
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceControlling ligand density and viscoelasticity in synthetic biomimetic polyisocyanide hydrogels for studying cell behaviours: the key to truly biomimetic hydrogels†
Nicholas J.
Westra van Holthe‡
 *ac,
Zhao
Wang‡
a,
Jan
Lauko
a,
Elliot P.
Gilbert
b,
Vishaak
Gangasandra
a and
Alan E.
Rowan
*ac
*ac,
Zhao
Wang‡
a,
Jan
Lauko
a,
Elliot P.
Gilbert
b,
Vishaak
Gangasandra
a and
Alan E.
Rowan
*ac
aAustralian Institute for Bioengineering and Nanotechnology, The University of Queensland, Brisbane, Queensland 4072, Australia. E-mail: n.westravanholthe@uq.edu.au; alan.rowan@uq.edu.au
bAustralian Centre for Neutron Scattering, Australian Nuclear Science and Technology Organisation, Lucas Heights, NSW 2234, Australia
cARC Centre of Excellence in Quantum Biotechnology, St Lucia, Queensland, Australia
First published on 11th November 2024
Abstract
Many cell types within whole living organisms continuously sense external forces from their microenvironment through an extracellular matrix that orchestrates biophysically-cued cellular processes and mechanosensitive cell behaviours. Studying cells in 3D environments rather than in conventional 2D, is increasingly seen as a necessity for obtaining a comprehensive understanding of the dynamic and complex cell–matrix interactions and cellular responses to biophysical stimuli. While many synthetic hydrogels used in 3D cell culture applications do not provide a microenvironment that accurately mimics the structural and mechanical properties of native ECMs, ethylene-glycol grafted isocyanopeptide-based polymers (PIC) have become one of the most promising types of synthetic material for controllably mimicking endogenous ECM microenvironments based on their unique biomimetic strain-stiffening response. However, the precise effects of polymer modification through integrin-binding peptide addition and density on the nanoscale architecture of PIC polymer–peptide conjugates have not yet been comprehensively studied and the stress relaxation behaviours of PIC-based hydrogels have also yet to be comprehensively explored. Here we present new data on the structure–property relationship for PIC-based polymer–peptide conjugates by highlighting the effect of peptide addition and density on the PIC hydrogel physicochemical properties and nanoscale network structure. We show that the addition of a peptide-linker to polymer-bundle forming PIC polymers alters the polymer-bundle network structure, by potentially inhibiting the polymer-bundle formation and reducing the polymer-bundle diameter and density. Furthermore, we demonstrate the synergistic effects of ligand density, strain-stiffening responsiveness and viscoelasticity on fibroblast rapid cell spreading, morphological changes, and cytoskeleton alignment in comparison to a natural fibrin hydrogel. Cells cultured in a 6% (mol mol−1) ligand density PIC hydrogel, with a low critical stress point and fast stress relaxation, facilitates similar spreading kinetics, spreading phenotype and F-actin cytoskeleton alignment to fibroblast cells cultured in a naturally-derived fibrin hydrogel. We find that the viscoelastic high ligand density PIC–peptide conjugate facilitates an accurate replication of fibroblast cellular behaviours in a natural matrix and that this material composition shows considerable promise for directing the optimal design of bioinspired materials for mechanotherapy and 3D cell culture.
Introduction
Studying mammalian cells in conventional two-dimensional (2D) culture systems has been an imperative methodology for studying cellular function, behaviour, and response to external stimuli. However, 2D cell culture methods position cells within an artificial environment that is incomparable to the native three-dimensional (3D) heterogenous and dynamic extracellular matrix (ECM) that tissue cells inhabit within whole living organisms.1 This limitation makes 2D cell culture methods problematic for obtaining a comprehensive understanding of several dynamic and complex 3D cell behaviours, along with biophysical and mechanically-driven cell–matrix interactions.2–4 The native ECM is comprised of an array of macromolecules and proteins such as collagen, fibronectin, fibrin, hyaluronic acid, elastin and laminin, all of which self-assemble into fibrous or fibrillar networks with specific nano- and micro-scale architectures.5,6 The architectural properties of these natural matrices such as pore size, stiffness, stress-stiffening and stress-relaxation directly influence cell behaviours such as spreading, proliferation, migration, differentiation, and apoptosis through cellular mechano-sensing and mechanotransduction.7,8 Therefore, studying cells within a controlled 3D matrix system that definitively replicates the native ECM is crucial for acquiring detailed information about biophysically-cued cellular processes and mechanosensitive cell behaviours.9–11While various 3D cell culture systems using either naturally derived (e.g. Matrigel, collagen, etc.) or synthetic matrices have been reported over decades of research, several inevitable limitations such as lack of reproducibility or high batch-to-batch variability, ill-defined and variable composition, uncontrolled mechanical properties and ligand density, quasi-mimicry of biological matrix properties and the inability to precisely control the 3D architecture have hindered the acquisition of detailed and relevant cellular behaviour data from these systems.12,13 Additionally, mechanoresponsive properties of the matrix that are key for natural cellular behaviours and intracellular communication such as stress-stiffening and stress relaxation are rarely replicated by synthetic hydrogel systems.14–16 Ethylene-glycol grafted isocyanopeptide (or isocyanide)-based polymers have become one of the most promising synthetic materials for elucidating biophysically-driven cellular processes due to their biomimetic and controllable mechanical properties, and their tailored composition.17,18 Thus, polyisocyanopeptide (PIC)-based hydrogels have been widely used to facilitate the study of cellular behaviours and cell–matrix interactions in 3D including stress-stiffening-mediated stem cell differentiation, epithelial morphogenesis, mammary organoid formation, cell morphological organisation and cell-induced matrix stiffening and remodelling.18–22 PICs are one of the only known synthetic hydrogel-forming polymers that presents a strain-stiffening response within the biologically relevant stress window that is analogous to fibrin and collagen.17,18,23 Importantly, this unique biomimetic stress-stiffening behaviour is believed to result from the β-helical polymer backbone generated by the nickel-catalysed coordination polymerisation that is stabilised by hydrogen-bonded β-sheets along the helical architecture.24–26 In addition, PIC polymers display thermoresponsive behaviours where strain-stiffening hydrogels are formed at elevated temperatures above the lower critical solution temperature (LCST). The mechanism of PIC hydrogel formation is ascribed to the temperature dependant formation of polymer bundles above the LCST, where bundle formation is thought to be derived from an entropic dehydration process with increasing temperature driving water expulsion and trapping between the oligoethylene glycol side chains, leading to an interconnected polymer-bundle network structure.27,28
Fibrous proteins within the endogenous ECM present specific integrin-binding peptide components that facilitate adhesive interactions between cells and the ECM, allowing cells to bind to the matrix and respond to mechanical stimuli from the surrounding microenvironment.2,29 Thus, covalently incorporating specific peptide sequences within synthetic matrix systems is crucial for building accurate synthetic mimics of the natural ECM. While several PIC–peptide hydrogels have been presented previously as 3D cell culture systems, comprehensive characterisation of the polymer modification process and the resulting polymer–peptide conjugates is not typically presented, and a clear correlation between the peptide density and the hydrogel mechanical properties has not yet been systematically identified.19 While the nanoscale architecture of unmodified PIC-based hydrogels has been elucidated extensively, the effect of peptide addition and density on the nanoscale architecture of PIC polymer–peptide conjugates has not yet been comprehensively studied.16,27
In this manuscript, we present a revised peptide-linker synthetic methodology that provides enhanced control and accuracy of the addition of peptides to ECM-mimicking PIC polymers. New evidence is presented herein highlighting the effect of peptide addition and density on the hydrogel physicochemical properties and nanoscale network structure. We show that the addition of a peptide-linker to polymer-bundle forming PIC polymers alters the polymer-bundle network structure, potentially by inhibiting the polymer-bundle formation (illustrated in Fig. 1), thus reducing the polymer-bundle diameter and density. Importantly, we observe a clear relationship between the degree of peptide loading, the hydrogel mechanical properties and the polymer-bundle nanostructure. Moreover, we demonstrate the synergistic effects of ligand density, stiffness, strain stiffening and viscoelasticity on fibroblast rapid spreading, morphological changes, and cytoskeleton alignment in comparison to a natural fibrin hydrogel. We introduce a ligand density 2 times higher than previously achieved for PIC-based hydrogels and illustrate that higher ligand density, lower matrix stiffness, high strain stiffening responsiveness and faster stress relaxation synergistically facilitate fast cell spreading and F-actin cytoskeleton alignment in cultured fibroblasts. Cells cultured in the PIC system at 6% (mol mol−1) ligand density show a similar spreading phenotype and kinetics to fibroblast cells cultured in a naturally derived fibrin hydrogel. These results demonstrate the high sensitivity of fibroblast cells in particular towards ligand density and hydrogel mechanics and show promise for directing the optimal design of future biomaterial systems for specific cell culture and wider biomedical applications. To the best of our knowledge, this is the first synthetic biomimetic 3D cell culture hydrogel system that accurately replicates the biophysical properties of a natural fibrin matrix where the observed fibroblast cellular responses are similar between the synthetic PIC-based hydrogel and the natural fibrin matrix.
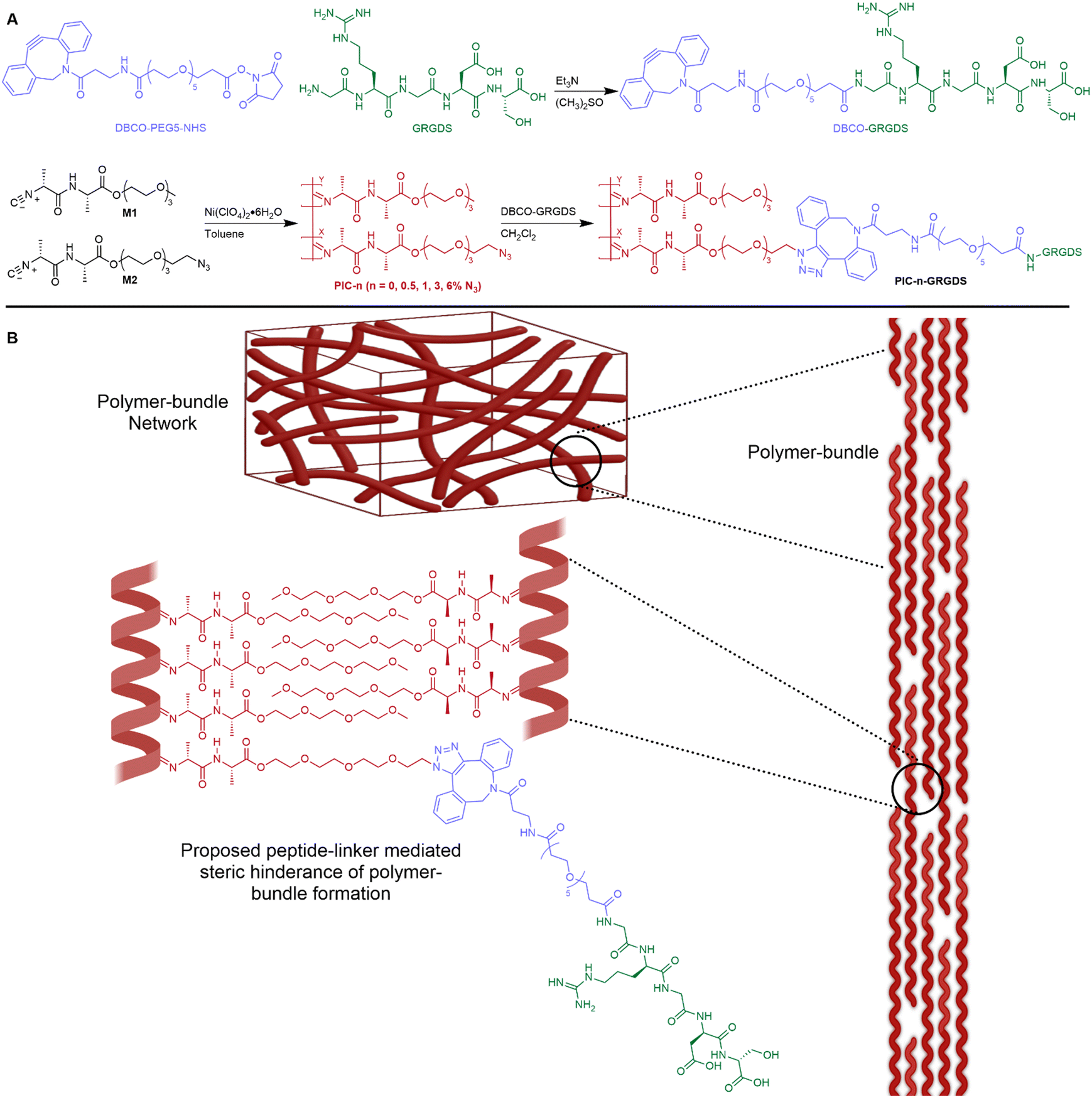 | ||
| Fig. 1 Polymer synthesis and polymer-bundle networks of PIC-GRGDS hydrogels. (A) Summary of synthesis procedure for the preparation of GRGDS-DBCO linker and PIC-GRGDS. (B) Illustration of PIC-GRGDS nano- and micro-scale architecture of PIC polymer bundles and LCST-driven polymer-bundle network formation. | ||
Experimental section
Materials
Unless otherwise stated, all reagents and solvents were purchased from Sigma Aldrich and used without further purification. 2-(2-(2-Methoxyethoxy)ethoxy)ethyl ((R)-2 isocyanopropanoyl)-L-alaninate (M1) was purchased from ChiroBlock GmBH, Germany and was vacuum-dried prior to use. 2-(2-(2-(2-Azidoethoxy)ethoxy)ethoxy)ethyl ((R)-2-isocyanopropanoyl)-L-alaninate (M2) was purchased from Synkola s.r.o., Slovakia.30 DBCO-PEG5-NHS was purchased from Click Chemistry Tools and used without further purification. The peptide sequence Gly–Arg–Gly–Asp–Ser (GRGDS) was purchased from GenScript with a reported purity of 99.1% and used without further purification. Deionised water with a resistivity of 18.2 MΩ cm was obtained from an Elga ultra-pure Milli-Q water system. Alexa Fluor Plus 647 Phalloidin, Hoechst 33342, Dulbecco's Modified Eagle Medium (DMEM), fetal bovine serum, penicillin–streptomycin, and cell culture polystyrene flasks (Nunc™ EasYFlask™) were all purchased from ThermoFisher Scientific. The fibrinogen protein (VeraSeal, #336837, ETHICON) used in this study was bought from Australian Therapeutic Goods Administration. Human prothrombin (#HCP-0010) was from Haematologic Technologies, USA. 15-Well chamber slides for 3D cell culture were purchased from Ibidi, USA.Synthesis of polyisocyanides
Polyisocyanide β-helical polymers were synthesised according to previously reported methodologies (Fig. 1).19,24,31 Isocyanide monomers M1 and M2 were combined to yield PIC(methoxy-co-azide) polymers with varying amounts of azide functional groups as presented in Table 1. Individual reaction procedures are described in the ESI.† An inert atmosphere was generated through 3 successive vacuum/argon cycles before the isocyanide monomers were dissolved in anhydrous toluene. The appropriate amount of Ni(ClO4)2·6H2O catalyst solution was combined with the monomer solution to initiate the polymerisation, and the mixture was stirred at room temperature for 48 h. Once the polymerisation was confirmed complete by FTIR–ATR, the solvent was removed in vacuo. The crude polymer was redissolved in CH2Cl2 and precipitated three times against cold diisopropyl ether. Purified polymer was dried under reduced pressure to yield a beige fibrous solid. The viscosity average molecular weight (Mv) of the polymers was measured by viscometric analysis.17,32 The intrinsic viscosity of polymer solutions was measured at 25 °C. The Mv was subsequently calculated using the empirical Mark–Houwink equation, [η] = KMav where [η] is the intrinsic viscosity of the polymer solution, K and a are constants that depend on the polymer and solvent characteristics. K and a were set to values that have been previously determined for similar rigid PICs.17K = 1.4 × 10−9, a = 1.75. Polymer length (L) was calculated from the Mv as previously described (see ESI†).17| Polymer | M1:M2% | M co (g mol−1) | M v (kg mol−1) | L (nm) | N3 spacing (nm) |
|---|---|---|---|---|---|
| PIC-0 | 0 | 316.3 | 368 | 146 | — |
| PIC-0.5 | 0.5 | 316.6 | 373 | 148 | 23.5 |
| PIC-1.0 | 1 | 317.0 | 375 | 148 | 11.7 |
| PIC-3.0 | 3 | 318.0 | 368 | 146 | 3.91 |
| PIC-6.0 | 6 | 319.6 | 371 | 147 | 1.95 |
Peptide-linker synthesis
The GRGDS-DBCO linker was synthesised via the following generalised procedure. DBCO-PEG5-NHS (0.055 g, 0.079 mmol) and GRGDS (0.054 g, 0.111 mmol) was dissolved in anhydrous DMSO (4 mL) and triethylamine (Et3N, 0.05 mL, 0.396 mmol). The reaction mixture was stirred at room temperature for 4 h protected from light. The efficacy of the peptide-linker synthesis and the ratio of product to starting material was determined by liquid chromatography-mass spectrometry (LC-MS) conducted using a Dionex HPLC module equipped with a 4.6 × 50 mm C18 Kintetex column and UV-Vis detector connected to a ThermoFisher TSQ Quantum Ultra QqQ MS. The crude mixture was separated through the C18 column with an eluent system starting at 95% A: 5% B and moving through a gradient to 100% B (A = 0.2% formic acid in H2O, B = 0.2% formic acid 80% MeCN in H2O). UV-Vis detector channels (220, 250 and 310 nm) were selected to probe the absorbance of both starting materials and the product. m/z: calculated for DBCO-GRGDS product C49H68N10O17 1068.48, found 1069.69 ([M + H]+).Post-polymerisation peptide addition
Each PIC(methoxy-co-azide) polymer (PIC-0.5, PIC-1.0, PIC-3.0, PIC-6.0, ≈200 mg) was dissolved in CH2Cl2 (75 mL) before an appropriate volume of the crude GRGDS-DBCO solution in DMSO was combined with the polymer solution. The GRGDS-DBCO linker was combined in 1.5 molar equivalents to the calculated moles of azide functional groups for each PIC methoxy-co-azide polymer as per the calculated molecular weight of the average repeating copolymer subunit (Mco) (Table 1). The reaction mixture was stirred at room temperature for 48 h protected from light before the solvent volume was reduced by rotary evaporation. The crude polymer was precipitated against cold diisopropyl ether (250 mL) and collected by centrifugation. The polymer was purified through a total of 3 precipitations against diisopropyl ether then dried under reduced pressure to reveal the purified PIC-GRGDS product as a beige/yellow powdered solid.Characterisation of hydrogels
Fourier-transform infrared (FTIR) spectroscopy was acquired on solid polymer samples using a Nicolet Nexus 870 FTIR spectrometer equipped with a Nicolet Smart Endurance single bounce, diamond ATR accessory. Rheological analysis was assessed using an Anton Paar MCR-502WESP rheometer (RheoCompass V1.23 software) equipped with a 25 mm parallel stainless-steel plate geometry set to a 0.5 mm gap. Polymers were dissolved in MilliQ water at a concentration of 6.32 mM at 4 °C on an intellimixer for 18–24 h before rheological analysis. Chilled polymer solutions were loaded onto the rheometer plate at a plate temperature of 5 °C. The storage (G′) and loss moduli (G′′) were recorded over a temperature ramp of 5–37 °C at a heating rate of 1 °C min−1 and an oscillating shear strain of 1% and frequency of 1 Hz. After equilibrating at 37 °C for 30 min a frequency sweep was performed at 37 °C to determine G′ and G′′ within a frequency range of 0.1–10 Hz at a shear strain of 1%. Subsequently, a nonlinear pre-stress protocol was applied to measure the elastic differential modulus (K′ = dσ/dγ) as previously described where σ and γ represent stress and strain respectively.33K′ data was analysed in GraphPad Prism 10.0. A linear regression (slope = 0) was applied to the linear viscoelastic regime at low strain to determine the gel bulk stiffness (G0). At higher strain values a power regression was applied to the data. The intercept of the linear and power functions was calculated as the critical stress (σc). Stress-relaxation of the hydrogel matrices was measured at 5% and 20% strain held constant while the stress and relaxation modulus were recorded as a function of time. A timescale (T1/2) was defined as the timescale at which the stress recording reaches half of its initial value to determine the scale of stress relaxation. For each polymer, one sample was analysed in triplicate (n = 3) and values are recorded as the mean of 3 measurements.Small-angle neutron scattering
Small-angle neutron scattering (SANS) was employed according to previous measurements of PIC-based hydrogels.34 PIC polymer samples were hydrated in D2O at 2 mg mL−1 at 4 °C for 48 hours before SANS detection. Upon measurement, 500 μL of each PIC sample was loaded to the cold SANS quartz cuvettes (2 mm). Samples were heated at 37 °C for at least 2 hours before the commencement of the SANS measurement to ensure the complete PIC network formation. SANS measurements were performed on the Quokka instrument at the OPAL reactor at the Australian Nuclear Science and Technology Organisation (ANSTO, Lucas Heights, Sydney, Australia).35,36 SANS measurements covered a Q range from 6 × 10−4 to 0.5 Å−1. The magnitude of scattering vector Q is defined as Q = 4π![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) sin(θ)/λ, in which θ is half of the scattering angle using a wavelength (λ) of 5 Å (and 8.1 Å for the focussing configuration) with 10% wavelength resolution. Three source-to-sample (SSD) and sample-to-detector distance (SDD) configurations including SSD = SDD = 20 m (standard and focussing), SSD = SDD = 8 m, and SSD = 4 m and SDD = 1.3 m were performed with the latter using 300 mm lateral detector offset. The source aperture diameter was 50 mm and the sample aperture diameter was 12.5 mm. All measurements were conducted at 37 °C. The data were reduced using NCNR SANS reduction macros modified for the QUOKKA instrument, using the Igor software package (Wavemetrics, Lake Oswego, OR) with data corrected for D2O background and detector sensitivity.37 The data were transformed to absolute scale using an attenuated direct beam transmission measurement.
sin(θ)/λ, in which θ is half of the scattering angle using a wavelength (λ) of 5 Å (and 8.1 Å for the focussing configuration) with 10% wavelength resolution. Three source-to-sample (SSD) and sample-to-detector distance (SDD) configurations including SSD = SDD = 20 m (standard and focussing), SSD = SDD = 8 m, and SSD = 4 m and SDD = 1.3 m were performed with the latter using 300 mm lateral detector offset. The source aperture diameter was 50 mm and the sample aperture diameter was 12.5 mm. All measurements were conducted at 37 °C. The data were reduced using NCNR SANS reduction macros modified for the QUOKKA instrument, using the Igor software package (Wavemetrics, Lake Oswego, OR) with data corrected for D2O background and detector sensitivity.37 The data were transformed to absolute scale using an attenuated direct beam transmission measurement.
Fitting and analysis were performed using the SASView software. Eqn (1)–(3) were based on the preset fitting models from SASView. The average bundle size (RB) of the PIC network was calculated using Guinier fitting model according to eqn (1) and (2), where I(Q) is the scattering intensity at scattering vector (Q) and Rg is the radius of gyration.
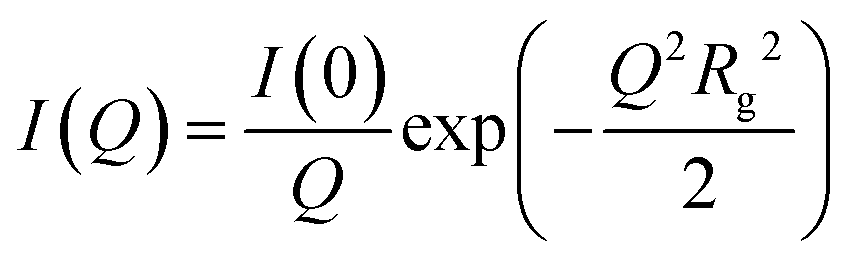 | (1) |
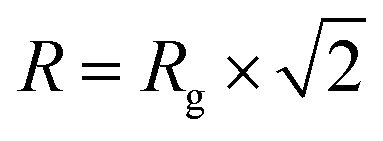 | (2) |
The average pore size (ξ) of the PIC network was extracted from eqn (3) based on a fractal model evaluating the network structure where Df is the fractal dimension, S(q) is the structure factor.38
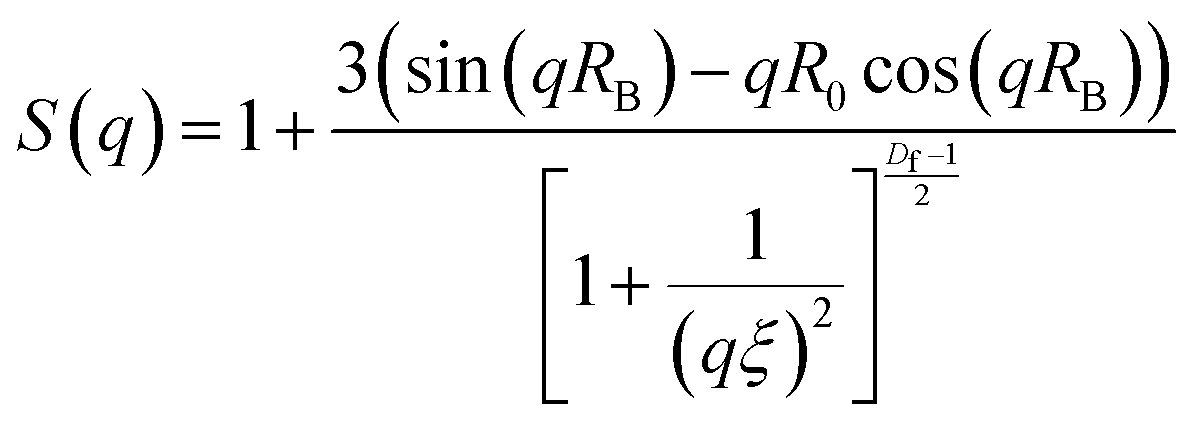 | (3) |
Cell growth and 3D cell culture
Human dermal fibroblasts (ATCC, #PCS-201-012) were cultured in the polystyrene flasks to passage 10–15 in the complete media (DMEM + 10% FBS + 1% pen/strep). For 3D cell culture in PIC-1.0 N3 or PIC-GRGDS matrices, 2.5 × 105 cells mL−1 fibroblasts were mixed with PIC polymer solutions on ice. The mixture was then gently transferred into 15-well chamber slides (20 μL per well) and heated to 37 °C. For the fibroblasts 3D cultured in fibrin, a similar ecarin-initiated fibrin formation protocol was applied as described previously.13 A fibrin network was initiated after mixing 4 mg mL−1 fibrinogen and 10−4 IU μL−1 prothrombin with 1 nM ecarin in the buffer containing 2 mM CaCl2, 10 μM textilinin (to inhibit fibrin degradation), 20 mm HEPES and 150 mM NaCl, pH 7.4. Fibroblasts (2.5 × 105 cells mL−1) were added to the initiated fibrinogen mixture and quickly transferred to the 15-well chamber slides (20 μL per well), flipping the chamber slides regularly to avoid cells dropping to the bottom of the wells until the fibrin network was formed within 2–3 min. 30 min after the initial gelation of matrices, complete media (containing 10 μM textilinin) was added on top of the gel in each well and changed 2 hours later. Cell culture media was later changed on day 1, 3, and 5 with 10 μM textilinin to maintain the fibrin network stability.Immunofluorescence staining
For the immunostaining of fibroblasts on day 7 after 3D culture in PIC matrices or fibrin networks, all the procedures were performed on a heating block at 37 °C to maintain the PIC matrix network. Cells within gels were fixed in 4% paraformaldehyde (PFA) for 1 hour before being permeabilised by 0.1% Triton X-100 for 30 min. Cells were then incubated with Alexa Fluor Plus 647 phalloidin (1:1000) and Hoechst 33342 (1:1000) for 4 hours and washed with PBS before conducting confocal microscopy.
Confocal laser scanning microscopy
Confocal laser scanning microscopy was conducted on a TCS SP8 WLL, Leica Microsystems equipped with a 20× (HC PL FLUOTAR L 20×/0.40 DRY) air objective. For each image, a 775 μm × 775 μm × 200 μm (2 μm per frame) areas were imaged 20–40 μm distant from the coverslip surface. Immunofluorescence of F-actin (excitation: 647 nm) and nuclear Hoechst staining (excitation: 420 nm) were imaged simultaneously. At least three randomly picked areas were imaged for each condition.Cell morphology analysis
Cell morphology including cell body morphology and nuclear morphology was analysed using the maximum intensity projected confocal images by FIJI ImageJ software. Each cell body and cell nuclei were contoured manually before the circularity, aspect ratio and 2D projected area of each cell body and nuclei were analysed.Results and discussion
Refining the peptide-linker synthesis methodology
Introducing peptide sequences within synthetic 3D cell culture to facilitate cell–matrix interactions is a crucial aspect of studying cells in truly biomimetic 3D cell culture systems.18 Yet, the effect of peptide addition and density on the mechanical properties and the nanoscale architecture of hydrogel-forming PIC polymers has not been comprehensively studied previously. To unveil the effect of peptide addition on the PIC physicochemical properties, the peptide-linker synthesis must first be delicately controlled and the covalent binding of the peptide to the DBCO-PEG5-NHS linker must be quantitative. The efficacy of the peptide-linker synthesis is vital to the accuracy of the final peptide ligand density within the hydrogel and therefore vital to the accuracy of the hydrogel mechanical properties and subsequent cell experiments. Historically, for the synthesis of peptide decorated PIC-based ECM mimicking hydrogels, the nucleophilic substitution reaction between the peptide and a linker (to form the strained alkyne terminated peptide-linker) has been conducted in an aqueous-based solvent, and the resultant peptide-linker reaction efficacy and product purity within the crude reaction mixture has not typically been assessed by quantitative means prior to polymer addition.39 Therefore, we have revised and refined the peptide-linker synthetic methodology and prepared the peptide-linker (DBCO-GRGDS) in an organic solvent with the addition of an organic base (Et3N) to yield a high efficacy nucleophilic substitution reaction between the NHS-activated ester of the linker (DBCO-PEG5-NHS) and the primary amine of the peptide. The resultant peptide-linker product purity within the crude reaction mixture was analysed by LC-MS (Fig. S2, ESI†) to reveal a peptide-linker conversion efficacy above 99%, validating that more than 99% of the DBCO units have a peptide attached using the non-aqueous methodology. In addition, we analysed the reaction efficacy of a peptide-linker synthesis conducted in the commonly reported aqueous conditions (DMSO + Borate buffer) by LC-MS and found that less than 50% peptide-linker conversion was achieved in the aqueous-based borate buffer solvent (Fig. S5, ESI†). Importantly, this result suggests that the nucleophilic substitution reaction between the peptide–NH2 and the NHS-activated ester conducted within aqueous conditions can result in less than 50% of the DBCO units having a covalently bound peptide, and subsequent peptide decorated hydrogel systems synthesised via this methodology could possibly consist of a far lower peptide concentration than anticipated.Polymer characterisation pre-peptide addition
Ethylene-glycol grafted isocyanopeptide-based polymers with a hydrogen-bonded β-helical architecture are one of very few hydrogel-forming synthetic polymers with a strain-stiffening response within the biologically relevant stress window, similar to biological ECM polymers such as actin, collagen, and fibrin.18,40 PIC hydrogel systems have been studied extensively in several cell culture applications and are seen as advantageous for the development of 3D cell culture systems to study cellular functions and responses, specifically in the realm of exploring mechanobiology and cellular function within wound healing.41 Polyisocyanopeptide-based polymers were synthesised here with varying molar ratios of methoxy- and azide-terminated isocyano-(D)-alanyl-(L)-alanine monomers to generate a series of polymers with increasing azide content from 0–6% N3 (mol mol−1). All polymers were synthesised using a specific catalyst-to-monomer ratio to maintain consistency in the viscosity average molecular weight and polymer length over the series (Table 1). The viscosity average molecular weight and thus polymer length was determined to be highly consistent over the series of polymers and polymer lengths were calculated from the Mv to range from 145–148 nm (see ESI†).17 Additionally, spacing of the azide-terminated side chains along one of the 4 hydrogen-bonded β-sheets alongside the polymer backbone was calculated based on a 4.7 Å average spacing between the side chains.24 Based on a 4.7 Å average spacing the theoretical spacing between azide groups was calculated to range from 23.5 nm for the 0.5% N3 to 1.95 nm for the 6% (Table 1).ATR-FTIR confirmed the presence of azide functional groups across the polymer series whereby the peak at 2106 cm−1 within the expected azide region increases across the polymer series which is most obvious at azide percentages of 3 and 6% (Fig. 2). In addition, the PIC(methoxy-co-azide) polymers were dissolved in water (2 mg mL−1) and subject to rheological analysis (Table 2) to determine the gelation temperature, bulk stiffness G0 and the differential modulus K′ as a function of strain (Fig. 3). We observed a clear correlation between the gelation temperature, the hydrogel storage modulus, and the azide content within the series of consistent-length polymers. Interestingly, we observed a significant decrease in the gelation temperature (Tgel) with increasing azide content, likely due to an increase in the polymer hydrophobicity and the number of charged units along the polymer, an analogous observation to increasing the salt concentration of PIC solutions.40,42 Simultaneously, G0 consistently increases in conjunction with the increasing azide content from 235 Pa in PIC-0 (L = 145 nm), to 563 Pa in PIC-6.0 (L = 146 nm), despite the consistent polymer length. To further explore this observation, we conducted small-angle neutron scattering (SANS) experiments that indicated an increase in the polymer-bundle diameter correlating with the increase in azide content (Table 3). These results suggest that the azide presence influences the nanoscale polymer-bundle architecture by increasing the polymer-bundle diameter and the number of polymers within each bundle, consequently increasing the hydrogel bulk stiffness.16,34
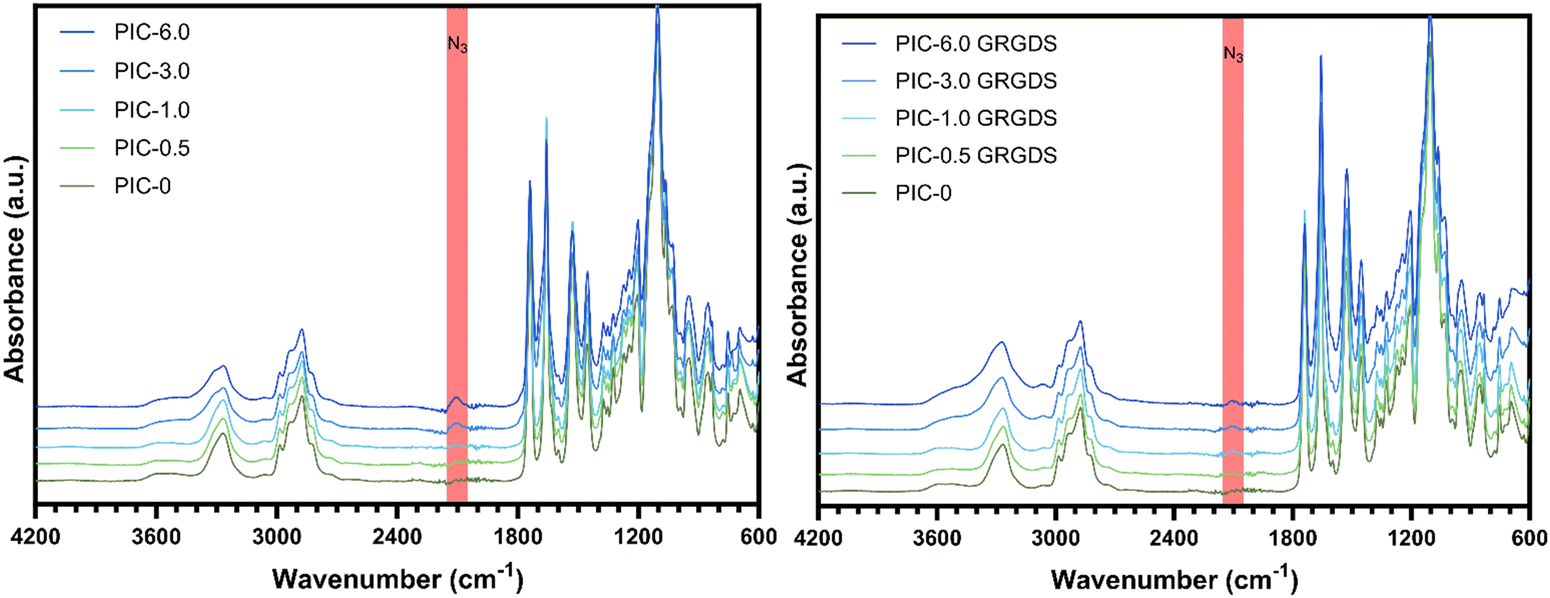 | ||
| Fig. 2 ATR-FTIR of PIC(methoxy-co-azide) polymers demonstrating an increase in the azide peak across the series of polymers with increasing azide co-monomer (left) and PIC-GRGDS demonstrating a reduction in the azide peak post-peptide addition (right). | ||
| Polymer | G 0 (Pa) | σ c (Pa) | Gelation temperature (°C) |
|---|---|---|---|
| PIC-0 | 235 | 22.0 | 21.0 |
| PIC-0.5 | 273 | 25.1 | 20.5 |
| PIC-1.0 | 288 | 27.9 | 20.5 |
| PIC-3.0 | 419 | 37.0 | 18.0 |
| PIC-6.0 | 559 | 43.6 | 16.0 |
| PIC-0.5 GRGDS | 177 | 23.7 | 20.5 |
| PIC-1.0 GRGDS | 120 | 17.7 | 21.5 |
| PIC-3.0 GRGDS | 33.6 | 6.52 | 24.0 |
| PIC-6.0 GRGDS | 19.7 | 3.81 | 27.0 |
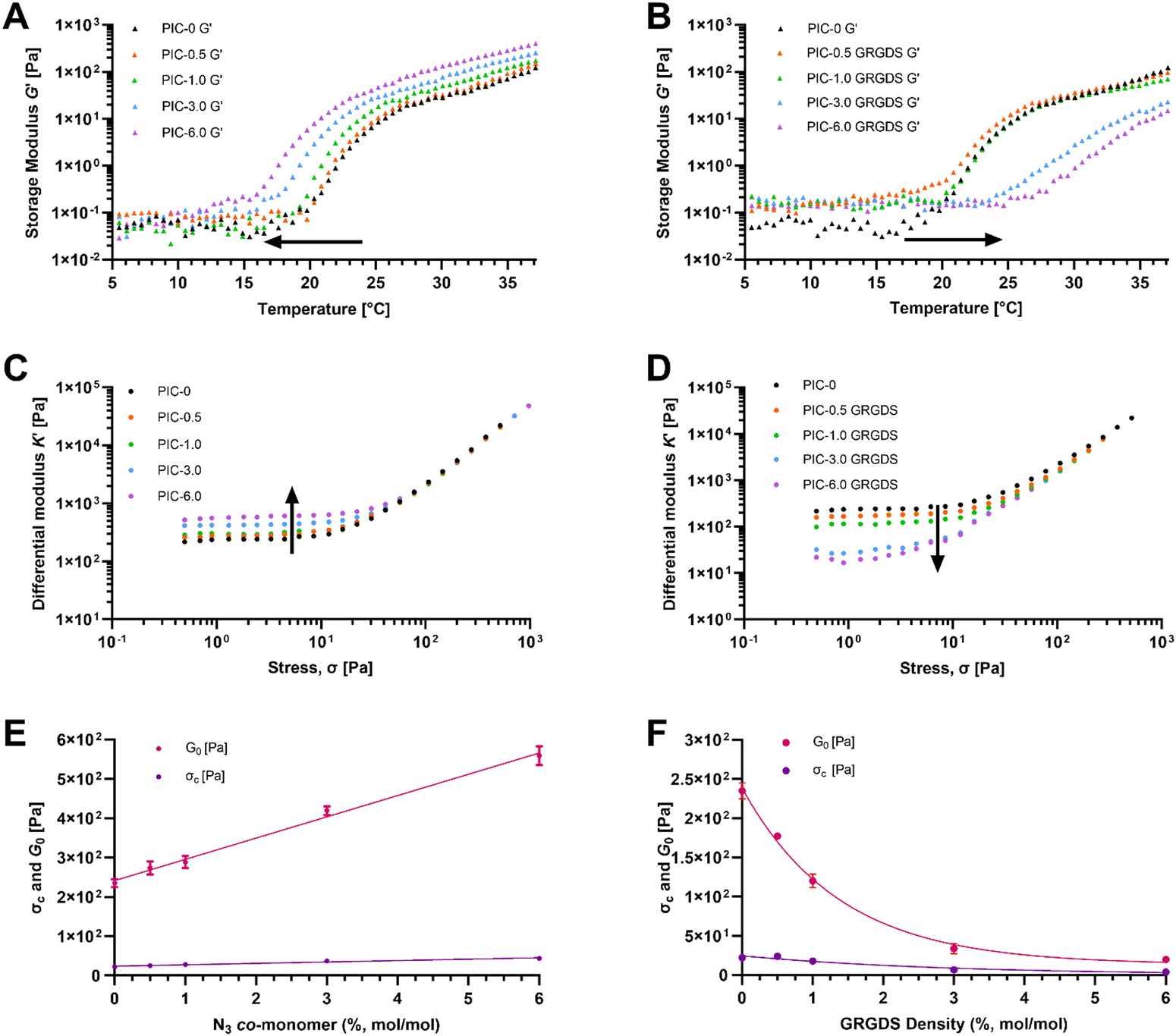 | ||
| Fig. 3 Hydrogel mechanical characterisation by rheological analysis. Temperature ramp: storage (G′) and loss (G′′) moduli as a function of temperature between 5–37 °C for (A) PIC(methoxy-co-azide) and (B) PIC-GRGDS. Arrows indicate the direction of the trend across the series of polymers. Differential modulus K′ as a function of applied stress (H2O, T = 37 °C) for gels: (C) PIC(methoxy-co-azide) and (D) PIC-GRGDS. Arrows indicate the direction of the trend across the series of polymers. (E) G0 and σc as a function of azide co-monomer percentage for PIC(methoxy-co-azide) with linear regression applied to both G0 and σc. (F) G0 and σc as a function of theoretical peptide density for PIC-GRGDS polymers with a one phase exponential decay applied to both G0 and σc. All gels c = 6.32 ± 0.225 mM in H2O. | ||
| Polymer | Bundle diameter (nm) | Pore size (nm) |
|---|---|---|
| PIC-0 | 11.0 ± 3.0 | 240 ± 11.1 |
| PIC-3.0 | 12.6 ± 3.0 | 287 ± 125 |
| PIC-6.0 | 18.2 ± 8.1 | Too large |
| PIC-1.0 GRGDS | 12.5 ± 2.6 | 259 ± 118 |
| PIC-3.0 GRGDS | 9.20 ± 1.0 | 169 ± 96.8 |
| PIC-6.0 GRGDS | 7.50 ± 1.5 | 173 ± 3.53 |
PIC-GRGDS polymer characterisation
PIC(methoxy-co-azide) polymers were modified with the GRGDS peptide-linker through a strain-promoted azide–alkyne cycloaddition (SPAAC) click reaction between the polymer-bound azide groups and the strained alkyne of the peptide-linker. Importantly, whilst SPAAC click reactions are well known for high efficacy under mild conditions, in the context of incorporating low concentrations of cell–matrix interaction peptides into viscous PIC polymers, the efficacy of the cycloaddition is not commonly measured and is often assumed to be quantitative regardless of the reaction conditions.43,44 Due to the typically low concentration of peptide, high viscosity, and room temperature LCST of the hydrogel-forming PIC polymers, direct detection and quantification of the peptide units remains challenging and a reliable method for analysing the addition of low concentrations of peptides to high viscosity polymers is needed.19,45 Whilst a reliable methodology to directly measure the peptide addition to the polymer structure remains a persistent challenge, we demonstrate here that the SPAAC reaction efficacy can be monitored by FTIR (Fig. 2) through the reduction in the azide peak (2106 cm−1) post-peptide addition. Normalising the FTIR spectrum of each PIC-GRGDS polymer to the corresponding PIC(methoxy-co-azide) spectrum reveals a reduction in the azide peak post-peptide addition which is most obvious in PIC-3.0 GRGDS and PIC-6.0 GRGDS. However, despite the reduction in the azide peak post-peptide addition, a small azide peak persists in the PIC-GRGDS polymers, particularly in the PIC-6.0 GRGDS spectrum and potentially suggests that the SPAAC reaction efficacy may have been slightly lower than 100% for each polymer despite the 1.5 molar excess of the alkyne. Although monitoring the reaction efficacy by FTIR can provide an indirect indication of the SPAAC reaction efficacy at azide percentages of 3% and above, the diamond ATR accessory typically produced high background signal within the 2100 cm−1 region that interferes with detection and quantification of the azide peak. A germanium plated ATR accessory may provide more clarity of the azide peak region of the spectra due to the lower interference within 2100 cm−1 region. As the ATR-FTIR methodology is less clear with the lower concentrations of azide, a technique that facilitates direct detection and quantification of the peptide units attached to the high molecular weight PIC polymers is still required.Recently, it has become increasingly common for PIC-based cell culture systems to present increased peptide density above the initially explored 1 molar percentage.46–48 Yet, a clearly defined relationship between the peptide density, the physicochemical properties and the nanoscale architecture of PIC-based polymer–peptide conjugate hydrogels, has yet to be identified.19 Here our analysis presents unambiguous evidence of a direct relationship between the peptide density and the hydrogel physicochemical/mechanical properties. We observe a substantial increase in Tgel with increasing peptide content (Fig. 3(B)). Importantly, we also observe Tgel to increase after peptide addition compared to the original PIC(methoxy-co-azide) gels when peptide loading is above 0.5% (mol mol−1). For example, at 1% (mol mol−1) peptide Tgel increases by 1 °C from 20.5 to 21.5 °C after peptide loading, at 3% Tgel increases by 6 °C, and at 6% Tgel increases by 12 °C. Therefore, based on the proportionality of the change in Tgel with the quantity of peptide loading, this result suggests that a change in Tgel post peptide addition at 1% and above is indicative of successful peptide conjugation. Rheological analysis of the PIC-GRGDS hydrogels with different GRGDS loading synthesised by our robust peptide-linker synthetic methodology, presented a clear inverse relationship between the amount of peptide covalently bound to the polymer and the hydrogel's G0 and σc. Importantly, Fig. 3(D) presents K′ as a function of applied stress for all PIC-GRGDS gels and demonstrates an opposing trend compared to the PIC(methoxy-co-azide) gels (Fig. 3(C)) where K′ (within the low strain regime) decreases across the PIC-GRGDS series. In addition, Fig. 3(F) presents the mean G0 and σc of the PIC-GRGDS gels as a function of peptide density and clearly identifies an inverse relationship. We also find that the higher peptide loading results in more stress-responsive hydrogels where the σc follows the same trend as a function of peptide density. A one phase exponential decay function was applied to G0 and σc data revealing a strong correlation for both mechanical parameters with R2 values of 0.99 and 0.94 respectively. Furthermore, rheology data clearly indicated that peptide loading results in a substantial decrease in the bulk stiffness compared to the former PIC(methoxy-co-azide) hydrogel (Table 2). Specifically, G0 of PIC-0.5 GRGDS reduces by 35% and PIC-1.0 GRGDS reduces by 58% post peptide addition. The reduction in G0 after peptide addition scales with peptide loading where G0 of PIC-3.0 GRGDS decreases by 92% and PIC-6.0 GRGDS decreases by 96%. This result indicates that a drop in the bulk stiffness between the base PIC(methoxy-co-azide) gel and the PIC–peptide gel is indicative of successful peptide-linker binding, and may suggest that reports showing no or very little change in the storage modulus post peptide addition could possibly have had a far lower peptide loading than anticipated.
Further analysis by SANS indicated that the polymer-bundle structure is affected by both the increasing azide content and the addition of peptides to the polymer structure. We observed an inverse relationship between the peptide density, the polymer-bundle diameter and the hydrogel pore size, further indicating that peptide addition alters the nanoscale network architecture. Our analysis shows a weaker scattering intensity of the PIC-GRGDS gels compared to the corresponding PIC(methoxy-co-azide) gels indicating thinner or less dense polymer bundles, which is in line with previous SANS analysis of PIC hydrogels.34 Specifically, the bundle thickness measured for PIC-3.0 and PIC-6.0 of 12.6 ± 3.0 and 18.2 ± 8.1 nm respectively, decreases after peptide addition to 9.2 ± 1.0 nm for PIC-3.0 GRGDS and 7.50 ± 1.5 nm for PIC-6.0 GRGDS. Furthermore, we found the bundle diameter to decrease consistently across the PIC-GRGDS series with increasing peptide density where the highest ligand density presented the thinnest bundle diameter. Consequently, the pore size measured by SANS changed in accordance with the bundle diameter where thinner bundles correlated with a smaller pore size, likely due to an increased number of fibrils. Importantly, PIC-6.0 with the thickest bundle structure of 18.2 ± 8.1 nm presented a very large pore size that could not be detected by SANS. However, PIC-6.0 would not be an ideal system for culturing cells, therefore the actual value of the pore size is less important than the observation that pore size and stiffness increase significantly with azide co-monomer concentration. The combination of the rheological and SANS data presented here highlights that peptide addition and density directly influences the hydrogel nanoscale bundle structure at physiologically relevant temperature. We hypothesise that the variation in the bundle diameter and stiffness across the gels post peptide addition is largely driven by a combination of the increase in solubility (LCST) of the PIC-GRGDS polymers and the peptide-linker sterically hindering individual polymer chains from coming together during bundle formation.28 We propose this to be crucial information for the future development of PIC-based hydrogel systems used in 3D cell culture, revealing a complex synergy between peptide density, polymer-bundle diameter and the nanoscale architecture.
Stress relaxation and peptide density
Stress relaxation of the matrix is another vital dynamic viscoelastic property present within the endogenous ECM. Viscoelastic properties such as stress relaxation can modulate biophysical cues and regulate cellular behaviour, and is a key property that synthetic biomimetic hydrogel systems must also exhibit.49 Time-dependant matrix viscoelasticity or matrix creep under loading allows for matrix deformation in response to cellular forces applied to the matrix, as the cells manipulate the matrix to generate space and reduce matrix confinement.1,15,50 ECM properties such as stiffness and stress relaxation vary between different soft tissue microenvironments and types of ECMs. The stress relaxation half-life, which is defined as the time point for the stress on the matrix to relax to half of its original value in response to constant strain, and is generally considered to vary between 10 to 1000 s over the various types of natural matrices and most soft tissue types.1,51 While the strain-stiffening behaviours of PIC-based hydrogels have been extensively studied, the stress relaxation or mechanical creep behaviours of different PIC-based hydrogels have yet to be comprehensively explored. Our analysis of the stress relaxation behaviours of the PIC polymer–peptide conjugates (Table S1, ESI†) revealed a similar trend to the mechanical properties G0 and σc as a function of peptide density where the relaxation half-life decreases exponentially across the series of hydrogels with increasing peptide density (Fig. 4). Importantly, the PIC-0 (L = 145 nm) is very slow to relax with a T1/2 of 8690 s under a constant 5% strain and a T1/2 of 1151 s under a constant 20% strain, which is approximately 9 times and 3 times slower respectively than the generally accepted T1/2 upper limit for the various types of natural ECMs. Moreover, the PIC-0.5 GRGDS and PIC-1.0 GRGDS hydrogels also displayed a stress relaxation rate higher than what is considered to be analogous to endogenous ECMs with half-lives of 3860 and 3424 s respectively with a 5% strain. Interestingly, the higher ligand density polymer–peptide conjugates PIC-3.0 GRGDS and PIC-6.0 GRGDS displayed much faster relaxation rates with a T1/2 of 649 and 621 s respectively under a 5% strain and a T1/2 of 433 and 349 s respectively under a constant 20% strain, which are both within the generally observed range for natural ECMs. Therefore, both the PIC-3.0 GRGDS and PIC-6.0 GRGDS hydrogels present stress relaxation properties that are analogous to the properties of some natural ECMs and are both likely to promote cell spreading and proliferation more so than the 1% GRGDS density.49,52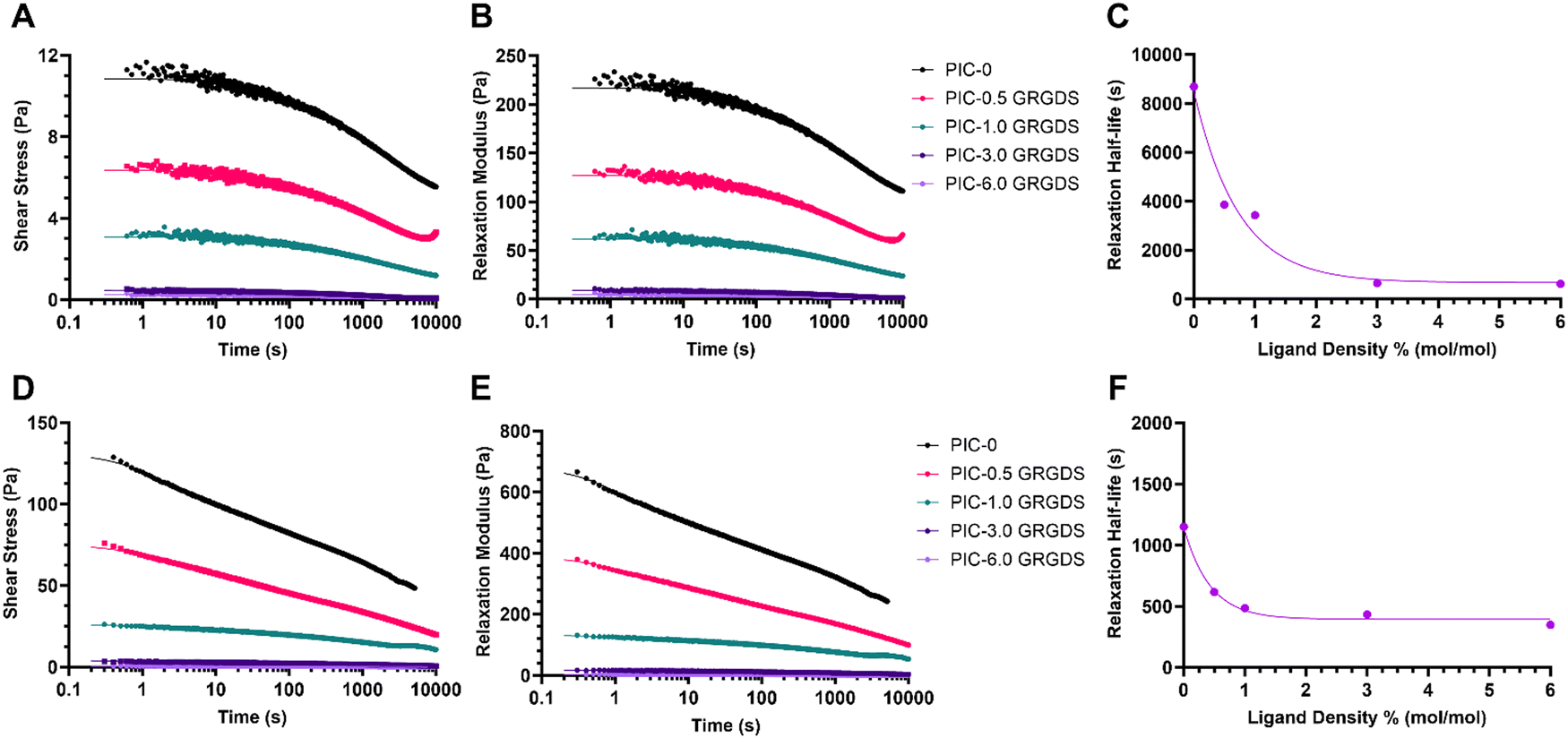 | ||
| Fig. 4 Viscoelasticity/stress relaxation measurements of PIC-based hydrogels PIC-0 and polymer–peptide conjugates (PIC GRGDS 0.5–6%). Stress relaxation data at 5% strain (A) shear stress, (B) relaxation modulus and (C) stress relaxation half-life (T1/2). Stress relaxation data at 20% strain (D) shear stress, (E) relaxation modulus and (F) stress relaxation half-life (T1/2). (A), (B), (D), (E) A two-phase exponential decay function was applied to each hydrogel data set. (C) and (F) Relaxation half-life as a function of theoretical GRGDS ligand density for with a one phase exponential decay function. | ||
Cell responses within high ligand density PIC are analogous to a natural fibrin
Several human primary cells have been 3D cultured in polyisocyanide matrices where high viability and tuneable functional outcomes have been observed. These observations include fibroblast differentiation, stem cell fate determination, endothelial angiogenesis and organoid organisation etc.18,20,53 Yet, the direct comparison of cellular responses within synthetic 3D PIC-based matrices and natural matrices has not been systematically presented previously. We chose human dermal primary fibroblasts, a type of cell that plays a crucial role in cutaneous wound healing, to investigate their responses towards the PIC matrices with increasing linear peptide densities as established above, and a natural fibrin matrix. Since the synthetic peptide sequence GRGDS mimics the RGD sequences in native fibrin matrix for cell attachment in the early wound healing stages, we 3D cultured fibroblasts in the PIC-GRGDS matrices at different concentrations (1.5–4.0 mg mL−1) and ligand densities (0–6%). Cells were mixed in the PIC-GRGDS polymer solution below the LCST before the mixture was switched to 37 °C to embed the cells within the PIC hydrogel. The subsequent responsive changes including spreading morphology, nuclear shape, and cytoskeleton alignment were monitored out to 7 days post cell embedment with cells also cultured in a purified human fibrin matrix for comparison.In a 3D context, cell spreading and the following membrane stretching-induced activation of mechanotransduction pathways are the prerequisites of cell behaviours and functions including cell proliferation and differentiation.54,55 Cell spreading kinetics and the accompanied enhanced cell volume adaptation have been found to regulate cell functions and are closely related to the structural and mechanical properties of the 3D matrix surrounding the cell.56,57 Here, we examined cell spreading kinetics from an early time point (∼12 hours) after cell embedment by monitoring cell morphological changes and comparing the differential responses observed between the synthetic PIC-GRGDS gels and the natural fibrin matrix. In comparison to some previous reports presenting 3D cultured fibroblasts, our fibroblasts in the natural fibrin displayed rapid cell spreading at ∼12 hours (Fig. S26, ESI†) while the spreading of cells in the PIC-GRGDS was dependent on the GRGDS density and matrix concentration. Cells in the PIC-GRGDS matrices with higher ligand densities and lower concentrations have a more elongated spreading morphology quantified with a lower cell body circularity, higher aspect ratio and larger single plane projected cell area. When the GRGDS ligand concentration is as high as 6% at a hydrogel concentration of 2 mg mL−1 resulting in a theoretical ligand concentration of 374 μM within the matrix, fibroblast cells present a similar spreading morphology to the cells cultured in the natural fibrin matrix at ∼12 hours. These results suggest that the higher ligand density PIC-GRGDS hydrogel composition (6%, 2 mg mL−1, 374 μM) promotes fibroblast cell spreading and facilitates cell spreading within the same timeframe as the natural fibrin matrix.
We observed the longer-term cellular responses within the PIC-GRGDS matrices with different ligand densities and concentrations along with the fibrin matrix at 7 days post cell embedment including, cell proliferation, spreading morphology, F-actin cytoskeleton alignment and nuclear shape by fluorescent immunostaining. As shown in Fig. 5(a), cells in PIC-1.0 N3 did not spread after 7 days, while a higher ligand density and a lower concentration facilitated cell spreading in the PIC hydrogels, a similar trend to the results at the earliest timepoint of ∼12 hours. Importantly, a more widespread cell morphology is accompanied by a higher F-actin expression. Thus, the fibroblasts cultured in the PIC-GRGDS with higher ligand density and a matrix concentration of 2 mg mL−1 presented a much higher total F-actin expression intensity and F-actin intensity per cell (Fig. 5(c) and (d)) in comparison to the other PIC matrices. Interestingly, the fibroblast cellular response at day 7 within the PIC-6.0 GRGDS (6%, 2 mg mL−1, 374 μM) and the PIC-3.0 GRGDS (3%, 4 mg mL−1, 374 μM) is different despite the theoretically consistent ligand concentration within the matrix, with lower cell viability and lower overall F–actin intensity within the PIC-3.0 GRGDS at 4 mg mL−1. While the mechanical properties may differ between the 3% at 4 mg mL−1 and 6% at 2 mg mL−1 hydrogels, the pore size and the overall space occupied by the hydrogel network also likely differs between the two hydrogels as the bundle dimensions are likely to remain constant irrespective of polymer concentration.16,27 Thus, these results potentially indicate that within these synthetic PIC hydrogels, fibroblast cell spreading and proliferation is not purely dependant on the ligand density alone but is influenced by the combination of high ligand density, high strain stiffening responsiveness (low σc), high viscoelasticity (fast stress relaxation), and low stiffness. The likely difference in pore size and possible difference in cell confinement between the 3% at 4 mg mL−1 and 6% at 2 mg mL−1 hydrogels is also possibly a contributing factor to the observed difference in cellular behaviours between the two systems with theoretically consistent ligand concentration. Moreover, compared to the fibroblast cells cultured in the fibrin matrix, fibroblasts cultured in the PIC-6.0 GRGDS (6%, 2 mg mL−1, 374 μM) matrix presented a comparable F-actin expression intensity as cells in the fibrin matrix after 7 days of culture. Meanwhile, we also compared cell proliferation in the different matrices by counting the number of cells within a certain volume. In the PIC-GRGDS matrices we found that both the 3% and 6% ligand densities at a matrix concentration of 2 mg mL−1 had a much higher cell number compared to the other PIC matrices, and that these two higher ligand density matrices were comparable to the natural fibrin matrix in terms of cell number at 7 days. Taken together, it is clear that GRGDS ligand density within the synthetic PIC-GRGDS hydrogels strongly influences fibroblast behaviours. This work provides strong evidence that the fibroblast cell response in the PIC-6.0 GRGDS matrix with the highest GRGDS ligand density (6%, 2 mg mL−1, 374 μM) along the polymer chain is analogous to the cell response in the natural fibrin matrix as it facilitates fibroblast early spreading, F-actin cytoskeleton alignment, proliferation and nuclear shape elongation. It should be noted that from this data it appears small changes in stiffness and ligand density can have significant effects on fibroblast cell structure and function, highlighting the importance of controlling the matrix synthesis. Furthermore, these results support the potential of synthetic PIC-based hydrogels in recapitulating and replacing a native ECM for a wide range of applications within 3D cell culture and biophysical/mechanobiology studies, tissue engineering, wound healing, and mechanotherapy.
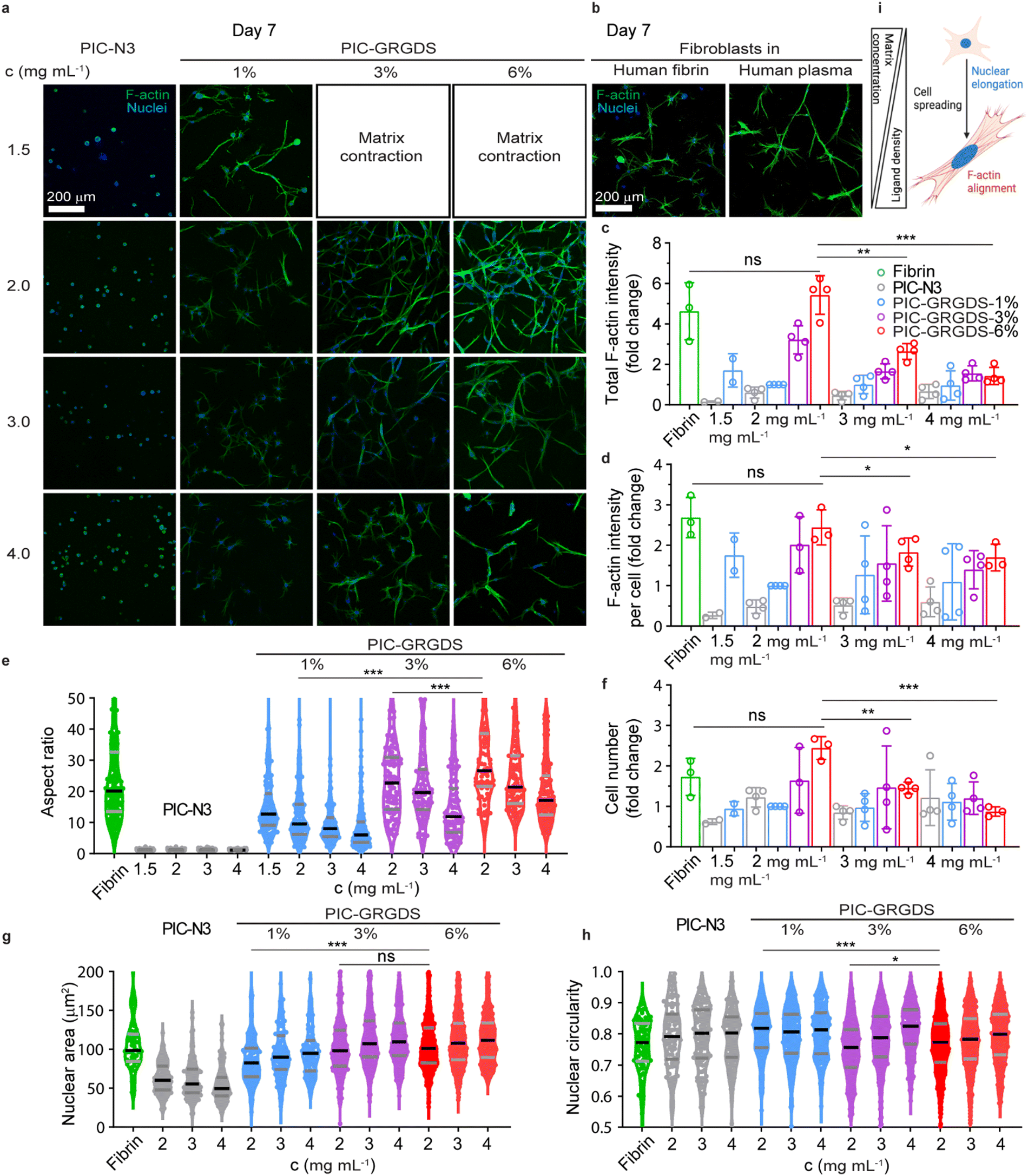 | ||
| Fig. 5 Fibroblast spreading, morphological changes cytoskeleton alignment, and nuclear changes in PIC-GRGDS matrices with different ligand densities compared with a natural fibrin matrix after 7 days of growth. F-actin (green) and nuclei (blue) immunostaining of fibroblasts from different PIC-GRGDS matrices (a) and human fibrin or plasma fibrin (b) imaged using CLSM. PIC-GRGDS concentrations at 2 mg mL−1 1% = 64 μM, 3% = 187 μM, 6% = 374 μM GRGDS. For each image, a 775 μm × 775 μm area with 200 μm thickness (2 μm per frame) was imaged and shown as the maximum projected image. Scale bar, 200 μm. PIC-GRGDS 3 and 6% at 1.5 mg mL−1 no images were obtained because of matrix contraction. Expression of F-actin in fibroblasts in different matrices quantified as total F-actin intensity (c) and F-actin intensity per cell (d) and displayed as relative fold changes. Quantification of fibroblast cell aspect ratio (e) and nuclear area (g), nuclear circularity (h) of individual cells in different matrices after 7 days (n ≥ 3, two-tailed Wilcoxon signed-rank test). In each violin plot, the solid black line indicates the median value, while the grey lines indicate the quartiles. The relative number of fibroblasts in each group quantified by nuclear staining was counted on day 7 and expressed as a relative percentage (f). For c, d, and f, one-way ANOVA statistical analysis. * = p < 0.05, ** = p < 0.01, *** = p < 0.001. ns = not significant. Illustration of characteristic fibroblast spreading and nuclear shape (i). Fibroblasts cultured in the PIC-GRGDS hydrogel with a lower polymer concentration and higher ligand density exhibit a more elongated cellular and nuclear morphology, higher aspect ratio, larger nuclear area, lower nuclear circularity, and enhanced F-actin cytoskeleton alignment. | ||
Conclusions
Whether the ultimate goal for biomimetic materials research is to comprehensively study mechanosensitive cellular behaviours in 3D and understand the environmental and mechanical factors that influence cellular processes and corresponding disease states, or to develop materials for mechanotherapy to directly manipulate cellular activity and facilitate specific outcomes.50 In either case, the multitude of complex structural and mechanical features of endogenous ECMs need to be holistically replicated and controlled in synthetic matrices to replicate certain endogenous ECM microenvironments. Integrin binding ligand density, stress stiffening responsiveness and viscoelasticity remain key properties that require modulation to generate truly biomimetic 3D cell culture hydrogels that provide the means to obtain a comprehensive understanding of dynamic and complex 3D cell behaviours and the biophysical influence of mechanically-driven cell–matrix interactions. The significance of this work from a materials chemistry perspective resides in the combination of (1) an optimised synthetic methodology for the inclusion of cell-adhesion peptides into PIC-based polymer–peptide conjugates, where the revised methodology provides enhanced control and accuracy of the peptide addition to the polymer structure compared to previously utilised methods; (2) comprehensive mechanical characterisation with stress relaxation measurements of the polymer–peptide conjugates highlighting the effect of peptide addition to the polymer structure and identifying a clear correlation between the peptide density and the hydrogel mechanical properties (G0, σc and T1/2); building upon previous knowledge of the structure–property relationship for PIC-based polymer–peptide conjugates; and (3) investigation of the nanoscale architecture of PIC-based polymer–peptide conjugates revealing the effect of peptide addition and density on the PIC polymer-bundle nanostructure where peptide addition alters the polymer-bundle network potentially by sterically inhibiting polymer-bundle formation and reducing the polymer-bundle diameter and density. We also demonstrate that human dermal primary fibroblasts cultured within the PIC-6.0 GRGDS (G0 = 20 Pa, σc = 4 Pa, T1/2 = 600 s) hydrogel present cell behaviours that are analogous to the behaviours observed when primary fibroblasts are cultured in a naturally derived fibrin hydrogel where the synthetic system facilitates fibroblast early spreading, F-actin cytoskeleton alignment, proliferation and nuclear shape elongation. Ultimately, this work further demonstrates that while hydrogel stiffness, strain responsiveness, viscoelasticity, ligand density, pore size and matrix architecture can be controlled, these parameters are synergistically intertwined for bundle forming PIC-based hydrogels. This work also demonstrates the collective complex influence of ligand density, strain stiffening responsiveness, viscoelasticity, stiffness and matrix architecture on cellular behaviours within 3D microenvironments. Controlling each of these parameters is likely key towards understanding exactly how different cell types respond to their microenvironment and how to guide cellular behaviours towards a specific desired outcome. We find that high ligand density PIC–peptide conjugate produces a microenvironment that facilitates an accurate replication of cellular behaviours observed within a natural hydrogel, and that this material composition shows considerable promise for directing the optimal design of materials for further cell studies, mechanotherapy and other biomedical applications.Author contributions
N. J. WvH. and Z. W. conceptualisation, design, methodology, investigation, formal analysis, validation, interpretation, writing – original draft, review & editing. J. L. investigation, interpretation, writing – review & editing, funding acquisition. E. P. G. methodology, investigation, formal analysis. V. G. formal analysis. A. E. R. interpretation, supervision, writing – review & editing, resources, funding acquisition.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
Research was funded by financial support from the Australian Research Council Laureate Fellowship (A. E. R.: FL160100139), Australian Nuclear Science and Technology Organisation (ANSTO) proposal grant no. 8534 and the Australian Research Council Centre of Excellence in Quantum Biotechnology (QUBIC, CE230100021). This work was performed in part at the Queensland node of the Australian National Fabrication Facility (ANFF). A company established under the National Collaborative Research Infrastructure Strategy to provide nano and microfabrication facilities for Australia's researchers. Figures were created with the assistance of BioRender.com.References
- A. Saraswathibhatla, D. Indana and O. Chaudhuri, Nat. Rev. Mol. Cell Biol., 2023, 24, 495 CrossRef CAS.
- C. Ligorio and A. Mata, Nat. Rev. Bioeng., 2023, 1, 518 CrossRef.
- M. Ravi, V. Paramesh, S. R. Kaviya, E. Anuradha and F. D. P. Solomon, J. Cell. Physiol., 2015, 230, 16 CrossRef CAS PubMed.
- C. Jensen and Y. Teng, Front. Mol. Biosci., 2020, 7, 33 CrossRef CAS.
- C. Frantz, K. M. Stewart and V. M. Weaver, J. Cell Sci., 2010, 123, 4195 CrossRef CAS PubMed.
- H. Zhou, Y. Zhu, B. Yang, Y. Huo, Y. Yin, X. Jiang and W. Ji, J. Mater. Chem. B, 2024, 12, 1748 RSC.
- B. Yang, K. Wei, C. Loebel, K. Zhang, Q. Feng, R. Li, S. H. D. Wong, X. Xu, C. Lau, X. Chen, P. Zhao, C. Yin, J. A. Burdick, Y. Wang and L. Bian, Nat. Commun., 2021, 12, 3514 CrossRef CAS PubMed.
- R. O. Hynes, Science, 2009, 326, 1216 CrossRef CAS PubMed.
- M. Tanaka, M. Nakahata, P. Linke and S. Kaufmann, Polym. J., 2020, 52, 861 CrossRef CAS.
- C. Ma, X. Duan and X. Lei, Front. Bioeng. Biotechnol., 2023, 11, 1136583 CrossRef.
- L. Rijns, M. B. Baker and P. Y. W. Dankers, J. Am. Chem. Soc., 2024, 146, 17539 CrossRef CAS.
- E. A. Aisenbrey and W. L. Murphy, Nat. Rev. Mater., 2020, 5, 539 CrossRef CAS PubMed.
- Z. Wang, J. Lauko, A. W. Kijas, E. P. Gilbert, P. Turunen, R. Yegappan, D. Zou, J. Mata and A. E. Rowan, Nat. Commun., 2023, 14, 1029 CrossRef CAS.
- R. C. Ollier, Y. Xiang, A. M. Yacovelli and M. J. Webber, Chem. Sci., 2023, 14, 4796 RSC.
- J. Lou and D. J. Mooney, Nat. Rev. Chem., 2022, 6, 726 CrossRef CAS PubMed.
- P. H. J. Kouwer, M. Koepf, V. A. A. Le Sage, M. Jaspers, A. M. van Buul, Z. H. Eksteen-Akeroyd, T. Woltinge, E. Schwartz, H. J. Kitto, R. Hoogenboom, S. J. Picken, R. J. M. Nolte, E. Mendes and A. E. Rowan, Nature, 2013, 493, 651 CrossRef CAS.
- M. Jaspers, M. Dennison, M. F. J. Mabesoone, F. C. MacKintosh, A. E. Rowan and P. H. J. Kouwer, Nat. Commun., 2014, 5, 5808 CrossRef.
- R. K. Das, V. Gocheva, R. Hammink, O. F. Zouani and A. E. Rowan, Nat. Mater., 2016, 15, 318 CrossRef CAS PubMed.
- K. Liu, S. M. Mihaila, A. Rowan, E. Oosterwijk and P. H. J. Kouwer, Biomacromolecules, 2019, 20, 826 CrossRef CAS.
- Y. Zhang, C. Tang, P. N. Span, A. E. Rowan, T. W. Aalders, J. A. Schalken, G. J. Adema, P. H. J. Kouwer, M. M. P. Zegers and M. Ansems, Adv. Sci., 2020, 7, 2001797 CrossRef CAS.
- Y. Zhang, M. M. P. Zegers, A. Nagelkerke, A. E. Rowan, P. N. Span and P. H. J. Kouwer, Adv. Sci., 2021, 8, 2003380 CrossRef CAS PubMed.
- H. Yuan, K. Liu, M. Cóndor, J. Barrasa-Fano, B. Louis, J. Vandaele, P. de Almeida, Q. Coucke, W. Chen and E. Oosterwijk, Proc. Natl. Acad. Sci. U. S. A., 2023, 120, e2216934120 CrossRef CAS PubMed.
- M. L. Ohnsorg, K. M. Mash, A. Khang, V. V. Rao, B. E. Kirkpatrick, K. Bera and K. S. Anseth, Adv. Mater., 2024, 36, 2403198 CrossRef CAS.
- J. J. L. M. Cornelissen, J. J. J. M. Donners, R. de Gelder, W. S. Graswinckel, G. A. Metselaar, A. E. Rowan, N. A. J. M. Sommerdijk and R. J. M. Nolte, Science, 2001, 293, 676 CrossRef CAS.
- E. Schwartz, M. Koepf, H. J. Kitto, R. J. M. Nolte and A. E. Rowan, Polym. Chem., 2011, 2, 33 RSC.
- M. Lago-Silva, M. Fernández-Míguez, R. Rodríguez, E. Quiñoá and F. Freire, Chem. Soc. Rev., 2024, 53, 793 RSC.
- M. Jaspers, A. C. H. Pape, I. K. Voets, A. E. Rowan, G. Portale and P. H. J. Kouwer, Biomacromolecules, 2016, 17, 2642 CrossRef CAS PubMed.
- E. P. van Dam, H. Yuan, P. H. J. Kouwer and H. J. Bakker, J. Phys. Chem. B, 2021, 125, 8219 CrossRef CAS PubMed.
- S. P. Massia and J. A. Hubbell, J. Cell Biol., 1991, 114, 1089 CrossRef CAS PubMed.
- S. Mandal, Z. H. Eksteen-Akeroyd, M. J. Jacobs, R. Hammink, M. Koepf, A. J. A. Lambeck, J. C. M. van Hest, C. J. Wilson, K. Blank, C. G. Figdor and A. E. Rowan, Chem. Sci., 2013, 4, 4168 RSC.
- M. Koepf, H. J. Kitto, E. Schwartz, P. H. J. Kouwer, R. J. M. Nolte and A. E. Rowan, Eur. Polym. J., 2013, 49, 1510 CrossRef CAS.
- A. Van Beijnen, R. Nolte, W. Drenth, A. Hezemans and P. Van de Coolwijk, Macromolecules, 1980, 13, 1386 CrossRef CAS.
- C. P. Broedersz, K. E. Kasza, L. M. Jawerth, S. Münster, D. A. Weitz and F. C. MacKintosh, Soft Matter, 2010, 6, 4120 RSC.
- M. Gavrilov, E. P. Gilbert, A. E. Rowan, J. Lauko and G. E. Yakubov, Macromol. Rapid Commun., 2020, 41, e2000304 CrossRef PubMed.
- E. P. Gilbert, J. C. Schulz and T. J. Noakes, Phys. B, 2006, 385–386, 1180 CrossRef CAS.
- K. Wood, J. P. Mata, C. J. Garvey, C.-M. Wu, W. A. Hamilton, P. Abbeywick, D. Bartlett, F. Bartsch, P. Baxter, N. Booth, W. Brown, J. Christoforidis, D. Clowes, T. d’Adam, F. Darmann, M. Deura, S. Harrison, N. Hauser, G. Horton, D. Federici, F. Franceschini, P. Hanson, E. Imamovic, P. Imperia, M. Jones, S. Kennedy, S. Kim, T. Lam, W. T. Lee, M. Lesha, D. Mannicke, T. Noakes, S. R. Olsen, J. C. Osborn, D. Penny, M. Perry, S. A. Pullen, R. A. Robinson, J. C. Schulz, N. Xionga and E. P. Gilbert, J. Appl. Crystallogr., 2018, 51, 294 CrossRef CAS.
- S. R. Kline, J. Appl. Crystallogr., 2006, 39, 895 CrossRef CAS.
- K. M. Weigandt, D. C. Pozzo and L. Porcar, Soft Matter, 2009, 5, 4321 RSC.
- J. Kumari, T. Wubs, A. P. M. van Caam, D. N. Dorst, F. A. D. T. G. Wagener and P. H. J. Kouwer, Adv. Ther., 2023, 6, 2200180 CrossRef CAS.
- H. Yuan, J. Xu, E. P. van Dam, G. Giubertoni, Y. L. A. Rezus, R. Hammink, H. J. Bakker, Y. Zhan, A. E. Rowan, C. Xing and P. H. J. Kouwer, Macromolecules, 2017, 50, 9058 CrossRef CAS.
- R. Sun, X. Jin, Y. Bao, Z. Cao, D. Gao, R. Zhang, L. Qiu, H. Yuan and C. Xing, Nano Lett., 2024, 24(10), 3257 CrossRef CAS PubMed.
- M. Jaspers, A. E. Rowan and P. H. J. Kouwer, Adv. Funct. Mater., 2015, 25, 6503 CrossRef CAS.
- R. Yegappan, J. Lauko, Z. Wang, M. F. Lavin, A. W. Kijas and A. E. Rowan, Adv. Healthcare Mater., 2022, 11, 2200574 CrossRef CAS.
- M. F. Debets, S. S. van Berkel, J. Dommerholt, A. J. Dirks, F. P. J. T. Rutjes and F. L. van Delft, Acc. Chem. Res., 2011, 44, 805 CrossRef CAS PubMed.
- K. Liu, J. Vandaele, D. Bernhagen, M. van Erp, E. Oosterwijk, P. Timmerman, S. Rocha and P. H. J. Kouwer, bioRxiv, 2022 DOI:10.1101/2022.02.01.478177.
- J. Kumari, F. A. D. T. G. Wagener and P. H. J. Kouwer, ACS Appl. Mater. Interfaces, 2022, 14, 19212 CrossRef CAS.
- Z. Zhang, W. Chen, D. M. Tiemessen, E. Oosterwijk and P. H. J. Kouwer, Adv. Healthcare Mater., 2022, 11, 2102389 CrossRef CAS.
- A. N. Gudde, M. J. J. van Velthoven, J.-P. W. R. Roovers, P. H. J. Kouwer and Z. Guler, Biomater. Adv., 2022, 141, 213104 CrossRef CAS.
- O. Chaudhuri, L. Gu, D. Klumpers, M. Darnell, S. A. Bencherif, J. C. Weaver, N. Huebsch, H.-P. Lee, E. Lippens, G. N. Duda and D. J. Mooney, Nat. Mater., 2016, 15, 326 CrossRef CAS.
- Y. Shou, X. Y. Teo, K. Z. Wu, B. Bai, A. R. K. Kumar, J. Low, Z. Le and A. Tay, Adv. Sci., 2023, 10, 2300670 CrossRef CAS.
- O. Chaudhuri, J. Cooper-White, P. A. Janmey, D. J. Mooney and V. B. Shenoy, Nature, 2020, 584, 535 CrossRef CAS PubMed.
- J. Lou, R. Stowers, S. Nam, Y. Xia and O. Chaudhuri, Biomaterials, 2018, 154, 213 CrossRef CAS PubMed.
- C. Ma, K. Liu, Q. Li, Y. Xiong, C. Xu, W. Zhang, C. Ruan, X. Li and X. Lei, Bioengineering, 2022, 9, 453 CrossRef CAS.
- H. Liu, M. Wu, Y. Jia, L. Niu, G. Huang and F. Xu, NPG Asia Mater., 2020, 12, 45 CrossRef CAS.
- K. Sethi, E. J. Cram and R. Zaidel-Bar, Semin. Cell Dev. Biol., 2017, 71, 146 CrossRef CAS PubMed.
- H.-p Lee, R. Stowers and O. Chaudhuri, Nat. Commun., 2019, 10, 529 CrossRef.
- I. L. Chin, S. E. Amos, J. H. Jeong, L. Hool, Y. Hwang and Y. S. Choi, Mater. Today Bio, 2022, 16, 100391 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ma00793j |
| ‡ N. J. WvH. and Z. W. contributed to this work equally. |
| This journal is © The Royal Society of Chemistry 2024 |