
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInfluence of redox engineering on the trade-off relationship between thermopower and electrical conductivity in lanthanum titanium based transition metal oxides†
Mohammad
El Loubani
 a,
Gene
Yang
a,
Seyed Morteza Taghavi
Kouzehkanan
b,
Tae-Sik
Oh
b,
Santosh Kiran
Balijepalli
c and
Dongkyu
Lee
*a
a,
Gene
Yang
a,
Seyed Morteza Taghavi
Kouzehkanan
b,
Tae-Sik
Oh
b,
Santosh Kiran
Balijepalli
c and
Dongkyu
Lee
*a
aDepartment of Mechanical Engineering, University of South Carolina, Columbia, SC 29208, USA. E-mail: dongkyu@cec.sc.edu
bDepartment of Chemical Engineering, Auburn University, Auburn, AL 36849, USA
cOffice of the Vice President for Research, University of South Carolina, Columbia, SC 29208, USA
First published on 18th October 2024
Abstract
Discovery of new materials plays a critical role in developing advanced high-temperature thermoelectric (TE) applications. Transition metal oxides (TMOs) are one of the attractive candidates for high-temperature TE applications due to their thermal and chemical stability. However, the trade-off relationship between thermopower (S) and electrical conductivity (σ) limits the maximum attainable power factor (PF), thereby hindering improvements in TE conversion efficiency. To overcome this trade-off relationship, the emerging approach of the redox-driven metal exsolution in TMOs shows promise in improving both S and σ. However, the effect of metal exsolution with different particle sizes and densities on S and σ is still largely unexplored. This study demonstrates an unusually large enhancement in PF through the exsolution of Ni nanoparticles in epitaxial La0.7Ca0.2Ni0.25Ti0.75O3 (LCNTO) thin films. Metal exsolution leads to a decrease in the carrier concentration while increasing the carrier mobility due to energy filtering effects. In addition, the exsolved metal particles introduce high-mobility electron carriers into the low-mobility LCNTO matrix. Consequently, the exsolution of metal particles results in a significant enhancement in S along with a substantial increase in σ, compared to the pristine film. Overall, the TE power factor of LCNTO is dramatically enhanced by up to 8 orders of magnitude owing to the presence of exsolved metal particles. This enhancement is attributed to the selective filtering of carriers caused by energy band bending at the metal–oxide interfaces and the high-mobility carriers from the exsolved Ni particles with a high Ni0 fraction. This study unequivocally demonstrates the impact of metal exsolution on oxide TE properties and provides a novel route to tailor the interconnected physical and chemical properties of oxides, leading to enhanced TE power output.
Introduction
Due to the increasing demands for alternative green energy technologies, thermoelectric (TE) energy harvesting, which involves the direct conversion of waste or solar thermal energy into electrical energy, has attracted significant attention.1–3 In TE applications, materials play a critical role, as the TE conversion efficiency is determined by the interplay of TE properties and electronic correlations of the materials. While conventional TE materials, such as chalcogenides, exhibit good TE properties for low to intermediate temperature applications, they are compromised by their cost, toxicity, natural abundance, and high-temperature stability.Transition metal oxides (TMOs) are promising alternatives due to their excellent high-temperature stability, eco-friendliness, and cost-effectiveness.4,5 Furthermore, TMOs possess unique redox flexibility, allowing for precise manipulation of their defect chemistry through atmospheric interactions, thereby facilitating the tuning of their electrical properties. Since the discovery of large thermopower (S) combined with high electrical conductivity (σ) in strongly correlated TMOs, such as NaCo2O46 and Ca3Co4O9,7 efforts have focused on controlling the interplay between S and σ to achieve high-performance TE TMOs.
One of the most widely used approaches to improve the TE properties of TMOs is substituting other cations for the A- or B-site of TMOs to alter their carrier concentration.8,9 This carrier doping method led to increased PFs (PF = S2·σ) in La-doped SrTiO3 (STO)10,11 and rare-earth doped CaMnO3.12,13 However, the trade-off relationship between S and σ limits the enhancement of PF.14S and σ of a material are both influenced by its carrier concentration but are often interrelated according to the carrier diffusion model.15 This model suggests that as carrier concentration increases, σ also increases while S decreases, resulting in a limit to the maximum achievable PF at a certain carrier concentration.
To overcome such a limit, several studies have proposed nanostructuring approaches, such as the formation of epitaxial thin films16,17 and superlattices,18,19 in order to achieve the quantum confinement effect.20,21 Theoretically, quantum-well structures can enable the enhancement of PF by decreasing the thickness of quantum-well structures when the thickness is less than a few nanometers.22–24 Using this quantum confinement effect, the formation of an STO (barrier)/SrTi0.8Nb0.2O3 (well)/STO (barrier) superlattice resulted in an increase in S while maintaining σ in the two-dimensional electron gas layers.18 Epitaxial strain induced by the lattice mismatch between the film and the substrate also enables the manipulation of electronic structures, such as band gaps and density of states, which eventually influences both S and σ.25–28 The PF of epitaxial SrTi1−xNbxO3 thin films was successfully controlled by using different substrates that introduced different strain levels into the film.16 However, using the quantum confinement effect did not yield significantly enhanced PFs, primarily due to the negligible alteration of the electronic density of states as changes in strain are often subtle.8 Furthermore, the requirement of an ultrathin film (thickness on the order of a few nanometers) to induce the lattice strain is not practical.
The formation of heterointerfaces by embedding metallic particles in a TE material is another approach to modulating both S and σ. In the case of semiconductors containing metal nanoinclusions, energy-dependent scattering of electrons occurs due to band bending at metal/semiconductor interfaces, which results in an energy filtering effect, thereby leading to an increase of S.29 For example, incorporating Ag nanoparticles into the polycrystalline CdO matrix enhanced the PF of CdO resulting from the enhanced energy-dependent electron scattering at the heterointerface.30 However, embedding metallic particles to form heterointerfaces poses a significant challenge due to the inherent difficulty in maintaining the stability and security of metal nanoparticles in an oxide matrix.31 An emerging approach to overcoming these limitations is to utilize the unique redox flexibility and defect chemistry of oxides to form heterointerfaces by exsolving metal particles in TMOs.32–34 Exsolution is a controlled phase separation method used to uniformly grow nanoparticles on a support material.35,36 Under reducing conditions at elevated temperatures, targeted metals are exsolved as nanoparticles from an oxide lattice to an oxide surface. Previous studies reported that the substituted metal cations on the B-site of stoichiometric ABO3 perovskites can be exsolved on the surface or within the film as metal particles under reducing conditions.37–39 In particular, the exsolved metal particles were socketed into the oxide surface, enhancing cohesion between the metal nanoparticles and the supporting material.40
While the exsolution in the field of oxide thermoelectrics is still in the early stages of development, recent studies showed the potential of exsolution for enhancing the PFs of STO-based oxide bulk materials. The PF of porous Sr0.95[(Ti0.8Nb0.2)0.95Ni0.05]O3 was significantly enhanced up to 100 times by the formation of exsolved Ni nanoparticles41 and the exsolution of Mo particles also enabled the enhancement in the PF (up to 1.2 times) of Sr0.90La0.10Ti0.9Mo0.1O3.42 However, despite the enhancement of PFs by metal exsolution in both cases, there is a discrepancy in the primary factor responsible for the enhanced PFs resulting from metal exsolution. One hypothesis suggests that enhanced PF results from an increase in σ without an increase in S, whereas another explanation attributes the PF enhancement to an increase in S without an increase in σ. Thus, it is crucial to understand the impact of exsolution on the PFs of TMOs to facilitate the development of high-performance TE TMOs.
In this study, we investigated the effect of heterointerfaces formed by the exsolution of metal particles on the PF of epitaxial La0.7Ca0.2 Ni0.25Ti0.75O3−δ (LCNTO) thin films. Considering that the oxidation state of Ni is lower than that of the host Ti, the negative charge of the Ni2+ is expected to be compensated by the formation of oxygen vacancies, thereby resulting in the exsolution of Ni metal particles after reduction in LCNTO.43–47 A-site deficient perovskites are more prone to exsolve Ni dopants in the B sites than stoichiometric perovskites.44 In addition, to avoid the substrate shunt current from dominating the transport in thin film samples, it is necessary to use insulating substrates.48–50 Thus, (LaAlO3)0.3(Sr2TaAlO6)0.7 (LSAT) substrates are used to grow A-site deficient LCNTO thin films. After reducing the compressively strained LCNTO films (−1.47%) with an A-site deficiency under various conditions, we achieved the largest Ni0 fraction as compared to the total Ni in the LCNTO film. The exsolution of Ni metals increases both S and σ, leading to the improved PF of LCNTO comparable to other TMOs used in conventional TE modules. Moreover, we identified the relationship between metal exsolution and TE properties in LCNTO. Our work illustrates that forming heterointerfaces by exsolving metal particles is a simple yet powerful approach to overcoming the trade-off relationship between the TE properties and synergistically enhancing S and σ of TMOs, resulting in a significant enhancement of PF.
Experimental methods
Sample preparation
Given that titanate-based perovskite oxides exhibit high structure tolerance to doping and redox environments at elevated temperatures,51,52 LCNTO pulsed laser deposition (PLD) target pellets were synthesized to exsolve nickel (Ni) metal particles. The use of a La-rich (La, Ca) perovskite lattice was aimed to promote a greater degree of exsolution, as reported by previous studies.53 Furthermore, an A-site deficient perovskite structure was employed to enhance exsolution by providing a driving force for the expulsion of B-site cations.44,53 The XRD patterns of the PLD target matched the standard structure of LaTiO3 (PDF#841089). Epitaxial LCNTO thin films with a film thickness of 250 nm were grown using an LCNTO target with a 10% A-site deficiency by pulsed laser deposition (PLD) on single crystal (001) LSAT substrates. The LSAT substrate was affixed to the PLD substrate holder using a small amount of silver paint (Leitsilber 200, Ted Pella, USA) for thermal contact. PLD was performed using a KrF excimer laser at λ = 248 nm, 5 Hz pulse rate, and 1 J cm−2 pulse energy under an oxygen partial pressure of 100 mTorr at 700 °C. After completing the deposition, the samples were cooled down to room temperature for about 1 hour under a p(O2) of 100 torr. To form exsolved metal particles, pristine LCNTO thin films were reduced in a furnace by flowing dry H2 at 900 °C for varying durations (3 h, 4.5 h, 6 h, and 12 h). To explore the influence of reduction temperature on the exsolution process, films were reduced at temperatures ranging from 800 °C to 1000 °C for 3 hours. As shown in Fig. S1 (ESI†), the color of the film changed from light gray to dark gray as the reduction temperature increased. To determine if the exsolved particles are present only on the surface or within the film, the LCNTO thin film reduced at 900 °C for 3 hours was etched using HNO3 for 10 seconds.Characterization of physical and chemical properties
Phase purity and crystallography of the films were examined using high-resolution X-ray diffraction (HRXRD) with a four-circle diffractometer in both in-plane and out-of-plane configurations. Film thickness was characterized by X-ray reflectivity (XRR) measurements. To assess the strain state of the films, XRD reciprocal space mapping (RSM) was performed around the substrates’ 103 Bragg reflections. The LCNTO films were coherently grown on the substrates, indicating that the in-plane lattice constant is maintained coherently with the substrate up to a thickness of 250 nm. Microstructural information of the thin films was obtained using a field-emission scanning electron microscope (FE-SEM) and energy-dispersive X-ray (EDX) microanalysis. Atomic force microscopy (AFM) was also performed to examine the surface morphology. The chemical composition of the sample surfaces was measured by AXIS Ultra Delay-Line Detector (DLD) X-ray photoelectron spectroscopy (XPS, Kratos Analytical). XPS measurements were performed at a pressure of ∼1 × 109 torr. High-resolution core level spectra were acquired in the constant analyzer energy mode with a pass energy of 40 eV and a step size of 0.05 eV. The curve fitting procedure was carried out using the Casa XPS software,54 and the peak approximation was carried out by a combination of Gaussian–Lorentzian functions, with subtraction of Shirley-type background. Binding energies of all peaks were corrected with reference to the C 1s peak at 284.5 eV (hydrocarbon from contamination) and are given with an accuracy of ± 0.2 eV.Evaluation of thermoelectric properties
S was measured in the temperature range of 20–400 °C using the differential method55S = ΔV/ΔT, where ΔV is the thermoelectromotive force induced by the temperature gradient (ΔT). To measure ΔV as a function of ΔT in a high-temperature setup,56 two independent T-type thermocouples with exposed ends were placed at different positions on the sample surfaces and mechanically pressed for stable contacts. A temperature gradient was applied using a heating element. These thermocouples simultaneously monitored individual temperatures at each contact point, and the voltage difference between their contacting points, enabling measurement of S. Details of the measurement and extraction of S can be found in the ESI† (Fig. S2 and S3 respectively). σ was measured by a d.c. four-probe method with the van der Pauw electrode configuration in the temperature range of 20–400 °C. To confirm the repeatability, all properties were initially measured in air from 20 °C to 400 °C and then remeasured from 400 °C to 20 °C. Since measuring Hall voltages for all the films was challenging due to high measurement temperatures and low mobility, the weighted mobility (μw)57 was calculated. μw is well-known for providing nearly identical information about charge carrier mobility as the hall mobility, making it a powerful method for investigating charge carrier transport mechanisms in oxides.58–60 After obtaining μw, the weighted carrier properties, which include carrier concentration (nw),61 and relaxation time (τw)62 were calculated. Details about the weighted mobility and carrier property calculations can be found in the ESI.†Results and discussion
Influence of metal exsolution on structural changes in strained films
XRD θ–2θ data of the pristine and reduced LCNTO films collected at room temperature clearly revealed only 00l (l is an integer) peaks from LCNTO peaks, which indicates the epitaxial growth of the LCNTO film on (001) LSAT and the preservation of epitaxy after reduction (Fig. 1(a)). Interestingly, the c lattice constant of the LCNTO film decreased slightly after reduction with a noticeable decrease observed after 12 hours of reduction (Fig. 1(b)). Clear Kiessig fringes also assured a good structural quality of the thin films. Fig. 1(c) shows XRD reciprocal space maps (RSM) around the substrates’ 103 Bragg reflections. In addition to the decrease in the c lattice constant after reduction, the coherent growth of the LCNTO film on LSAT remained unaltered. This observation indicates that the in-plane lattice constants of all the samples are coherently maintained on the LSAT substrates (compressive strain ε = −1.47%). Under reducing conditions, oxygen atoms in the crystal lattice are removed, forming oxygen vacancies that lead to an increase in the expansion of unit cell volumes in perovskite oxides.32,44,63 However, our results demonstrate a contraction in the unit cell volume of the reduced LCNTO film. In the exsolution process, oxygen vacancies play a crucial role in supplying the sites for metal ions to nucleate and grow into larger particles. As the metal ions move to fill the oxygen vacancies, the oxygen vacancies are consumed, resulting in a decrease in the number of available oxygen vacancies.64 As metal exsolution occurs, there is a Schottky defect formation resulting in B-site cation vacancies, which contract the lattice.64 Considering the exsolution of metal particles, our results suggest that the oxygen vacancies created during the reduction process were consumed in the exsolution of metal particles. A similar contraction in the c lattice constant was observed previously in La0.2Sr0.8Ti0.9Ni0.1O3−δ powder after reduction, which resulted in Ni exsolution.65 This lattice contraction was also observed in La0.2Sr0.7Ni0.1Ti0.9O3−δ thin films grown on STO substrates, demonstrating that the reduction of Ni and subsequent exsolution of Ni metal particles removed oxygen vacancies.44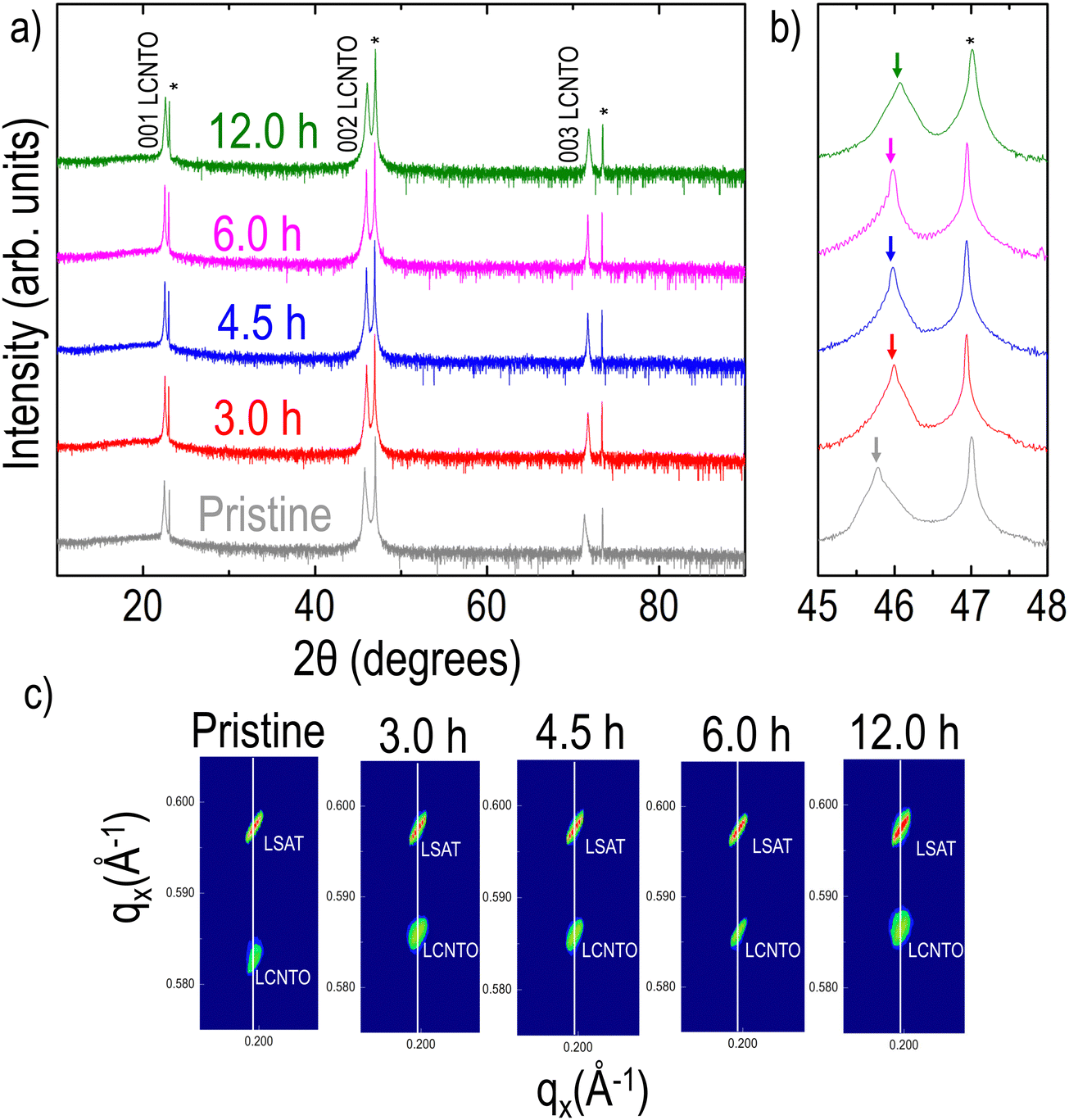 | ||
| Fig. 1 (a) XRD θ–2θ patterns of the LCNTO thin films before and after reduction at 900 °C with varying reduction times (substrate peaks are indicated with *). (b) XRD θ–2θ patterns around the 002 peaks (the 002 Bragg reflections from the LCNTO thin film and the substrates are denoted as arrows and asterisks, respectively). (c) XRD RSMs are shown around the asymmetric 103 reflection of the films and substrates. | ||
Tunable exsolution in the strained LCNTO films
To explore the formation of exsolved metal particles, FE-SEM analysis (Fig. 2(a–e)) was conducted on both pristine and reduced films. As shown in Fig. 2(a), the pristine LCNTO exhibited a smooth surface whereas exsolved metal particles were observed over the surface of the reduced LCNTO films under all reduction conditions (Fig. 2(b–e)). It is noted that the size of particles on the reduced films decreased with increasing reduction time, whereas the particle density increased (Fig. 2f). After the 3-hour reduction (Fig. 2b), the average particle size was found to be ∼80 nm. As the reduction time increased, the particle size decreased to ∼40 nm (Fig. 2c), followed by the onset of agglomeration (Fig. 2d). In the film reduced for 12 hours, particles of various shapes and sizes were observed, indicating a more heterogeneous distribution (Fig. 2e). This finding is consistent with AFM measurements (Fig. S4, ESI†), which showed a significantly larger root-mean-square (RMS) roughness value for the reduced film for 12 hours compared to the pristine sample. The exsolution process in TMOs involves a series of reaction steps, including the continuous reduction and migration of oxygen ions and metal cations, followed by nucleation and growth.52 Exsolving transition metal cations initially diffuse from the bulk to the surface and subsequently reduce to their metallic state (e.g., Mn+ to M0). The reduced metals then nucleate and grow over the treatment time, resulting in the formation of nanosized metal particles.40,66 Consequently, the growth of metal particles is governed by the diffusion of transition metal ions to the nanoparticle and their subsequent reduction to the metallic state. Based on a recent study, the exsolution kinetics are controlled by the reduction process that is limited by the rate of surface oxygen exchange reactions rather than that of bulk oxygen diffusion in the case of oxide thin films.67 Considering the exsolution kinetics in oxide thin films, we hypothesized that the observed trend in particle size under varying reduction conditions can be attributed to the slow kinetics of surface oxygen exchange reactions. It is known that oxygen vacancies at the surface lower the surface energy compared to flat surfaces, resulting in preferential sites for nucleation,52 where further metal cluster growth and reduction take place.68 For example, the 3-hour reduction yields relatively fewer metal cations (Mn+) that are reduced to their metallic state. With fewer reduced cations available, fewer nucleation sites form, leading to larger particles due to the coalescence of growing nuclei, resulting in low-density larger particles. In the 4.5-hour reduction process, more metal cations are gradually reduced, but the reduction process limits the overall number of reduced cations available for nucleation and growth. A prolonged period can lead to more nucleation, but the growth of particles is limited by the reduced cation availability, resulting in smaller particles with a higher density. With longer reduction times, the metal supply becomes exhausted with a large number of nucleation sites, which in turn results in the preferential migration of the particles for growth. In the case of the 12-hour reduction, a significantly increased particle density with various particle shapes, such as the nanorod-type, and nanoparticle cluster-types was observed on the surface. Considering that thin films are more susceptible to decomposition compared to their bulk counterpart, this phenomenon stems from the combination of particle agglomeration and decomposition of the film under harsh post-annealing conditions. The effect became more pronounced with increasing reduction temperature (Fig. S5a, ESI†), where a distinct Ni metal XRD peak was observed (Fig. S5b, ESI†). A similar observation was also previously reported for La0.8Ce0.1Ni0.4Ti0.6O3,69 which showed Ni metal XRD peaks after reduction at 1000 °C for 10 hours. Additionally, the film post-annealed in air at 1200 °C for 12 hours exhibited cracks and agglomerated residue (Fig. S6, ESI†). This suggests that under harsh post-annealing conditions, the deteriorated film interacts with the exsolved metal particles, leading to the formation of agglomerated particles. Several references also reported the formation of oxide nanowires under harsh reduction conditions.40,68,70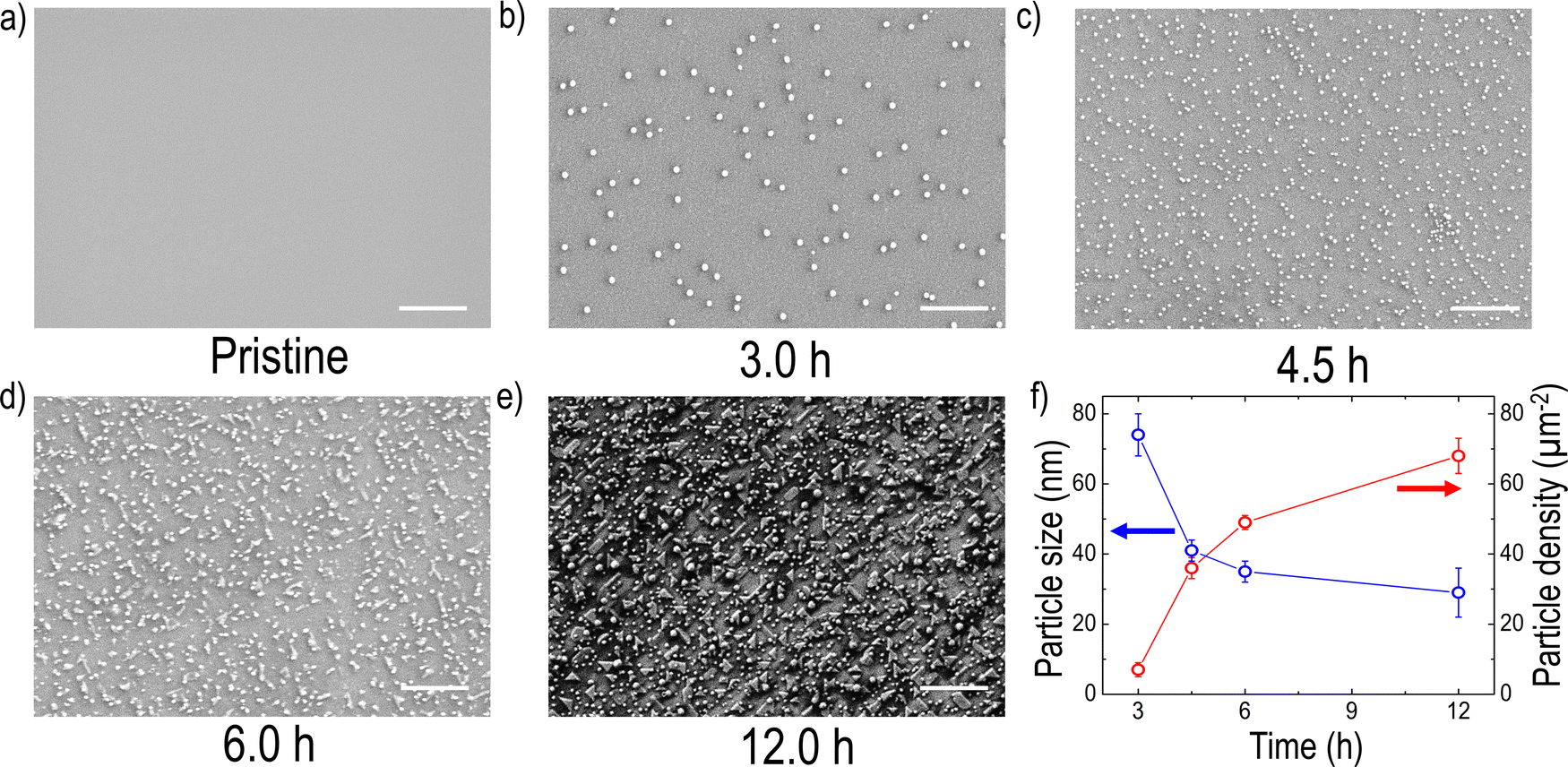 | ||
| Fig. 2 SEM images of the surface of LCNTO thin films: (a) before exsolution and (b) after exsolution at 900 °C for 3.0 h, (c) 4.5 h, (d) 6.0 h, and (e) 12.0 h. (f) The relationship between particle size and density of the exsolved samples, obtained by processing the SEM images using ImageJ software. The scale bar indicates 1 μm. | ||
As discussed earlier, Ni is theoretically exsolved due to its lower oxidation state compared to the host Ti. EDX mapping performed on the sample after the 3-hour reduction (Fig. S7, ESI†) confirmed that the exsolved particles are Ni metal. To analyze the phase of the particles of the reduced samples more accurately, elemental analysis was conducted using XPS on samples both before and after reduction (Fig. 3). For quantitative analysis, each spectrum was deconvoluted using Lorentzian–Gaussian curves after background subtraction. Before the exsolution process, the Ni 3p spectra could be assigned to the Ni2+ 3p3/2 (Eb ≈ 68.0 eV) or the Ni2+ 3p1/2 peak (Eb ≈ 69.1 eV), indicating that the Ni ions were in the LCNTO lattice.71 As the exsolution proceeded, the intensities of the deconvoluted Ni0 3p3/2 (Eb ≈ 66.3 eV) and Ni0 3p1/2 (Eb ≈ 67.2 eV) peaks increased at the expense of the Ni2+ 3p3/2 and Ni2+ 3p1/2 peaks, which directly shows the emergence of metallic Ni nanoparticles from the reduction of Ni2+.44,52,71 The fraction of the metallic Ni0 3p peak decreased from 90.77% to 35.5% as the exsolution time increased, indicating that the 3-hour reduction produced the largest Ni metal particles in LCNTO. Despite the reducing treatments and Ni migration, the Ti oxidation states did not show a substantial change. These XPS results clearly demonstrate the exsolution of Ni metal particles. Furthermore, these data support our previous assumption based on analysis of the XRD lattice-volume changes.
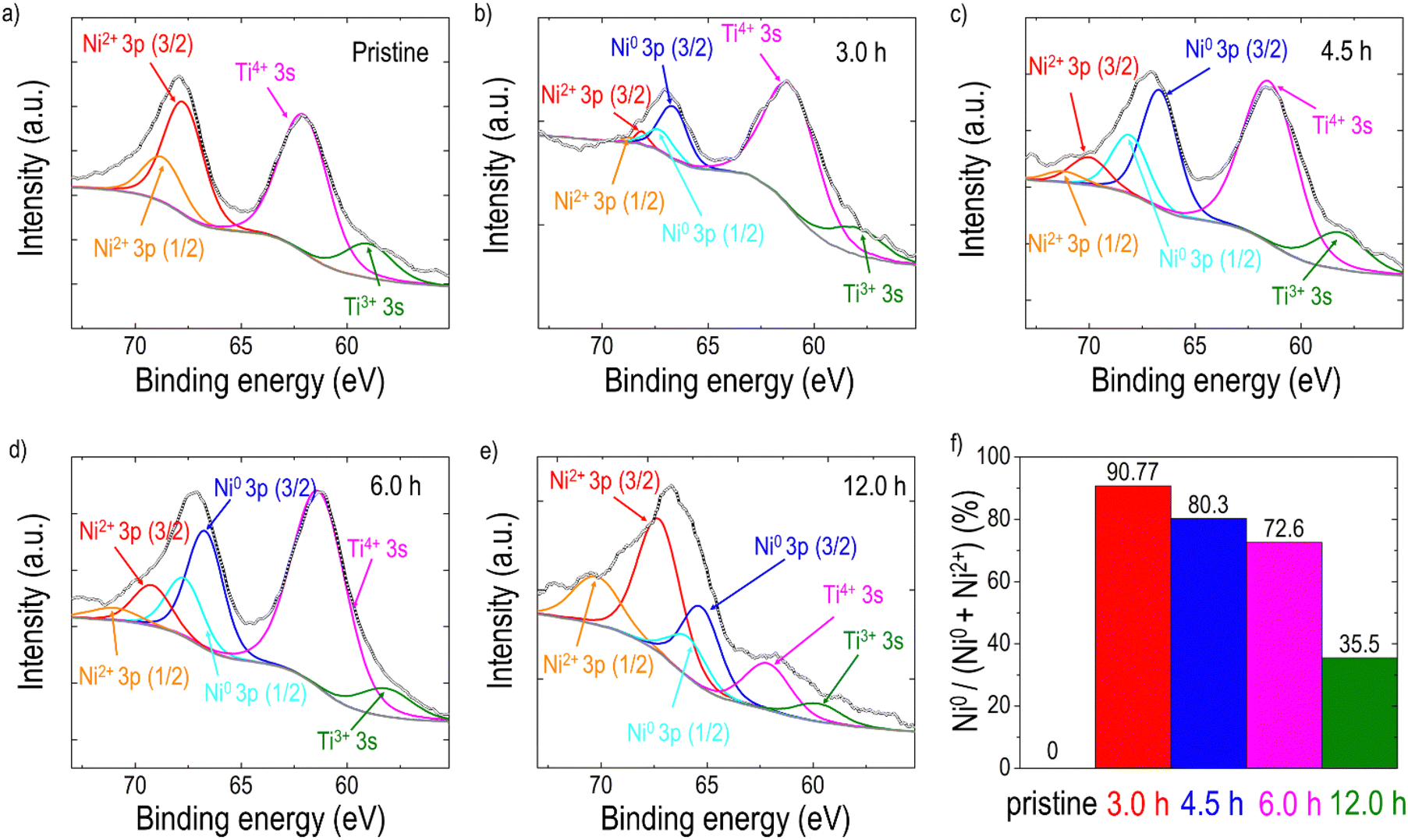 | ||
| Fig. 3 Deconvoluted XPS spectra for Ni 3p and Ti 3s of LCNTO thin films: (a) before exsolution and (b) after exsolution at 900 °C for 3.0 h, (c) 4.5 h, (d) 6.0 h, and (e) 12.0 h. (f) Variation in the Ni0 fraction of LCNTO films as a function of reduction time. | ||
Effect of metal exsolution on the TE properties of the strained LCNTO films
As mentioned earlier, the LCNTO films were grown on LSAT substrates to avoid any effects of the substrate on the measurements. As shown in Fig. S8 (ESI†), the TE properties of the LSAT substrate after reduction at 900 °C for 3 hours were comparable to those of the bare LSAT substrate, indicating that the LSAT substrate did not influence the measurements of the film's TE properties. Fig. 4(a) shows the temperature-dependence of S of the pristine and reduced LCNTO films at various reduction times measured along the in-plane direction. A negative sign of S was observed in this study, indicating that electrons are the majority carriers in LCNTO. The pristine LCNTO film exhibited very low S values (∼−21 μV K−1), which remained nearly constant across different temperatures. However, after reduction at 900 °C, the S values of the LCNTO film increased significantly by up to 8 times at elevated temperatures. No clear trend was observed between the reduction time and S. In addition, the change in S was not substantial as a function of reduction time. Fig. 4(b) presents the temperature-dependent σ of the LCNTO thin films before and after reduction measured along the in-plane direction. Interestingly, σ was significantly influenced by the reduction time. Notably, the largest enhancement of σ was achieved with the 3-hour reduction, which yielded the largest Ni0 fraction as shown in Fig. 3. According to the aforementioned carrier diffusion model,15 an increase in carrier concentration results in a higher σ but a lower S. Consequently, this trade-off between S and σ typically leads to local maxima of the PF at a certain carrier concentration. However, our results demonstrate a different trend: a synergistic enhancement in both S and σ.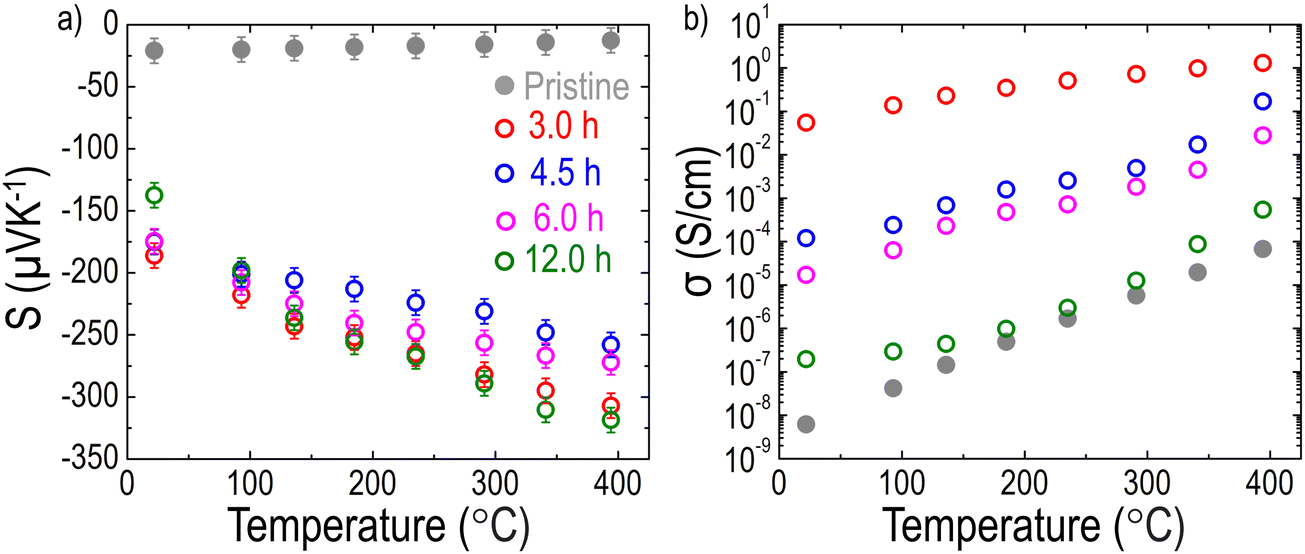 | ||
| Fig. 4 Temperature dependencies of (a) S and (b) σ of the LCNTO thin films before and after reduction at 900 °C with varying reduction times measured from 20 to 400 °C. | ||
Fig. 5 shows the temperature dependence of carrier transport properties for the LCNTO films before and after reduction. In the pristine film, the temperature dependence of nw exhibits typical semiconductor behavior,72 characterized by an increase in nw with increasing temperature (Fig. 5a). This behavior aligns well with the results obtained from σ measurements (Fig. 4b). In contrast, the reduction process led to a significant decrease in nw with minimal variation observed regardless of the reduction time. This change in nw does not correlate with the observed trend in σ after reduction. Instead, these results suggest that the presence of exsolved Ni particles enhanced S by reducing nw. Interestingly, the temperature dependence of μw showed a strong dependence on the reduction time (Fig. 5b). Following the reduction, a substantial increase in μw was observed. Specifically, μw increased markedly after 3 hours of reduction but subsequently decreased with further extension of the reduction time. This trend is consistent with the behavior of σ, suggesting that the increased μw contributes to the enhancement of σ after reduction.
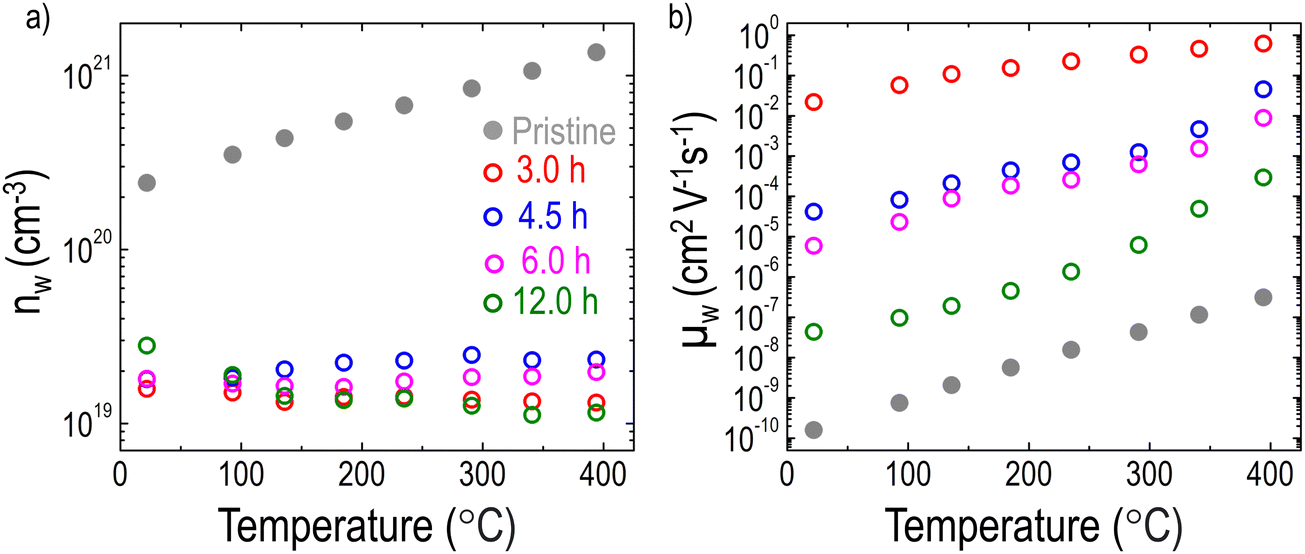 | ||
| Fig. 5 Temperature dependencies of (a) nw and (b) μw of the LCNTO thin films before and after reduction at 900 °C with varying reduction times. | ||
Proposed origin for the enhanced TE properties of the strained LCNTO films
Heterointerfaces formed by metal exsolution enable the manipulation of the band alignment between the metal and the semiconductor, creating suitable charge carrier traps or barriers.73 As a result, low-energy charge carriers are filtered out by a potential barrier existing between the semiconductor and the metal in a nanocomposite material.14,74,75 Considering that the work function of Ni (5.22 eV)76 is higher than that of the n-type semiconductor, LaTiO3 (2.99 eV),77 a Schottky barrier is likely to form at the interface between the LCNTO matrix and the exsolved Ni particles.78 Consequently, electrons with energy below ΔE = 2.23 eV are filtered out, flowing from the LCNTO to Ni. This selective transport of higher-energy charge carriers is responsible for the significant reduction of nw by metal exsolution shown in Fig. 5a. To confirm that the enhanced S is attributed to the energy filtering effect, a single-band parabolic (SPB) model is used to predict the trend of S as a function of nw.3 The Pisarenko plot at room temperature is shown in Fig. S9 (ESI†). We note that S for the reduced samples is higher than S predicted by the Pisarenko line. Zou et al.79 reported that the S of nanocomposites must lie on the Pisarenko line if there is no energy filtering effect. Hence, this strengthens the notion of an enhanced energy filtering effect.80 Conclusively, the reduced carrier density enhances S, which depends on the average excess energy of the charge carriers relative to the Fermi energy level (E–Ef).81 A previous study29 also demonstrated the enhancement of S by varying the potential height. However, due to the Pisarenko trade-off relationship,82 the reduction of nw typically results in a decrease in σ, which differs from the trend observed in σ after metal exsolution in Fig. 4b.It is known that as the density of charge carriers increases, the frequency of collisions and interactions among them also rises. Consequently, a higher nw leads to a lower μw, which in turn shortens τw.83 As shown in Fig. S10 (ESI†), the reduced LCNTO films exhibited a longer τw compared to the pristine LCNTO film. This finding is in good agreement with the observed trend in μw, indicating that the enhanced mobility in the reduced films compensates for the reduction in nw, thereby increasing σ. Furthermore, the energy filtering mechanism often does not act alone,84 but coexists with other mechanisms such as high-mobility electron injection. High-mobility electron injection involves high-mobility carriers from metal particles being injected into the low-mobility oxide matrix, thereby enhancing the overall carrier mobility.85,86 In this study, metal exsolution led to the formation of Ni metal particles in the LCNTO films. Considering the electron mobility of Ni metal (171.8 cm2 V−1 s−1)87 and NiO (0.01 cm2 V−1 s−1),88 LCNTO with a more Ni metal is expected to have a higher mobility compared to that with a more NiO. As shown in Fig. 3f, the Ni0 fraction decreased with increasing the reduction time. This finding is in good agreement with the trends of μw and σ as a function of the reduction time.
Ultimately, stemming from the unusual simultaneous increase in S and σ, the PF of LCNTO was dramatically enhanced by up to 8 orders of magnitude after reduction (Fig. 6). As discussed, metal exsolution induced both the energy filtering effect and the high-mobility electron injection effect, enabling the simultaneous increase in S and σ. A similar trend was reported in a previous study on half-Heusler Ti(Co,Fe)Sb–InSb nanocomposites,85 where the PF was enhanced by increasing S and σ via a synergistic combination of high-mobility electron injection and energy filtering effect. Despite several attempts to enhance the PFs of TMOs via energy filtering induced by metal exsolution,41,42 no successful results demonstrated the simultaneous increase of both S and σ. To achieve the energy filtering effect, the characteristic size of the nanostructures should be comparable to the electron mean free path.84,89 Hence, the nanostructure size is required to be around 20–80 nm.90 Therefore, these conditions must be carefully considered to successfully realize the energy filtering effect.
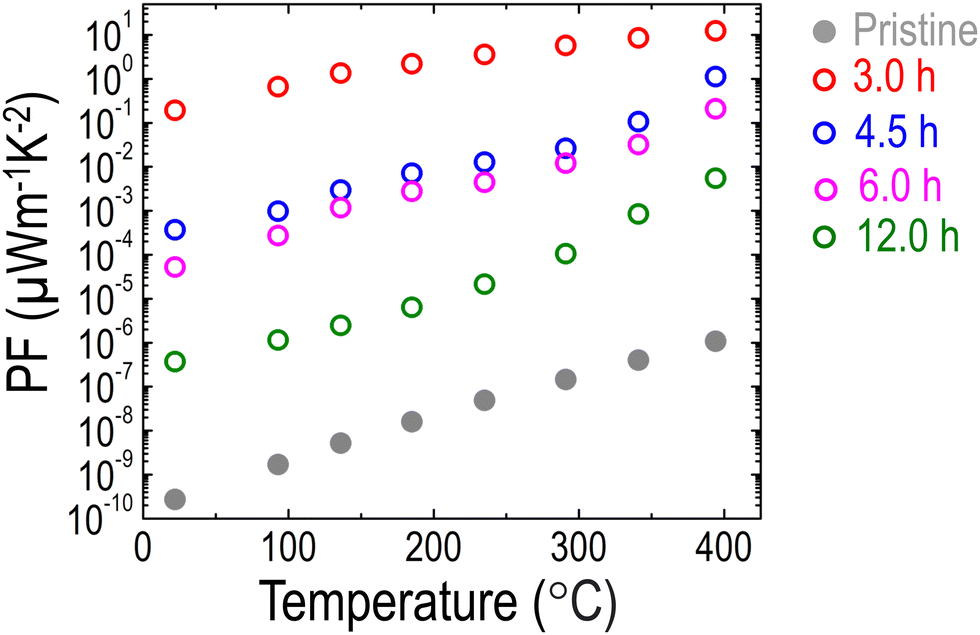 | ||
| Fig. 6 Temperature dependencies of the PF of the LCNTO thin films before and after reduction at 900 °C with varying reduction times. | ||
As shown in Fig. 2 and 3, the formation of Ni metal particles on the film surface after reduction was confirmed. Although various densities of the Ni metal particles were observed depending on the reduction time, a key question remains: are the exsolved Ni particles on the surface sufficient to account for the substantial enhancement in PF? While there is ongoing debate about whether metal particles exist solely on the surface or also within the bulk,91,92 several studies reported that exsolved particles can also be found within the bulk of the films.37,38,69,93 To investigate whether particles are present within the bulk, the LCNTO film reduced at 900 °C for 3 hours was etched in nitric acid for 10 seconds, which is sufficient to remove the nickel particles without compromising the film quality.37 After etching and cleaning the sample with distilled water, the surface of the sample was examined using SEM. Fig. 7a shows pits on the surface, indicating most of the exsolved particles were removed. Etching did not affect the crystallinity of the sample, as confirmed by the XRD data (Fig. 7b). Notably, both S and σ of the etched film were intermediate between those of the pristine and reduced samples (Fig. 7c and d). This observation confirms that the exsolved Ni metal particles within the bulk of the film also contribute to the enhancement of S and σ. In addition, this result is supported by the cross-sectional SEM image of the reduced LCNTO film, where Ni particles were observed within the bulk of the film (Fig. S11, ESI†).
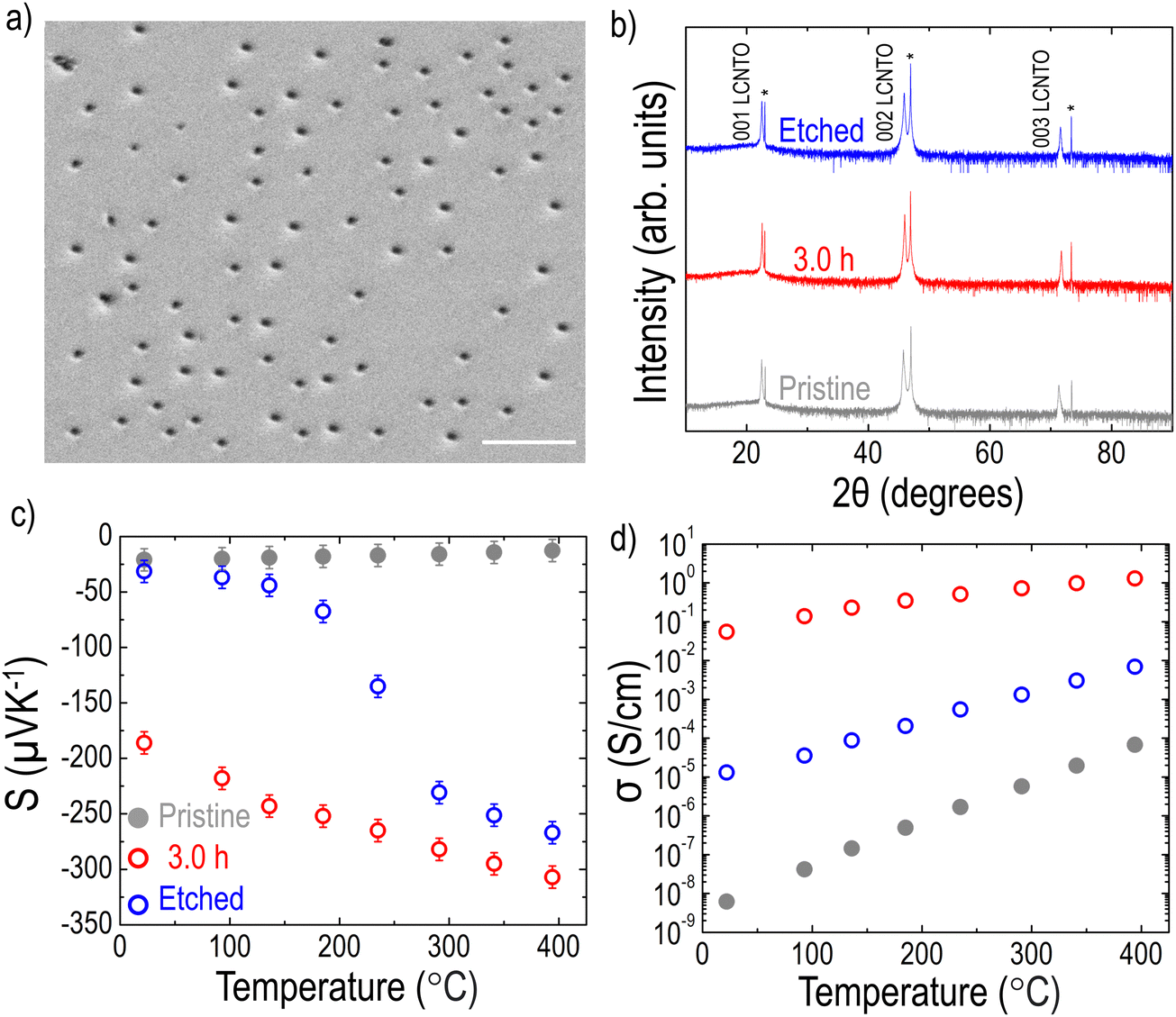 | ||
| Fig. 7 (a) SEM image of the LCNTO sample reduced at 900 °C 3.0 h after etching in nitric acid (scale bar: 1 μm). (b) XRD θ–2θ patterns of the pristine, reduced, and etched LCNTO thin films (substrate peaks are indicated with *). Temperature dependencies of (c) S and (d) σ of the pristine, reduced, and etched LCNTO thin films measured from 20 to 400 °C. | ||
Conclusions
In summary, we successfully demonstrated the effect of exsolution on the TE properties of LCNTO thin films with a 10% A-site deficiency. By systematically changing the reduction conditions, we controlled the density and size of the exsolved Ni nanoparticles. The exsolved Ni metal particles with a higher Ni0 fraction resulted in a dramatic enhancement in both S and σ, leading to an increase in PF by up to 8 orders of magnitude compared to the pristine film. This unusual simultaneous increase in S and σ was attributed to the combined effects of energy filtering, due to band bending at the interface between the exsolved Ni particles and the LCNTO, and high-mobility electron injection from the high fraction of exsolved Ni metal particles. The combined effects of energy filtering and high-mobility electron injection resulted in changes of nw and μw. Consequently, a significant decrease in σ was prevented while S was enhanced. This study highlights the potential of metal exsolution as a nanostructuring approach for tailoring the TE properties of TMOs, facilitating the development of high-performance oxide TE materials. Moreover, our findings pave the way for creating multifunctional materials that combine excellent thermoelectric properties with catalytic and electrocatalytic activities.Author contributions
Mohammad El Loubani: investigation (lead) and writing – original draft (lead). Gene Yang: investigation (supporting) and writing – original draft (supporting). Seyed Morteza Taghavi Kouzehkanan: investigation (supporting). Tae-Sik Oh: conceptualization (supporting); funding acquisition (equal); and writing – review & editing (supporting). Santosh Kiran Balijepalli: investigation (supporting). Dongkyu Lee: conceptualization (lead); funding acquisition (equal); supervision (lead); and writing – review & editing (lead).Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This research was funded by the National Science Foundation under NSF Award Number DMR-2340234 and ECCS-2110033. S. T. and T. O. acknowledge financial support from the Korea Institute of Energy Research (research and development program, C2-2466). FE-SEM and AFM measurements were conducted as part of a user project at the Center for Nanophase Materials Sciences (CNMS), which is a US Department of Energy, Office of Science User Facility at Oak Ridge National Laboratory. The XPS Research facility receives financial support from the Office of the Vice President for Research, University of South Carolina.References
- F. J. DiSalvo, Science, 1999, 285, 703–706 CrossRef.
- G. Chen, M. Dresselhaus, G. Dresselhaus, J.-P. Fleurial and T. Caillat, Int. Mater. Rev., 2003, 48, 45–66 CrossRef.
- G. J. Snyder and E. S. Toberer, Nat. Mater., 2008, 7, 105–114 CrossRef PubMed.
- J. He, Y. Liu and R. Funahashi, J. Mater. Res., 2011, 26, 1762–1772 CrossRef.
- S. Walia, S. Balendhran, H. Nili, S. Zhuiykov, G. Rosengarten, Q. H. Wang, M. Bhaskaran, S. Sriram, M. S. Strano and K. Kalantar-zadeh, Prog. Mater. Sci., 2013, 58, 1443–1489 CrossRef.
- I. Terasaki, Y. Sasago and K. Uchinokura, Phys. Rev. B: Condens. Matter Mater. Phys., 1997, 56, R12685 CrossRef.
- A. Masset, C. Michel, A. Maignan, M. Hervieu, O. Toulemonde, F. Studer, B. Raveau and J. Hejtmanek, Phys. Rev. B: Condens. Matter Mater. Phys., 2000, 62, 166 CrossRef.
- K. Koumoto, Y. Wang, R. Zhang, A. Kosuga and R. Funahashi, Annu. Rev. Mater. Res., 2010, 40, 363–394 CrossRef CAS.
- X.-L. Shi, H. Wu, Q. Liu, W. Zhou, S. Lu, Z. Shao, M. Dargusch and Z.-G. Chen, Nano Energy, 2020, 78, 105195 CrossRef.
- S. Ohta, T. Nomura, H. Ohta and K. Koumoto, J. Appl. Phys., 2005, 97, 034106 CrossRef.
- T. Okuda, K. Nakanishi, S. Miyasaka and Y. Tokura, Phys. Rev. B: Condens. Matter Mater. Phys., 2001, 63, 113104 CrossRef.
- D. Flahaut, T. Mihara, R. Funahashi, N. Nabeshima, K. Lee, H. Ohta and K. Koumoto, J. Appl. Phys., 2006, 100, 084911 CrossRef.
- M. Ohtaki, H. Koga, T. Tokunaga, K. Eguchi and H. Arai, J. Solid State Chem., 1995, 120, 105–111 CrossRef.
- T. Zhu, Y. Liu, C. Fu, J. P. Heremans, J. G. Snyder and X. Zhao, Adv. Mater., 2017, 29, 1605884 CrossRef.
- C. Wood, Rep. Prog. Phys., 1988, 51, 459 CrossRef.
- S. Bhansali, W. Khunsin, A. Chatterjee, J. Santiso, B. Abad, M. Martín-González, G. Jakob, C. S. Torres and E. Chávez-Angel, Nanoscale Adv., 2019, 1, 3647–3653 RSC.
- A. Chatterjee, Z. Lan, D. V. Christensen, F. Bauitti, A. Morata, E. Chavez-Angel, S. Sanna, I. E. Castelli, Y. Chen and A. Tarancon, Phys. Chem. Chem. Phys., 2022, 24, 3741–3748 RSC.
- H. Ohta, S. Kim, Y. Mune, T. Mizoguchi, K. Nomura, S. Ohta, T. Nomura, Y. Nakanishi, Y. Ikuhara and M. Hirano, Nat. Mater., 2007, 6, 129–134 CrossRef CAS.
- Y. Mune, H. Ohta, K. Koumoto, T. Mizoguchi and Y. Ikuhara, Appl. Phys. Lett., 2007, 91, 192105 CrossRef.
- L. D. Hicks and M. S. Dresselhaus, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 16631 CrossRef CAS.
- G. Mahan and J. Sofo, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 7436–7439 CrossRef CAS.
- L. D. Hicks and M. S. Dresselhaus, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 12727 CrossRef CAS PubMed.
- M. Dresselhaus, G. Dresselhaus, X. Sun, Z. Zhang, S. Cronin, T. Koga, J. Ying and G. Chen, Microscale Thermophys. Eng., 1999, 3, 89–100 CrossRef CAS.
- M. S. Dresselhaus, G. Chen, M. Y. Tang, R. Yang, H. Lee, D. Wang, Z. Ren, J. P. Fleurial and P. Gogna, Adv. Mater., 2007, 19, 1043–1053 CrossRef CAS.
- P. Bach, J. M. Vila-Fungueiriño, V. Leborán, E. Ferreiro-Vila, B. Rodríguez-González and F. Rivadulla, APL Mater., 2013, 1, 021101 CrossRef.
- P. Brinks, B. Kuiper, E. Breckenfeld, G. Koster, L. W. Martin, G. Rijnders and M. Huijben, Adv. Energy Mater., 2014, 4, 1301927 CrossRef.
- D. Kan and Y. Shimakawa, Appl. Phys. Lett., 2019, 115 Search PubMed.
- B. Geisler and R. Pentcheva, Phys. Rev. Mater., 2018, 2, 055403 CrossRef CAS.
- S. V. Faleev and F. Léonard, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 77, 214304 CrossRef.
- L. Gao, S. Wang, R. Liu, X. Zha, N. Sun, S. Wang, J. Wang and G. Fu, Dalton Trans., 2016, 45, 12215–12220 RSC.
- K. Kousi, C. Tang, I. S. Metcalfe and D. Neagu, Small, 2021, 17, 2006479 CrossRef PubMed.
- D. Neagu, G. Tsekouras, D. N. Miller, H. Ménard and J. T. Irvine, Nat. Chem., 2013, 5, 916–923 CrossRef PubMed.
- O. Kwon, S. Joo, S. Choi, S. Sengodan and G. Kim, J. Phys.: Energy, 2020, 2, 032001 Search PubMed.
- H. Jeong, Y. H. Kim, B.-R. Won, H. Jeon, C.-H. Park and J.-H. Myung, Chem. Mater., 2023, 35, 3745–3764 CrossRef.
- S.-K. Otto, K. Kousi, D. Neagu, L. Bekris, J. R. Janek and I. S. Metcalfe, ACS Appl. Energy Mater., 2019, 2, 7288–7298 CrossRef.
- V. Kyriakou, R. K. Sharma, D. Neagu, F. Peeters, O. De Luca, P. Rudolf, A. Pandiyan, W. Yu, S. W. Cha and S. Welzel, Small Methods, 2021, 5, 2100868 CrossRef CAS.
- D. Neagu, T.-S. Oh, D. N. Miller, H. Ménard, S. M. Bukhari, S. R. Gamble, R. J. Gorte, J. M. Vohs and J. T. Irvine, Nat. Commun., 2015, 6, 8120 CrossRef PubMed.
- K. J. Kim, H. Han, T. Defferriere, D. Yoon, S. Na, S. J. Kim, A. M. Dayaghi, J. Son, T.-S. Oh and H. M. Jang, J. Am. Chem. Soc., 2019, 141, 7509–7517 CrossRef PubMed.
- D. Neagu, J. T. Irvine, J. Wang, B. Yildiz, A. K. Opitz, J. Fleig, Y. Wang, J. Liu, L. Shen and F. Ciucci, J. Phys.: Energy, 2023, 5, 031501 Search PubMed.
- D. Neagu, V. Kyriakou, I.-L. Roiban, M. Aouine, C. Tang, A. Caravaca, K. Kousi, I. Schreur-Piet, I. S. Metcalfe and P. Vernoux, ACS Nano, 2019, 13, 12996–13005 CrossRef PubMed.
- S. Hirata, M. Ohtaki and K. Watanabe, Ceram. Int., 2020, 46, 25964–25969 CrossRef.
- A. V. Kovalevsky, K. V. Zakharchuk, M. H. Aguirre, W. Xie, S. G. Patrício, N. M. Ferreira, D. Lopes, S. A. Sergiienko, G. Constantinescu and S. M. Mikhalev, J. Mater. Chem. A, 2020, 8, 7317–7330 RSC.
- J.-h Myung, D. Neagu, D. N. Miller and J. T. Irvine, Nature, 2016, 537, 528–531 Search PubMed.
- H. Han, J. Park, S. Y. Nam, K. J. Kim, G. M. Choi, S. S. Parkin, H. M. Jang and J. T. Irvine, Nat. Commun., 2019, 10, 1471 CrossRef PubMed.
- Z. Li, M. Peng, Y. Zhu, Z. Hu, C.-W. Pao, Y.-C. Chang, Y. Zhang, Y. Zhao, J. Li and Y. Sun, J. Mater. Chem. A, 2022, 10, 20350–20364 RSC.
- Z. Sun, W. Fan and Y. Bai, Adv. Sci., 2022, 9, 2200250 CrossRef CAS PubMed.
- X. Yue, A. Pukhova, S. He and N. Zhang, J. Electrochem. Soc., 2021, 168, 104514 CrossRef.
- P. Yordanov, P. Wochner, S. Ibrahimkutty, C. Dietl, F. Wrobel, R. Felici, G. Gregori, J. Maier, B. Keimer and H.-U. Habermeier, Appl. Phys. Lett., 2017, 110 Search PubMed.
- D. Li, P. Chen, Z. Huang, H. Liu and S. Chen, Int. J. Solids Struct., 2023, 267, 112160 Search PubMed.
- A. Riss, M. Stöger, M. Parzer, F. Garmroudi, N. Reumann, B. Hinterleitner, T. Mori and E. Bauer, Phys. Rev. Appl., 2023, 19, 054024 Search PubMed.
- H. Khalid, A. u Haq, B. Alessi, J. Wu, C. D. Savaniu, K. Kousi, I. S. Metcalfe, S. C. Parker, J. T. Irvine and P. Maguire, Adv. Energy Mater., 2022, 12, 2201131 Search PubMed.
- Y. Gao, D. Chen, M. Saccoccio, Z. Lu and F. Ciucci, Nano Energy, 2016, 27, 499–508 CrossRef.
- Y. H. Kim, Y. Kang, S. Jo, H. Jeong, D. Neagu and J.-H. Myung, Chem. Eng. J., 2022, 441, 136025 CrossRef.
- N. Fairley, V. Fernandez, M. Richard-Plouet, C. Guillot-Deudon, J. Walton, E. Smith, D. Flahaut, M. Greiner, M. Biesinger and S. Tougaard, Appl. Surf. Sci. Adv., 2021, 5, 100112 CrossRef.
- D. Han, R. Moalla, I. Fina, V. M. Giordano, M. d’Esperonnat, C. Botella, G. Grenet, R. Debord, S. Pailhès and G. Saint-Girons, ACS Appl. Electron. Mater., 2021, 3, 3461–3471 CrossRef.
- D. Lee, J. Zhou, G. Chen and Y. Shao-Horn, Adv. Electron. Mater., 2019, 5, 1800624 CrossRef.
- G. J. Snyder, A. H. Snyder, M. Wood, R. Gurunathan, B. H. Snyder and C. Niu, Adv. Mater., 2020, 32, 2001537 CrossRef CAS PubMed.
- T. Katase and T. Kamiya, J. Ceram. Soc. Jpn., 2023, 131, 343–349 CrossRef.
- M. J. KP, A. Rengarjan, N. Mani and H. Santhana Krishnan, J. Phys. Chem. C, 2023, 127, 11707–11711 CrossRef.
- Y. Zheng, H. Chen, Z. Zhou, Y. Yang, M. Zou, W. Zhang, B. Wei, J. Cai, J. L. Lan and D. Yi, Adv. Funct. Mater., 2023, 2301815 CrossRef.
- T. Katase, X. He, T. Tadano, J. M. Tomczak, T. Onozato, K. Ide, B. Feng, T. Tohei, H. Hiramatsu and H. Ohta, Adv. Sci., 2021, 8, 2102097 CrossRef PubMed.
- M. Massetti, F. Jiao, A. J. Ferguson, D. Zhao, K. Wijeratne, A. Würger, J. L. Blackburn, X. Crispin and S. Fabiano, Chem. Rev., 2021, 121, 12465–12547 CrossRef PubMed.
- K. Kim, B. Koo, Y.-R. Jo, S. Lee, J. K. Kim, B.-J. Kim, W. Jung and J. W. Han, Energy Environ. Sci., 2020, 13, 3404–3411 RSC.
- J. Wang, J. Yang, A. K. Opitz, W. Bowman, R. Bliem, G. Dimitrakopoulos, A. Nenning, I. Waluyo, A. Hunt and J.-J. Gallet, Chem. Mater., 2021, 33, 5021–5034 CrossRef.
- B. H. Park and G. M. Choi, Solid State Ionics, 2014, 262, 345–348 CrossRef.
- D. C. Ghosh and R. Biswas, Int. J. Mol. Sci., 2002, 3, 87–113 CrossRef.
- J. Wang, D. Kalaev, J. Yang, I. Waluyo, A. Hunt, J. T. Sadowski, H. L. Tuller and B. Yildiz, J. Am. Chem. Soc., 2023, 145, 1714–1727 CrossRef PubMed.
- E. Calì, M. P. Thomas, R. Vasudevan, J. Wu, O. Gavalda-Diaz, K. Marquardt, E. Saiz, D. Neagu, R. R. Unocic and S. C. Parker, Nat. Commun., 2023, 14, 1754 CrossRef PubMed.
- K. Kousi, D. Neagu, L. Bekris, E. I. Papaioannou and I. S. Metcalfe, Angew. Chem., 2020, 132, 2531–2540 CrossRef.
- R. Thalinger, M. Gocyla, M. Heggen, B. Klötzer and S. Penner, J. Phys. Chem. C, 2015, 119, 22050–22056 CrossRef PubMed.
- N. McIntyre and M. Cook, Anal. Chem., 1975, 47, 2208–2213 CrossRef.
- Y. P. Varshni, Physica, 1967, 34, 149–154 CrossRef.
- S. H. Zaferani, R. Ghomashchi and D. Vashaee, ACS Appl. Energy Mater., 2021, 4, 3573–3583 CrossRef.
- A. M. Dehkordi, M. Zebarjadi, J. He and T. M. Tritt, Mater. Sci. Eng., R, 2015, 97, 1–22 CrossRef.
- J. Wang, X. Ning, Z. Gao, J. Wang, X. San and S. Wang, Acta Mater., 2022, 227, 117692 CrossRef CAS.
- B. Baker, B. Johnson and G. Maire, Surf. Sci., 1971, 24, 572–586 CrossRef CAS.
- R. Jacobs, J. Booske and D. Morgan, Adv. Funct. Mater., 2016, 26, 5471–5482 Search PubMed.
- S. M. Sze, Y. Li and K. K. Ng, Physics of semiconductor devices, John Wiley & Sons, 2021 Search PubMed.
- T. Zou, X. Qin, Y. Zhang, X. Li, Z. Zeng, D. Li, J. Zhang, H. Xin, W. Xie and A. Weidenkaff, Sci. Rep., 2015, 5, 17803 CrossRef PubMed.
- B. Madavali, H.-S. Kim, K.-H. Lee and S.-J. Hong, Intermetallics, 2017, 82, 68–75 CrossRef.
- C. Soumya and P. Pradyumnan, J. Mater. Sci., 2023, 58, 16771–16784 CrossRef.
- Y. Pei, A. LaLonde, S. Iwanaga and G. J. Snyder, Energy Environ. Sci., 2011, 4, 2085–2089 RSC.
- M. E. Levinshtein, S. L. Rumyantsev and M. S. Shur, Properties of Advanced Semiconductor Materials: GaN, AIN, InN, BN, SiC, SiGe, John Wiley & Sons, 2001 Search PubMed.
- C. Gayner and Y. Amouyal, Adv. Funct. Mater., 2020, 30, 1901789 CrossRef.
- W. Xie, Y. Yan, S. Zhu, M. Zhou, S. Populoh, K. Gałązka, S. J. Poon, A. Weidenkaff, J. He and X. Tang, Acta Mater., 2013, 61, 2087–2094 CrossRef.
- C. Jia, B. Zhu, C. Pang, C. Yuan, P. Xu, B. Xu, J. Bai, L. Tao, F. Xue and G. Tang, Mater. Today Phys., 2023, 33, 101039 CrossRef.
- G. Adesakin, T. Akande, O. Olubosede, O. Edema, A. Akinbolusere, E. Aliyu, M. Adekoya and A. Fatigun, J. Adv. Res. Dyn. Control Syst., 2019, 11(6), 1986–1995 Search PubMed.
- A. Liu, Y. Meng, H. Zhu, Y.-Y. Noh, G. Liu and F. Shan, ACS Appl. Mater. Interfaces, 2017, 10, 25841–25849 CrossRef PubMed.
- T.-H. Liu, J. Zhou, M. Li, Z. Ding, Q. Song, B. Liao, L. Fu and G. Chen, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 879–884 CrossRef CAS.
- G. Korotcenkov, V. Brinzari and M.-H. Ham, Crystals, 2018, 8, 14 CrossRef.
- A. J. Carrillo, A. López-García, B. Delgado-Galicia and J. M. Serra, Chem. Commun., 2024, 60, 7987 RSC.
- Y. H. Kim, H. Jeong, B.-R. Won, H. Jeon, C.-H. Park, D. Park, Y. Kim, S. Lee and J.-H. Myung, Nano-Micro Lett., 2024, 16, 33 CrossRef PubMed.
- T.-S. Oh, E. K. Rahani, D. Neagu, J. T. Irvine, V. B. Shenoy, R. J. Gorte and J. M. Vohs, J. Phys. Chem. Lett., 2015, 6, 5106–5110 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ma00829d |
| This journal is © The Royal Society of Chemistry 2024 |