
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A novel characterization technique for hydrogels – in situ rheology-Raman spectroscopy for gelation and polymerization tracking†
Sina
Lambrecht
 a,
Marek
Biermann
b,
Selin
Kara
cd,
Stefan
Jopp
a and
Johanna
Meyer
*d
a,
Marek
Biermann
b,
Selin
Kara
cd,
Stefan
Jopp
a and
Johanna
Meyer
*d
aDepartment Life, Light & Matter, University of Rostock, Albert-Einstein-Str. 25, 18059 Rostock, Germany
bAnton Paar OptoTec GmbH, Lise-Meitner-Str. 6, 30926 Seelze-Letter, Germany
cDepartment of Biological and Chemical Engineering, Aarhus Universtity, Gustav Wieds Vej 10, 8000 Aarhus, Denmark
dInstitute of Technical Chemistry, Leibniz University Hannover, Callinstraße 5, 30167 Hannover, Germany. E-mail: johanna.meyer@iftc.uni-hannover.de
First published on 6th August 2024
Abstract
The interest in hydrogels has grown considerably across a number of disciplines, including but not limited to the immobilization of (bio)catalysts in matrices and in the medical sector, for example, in drug delivery systems, contact lenses, biosensors, electrodes, and tissue engineering. Consequently, the characterization of these materials is frequently the subject of cutting-edge research. However, hydrogels are often insoluble, which precludes the use of many analytical methods, such as nuclear magnetic resonance (NMR). Consequently, other established analytical techniques, such as attenuated total reflection (ATR), infrared spectroscopy (IR), Raman spectroscopy, or rheological measurements, are frequently employed. These methods are generally straightforward to use and can be completed rapidly. However, IR spectroscopy, for instance, is inherently limited by the interference of water's vibrational bands. In this study, we present a method for the characterization of hydrogels that can simultaneously observe the gelation and polymerization of hydrogels.
Introduction
Hydrogels are three-dimensional (3D) networks, consisting of hydrophilic polymer structures that do not dissolve in water but, conversely, absorb large amounts of water or aqueous media. While swelling, hydrogels retain their 3D shape, while increasing their volume.1–4 These polymers are well established in various disciplines, such as biotechnology (stent-coating),5–7 biomedicine (drug-delivery, tissue engineering),8,9 agriculture (water storage),10 and catalysis (immobilization matrix).11,12 The versatility of hydrogels in terms of their chemical structure and properties allows them to be applied in this wide range of possible applications.13 The specific type of hydrogel employed in this study was a semi-synthetic carbohydrate-based ionic hydrogel.These hydrogels were synthesized via free radical polymerization and with different types and amounts of crosslinkers, including polyethylene glycol diacrylate (PEGDA) (Scheme 1).
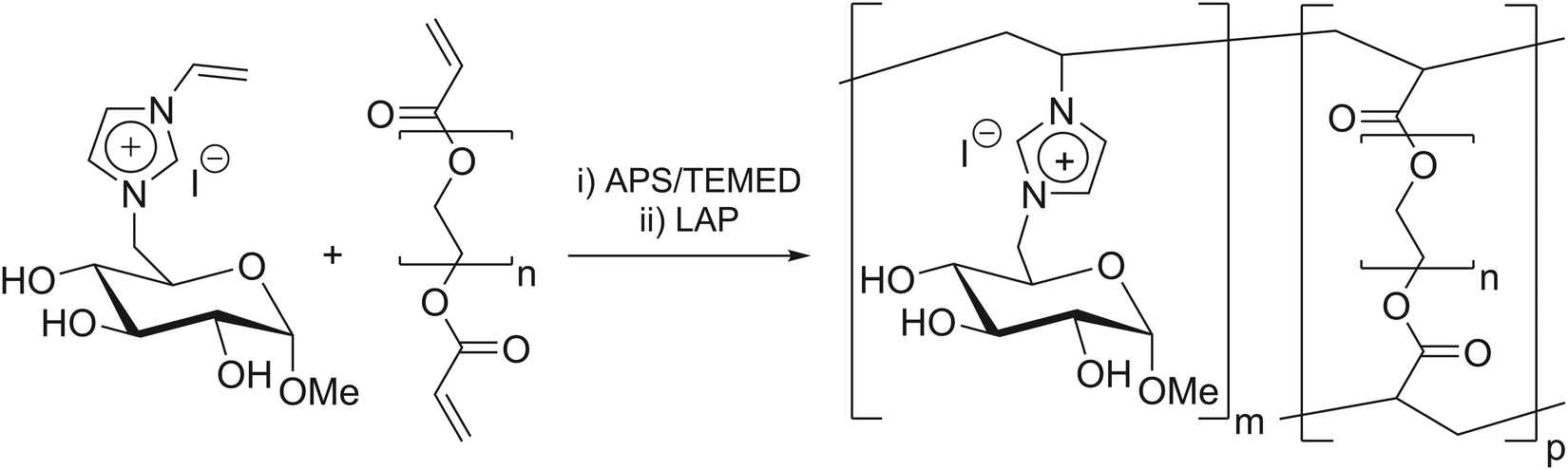 | ||
| Scheme 1 Synthesis of polyGVIM-I via radical polymerization with PEGDA as crosslinker. | ||
Various methods have already been established to analyze the structure of hydrogels and their precursors, such as monomers or polymer chains that need to be crosslinked in 3D. These methods include microscopy (such as scanning electron microscopy (SEM), transmission electron microscopy (TEM) and related techniques) to determine the local hydrogel structure, (solid-state) nuclear magnetic resonance (NMR) as an analytical technique to analyze the chemical structure, as well as Raman spectroscopy as a technique to measure the molecular interaction. In addition, Fourier transform infra-red (FT-IR) spectroscopy and attenuated transmission reflectance Fourier transform infra-red (FT-IR) spectroscopy (ATR–FTIR) can be employed to quantify the chemical composition of a given sample by measuring the vibrations that occur as a result of the sample's composition. These techniques are effective, simple, and rapid. The thermal stability of hydrogels can be investigated through the use of thermal gravimetric analysis (TGA) and differential scanning calorimetry (DSC). The characterization of the mechanical properties of hydrogels is best achieved through the utilization of rheological measurements. The synthesis and characterization of hydrogels is a broad research field. As this article covers only provides a brief overview of a few methods, we would like to direct interested readers to a comprehensive review article by Raghuwanshi and Garnier, presenting a good overview of the mentioned methods for characterizing hydrogels with the respective advantages and drawbacks, and the expansion of this article by Patel and Thareja.14,15
On the one hand, NMR spectroscopy is usually performed in solution and is therefore generally not applicable to insoluble hydrogels. It can be used for the characterization of the monomers and crosslinkers prior to hydrogel formation. On the other hand, solid state NMR can be used to determine the chemical structure of hydrogels, as recently shown by our group.16
SEM and associated variance techniques provide information about the surface morphology, additives in the gel, and inter-connecting pores. It is, however, essential that the sample must be prepared by drying, as the presence of water significantly disturbs the vacuum and impedes the imaging process during the measurement. Two further options are available for sample preparation: air drying and freeze drying. However, these can lead to the formation of artifacts and impair the morphology of the sample.17–19 An alternative is supercritical drying with liquid CO2. This protects a gel from pore collapse and the structure remains the same in the dried state as in the wet state.20
IR spectroscopy is a simple, non-destructive, and sensitive characterization technique that requires minimal sample preparation. Das et al. employed the FTIR technique to analyze the functional groups of their cellulose materials, thereby confirming the reaction between the hydroxyl and carboxyl groups during the crosslinking process due to the diminution of the hydroxyl group's stretching vibration.21 Mao et al. and Azam et al. have recently used FTIR to identify the chemical composition and structural characteristics of their alginate hydrogels, to cite a few examples.22,23 Nevertheless, this technique is not without significant limitations. The strong bands of water present in the hydrogel may overlap with the bands from the polymer phase, thereby dominating them due to their intense nature.15,24–26
Raman spectroscopy, which is complementary in nature to IR spectroscopy, can be used in addition to it, but also be employed as its own characterization method.27–29 The water bands are much less dominant than in IR spectroscopy and do hardly interfere with the Raman bands. Nie et al. employed IR and Raman spectroscopy to investigate the effect of calcium ions on the structure of their cellulose-based hydrogel beads.30 Affatato et al. demonstrated, using Raman spectroscopy, that C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bonds were not fully converted and determined the degree of crosslinking. Additionally, it was observed that the washing of the hydrogels in an aqueous medium resulted in the leaching of the unreacted crosslinker.31
C double bonds were not fully converted and determined the degree of crosslinking. Additionally, it was observed that the washing of the hydrogels in an aqueous medium resulted in the leaching of the unreacted crosslinker.31
Rheological measurements are crucial for characterizing hydrogels. One important parameter derived from rheological investigations is the viscoelastic response.32 The measurement of the shear strain provides insight into the stiffness and stability of the hydrogel against shear stress, enabling the monitoring of its formation. An understanding of the rheological behavior of hydrogels is essential. The rheological parameters, such as viscosity, thixotropy, and viscoelastic behavior have a significant impact on the success of 3D-printing, as demonstrated by recent studies.33–38 These parameters are also relevant in other fields such as the food industry,39,40 cell culture, or tissue engineering.41,42
One of the mentioned methods alone cannot fully characterize the chemical structure and the properties of a sample. It is always necessary to combine several methods.14,29 One option for using two methods simultaneously is the rheology-Raman combination, which enables the recording of Raman spectra and simultaneously investigate the rheological properties of a sample. Völker-Pop's group has used the rheology-Raman combination to characterize polymer melts and the curing behavior of epoxy resins. The transition to the solid phase, as observed by rheological measurements, was found to occur at a higher temperature than the crystallization indicated by the Raman data.43 Kida et al. investigated the flow-induced crystallization (FIC) of high-density polyethylene. The effect of shear rate, shear-flow time, and molecular weight on the time dependencies of the different conformer mass fractions of the molecular chains during crystallization was analyzed, and it was found that the trans chains formed during isothermal crystallization were strongly dependent on the FIC conditions.44 In this work, the rheology-Raman combination is introduced as a powerful tool for characterizing hydrogels, and a comparison is made between this method and established methods.
Experimental
Materials
The chemicals methyl-α-D-glucopyranoside (99%), triphenylphosphine (99%), imidazole (99%), N-vinylimidazole (99%), ethyleneglycole diacrylate (EGDA; >90%) and ammonium persulfate (APS; 98%) have been supplied by Thermo Fisher Scientific and were used as received. Polyethylene glycol diacrylate (PEGDA; Mn = 250, 575 and 700 g mol−1), N,N′-methylene bisacrylamide (MBAA; 99%) and N,N,N′,N′-tetramethylethylenediamine (TEMED; >99%) were supplied by Sigma-Aldrich (Germany) and were used as received. Iodine (>99.5%) was supplied by Carbolution. Lithium phenyl(2,4,6-trimethylbenzoyl) phosphinate (LAP; >98%) was supplied by TCI and was used as received. The solvents THF (99.9%) and ethyl acetate (99.7%) were supplied by Honeywell Riedel–de-Haën and the solvents chloroform (>99.8%), methanol (99.8%) and DMF (99.5%) were supplied by Thermo Fisher Scientific and were used as received. Column chromatography was performed with silica gel (230–400 mesh particle size) supplied by Supelco (Germany). Additionally, phosphate-buffered saline (PBS, Thermo Fisher Scientific Inc., Waltham, USA) was used as received.Procedure for the syntheses of the 1-(methyl-α-D-glucopyranosid-6-yl)-3-vinylimidazolium iodide hydrogels (polyGVIM-I)
The method for synthesizing the monomer 1-(methyl-α-D-glucopyranosid-6-yl)-3-vinylimidazolium iodide (GVIM-I) has been previously described by our group.16 Furthermore, the method and analytics are outlined in the ESI.† The hydrogels in this study were produced using two different methods: (i) with APS and TEMED as the initiator system and (ii) with LAP as the photoinitiator. In the initial approach, GVIM-I (1.25 mol L−1) was dissolved in PBS and the appropriate weight-percent of the crosslinker was added. Radical polymerization (Scheme 1) was initiated by adding the APS solution and TEMED. Once the solution had been thoroughly mixed for 10 s, the gelation took place within seconds or a few minutes. In the second method, GVIM-I (1.25 mol L−1), crosslinker (10, 15 or 20 mol%) and the appropriate weight-percent of LAP were dissolved in PBS (pH = 7.4). The monomer solution was photopolymerized with an UV intensity (λ = 365 nm, Biolinker, VILBER, Collégien, France) of 1.2 J cm2 and 2.4 J cm2, respectively.Characterization of the monomers and hydrogels
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) s and the probe was immersed again.
s and the probe was immersed again.
Results and discussion
Hydrogel characterization with ATR-FTIR spectroscopy
The ATR-FTIR spectra of GVIM-I (red), the reaction solution with GVIM-I solved in PBS containing crosslinker and APS (blue) and a freeze-dried polyGVIM-I with PEGDA 575 as crosslinker and APS/TEMED as initiator system (black) are shown in Fig. 1A. The broad band observed in the range of 3150 to 3600 cm−1 is attributed to –OH stretching vibrations. The various C–H vibrations are observed within the range of 3088 cm−1 to 2833 cm−1. The typical vibrations of the imidazole ring are present in the range of 1550–1570 cm−1 (CN vibration) and at 1450 cm−1 (deformation vibration of the –CH backbone). In the fingerprint region, the C–O–C stretching vibration can be observed at 1133 cm−1 and 896 cm−1, while the stretching and deformation vibration of CH from the imidazole ring are found at 1092 cm−1. The CCH2 vibrations are observed in the range from 920 to 956 cm−1.45,46 The spectra of the GVIM-I and the hydrogel polyGVIM-I differ significantly in the visible vibrations of the CC double bond in the monomer spectrum (highlighted in grey). The band at 1605 cm−1 can be attributed to the stretching vibrations, while the band at 927 cm−1 can be associated with the bending vibrations of the vinyl group.
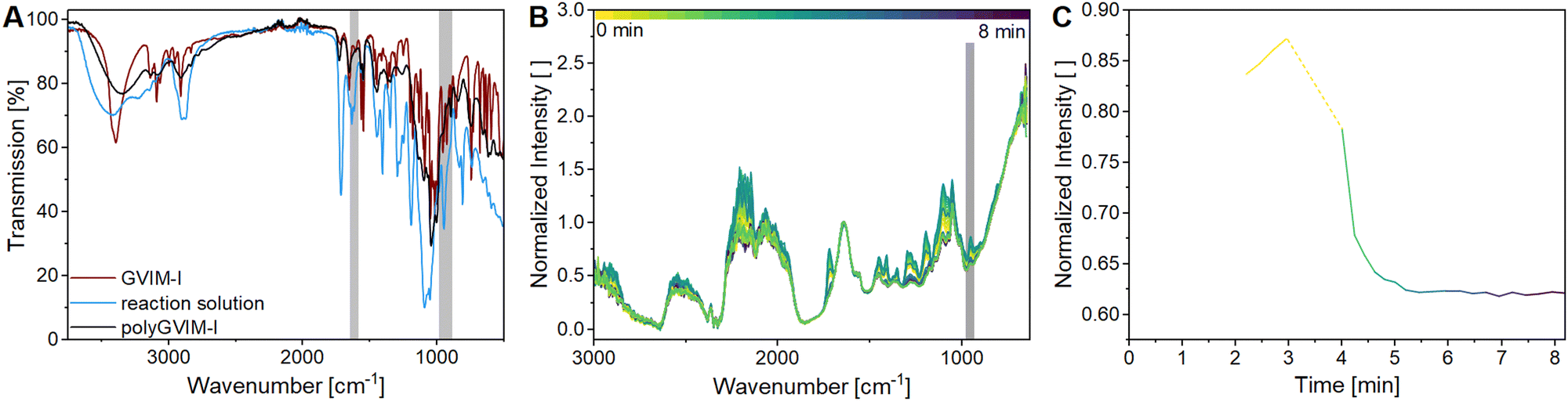 | ||
| Fig. 1 IR spectra of the carbohydrate-based hydrogels via ATR-FTIR spectroscopy and in situ IR spectra: (A) ATR-FTIR spectra of GVIM-I, the reaction solution (containing GVIM-I, PEGDA 575 and APS solved in PBS) and polyGVIM-I with PEGDA 575 as crosslinker (freeze-dried). Grey highlighted are the significant vibration bands of CC double bonds (1605 cm−1 and 927 cm−1), showing only in the IR spectrum of GVIM-I and the reaction solution, but not in polyGVIM-I. (B) In situ IR spectra for polymerization tracking of polyGVIM-I with PEGDA 575 as crosslinker and APS/TEMED as an initiator system. Grey highlighted is one of the significant vibration bands of the CC double bond (955 cm−1), spectrum was normalized on the band at 1637 cm−1. (C) Time trace of the 955 cm−1 band during polymerization tracking of the hydrogel (from B highlighted in grey from 969 to 917 cm−1). The dashed line represents the progression of the intensity from initial intensity without TEMED until the addition of the radical starter TEMED and consequently the start of the radical polymerization (cGVIM-I = 1.25 mol L−1, cPEGDA575 = 10.4 mol%). | ||
Hydrogel characterization with in situ IR spectroscopy
In order to gain a deeper insight into the polymerization process of the hydrogel and to monitor the bands of interest, an in situ IR measurement was conducted. The polymerization was tracked for a total of 90 min. The initial 8 min of the measurement are presented in this manuscript, as no change occurred afterwards (Fig. 1B). In this experiment, APS and TEMED were used as the initiator system. Additionally, all bands above 2000 cm−1 are indicative of noise due to the resolution of the Mettler Toledo React-IR 15 used in this experiment, and the fact that the baseline is not flat, the spectra are difficult to interpret. Besides, several bands do not correspond to those depicted in Fig. 1A. This discrepancy may be due to the fact that the samples measured in situ are in an aqueous solution and consist of a mixture of the crosslinking components (GVIM-I, PEGDA, APS, and TEMED). Despite this, the bending vibration bands of the CC double bond at 955 cm−1 could be detected and are highlighted in grey in Fig. 1B, whereas the tracing of the intensity decrease is shown in Fig. 1C. The initial intensity of the reaction solution and the intensity of the first measurement in the gelation process are connected by a dashed line. During this interval, the reaction solution was supplemented with TEMED, thereby initiating the polymerization of the monomer to form the hydrogel. The graph shows that the intensity of the band of CC double bonds is high before the addition of TEMED and decreases rapidly as soon as it is added to the reaction solution. During polymerization, the CC double bonds of GVIM-I and the crosslinker react, causing the intensity to decrease. After about 7 minutes, the intensity of the band reaches a steady state, indicating that the polymerization is complete.
Hydrogel characterization with rheology
A comprehensive understanding of the relationship between the chemical structure and physical behavior of hydrogels is crucial for possible applications.13,47,48 For example, the migration, proliferation and differentiation of cells are closely linked to the mechanical properties of hydrogels. Various levels of stiffness lead to stem cells differentiating into different cell types.49,50 The stiffness and other relevant properties of a hydrogel can be determined using rheological measurements. Furthermore, in the production of ceramic inks for direct ink writing, an understanding of rheological properties is crucial for the efficient design of ceramic inks with enhanced performance.51 In 3D printing, there are many different areas where understanding these properties is of great economic importance. For example, in the development of inks for extrusion-based 3D bioprinting to obtain bioinks with high structural fidelity or in 3D printing in the food industry for the production and improvement of bigels and edible gels.52–56In this work, the rheological behaviour of the hydrogels was determined via gelation tracking and amplitude sweep measurements. The gelation tracking method is of particular interest for the hydrogel characterization, to estimate an optimal gelation time.
Fig. 2A shows the formation of a hydrogel following the shear storage modulus G′ and the loss modulus G′′ as a function of time. G′ represents the elastic part of the viscoelastic behaviour and stands for the stored deformation energy. In contrast G′′ characterises the viscous part of the viscoelastic behaviour, representing the deformation energy that is lost due to internal friction during flow.57,58
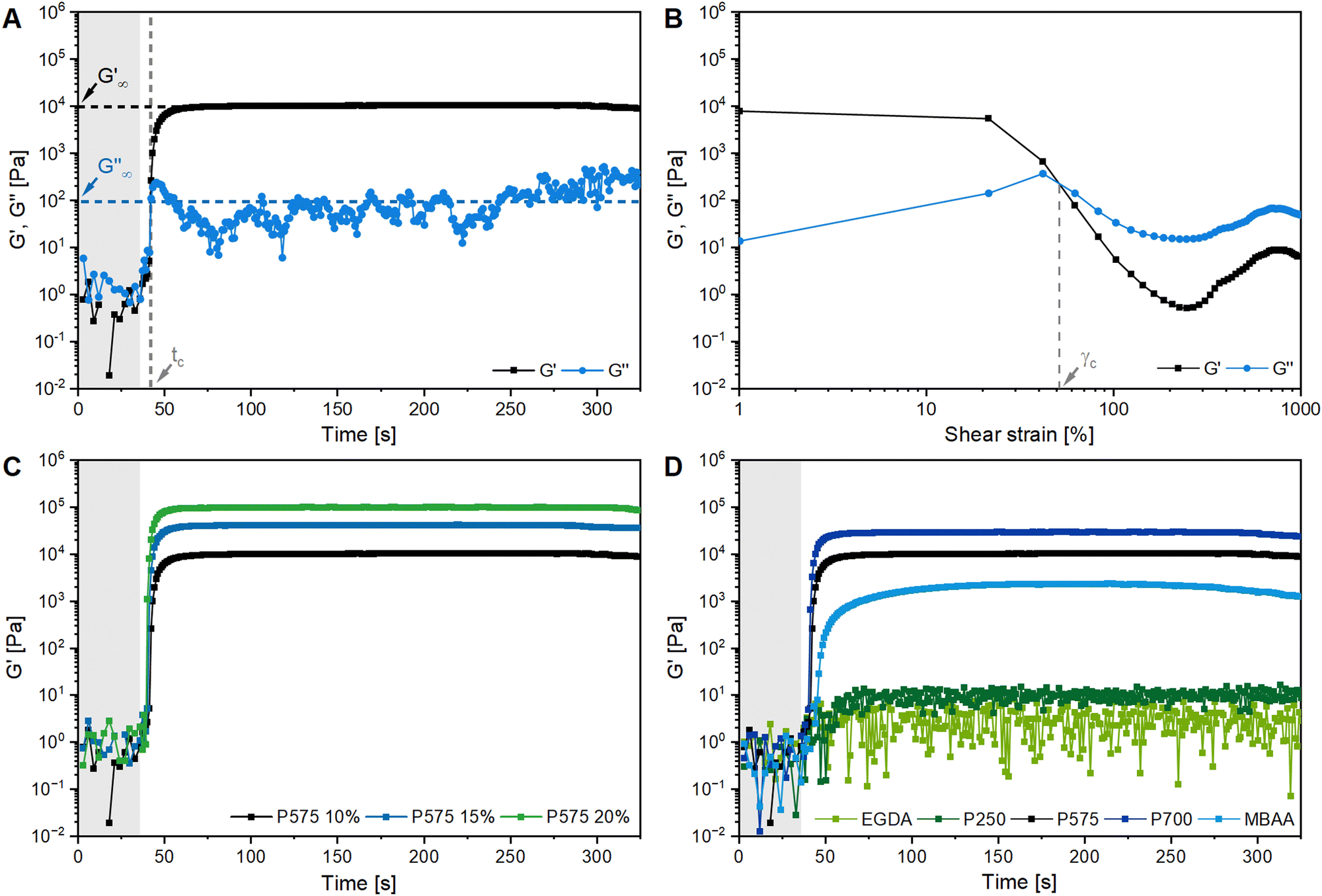 | ||
| Fig. 2 Rheological characterization of polyGVIM-I hydrogels with LAP as photoinitiator: (A) gelation tracking of polyGVIM-I with PEGDA 575 (10 mol%) as crosslinker and (B) the following amplitude sweep measurement of the same hydrogel, with G′ and G′′ plotted against the shear strain. Gelation tracking of polyGVIM-I hydrogels with (C) PEGDA 575 in three different concentrations (10, 15, and 20 mol%) and (D) five different crosslinker structures (ccrosslinker = 10 mol%). The values of the storage modulus G′, the loss modulus G′′ and the crossing point tc were read of each diagram, according to A and listed in Table 1. At the beginning of the measurement, the UV lamp of the rheometer remains switched off (highlighted in grey). (The oscillating frequency was 0.1 Hz at 37 °C. PEGDA was abbreviated to P due to space constraints in the figures. cGVIM-I = 1.25 mol L−1.) | ||
At the start of the measurement, the value of G′′ is greater than G′, because the monomer solution is a liquid. As soon as the UV lamp is switched on, gelation begins. This is clearly shown by the increase in G′. The point where G′ = G′′ is referred to as the crossing point or gel point and is labelled tc in the diagram. It indicates the transition from a liquid-like behaviour to a solid-like behavior.57 The value of G′ increases furthermore until it reaches a steady state plateau. This is marked as 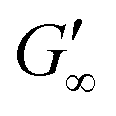 in the diagram. The gelation time, the crossing point of G′ and G′′, and
in the diagram. The gelation time, the crossing point of G′ and G′′, and 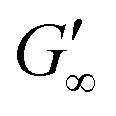 value depend on the polymerization rate, crosslinking density, crosslinker structure, and type of crosslinking.59,60
value depend on the polymerization rate, crosslinking density, crosslinker structure, and type of crosslinking.59,60
Fig. 2C shows the results of testing three samples with the same monomer concentration (1.25 mol L−1) but different crosslinker concentrations (10, 15 and 20 mol%). The values of G′ for all three samples increased rapidly as soon as the UV lamp was switched on, with 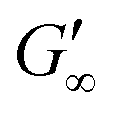 values being reached after approximately 100 s. The stiffness of the sample is directly proportional to its G′ value. As expected, the hydrogel became stiffer with increasing crosslinker concentration, which was confirmed by this measurement (Table 1).
values being reached after approximately 100 s. The stiffness of the sample is directly proportional to its G′ value. As expected, the hydrogel became stiffer with increasing crosslinker concentration, which was confirmed by this measurement (Table 1).
| Crosslinker |
|
|
t c [s] | γ L [%] | γ c [%] |
|---|---|---|---|---|---|
| MBAA | 2243.3 ± 53.4 | 45.3 ± 24.3 | 46.2 ± 0 | 1.0 ± 0 | 62.2 ± 0 |
| EGDA | ND | ND | 40.5 ± 1.7 | 48.7 ± 67.4 | 660.3 ± 51.5 |
| PEGDA 250 | 13.1 ± 1.9 | 4.6 ± 1.5 | 47.2 ± 5.9 | 21.4 ± 16.7 | 931.7 ± 38.7 |
| PEGDA 575 10 mol% | 10116.7 ± 88.1 |
59.8 ± 69.9 | 42.2 ± 0 | 1.0 ± 0 | 69.2 ± 9.7 |
| PEGDA 575 15 mol% | 41106.7 ± 411.0 |
309.4 ± 50.7 | 41.2 ± 0 | 1.0 ± 0 | 62.4 ± 0 |
| PEGDA 575 20 mol% | 97766.7 ± 436.5 |
1217.7 ± 92.4 | 40.2 ± 0 | 1.0 ± 0 | 69.2 ± 9.7 |
| PEGDA 700 | 29256.7 ± 125.0 |
306.3 ± 61.7 | 37.8 ± 1.8 | 1.0 ± 0 | 28.3 ± 9.7 |
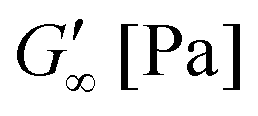
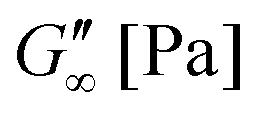
The gelation behaviour can be influenced by varying the molar mass and the resulting chain length of the crosslinkers (Fig. 2D). Samples with lower molar mass crosslinkers (PEGDA 250) only show an increase in 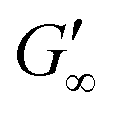 to 13.1 Pa, while the
to 13.1 Pa, while the 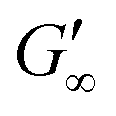 for PEGDA 700 is 29256.7 Pa. This indicates that the stiffness of the hydrogels increases with longer crosslinker chains. Additionally, an acrylamide crosslinker (MBAA) was also used in this work. MBAA crosslinked gels gel slower than PEGDA 575 and only reach a
for PEGDA 700 is 29256.7 Pa. This indicates that the stiffness of the hydrogels increases with longer crosslinker chains. Additionally, an acrylamide crosslinker (MBAA) was also used in this work. MBAA crosslinked gels gel slower than PEGDA 575 and only reach a 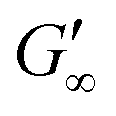 of 2243.3 Pa, which is significantly softer than the PEGDA 575 at 10116.7 Pa (Table 1). This difference in stiffness is probably due to the structure of the crosslinker. MBAA, as an amide, is more electron-rich than the corresponding ester of PEGDA, leading to better stabilization of the formed radical during gelation. This increased stability of the radical in case of MBAA results in slower overall gelation due to its lower reactivity.61
of 2243.3 Pa, which is significantly softer than the PEGDA 575 at 10116.7 Pa (Table 1). This difference in stiffness is probably due to the structure of the crosslinker. MBAA, as an amide, is more electron-rich than the corresponding ester of PEGDA, leading to better stabilization of the formed radical during gelation. This increased stability of the radical in case of MBAA results in slower overall gelation due to its lower reactivity.61
In addition to gelation tracking, the amplitude sweep measurement provides useful information about the stiffness of a hydrogel and its stability to shear stress. This measurement shows the linear viscoelastic (LVE) range of a sample, where G′ is independent of the shear strain (γL in Table 1). This indicates that the deformation behaviour of the hydrogel is in the non-destructive range (±5% of the initial value of G′). If the LVE range limit is exceeded, the sample will begin to form microcracks and exhibit brittle fracturing behaviour. The value of G′ is greater than G′′, indicating a gel-like structure and the behaviour of a viscoelastic solid. The microcracks develop into macrocracks within the hydrogel. After the crossing point (G′ = G′′, γc in Fig. 2B), G′′ is significantly larger than G′, indicating the behaviour of a viscoelastic liquid.4 The hydrogel is more stable against shear stress and deformation with a longer LVE region.32
All samples with a high G′ value in gelation tracking have a very short LVE range (Table 1). Hydrogels containing MBAA and PEGDA, without PEGDA 250, become brittle after only 1% of shear strain, indicating instability to deformation.32 Hydrogels containing EGDA and PEGDA 250 as crosslinkers have a longer LVE of 48.7 ± 67.4% and 21.4 ± 16.7%, respectively. The crossing point of these two samples (660.3 ± 51.5% and 931.7 ± 38.7%) occurs at a much higher shear strain than all other samples (28–69%).
A wide range of γL and γc values is also reported in the literature. For example, Zhang et al. prepared self-healing hydrogels based on xanthan gum. These hydrogels had critical strains ranging from 24.2 to 29.8% and showed G′ recovery rates ranging from 91.36 to 93.69%.62 In another study, Jastram et al. synthesized hydrogels that also contained a vinylimidazole unit and were crosslinked with MBAA.4 These hydrogels showed γL values of 91 ± 2 to 92 ± 7%, which is significantly higher than the values achieved in this work.
The hydrogels, with the exception of EGDA and PEGDA 250, exhibit high stiffness and low stability against shear stress, indicating their rigidity. Therefore, they need to be adapted for medical applications, such as stent coating or in tissue engineering. Copolymerisation with a long-chain monomer or polymer could increase their stability to deformation stress and generally improve the mechanical properties. The use of PEGDA with molar masses of 3000 to 20000 g mol−1 would be a promising approach here.63,64
Hydrogel characterization with Raman spectroscopy
Raman spectroscopy is another option for characterizing hydrogel structure. Fig. 3A shows the Raman-spectra of the monomer GVIM-I (red spectra), the reaction mixture without TEMED (since polymerization occurs within seconds of adding the initiator) (blue spectra), and the dried polyGVIM-I hydrogel (black spectra). The Raman shift at 750 cm−1 indicates the band from C–O–C, which belongs to the GVIM-I monomer and the crosslinker PEGDA. The Raman shifts in the range from 850 to 1150 cm−1 are attributed to C–C stretching vibrations originating from the GVIM-I sugar backbone, but also from the backbone of the polymer chain. This explains why the Raman shifts in the hydrogel have a higher normalized intensity, as the backbone forms during gelation and the number of C–C bonds increases. The Raman shift at 1426 cm−1 indicates the deformation vibration of CH2 at the C-6 position in the sugar. This band was used for normalization, as it does not change during polymerization. The vibrations of the CC double bonds can be seen in the spectra at 1656 cm−1. The normalized intensity of the band is significantly lower after the hydrogel formation than in the monomer solution. During the formation of the hydrogel, the CC groups of the crosslinker are linked to the vinyl group of the monomer, which explains the decrease in intensity of the band.
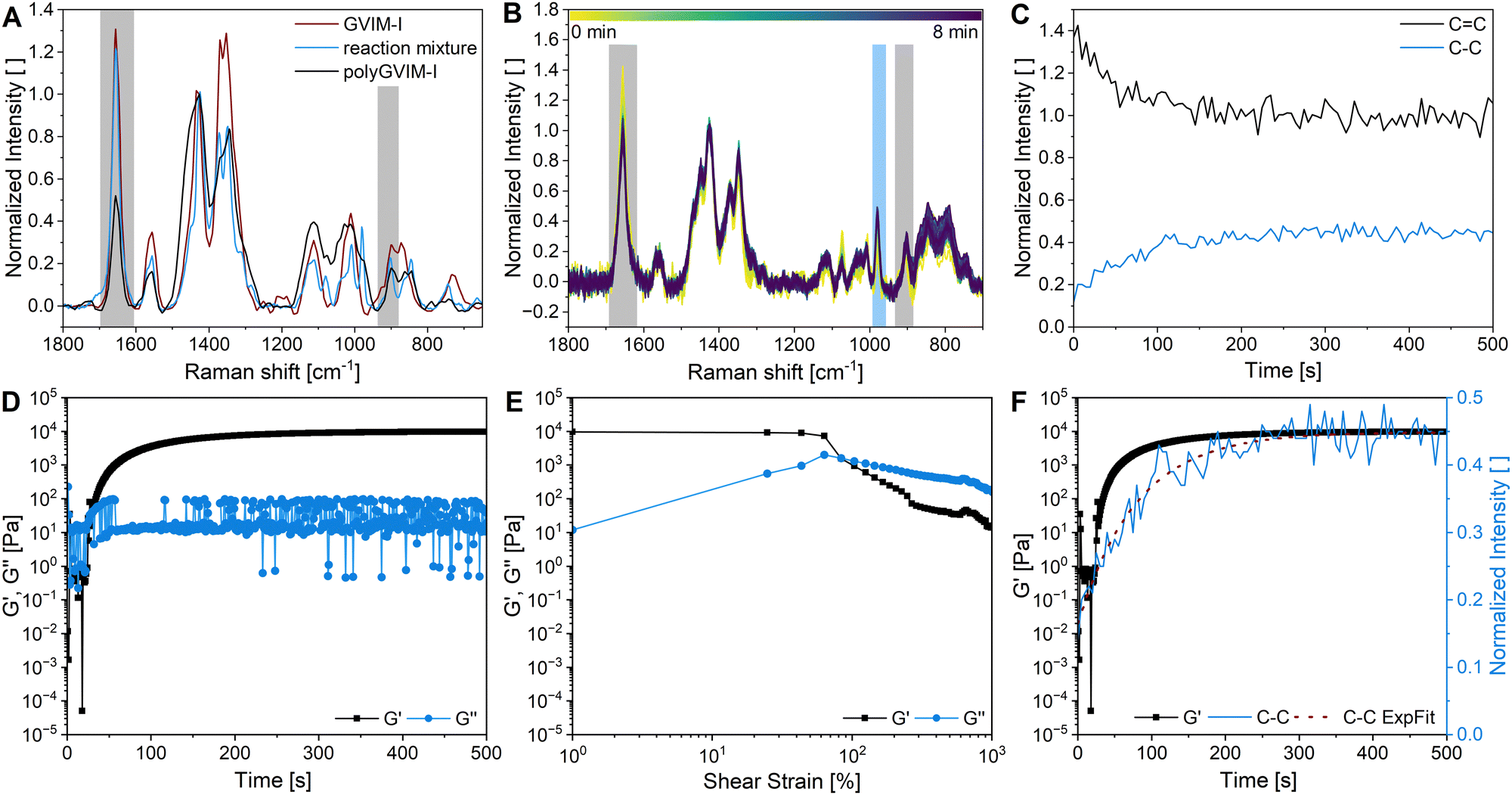 | ||
| Fig. 3 Characterization of polyGVIM-I hydrogels via (A) Raman spectroscopy, as well as (B)–(F) in situ rheology-Raman spectroscopy: (A) Raman spectra at a 1064 nm wavelength of the monomer GVIM-I (red), the reaction solution in PBS with PEGDA 575 as crosslinker without TEMED at a wavelength of 785 nm (blue) and the dried corresponding polyGVIM-I hydrogel at a wavelength of 1064 nm (black). Grey highlighted are the significant bands for CC stretching vibrations (1656 cm−1) and CC bending vibrations (900 cm−1). The spectra were height normalized on the 1426 cm−1 band. (B) In situ Raman spectra at 785 nm wavelength for polymerization tracking over 8 min of polyGVIM-I with PEGDA 575 as a crosslinker and APS/TEMED as an initiator system. Grey highlighted are the significant bands for CC stretching vibrations (1656 cm−1) and CC bending vibrations (900 cm−1). Blue highlighted is the significant band for C–C stretching vibrations (980 cm−1). The spectra were normalized on the 1428 cm−1 band. (C) Time trace of the CC consumption (hight of the band at 1656 cm−1) (black) and C–C formation (height of the band at 980 cm−1) (blue) during polymerization, corresponding to the in situ Raman spectra (B). (D) Gelation tracking of polyGVIM-I with PEGDA 575 as crosslinker and ASP/TEMED as initiator system and (E) the corresponding amplitude sweep measurement. (F) Overlay of the G′ during gelation and the C–C formation during polymerization (approximated with an exponential fit in red) of the in situ rheology-Raman spectroscopy. | ||
Hydrogel characterization with in situ rheology-Raman spectroscopy
The stiffness and the LVE region of hydrogels as well as the gelation time of a reaction mixture to a hydrogel, can be determined by rheological measurements, as shown in two previous chapters. The formation of a polyGVIM-I hydrogel with PEGDA 575 as crosslinker and APS/TEMED as initiator system was followed with the rheometer-Raman combination, which means an in situ Raman measurement and simultaneous gelation tracking on the rheometer. Fig. 3B shows the Raman spectra of the in situ Raman measurement. The normalized intensity of the band at 1656 cm−1 decreases during the measurement and the band at 980 cm−1 increases. To better visualize the change in intensity of these two bands, the intensities were plotted against time (Fig. 3C). It can clearly be seen that the intensity of the vibration of the CC double bond (black) decreases sharply within the first 200 s and then reaches a steady state at 300 s, while the intensity of the vibration of the C–C single bond (blue) increases and then also reaches a steady state at the same time. The graph shows that the breaking of the CC double bonds occurs simultaneously with the formation of the single bonds of the polymer backbone. The rheological measurement taken during gel formation is shown in Fig. 3D. Here, as already shown in the previous chapter, G′ and G′′ have been plotted against time. At the start of the data recording, the value of G′ is less than G′′, which means that the monomer solution behaves like a viscoelastic fluid, as expected. The initiator TEMED was added just before the start of the data recording. It can be seen, that the crossing point of G′ and G′′ occurred after 30 s, indicating a very fast gelation. After the crossing point, the value of G′ rises rapidly, then flattens out until it reaches a plateau at about 200 s. The plateau indicates that the formation of the three-dimensional hydrogel structure is complete. Fig. 3E shows the subsequent amplitude sweep measurement of the same hydrogel. The LVE range of this hydrogel is 24.7%, which is much higher than the same hydrogels with LAP as initiator instead of APS/TEMED (see Table 1). The value of γc with 83.7% is at a significantly higher shear strain, indicating an overall more stable hydrogel. Fig. 3F compares the progression of G′ of the gelation tracking with the intensity of the C–C single bond vibration of the in situ Raman measurement. By overlaying the two data sets, conclusions can be drawn about the gelation and polymerization processes. Gelation i.e. the transformation of the sample from liquid-like to solid-like behavior, can be seen in the rheometer data after 30 s. The value of G′ is greater than G′′ from this point onwards. The Raman data shows in direct comparison that only a small proportion of C–C bonds have formed after 30 s. At the gelation point of the gel (crossing point of G′ and G′′) an increase in the intensity of the C–C bond from 0.15 to 0.25, which can be correlated with a conversion of the double bonds of 33%, if the normalized Raman intensity of 0.45 is attributed to a conversion of around 100%. The formation of C–C single bonds, i.e. the structural change of the sample, is the polymerization. It can be assumed that complete polymerization occurred approximately 300 seconds after the start of the experiment, as evidenced by a normalized intensity of the C–C vibration of 0.45 in the Raman spectrum (100% conversion of the CC bond in the Raman spectrum). However, the value of G′ continued to increase (from 7413.9 Pa to 9782.0 Pa at the end of the measurement). This could be caused not only by incomplete gelation but also by the drying effects of the hydrogel.
Conclusion
In the present study, we have successfully characterized carbohydrate-based hydrogels, before, after and during gelation or polymerization, with different formulations using established methods and have demonstrated a new alternative technique for hydrogel characterization.We were able to show that ATR-FTIR is initially suitable for spectroscopic characterization to get a first impression of the structure of the monomer (GVIM-I) and the hydrogel. The advantage of ATR-FTIR is that it requires little sample preparation, and is often accessible and inexpensive. Nevertheless, the water present in the reaction solution can lead to the formation of broad bands which may mask signals of interest. This issue can be avoided by pre-drying the samples. The in situ IR measurement shows strong noise of signals above 1900 cm−1 which can be attributed to the device setup (quartz glass), resulting in superposition of the signals of interest. Furthermore, the results of the CC double bond vibration tracking should be treated with caution. In addition to the intensity of the band at 955 cm−1, numerous other band intensities that should remain constant, also decrease. This indicates that the polymerization monitoring by IR was unsuccessful in this setup.
These limitations do not apply to Raman spectroscopy. Water signals do not interfere with the measurements and do not overlay the bands of interest, giving a clearer resolution than the IR spectra. The in situ Raman measurement allowed the monitoring of the intensity decrease of the CC double bonds and the intensity increase of the C–C bonds.
In addition to spectroscopic methods for monitoring polymerization, rheological techniques can be employed to track the gelation. For the additional determination of gelation time, it is also possible to identify other characteristics, such as stiffness and LVE region of hydrogels with different crosslinker types and concentrations.
The in situ rheology-Raman spectroscopy, a novel methodology for the characterization of hydrogels, was presented for the first time in this study and compared with conventional methods. This method has so far been limited to radical polymerizing hydrogels, although initial experiments have already investigated the curing of epoxy resins using rheometer-Raman.43 Furthermore, this technique could be applied in the future to hydrogels based on click chemistry, for example Diels–Alder crosslinking. However, it should be noted that this method is not suitable for hydrogel gelation based on ionic interaction.
The rheometer-Raman combination offers several advantages for hydrogels with already described polymerization forms, such as radical polymerization. These advantages include the ability to track the Raman bands corresponding to the CC double bond and the C–C bond, as well as the direct comparison to the gelation of the rheological measurement and the aforementioned bands. All in all, the combination of rheology and Raman spectroscopy, has led to a significant expansion of knowledge in a relatively short period by running the methods simultaneously, enabling the detection of differences between polymerization and gelation.
Author contributions
Sina Lambrecht: formal analysis, investigation & writing – original draft; Marek Biermann: formal analysis, investigation & writing – review & editing; Selin Kara: funding acquisition, writing – review & editing; Stefan Jopp: writing – review & editing; Johanna Meyer: conceptualization, methodology, formal analysis, investigation & writing – original draft. All authors approved the final version of the manuscript.Data availability
The authors confirm that the data supporting the findings of this study are available within the article and its supporting information. Raw data that support the findings of this study are available from the corresponding author.Conflicts of interest
There are no conflicts to declare.Acknowledgements
SL and SJ thanks the German Research Center (Deutsche Forschungsgemeinschaft, DFG) for the financial support (NFDI4Cat; DFG grant no 441926934). SK thanks the Ministry for Science and Culture for Lower Saxony for the Holen & Halten starting grant (grant no. 12.5-76251-17-9/20). We would like to thank Tabea Thiel from the Leibniz Institute of Catalysis (Rostock) for providing the in situ IR device. We also like to thank Martin Pähler, Martina Weiß, Caroline Müller and Enes Yayci from the Institute of Technical Chemistry (Leibniz University Hannover), and Sandra Diederich from the Institute of Chemistry (University of Rostock) for their support on the day-to-day laboratory routine.References
- P. Calvert, Hydrogels for Soft Machines, Adv. Mater., 2009, 21, 743–756, DOI:10.1002/adma.200800534.
- K. Y. Lee and D. J. Mooney, Hydrogels for tissue engineering, Chem. Rev., 2001, 101, 1869–1879, DOI:10.1021/cr000108x.
- J. Claus, F. O. Sommer and U. Kragl, Ionic liquids in biotechnology and beyond, Solid State Ionics, 2018, 314, 119–128, DOI:10.1016/j.ssi.2017.11.012.
- A. Jastram, J. Claus, P. A. Janmey and U. Kragl, Rheological properties of hydrogels based on ionic liquids, Polym. Test., 2021, 93, 106943, DOI:10.1016/j.polymertesting.2020.106943.
- P. Shah and S. Chandra, Review on emergence of nanomaterial coatings in bio-engineered cardiovascular stents, J. Drug Delivery Sci. Technol., 2022, 70, 103224, DOI:10.1016/j.jddst.2022.103224.
- Y. Wang, G. Li, L. Yang, R. Luo and G. Guo, Development of Innovative Biomaterials and Devices for the Treatment of Cardiovascular Diseases, Adv. Mater., 2022, 34, e2201971, DOI:10.1002/adma.202201971.
- M. Pacheco, B. Domingues, E. Lima, R. L. Reis and A. Barros, Hydrogels for Tissue Engineering and Regenerative Medicine, Elsevier, 2024, pp. 467–482 Search PubMed.
- J. Claus, T. Eickner, N. Grabow, U. Kragl and S. Oschatz, Ion Exchange Controlled Drug Release from Polymerized Ionic Liquids, Macromol. Biosci., 2020, 20, e2000152, DOI:10.1002/mabi.202000152.
- Y. Zhao, S. Song, X. Ren, J. Zhang, Q. Lin and Y. Zhao, Supramolecular Adhesive Hydrogels for Tissue Engineering Applications, Chem. Rev., 2022, 122, 5604–5640, DOI:10.1021/acs.chemrev.1c00815.
- F. Ai, X. Yin, R. Hu, H. Ma and W. Liu, Research into the super-absorbent polymers on agricultural water, Agric. Water Manage., 2021, 245, 106513, DOI:10.1016/j.agwat.2020.106513.
- A. Grollmisch, U. Kragl and J. Großeheilmann, Enzyme Immobilization in Polymerized Ionic Liquids-based Hydrogels for Active and Reusable Biocatalysts, SynOpen, 2018, 02, 192–199, DOI:10.1055/s-0037-1610144.
- J. Meyer, L.-E. Meyer and S. Kara, Enzyme immobilization in hydrogels: A perfect liaison for efficient and sustainable biocatalysis, Eng. Life Sci., 2022, 22, 165–177, DOI:10.1002/elsc.202100087.
- G. Stojkov, Z. Niyazov, F. Picchioni and R. K. Bose, Relationship between Structure and Rheology of Hydrogels for Various Applications, Gels, 2021, 7(4), 255, DOI:10.3390/gels7040255.
- V. S. Raghuwanshi and G. Garnier, Characterisation of hydrogels: Linking the nano to the microscale, Adv. Colloid Interface Sci., 2019, 274, 102044, DOI:10.1016/j.cis.2019.102044.
- P. Patel and P. Thareja, Hydrogels differentiated by length scales: A review of biopolymer-based hydrogel preparation methods, characterization techniques, and targeted applications, Eur. Polym. J., 2022, 163, 110935, DOI:10.1016/j.eurpolymj.2021.110935.
- S. Lambrecht, H. Schröter, H. Pohle and S. Jopp, Swelling Behavior of Novel Hydrogels Produced from Glucose-Based Ionic Monomers with Varying Cross-Linkers, ACS Omega, 2024, 9, 5418–5428, DOI:10.1021/acsomega.3c06804.
- C. Marmorat, A. Arinstein, N. Koifman, Y. Talmon, E. Zussman and M. Rafailovich, Cryo-Imaging of Hydrogels Supermolecular Structure, Sci. Rep., 2016, 6, 25495, DOI:10.1038/srep25495.
- S. Liu and L. Li, Thermoreversible gelation and scaling behavior of Ca2 + -induced κ-carrageenan hydrogels, Food Hydrocolloids, 2016, 61, 793–800, DOI:10.1016/j.foodhyd.2016.07.003.
- D. J. Adams, Does Drying Affect Gel Networks?, Gels, 2018, 4(2), 32, DOI:10.3390/gels4020032.
- C. A. García-González, M. C. Camino-Rey, M. Alnaief, C. Zetzl and I. Smirnova, Supercritical drying of aerogels using CO2: Effect of extraction time on the end material textural properties, J. Supercrit. Fluids, 2012, 66, 297–306, DOI:10.1016/j.supflu.2012.02.026.
- D. Das, P. Prakash, P. K. Rout and S. Bhaladhare, Synthesis and Characterization of Superabsorbent Cellulose-Based Hydrogel for Agriculture Application, Starch, 2021, 73, 1900284, DOI:10.1002/star.201900284.
- S. Mao, Y. Ren, S. Chen, D. Liu, X. Ye and J. Tian, Development and characterization of pH responsive sodium alginate hydrogel containing metal-phenolic network for anthocyanin delivery, Carbohydr. Polym., 2023, 320, 121234, DOI:10.1016/j.carbpol.2023.121234.
- F. Azam, F. Ahmad, S. Ahmad, M. S. Zafar and Z. Ulker, Synthesis and characterization of natural fibers reinforced alginate hydrogel fibers loaded with diclofenac sodium for wound dressings, Int. J. Biol. Macromol., 2023, 241, 124623, DOI:10.1016/j.ijbiomac.2023.124623.
- Q. Wu, J. Wei, B. Xu, X. Liu, H. Wang, W. Wang, Q. Wang and W. Liu, A robust, highly stretchable supramolecular polymer conductive hydrogel with self-healability and thermo-processability, Sci. Rep., 2017, 7, 41566, DOI:10.1038/srep41566.
- L. Mendoza, T. Gunawardhana, W. Batchelor and G. Garnier, Nanocellulose for gel electrophoresis, J. Colloid Interface Sci., 2019, 540, 148–154, DOI:10.1016/j.jcis.2019.01.017.
- D. Das, A. P. Rameshbabu, P. Patra, P. Ghosh, S. Dhara and S. Pal, Biocompatible amphiphilic microgel derived from dextrin and poly(methyl methacrylate) for dual drugs carrier, Polymer, 2016, 107, 282–291, DOI:10.1016/j.polymer.2016.11.029.
- D. K. Patel, S. D. Dutta, W.-C. Shin, K. Ganguly and K.-T. Lim, Fabrication and characterization of 3D printable nanocellulose-based hydrogels for tissue engineering, RSC Adv., 2021, 11, 7466–7478, 10.1039/d0ra09620b.
- M. Jakubowski, A. Domke, M. Ratajczak, J. Szczuka, T. Buchwald, A. Voelkel and M. Sandomierski, Chitosan modified with lanthanum ions as implantable hydrogel for local delivery of bisphosphonates, Int. J. Biol. Macromol., 2023, 230, 123429, DOI:10.1016/j.ijbiomac.2023.123429.
- G. Tatrari, C. Tewari, M. Pathak, D. Bhatt, M. Karakoti, S. Pandey, D. S. Uniyal, F. U. Shah and N. G. Sahoo, 3D-graphene hydrogel and tungsten trioxide-MnO2 composite for ultra-high-capacity asymmetric supercapacitors: A comparative study, J. Energy Storage, 2023, 68, 107830, DOI:10.1016/j.est.2023.107830.
- G. Nie, Y. Zang, W. Yue, M. Wang, A. Baride, A. Sigdel and S. Janaswamy, Cellulose-based hydrogel beads: Preparation and characterization, Carbohydr. Polym. Technol. Appl., 2021, 2, 100074, DOI:10.1016/j.carpta.2021.100074.
- S. Affatato, D. Trucco, P. Taddei, L. Vannozzi, L. Ricotti, G. D. Nessim and G. Lisignoli, Wear Behavior Characterization of Hydrogels Constructs for Cartilage Tissue Replacement, Materials, 2021, 14(2), 428, DOI:10.3390/ma14020428.
- M. Cofelice, M. C. Messia, E. Marconi, F. Cuomo and F. Lopez, Effect of the xanthan gum on the rheological properties of alginate hydrogels, Food Hydrocolloids, 2023, 142, 108768, DOI:10.1016/j.foodhyd.2023.108768.
- S. Bom, R. Ribeiro, H. M. Ribeiro, C. Santos and J. Marto, On the progress of hydrogel-based 3D printing: Correlating rheological properties with printing behaviour, Int. J. Pharm., 2022, 615, 121506, DOI:10.1016/j.ijpharm.2022.121506.
- S. A. Khan, A. N. Malik, B. Velioglu, S. Gul, I. H. Kavakli and I. Lazoglu, A novel smart disinfection system using 3D-printed and electrically conductive composite hydrogel, Emergent Mater., 2024 DOI:10.1007/s42247-024-00632-1.
- Q. Zhong, Y. Chen, X. Zhang, G. Yang, W. Jin, D. Peng and Q. Huang, Correlation between 3D printability and rheological properties of biopolymer fluid: A case study of alginate-based hydrogels, J. Food Eng., 2024, 370, 111970, DOI:10.1016/j.jfoodeng.2024.111970.
- D. Bhardwaj, R. Singhmar, M. Garg, D. Gupta, A. Dhiman, S. Soo Han and G. Agrawal, Designing advanced hydrogel inks with direct ink writing based 3D printability for engineered biostructures, Eur. Polym. J., 2024, 205, 112736, DOI:10.1016/j.eurpolymj.2023.112736.
- M. Nadgorny, Z. Xiao and L. A. Connal, 2D and 3D-printing of self-healing gels: design and extrusion of self-rolling objects, Mol. Syst. Des. Eng., 2017, 2, 283–292, 10.1039/c7me00023e.
- S. Vaupel, R. Mau, S. Kara, H. Seitz, U. Kragl and J. Meyer, 3D printed and stimulus responsive drug delivery systems based on synthetic polyelectrolyte hydrogels manufactured via digital light processing, J. Mater. Chem. B, 2023, 11, 6547–6559, 10.1039/d3tb00285c.
- B. Ozel and M. H. Oztop, Advances in Food Rheology and Its Applications, Elsevier, 2023, pp. 661–688 DOI:10.1016/B978-0-12-823983-4.00018-2.
- S. M. R. Mousavi, A. Rafe and S. Yeganehzad, Structure-rheology relationships of composite gels: Alginate and Basil seed gum/guar gum, Carbohydr. Polym., 2020, 232, 115809, DOI:10.1016/j.carbpol.2019.115809.
- J. M. Zuidema, C. J. Rivet, R. J. Gilbert and F. A. Morrison, A protocol for rheological characterization of hydrogels for tissue engineering strategies, J. Biomed. Mater. Res., Part B, 2014, 102, 1063–1073, DOI:10.1002/jbm.b.33088.
- J. Romischke, A. Scherkus, M. Saemann, S. Krueger, R. Bader, U. Kragl and J. Meyer, Swelling and Mechanical Characterization of Polyelectrolyte Hydrogels as Potential Synthetic Cartilage Substitute Materials, Gels, 2022, 8(5), 296, DOI:10.3390/gels8050296.
- L. Völker-Pop, J. A. R. Agudo and C. Giehl, Novel Trends in Rheology IX, AIP Publishing, 2023, p. 30001 Search PubMed.
- T. Kida, K. Janchai, K. Tokumitsu and M. Yamaguchi, Flow-induced crystallization behavior of high-density polyethylene evaluated by Rheo-Raman spectroscopic system, Polym. J., 2023, 55, 1141–1150, DOI:10.1038/s41428-023-00821-5.
- M. Hesse, H. Meier and B. Zeeh, Spektroskopische Methoden in der organischen Chemie. 102 Tabellen, Thieme, Stuttgart, 7th edn, 2005 Search PubMed.
- J. L. Lippert, J. A. Robertson, J. R. Havens and J. S. Tan, Structural studies of poly(N-vinylimidazole) complexes by infrared and Raman spectroscopy, Macromolecules, 1985, 18, 63–67, DOI:10.1021/ma00143a010.
- A. Lavrentieva, T. Fleischhammer, A. Enders, H. Pirmahboub, J. Bahnemann and I. Pepelanova, Fabrication of Stiffness Gradients of GelMA Hydrogels Using a 3D Printed Micromixer, Macromol. Biosci., 2020, 20, e2000107, DOI:10.1002/mabi.202000107.
- M. Kirsch, L. Birnstein, I. Pepelanova, W. Handke, J. Rach, A. Seltsam, T. Scheper and A. Lavrentieva, Gelatin-Methacryloyl (GelMA) Formulated with Human Platelet Lysate Supports Mesenchymal Stem Cell Proliferation and Differentiation and Enhances the Hydrogel’s Mechanical Properties, Bioengineering, 2019, 6(3), 76, DOI:10.3390/bioengineering6030076.
- A. J. Engler, S. Sen, H. L. Sweeney and D. E. Discher, Matrix elasticity directs stem cell lineage specification, Cell, 2006, 126, 677–689, DOI:10.1016/j.cell.2006.06.044.
- Y.-H. Tsou, J. Khoneisser, P.-C. Huang and X. Xu, Hydrogel as a bioactive material to regulate stem cell fate, Bioact. Mater., 2016, 1, 39–55, DOI:10.1016/j.bioactmat.2016.05.001.
- L. del-Mazo-Barbara and M.-P. Ginebra, Rheological characterisation of ceramic inks for 3D direct ink writing: A review, J. Eur. Ceram. Soc., 2021, 41, 18–33, DOI:10.1016/j.jeurceramsoc.2021.08.031.
- P. A. Amorim, M. A. d’Ávila, R. Anand, P. Moldenaers, P. van Puyvelde and V. Bloemen, Insights on shear rheology of inks for extrusion-based 3D bioprinting, Bioprinting, 2021, 22, e00129, DOI:10.1016/j.bprint.2021.e00129.
- A. S. Fernandes, B. V. Neves, T. M. Mazzo, E. Longo, E. Jacob-Lopez, L. Q. Zepka and V. V. de Rosso, Bigels as potential inks for extrusion-based 3d food printing: Effect of oleogel fraction on physical characterization and printability, Food Hydrocolloids, 2023, 144, 108986, DOI:10.1016/j.foodhyd.2023.108986.
- R. Qiu, K. Wang, H. Tian, X. Liu, G. Liu, Z. Hu and L. Zhao, Analysis on the printability and rheological characteristics of bigel inks: Potential in 3D food printing, Food Hydrocolloids, 2022, 129, 107675, DOI:10.1016/j.foodhyd.2022.107675.
- J. M. H. Rahman, M. N. I. Shiblee, K. Ahmed, A. Khosla, M. Kawakami and H. Furukawa, Rheological and mechanical properties of edible gel materials for 3D food printing technology, Heliyon, 2020, 6, e05859, DOI:10.1016/j.heliyon.2020.e05859.
- H. Jiang, L. Zheng, Y. Zou, Z. Tong, S. Han and S. Wang, 3D food printing: main components selection by considering rheological properties, Crit. Rev. Food Sci. Nutr., 2019, 59, 2335–2347, DOI:10.1080/10408398.2018.1514363.
- T. G. Mezger, The Rheology Handbook. 4th Edition, Vincentz Network, Hannover, 4th edn, 2014 Search PubMed.
- H. M. Wyss, in Fluids, colloids, and soft materials, ed. A. Fernandez-Nieves and A. M. Puertas, John Wiley & Sons Inc, Hoboken, New Jersey, 2016, pp. 149–164 Search PubMed.
- N. Sahiner, M. Singh, D. de Kee, V. T. John and G. L. McPherson, Rheological characterization of a charged cationic hydrogel network across the gelation boundary, Polymer, 2006, 47, 1124–1131, DOI:10.1016/j.polymer.2005.10.129.
- F. Chambon and H. H. Winter, Linear Viscoelasticity at the Gel Point of a Crosslinking PDMS with Imbalanced Stoichiometry, J. Rheol., 1987, 31, 683–697, DOI:10.1122/1.549955.
- N. Kanovsky and S. Margel, Fabrication of Transparent Silica/PEG Smooth Thin Coatings on Polymeric Films for Antifogging Applications, ACS Omega, 2022, 7, 20505–20514, DOI:10.1021/acsomega.1c07293.
- R. Zhang, Y. Tao, Q. Xu, N. Liu, P. Chen, Y. Zhou and Z. Bai, Rheological and ion-conductive properties of injectable and self-healing hydrogels based on xanthan gum and silk fibroin, Int. J. Biol. Macromol., 2020, 144, 473–482, DOI:10.1016/j.ijbiomac.2019.12.132.
- M. Jamadi, P. Shokrollahi, B. Houshmand, M. D. Joupari, F. Mashhadiabbas, A. Khademhosseini and N. Annabi, Poly (Ethylene Glycol)-Based Hydrogels as Self-Inflating Tissue Expanders with Tunable Mechanical and Swelling Properties, Macromol. Biosci., 2017, 17(8), 1600478, DOI:10.1002/mabi.201600479.
- N. Rekowska, D. Arbeiter, H. Seitz, R. Mau, A. Riess, T. Eickner, N. Grabow and M. Teske, The influence of PEGDA's molecular weight on its mechanical properties in the context of biomedical applications, CDBME, 2022, 8, 181–184, DOI:10.1515/cdbme-2022-1047.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ma00543k |
| This journal is © The Royal Society of Chemistry 2024 |