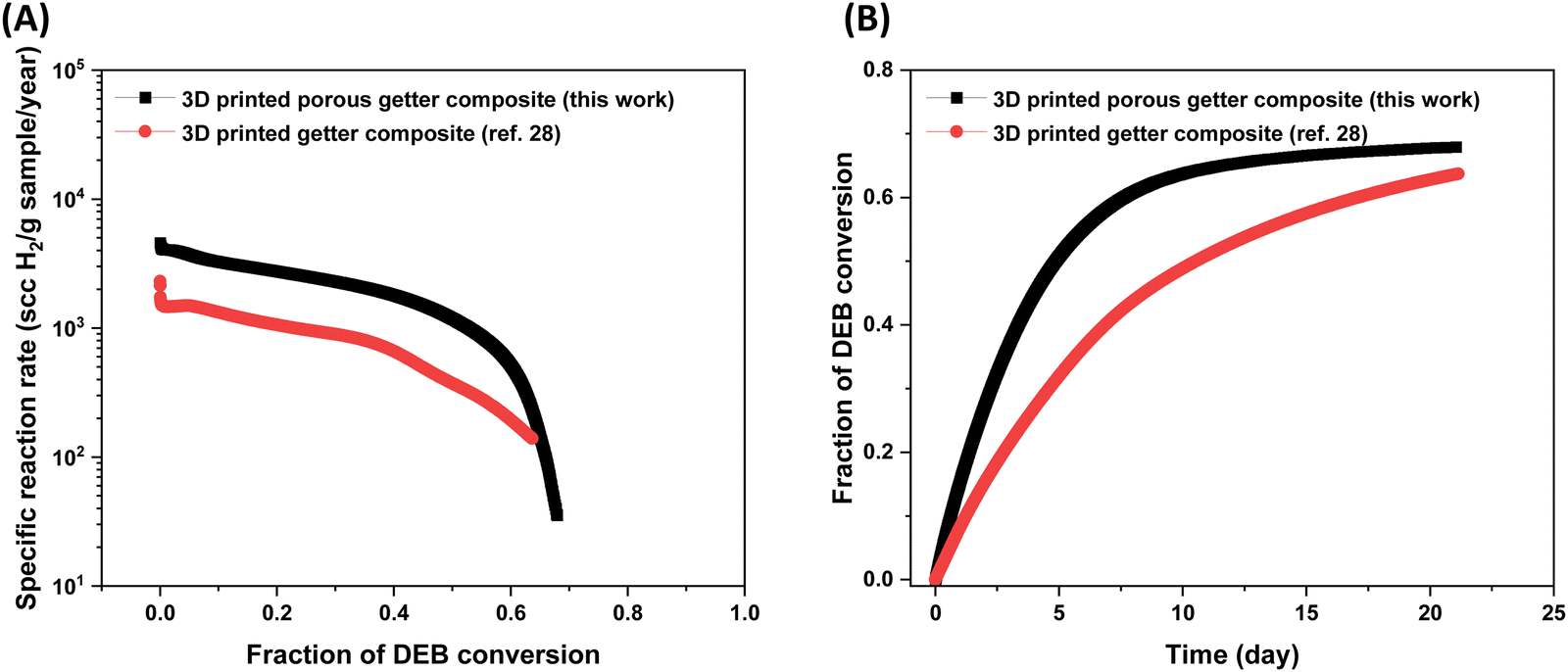 | ||
| Fig. 9 Plot of (A) reaction rate vs. fraction of DEB conversion and (B) fraction of DEB conversion vs. time for printed getter composites with and without porosity in the printed strands. The data for the 3D printed getter composite without porosity in the printed strand was taken from our previous work for comparison.28 Note that the hydrogen absorption studies were run for 21 days. | ||
| Sample | Amount of DEB–Pd/C | Initial reaction rate (scc H2 per g sample per year) × 103 | Normalized DEB conversion after 21 days | Ref. |
|---|---|---|---|---|
| 3D printed porous getter composite | 20 wt% | 4.31 | 0.67 | This work |
| 3D printed getter composite | 20 wt% | 1.56 | 0.63 | 28 |
4. Conclusions
We report a novel method to develop DIW resin by grinding NaCl with silicone pre-polymers in order to reduce the NaCl particle size and to facilitate the DIW printing with functional fillers to expand functionality of 3D printed porous materials for different applications. NaCl was ground with silicone prepolymer to mean particle size of 8.8 μm, which enabled successful DIW printing and, after organic solvent free salt leaching, introduced microscale porosity into the printed strands in addition to the macroscale porosity present within the FCT lattice structures. Functional fillers (fumed silica and CNFs) were added to the silicone polymer system, where they demonstrated tunable rheology within a printable range for DIW feedstocks. While the porous base polymer had ∼0.4 MPa tensile strength and ∼130% tensile strain, the tested fillers tuned the strain to between 90% and 100%. Fumed silica resulted in an ∼25% increase in tensile strength, while the CNF filler resulted in an increase of 130%, likely because of the high reinforcing effect due to the high aspect ratio of CNFs. The uniaxial compressive behavior was found to be less dependent on filler: strains at 0.6 MPa increased between 10% and 17% after salt extraction, and all foams compressed around 50% after extraction of NaCl. The CHIP Foam model validated the experimental compression behavior of these porous polymer with fillers. Water droplet contact angles on fumed silica strands ranged between 87° and 120°, demonstrating capabilities for both hydrophobic and hydrophilic applications of the porous silicone polymer composites. The CNF foam demonstrated the highest hydrophobicity (123° contact angle), and it demonstrated effective oil/water separation capability with both chloroform (450% absorption efficiency) and toluene (330% absorption efficiency). The functional potential for these filled porous silicone polymer composites is further demonstrated with the addition of a hydrogen getter (DEB–Pd/C). The hydrogen getter composite with hierarchical porosity in the FCT structure has a higher specific reaction rate per fraction of DEB conversion and has a higher fraction of DEB conversion over time, compared to the same material composition in an FCT structure without porosity in the strands. Because these promising applications have been demonstrated for this limited selection of fillers, it would be of interest to continue expanding DIW feedstock capabilities by exploring additional fillers and concentration ratios to expand the diversity of impactful, functional applications.Author contributions
Credit: Santosh Adhikari conceptualization (lead), investigation (lead), data curation (lead), formal analysis (lead), methodology (lead), writing – original draft (lead), writing – review & editing (equal); Xavier M. Torres investigation (equal), formal analysis (equal), methodology (equal), writing – original draft (supporting), writing – review & editing (supporting); John R. Stockdale investigation (equal), data curation (equal), methodology (equal), formal analysis (equal), writing – review & editing (equal); Shelbie A. Legett data curation (supporting); Lindsey B. Bezek data curation (supporting), writing – review & editing (equal); Jesus A. Guajardo data curation (supporting), Adam Pacheco data curation (supporting), Karthik Ramasamy data curation (supporting), formal analysis (supporting), writing – review & editing (supporting), Bart Benedikt data curation (supporting), formal analysis (supporting), writing – original draft (supporting), Matthew Lewis data curation (supporting), Andrea Labouriau funding acquisition (lead), supervision (lead), writing – review & editing (equal).Data availability
The data supporting the findings of this work are available within the article and its ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was performed under the U.S. Department of Energy through the Los Alamos National Laboratory. Los Alamos National Laboratory is operated by Triad National Security, LLC, for the National Nuclear Security Administration of U.S. Department of Energy (Contract No. 89233218CNA000001). It was also partially supported by the Institute of Material Science, Los Alamos National Laboratory.References
- D. Wu, F. Xu, B. Sun, R. Fu, H. He and K. Matyjaszewski, Chem. Rev., 2012, 112, 3959–4015 CrossRef CAS PubMed.
- J. Wu, F. Xu, S. Li, P. Ma, X. Zhang, Q. Liu, R. Fu and D. Wu, Adv. Mater., 2019, 31, 1802922 CrossRef PubMed.
- W. Lu, D. Yuan, D. Zhao, C. I. Schilling, O. Plietzsch, T. Muller, S. Bräse, J. Guenther, J. Blümel, R. Krishna, Z. Li and H.-C. Zhou, Chem. Mater., 2010, 22, 5964–5972 CrossRef CAS.
- R.-X. Yang, T.-T. Wang and W.-Q. Deng, Sci. Rep., 2015, 5, 10155 CrossRef CAS PubMed.
- B. Zhang, W. Wang, L. Liang, Z. Xu, X. Li and S. Qiao, Coord. Chem. Rev., 2021, 436, 213782 CrossRef CAS.
- J. Mandal, Y. Fu, A. C. Overvig, M. Jia, K. Sun, N. N. Shi, H. Zhou, X. Xiao, N. Yu and Y. Yang, Science, 2018, 362, 315–319 CrossRef CAS PubMed.
- T. Zhang, G. Xing, W. Chen and L. Chen, Mater. Chem. Front., 2020, 4, 332–353 RSC.
- Y. Zhu, P. Xu, X. Zhang and D. Wu, Chem. Soc. Rev., 2022, 51, 1377–1414 RSC.
- Y. Mi, Z. Zhao, H. Wu, Y. Lu and N. Wang, Polymers, 2023, 15, 4383 CrossRef CAS PubMed.
- D. Zhu, S. Handschuh-Wang and X. Zhou, J. Mater. Chem. A, 2017, 5, 16467–16497 RSC.
- S. Hong, H. Kim, N. Qaiser, P. Baumli and B. Hwang, J. Nat. Fibers, 2023, 20, 2264497 CrossRef.
- X. Zhao, L. Li, B. Li, J. Zhang and A. Wang, J. Mater. Chem. A, 2014, 2, 18281–18287 RSC.
- I. Miranda, A. Souza, P. Sousa, J. Ribeiro, E. M. S. Castanheira, R. Lima and G. Minas, J. Funct. Biomater., 2021, 13, 2 CrossRef PubMed.
- S. Masihi, M. Panahi, D. Maddipatla, A. J. Hanson, A. K. Bose, S. Hajian, V. Palaniappan, B. B. Narakathu, B. J. Bazuin and M. Z. Atashbar, ACS Sens., 2021, 6, 938–949 CrossRef CAS PubMed.
- S. Wu, J. Zhang, R. B. Ladani, A. R. Ravindran, A. P. Mouritz, A. J. Kinloch and C. H. Wang, ACS Appl. Mater. Interfaces, 2017, 9, 14207–14215 CrossRef CAS PubMed.
- C. Yu, C. Yu, L. Cui, Z. Song, X. Zhao, Y. Ma and L. Jiang, Adv. Mater. Interfaces, 2017, 4, 1600862 CrossRef.
- C. Parameswaran, R. P. Chaudhary, S. Hosangadi Prutvi and D. Gupta, ACS Appl. Polym. Mater., 2022, 4, 2047–2056 CrossRef CAS.
- M. M. Keshtiban, M. M. Zand, A. Ebadi and Z. Azizi, Biomed. Mater., 2023, 18, 045012 CrossRef PubMed.
- E. Pedraza, A.-C. Brady, C. A. Fraker and C. L. Stabler, J. Biomater. Sci., Polym. Ed., 2013, 24, 1041–1056 CrossRef CAS PubMed.
- J. Zhao, G. Luo, J. Wu and H. Xia, ACS Appl. Mater. Interfaces, 2013, 5, 2040–2046 CrossRef CAS PubMed.
- M. Abshirini, M. C. Saha, M. Cengiz Altan and Y. Liu, Mater. Des., 2021, 212, 110194 CrossRef CAS.
- M. Abshirini, M. C. Saha, L. Cummings and T. Robison, Polym. Eng. Sci., 2021, 61, 1943–1955 CrossRef CAS.
- H. Wang, R. Zhang, D. Yuan, S. Xu and L. Wang, Adv. Funct. Mater., 2020, 30, 2003995 CrossRef CAS.
- D. Ortiz-Acosta, T. Moore, D. J. Safarik, K. M. Hubbard and M. Janicke, Adv. Funct. Mater., 2018, 28, 1707285 CrossRef.
- Z. He, N. Wang, L. Mu, Z. Wang, J. Su, Y. Chen, M. Luo, Y. Wu, X. Lan and J. Mao, Front. Bioeng. Biotechnol., 2023, 11, 1272565 CrossRef PubMed.
- L. A. Connal and G. G. Qiao, Adv. Mater., 2006, 18, 3024–3028 CrossRef CAS.
- R. Zheng, Y. Chen, H. Chi, H. Qiu, H. Xue and H. Bai, ACS Appl. Mater. Interfaces, 2020, 12, 57441–57449 CrossRef CAS PubMed.
- S. Adhikari, D. J. Safarik, J. R. Stockdale, X. M. Torres, A. Pacheco, S. A. Legett and A. Labouriau, ACS Omega, 2024, 9, 15547–15555 CrossRef CAS PubMed.
- J. R. Stockdale, S. A. Legett, X. M. Torres, A. Pacheco, S. Adhikari, L. Bezek and A. Labouriau, J. Appl. Polym. Sci., 2024, 141, e55236 CrossRef CAS.
- Q. Chen, J. Zhao, J. Ren, L. Rong, P.-F. Cao and R. C. Advincula, Adv. Funct. Mater., 2019, 29, 1900469 CrossRef.
- C. K. Loeb, D. T. Nguyen, T. M. Bryson, E. B. Duoss, T. S. Wilson and J. M. Lenhardt, Addit. Manuf., 2022, 55, 102837 CAS.
- M. Lewis, Eur. J. Eng. Mech., 2019, 36, 88–101 Search PubMed.
- M. Danielsson, D. M. Parks and M. C. Boyce, Mech. Mater., 2004, 36, 347–358 CrossRef.
- S. A. Legett, X. Torres, A. M. Schmalzer, A. Pacheco, J. R. Stockdale, S. Talley, T. Robison and A. Labouriau, Polymers, 2022, 14, 4661 CrossRef CAS PubMed.
- K. L. Klein, A. V. Melechko, T. E. McKnight, S. T. Retterer, P. D. Rack, J. D. Fowlkes, D. C. Joy and M. L. Simpson, J. Appl. Phys., 2008, 103, 061301 CrossRef.
- X. M. Torres, J. R. Stockdale, S. Adhikari, S. A. Legett, A. Pacheco, J. A. Guajardo and A. Labouriau, Polymers, 2023, 15, 4334 CrossRef CAS PubMed.
- F. Chen, Y. Lu, X. Liu, J. Song, G. He, M. K. Tiwari, C. J. Carmalt and I. P. Parkin, Adv. Funct. Mater., 2017, 27, 1702926 CrossRef.
- H. Dong, Y. Wang, H. Fu, M. Ye, G. Tang, J. Pan and X. Xia, ACS Appl. Polym. Mater., 2020, 2, 3243–3250 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ma00457d |
| This journal is © The Royal Society of Chemistry 2024 |