
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Investigating the role of mixed-cation ionic liquid electrolytes in sodium battery efficiency and stability†
Lixu
Huang
 ,
Faezeh
Makhlooghiazad
,
Luke A.
O'Dell
,
Patrick C.
Howlett
and
Maria
Forsyth
*
,
Faezeh
Makhlooghiazad
,
Luke A.
O'Dell
,
Patrick C.
Howlett
and
Maria
Forsyth
*
Institute for Frontier Materials (IFM), Deakin University, Burwood, Victoria 3125, Australia. E-mail: maria.forsyth@deakin.edu.au; Tel: +61 3 92446821
First published on 17th July 2024
Abstract
This study explores the influence of mixed-cation ionic liquid (IL)-NaFSI based electrolyte systems on their physicochemical and electrochemical properties. Utilising two ionic liquids with distinct cation chemistries, (trimethyl isobutyl phosphonium) P111i4+ and (N-methyl-N-propylpyrrolidinium) C3mpyr+, combined in various ratios with either a constant 42 mol% NaFSI salt concentration or a saturated NaFSI concentration, we examined their thermal behaviour, ionic conductivities, and electrochemical properties over a range of temperatures. We discovered that NaFSI addition disrupts in the neat IL crystallization in both salt concentrations (42 mol% and saturated), resulting in only glass transition temperatures (Tg) that were lower for P111i4/NaFSI than in C3mpyrFSI/NaFSI systems indicating an influence of the cation type on the thermal properties of these IL electrolytes. In both the 42 mol% NaFSI and saturated NaFSI systems, Tg in C3mpyrFSI/P111i4FSI/NaFSI mixtures slightly decreased as the P111i4FSI content increased. This was consistent with a slight increase in ionic conductivity and in cation and anion diffusion coefficients with higher P111i4FSI content and temperature. Despite modest changes in ionic conductivity and diffusion coefficients, the electrochemical behaviour shows increased current density and earlier sodium plating initiation with increasing P111i4FSI concentration, highlighting the potential of cation mixing to enhance electrochemical properties suggesting that cation mixing shows potential for improved electrochemical properties through optimized IL electrolyte composition. This work underscores the feasibility of optimizing IL electrolyte compositions for improved performance and stability in sodium batteries, paving the way for future research on cation chemistry effects and practical applications in high-temperature (50 °C) Na–metal-based batteries.
1. Introduction
Fossil fuels, such as coal, petroleum, and natural gas, have long served as the primary global energy sources, driving economic growth but also causing environmental concerns such as the greenhouse effect and climate change. To mitigate these issues, there is an urgent need to transition from fossil fuels to renewable energy sources. Rechargeable batteries emerge as the most competitive option due to their high energy efficiency, low maintenance requirements, and extended cycle life.1 While lithium-ion batteries currently dominate, their sustainability is challenged by uneven distribution and limited supply of lithium resources, as well as rising prices for related elements such as cobalt.2 As an alternative, sodium (Na) batteries present a promising solution for some applications, utilizing abundant and cost-effective sodium resources.3In the current sodium battery research, the focus is mainly on the exploration of positive and negative electrode materials, and electrolyte materials only account for a small proportion of academic discussions.4,5 Despite the widespread use of commercial organic materials as solvents in electrolytes, their poor thermal stability raises serious safety concerns.6,7 Therefore, developing safer electrolyte materials has become a key task in promoting the widespread adoption of sodium batteries in energy storage applications. Ionic liquids (ILs), composed of large organic cations and high-charge delocalized anions, present an intriguing alternative.8 With unique properties such as negligible vapor pressure, low flammability, and high thermal stability, ILs offer safe alternatives to conventional organic electrolytes.9 An advantage of ionic liquids is that they are designable by the combination of cations and anions, which is also achieved by mixing different ionic liquids to tune the physicochemical and electrochemical properties of electrolytes.10
The most commonly studied ionic liquid electrolytes are predominantly based on imidazolium or pyrrolidinium cations. By making slight modifications to their structure, it is possible to introduce a greater variety of cationic species in ionic liquids than anionic ones. Typically, introducing bulky or long substituents lowers the melting point, while the viscosity increases and the ionic conductivity tends to decrease. As a result, asymmetric cations with short alkyl chains, such as [C3mpyr]+ and 1-ethyl-3-methylimidazolium [EMIM]+, are often preferred.3 Compared to their imidazole-based counterparts, non-aromatic pyrrolidine-based cations exhibit greater reductive stability, making them a popular choice for use in lithium and sodium batteries.11
The C3mpyrFSI IL with different molar ratios of NaFSI was characterized by Hagiwara's group, who reported an ionic conductivity of 3.2 × 10−3 S cm−1 at 25 °C with an IL:Na ratio of 8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2.12 The Na cells with C3mpyrFSI and 20 mol% NaFSI showed a stable charge–discharge behaviour, delivering capacities of 92 and 106 mA h g−1 at 25 and 80 °C, respectively, using a current of 20 mA g−1. Research on the use of different concentrations of NaFSI salt with C3mPyrFSI has also been systematically conducted by Forsyth et al. They demonstrated that the highest concentration of NaFSI (50 mol% in C3mpyrFSI) showed the most stable Na cycling at higher current densities (1 mA cm−2) relative to the lower concentrations, while the interfacial resistance decreased with high NaFSI salt concentration, as determined by impedance measurements, resulting in faster charge transfer at the interface.13 Both research groups proposed that the higher transference number for the Na cation in mixtures with higher concentrations of ILs is the reason for the improved performance in Na batteries. This was also recently illustrated by an in-depth modelling study of the dynamics of concentrated IL ions (C3mpyrFSI + NaFSI) which demonstrated that the structural rearrangement in the IL structure promotes the decoupling motion of Na+ with increasing salt addition.14,15
:2.12 The Na cells with C3mpyrFSI and 20 mol% NaFSI showed a stable charge–discharge behaviour, delivering capacities of 92 and 106 mA h g−1 at 25 and 80 °C, respectively, using a current of 20 mA g−1. Research on the use of different concentrations of NaFSI salt with C3mPyrFSI has also been systematically conducted by Forsyth et al. They demonstrated that the highest concentration of NaFSI (50 mol% in C3mpyrFSI) showed the most stable Na cycling at higher current densities (1 mA cm−2) relative to the lower concentrations, while the interfacial resistance decreased with high NaFSI salt concentration, as determined by impedance measurements, resulting in faster charge transfer at the interface.13 Both research groups proposed that the higher transference number for the Na cation in mixtures with higher concentrations of ILs is the reason for the improved performance in Na batteries. This was also recently illustrated by an in-depth modelling study of the dynamics of concentrated IL ions (C3mpyrFSI + NaFSI) which demonstrated that the structural rearrangement in the IL structure promotes the decoupling motion of Na+ with increasing salt addition.14,15
Phosphonium-based ILs and their solid-state counterparts, known as organic ionic plastic crystals (OIPCs), have been reported to offer significant advantages compared to their nitrogen-based IL correspondents. These advantages include higher ionic conductivity and enhanced electrochemical stability.16 Hilder and co-workers explored the physicochemical and electrochemical properties of FSI− anion-based ILs paired with three different cations: two small alkyl phosphonium cations: P111i4+ and P1i4i4i4+ and an alkoxy ammonium counter cations: N2(2O2O1)3+. These ILs were mixed with NaFSI salt to near saturation (at approximately 1:1 mol ratio). At 50 °C, the ionic conductivity of the high salt content IL mixture (P111i4FSI–NaFSI) was measured to be 4.4 mS cm−1 which is the highest among the electrolytes studied, whereas the N2(2O2O1)3FSI–NaFSI system exhibited the lowest ionic conductivity. Interestingly, despite its lower ionic conductivity, the N2(2O2O1)3FSI–NaFSI mixture demonstrated excellent reversible sodium reduction/oxidation in CV measurements, with a maximum peak current density of 10 mA cm−2, comparable to 17 mA cm−2 of the P111i4FSI–NaFSI system, which had the highest ionic conductivity. Na symmetric cycling revealed that sodium stripping/plating in the N2(2O2O1)3FSI–NaFSI mixture was as stable as in the P111i4FSI–NaFSI electrolyte underscoring the critical role of the solid–electrolyte interphase (SEI) structure and morphology on the surface of sodium metal in cycling performance. Rakov et al. reported the formation of a Nax(FSI)y molten salt-like nanostructured interfacial layer at the surface of the electrode, preconditioned with a high-current-density protocol in a high sodium salt content C3mpyrFSI electrolyte containing 50 mol% NaFSI.17 In a comparative surface study, Ferdousi et al. highlighted that phosphonium IL-based electrolytes demonstrated higher cycling stability and capacity (up to 4 mA h cm−2 at current densities of 1, 2, and 4 mA h cm−2) compared to the rapid failure of C3mpyrFSI electrolytes (at 1 mA h cm−2 for 4 mA h cm−2). Surface characterization revealed that the addition of water significantly improved the SEI of the phosphonium cation IL, evidenced by a greater proportion of a Na complex and a NaF-rich surface compared to that with C3mpyrFSI.18
It has been reported that the properties of ILs, such as ionic conductivity, viscosity, and cost, can be adjusted by mixing ILs with different anions. The addition of NaDCA, NaFTFSI, NaTFSI and NaFSI influences the morphology of the Na surface after cycling in C3mpyrDCA IL. NaFSI was found to form a more stable SEI layer, as indicated by prolonged symmetrical cell cycling. In contrast, both the TFSI and FTFSI salts led to the formation of thicker, highly passivating surfaces. The SEI layer in the C3mpyrDCA–NaFSI system was primarily composed of NaF, which facilitated stable cycling with the lowest overpotential for more than 100 cycles.19
This study demonstrated that, in principle, utilizing a low-cost IL can retain the benefit of safety and recyclability while simultaneously providing the desirable SEI forming features required for effective battery cycling. While most research has focused on the effect of mixed anion ILs on Na anodes, only minimal attention has been given to the effect of mixed cations and the implications on bulk properties and electrochemical behaviour.
In this study, we investigate the influence of mixed-cation IL electrolyte systems on their physicochemical and electrochemical properties, focusing on P111i4+ and C3mpyr+ as cation chemistry. P111i4+ is recognized for its high ionic conductivity and chemical electrochemical stability; however, its synthesis pathway results in high cost which would currently limit its practical application. Conversely, C3mpyr+ emerges as a more cost-effective alternative with promising properties. By mixing these ILs, we aim to achieve a balance between superior performance and economic feasibility. We select the FSI− anion for its ability to form a reliable solid electrolyte interphase (SEI). Our methodology involves two strategies: initially, mixing varying ratios of C3mpyrFSI/P111i4FSI with a saturated NaFSI to maintain a high salt concentration, beneficial for uniform metal deposition and dendrite prevention on the metal anode during cycling. Second, we employed a fixed 42 mol% NaFSI concentration – saturated in the P111i4FSI IL – to isolate the effects of cation chemistry on the mixed IL electrolyte's properties. Our findings reveal a direct correlation between C3mpyrFSI proportion and physicochemical properties in the 42 mol% NaFSI system, as well as the significant impact of sodium salt concentration on these properties.
2. Experimental and/or theoretical methods
2.1. Sample preparation
Ionic liquids P111i4FSI and C3mpyrFSI were purchased from Boron Molecular company with 99.9% purity, and sodium bis(fluorosulfonyl)imide (NaFSI, 99.9%) was purchased from Solvionic with 99.9% purity. The structures of the two ionic liquids are presented in Fig. S1 (ESI†). To eliminate the effect of water, the two ILs were dried at 50 °C under vacuum for 48 hours and their water content was reduced to less than 50 ppm as confirmed by Karl Fischer methods (831 Karl Fisher Coulometer with Hydranal® Coulomat AG as the titrant). As shown in Table 1, C3mpyrFSI and P111i4FSI were mixed at different ratios, and NaFSI salt was added until near saturation, all within an argon-atmosphere glove box. Finally, all electrolytes were vacuum-sealed and stored in sealed vials under argon inside the glove box.| Expt. no. | Electrolyte composition (mol%) | IL composition (mol%) | ||
|---|---|---|---|---|
| NaFSI salt | ILs | C3mpyrFSI | P111i4FSI | |
| 1 | 50 | 50 | 100 | 0 |
| 2 | 49 | 51 | 75 | 25 |
| 3 | 47 | 53 | 45 | 55 |
| 4 | 45 | 55 | 15 | 85 |
| 5 | 36 | 64 | 0 | 100 |
C3mpyrFSI and P111i4FSI were also mixed at different ratios, and a fixed NaFSI concentration (42 mol%) was added as shown in Table 2.
| Expt. no. | Electrolyte composition (mol%) | IL composition (mol%) | ||
|---|---|---|---|---|
| NaFSI salt | ILs | C3mpyrFSI | P111i4FSI | |
| 1 | 42 | 58 | 100 | 0 |
| 2 | 42 | 58 | 80 | 20 |
| 3 | 42 | 58 | 50 | 50 |
| 4 | 42 | 58 | 20 | 80 |
| 5 | 42 | 58 | 0 | 100 |
2.2. Differential scanning calorimetry (DSC)
DSC measurements were conducted to investigate the thermal behaviour of the electrolytes, such as melting temperature, glass transition temperature, transition entropies and enthalpies. The instrument used for DSC was a NETZSCH DSC 214 Polyma. Three heating and cooling cycles were performed at a scan rate of 10 °C min−1 from −120 to 30 °C. The first scan was affected by the thermal history of the sample; therefore, the reproducible data from the second and third scans are reported. The onset temperature was taken as the glass transition temperature. Cyclohexane from Sigma Aldrich was used as an analytical standard for temperature correction.2.3. Ionic conductivity
A BioLogic MTZ-35 impedance analyzer was used to determine ionic conductivity through EIS measurements. In this study, a custom-built dip-cell was used for conductivity measurements of the electrolyte solutions. This cell contained two platinum wires sheathed in glass sealed with a rubber O-ring and fitted into the cavity of a brass block that was connected to a Eurotherm 2204 temperature controller. Temperature ramping was set at 0.5 °C min−1 until the desired temperature was obtained (±0.3 °C for 20 min). Electrolyte resistivity was determined from the touch down point of the Nyquist plot (x-axis). This resistance was used to calculate conductivity by eqn (1). | (1) |
| G* = R × σ | (2) |
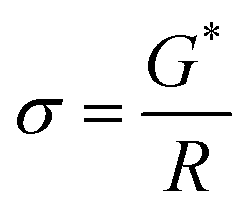 | (3) |
2.4. Nuclear magnetic resonance spectroscopy (NMR) and ion diffusion measurements
Pulsed-field gradient stimulated echo (PFG-STE) diffusion measurements were conducted for 1H, 19F, and 31P using a Bruker Avance III 500 MHz wide-bore spectrometer equipped with a 5 mm PFG probe. The gradient pulse time (δ = 2 ms) and the diffusion time (Δ = 20 ms) were employed, following the procedure described by Bayley et al.21 The diffusion coefficients for the C3mpyr+ cation, the FSI− anion, and the P111i4+ cation were determined using the respective 1H, 19F, and 31P nuclei. Sodium diffusion could not be obtained due to the short relaxation time of the 23Na nucleus in these samples. To minimize the effects of convection, the samples were filled to a height of 50 mm in 3 mm NMR tubes within an argon-filled glovebox, flame-sealed, and then water-bathed inside 5 mm NMR tubes sealed with Teflon tape and a cap. The temperature range for these experiments spanned from 20 to 80 °C.2.5. Cyclic voltammetry
Cyclic voltammetry (CV) experiments were conducted using a Biologic VMP3 potentiostat with a coin cell setup. The coin cells (components CR2032) were purchased from Hoshen Corporation, Japan. The working electrode is made of copper (12.7 mm diameter), and the counter and pseudo-reference electrode is made of sodium metal (8 mm diameter). The copper electrode was punched and washed with 1 M HCl solution for 2 minutes followed by washing with deionized (DI) water for 2 minutes and finally with acetone for 2 minutes. The electrodes were then vacuum-dried for 24 hours at 100 °C and transferred into the glovebox. A 16 mm diameter polyethylene separator (gratis, Lydall, 7P03A, 50 mm thickness, 85% 0.3 mm porosity) soaked with the electrolyte solution was used. A 0.5 mm spacer and a 1.4 mm spring were used to ensure contact. All the measurements were carried out at 50 °C and at a scan rate of 20 mV s−1. The onset potential was determined by intersection of the tangent to the exponentially increasing current part of the current curve with the linear extrapolation of the baseline current.3. Results and discussion
3.1. Mixed cations with a saturated NaFSI system
The utilization of a high salt concentration in the electrolyte proves beneficial in achieving uniform metal deposition and preventing dendrite formation at the metal anode during cycling, as reported in previous studies.17 Consequently, we initially chose the saturation salt concentration for each of the mixed IL electrolytes to compare what we considered to be the optimum salt composition in each case. From the literature, the saturation limits of NaFSI in pure P111i4FSI and C3mpyrFSI are reported to be 42 mol% and 50 mol%, respectively.16,22 Results from solubility tests showed that the saturation limit of NaFSI in 15:85, 45:55, and 75:25 C3mpyrFSI:P111i4FSI was measured to be 45 mol%, 47 mol%, and 49 mol%, respectively. The properties of these systems were initially investigated.
 | ||
| Fig. 1 (a) DSC heating traces of the mixed P111i4FSI and C3mpyrFSI with a saturated NaFSI system. (b) Glass transition temperature as a function of C3mpyrFSI proportion. | ||
In cases where mixed ILs with various ratios were combined with saturated NaFSI, only Tgs were detected. Notably, the onset of these Tgs increases with higher C3mpyrFSI content, which also contains higher salt content. Specifically, a significant increase in Tg was observed when the content of C3mpyrFSI increased from 0 to 15 mol% from −74.95 °C to −68.66 °C. The thermal behaviour of the mixtures is more similar to that of C3mpyrFSI even with only 15 mol% of C3mpyrFSI in the mixture. Interestingly, ΔTg (the temperature range between the onset and end of the Tg peak) is larger for the neat P111i4FSI IL indicating less fragility of this IL compared to both C3mpyrFSI and mixtures of the two ILs.25 The ion mobility in the mixtures is discussed in the following sections.
The ionic conductivity of the mixtures as shown in Fig. 2 increases with the addition of P111i4FSI to the C3mpyrFSI system. In the lower temperature ranges (−20 to 0 °C), adding 25% P111i4FSI significantly increases the ionic conductivity of the neat C3mpyrFSI/NaFSI system. Here, P111i4FSI emerges as the dominant contributor to the enhancement of the ionic conductivity, as the mixtures exhibit ionic conductivities more closely aligned with those of pure P111i4FSI as illustrated in Fig. 2b.
 | ||
| Fig. 2 (a) Temperature-dependent ionic conductivity, (b) the ionic conductivity at −20 °C, (c) the ionic conductivity at 50 °C, (d) the ionic conductivity at 120 °C. | ||
At higher temperatures (above 0 °C), the trend in ionic conductivity appears nearly linear as depicted in Fig. 2c and d. It is noteworthy that the increase in ionic conductivity could be due to either a reduction in NaFSI salt concentration, which lowers the viscosity, or an increased proportion of P111i4FSI, as the NaFSI salt concentration was increased from 42 mol% in P111i4FSI to 50 mol% in C3mpyrFSI.
Fig. 3 presents the diffusion coefficients of ions as a function of the C3mpyrFSI proportion within the temperature range of 20 to 50 °C. Fig. 3a–c show a consistent increase in the diffusion coefficients for both cations and anions with higher P111i4FSI content and higher temperature. These trends correspond to the observed decrease in glass transition temperature and ionic conductivity. The anion diffusion coefficient is slightly higher than that of the cation. This is likely due to the anion's smaller size, which may allow it to diffuse faster. Fig. 3d and e depict almost linear trends in the diffusion coefficients for cations and anions as the C3mpyrFSI proportion changes. A significant observation is a distinct drop in the diffusion coefficient of C3mpyr+ at 20 °C.
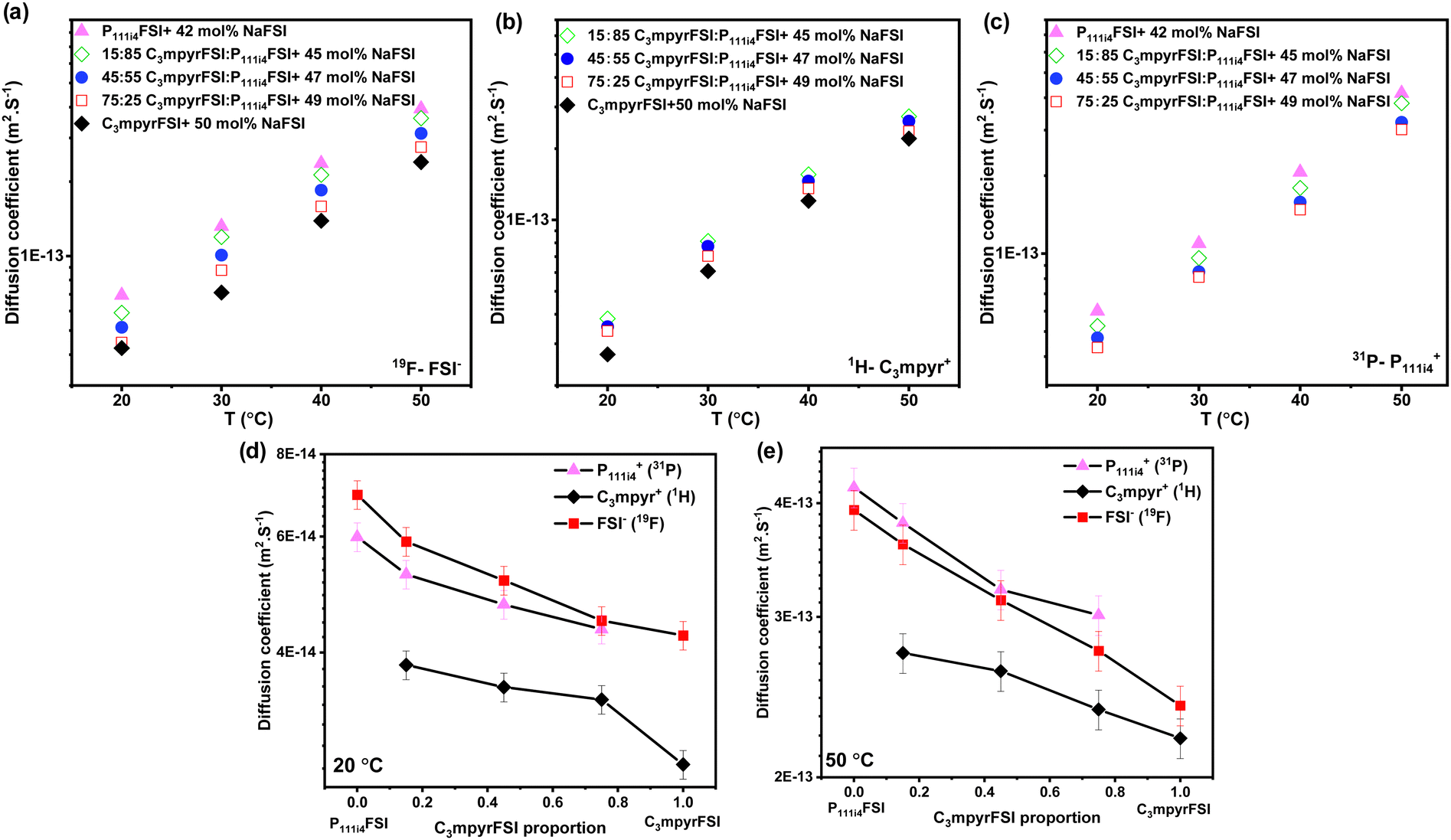 | ||
| Fig. 3 Diffusion coefficients of (a) anion (19F), (b) C3mpyr cation (1H), (c) P111i4 cation (31P) as a function of C3mpyrFSI proportion, (d) diffusion coefficients at 20 °C (e) at 50 °C. | ||
Three-electrode cyclic voltammetry was employed to characterize the electrochemical properties of the electrolytes, as shown in Fig. 4, using a Cu working electrode which has previously been reported to facilitate plating/stripping of sodium metal.19,26 The current density of the reduction peak follows the ratio of two mixed ILs, C3mpyrFSI < 75:25 C3mpyrFSI:P111i4FSI < 45:55 C3mpyrFSI:P111i4FSI < 15:85 C3mPyrFSI:P111i4FSI < P111i4FSI. At the same potential, the higher current density is likely associated with a lower overall resistance (including bulk electrolyte, charge transfer and SEI resistance). The P111i4FSI certainly has the highest ionic conductivity in the bulk and has previously been suggested to have an improved SEI.18 The current density of the mixtures are closer to the value for C3mpyrFSI, possibly due to the closer ionic conductivity of the mixtures to C3mpyrFSI at 50 °C and may also reflect the dominance of the C3mpyr cation on the interfacial properties. The enlarged reduction process presented in Fig. S4 (ESI†) shows the reductive decomposition of the electrolyte which is also dependent on the electrolyte composition and appears to be more extensive in the C3mpyrFSI case.
 | ||
| Fig. 4 Cyclic voltammetry curves for the mixed P111i4FSI and C3mpyrFSI with a saturated NaFSI system during the 1st cycle. | ||
3.2. Mixed cations with a constant concentration of NaFSI (42 mol%)
As the saturated system involves two variables – the cation ratio and the NaFSI salt concentration – the specific influence of the cation ratio on the physicochemical properties remains uncertain. To investigate the impact of cations on both physicochemical and electrochemical properties, a fixed NaFSI concentration (42 mol%) system was used. | ||
| Fig. 5 (a) Thermal phase behaviour of the mixed P111i4FSI and C3mpyrFSI with 42 mol% NaFSI system. (b) Glass transition temperature as a function of C3mpyrFSI proportion. | ||
Incorporating P111i4FSI into C3mpyrFSI/42 mol% NaFSI led to a slight increase in ionic conductivity as shown in Fig. 6. This figure demonstrates a clear linear relationship between the ionic conductivity and the C3mpyrFSI contents. However, as the temperature increases from −20 to 120 °C (Fig. 6b–d), the increase in ionic conductivity becomes less pronounced. This reduced rate of increase in ionic conductivity could be attributed to factors such as modifications in molecular interactions as temperature rises.
 | ||
| Fig. 6 (a) Temperature-dependent ionic conductivity of the mixed P111i4FSI and C3mpyrFSI with 42 mol% NaFSI system, (b) the ionic conductivity at 20 °C, (c) the ionic conductivity at 50 °C, (d) the ionic conductivity at 80 °C. | ||
 | ||
| Fig. 7 Diffusion coefficient of (a) anion (19F), (b) C3mpyr cations (1H), (c) P111i4 cations (31P) as a function of C3mpyrFSI proportion, (d) diffusion coefficients at 20 °C (e) at 50 °C (f) at 80 °C. | ||
The findings from the experiments with mixed cations so far indicate that the physicochemical characteristics of the C3mpyrFSI/42 mol% NaFSI mixture remain largely unaffected by the addition of P1111i4FSI. In the next section, we will explore the electrochemical behaviours of both of the individual ILs/42 mol% NaFSI and their mixtures as electrolytes, using cyclic voltammetry for our analysis.
Fig. 8a shows the comparison of the cyclic voltammetry curves of the different mixtures from the 1st cycle. As the concentration of P111i4FSI increases, changes in the electrochemical behaviour become evident. Specifically, the current density increases, indicating a corresponding decrease in resistance within the system, which may contribute to the high ionic conductivity in the bulk, charge transfer and SEI on the electrode surface. It is worth noting that before the sodium plating (Fig. 8b), two distinct cathodic peaks were observed in all 42 mol% NaFSI system. The peak at 0.2 V corresponds to the decomposition of the FSI− anion, while the peak at 0.8 V is strongly dependent on the substrate and water content.27 The increasing onset potential, as depicted in Fig. 8c, indicates that sodium plating initiates earlier with higher P111i4FSI content, i.e., sodium is more easily plated in higher P111i4FSI content systems.
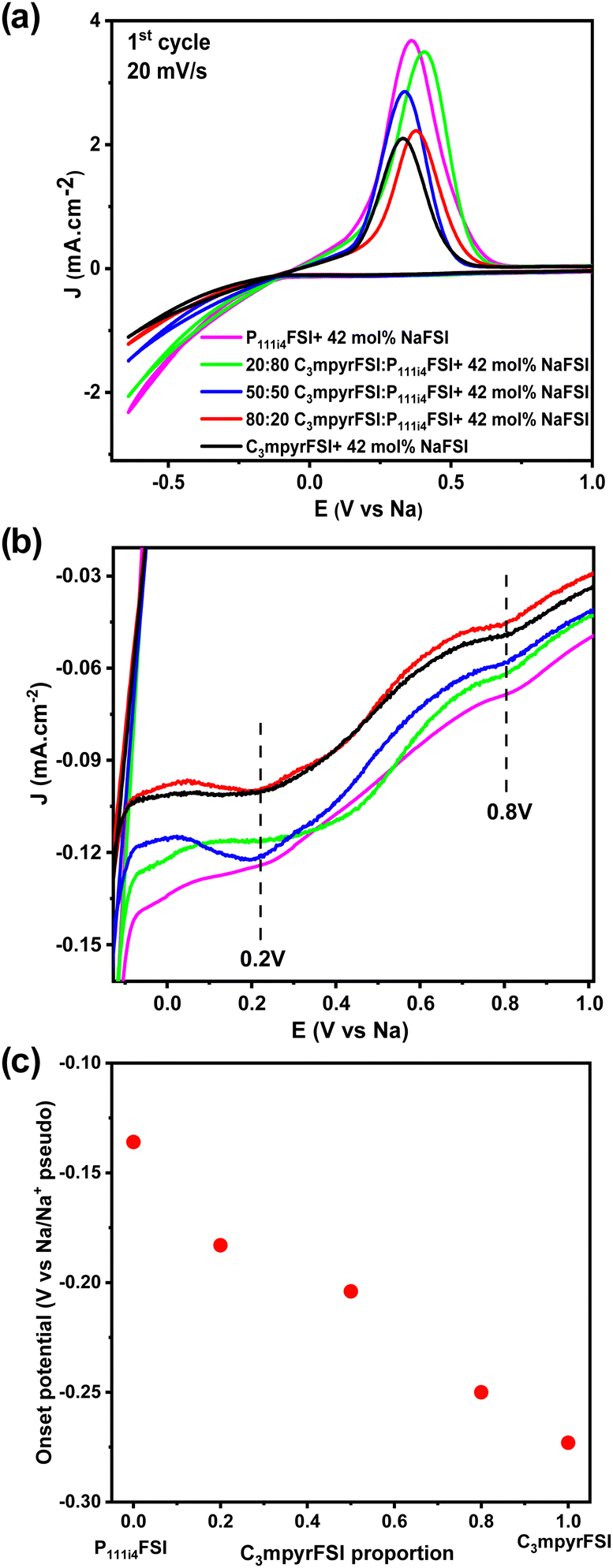 | ||
| Fig. 8 (a) Cyclic voltammetry curves for the mixed P111i4FSI and C3mpyrFSI with 42 mol% NaFSI system at 1st cycle, (b) the enlarged reduction process, and (c) the onset potential as a function of C3mpyrFSI proportion. | ||
Fig. 9a shows, a comparison of the 1st, 2nd, and 5th cycles for C3mpyrFSI + 42 mol% NaFSI highlighting an increase in onset potential from −0.27 V in the 1st cycle to −0.048 V in the 3rd cycle. This suggests that sodium is more easily plated over successive cycles. Additionally, the slope of the CV curve on plating sodium becomes sharper with cycling, suggesting an accelerated rate of sodium deposition. Following the sharp increase, there is a subsequent decrease in current density, which may be attributed to the complete utilization of sodium ions on the electrode surface, resulting in a decline in the rate of sodium deposition. Fig. 9b shows with increasing cycle number, the coulombic efficiency increases with cycling. This behaviour could be related to the formation of solid electrolyte interface film on the electrode that is usually formed during the first charge cycle.7,28 Notably, the C3mpyr and P111i4 CE values exhibit a reverse trend with cycling, with C3mpyr showing the highest 1st cycle CE value (78%) and the lowest in the 3rd cycle (89%) with the opposite occurring for the higher P111i4 compositions (P111i4FSI 1st cycle 71% and 3rd cycle 93%). This suggests more substantial and complete SEI formation in the 1st cycles when the P111i4 cation is introduced compared to less substantial and complete SEI formation for the C3mpyrFSI dominant compositions, which achieve lower efficiency overall.
 | ||
| Fig. 9 (a) Cyclic voltammetry curves for C3mpyrFSI + 42 mol% NaFSI from 1st, 2nd and 5th cycle. (b) Coulombic efficiencies as a function of C3mpyrFSI proportion. | ||
4. Conclusions
This study focused on exploring the impact of mixed cations on the physicochemical properties of electrolytes using both saturated and fixed NaFSI concentration systems. In the saturated system, we observed significant changes in thermal behaviour and ionic conductivity even though the salt content variation was not particularly high. The addition of NaFSI disrupted the crystallization process in ILs and resulted in an increase in Tg with higher C3mpyrFSI contents. Additionally, diffusion coefficients for both cations and anions showed consistent increases with elevated P111i4FSI content and higher temperature, corresponding to the rise in Tg and ionic conductivity. Notably, a drop in the diffusion coefficient of C3mpyr/NaFSI at 20 °C was observed.In the fixed 42 mol% NaFSI system, we found that Tg slightly decreased with increasing P111i4FSI content. However, the observed change was less than 5 °C, indicating a modest impact. Similarly, the addition of P111i4FSI to C3mpyrFSI/42 mol% NaFSI led to a slight increase in ionic conductivity, showing a linear relationship with the C3mpyrFSI ratio. In terms of diffusion, while there was a slight increase in FSI-diffusion, changes in cation diffusion coefficients were not pronounced. Notably, the CV analysis revealed changes in electrochemical behaviour with increasing P111i4FSI content, including an elevated onset potential for sodium plating and changes in current density and coulombic efficiency over cycling, suggesting an accelerated rate of sodium deposition followed by a decline in deposition rate. This work suggests that the approach of mixing IL cations to control physicochemical properties and electrochemical behaviour is feasible and future work will investigate the effect of different cation chemistries. A more detailed study of the electrochemical cycling behaviour in these mixed IL systems is also currently underway.
Author contributions
The manuscript was written through the contributions of all authors. All authors have approved the final version of the manuscript. L. H. – experimental design, methodology, investigation, data analysis, writing original draft. F. M. – supervising, scientific discussion and understanding concept. L. A. O – NMR supervision and scientific discussion. P. H. – conceptualization, supervising, scientific discussion and understanding concept. M. F. – conceptualization, supervising, scientific discussion and understanding concept.Data availability
1H spectra in NMR, cyclic voltammetry curves for the saturated system, repeated mixtures, and 2nd cycle studied in this work.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The authors thank the ARC (Australian Research Council) for funding through Discovery Project funding DP210101172 and Future Energy Storage Technologies (StorEnergy) IC180100049 and Deakin University Battery Research and Innovation Hub facilities.References
- Y. Hu, X. Zhu and L. Wang, Two-Dimensional Material-Functionalized Separators for High-Energy-Density Metal-Sulfur and Metal-Based Batteries, ChemSusChem, 2020, 13(6), 1366–1378 CrossRef CAS PubMed.
- X. Zhu and L. Wang, Advances in materials for all-climate sodium-ion batteries, EcoMat, 2020, 2(3), e12043 CrossRef CAS.
- K. Matsumoto, J. Hwang, S. Kaushik, C.-Y. Chen and R. Hagiwara, Advances in sodium secondary batteries utilizing ionic liquid electrolytes, Energy Environ. Sci., 2019, 12(11), 3247–3287 RSC.
- D. Kundu, E. Talaie, V. Duffort and L. F. Nazar, The emerging chemistry of sodium ion batteries for electrochemical energy storage, Angew. Chem., Int. Ed., 2015, 54(11), 3431–3448 CrossRef CAS PubMed.
- V. Palomares, P. Serras, I. Villaluenga, K. B. Hueso, J. Carretero-González and T. Rojo, Na-ion batteries, recent advances and present challenges to become low cost energy storage systems, Energy Environ. Sci., 2012, 5(3), 5884–5901 RSC.
- A. Ponrouch, D. Monti, A. Boschin, B. Steen, P. Johansson and M. R. Palacín, Non-aqueous electrolytes for sodium-ion batteries, J. Mater. Chem. A, 2015, 3(1), 22–42 RSC.
- G. G. Eshetu, G. A. Elia, M. Armand, M. Forsyth, S. Komaba, T. Rojo and S. Passerini, Electrolytes and Interphases in Sodium-Based Rechargeable Batteries: Recent Advances and Perspectives, Adv. Energy Mater., 2020, 10(20), 2000093 CrossRef CAS.
- T. Nestler, E. Roedern, N. F. Uvarov, J. Hanzig, G. Antonio Elia and M. de Vivanco, Separators and electrolytes for rechargeable batteries: Fundamentals and perspectives, Phys. Sci. Rev., 2019, 4(4), 20170115 Search PubMed.
- K. Dong, X. Liu, H. Dong, X. Zhang and S. Zhang, Multiscale Studies on Ionic Liquids, Chem. Rev., 2017, 117(10), 6636–6695 CrossRef CAS PubMed.
- S. Cha and D. Kim, Anion exchange in ionic liquid mixtures, Phys. Chem. Chem. Phys., 2015, 17(44), 29786–29792 RSC.
- Z. Xue, L. Qin, J. Jiang, T. Mu and G. Gao, Thermal, electrochemical and radiolytic stabilities of ionic liquids, Phys. Chem. Chem. Phys., 2018, 20(13), 8382–8402 RSC.
- C. Ding, T. Nohira, K. Kuroda, R. Hagiwara, A. Fukunaga, S. Sakai, K. Nitta and S. Inazawa, NaFSA–C1C3pyrFSA ionic liquids for sodium secondary battery operating over a wide temperature range, J. Power Sources, 2013, 238, 296–300 CrossRef CAS.
- M. Forsyth, H. Yoon, F. Chen, H. Zhu, D. R. MacFarlane, M. Armand and P. C. Howlett, Novel Na+ Ion Diffusion Mechanism in Mixed Organic–Inorganic Ionic Liquid Electrolyte Leading to High Na+ Transference Number and Stable, High Rate Electrochemical Cycling of Sodium Cells, J. Phys. Chem. C, 2016, 120(8), 4276–4286 CrossRef CAS.
- F. Chen, P. Howlett and M. Forsyth, Na-Ion Solvation and High Transference Number in Superconcentrated Ionic Liquid Electrolytes: A Theoretical Approach, J. Phys. Chem. C, 2018, 122(1), 105–114 CrossRef CAS.
- K. Matsumoto, Y. Okamoto, T. Nohira and R. Hagiwara, Thermal and Transport Properties of Na[N(SO2F)2]− [N-Methyl-N-propylpyrrolidinium][N(SO2F)2] Ionic Liquids for Na Secondary Batteries, J. Phys. Chem. C, 2015, 119(14), 7648–7655 CrossRef CAS.
- M. Hilder, P. C. Howlett, D. Saurel, E. Gonzalo, A. Basile, M. Armand, T. Rojo, M. Kar, D. R. MacFarlane and M. Forsyth, The effect of cation chemistry on physicochemical behaviour of superconcentrated NaFSI based ionic liquid electrolytes and the implications for Na battery performance, Electrochim. Acta, 2018, 268, 94–100 CrossRef CAS.
- D. A. Rakov, F. Chen, S. A. Ferdousi, H. Li, T. Pathirana, A. N. Simonov, P. C. Howlett, R. Atkin and M. Forsyth, Engineering high-energy-density sodium battery anodes for improved cycling with superconcentrated ionic-liquid electrolytes, Nat. Mater., 2020, 19(10), 1096–1101 CrossRef CAS PubMed.
- S. A. Ferdousi, L. A. O'Dell, J. Sun, Y. Hora, M. Forsyth and P. C. Howlett, High-Performance Cycling of Na Metal Anodes in Phosphonium and Pyrrolidinium Fluoro(sulfonyl)imide Based Ionic Liquid Electrolytes, ACS Appl. Mater. Interfaces, 2022, 14(13), 15784–15798 CrossRef CAS PubMed.
- M. Forsyth, M. Hilder, Y. Zhang, F. Chen, L. Carre, D. A. Rakov, M. Armand, D. R. Macfarlane, C. Pozo-Gonzalo and P. C. Howlett, Tuning Sodium Interfacial Chemistry with Mixed-Anion Ionic Liquid Electrolytes, ACS Appl. Mater. Interfaces, 2019, 11(46), 43093–43106 CrossRef CAS PubMed.
- E. M. Barsoukov and J. R. Macdonald, Impedance Spectroscopy Theory Experiment And Applications, 2004 Search PubMed.
- P. M. Bayley, G. H. Lane, N. M. Rocher, B. R. Clare, A. S. Best, D. R. MacFarlane and M. Forsyth, Transport properties of ionic liquid electrolytes with organic diluents, Phys. Chem. Chem. Phys., 2009, 11(33), 7202–7208 RSC.
- S. A. Ferdousi, M. Hilder, A. Basile, H. Zhu, L. A. O'Dell, D. Saurel, T. Rojo, M. Armand, M. Forsyth and P. C. Howlett, Water as an Effective Additive for High-Energy-Density Na Metal Batteries? Studies in a Superconcentrated Ionic Liquid Electrolyte, ChemSusChem, 2019, 12(8), 1700–1711 CrossRef CAS PubMed.
- G. M. A. Girard, M. Hilder, H. Zhu, D. Nucciarone, K. Whitbread, S. Zavorine, M. Moser, M. Forsyth, D. R. Macfarlane and P. C. Howlett, Electrochemical and physicochemical properties of small phosphonium cation ionic liquid electrolytes with high lithium salt content, Phys. Chem. Chem. Phys., 2015, 17(14), 8706–8713 RSC.
- H. Yoon, H. Zhu, A. Hervault, M. Armand, D. R. Macfarlane and M. Forsyth, Physicochemical properties of N-propyl-N-methylpyrrolidinium bis(fluorosulfonyl)imide for sodium metal battery applications, Phys. Chem. Chem. Phys., 2014, 16(24), 12350–12355 RSC.
- J. J. Moura Ramos, C. A. M. Afonso and L. C. Branco, Glass transition relaxation and fragility in two room temperature ionic liquids, J. Therm. Anal. Calorim., 2003, 71, 659–666 CrossRef CAS.
- A. Basile, F. Makhlooghiazad, R. Yunis, D. R. MacFarlane, M. Forsyth and P. C. Howlett, Extensive Sodium Metal Plating and Stripping in a Highly Concentrated Inorganic−Organic Ionic Liquid Electrolyte through Surface Pretreatment, ChemElectroChem, 2017, 4(5), 986–991 CrossRef CAS.
- P. C. Howlett, E. I. Izgorodina, M. Forsyth and D. R. MacFarlane, Electrochemistry at Negative Potentials in Bis(trifluoromethanesulfonyl)amide Ionic Liquids, Z. Phys. Chem., 2006, 220(10), 1483–1498 CrossRef CAS.
- Z. W. Seh, J. Sun, Y. Sun and Y. Cui, A Highly Reversible Room-Temperature Sodium Metal Anode, ACS Cent. Sci., 2015, 1(8), 449–455 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: 1H spectra in NMR, cyclic voltammetry curves for the saturated system, repeated mixtures, and 2nd cycle studied in this work. See DOI: https://doi.org/10.1039/d4ma00533c |
| This journal is © The Royal Society of Chemistry 2024 |