
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Solid-state and aggregation-induced emission of novel bicyclic and tricyclic difluoroboron heterocycles†
Martina
Žabenská
a,
Chiara
Capolungo
b,
Chiara
Mariani
b,
Damiano
Genovese
 b,
Tomáš
Mikysek
c,
Jiří
Váňa
a,
Aleš
Růžička
d,
František
Josefík
a,
Markéta
Svobodová
*a and
Petr
Šimůnek
*a
b,
Tomáš
Mikysek
c,
Jiří
Váňa
a,
Aleš
Růžička
d,
František
Josefík
a,
Markéta
Svobodová
*a and
Petr
Šimůnek
*a
aInstitute of Organic Chemistry and Technology, University of Pardubice, Faculty of Chemical Technology, Studentská 573, CZ 532 10, Pardubice, Czech Republic. E-mail: marketa.svobodova@upce.cz; petr.simunek@upce.cz
bDipartimento di Chimica “Giacomo Ciamician”, Università di Bologna, via Selmi 2, 40126, Bologna, Italy
cDepartment of Analytical Chemistry, University of Pardubice, Faculty of Chemical Technology, Studentská 573, CZ 532 10, Pardubice, Czech Republic
dDepartment of General and Inorganic Chemistry, University of Pardubice, Faculty of Chemical Technology, Studentská 573, CZ 532 10, Pardubice, Czech Republic
First published on 2nd September 2024
Abstract
Six novel bicyclic and tricyclic difluoroboron NBN and OBN heterocycles were designed here in the quest for novel luminogenic molecular architectures because of their strong application potential as active layers in optoelectronics and as responsive units in sensing. They were prepared and then characterized with spectral (UV-vis, NMR, and luminescence) and electrochemical methods, and assessed via theoretical and X-ray investigations. Most of the compounds are non-emissive in solution, but luminescent in the solid state (red or yellow-green luminescence), and they are AIE active, offering excellent contrast for sensing schemes. Keeping this in view, two compounds were successfully embedded in Pluronic-silica nanoparticles (PluS NPs), coupling the AIEgenic properties of these compounds with the exceptional colloidal stability and functional surface of this type of nanostructure.
Introduction
Nowadays, organic luminescent materials (OLM) that are highly bright in the solid state may have great potential for application in optoelectronic technologies and responsive and smart materials. Due to their interesting properties, trivalent and tetravalent organoboron compounds have become an important class of OLM.1 Although known for decades, whereby many of them have been prepared and characterized, boron-containing luminescent polymers,1f,2 such as tetra-coordinated boron complexes, remain valuable targets for material chemistry due to their good emissive, AIE, CIE, and MFC properties.1f,2 They are thus the subjects for various structural modifications. The potential of tetra-coordinated boron compounds for the design of TADF emitters with high external quantum efficiency (EQE) has also been proved.1a,b,d Recently, boron-thioketonates were prepared and characterized3a and some tetra-coordinated spiroborates (SBOX) were used as the host materials for blue OLEDs.3b Both NBN and – more recently – OBN heterocycles were synthesized and proposed for use in optical data encryption,3c OLED,2g and stimuli-responsive materials.3d The advantages of tetra-coordinated boron compounds are their stability towards both oxygen and moisture, as well as the ease of their preparation. These compounds can also be easily chemically modified allowing their properties to be tuned up.The chemistry of tetra-coordinated boron complexes is very diverse. They may contain various ring sizes and heteroatom combinations, making their classification complex. Probably the simplest and clearest classification is the one based on the denticity of the ligand to which the boron atom is complexed, published by Tanaka et al.1f They divided the tetra-coordinated boron complexes into two large groups: with bidentate and tridentate ligands. Some representative general structures are shown in Chart 1.
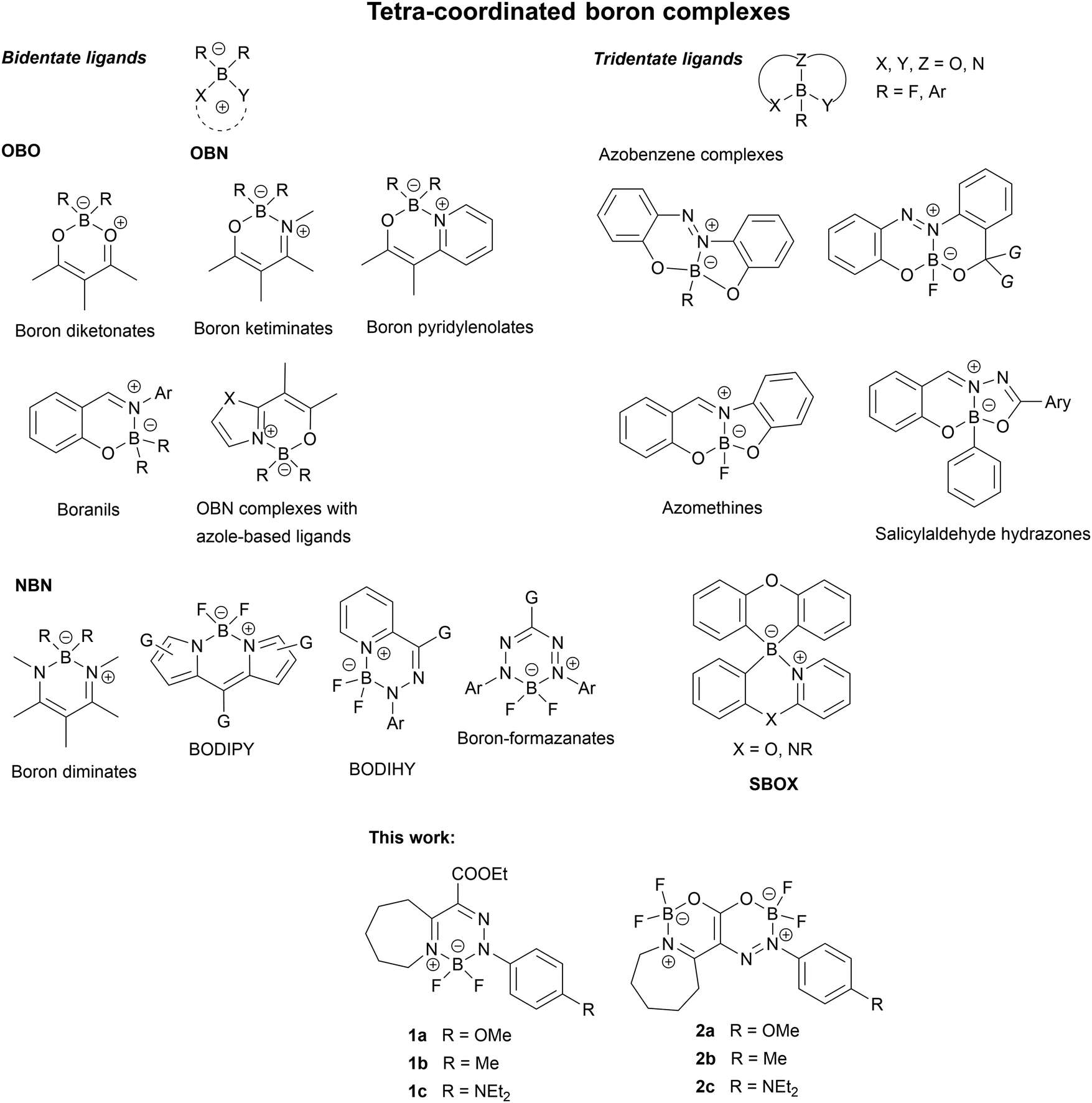 | ||
| Chart 1 Bidentate tetra-coordinated boron complexes and compounds studied in this work. | ||
A deep understanding of the structural properties and their relationships in OLMs is essential for tuning the characteristics of OLED devices in which they are used. Therefore, the synthesis and characterization of novel materials is an important and timely task for material organic chemistry.
In this work, we describe the synthesis, as well as the spectral, luminescence, and electrochemical properties of novel bicyclic and tricyclic tetra-coordinated NBN and OBN heterocycles 1 and 2 (see Chart 1). These compounds are important to realise our interest in the synthesis and characterization of polynitrogen boron-containing heterocycles.4 Seven-membered rings are incorporated in their structures, giving them the potential for RIV (restricted intramolecular vibration), and hence, making them AIEgens.5a This medium-ring strategy has been recently used to suppress aggregation-caused quenching (ACQ) at boron-based MR emitters.5b The presence of target functional groups allows us to unravel the relationships between the structure and physical properties of these NBN and OBN heterocycles, particularly highlighting the role of electron-donating substituents and the acid–base active pendants on the optical properties of these novel compounds.
Results and discussion
The target compounds were prepared according to Scheme S1. For experimental details and synthetic procedures see ESI.† The suggested mechanism for the synthesis of 1 and 2 is depicted in Scheme S2 (ESI†).NMR spectroscopy
Boron-11 and fluorine-19 NMR spectroscopy are useful tools for the structural study of BF2-containing compounds. Both boron and fluorine NMR parameters sensitively reflect the electronic and steric surroundings.6 Fluorine-19 NMR spectra of compound 1 consist of one 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1:1 quartet due to the coupling with a single boron-11 nucleus (I = 3/2). It reveals that both the fluorine atoms are symmetrically equivalent. The equivalence could be due to either the planarity of the chelate ring or the exchange-average structure due to phenomena, such as fast ring-flipping. Therefore, the boron-11 signal is a simple triplet.
:1:1:1 quartet due to the coupling with a single boron-11 nucleus (I = 3/2). It reveals that both the fluorine atoms are symmetrically equivalent. The equivalence could be due to either the planarity of the chelate ring or the exchange-average structure due to phenomena, such as fast ring-flipping. Therefore, the boron-11 signal is a simple triplet.
The analysis of boron-11 NMR spectra of compound 2 reveals a couple of triplets, indicating the presence of two different BF2 fragments. Both the fluorine atoms within each fragment are equivalent, probably due to fast ring-flipping. Therefore, the fluorine-19 spectra consist of two broad signals, one for each BF2 fragment. In contrast to 1, fine splitting is not observable here, probably due to the molecular dynamics leading to line-broadening. All the NMR spectra are deployed in ESI.†
Electrochemistry
Voltammetric techniques (cyclic voltammetry, rotating disk voltammetry) were employed to study the electrochemical properties of selected boron heterocycles in acetonitrile containing 0.1 M Bu4N+ PF6−. The electrochemical data are summarized in Table 1. Furthermore, representative cyclic voltammograms can be found in the ESI† (Fig. S64–S69, ESI†). The first oxidation of all the studied compounds proceeded at potentials from +1.81 to +0.66 V (vs. SCE) as a one-electron (quasi)reversible process (cathodic-anodic peak separation from 78 to 130 mV). The only exception was compound 2b, where the (quasi)reversibility was observed at a higher scan rate (500 mV s−1). Changes in the substitution significantly affected the oxidation of the boron heterocycle. In comparison of 1b with 2b and 1a with 2a, the addition of another borinine ring (i.e., another electron acceptor) into the molecule caused the shift of the first oxidation potential by about 250 mV to more positive values. The same shift, but in the opposite direction (i.e., towards less positive values), can be observed upon changing methyl (with +I effect) to a stronger electron-donating methoxy or diethylamino group. Furthermore, the first reduction of 1 and 2 proceeds at potentials from −0.86 to −1.42 V (vs. SCE) as the one-electron reversible process by the formation of a stable radical anion. Concerning different substitutions on the borinine core, the addition of another borinine ring (another redox center) extends the acceptor moiety. When comparing compound 1b with 2b (or 1a with 2a), the shift in the first reduction potential by approximately 440 mV to less negative values can be observed. On the other hand, the change in the electron-donating group within the series has a negligible effect on the first reduction potential (shift by about 50 mV). Moreover, the reduction in the second borinine ring can be found at ca −2 V. Last but not least, the difference between the first oxidation and reduction potential (see Table 1) reflects the delocalization of electron density in the molecule and is also aligned with the HOMO–LUMO gap (see Table 1).| Compound | λ max (nm) | ε (dm−3 cm−1 mol−1) | λ A(max) calc (nm) | f | Transition | Main orbital transition | E oF (ox1) (V) vs. SCE | E oF (red1) (V) vs. SCE | E ox1 oF–Ered1oF (V) | HOMO–LUMO gap (eV) |
|---|---|---|---|---|---|---|---|---|---|---|
| 1a | 401 | 9577 | 412 | 0.54 | S0 → S1 | HOMO→LUMO | 1.32 | −1.34 | 2.66 | 3.44 |
| 1b | 389 | 10023 |
386 | 0.57 | S0 → S1 | HOMO→LUMO | 1.56 | −1.29 | 2.85 | 3.66 |
| 1c | 472 | 9499 | 495 | 0.57 | S0 → S1 | HOMO→LUMO | 0.66 | −1.42 | 2.08 | 2.86 |
| 2a | 409 | 24813 |
398 | 0.69 | S0 → S1 | HOMO→LUMO | 1.56 | −0.90 | 2.46 | 3.25 |
| 2b | 389 | 24890 |
432 | 0.69 | S0 → S1 | HOMO→LUMO | 1.81 | −0.86 | 2.67 | 3.53 |
| 2c | 517 | 13449 |
521 | 0.76 | S0 → S1 | HOMO→LUMO | 0.84 | −1.00 | 1.84 | 2.63 |
Electronic spectra and theoretical studies
Electronic spectra of compound 1 measured in acetonitrile (Fig. 1) consist of one broad band across 389–472 nm. The wavelength of the band correlates with the electronic properties of the substituents and increases in the order Me < OMe < NEt2. The intensity of the band is comparable for all the compounds with ε of approximately 10000. These bands correspond to the π–π* transitions. The less intense bands containing multiple peaks with maxima of ∼250 nm correspond to benzenoid bands (B bands).
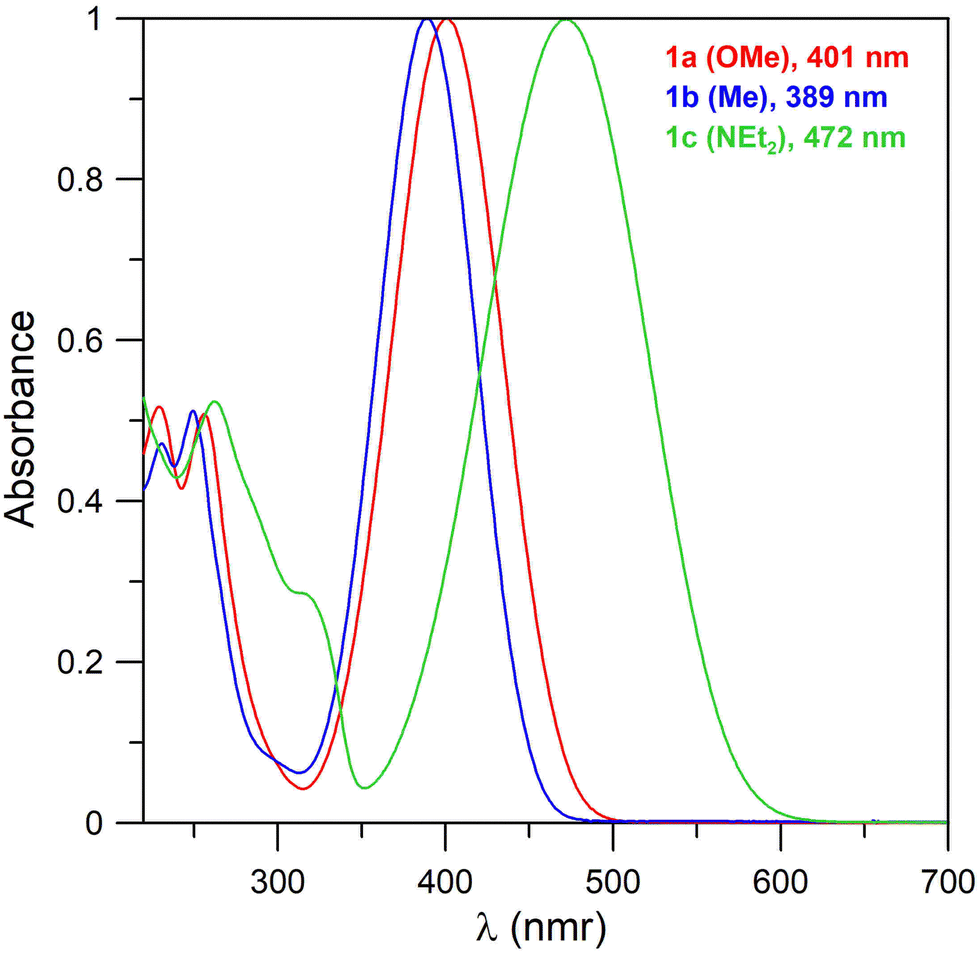 | ||
| Fig. 1 Normalized UV-vis spectra of triazaborines 1a–c in acetonitrile. | ||
UV-vis spectra of bis-boron derivatives 2 (Fig. 2) also consist of broad bands across 389–517 nm, corresponding to the π–π* transitions. The value for methyl derivative 2a (389 nm) is the same as for its 1a analogue. The maxima of the bands for the other two compounds are shifted bathochromically in comparison with their mono-boron analogues (401 → 409 nm for the OMe derivative and 472 → 517 nm for the NEt2 derivative). The order of the maxima also corresponds with the electronic properties of the substituents. The absorption properties thus appear to be more strongly dependent on the nature of the substituents rather than on the mono- or bis-boron heterocyclic structure. This also illustrates the localisation of HOMO orbitals (Fig. 3).
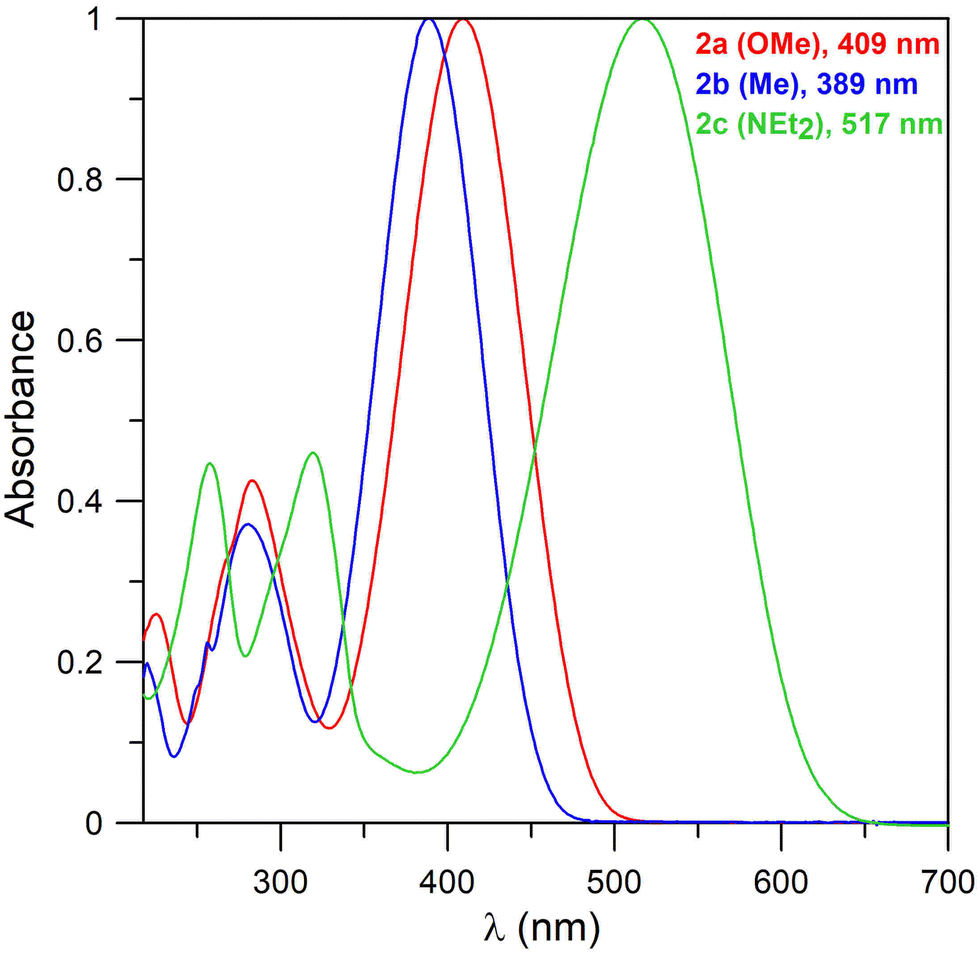 | ||
| Fig. 2 Normalized UV-vis spectra of compounds 2a–c in acetonitrile. | ||
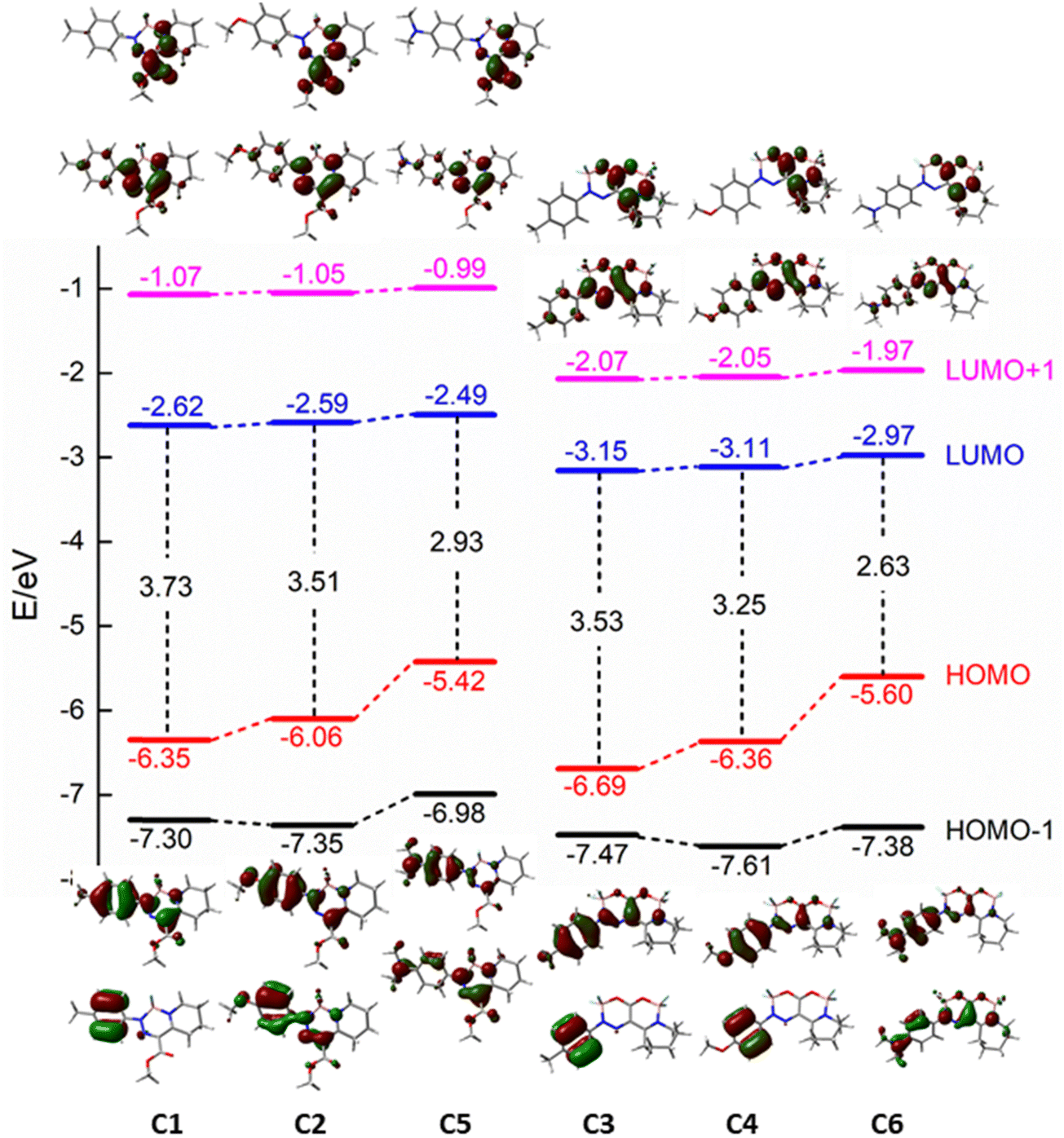 | ||
| Fig. 3 Molecular orbital energy diagram and iso-density surface (contour value 0.04) plots of the HOMO−1, HOMO, LUMO, and LUMO+1 orbitals calculated at the B3LYP/6-311+G(d,p) level of theory in CH3CN. | ||
Wavelengths of the main absorption bands, together with their absorption coefficients, for compounds 1 and 2 are summarized in Table 1.
For a deeper insight into the absorption properties, the TD-DFT calculations (B3LYP-6311+G(d,p)) were performed with acetonitrile as the solvent (CPCM). For the simplification of the calculations, the ethyl groups were substituted by methyl groups. The energy levels of HOMO−1, HOMO, LUMO, and LUMO+1 together with the orbital locations are depicted in Fig. 3. Results of the calculations are summarized in Table 1 to show that the red emission of both NEt2 derivatives is largely due to the increase in the energy of the HOMO, resulting in strong destabilization by the charged character introduced by the nitrogen atom.
Luminescence and AIE properties
The emission of diluted solutions of compounds 1a, 1b, 2a, and 2b in acetonitrile (concentration 5 μM) was very low, with PLQY below 0.01, as desired for high contrast luminogenic properties in AIE compounds. The emission spectra of the same compounds in the solid state (powder) measured with an integrating sphere, revealed intense yellow-orange photoluminescence upon excitation at λexc = 400 nm, with broad non-structured emission bands peaking at 560 nm (compounds 1a and 2a) or 525 nm (compounds 1b and 2b). Photoluminescence quantum yield (PLQY) was estimated to be in the range 0.18–0.5 for the four compounds (see Table 2 and Fig. 4). In addition, compound 1c showed a good enhancement of PLQY in the solid state compared to diluted acetonitrile solution (from 0.017 to 0.15), while compound 2c has only a low and constant PLQY (∼0.02) at both conditions. Interestingly, compound 1c is a bright solid-state emitter in the red spectral region. Compound 2c shows a narrow and bathochromically shifted emission band in the solid state (compared to the solution, Fig. S58, ESI†), which may suggest the onset of J-aggregates for this compound. On the contrary, all other compounds in the solid state show emissions in the same wavelength range as in diluted acetonitrile solution.| Compound | solid state | Aggregate | ||||
|---|---|---|---|---|---|---|
| PLQY | τ (ns) | λ em.max (nm) | PLQY | τ (ns) | λ em.max (nm) | |
| a Average lifetime (by intensity) of a multiexponential decay. | ||||||
| 1a | 0.13 | 1.70 | 568 | 0.15 | 1.98 | 560 |
| 1b | 0.51 | 3.33 | 521 | 0.27 | 3.08 | 526 |
| 1c | 0.15 | 1.84 | 620 | 0.10 | 0.19a | 625 |
| 2a | 0.18 | 2.62 | 568 | 0.22 | 1.89a | 560 |
| 2b | 0.47 | 2.72 | 527 | 0.31 | 1.85a | 521 |
| 2c | 0.014 | 0.71a | 716 | 0.002 | 0.33a | 645 |
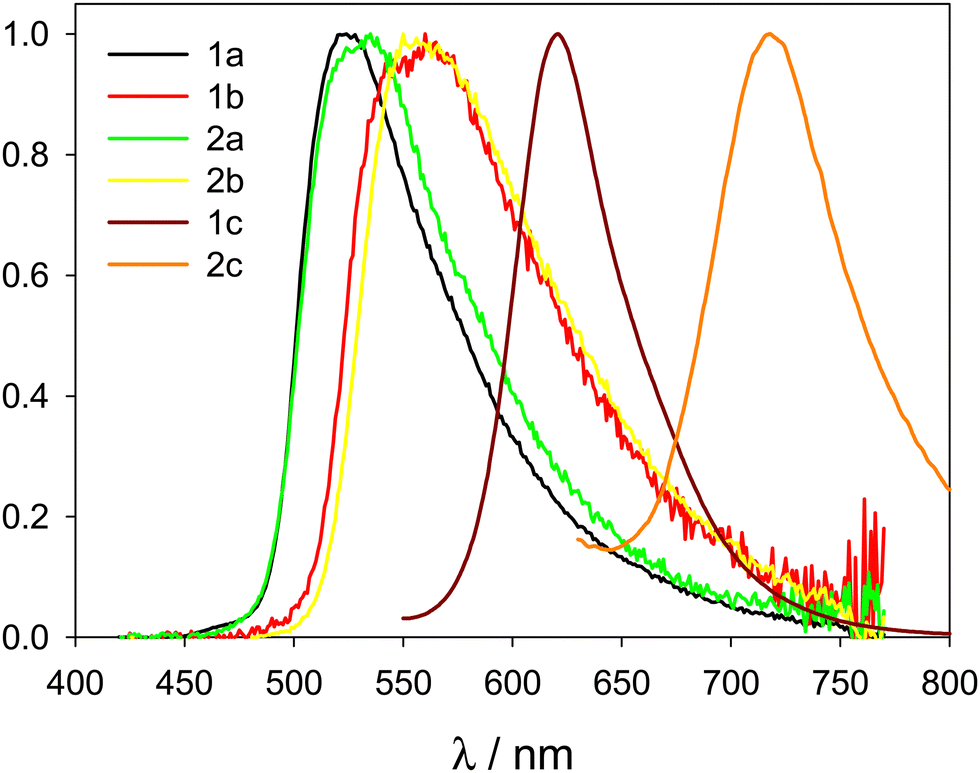 | ||
| Fig. 4 Normalized emission spectra of compounds 1a, b, c and 2a, b, c in the solid state (λexc = 400 nm). | ||
Triggered by this observation, we investigated the aggregation-induced emission (AIE) of these compounds by preparing solutions in acetonitrile/water mixtures with increasing water content. Formation of emissive aggregates was observed for all compounds except 2c, at a water content of at least 80% v/v (Fig. 5). In acetonitrile, the solutions are stable with no sign of aggregation even in the millimolar range. In water, instead, aggregates are formed readily even at the lowest concentration of 5 μM with clear AIE. Compound 1a only forms aggregates at a concentration of at least 20 μM in water with a peculiar kinetic activation (Fig. S61, ESI†). These aggregates are roughly as emissive as the solid powder with a similar lifetime for 1a and 1b and a shorter and multiexponential lifetime in the case of 1c, 2a, and 2b (Table 2). The multiexponential lifetime is ascribable to a distribution of the dyes (see below FLIM results). The aggregates also feature different colloidal stability: 2a and 2b form rather stable aggregates in water (Fig. 5D), while 1a and 1b have a clear tendency to sediment and stick at interfaces (microscopy images in Fig. S63, ESI†). Compounds 1c and 2c are soluble in acidic water due to the protonation of the tertiary amine, which results in blue-shifted absorption and emission (see Fig. S57 and S58 in ESI†). The non-protonated species, on the contrary, are much less water-soluble and form aggregates, while 1c shows a large enhancement in its luminescence upon deprotonation (>10 fold) due to the onset of AIE triggered by the lower solubility of the non-charged species. On the contrary, 2c does not show a significant increase in its brightness upon deprotonation and forms aggregates. For instance, in the solid state, its quantum yield remains very low. It is noteworthy that the acid–base nature of compound 1c makes it a good candidate for application as a pH-responsive AIE luminogen.
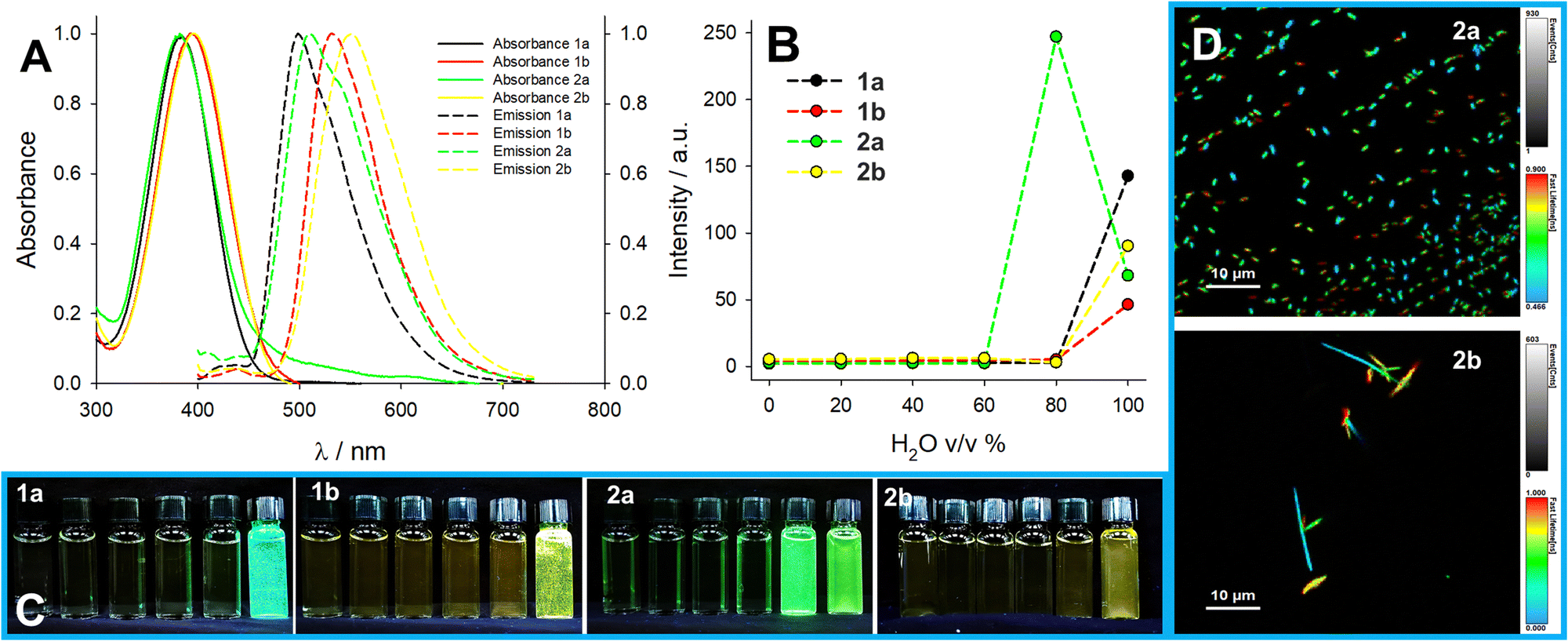 | ||
| Fig. 5 AIE properties: (A) Normalized absorbance and emission spectra of compounds 1a, 1b, 2a, and 2b in water (20 μM) and (B) trends of emission intensity at peak (λexc = 360 nm) in acetonitrile:water mixtures at increasing water content (full absorbance and emission spectra in Fig. S59 and S60, ESI†). (C) The photographs under UV light of compounds 1a, 1b, 2a, and 2b in acetonitrile:water mixtures at increasing water content. (D) FLIM images of samples 2a and 2b in water. | ||
Finally, we measured the emission decay after deoxygenation (purged with N2) to reduce the quenching of the triplet and to observe eventual TADF. Yet, significantly delayed emissions were not observed for long lifetime components, even upon removal of oxygen, neither in the solid state nor in the aggregates in water.
The aggregates formed in water were further analysed with confocal microscopy and fluorescence lifetime imaging (FLIM). Aggregates of compounds 1a and 1b, which were the most unstable in water, were difficult to find and could only be observed in large masses of aggregates at the boundary of the water droplet. In contrast, the aggregates of compounds 2a and 2b were distributed in the water droplet and on the surface of the cover glass. In particular, the aggregates of 2b were homogeneous in size (Fig. 5). Interestingly, FLIM images confirm that the emission decay of these aggregates is shorter than the emission in the solid state and multiexponential. Furthermore, FLIM of 2b reveals that aggregates, despite being homogeneous in size, are surprisingly polydisperse in terms of their emission lifetime, suggesting that during the formation of the aggregate, the dyes can assume different conformations with slightly different photophysical properties.
The reproducibility and long-term stability of these water-dispersed aggregates are unsolved issues and limit the application potential of their luminescence. To enhance the colloidal and photophysical stability of the AIE aggregates in water, we attempted to physically entrap the compounds as small, monodisperse, and water-soluble nanoparticles (NPs), composed of a silica core and PEG shell.7 The synthesis of these nanoparticles is based on a well-established procedure7 using tetraethoxysilane (TEOS) as the monomeric precursor and Pluronic F127 micelles as the template, which yields homogeneous particles of typical core and shell diameters of 10 and 25 nm, respectively.
We prepared NPs with encapsulated AIE-active compounds 1a, 1b, 2a, and 2b at relatively low degrees of doping (0.05% mol mol−1 TEOS) to minimize their interference with the formation of nanoparticles. Compounds 1c and 2c were not included owing to their water solubility in an acidic environment, which hinders the entrapment in hydrophobic micelles, and thus, in NPs. A clear peak was observed at ∼25 nm with DLS measurements for all prepared NPs; this is expected for this kind of NPs, but compounds 2a and 2b resulted in an additional peak at a very large diameter, probably due to the formation of dye aggregates independently from the formation of NPs, resulting in large average diameter and polydispersity index (PdI, see Table 3). Nonetheless, the DLS of NPs prepared in the presence of compounds 1a and 1b revealed the presence of a peak at ∼25 nm, suggesting a successful formation of dye-doped NPs.
| NPs | ϕ av (nm), PdIa | % Dopingb | PLQY | τ (ns) | B | χ 2 | τ av (ns) |
|---|---|---|---|---|---|---|---|
| a Obtained from DLS. b Obtained from absorbance spectra. c The average lifetimes (τAV) are calculated on the values of the pre-exponential coefficient B. | |||||||
| 1a | 22.4; 0.140 | 0.046 | 0.070 | 0.41 | 23100 |
0.868 | 0.71 |
| 1.29 | 10500 |
||||||
| 4.90 | 240 | ||||||
| 1b | 30.2; 0.229 | 0.048 | 0.040 | 0.30 | 25200 |
0.922 | 0.51 |
| 1.15 | 7100 | ||||||
| 5.22 | 170 | ||||||
| 2a | 289; 0.343 | ||||||
| 2b | 483; 0.630 | ||||||
The absorbance spectra of the prepared nanoparticles (Fig. 6) with AIE dyes embedded in them confirm the presence of large scattering aggregates for 2b@NPs, while the band of 2a@NPs is not present because of the precipitation of the aggregates before the measurement, also confirming that 2a was not efficiently incorporated in the NPs.
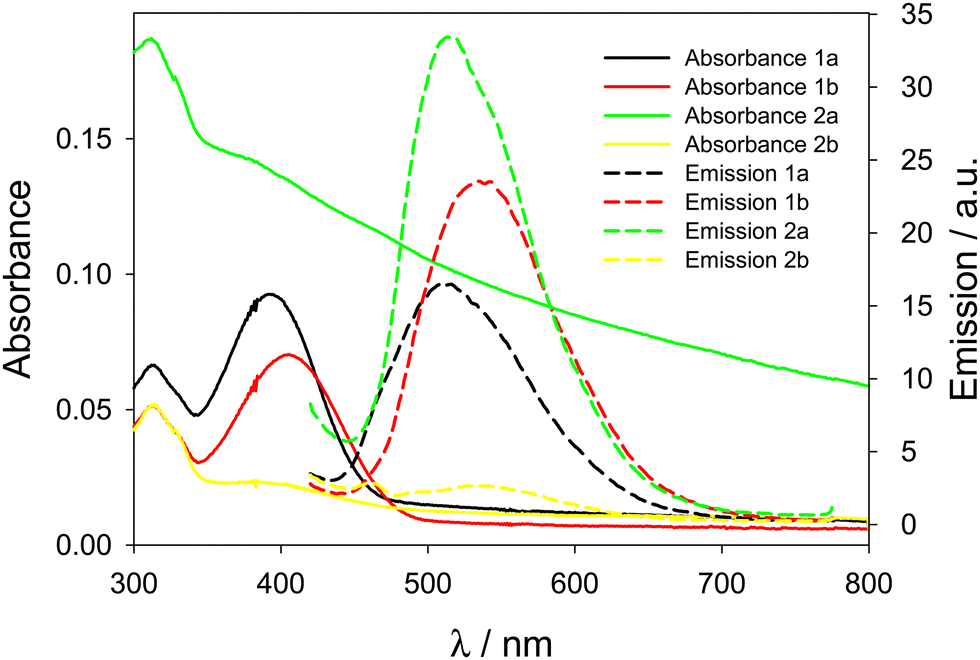 | ||
| Fig. 6 Absorbance (solid lines) and emission spectra (dashed lines, λexc = 400 nm and cut-off filter at 435 nm) of NPs embedding compounds 1a, 1b, 2a, and 2b. | ||
On the contrary, compounds 1a and 1b are efficiently incorporated in the nanoparticles: scattering of the solutions is low as expected for these small NPs, and the absorbance peaks confirm the presence of dyes 1a and 1b. From the absorbance spectra, from the known molar extinction coefficient of the dyes and the concentration of NPs (known and reproducible for this preparation method7), we calculated the effective doping degree of the dyes in the nanoparticles, confirming an efficient encapsulation of these two dyes (Table 3). Finally, the PLQY of 1a@NPs and 1b@NPs is lower compared to the aggregates in water, suggesting non-optimal AIE properties in this type of NPs, possibly due to residual molecular mobility. The analysis of the emission decays reveals short and multiexponential lifetimes for 1a@NPs and 1b@NPs in NPs, further corroborating this hypothesis (Table 3).
Crystallography
As for the crystallographic properties of these compounds, both complexes 1a and 2a crystallize in the monoclinic space group (P21/c) with four molecules in the crystal lattice. All the core six-membered rings, which contain four and three heteroatoms, respectively, are essentially planar with highly conjugated systems of π-electrons. In addition, the 4-methoxyphenyl substituents in 1a are nearly coplanar with that system and only slightly deviated (27.5°) in 2a. Propylene parts of the tetrahydroazepine substituent located opposite the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N connectors are screwed out of the central plane of both molecules. The interatomic distances and angles around the boron atoms (see Fig. 7–10) are in line with previously found values for single bonds. On the other hand, the azenyl parts of the molecules exhibit bigger differences when compared mutually and to the values found for NN bonds too. In 1a, the conjugation of the six-membered system causes the elongation of this bond up to 1.3128 (16) Å, but the same type of bond is much shorter for N1–N2, i.e., 1.2882 (15) Å, in 2a. In the same complex, the arrangement within the carboxylate unit bridging two borate anions (C2–C1(–O1)–O2) exhibits extremely short distances between involved atoms. Such a system, which is presumably described as a geminal diolate with a double bond between the C2 and C1 atoms has nearly equidistant C–O separations that are smaller than 1.28 Å, and thus, are more likely approaching an extreme situation in isobidentate carboxylates. Similar distances were found only in structures with a complex ionic-zwitterionic structure containing tetramethyl and trimethylphosphonium moieties or bridging carboxylate groups.7,8
N connectors are screwed out of the central plane of both molecules. The interatomic distances and angles around the boron atoms (see Fig. 7–10) are in line with previously found values for single bonds. On the other hand, the azenyl parts of the molecules exhibit bigger differences when compared mutually and to the values found for NN bonds too. In 1a, the conjugation of the six-membered system causes the elongation of this bond up to 1.3128 (16) Å, but the same type of bond is much shorter for N1–N2, i.e., 1.2882 (15) Å, in 2a. In the same complex, the arrangement within the carboxylate unit bridging two borate anions (C2–C1(–O1)–O2) exhibits extremely short distances between involved atoms. Such a system, which is presumably described as a geminal diolate with a double bond between the C2 and C1 atoms has nearly equidistant C–O separations that are smaller than 1.28 Å, and thus, are more likely approaching an extreme situation in isobidentate carboxylates. Similar distances were found only in structures with a complex ionic-zwitterionic structure containing tetramethyl and trimethylphosphonium moieties or bridging carboxylate groups.7,8
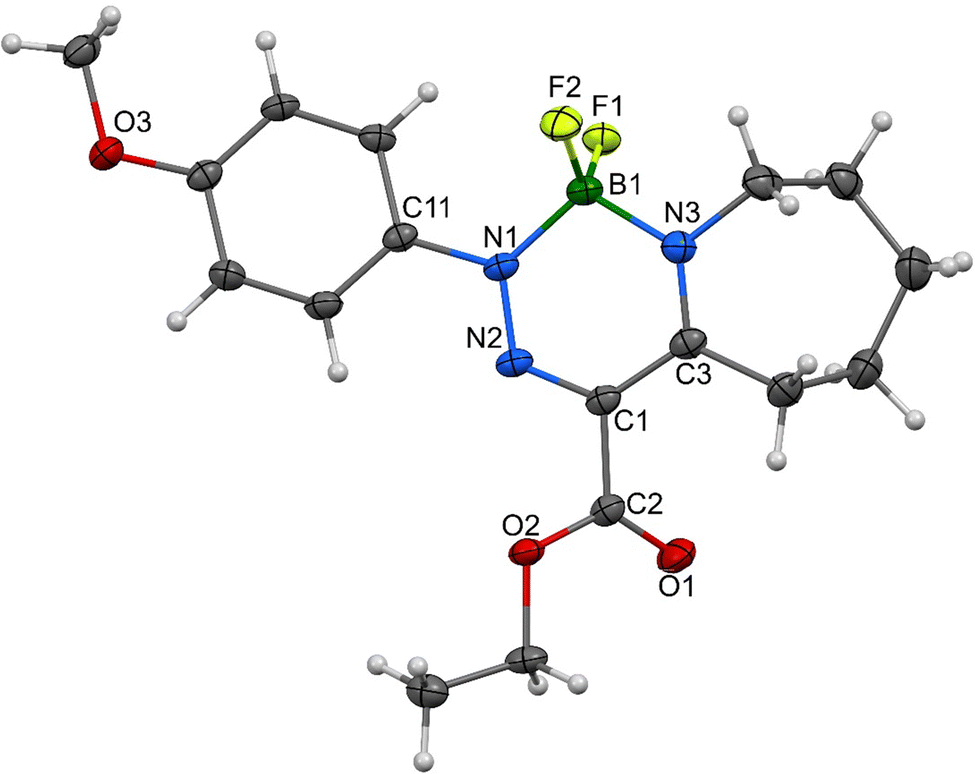 | ||
| Fig. 7 Molecular structure of 1a, ORTEP view, 50% probability level; selected interatomic distances [Å] and bond angles [°] are given in Table S5 (ESI†). | ||
 | ||
| Fig. 8 Molecular structure of 2a, ORTEP view, 50% probability level; selected interatomic distances [Å] and bond angles [°] are given in Table S6 (ESI†). | ||
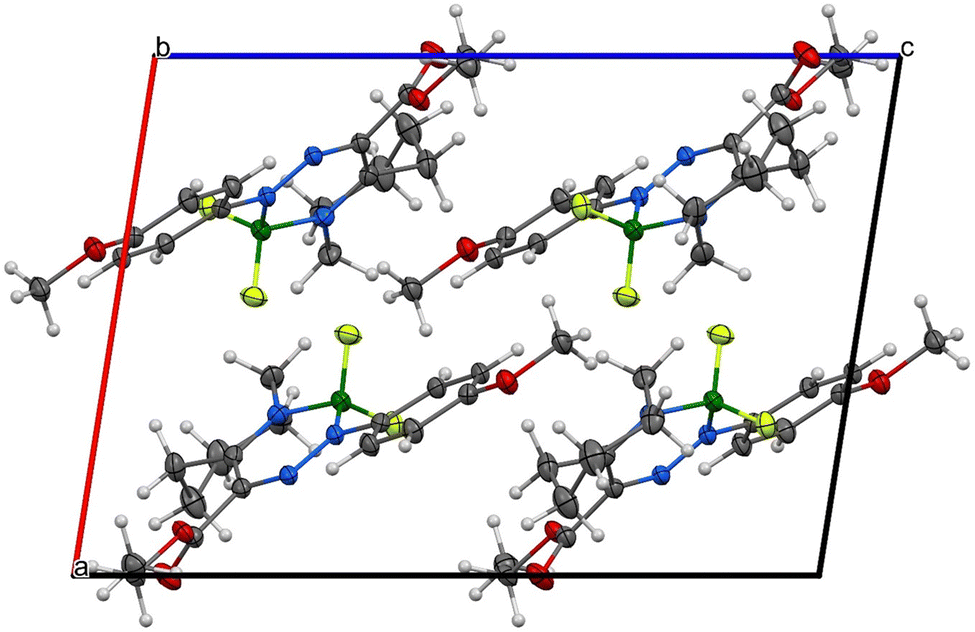 | ||
| Fig. 9 Supramolecular architecture/crystal packing of 1a, view along the b-axis. | ||
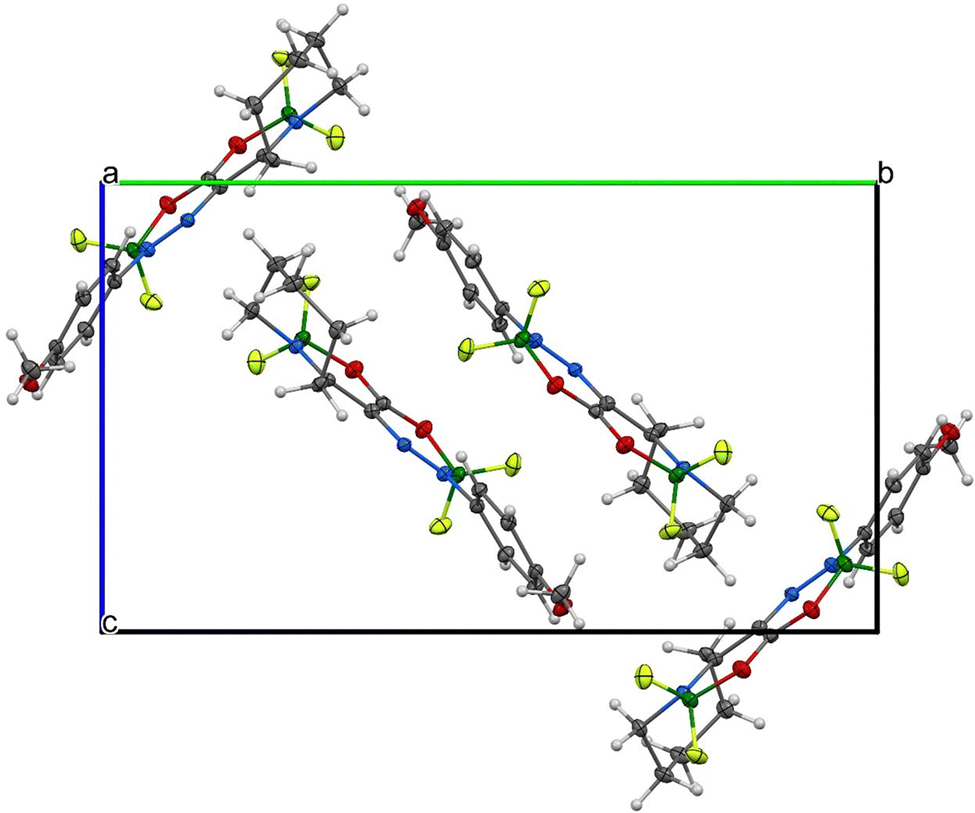 | ||
| Fig. 10 Supramolecular architecture/crystal packing of 2a, viewed along the a-axis. | ||
Due to the presence of the non-planar tetrahydroazepine substituent, both complexes cannot adopt π–π stacking, and only corrugated layers of centrosymmetrically related molecule pairs are found. Efforts of molecules to planarize in the solid state are more pronounced in 1a than in 2a (see Fig. 7–10). For experimental details, see Tables S3 and S4 (ESI†). For bond lengths and angles, see Tables S5 and S6 (ESI†).
Conclusions
Six novel difluoroboron heterocycles having either N–B–N or O–B–N arrangements were prepared and characterized using electrochemical and spectral (NMR, UV-vis, luminescence) methods. For two cases analysed here, X-ray diffraction analysis was also successfully performed. The results obtained were corroborated by DFT calculations.Except for 2c, all the prepared compounds show negligible luminescence in acetonitrile solutions and turn intensely luminescent in the solid state. Compounds 1a, b and 2a, b present a bright yellow-orange luminescence, whereas the luminescence of 1c is red. Therefore, the functionalization with ethers or tertiary amines is a good strategy for tuning the emissive properties of NBN and OBN compounds. These compounds are AIE active in a dioxan–water mixture.
Encapsulating 1a and 1b dyes in silica-PEG nanoparticles can represent an effective way to stabilize AIE aggregates and obtain increased emission in comparison with the solution phase, despite the PLQY enhancement being limited by residual mobility in this kind of NP. The average lifetimes are similar to the ones obtained for aggregates in water and lower than the ones measured for the solid state (0.51–0.74 ns).
Overall, the functionalization of mono-boron heterocycles containing the NEt2 group has proven to be a promising strategy to introduce pH responsiveness in this class of AIE-active compounds: AIE can be activated or the emissive aggregate disrupted upon deprotonation and protonation, respectively.
Experimental
Synthetic procedures, instrument details, as well as other characterization data are included in the ESI.†Data availability
All the data supporting the conclusions made in this manuscript are a part of the ESI.†Conflicts of interest
There are no conflicts to declare.References
- (a) S. S. Kothavale and J. Y. Lee, Adv. Opt. Mater., 2020, 2000922 CrossRef CAS; (b) H. Lee, D. Karthik, R. Lampande, J. H. Ryu and J. H. Kwon, Front. Chem., 2020, 8, 373 CrossRef CAS PubMed; (c) M. Hayakawa, M. Kameda, R. Kawasumi, S. Nakatsuka, N. Yasuda and T. Hatakeyama, Angew. Chem., Int. Ed., 2023, e202217512 CAS; (d) G. Li, W. Lou, D. Wang, C. Deng and Q. Zhang, ACS Appl. Mater. Interfaces, 2019, 11, 32209 CrossRef CAS; (e) A. Chandrasekar Murali, P. Nayak, S. Nayak, S. Das, S. P. Senanayak and K. Venkatasubbaiah, Angew. Chem., Int. Ed., 2023, e202216871 Search PubMed; (f) K. Tanaka, M. Gon, S. Ito, J. Ochi and Y. Chujo, Coord. Chem. Rev., 2022, 472, 214779 CrossRef CAS.
- (a) Y.-L. Rao and S. Wang, Inorg. Chem., 2011, 50, 12263 CrossRef CAS PubMed; (b) D. Li, H. Zhang and Y. Wang, Chem. Soc. Rev., 2013, 42, 8416 RSC; (c) D. Frath, J. Massue, G. Ulrich and R. Ziessel, Angew. Chem., Int. Ed., 2014, 53, 2290 CrossRef CAS PubMed; (d) S. S. Kothavale and J. Y. Lee, Adv. Opt. Mater., 2020, 2000922 CrossRef CAS; (e) H. Lee, D. Karthik, R. Lampande, J. H. Ryu and J. H. Kwon, Front. Chem., 2020, 8, 373 CrossRef CAS PubMed; (f) S. K. Mellerup and S. Wang, Chem. Soc. Rev., 2019, 48, 3537 RSC; (g) S. Mukherjee and P. Thilagar, J. Mater. Chem. C, 2016, 4, 2647 RSC.
- (a) A. Chandrasekar Murali, P. Nayak, S. Nayak, S. Das, S. P. Senanayak and K. Venkatasubbaiah, Angew. Chem., Int. Ed., 2023, e202216871 Search PubMed; (b) M. Hayakawa, M. Kameda, R. Kawasumi, S. Nakatsuka, N. Yasuda and T. Hatakeyama, Angew. Chem., Int. Ed., 2023, e202217512 CAS; (c) X. Zhu, R. Liu, Y. Li, H. Huang, Q. Wang, D. Wang, X. Zhu, S. Liu and H. Zhu, Chem. Commun., 2014, 50, 12951 RSC; (d) G. Tan, I. Maisuls, F. Strieth-Kalthoff, X. Zhang, C. Daniliuc, C. A. Strassert and F. Glorius, Adv. Sci., 2021, 8, 2101814 CrossRef CAS PubMed.
- (a) M. Svobodová, J. Bárta, P. Šimůnek, V. Bertolasi and V. Macháček, J. Organomet. Chem., 2009, 694, 63 CrossRef; (b) F. Josefík, M. Svobodová, V. Bertolasi, P. Šimůnek, V. Macháček, N. Almonasy and E. Černošková, J. Organomet. Chem., 2012, 699, 75 CrossRef; (c) M. Svobodová, P. Šimůnek, V. Macháček, L. Štruncová and A. Růžička, Tetrahedron, 2012, 68, 2052 CrossRef; (d) F. Josefík, T. Mikysek, M. Svobodová, P. Šimůnek, H. Kvapilová and J. Ludvík, Organometallics, 2014, 33, 4931 CrossRef.
- (a) J. Mei, N. L. C. Leung, R. T. K. Kwok, J. W. Y. Lam and B. Z. Tang, Chem. Rev., 2015, 115, 11718 CrossRef CAS PubMed; (b) B. Lei, Z. Huang, S. Li, J. Liu, Z. Bin and J. You, Angew. Chem., Int. Ed., 2023, e202218405 CAS.
- (a) F. P. Macedo, C. Gwengo, S. V. Lindeman, M. D. Smith and J. R. Gardinier, Eur. J. Inorg. Chem., 2008, 3200–3211 CrossRef CAS; (b) H. Doušová, P. Šimůnek, N. Almonasy and Z. Růžičková, J. Organomet. Chem., 2016, 802, 60 CrossRef; (c) H. Doušová, N. Almonasy, T. Mikysek, J. Váňa, M. Nepraš, B. Frumarová, M. Dvořák, Z. Růžičková and P. Šimůnek, Monatsh. Chem., 2018, 149, 1975 CrossRef.
- (a) F. Palomba, D. Genovese, L. Petrizza, E. Rampazzo, N. Zaccheroni and L. Prodi, Sci. Rep., 2018, 17095 CrossRef; (b) D. Genovese, M. Montalti, L. Prodi, E. Rampazzo, N. Zaccheroni, O. Tosic, A. Altenhöner, F. May and J. Mattay, Chem. Commun., 2011, 47, 10975 RSC; (c) D. Genovese, E. Rampazzo, N. Zaccheroni, M. Montalti and L. Prodi, Eur. J. Inorg. Chem., 2017, 5094 CrossRef CAS; (d) E. Rampazzo, S. Bonacchi, D. Genovese, R. Juris, M. Montalti, V. Paterlini, N. Zaccheroni, C. Dumas-Verdes, G. Clavier, R. Méallet-Renault and L. Prodi, J. Phys. Chem. C, 2014, 118, 9261 CrossRef CAS.
- (a) M. Radius, E. Sattler, H. Berberich and F. Breher, Chem. – Eur. J., 2019, 25, 12206 CrossRef CAS; (b) H. Binder, W. Matheis, H.-J. Doiseroth and H. Fu-Son, Z. Naturforsch., B: J. Chem. Sci., 1983, 38, 554 CrossRef; (c) H. Binder, W. Matheis, H.-J. Deiseroth and H. Fu-Son, Z. Naturforsch., B: J. Chem. Sci., 1984, 39, 1717 CrossRef; (d) H. Binder, W. Matheis, G. Heckmann, H.-J. Deiseroth and H. Fu-Son, Z. Naturforsch., B: J. Chem. Sci., 1985, 40, 934 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2189206 and 2189207. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4ma00653d |
| This journal is © The Royal Society of Chemistry 2024 |