
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnhanced fast charging capabilities in natural graphite/iron cross-linked pectin electrodes for lithium-ion batteries†
Chin-Yi
Chung
abc,
Wei-Ming
Chen
abc,
Yan-Ruei
Chen
 abc,
Liang-Yu
Chen
a,
Yu-Hsuan
Su
abc,
Po-Wei
Chi
*a,
Phillip M.
Wu
*ad,
Kuei-Shu
Chang-Liao
c,
Hong-Yi
Tang
e and
Maw-Kuen
Wu
a
abc,
Liang-Yu
Chen
a,
Yu-Hsuan
Su
abc,
Po-Wei
Chi
*a,
Phillip M.
Wu
*ad,
Kuei-Shu
Chang-Liao
c,
Hong-Yi
Tang
e and
Maw-Kuen
Wu
a
aInstitute of Physics, Academia Sinica 128, Section 2, Academia Road, Taipei 11529, Taiwan. E-mail: philwu@gmail.com; poweichi@gate.sinica.edu.tw
bNano Science and Technology Program, Taiwan International Graduate Program, Academia Sinica and National Tsing Hua University 128, Section 2, Academia Road, Taipei 11529, Taiwan
cDepartment of Engineering and System Science, National Tsing Hua University 101, Section 2, Kuang-Fu Road, Hsinchu 300044, Taiwan
dCollege of Science, National Chung Hsing University 145, Xingda Road, South District, Taichung 402, Taiwan
eDepartment of Applied Chemistry, National Chi-Nan University 1 University Road, Puli, Nantou, 545301, Taiwan
First published on 25th July 2024
Abstract
Rapid charging capabilities are required for the broader adoption of lithium-ion batteries in emerging technologies such as electric vehicles. Although commercial graphite anodes provide a high energy density, their limitations in quick charging warrant the exploration of novel materials. This study extends prior research on the use of pectin, a natural polysaccharide, as an organic binder for graphite anodes. By introducing iron into pectin, we propose a unique ion-crosslinking mechanism that causes the graphite anode to exhibit pseudocapacitive Li storage. Our findings, corroborated by 3D Bode analysis and AC impedance spectra, revealed a positive correlation between the iron ion and the pseudocapacitive vertex in the graphite reaction. Compared with conventional binders, graphite electrodes utilizing iron-doped pectin demonstrated superior initial and stable capacities, reaching 450 mA h g−1 after 100 cycles at 1C. This study highlights the synergistic effect of pectin and iron ions, offering a new avenue for high-performance, rapid-charging, and environmentally sustainable lithium-ion batteries.
Introduction
The adoption of lithium-ion batteries (LIBs) in portable electronics, electric vehicles (EVs), and renewable energy storage1,2 becomes essential to our daily activities. The attributes that have catapulted LIBs into technological prominence include a high energy density, long cycle life, and minimal memory effects. Graphite anodes have been the main choice for LIBs and other metal-ion batteries, owing to their proven track record in terms of energy density and stability.3 As our reliance on LIBs continues to grow, there is an urgent need for quicker charging capabilities, increased lifecycles, and eco-friendly components.4 This necessitates a systematic investigation of alternative materials and mechanisms that could augment anode performance. Although graphite anodes deliver a high energy density, their conventional binders, such as polyvinylidene fluoride (PVDF), pose limitations. These synthetic binders contribute to the environmental footprint of LIBs and act as bottlenecks for their fast-charging capabilities.5 Additionally, the use of PVDF presents significant drawbacks due to the necessity of employing a toxic organic solvent, N-methyl-2-pyrrolidone (NMP), to generate slurries containing active materials and additives for casting the electrode. The graphite anodes lead to safety concerns owing to the exothermic formation of F-containing species at elevated temperatures, which arises from the interaction between PVDF and LiC6, and subsequently has the potential to initiate thermal runaway.6 Besides, graphite anodes often suffer from inferior Li+ intercalation kinetics during the fast-charging process. Under high current densities, this can easily lead to lithium plating, which decreases overall battery performance and raises safety concerns.7 To address this challenge, the construction of an artificial solid electrolyte interphase (SEI) using water-based binders has been proposed as a potential solution.8,9 Furthermore, the recycling processes for PVDF-containing electrode are not eco-friendly. Therefore, there is a crucial need for a transition towards more sustainable and efficient binders for LIB electrodes. Although recent advances, such as those by Zheng et al.,10 who developed a rapid thermal shock method to improve the efficiency of recycling spent LIBs, offer promising solutions, the high cost remains a barrier to commercial viability.In recent years, there were many studies to search for fluorine-free, environmentally friendly, and water-soluble safer binders as alternatives to PVDF. This research focus has gained substantial momentum, extending its impact to both cathode and anode applications.11–13 Among the promising candidates is pectin, a natural polysaccharide abundant in the cell walls of fruits and vegetables.14,15 Pectin has emerged as an eco-friendly binder material, and preliminary studies have demonstrated its potential for effective binding to enhance the electrochemical performance of anodes in LIBs.16 Our previous studies on pectin as a water-soluble binder for both natural graphite and Li3VO4 anodes revealed a remarkable improvement in the electrochemical performance, surpassing that of PVDF and other water-soluble binders such as CMC.17,18 We found that the –OH groups on the pectin surface hindered the movement of Li+ ions towards Li3VO4, resulting in the accumulation of Li+ ions near or on the electrode surface and contributing to the pseudocapacitive lithium storage behavior; thus, the rate performance increased by 60%. On the other hand, the 10% capacity of natural graphite was improved when pectin was used as the binder.
Recent studies have provided hints on how the incorporation of metal ions into organic binders can enhance the electrochemical performance.19–23 For example, Liu et al.24 and Yoon et al.25 in their works using calcium-crosslinked alginate as a novel binder for Si/C composite anodes, demonstrated that crosslinking in alginate improved the mechanical properties of the binder compared to sodium alginate and other commercial binders. This enhancement ultimately led to an increased capacity in the Si/C anode while maintaining a stable cycling ability. Such improvements can facilitate rapid Li-ion migration, thereby enabling quicker battery charging. In our earlier study,18 we not only noted an electrochemical performance improvement in graphite with pectin binder, but also observed that the capacity stabilized at a higher value over extended cycles in the case of Fe-added pectin binder, especially after high current density activation. Thus, it is important to better understand the mechanism behind the enhanced electrochemical performance of graphite anodes with Fe-added pectin binders.
This study aims to advance the current understanding of the role of pectin as an anode binder by introducing various concentrations of iron ions. This study explores the impacts of ion crosslinking between pectin on the anode performance, with a specific focus on the charging speed. Consequently, Li-ion electrochemical kinetic analyses are required to elucidate the underlying mechanisms. Typically, kinetic analyses, which depend on the scan rate influence on the voltammetric current, are important for understanding the behavior of materials that store Li-ion charges.26 Transport assessments seek to gain insight into charge storage by determining the rate-limiting steps for diffusion-controlled or surface-limited processes.27 When using the Randles–Sevcik relation (i = avb) to examine how the current changes with the scan rate, the slope of the log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) i vs. logv plot indicates the primary factor limiting the process.28 To further enhance our understanding of the dynamic characteristics of Li-ion storage, AC impedance techniques commonly used in kinetic studies of double-layer processes can be employed to distinguish between diffusion-controlled and surface-limited processes. Expanding on the groundwork of Bai and Conway,29,30 Ko et al. applied similar analyses to unveil the intricate electrochemical responses of LiMn2O4@carbon nanofibers and MnOx@carbon31 electrodes across the potential, frequency, and capacitance (or phase angle). This enabled a clear distinction between double-layer, pseudocapacitive, and battery-like processes. Furthermore, other reports have extended the analysis to three materials chosen as model systems, each known to predominantly manifest a specific charge-storing process: active carbon (YP50F, double-layer), niobium oxide (Nb2O5, pseudocapacitive),32 and lithium iron phosphate (LiFePO4, battery-like).
i vs. logv plot indicates the primary factor limiting the process.28 To further enhance our understanding of the dynamic characteristics of Li-ion storage, AC impedance techniques commonly used in kinetic studies of double-layer processes can be employed to distinguish between diffusion-controlled and surface-limited processes. Expanding on the groundwork of Bai and Conway,29,30 Ko et al. applied similar analyses to unveil the intricate electrochemical responses of LiMn2O4@carbon nanofibers and MnOx@carbon31 electrodes across the potential, frequency, and capacitance (or phase angle). This enabled a clear distinction between double-layer, pseudocapacitive, and battery-like processes. Furthermore, other reports have extended the analysis to three materials chosen as model systems, each known to predominantly manifest a specific charge-storing process: active carbon (YP50F, double-layer), niobium oxide (Nb2O5, pseudocapacitive),32 and lithium iron phosphate (LiFePO4, battery-like).
In this study, we also used electroanalytical methods, including voltammetry and impedance analysis to determine the charge storage behavior of Li ions in the graphite/pectin:Fe electrode. Herein, we investigate the unique ion-crosslinking mechanism, by introducing iron into pectin with molar ratios 2:1, which enables the graphite anode to exhibit enhanced characteristics for lithium storage. We have calculated the methyl esterification degree (MED) of the pectin–Fe samples with varying concentrations of iron ions, based on the FTIR spectrum,18 following the method by Manrique et al.33 Our results indicate the following MED values, for pure pectin, the MED is 77.7%; for pectin–Fe 8:1 ratio, it is 37%; for a 4:1 ratio, it is 33%; and for a 2:1 ratio, it is 12%. As previous studies have shown,34–36 the degree of esterification significantly affects the limited transportation of Li+ cations in electrodes. Our data reveals that the pectin–Fe 2:1 sample, with the lowest MED, shows significant promise for further investigation. This focus on the pectin–Fe 2:1 sample is reinforced by similar findings in our recent publication.37 These results, supported by the 3D Bode analysis and AC impedance spectra, revealed a positive correlation between the iron ion and the pseudocapacitive vertex in the graphite reaction. This study emphasizes the synergistic effect of pectin and iron ions and offers a new avenue for the development of high-performance, rapid-charging, and environmentally sustainable lithium-ion batteries.
Experiment section
Material preparation
The adhesive binders synthesized for this study included polyvinylidene fluoride (PVDF), pectin, and iron-doped pectin (pectin–Fe). PVDF was dissolved in N-methyl-2-pyrrolidone (NMP) to achieve a 7% solution, ensuring homogeneity through continuous stirring. The pectin binder was prepared by mixing with 5% deionized (DI) water and stirring for 24 h to guarantee full dissolution. The pectin:Fe binder was created by blending pectin with ferrous chloride (FeCl2) in a 2:1 weight ratio, subsequently diluted to a 5% concentration with DI water, and stirred for 24 h to form a homogeneous solution.
Electrode preparation
Electrode slurries were formulated by combining 80% commercial natural graphite, 10% carbon black (Super P) for conductivity, and 10% of one of the three binders (PVDF, pectin, or pectin–Fe) by weight. The slurry was coated onto copper (Cu) foil and dried at 110 °C to remove the solvents effectively. The dried coated foils were punched into 14 mm diameter discs for assembly into lithium half-cells.Half cell assemble
Lithium half-cells were assembled in an argon-filled glovebox using 14 mm natural graphite electrodes as the working electrode and lithium metal foil as both the counter and reference electrodes. A microporous polypropylene separator soaked in a 1 M LiPF6 electrolyte solution, mixed in equal volumes of ethylene carbonate (EC) and dimethyl carbonate (DMC), ensured separation between the electrodes.Electrochemical characterization was performed using a PARSTAT MC 200 workstation. Cyclic voltammetry was executed at a scan rate of 0.1 mV s−1 across a potential range of 0.01 to 3 V, enabling detailed analysis of the redox processes. Lithium plating and stripping were conducted within a constant voltage range of −0.5 to 1.0 V (vs. Li/Li+) at a constant current density of 1 mA cm−2. Additionally, electrochemical impedance spectroscopy (EIS) measurements were carried out on this system, employing an AC amplitude of 10 mV across a frequency range of 100 kHz to 0.5 mHz.
Characterization
Microstructural investigations of the prepared samples were conducted by scanning electron microscopy (SEM). SEM analysis was performed using a JEOL-Japan JXA-840A instrument. Attenuated total reflectance Fourier transform infrared (ATR-FTIR) spectroscopy was employed to examine the chemical composition of PVDF, pectin, and pectin:Fe. ATR-FTIR spectra were recorded with a resolution of 4 cm−1 in absorption mode at room temperature. The spectral range for the measurement was extended from 600 to 4000cm−1. The electrolyte wettability of all prepared samples was evaluated by measuring the contact angle (CA, θ) of the electrolyte (1 M LiPF6 in EC:DMC 1:1) droplets on each sample surface. The contact angle measurements were conducted on five samples, as shown in Fig. S1 (ESI†). For each measurement, 5 μl droplets were carefully deposited onto the surface of the samples, and digital photographs of the droplets were taken using a digital camera. The average error in the electrolyte CA measurement, which was influenced by the image quality and built-in curve-fitting function, was estimated to be ±2°.
Results and discussion
Our previous work on the cycling performance of graphite electrodes with PVDF, pectin, and pectin:Fe binders, including the capacity at different C-rates, showed that the capacity remained stable for up to 100 cycles for all electrodes, as shown in Fig. S2 (ESI†). However, we observed that the capacities in cells using pectin and pectin:Fe binders were about 10% higher than that of the PVDF binder. Additionally, anomalous C-rate performance was observed, characterized by a presumed capacity switch occurring at 3C in graphite–pectin and graphite–pectin:Fe electrodes.18 Specific ligands form on the pectin surface upon contact with the electrolyte, including C–O–Li bonds from the Li–salt and –CH2–O bonds from diethyl carbonate. These ligands can act as nucleophilic or electrophilic sites, facilitating the formation of conducting filaments, which reduces the resistance of pectin and enhances electron mobility. In contrast, these ligands are not observed in the PVDF electrode. Furthermore, the pseudocapacitive behavior in our system induces an interfacial surface charge between the active material and the pectin. This interfacial polarization leads to charge redistribution, which contributes to the overall capacity during the charge process. Therefore, to better understand the details of the Li charge storage dynamics, we initiated our analysis using CV at various scanning rates (v) to clarify the Li storage mechanism, as shown in Fig. 1(a)–(c). In all samples, primary redox peaks at 0.02 V and 0.15 V, denoted as peak G, appear in the CV curve during cathodic/anodic scans at low scan rates. These peaks signify the LiCx phase transformation during the lithiation and delithiation processes.38 Notably, in graphite–pectin:Fe electrodes, additional redox peaks at 0.5 V and 1.5 V, named peak F, become more prominent at higher CV scan rates. Based on the Randles–Sevcik relation,39 the identification of Li storage mechanisms involving surface-controlled and diffusion-limited processes is typically achieved by plotting the peak currents of the voltammetric curves against the corresponding scan rates on a logarithmic scale for both peak G and peak F. Fig. 1(d)–(f) present the computed slopes, or b-values, for the cathodic and anodic of peak G, specifically 0.53/0.58, 0.47/0.51, and 0.44/0.56 for graphite–PVDF, graphite–pectin, and graphite–pectin:Fe electrodes, respectively. The b-values close to 0.5 for all the samples suggest that diffusion-limited processes primarily governed their current responses during the measurement period. In contrast, a surface-controlled current response is evident at the cathodic and anodic regions of peak F in the graphite–pectin:Fe electrode, confirmed by a b-value of 0.9/0.92, as shown in the inset of Fig. 1(f). Additionally, the contributions of surface-controlled and diffusion-limited processes were estimated for all samples, as shown in Fig. 1(g)–(i). The surface-controlled contribution of the graphite–PVDF electrode exhibited a relatively slower growth with increasing rate, reaching a maximum value of 42.6% at a high scan rate of 1 mV s−1. On the other hand, the surface-controlled contribution of the pectin-based electrode is much higher, particularly in graphite–pectin:Fe electrodes, where it expands from 46.8% to 77.5% as the scan rate increases from 0.05 to 1 mV s−1. In addition, the estimation of the relative electrochemical activity of the graphite–PVDF, graphite–pectin, and graphite–pectin:Fe electrodes can be derived from the correlation between the peak current and scan rate in the CV. As shown in Fig. S3(a)–(c) (ESI†), the slope of the i versus v−1/2 profile is proportional to the Li+ reactive surface area because the DLi+ values are similar between the electrodes. The anodic/cathodic values for the graphite–PVDF, graphite–pectin, and graphite–pectin:Fe electrodes were 0.42/0.44, 2.36/0.78, and 5.44/2.90, respectively. Thus, the relative electrochemical activity of graphite–pectin:Fe is almost 12 times that of graphite–PVDF. The cathodic value of the pectin-based electrodes was found to be less than the anodic value, which can be attributed to the presence of surface termination groups on the pectin surface. The –OH groups on the pectin surface impede the movement of Li+ ions towards the transition metal, causing significant diffusion barriers and the accumulation of Li+ ions near or on the electrode surface. This phenomenon increases the storage capacity of Li+ ions in the electrode.40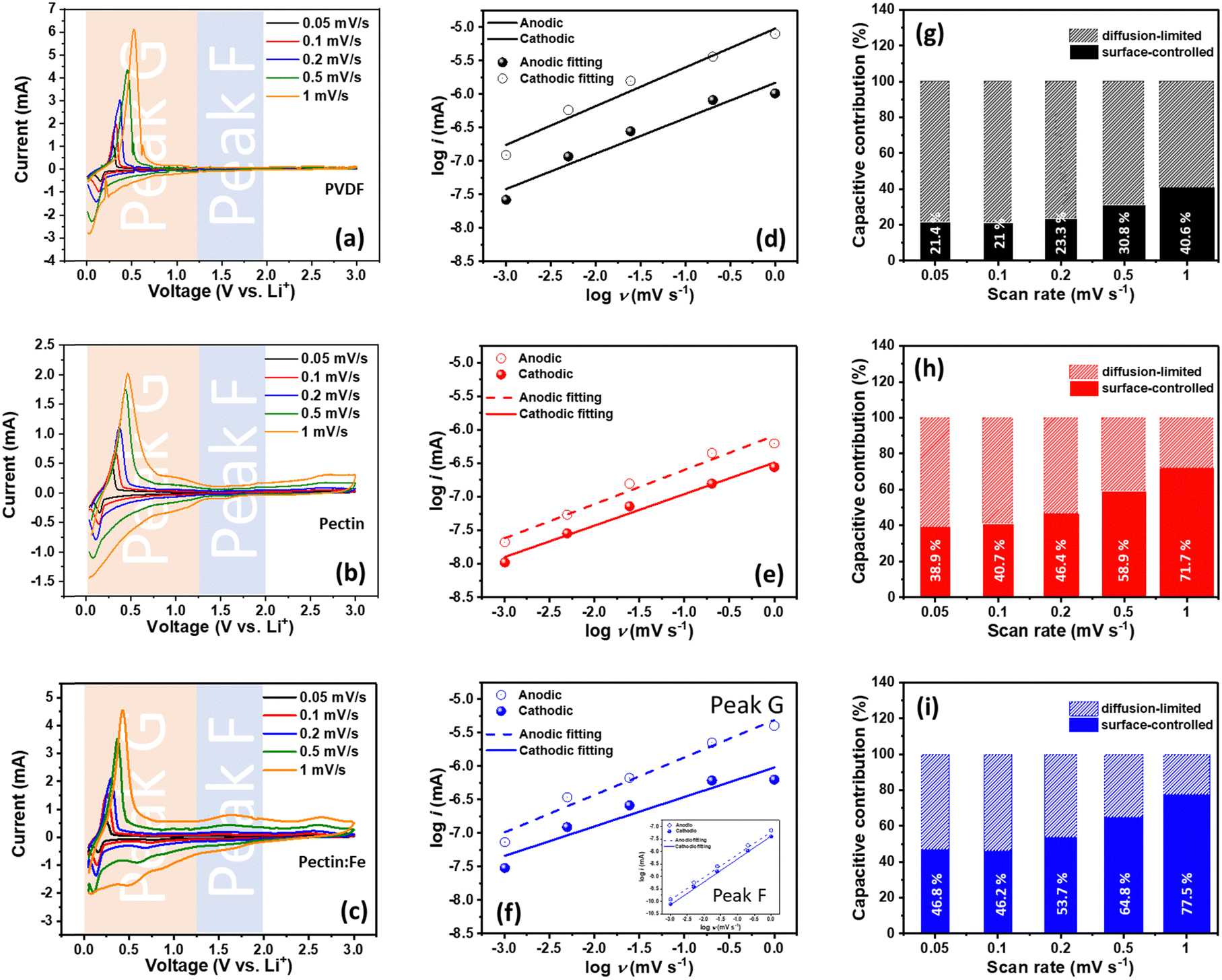 | ||
| Fig. 1 CV curves for (a) graphite–PVDF, (b) graphite–pectin, and (c) graphite–pectin:Fe electrodes at different voltage scan rates, ranging from 0.05 to 1 mV s−1. Determination of the b value for (d) graphite–PVDF, (e) graphite–pectin, and (f) graphite–pectin:Fe electrodes using the relationship between the peak current and sweep rate. (g)–(i) show the percentage of capacitive contribution at different voltage scan rates for all the samples. | ||
To further delve into the identification of the rate-limiting step for charge storage in terms of Li+ transport dynamics in graphite–PVDF, graphite–pectin, and graphite–pectin:Fe electrodes, we analyzed the impedance data (Fig. S4, ESI†) using a 3D Bode plot, as shown in Fig. 2(a)–(c), in which the real-part capacitance (C′) and phase angle (ϕ) serve as dependent variables, while the frequency (f) and cell voltage are independent variables. The 3D Bode plot of graphite–PVDF, graphite–pectin, and graphite–pectin:Fe electrodes reveals a notable signal at 0.5 V at low f (0.5 mHz). Upon comparison, the signal in the pectin-based electrodes was stronger than that in the graphite–PVDF electrodes. Moreover, at 0.5 mHz, the highest C′ value of 2 F cm−2 was observed in the graphite–pectin:Fe electrodes. This outcome demonstrates that pectin-based electrodes exhibit multiple charge storage dynamics involving both kinetically fast and reversible redox reactions. In contrast, for the graphite–PVDF electrode, C′ is lower than that of both graphite–pectin and graphite–pectin:Fe electrodes at a frequency of 0.5 mHz, suggesting the presence of rate-limiting behavior characteristic of a traditional battery-like response.
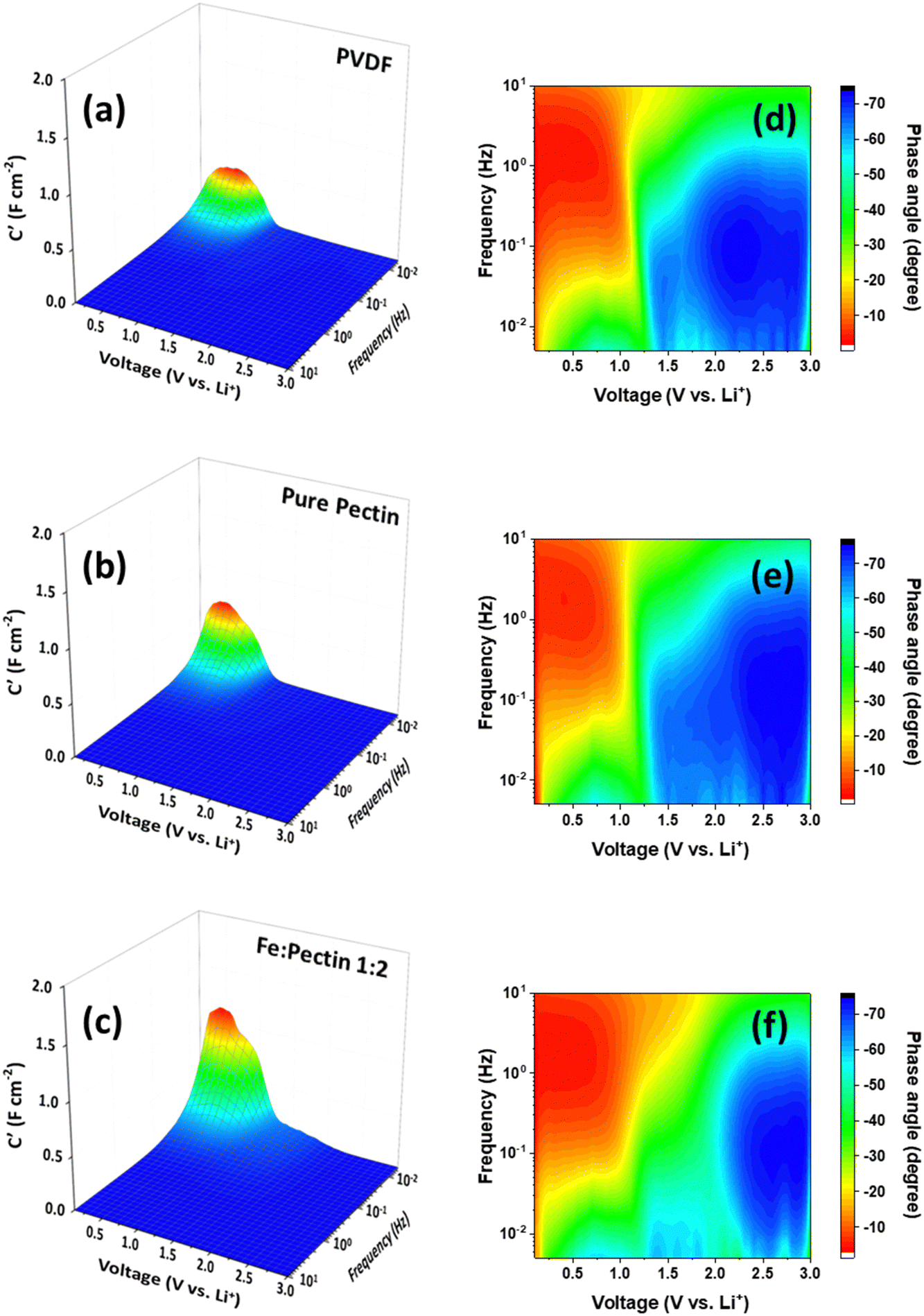 | ||
| Fig. 2 3D Bode plots representing the real area-normalized capacitance (C′) versus frequency versus voltage for (a) PVDF, (b) pectin, and (c) pectin:Fe. The corresponding representations of the phase angle (ϕ) versus frequency and voltage are shown for (d) PVDF, (e) pectin, and (f) pectin:Fe. | ||
Another parameter used to observe the rate-limiting kinetics of charge storage is the phase angle (ϕ). This parameter determines the balance between the capacitive and resistive responses in the series circuit, offering important insights into rate-limiting kinetics. The analysis of ϕ, where ϕ = 0° corresponds to a pure resistor, ϕ = 45° indicates a diffusion-limited response, and ϕ = 90° signifies a purely capacitive response, provides a more refined understanding of the system. As shown in Fig. 2(d)–(f), all electrodes exhibit ϕ nearly at 45° (in the green region) below 1.5 V at low f (<0.1 Hz), indicating a diffusion-controlled reaction. This behavior arises from the formation of a graphite intercalation compound, LiC6, during the process of Li+ extraction/insertion.41–43 However, at low f (0.5 mHz) below 0.5 V, a high ϕ (light blue) is observed, signaling a capacitor-like response. These findings indicate that pectin-based electrodes exhibit multiple charge storage dynamics involving both diffusion-limited and capacitor-like responses. Obviously, the capacitor-like response is more pronounced in the graphite–pectin:Fe electrode. The observed enhancement in the graphite–pectin:Fe electrode is possibly related to the presence of both Fe2+ and Fe3+ ions.18 This dual valence state of iron ions enhances the faradaic processes related to surface redox reactions. Similar effects have been reported in Fe-based supercapacitors.44
It has been established that electrodes featuring sophisticated architectures or complementary functionalities can fine-tune the surface/interface chemistry, thereby enhancing the intrinsic charge storage capabilities.45,46 Hence, the surface morphologies of the investigated electrodes were examined using SEM, as shown in Fig. 3(a)–(c). It is interesting that the films cast on the Cu substrates exhibit different morphologies. A granular structure was observed in the PVDF film. This can be attributed to the absence of active functional groups in PVDF, leading to mechanical or physical adsorption of the binder onto the surface.47 Pectin film displays a smooth and continuous surface, while pectin–Fe exhibits a network-like structure. Pectin contains reactive functional groups such as hydroxyl (–OH) and carboxylic acid (–COOH) groups, which are present on the polymer backbone, providing opportunities for bonding with each other. With the introduction of ferrous chloride (FeCl2), coordinated bonds were formed between the COOH units on the polymers. This facilitates stress dissipation in the film and leads to the formation of a network-like structure.48
 | ||
| Fig. 3 (a)–(c) SEM images of PVDF, pectin, and pectin:Fe electrodes, respectively. (d)–(f) Contact angle images of PVDF, pectin, and pectin:Fe electrodes. FTIR spectra comparison of (g) PVDF, (h) pectin, and (i) pectin:Fe electrodes, in the initial state (dark line) and after soaking in electrolyte for 12 h (light line). | ||
Furthermore, the wetting behavior between these solid surfaces at the electrode/electrolyte interface is an important factor in electrochemical interface dynamics. This influences the transport of electrolyte ions within the electrode channels and the distribution of charge on the surface of the electrode, both of which significantly affect the energy storage performance of the electrode in Li-ion batteries. As shown in Fig. 3(d)–(f), a contact angle of 15.6 ± 2° is observed in the PVDF film, which is higher than that of the pectin (8.1 ± 2°) and pectin:Fe (9.5 ± 2°) films. The improvement in electrolyte wettability is attributed to the polar groups (–OH and –COOH) of pectin, which result in high binding energy with electrolyte ions from organic electrolytes.49 Additionally, the unique surface charge distribution of the polar groups of pectin enables negatively charged ions to attract cations, and vice versa for positively charged ions in organic electrolyte solutions. Thus, pectin provides good electrolyte wettability and numerous ion transporters. This result is consistent with the findings of the FTIR spectrum, as shown in Fig. 3(g)–(i). Following a 12-hour immersion in the electrolyte, a transformation occurred in the PVDF, pectin, and pectin:Fe films. Specifically, the C–O absorption band of the pectin-based film reacts with the electrolyte, leading to the formation of a C–O–Li bond from the Li salt and a –CH2–O bond from diethyl carbonate.50 In contrast, the observed ligands were not present in the PVDF electrode. Instead, only a strong peak at 840 cm−1 was observed in the FTIR spectrum, originating from the νP–F stretching mode associated with PF6−, as shown in Fig. 3(g). This peak is commonly observed on the surface of PVDF-based electrodes.51 Therefore, the interfacial surface charge between the active material and pectin induces interfacial polarization, resulting in charge redistribution, which contributes to the overall capacity during the charge process. A noticeable shift from 1741 cm−1 to 1777 cm−1 in the absorption band of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching was observed when comparing the FTIR spectra of the pectin electrode with that of the pectic:Fe electrode. This shift indicates an interaction between the Fe ion and –COO groups of pectin.52 Such interactions further support the formation of the network-like structure observed by SEM. Furthermore, the bond length of the –COO groups in pectin:Fe electrode was determined to be 1.10 Å, which is shorter than the reported value of 1.21 Å in the literature.53 This shorter length indicates a higher bond energy. This observation is consistent with the bond energy calculated from the FTIR analysis, as shown in Fig. S5 (ESI†). This could be the reason why the surface-controlled process is more dominant in the pectin–Fe electrode.
O stretching was observed when comparing the FTIR spectra of the pectin electrode with that of the pectic:Fe electrode. This shift indicates an interaction between the Fe ion and –COO groups of pectin.52 Such interactions further support the formation of the network-like structure observed by SEM. Furthermore, the bond length of the –COO groups in pectin:Fe electrode was determined to be 1.10 Å, which is shorter than the reported value of 1.21 Å in the literature.53 This shorter length indicates a higher bond energy. This observation is consistent with the bond energy calculated from the FTIR analysis, as shown in Fig. S5 (ESI†). This could be the reason why the surface-controlled process is more dominant in the pectin–Fe electrode.
We also conducted Li plating and stripping experiments to observe the deposition/dissolution behavior between Li and the electrodes. As shown in Fig. 4, a fixed amount (1 mA h cm−2) of Li was first deposited onto the PVDF, pectin, and pectin:Fe electrodes from Li foil, and the as-deposited Li was stripped away. Remarkable dendrite-induced short circuits were observed for PVDF and pectin after 22 and 30 plating/stripping cycles, respectively. However, pectin:Fe exhibited a short circuit after 60 plating/stripping cycles. These results suggest that dendrite formation is much more pronounced in the PVDF electrode than in the pectin-based electrode. On the other hand, polarization voltages were observed from the voltage profiles, measured at 270, 260, and 150 mV for the PVDF, pectin, and pectin:Fe electrodes, respectively. The lower polarization voltage of the pectin-based electrode is attributed to its rapid Li+ transfer and uniform Li+ adsorption/desorption.54 Furthermore, the average coulombic efficiency was measured to determine the presence of excess Li in the electrode. The Coulombic efficiency effectively reflects the loss of Li on the working electrode owing to its reaction with the electrolyte or dead lithium. As shown in Fig. S6 (ESI†), after cycling for 50 hours, average coulombic efficiencies of 37, 49.9, and 54.9% were achieved for the PVDF, pectin, and pectin:Fe electrodes, respectively.
 | ||
| Fig. 4 Voltage profiles of the Li plating/stripping process for (a) PVDF, (b) pectin, and (c) pectin:Fe electrodes. Selected magnified voltage–time profiles of PVDF, pectin, and pectin:Fe electrodes are shown in (d)–(i). | ||
To illustrate the positive impact of the modified coating on the lithium deposition behavior, we investigated the overpotential of Li deposition in the first cycle. This overpotential is defined as the difference between the peak voltage (lithium nucleation overpotential) and the subsequent stable plateau voltage (mass-transfer overpotential) observed in the voltage curve during the discharge process. As shown in Fig. 5(a) and (b), the nucleation and mass transport overpotentials in the first cycle for PVDF were −281 mV and −13 mV, respectively. Therefore, the lithium deposition overpotential was calculated to be −268 mV. In contrast, for the other samples, the nucleation and mass transport overpotentials were −171 mV and −72 mV for pectin, and −169 mV and −58 mV for pectin:Fe, respectively. Consequently, the corresponding lithium deposition overpotentials are only −99 mV and −111 mV for pectin and pectin:Fe, respectively, which are significantly lower than those observed in the PVDF sample. The significant decrease in the lithium deposition overpotential demonstrated that there was no additional deposition barrier for lithium ions in the pectin-based samples. Moreover, based on the Tafel profile (Fig. 5(c)), a significantly higher exchange current density of 0.036 mA cm−2 was observed in cells employing pectin and pectin:Fe electrodes, which exceeded that of the PVDF electrode (0.015 mA cm−2). This observation confirms the enhanced Li+ ionic transmission and adsorption/desorption at the electrode interface in the pectin-based electrodes. Additionally, the CV curves of the Li/PVDF, Li/pectin, and Li/pectin:Fe half-cells within the third cycle range of −0.2 to 0.5 V are shown in Fig. 5(d)–(f). Appearance of redox peaks near 0 V (vs. Li+/Li) is characteristic of the lithium deposition/dissolution process, suggesting that these three types of binders do not disrupt the typical deposition and dissolution of lithium. Furthermore, in the third cycle, the pectin:Fe electrode exhibited a higher peak height (2.15 mA) than that of the PVDF electrode (1.06 mA). The increased peak height in the CV curves of the cell indicates faster lithium diffusion on the electrode surface.55 All pectin-based electrodes display considerably larger redox areas, indicating better Li+ adsorption/desorption kinetics. The significantly increased redox areas of the pectin-based electrodes are likely attributed to the more favorable surface chemistry for Li+ ion transfer. As evidenced by the Li nucleation overpotentials shown in Fig. 5(b), the pectin-based electrodes exhibited significantly lower values than the PVDF electrode, indicating a stronger Li+ ion binding energy and higher Li affinity on the pectin surfaces. The higher binding energy of Li atoms can lead to better adsorption of Li+ ions. Therefore, Li+ ions are expected to preferentially adsorb and plate on pectin surfaces compared to PVDF, resulting in better electrochemical performance.
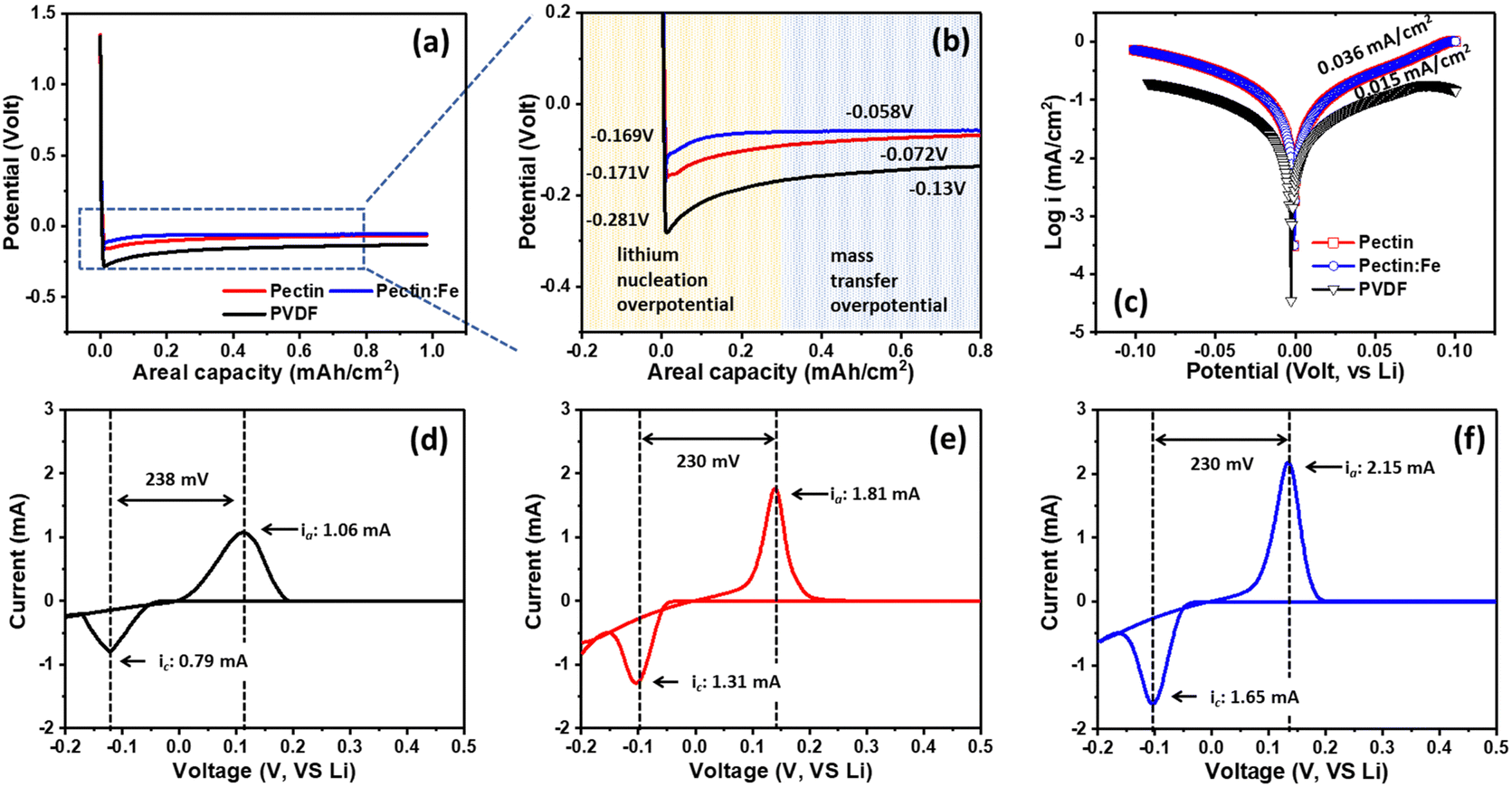 | ||
| Fig. 5 (a) Voltage–capacity curve for PVDF, pectin, and pectin:Fe electrodes during the first discharge cycle. (b) Enlarged view of the voltage range from 0.2 to 0.8 V. (c) Tafel profile for PVDF, pectin, and pectin:Fe electrodes. (d)–(f) CV curves for PVDF, pectin, and pectin:Fe electrodes. | ||
Conclusion
Our study on graphite anodes with PVDF, pectin, and pectin:Fe binders provides valuable insights into their electrochemical behavior in terms of cycling performance and charge-storage dynamics. The CV analysis showed that at low scan rates, all samples had primary redox peaks linked to the LiCx phase transformation. When the scan rates were increased, the graphite–pectin:Fe electrodes displayed extra redox peaks. Using power-law analysis, we found that diffusion-limited processes were dominant across all samples. However, in the graphite–pectin-based electrodes, surface-controlled processes played a significant role. This effect was even more pronounced when Fe ions were introduced into the pectin binder. This finding aligns with observations from the impedance analysis. The enhancement seen in the graphite–pectin:Fe electrode is likely due to the presence of both Fe2+ and Fe3+ ions. The dual valence states of iron ions are known to enhance faradaic processes, contributing to more robust surface redox reactions. Moreover, SEM analysis showed that pectin films have a smooth, continuous surface, while pectin–Fe films exhibit a network-like structure. This difference is due to the reactive hydroxyl and carboxylic acid groups in pectin. These groups enable bonding, and when Fe ion is added, coordinated bonds form between the COOH groups and Fe ions. This bonding creates a network-like structure in the pectin–Fe film, making stress dissipation and enhancing mechanical stability. Li plating and stripping experiments revealed the better performance of pectin-based electrodes with reduced dendrite-induced short circuits and lower polarization voltages, indicating rapid Li+ transfer and uniform adsorption/desorption. Furthermore, the pectin-based electrode demonstrated a significant decrease in the lithium deposition overpotential and enhanced Li+ ionic transmission, confirming it has the potential to prevent undesired reactions with lithium at the interface. Our study enhances the understanding of pectin, and pectin with additives, as an anode binder in lithium-ion batteries. It demonstrates their potential as viable candidates for applications requiring high power capability.Author contributions
P. M. Wu, P. W. Chi and M. K. Wu were responsible for providing the original concept and contributing to the design of the study and the final review of this document. C. Y. Chung, W. M. Chen, Y. R. Chen, and Y. H. Su performed the experiments. C. Y. Chung, W. M. Chen, and P. W. Chi analyzed the data. C. Y. Chung, P. W. Chi, P. M. Wu, and M. K. Wu collaborated on writing the manuscript and creating the figures. K. S. Chang-Liao and H. Y. Tang reviewed the results and provided feedback. P. M. Wu and P. W. Chi coordinated the teamwork. All authors have thoroughly revised the manuscript.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
The authors have no conflicts of interest to declare.Acknowledgements
The authors greatly appreciate the financial support from the Executive Yuan through its Forward-Looking Research Grant Number AS-FLI-110-LI. Phillip M. Wu acknowledges the support from the “Innovative Center on Sustainable Negative-Carbon Resources” from The Featured Areas Research Center Program within the framework of the Higher Education Sprout Project by the Ministry of Education (MOE) in Taiwan and National Science and Technology Council of Taiwan grant number NSTC 113-2112-M-001-022-MY2.References
- E. A. Olivetti, G. Ceder, G. G. Gaustad and X. Fu, Joule, 2017, 1, 229–243 CrossRef.
- Y. Ding, Z. P. Cano, A. Yu, J. Lu and Z. Chen, Electrochem. Energy Rev., 2019, 2, 1–28 CrossRef CAS.
- Z. Mao, X. Shi, T. Zhang, Z. Zheng, X. Liang, R. Wang, J. Jin, B. He, Y. Gong and H. Wang, Small, 2023, 19, 2302987 CrossRef CAS PubMed.
- K. K. Jena, A. AlFantazi and A. T. Mayyas, Energy Fuels, 2021, 35, 18257–18284 CrossRef CAS.
- F. Zou and A. Manthiram, Adv. Energy Mater., 2020, 10, 2002508 CrossRef CAS.
- A. Du Pasquier, F. Disma, T. Bowmer, A. S. Gozdz, G. Amatucci and J. M. Tarascon, J. Electrochem. Soc., 1998, 145, 472 CrossRef.
- H. Zhang, Z. Song, J. Fang, K. Li, M. Zhang, Z. Li, L. Yang and F. Pan, J. Phys. Chem. C, 2023, 127, 2755–2765 CrossRef CAS.
- S. Komaba, N. Yabuuchi, T. Ozeki, K. Okushi, H. Yui, K. Konno, Y. Katayama and T. Miura, J. Power Sources, 2010, 195, 6069–6074 CrossRef CAS.
- H. Wang, N. Qin, Y. Li, Z. Li, F. Zhang, W. Luo, C. Zeng, Z. Lu and H. Cheng, Carbon, 2023, 205, 435–443 CrossRef CAS.
- S. H. Zheng, X. T. Wang, Z. Y. Gu, H. Y. Lü, S. Li, X. Y. Zhang, J. M. Cao, J. Z. Guo and X. L. Wu, J. Colloid Interface Sci., 2024, 667, 111–118 CrossRef CAS PubMed.
- H. Chen, M. Ling, L. Hencz, H. Y. Ling, G. Li, Z. Lin, G. Liu and S. Zhang, Chem. Rev., 2018, 118, 8936–8982 CrossRef CAS PubMed.
- T. Kwon, J. W. Choi and A. Coskun, Chem. Soc. Rev., 2018, 47, 2145–2164 RSC.
- S. Huang, J. Ren, R. Liu, M. Yue, Y. Huang and G. Yuan, Int. J. Energy Res., 2018, 42, 919–935 CrossRef CAS.
- A. Roman-Benn, C. A. Contador, M. W. Li, H. M. Lam, K. Ah-Hen, P. E. Ulloae and M. C. Ravanal, Food Chem. Adv., 2023, 100192 CrossRef.
- C. Lara-Espinoza, C. Lara-Espinoza, E. Carvajal-Millán, R. Balandrán-Quintana, Y. López-Franco and A. Rascón-Chu, Molecules, 2018, 23, 942 CrossRef PubMed.
- D. E. Yoon, C. Hwang, N. R. Kang, U. Lee, D. Ahn, J. Y. Kim and H. K. Song, ACS Appl. Mater. Interfaces, 2016, 8, 4042–4047 CrossRef CAS PubMed.
- Y. H. Su, C. Y. Chung, Y. R. Chen, F. Y. Wu, Y. H. Lin, P. W. Chi, P. M. Wu, T. Paul, H. E. Lin, K. S. Chang-Liao, S. F. Wang and M. K. Wu, J. Energy Storage, 2023, 72, 108454 CrossRef.
- P. M. Wu, C. Y. Chung, Y. R. Chen, Y. H. Su, K. S. Chang-Liao, P. W. Chi, T. Paul, Y. J. Chen, Y. L. Chen, S. F. Wang, P. Badgujar, B. N. Chen, C. L. Cheng and M. K. Wu, PNAS Nexus, 2022, 1, pgac127 CrossRef PubMed.
- B. Gendensuren and E. S. Oh, J. Power Sources, 2018, 384, 379–386 CrossRef CAS.
- A. Afif, S. M. Rahman, A. T. Azad, J. Zaini, M. A. Islan and A. K. Azad, J. Energy Storage, 2019, 25, 100852 CrossRef.
- H. E. Lin, Y. Kubota, Y. Katayanagi, T. Kishi, T. Yano and N. Matsushita, Electrochim. Acta, 2019, 323, 134794 CrossRef CAS.
- C. Choi, D. S. Ashby, D. M. Butts, R. H. DeBlock, Q. Wei, J. Lau and B. Dunn, Nat. Rev. Mater., 2020, 5, 5–19 CrossRef.
- D. Malavekar, S. Pujari, S. Jang, S. Bachankar and J. H. Kim, Small, 2024, 2312179 CrossRef PubMed.
- J. Liu, Q. Zhang, Z. Y. Wu, J. T. Li, L. Huang and S. G. Sun, ChemElectroChem, 2015, 2, 611–616 CrossRef CAS.
- J. Yoon, D. X. Oh, C. Jo, J. Lee and D. S. Hwang, Phys. Chem. Chem. Phys., 2014, 16, 25628–25635 RSC.
- J. S. Ko, C. H. Lai, J. W. Long, D. R. Rolison, B. Dunn and J. N. Weker, ACS Appl. Mater. Interfaces, 2020, 12, 14071–14078 CrossRef CAS PubMed.
- A. Das, B. Raj, M. Mohapatra, S. M. Andersen and S. Basu, Wiley Interdiscip. Rev.: Energy Environ., 2022, 11, e414 CAS.
- M. Wang, J. Wang, J. Xiao, N. Ren, B. Pan, C. S. Chen and C. H. Chen, ACS Appl. Mater. Interfaces, 2022, 14, 16279–16288 CrossRef CAS PubMed.
- L. Bai and B. E. Conway, J. Electrochem. Soc., 1991, 138, 2897 CrossRef CAS.
- L. Bai and B. E. Conway, Electrochim. Acta, 1993, 38, 1803–1815 CrossRef CAS.
- J. S. Ko, M. B. Sassin, D. R. Rolison and J. W. Long, Electrochim. Acta, 2018, 275, 225–235 CrossRef CAS.
- J. W. Kim, V. Augustyn and B. Dunn, Adv. Energy Mater., 2012, 2, 141–148 CrossRef CAS.
- G. D. Manrique and F. M. Lajolo, Postharvest Biol. Technol., 2002, 25, 99–107 CrossRef CAS.
- J. Shim and K. A. Striebel, J. Power Sources, 2003, 119, 955–958 CrossRef.
- N. Lingappan, L. Kong and M. Pecht, Renewable Sustainable Energy Rev., 2021, 147, 111227 CrossRef CAS.
- S. F. Lux, F. Schappacher, A. Balducci, S. Passerini and M. Winter, J. Electrochem. Soc., 2010, 157(3), A320 CrossRef CAS.
- Y. R. Chen, L. Y. Chen, C. Y. Chung, Y. H. Su, F. Y. Wu, T. M. Hsu, P. W. Chi, P. M. Wu, K. S. Chang-Liao, H. Y. Tang and M. K. Wu, J. Power Sources, 2024, 613, 234861 CrossRef CAS.
- F. Ding, W. Xu, D. Choi, W. Wang, X. Li, M. H. Engelhard, X. Chen, Z. Yang and J. G. Zhang, J. Mater. Chem., 2012, 22, 12745–12751 RSC.
- C. Ding, Y. Zhao, D. Yan, D. Su, Y. Zhao, H. Zhou, J. Li and H. Jin, Electrochim. Acta, 2017, 251, 129–136 CrossRef CAS.
- Q. Tang, Z. Zhou and P. Shen, J. Am. Chem. Soc., 2012, 134, 16909–16916 CrossRef CAS PubMed.
- V. Dusastre, Nature, 2001, 414, 331 CrossRef.
- H. Zhang, Y. Yang, D. Ren, L. Wang and X. He, Energy Storage Mater., 2021, 36, 147–170 Search PubMed.
- L. Zhao, B. Ding, X. Y. Qin, Z. Wang, W. Lv, Y. B. He, Q. H. Yang and F. Kang, Adv. Mater., 2022, 34, 2106704 Search PubMed.
- H. E. Lin, Y. Kubota, Y. Katayanagi, T. Kishi, T. Yano and N. Matsushita, Electrochim. Acta, 2019, 323, 134794 CrossRef CAS.
- J. Jiang, Y. Li, J. Liu, X. Huang, C. Yuan and X. W. Lou, Adv. Mater., 2012, 24, 5166–5180 CrossRef CAS PubMed.
- L. F. Cui, Y. Yang, C. M. Hsu and Y. Cui, Nano Lett., 2009, 9, 3370–3374 CrossRef CAS PubMed.
- N. Lingappan, L. Kong and M. Pecht, Renewable Sustainable Energy Rev., 2021, 147, 111227 Search PubMed.
- M. Jiang, P. Mu, H. Zhang, T. Dong, B. Tang, H. Qiu, Z. Chen and G. Cui, Nano-Micro Lett., 2022, 14, 87 CrossRef CAS PubMed.
- B. Sun, P. Li, J. Zhang, D. Wang, P. Munroe, C. Wang, P. H. L. Notten and G. Wang, Adv. Mater., 2018, 30, 1801334 CrossRef PubMed.
- P. N. Ross, ECS Trans., 2006, 1, 161 Search PubMed.
- M. Nie, D. P. Abraham, D. M. Seo, Y. Chen, A. Bose and B. L. Lucht, J. Phys. Chem. C, 2013, 117, 25381–25389 CrossRef CAS.
- R. Niu, Z. Qin, F. Ji, M. Xu, X. Tian, J. Li and F. Yao, Soft Matter, 2017, 13, 9237–9245 RSC.
- J. Demaison and A. G. Csaszar, J. Mol. Struct., 2012, 1023, 7–14 CrossRef CAS.
- F. Ren, Z. Lu, H. Zhang, L. Huai, X. Chen, S. Wu, Z. Peng, D. Wang and J. Ye, Adv. Funct. Mater., 2018, 28, 1805638 Search PubMed.
- T. T. Beyene, B. A. Jote, Z. T. Wondimkun, B. W. Olbassa, C. J. Huang, B. Thirumalraj, C. H. Wang, W. N. Su, H. Dai and B. J. Hwang, ACS Appl. Mater. Interfaces, 2019, 11, 31962–31971 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Fig. S1–S6. See DOI: https://doi.org/10.1039/d4ma00584h |
| This journal is © The Royal Society of Chemistry 2024 |