
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Perylene diimide architecture-based electromechanical sensors: a systematic experimental and theoretical framework for the comparative analysis and study of the transduction mechanism†
Aditya
Tiwari
a,
Vivek
Adepu
b,
Rikitha S.
Fernandes
c,
Nilanjan
Dey
*c,
Parikshit
Sahatiya
 *bd and
Sayan
Kanungo
*bd
*bd and
Sayan
Kanungo
*bd
aDepartment of Electronics and Communication Engineering, Amrita School of Engineering, Amrita Vishwa Vidyapeetham, Bengaluru 560035, India
bDepartment of Electrical and Electronics Engineering, Birla Institute of Technology and Science Pilani, Hyderabad Campus, Hyderabad 500078, India. E-mail: parikshit@hyderabad.bits-pilani.ac.in; sayan.kanungo@hyderabad.bits-pilani.ac.in
cDepartment of Chemistry, Birla Institute of Technology and Science Pilani, Hyderabad Campus, Hyderabad 500078, India. E-mail: nilanjan@hyderabad.bits-pilani.ac.in
dMaterials Center for Sustainable Energy & Environment, Birla Institute of Technology and Science Pilani, Hyderabad Campus, Hyderabad 500078, India
First published on 1st November 2024
Abstract
This work presents a detailed comparative study on the effects of functional groups on engineered PDI (perylene diimide) compounds for pressure and breath sensing applications using experimental findings and density functional theory (DFT)-based theoretical calculations. The results demonstrate that the deposition of N-substituted perylene-3,4-dicarboxylic acid imide derivatives (PDI-1, PDI-2, PDI-3, and PDI-4) with different functional groups (3-aminopentane, 2,5-di-tert-butylaniline, 1-phenylethylamine, etc.) on the paper substrate forms a moderately conducting percolating molecular network with enhanced pressure and breath-sensing performances. The determined pressure sensitivity value for PDI-1 was 0.315 kPa−1, for PDI-2 was 1.266 kPa−1, for PDI-3 was 0.749 kPa−1, and for PDI-4 was 2.120 kPa−1. Among all the fabricated PDI-based pressure sensors, PDI-4 displayed maximum sensitivity owing to the inherent asymmetric nature of the compound with two different terminal substituents. The sensor displayed a steady response of up to ∼8000–10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cycles, confirming the mechanical sturdiness of fabricated PDI-based pressure sensors. The DFT-based theoretical analysis offers detailed insight into the transduction mechanism of pressure and breath sensing for different PDI molecules, wherein it can be surmised that both the structural configuration and electronic properties of PDI-4 (PDI-1) are suitable (undesirable) to ensure a large increase in intermolecular tunneling components and, thereby, in the overall conductivity of the percolating network under applied pressure. Hence, PDI-4 (PDI-1) is the most (least) favorable PDI molecule for pressure sensing applications. In contrast, a moderate response can be expected in PDI-2 and PDI-3 during pressure sensing as two competing factors influence the overall efficacy of transduction in these cases.
000 cycles, confirming the mechanical sturdiness of fabricated PDI-based pressure sensors. The DFT-based theoretical analysis offers detailed insight into the transduction mechanism of pressure and breath sensing for different PDI molecules, wherein it can be surmised that both the structural configuration and electronic properties of PDI-4 (PDI-1) are suitable (undesirable) to ensure a large increase in intermolecular tunneling components and, thereby, in the overall conductivity of the percolating network under applied pressure. Hence, PDI-4 (PDI-1) is the most (least) favorable PDI molecule for pressure sensing applications. In contrast, a moderate response can be expected in PDI-2 and PDI-3 during pressure sensing as two competing factors influence the overall efficacy of transduction in these cases.
Introduction
Electromechanical sensors have steadily gained viability for commercial use in the last few decades owing to their superior sensitivity and mechanical durability. They are attractive for the fields of smart electronic devices, Internet of Things (IoT), wearable applications, and robotics.1–3 Generally, electromechanical sensors are of resistive, capacitive, and optical types. Some of the most desirable attributes of electromechanical sensors includes mechanical durability, conductivity, and functional fillers with flexible soft materials. Materials with piezoelectric resistance or other changing electrical signals can be used to precisely measure deformation in electromechanical sensors.4–6 While many factors are essential to consider when constructing these sensors, the most important ones are their sensitivity, scalability, linearity, stretchability, and durability.7 Unfortunately, optimizing all these characteristics in a single physical sensor is difficult. Studies have demonstrated that there is a strong correlation between sensitivity, stretchability, and linearity of electrochemical sensors and their electromechanical sensing. For example, highly flexible sensors cannot be highly linear. The mechanisms that affect the sensitivity of a piezoresistive electromechanical sensor are its innate resistivity, disconnection mechanisms, crack formation in conductive coatings, geometrical modifications, and tunneling resistance variations.8,9 Tunneling effects are thought to impact the conductivity of the composite close to filler percolation. A minor variation in strain can significantly impact the tunneling resistance, increasing the sensitivity. Inorganic materials, such as graphene, TMDs, and MXenes, are mostly utilized for electromechanical sensors.10–15 Furthermore, sensors made of biodegradable, eco-friendly, self-healing, and organic materials with improved electromechanical properties can ensure long-term usage, leading to cost-effective medical diagnostics and beneficial clinical applications.16,17 Hence, there is a broad scope for exploring electromechanical sensors made of organic materials, and there is an immediate need to explore them.In this context, a small organic molecule-based pressure sensor design has recently drawn significant attention of the flexible and wearable electronics community. Usually, the synthesis of small molecular probes from low-cost precursor materials are easy, controlled, and cost-effective.18–21 Simultaneously, the significant structural diversity and chemically tunable electronic/optical properties of these tiny molecular probes are also considered highly desirable attributes for pressure-sensing applications.22,23 Consequently, the substantial flexibility towards the molecular engineering of probe molecules ensures low-cost and controlled synthesis of tailor-made probe molecules optimized for efficient electromechanical transduction during pressure sensing.
Based on these studies, in this study, a durable perylene diimide (PDI) organic material was utilized as the active material.24 In 1913, Kardos discovered that perylene diimide (PDI), an n-type organic semiconductor.25 Initially, it was utilized as a textile dye exhibiting high performance as a pigment that appeared in violet, red, and near-black shades.26 PDIs possess superior photostability and thermal steadiness under normal atmospheric conditions,16 making them feasible for self-assembly handling and diverse electronic device applications.27 Well-shaped supramolecular nanostructures were prepared using numerous PDI molecules. These developments have also inspired the field of self-assembly of various other n-type organic semiconductor molecules.28 The subsistence of electron-extracting imide splits in PDIs allows them to be minimally reduced electrochemically/chemically, modeling radical anions; however, they remain steady to oxidation.29 Moreover, the resistance to oxidation capability of PDIs makes them act as durable organic materials of n-type.30 PDIs have been found as potential entrants for formulating sensors and organic optoelectronic materials and devices owing to their intriguing properties, such as simplistic functionalization of chemical structures and exceptional light-absorption capabilities in visible region. However, PDI scaffold physical/chemical properties demonstrate high sensitivity towards functional group engineering, leading to diverse electronic and electrochemical properties in different PDI derivatives. Therefore, a systematic approach for investigating, designing, optimizing, and screening different PDI derivatives is necessary to harvest the complete potential of these probe molecules for flexible and wearable pressure sensing applications. In this context, the density functional theory (DFT)-based first principle investigation plays a pivotal role in exploring the relative properties of different probe molecules and developing detailed physical insight into the transduction mechanism associated with particular sensing applications.21,22,31 The physical insight developed through such a theoretical study complements the experimental findings and can guide the experimental exploration focusing on small molecular probe design and screening. However, to the best of our knowledge, no such systematic comparative analysis has been reported in the literature.
Consequently, in this work, we employed a DFT-based detailed theoretical analysis to study the electronic properties of the N-substituted perylene-3,4-dicarboxylic acid imide derivatives with different functional groups (3-aminopentane, 2,5-di-tert-butylaniline, 1-phenylethylamine, etc.) and subsequently supported the mechanism for pressure/breath sensing in validation with the experimental studies. The determined sensitivity value for PDI-1 was 0.315 kPa−1, PDI-2 was 1.266 kPa−1, PDI-3 was 0.749 kPa−1, and PDI-4 was 2.120 kPa−1. Among all the PDI-based pressure sensors fabricated, PDI-4 displayed maximum sensitivity owing to the inherent asymmetric nature of the compound with two different terminal substituents. The sensor displayed a steady response of up to ∼8000–10000 cycles, confirming the mechanical sturdiness of all fabricated PDI-based pressure sensors. From the theoretical study, it can be summarized that both the structural configuration and electronic properties of PDI-4 (PDI-1) are suitable to ensure a large increase in intermolecular tunneling components and, thereby, in the overall conductivity of the percolating network under applied pressure. Hence, PDI-4 (PDI-1) is the most (least) favorable PDI molecule for pressure sensing applications. In contrast, a moderate response can be expected in PDI-2 and PDI-3 during pressure sensing; in these cases, two factors have competing influences on the overall efficacy of transduction. In addition, we examined the impact of functional group modifications on the electronic properties and conductivity of the percolating network under pressure. The structural configuration of PDI-4 boosts intermolecular tunneling, enhancing conductivity, while PDI-1 shows limited tunneling and lower conductivity. Functionalizing PDI-4 improves electron transfer and conductivity, particularly under stress, making it suitable for pressure and breath sensing. Oxygen-containing groups, for instance, enhance adsorption and response speed but slow recovery. This highlights the importance of functional group engineering in balancing response and recovery for optimal sensor performance.
Results and discussion
Synthesis and characterization
The N-substituted perylene-3,4-dicarboxylic acid imide derivatives (PDI-1, PDI-2, PDI-3, and PDI-4) with different functional groups (3-aminopentane, 2,5-di-tert-butylaniline, 1-phenylethylamine, etc.) were synthesized. The detailed synthetic procedure and characterization data are described in the ESI.† In all cases, the pure compounds were obtained after silica gel column chromatography purification. The purified compounds were thoroughly characterized by 1H/13C-NMR, FT-IR, and mass spectroscopy. Among these, only compound PDI-4 was asymmetric with two different terminal substituents, as shown in Fig. 1(a).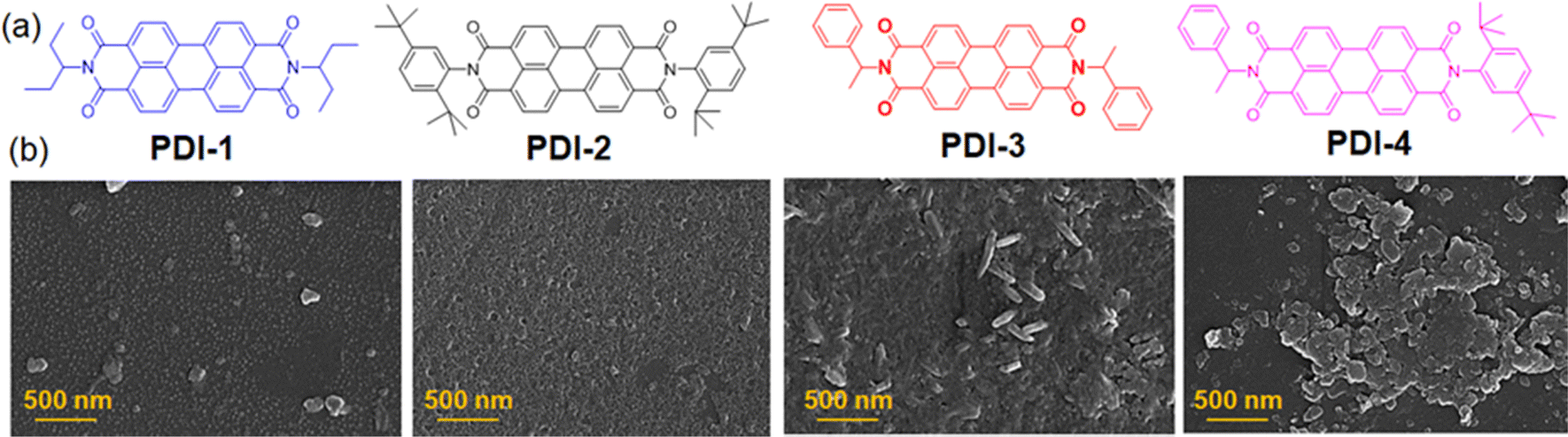 | ||
| Fig. 1 (a) Structures of N-substituted PDI derivatives involved in the present study (PDI-1, PDI-2, PDI-3 and PDI-4). (b) FESEM images of the PDI derivatives. | ||
Investigation of spectral properties
The UV-vis spectra of all four compounds (PDI-1, PDI-2, PDI-3, and PDI-4) in the THF medium showed the typical perylene characteristic absorption peaks. Three absorption maxima are observed in the 455, 487, and 522 nm regions, which could be assigned to the 0 → 2, 0 → 1, and 0 → 0 transitions, respectively, according to the literature reports, as depicted in Fig. 2(a).32,33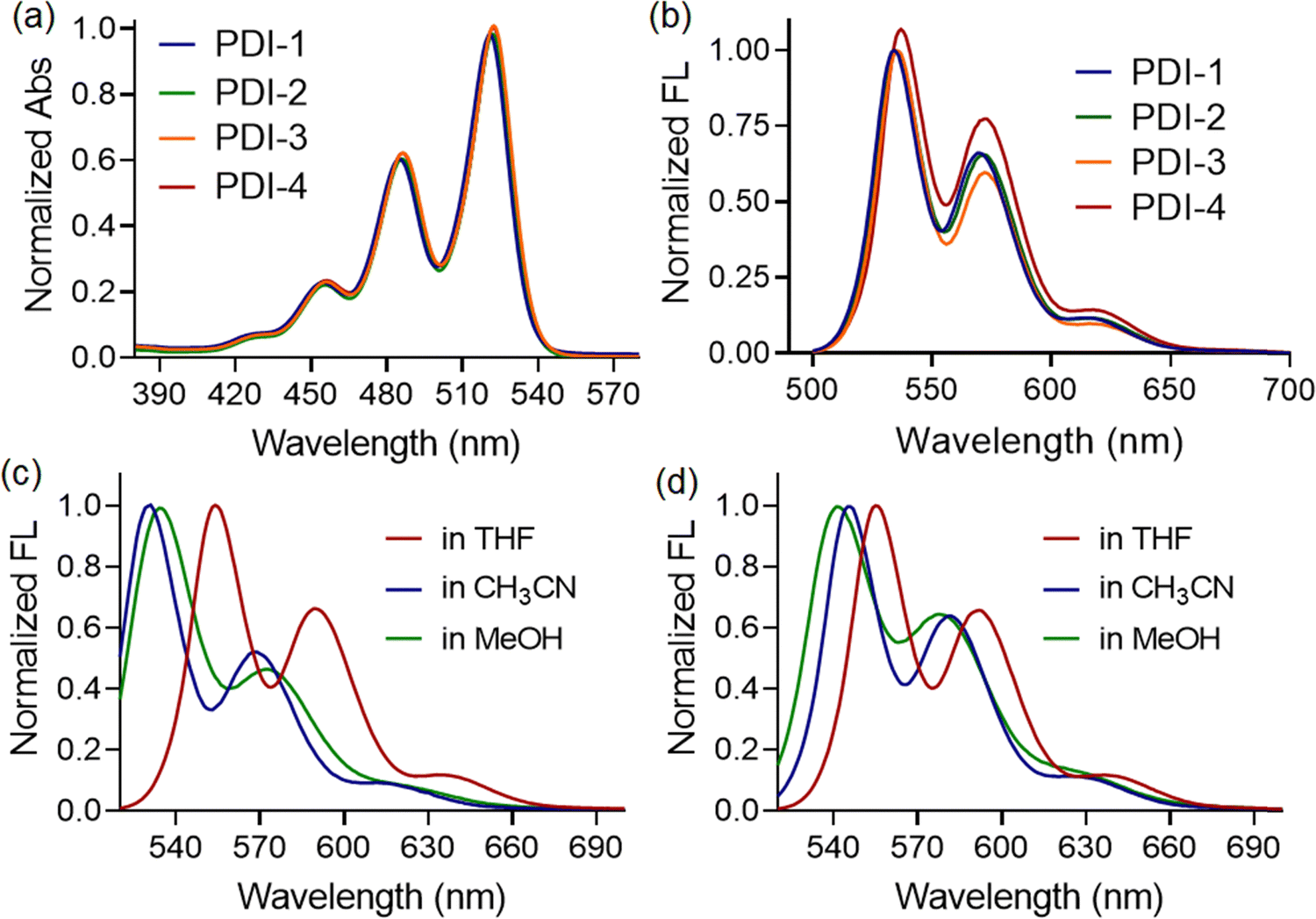 | ||
| Fig. 2 (a) Normalized absorption spectra of N-substituted PDI derivatives (10 μM) in THF medium. (b) Normalized fluorescence spectra of N-substituted PDI derivatives (10 μM, λex = 450 nm) in THF medium. (c) Normalized fluorescence spectra of PDI-1 (10 μM, λex = 450 nm) in different organic media. (d) Normalized fluorescence spectra of PDI-2 (10 μM, λex = 450 nm) in different organic media. | ||
No significant difference in spectral shape or peak positions was observed when the PDI compounds with different terminal substituents were investigated. These features indicate the negligible effects of the substituents and that the perylene diimide moiety dominates the visible spectrum. Similarly, the fluorescence spectra of compounds in the THF medium showed the presence of three emission bands centered at 554, 592, and 640 nm, respectively (λex = 450 nm) in the THF medium, as depicted in Fig. 2(b). Here, substituents also exhibited negligible influence on the spectral shape and emission maxima. However, blue shifts in emission maxima were witnessed when we recorded the fluorescence spectra of compounds in relatively polar solvents, such as acetonitrile or methanol. The observation was the same for both PDI-1 (as shown in Fig. 2(c)) and PDI-2 (as shown in Fig. 2(d)) compounds, irrespective of the functional groups present at the terminal positions. Such kind of blueshifts in emission maxima, along with intensity quenching, indicated the formation of H-type aggregates in polar solvents. The formation of π-stacked aggregated structures was observed in methanol (polar protic solvents) and acetonitrile medium (polar aprotic solvents), as evidenced by a very similar shift in emission maxima. The enhancement of the 0–1 vibronic peak compared to the 0–0 vibronic peak is a characteristic of an H-type aggregate.34 A weak hypsochromic shift in the absorption and emission spectra with increasing solvent polarity (negative solvatochromism) was observed, demonstrating that polar solvents stabilize the ground state of the molecules relative to the excited state. This indicates that the ground state of PDI molecules is more polar than its excited state.35 This conclusion is further strengthened by the fact that the above distinctive changes in the UV/vis spectra are accompanied by the strong quenching of the fluorescence emission signal, indicating a characteristic feature of H-type aggregates.36
However, the UV-vis spectra of PDI-1, PDI-2, PDI-3, and PDI-4 in MeOH and CH3CN showed characteristic absorption peaks at 455, 487, and 522 nm, typical of perylene derivatives (π–π* transitions). These correspond to the 0–2, 0–1, and 0–0 vibrational transitions, as reported. PDI-3 also displayed a broad band at 566–675 nm, more pronounced in polar MeOH. No significant differences in spectral profiles or peak positions were observed across the compounds, indicating that the substituents have minimal impact on the electronic absorption, with the perylene diimide core dominating the visible spectrum.
Further, we compare the fluorescence spectra of all four compounds in a polar medium. Interestingly, the emission maxima of compounds PDI-1 and PDI-4 were found to be blue-shifted (∼10 nm) than that observed for PDI-2 and PDI-3, as shown in Fig. 3(a). Along with the blueshift, the relative fluorescence intensities at different vibronic bands also changed depending on the substituents present. Thus, we can conclude that compounds with 1-phenylethylamine (PDI-4) and 3-aminopentane (PDI-1) as terminal substituents showed a higher propensity to form aggregated structures in a polar solvent, such as methanol, than those observed with PDI-2 and PDI-3. We believe that the presence of bulky substituents, such as 2,5-di-tert-butyl at terminal positions, resists approaching two PDI entities beyond a certain distance, making the situation difficult for the formation of π-stacked structures. To illustrate the nature of the aggregates, we also recorded the fluorescence excitation spectra of all the compounds in a methanol medium. The λmax values for PDI-I and PDI-3 compounds were found to be blue-shifted compared to those for PDI-2 and PDI-4, as depicted in Fig. 3(b). This again confirms that the nanoscopic aggregates formed in this case are H-type (face-to-face) in nature. In addition, the intensity ratio of the vibronic absorption transitions A0–0 and A0–1 serves as a spectral marker for estimating the degree of aggregation, providing insight into the extent of excitonic coupling. The reduced vibronic peak ratio (A0–0/A0–1) observed for PDI-2 and PDI-4 suggests the presence of relatively weak H-type coupling between the two PDI units. In contrast, PDI-I and PDI-3 demonstrate more pronounced H-aggregation, as evidenced by an increase in the A0–0/A0–1 peak ratio and a corresponding blue shift in the absorption peaks.37
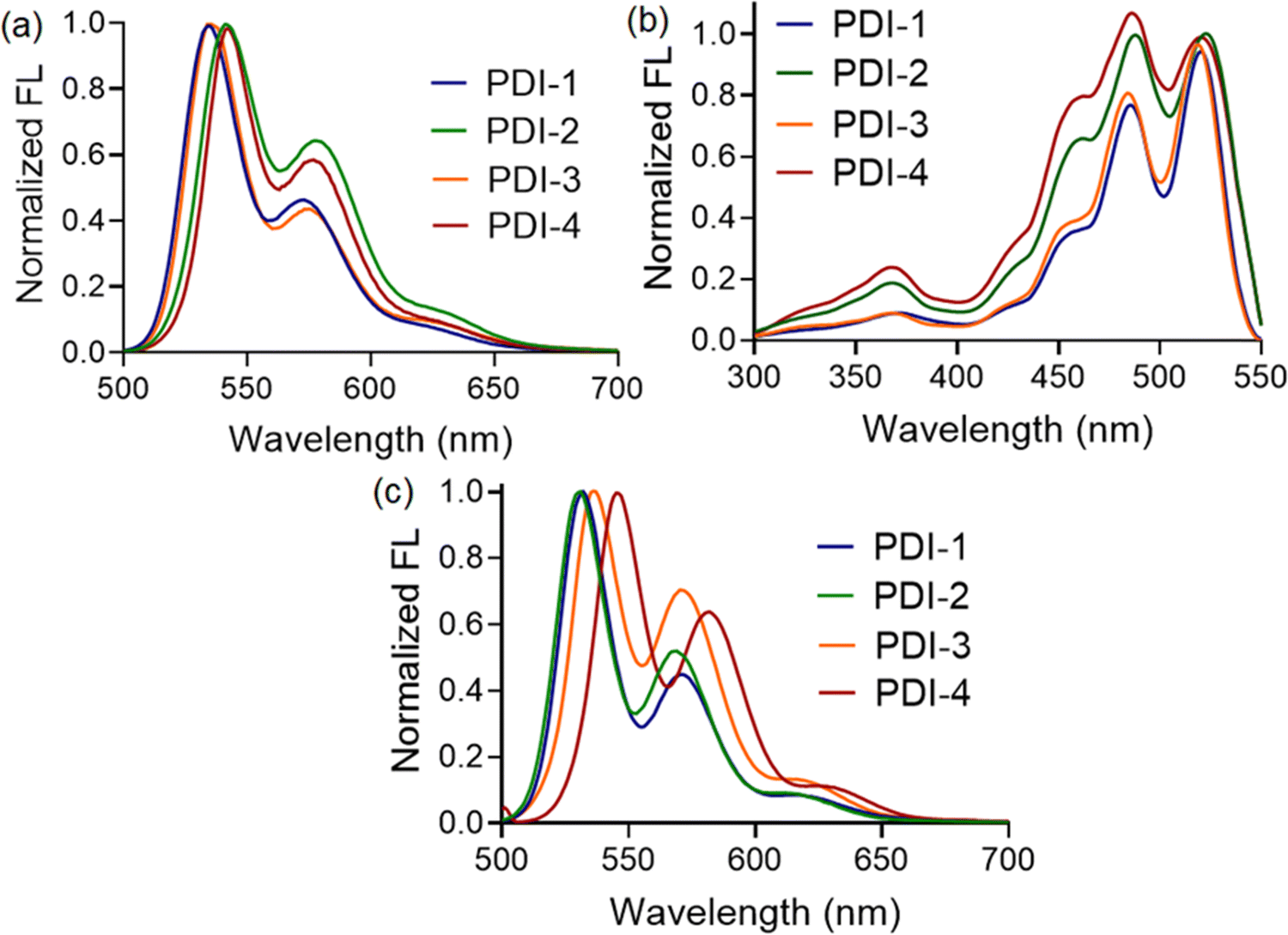 | ||
| Fig. 3 (a) Normalized fluorescence spectra of N-substituted PDI derivatives (10 μM, λex = 450 nm) in methanol medium. (b) Normalized fluorescence excitation spectra of N-substituted PDI derivatives (10 μM) in methanol medium. (c) Normalized fluorescence spectra of N-substituted PDI derivatives (10 μM, λex = 450 nm) in acetonitrile medium. | ||
To determine whether such aggregate formation is driven by hydrogen bonding interaction, we also investigated the fluorescence spectra of compounds in an acetonitrile medium. Despite the absence of solvent-mediated hydrogen bonding interaction. Here, we also observed similar blue-shifted emission maxima for the PDI-I and PDI-3 compounds, as shown in Fig. 3(c). The scanning electron microscopic images of the compounds (PDI-1, PDI-2, PDI-3, and PDI-4) were recorded using the drop-casted samples on the solid surface. In variably, in all cases, we observed spherical aggregates that were subsequently connected to form extended supramolecular structures, as depicted in Fig. 1(b).
Device fabrication
By utilizing the as-synthesized PDI with different functional groups, the flexible sensor was fabricated by following the procedure shown in Fig. 4. First, the synthesized PDI powder, which is an N-substituted perylene-3,4-dicarboxylic acid imide derivative with different functional groups (i.e., by employing the procedure explained in the synthesis and characterization section above), was dissolved in a dimethyl sulfoxide (DMSO) solvent to make a 0.1% (wt/v) concentration solution. Consequently, the solution was deposited using vacuum filtration on a cellulose paper substrate. Later, hot air oven drying was used (∼15 minutes) to dry cellulose paper with a PDI thin film deposit. The Cu contacts were made of cellulose paper with a PDI deposit using adhesive silver paste and used as a breath sensor. | ||
| Fig. 4 Schematic for synthesizing PDI-based compounds and device fabrication of breath and pressure sensors. | ||
The encapsulation of SYLGARD®184 silicone-based elastomer PDMS (polydimethylsiloxane) with a 10:1 base-to-elastomer ratio was done to obtain a PDI-based pressure sensor. Finally, the fabricated PDI-polymer-based sensor was applied for device characterization using the Keithley 2450 source meter. A schematic illustration of the synthesis of PDI-based compounds and device fabrication is shown in Fig. 4.
Device characterization
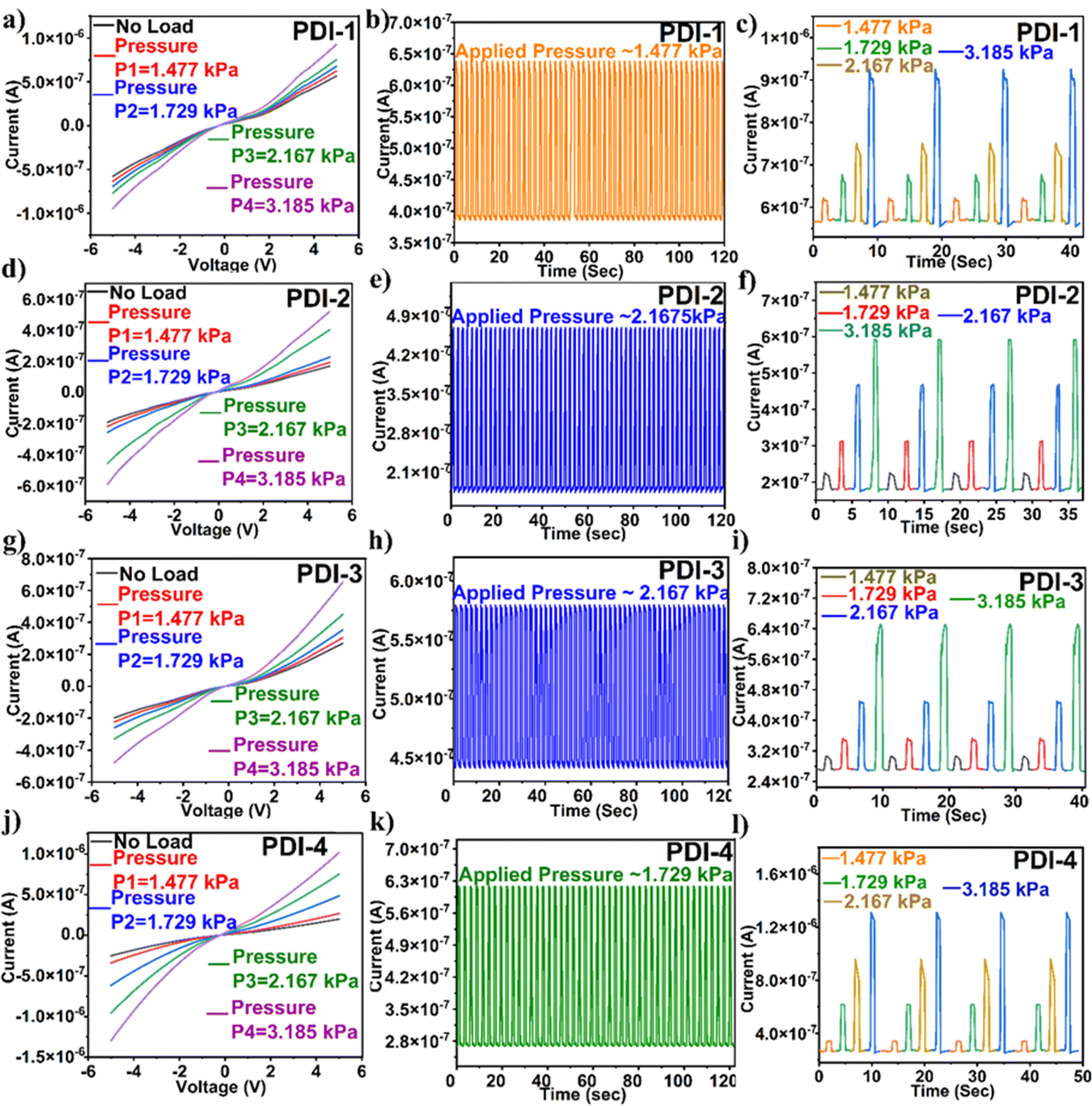 | ||
| Fig. 5 (a), (d), (g) and (j) IV characteristics, (b), (e), (h) and (k) temporal response plots at constant applied pressure and (c), (f), (i) and (l) temporal response plots at different applied pressure loads for the PDI-based flexible pressure sensors. | ||
The alteration in the slope of an electrical signal, i.e., current vs. pressure, indicates the pressure sensor sensitivity. The sensitivity of the PDI-based pressure sensor calculated sensitivity = (ΔI/Io)/P, where ΔI is the difference in current and Io is the original sensor current before the pressure application. The determined sensitivity value for PDI-1 was 0.315 kPa−1, PDI-2 was 1.266 kPa−1, PDI-3 was 0.749 kPa−1, and PDI-4 was 2.120 kPa−1. Among all the PDI-based pressure sensors fabricated, as shown in Fig. 6(a), (d), (g) and (j), PDI-4 displayed maximum sensitivity owing to the inherent asymmetric nature of the compound with two different terminal substituents (as shown in Fig. 1(a)).
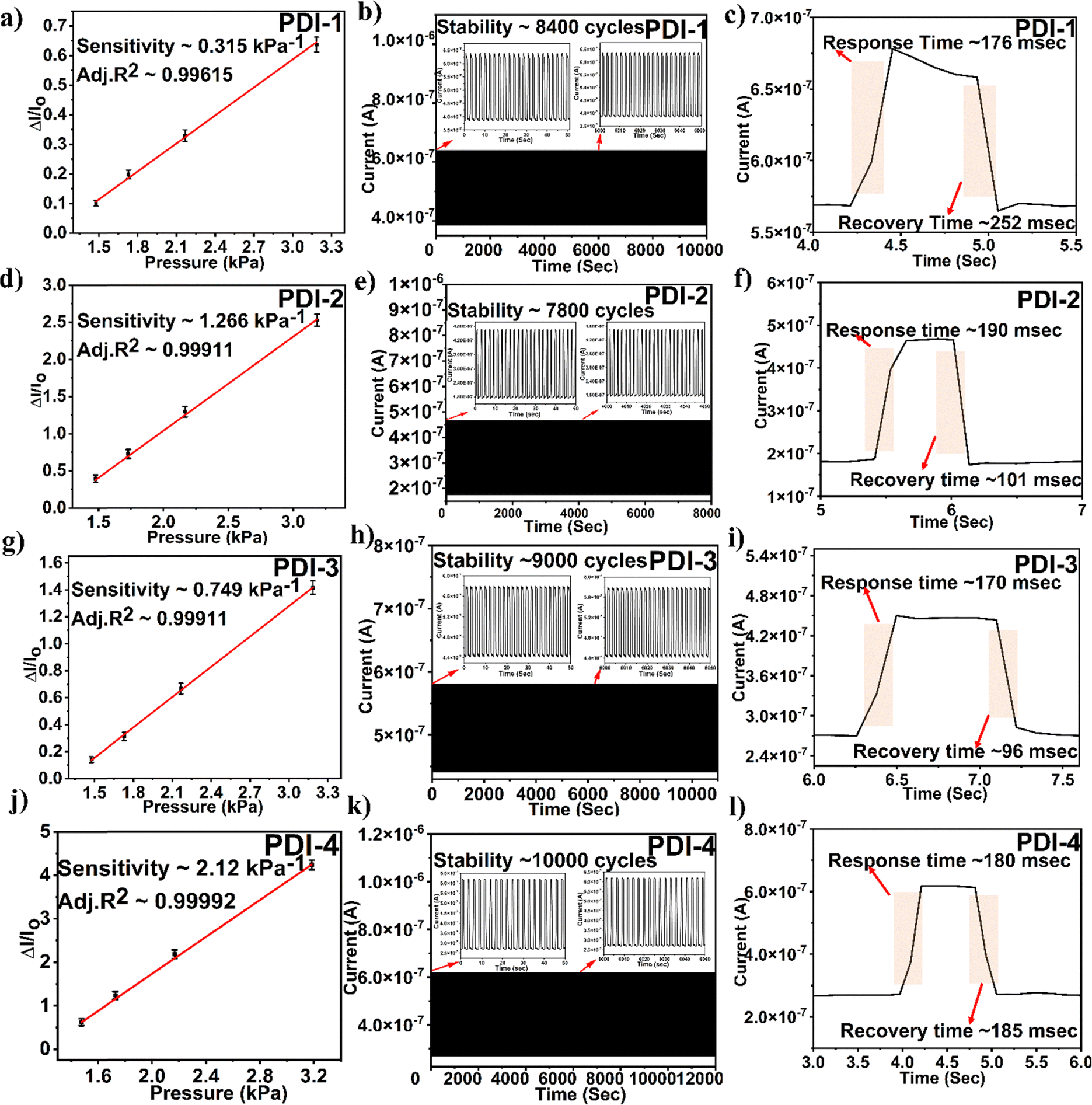 | ||
| Fig. 6 (a), (d), (g) and (j) Normalized current vs. applied pressure to attain sensitivity, (b), (e), (h) and (k) endurances of the fabricated sensors, and (c), (f), (i) and (l) response and recovery times for fabricated PDI-based pressure sensors. | ||
In addition, to examine the mechanical sturdiness of the fabricated PDI-based pressure sensors, the relative resistance variations in the sensors in the form of normalized current were measured when unceasing pressure loading and unloading cycles were applied for ∼8000–10000 cycles, as shown in Fig. 6(b), (e), (h) and (k). A negligible alteration in the sensor current was observed after the stability test, indicating that the fabricated pressure sensor was sturdy and consistent. The response and recovery time are the key constraints of the pressure sensor, which is shown in Fig. 6(c), (f), (i) and (l). The Δt over which the current goes to 90% of the absolute value and lowers to 10% of the saturated value is known as the response and recovery times. The obtained response and recovery times for all PDI-based pressure sensors were 176 and 252 s for PDI-1, 190 and 101 ms for PDI-2, 170 and 96 ms for PDI-3, and 180 and 185 ms for PDI-4.
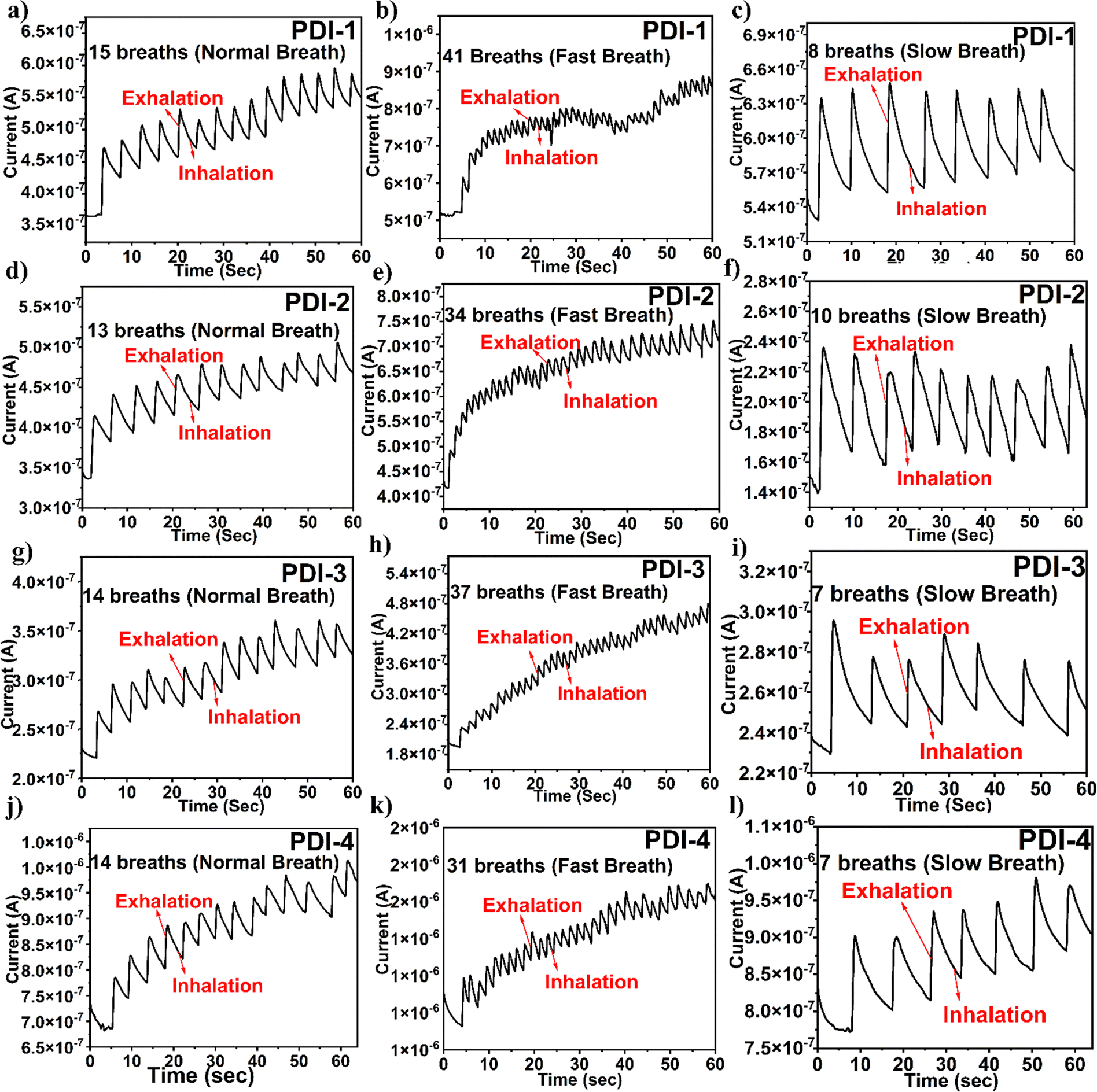 | ||
| Fig. 7 (a), (d), (g) and (j) Non-stop time response (temporal response plot) depicting PDI-based sensor under normal breath, (b), (e), (h) and (k) response under fast breath, and (c), (f), (i) and (l) response under slow breath. | ||
It is significant to observe that the terminal functionality of the PDI-based breath sensor does not reach its primary baseline current in the temporal plots shown in Fig. 7. This is because the adsorption of water molecules is a quicker method, and desorption is a gradual method. However, the unstable baseline is not relevant in the analysis as the respiration pattern depends on the number of peaks. Even though the unstable baseline is not a substantial point for exhibiting breath sensing, the number of peaks detected in the response of the plots shown in Fig. 7 displays a definite part in analyzing various breath patterns. The response and recovery time of the fabricated breath sensor are depicted in the ESI† in Fig. S5.
Finally, the experimental state-of-the-art on PDI-based sensors is duly compared with the present work and is presented in the ESI† (Table S1).
Sensing mechanism
Computational methods and theoretical findings
In this work, an ab initio theoretical investigation is performed to identify the primary transduction mechanisms associated with breath sensing and pressure sensing using the synthesized PDI molecules, and the relative performance of different PDI molecules is also assessed from these aspects. In their previous work, the authors have established that the synthesized small molecules form a percolating network after deposition, wherein the overall conductivity of the network depends on both intra-molecular and intermolecular conductivity.22 For both breath and pressure sensing performed in this work, the detection is achieved in terms of resistance modulations. Subsequently, the associated transduction mechanism is closely related to the overall conductivity modulation of the percolating network of PDI molecules in the presence of breath and applied pressure.To model the transduction mechanism related to breath sensing, the vapor (H2O) adsorptions on the different probable sites of PDI molecules are considered, as the H2O is the most pre-dominant constituent of breath. The strength of adsorption and the efficacy of transduction are assessed in terms of binding energy (Ebinding) and molecular charge transfer (QTransfer), respectively.
The H2O molecule binding energy on specific probes, i.e., PDI molecules, is defined by40,41
| EBinding = EGas_and_Host − (EGas + EHost), | (1) |
However, the charge (electron) transfer, i.e., QTransfer between the adsorbed H2O and probe PDI molecules is calculated using Mulliken charge analysis42 and defined as follows:21,22
| QTransfer = QGas_before_adsorption − QGas_after_adsorption. | (2) |
The definition of QTransfer in eqn (2) suggests that when QTransfer > 0, the adsorbed H2O molecules donate electrons to the probe PDI molecule. In contrast, QTransfer < 0 represents acceptance of electrons by the adsorbed H2O molecules from the probe PDI molecule.
Moreover, applied pressure on the percolating network of PDI molecules is expected to influence both intra- and inter-molecular electronic transport and, thus, the corresponding conductivities. In this context, it is worth noting that the intermolecular quantum mechanical tunneling dominates the intermolecular charge transport within a percolating network of inorganic/organic nanostructures, including small molecular systems, such as PDI.22,43–47 Consequently, to model the transduction mechanism related to pressure sensing, two identical PDI molecules are considered at different distances, and the effects of relative proximity are analyzed on the molecular energy spectrums and correlate with the electronic transport properties.
The density functional theory (DFT)-based ab initio calculation of this study is performed using the commercially available Virtual Nano Lab (VNL) and Atomistix Tool Kit (ATK) and simulation packages from Synopsis.34 This work performs the DFT calculations in a linear combination of an atomic orbital (LCAO) double-zeta polarized basis set with a density mesh cut-off energy of 125 Hartree. Moreover, a Monkhorst Pack grid of 12 × 12 × 1 is considered for sampling the Brillouin zone.
The four PDI molecules were initially built using an ATK VNL molecular builder. Next, the molecules are relaxed at their minimum energy and minimum force/atom configuration. The structural relaxation/geometry optimization is performed using the Limited memory Broyden Fletcher Goldfarb Shanno (LBFGS) algorithm with a force tolerance of 0.01 eV Å−1. For both geometry optimization and electronic property/energetic calculation, the Generalized Gradient Approximation (GGA) DFT method is considered with the Purdew Burke and Ernzerhof (PBE) exchange–correlation functional. Moreover, for calculating the ground state energy H2O/PDI (during breath sensing) or PDI/PDI (during pressure sensing) conjugate system, the Grimmie's DFT-D3 method, and counterpoise (CP) correction were used to incorporate van der Waals (vdW) interactions, and address the inherent Basis Set Superposition Error (BSSE), respectively. It is worth mentioning that compared to Grimmie's DFT-D3 method, Grimmie's DFT-D2 method usually tends to overestimate the vdW interactions between two molecular subsystems. Consequently, in this work, Grimmie's DFT-D3 method is considered more reliable to account for the possible vdW interactions between PDI and water molecules.48
The relaxed PDI molecules are depicted in Fig. 8. It is noteworthy that for the breath sensing study, the H2O molecules are introduced near the side oxygen (O) atom and the base nitrogen (N) atom of the PDI core, and these adsorption sites are termed site-1 and site-2, respectively, as illustrated in Fig. 8. The PDI molecules in the present study have mostly aromatic substitutions at the terminal positions with no hydrogen bonding motif available. This rules out the possibility of intra-molecular hydrogen bonding between the side-O (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) atom and the adjacent functional group. Therefore, the O (CO) atom is easily involved in the intermolecular hydrogen bonding interaction with H2O molecules. Therefore, the interaction of the H2O molecule with other molecular probes is usually expected to be dominated by the hydrogen bonding mechanism, where the H atom of H2O is expected to act as a hydrogen bond donor. Consequently, the H atom of H2O should interact with highly electronegative atoms that can efficiently receive the hydrogen bond, wherein the side-O (CO) and base-N atoms of probe PDI molecules are considered the most probable interaction sites for H2O. However, due to steric crowding, the interaction of H2O is subdued near the base N-atom. In each of these adsorption sites, the H2O is introduced with different molecular orientations, and the molecular orientation corresponding to the highest binding energy is considered for the rest of the study.
O) atom and the adjacent functional group. Therefore, the O (CO) atom is easily involved in the intermolecular hydrogen bonding interaction with H2O molecules. Therefore, the interaction of the H2O molecule with other molecular probes is usually expected to be dominated by the hydrogen bonding mechanism, where the H atom of H2O is expected to act as a hydrogen bond donor. Consequently, the H atom of H2O should interact with highly electronegative atoms that can efficiently receive the hydrogen bond, wherein the side-O (CO) and base-N atoms of probe PDI molecules are considered the most probable interaction sites for H2O. However, due to steric crowding, the interaction of H2O is subdued near the base N-atom. In each of these adsorption sites, the H2O is introduced with different molecular orientations, and the molecular orientation corresponding to the highest binding energy is considered for the rest of the study.
 | ||
| Fig. 8 Schematic of relaxed isolated PDI-1, PDI-2, PDI-3, and PDI-4, and H2O molecules from the top view. | ||
Next, the H2O adsorption on different PDI molecules and the subsequent Ebinding and QTransfer values are depicted in Fig. 9.
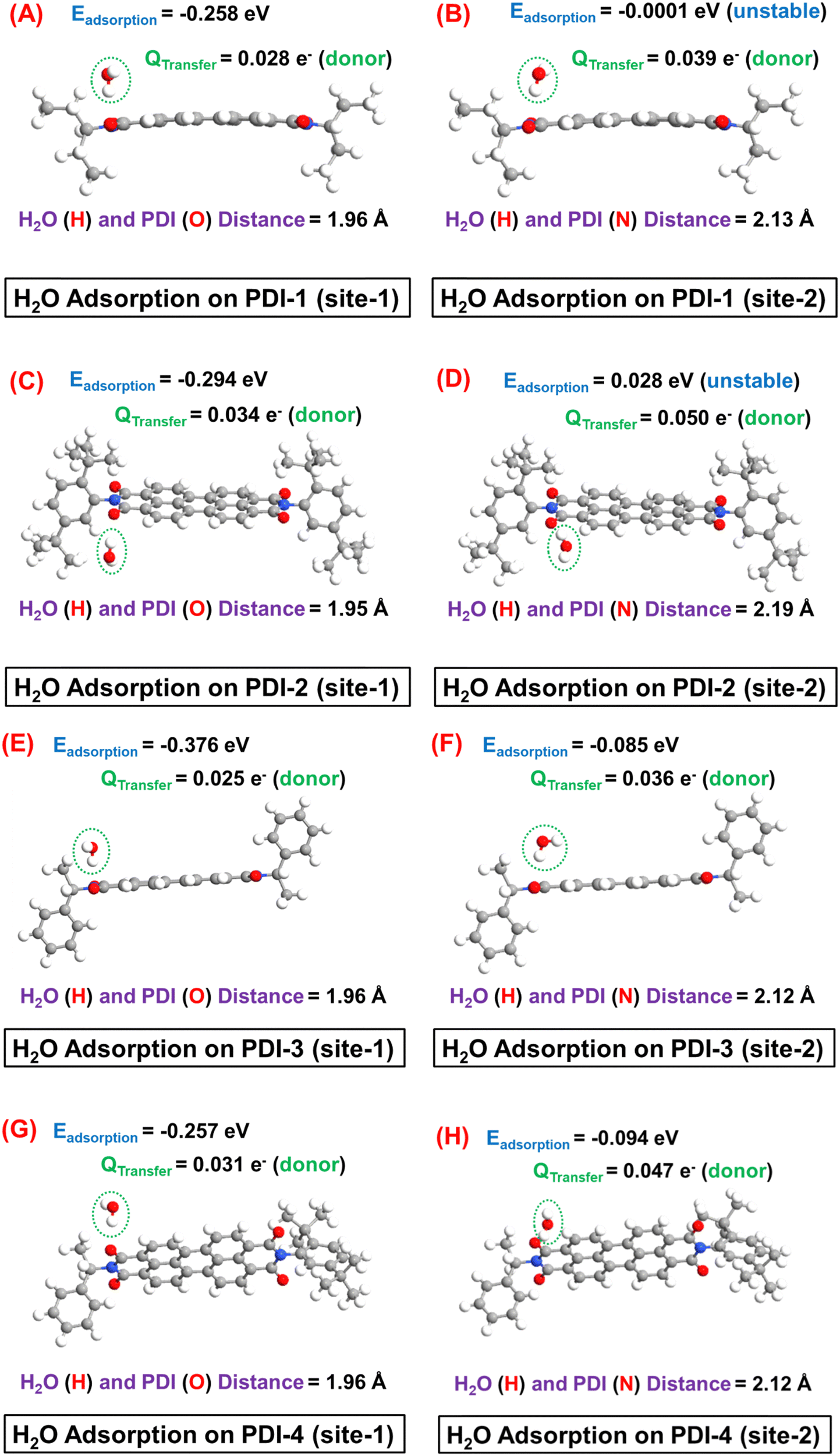 | ||
| Fig. 9 Schematic representation of H2O adsorption on (A) site-1 and (B) site-2 of PDI-1, (C) site-1 and (D) site-2 of PDI-2, (E) site-1 and (F) site-2 of PDI-3, (G) site-1 and (H) site-2 of PDI-4 from the side-view. | ||
It is interesting to observe that the H2O molecule is preferentially adsorbed near the edge O (CO) atom, where the H (of H2O) and O (of PDI) distance (1.95–1.96 Å) suggest a hydrogen bond formation. Specifically, during this interaction, the H atom of H2O acts as a hydrogen bond donor to the edge O atom. Consequently, owing to the relatively weaker nature of hydrogen bond-mediated molecular interaction, a moderate adsorption strength with the Ebinding ∼ 0.26–0.38 eV can be observed in site-1 in all cases. In contrast, a metastable (Ebinding ∼ KT at T = 300 K) or even unstable H2O adsorption can be observed near the base N atom, which can be further corroborated by the notable larger (2.12–2.19 Å) distance between H (of H2O) and N (of PDI) atoms in this case. Therefore, the H2O molecular adsorption study clearly suggests that H2O forms an intermolecular hydrogen bond with the probe PDI molecules (with a typical hydrogen band distance in the range of 1.8–2 Å) during its adsorption. The relatively highly electronegative side-O (CO) atom acts as the most favourable side for such intermolecular hydrogen-bond formation and receives the hydrogen bond from the hydrogen atom of H2O.
In the adsorption site-1, a moderate (Qtransfer ∼ 0.025–0.034 e−) donor-type charge transfer can be observed after H2O adsorption in each PDI molecule. However, it is worth mentioning that each PDI molecule offers four identical adsorption sites near the edge O atom, which should result in an even higher electron transfer from adsorbed H2O to PDI. The electron transfer from H2O to PDI is expected to enhance the overall electronic conductivity of the percolating network of PDI molecules. Consequently, in the experimental study, a notable increase in current is observed in each sensor prototype, which is consistent with the theoretical study of breath sensing. Finally, it is worth noting that the edge O (CO) atom of the base PDI molecule offers the most stable adsorption sites for H2O molecule, where the specification of the side functional group has a negligible influence on the strength of H2O molecule adsorption in PDI. Because the molecular structures of individual probe molecules considered in this study differ only by the side functional group specifications associated with base PDI molecules, this leads to comparable Ebinding and Qtransfer magnitudes and gas–host distance in each case.
Next, the pressure sensing mechanism is investigated by considering a pair of each PDI molecule in identical orientations far apart (distance between the base planes ∼25 Å) to represent the case without applied pressure and in proximity (distance between the base planes ∼10 Å) to represent the case with applied pressure, as depicted in Fig. 10. In practical situations, various relative molecular orientations are possible in any percolating network of PDI molecules. However, for the theoretical modeling in this work, an ideal representation is considered by keeping the identical molecular orientations between the PDI molecule, which should suffice to qualitatively analyze the underlying transduction mechanism. The molecular energy spectrum (MES) of PDI molecules without (distance between the base plane ∼25 Å) and with (distance between the base planes ∼10 Å) applied pressure, and the electron density profiles of PDI molecules under applied pressure are also illustrated in Fig. 10. It is noteworthy that the 25 Å distance between the base planes is sufficient to ensure notable intermolecular tunneling49–51 in the absence of applied pressure in the percolating network of PDI molecules. Furthermore, the 10 Å distance is considered following the fact that this is the common minimum distance between each pair of PDI molecules, where the individual molecules retain their conformal integrity. It should be noted that any conformation modification is an energy-expensive process. In any percolating network, the constituent PDI molecules can reorient themselves and, therefore, are less likely to undergo conformation changes under applied pressure.
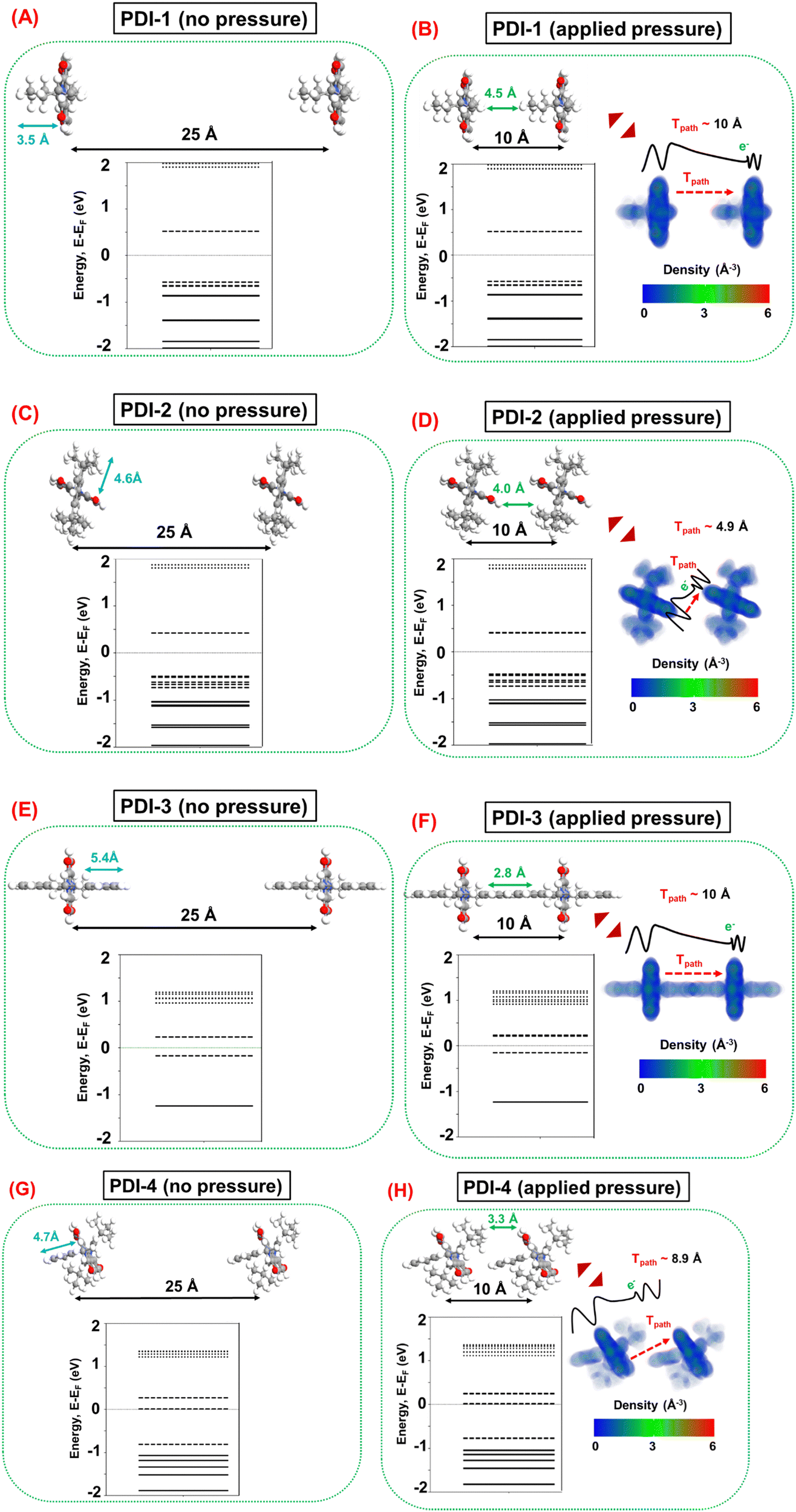 | ||
| Fig. 10 Schematic of molecular pair conjugates (A) before and (B) after applying pressure in PDI-1; (C) before and (D) after applying pressure in PDI-2; (E) before and (F) after applying pressure in PDI-3; (G) before and (H) after applying pressure in PDI-4 from the side-view. | ||
Fig. 10 illustrates a suggestion that at ∼10 Å distance between the base planes, there are negligible electron cloud overlaps between two adjacent PDI molecules, which results in marginal influence on the MES of the molecular systems in all cases before and after applying pressure. This finding suggests that the intramolecular conductivity is less likely to be influenced by the applied pressure, wherein intermolecular conductivity modulation dominates the transduction mechanism during pressure sensing. Subsequently, this analysis emphasizes the relative effects of pressure on the intermolecular tunneling mechanism, wherein the applied pressure predominantly reduces the tunneling distance between two adjacent molecules and is expected to enhance the intermolecular conductivity. However, the relative increase in intermolecular tunneling depends on several factors associated with the particular molecular system under question.
Intermolecular tunneling depends on the cross-sectional area in the tunneling path, which suggests that the dominant tunneling contribution can be expected between the base planes of two adjacent PDI molecules. In this context, it is interesting to note that both PDI-2 and PDI-4 side functional groups are vertically twisted with respect to their base planes, as shown in Fig. 8. In contrast, the side functional groups exhibit no vertical rotations in PDI-1 and PDI-3. Consequently, in PDI-2 and PDI-4, the presence of such a twisted side chain is generally more favorable for the molecular orientations where the base planes of the adjacent molecules are in proximity to each other, as depicted in Fig. 10. Therefore, PDI-2 and PDI-4 are relatively more favorable for increasing the tunneling component under applied pressure. However, intermolecular tunneling also strongly depends on the electronic properties of the PDI molecules, i.e., the molecular electronic state density near the Fermi level, where more electronic states at closer proximity to the Fermi level demonstrate a relatively larger number of electrons that are available for tunneling. In this context, Fig. 10 shows that PDI-4 has molecular states at the Fermi level, suggesting that it is the most suitable PDI molecule for intermolecular tunneling. The next best candidate is PDI-3, demonstrating molecular states energetically closer to the Fermi level. However, the presence of the molecular states sufficiently away from the Fermi level in PDI-1 suggests that it is the least suitable PDI molecule for intermolecular tunneling.
Therefore, from the theoretical study, it can be surmised that both the structural configuration and electronic properties of PDI-4 (PDI-1) are suitable (undesirable) to ensure a large increase in intermolecular tunneling components and, thereby, in the overall conductivity of the percolating network under applied pressure. Hence, the PDI-4 (PDI-1) is the most (least) favorable PDI molecule for pressure sensing applications. In contrast, a moderate response can be expected in PDI-2 and PDI-3 during pressure sensing; in these cases, two factors have competing influences on the overall efficacy of transduction. Finally, it is worth mentioning that the experimentally observed relative sensing performances of the fabricated molecular strain sensor devices are in good agreement with the prediction of the theoretical analysis.
Finally, the key theoretical findings of this work are duly summarized in Table 1. It is noteworthy that for the H2O adsorption study, only the highest Ead configurations are considered in the table.
| Sensing specifications | Probe molecule | Adsorption energy (eV) | Charge transfer (e−) | Hydrogen bond [H2O (H)–PDI (O)] distance (Å) | Effective tunnelling length (Å) | Change in molecular energy spectrum |
|---|---|---|---|---|---|---|
| Breath sensing (H2O adsorption) | PDI-1 | −0.258 | 0.028 (donor) | 1.96 | — | — |
| PDI-2 | −0.294 | 0.034 (donor) | 1.95 | — | — | |
| PDI-3 | −0.376 | 0.025 (donor) | 1.96 | — | — | |
| PDI-4 | −0.257 | 0.031 (donor) | 1.96 | — | — | |
| Pressure sensing | PDI-1 | — | — | — | 10.0 | No |
| PDI-2 | — | — | — | 4.9 | Yes (HOMO) | |
| PDI-3 | — | — | — | 10.0 | Yes (LUMO) | |
| PDI-4 | — | — | — | 8.9 | Yes (LUMO) |
Conclusion
In summary, this study reveals the PDI (perylene diimide) compounds fabricated using an economically viable vacuum filtration deposition method based on pressure and breath sensors. The deposition of the N-substituted perylene-3,4-dicarboxylic acid imide derivatives (PDI-1, PDI-2, PDI-3, and PDI-4) with different functional groups (3-aminopentane, 2,5-di-tert-butylaniline, 1-phenylethylamine, etc.) on the paper substrate forms a moderately conducting percolating molecular network, demonstrating enhanced pressure/breath-sensing performance. The determined sensitivity value for PDI-1 was 0.315 kPa−1, PDI-2 was 1.266 kPa−1, PDI-3 was 0.749 kPa−1, and PDI-4 was 2.120 kPa−1. Amid all the PDI-based pressure sensors fabricated, PDI-4 displayed maximum sensitivity owing to the inherent asymmetric nature of the compound with two different terminal substituents. The sensor displayed a steady response of up to ∼8000–10000 cycles, confirming the mechanical sturdiness of fabricated PDI-based pressure sensors. The experimental findings are supplemented by density functional theory (DFT)-based theoretical analysis of the pressure and breath sensing transduction mechanisms for different PDI molecules. From the theoretical study, it can be surmised that both the structural configuration and electronic properties of PDI-4 (PDI-1) are suitable (undesirable) to ensure a large increase in intermolecular tunneling components and, thereby, in the overall conductivity of the percolating network under applied pressure. Hence, the PDI-4 (PDI-1) is the most (least) favorable PDI molecule for pressure sensing applications. In contrast, a moderate response can be expected in PDI-2 and PDI-3 during pressure sensing; in these cases, two competing factors influence the overall efficacy of transduction. This demonstration of pressure and breath-sensing using PDI-based flexible sensors opens innovative perspectives for fabricating highly responsive, cost-effective electromechanical sensors.
Data availability
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The authors thank Central Analytical Laboratory, BITS Pilani Hyderabad Campus, for the assistance in material characterization.References
- K. Kamath, V. Adepu, V. Mattela and P. Sahatiya, Development of Ti3C2Tx/MoS2xSe2(1–x) Nanohybrid Multilayer Structures for Piezoresistive Mechanical Transduction, ACS Appl. Electron. Mater., 2021, 3, 4091–4104 CrossRef CAS.
- H. Souri, H. Banerjee, A. Jusufi, N. Radacsi, A. A. Stokes, I. Park, M. Sitti and M. Amjadi, Wearable and stretchable strain sensors: materials, sensing mechanisms, and applications, Adv. Intell. Syst., 2020, 2, 2000039 CrossRef.
- M. A. U. Khalid and S. H. Chang, Flexible strain sensors for wearable applications fabricated using novel functional nanocomposites: A review, Compos. Struct., 2022, 284, 115214 CrossRef CAS.
- M. Ju, Z. Dou, J. W. Li, X. Qiu, B. Shen, D. Zhang and K. Wang, Piezoelectric materials and sensors for structural health monitoring: fundamental aspects, current status, and future perspectives, Sensors, 2023, 23, 543 Search PubMed.
- V. Adepu, M. Tathacharya, C. S. Raghuram and P. Sahatiya, MXene (Ti3C2Tx)/TMD (ReSe2) nanohybrid-based flexible electromechanical sensors for cervical collar strain and shoulder load detection applications, J. Micromech. Microeng., 2023, 33, 115007 CrossRef CAS.
- V. Adepu, M. Tathacharya, V. Mattela and P. Sahatiya, Development of WS2/MXene (Ti3C2Tx) nanohybrid based multi-functional textronic sensor for non-invasive personal healthcare monitoring, Flex. Print. Electron., 2023, 8, 015001 CrossRef.
- S. Xu, Z. Xu, D. Li, T. Cui, X. Li, Y. Yang and T. Ren, Recent advances in flexible piezoresistive arrays: materials, design, and applications, Polymers, 2023, 15, 2699 CrossRef CAS PubMed.
- X. Wang, L. Dong, H. Zhang, R. Yu, C. Pan and Z. L. Wang, Recent progress in electronic skin, Adv. Sci., 2015, 2, 1500169 CrossRef PubMed.
- Z. Liu, T. Zhu, J. Wang, Z. Zheng, Y. Li, J. Li and Y. Lai, Functionalized fiber-based strain sensors: pathway to next-generation wearable electronics, Nano-Micro Lett., 2022, 14, 61 Search PubMed.
- V. Adepu, A. Kunchur, M. Tathacharya, V. Mattela and P. Sahatiya, SnS/Ti3C2T x (MXene) Nanohybrid-Based Wearable Electromechanical Sensors for Sign-to-Text Translation and Sitting Posture Analysis, ACS Appl. Electron. Mater., 2022, 4, 1756–1768 CrossRef CAS.
- S. E. Zhu, M. Krishna Ghatkesar, C. Zhang and G. C. A. M. Janssen, Smart paper from graphene coated cellulose for high-performance humidity and piezoresistive force sensor, Appl. Phys. Lett., 2013, 102, 161907 CrossRef.
- V. Adepu, K. Kamath, V. Mattela and P. Sahatiya, Laser-assisted Gaussian microstructure patterned PDMS encapsulated Ti3C2Tx (MXene)-based pressure sensor for object and touch detection, IEEE Sens. J., 2021, 21, 16547–16553 CAS.
- J. Guan, D. Zhang and T. Li, Flexible pressure sensor based on molybdenum diselide/multi-walled carbon nanotubes for human motion detection, IEEE Sens. J., 2021, 21, 10491–10497 CAS.
- V. Adepu, M. Tathacharya, R. CS, V. Mattela and P. Sahatiya, TeNWs/Ti3C2Tx Nanohybrid-Based Flexible Pressure Sensors for Personal Safety Applications Using Morse Code, ACS Appl. Nano Mater., 2022, 5, 18209–18219 CrossRef CAS.
- V. Adepu, V. Mattela and P. Sahatiya, A remarkably ultra-sensitive large area matrix of MXene based multifunctional physical sensors (pressure, strain, and temperature) for mimicking human skin, J. Mater. Chem. B, 2021, 9, 4523–4534 RSC.
- H. Yao, J. Ge, C. F. Wang, X. Wang, W. Hu, Z. J. Zheng, Y. Ni and S. H. Yu, A flexible and highly pressure-sensitive graphene–polyurethane sponge based on fractured microstructure design, Adv. Mater., 2013, 25, 6692–6698 CrossRef CAS PubMed.
- L. Pan, A. Chortos, G. Yu, Y. Wang, S. Isaacson, R. Allen, Y. Shi, R. Dauskardt and Z. Bao, An ultra-sensitive resistive pressure sensor based on hollow-sphere microstructure induced elasticity in conducting polymer film, Nat. Commun., 2014, 5, 3002 CrossRef PubMed.
- M. L. Tang, J. H. Oh, A. D. Reichardt and Z. Bao, Chlorination: a general route toward electron transport in organic semiconductors, J. Am. Chem. Soc., 2009, 131, 3733–3740 CrossRef CAS PubMed.
- J. H. Oh, S.-L. Suraru, W.-Y. Lee, M. Könemann, H. W. Höffken, C. Röger, R. Schmidt, Y. Chung, W. C. Chen, F. Würthner and Z. Bao, High-Performance Air-Stable n-Type Organic Transistors Based on Core-Chlorinated Naphthalene Tetracarboxylic Diimides, Adv. Funct. Mater., 2010, 20, 2148–2156 CrossRef CAS.
- W. Wang, A. D. Shaller and A. D. Li, Twisted perylene stereodimers reveal chiral molecular assembly codes, J. Am. Chem. Soc., 2008, 130, 8271–8279 Search PubMed.
- A. Tiwari, R. S. Fernandes, N. Dey and S. Kanungo, Site-specific ammonia adsorption and transduction on a naphthalimide derivative molecule–a complementary analysis involving ab initio calculation and experimental verification, Phys. Chem. Chem. Phys., 2023, 25, 17021–17033 RSC.
- V. Adepu, M. Tathacharya, R. S. Fernandes, A. Tiwari, S. Siraj, S. Kanungo, N. Dey and P. Sahatiya, Perylene Diimide (PDI) based Flexible Multifunctional Sensor Design for Personal Healthcare Monitoring-A Complementary Approach Involving Experimental and Theoretical Investigations, Adv. Mater. Technol., 2023, 8, 2201633 CrossRef CAS.
- N. Wu, Y. Zhang, C. Wang, P. M. Slattum, X. Yang and L. Zang, Thermoactivated electrical conductivity in perylene diimide nanofiber materials, J. Phys. Chem. Lett., 2017, 8, 292–298 CrossRef CAS PubMed.
- D. Zhao, Q. Wu, Z. Cai, T. Zheng, W. Chen, J. Lu and L. Yu, Electron acceptors based on α-substituted perylene diimide (PDI) for organic solar cells, Chem. Mater., 2016, 28, 1139–1146 Search PubMed.
- E. Kozma and M. Catellani, Perylene diimides based materials for organic solar cells, Dyes Pigm., 2013, 98, 160–179 CrossRef CAS.
- High Performance Pigments, ed. E. B. Faulkner and R. J. Schwartz, John Wiley & Sons, 2009 Search PubMed.
- X. Zhan, A. Facchetti, S. Barlow, T. J. Marks, M. A. Ratner, M. R. Wasielewski and S. R. Marder, Rylene and related diimides for organic electronics, Adv. Mater., 2011, 23, 268–284 Search PubMed.
- L. Zang, Y. Che and J. S. Moore, One-dimensional self-assembly of planar π-conjugated molecules: adaptable building blocks for organic nanodevices, Acc. Chem. Res., 2008, 41, 1596–1608 CrossRef CAS PubMed.
- F. Würthner, Perylene bisimide dyes as versatile building blocks for functional supramolecular architectures, Chem. Commun., 2004, 1564–1579 RSC.
- C. Huang, S. Barlow and S. R. Marder, Perylene-3, 4, 9, 10-tetracarboxylic acid diimides: synthesis, physical properties, and use in organic electronics, J. Org. Chem., 2011, 76, 2386–2407 Search PubMed.
- R. S. Fernandes, A. Tiwari, S. Kanungo and N. Dey, Formation of stable naphthalenediimide radical anion: Substituent-directed synergistic effects of hydrogen bonding and charge transfer interactions on chromogenic response towards hydrazine, J. Mol. Liq., 2023, 387, 122238 Search PubMed.
- S. Naqvi, M. Kumar and R. Kumar, Facile synthesis and evaluation of electron transport and photophysical properties of photoluminescent PDI derivatives, ACS Omega, 2019, 4, 19735–19745 Search PubMed.
- A. K. Dwivedi, M. Pandeeswar and T. Govindaraju, Assembly modulation of PDI derivative as a supramolecular fluorescence switching probe for detection of cationic surfactant and metal ions in aqueous media, ACS Appl. Mater. Interfaces, 2014, 6, 21369–21379 CrossRef CAS PubMed.
- X. Xu, A. Austin, S. E. Mylon, J. Plenge and J. M. Szarko, Improving the quantum yields of perylene diimide aggregates by increasing molecular hydrophobicity in polar media, ChemPhysChem, 2017, 18, 2430–2441 CrossRef CAS PubMed.
- V. Karunakaran, D. D. Prabhu and S. Das, Optical investigation of self-aggregation of a tetrazole-substituted diphenylacetylene derivative: steady and excited state dynamics in solid and solution state, J. Phys. Chem. C, 2013, 117, 9404–9415 Search PubMed.
- G. Pistolis, V. S. Petrakis, F. Kournoutas, N. Karakostas, E. Martinou, E. K. Efthimiadou, M. Fakis and I. M. Mavridis, Cooperative self-assembly enables two-dimensional H-type aggregation of a sterically crowded perylene-bisimide dimer, Cryst. Growth Des., 2019, 19, 4252–4263 Search PubMed.
- J. Gorman, S. R. Orsborne, A. Sridhar, R. Pandya, P. Budden, A. Ohmann, N. A. Panjwani, Y. Liu, J. L. Greenfield, S. Dowland and V. Gray, Deoxyribonucleic acid encoded and size-defined π-stacking of perylene diimides, J. Am. Chem. Soc., 2021, 144, 368–376 CrossRef PubMed.
- I. Smith, J. Mackay, N. Fahrid and D. Krucheck, Respiratory rate measurement: a comparison of methods, Br. J. Healthc. Assist., 2011, 5, 18–23 CrossRef.
- I. Smith, J. Mackay, N. Fahrid and D. Krucheck, Respiratory rate measurement: a comparison of methods, Br. J. Healthc. Assist., 2011, 5, 18–23 Search PubMed.
- A. Tiwari, J. Palepu, A. Choudhury, S. Bhattacharya and S. Kanungo, Theoretical analysis of the NH3, NO, and NO2 adsorption on boron-nitrogen and boron-phosphorous co-doped monolayer graphene-A comparative study, FlatChem, 2022, 34, 100392 Search PubMed.
- A. Tiwari, N. Bahadursha, S. Chakraborty, S. Das and S. Kanungo, Carbon monoxide adsorption on different sub-lattice sites of nitrogen and phosphorous doped and Co-doped germanene-a first principle study, Physica E, 2023, 151, 115746 CrossRef CAS.
- S. Smidstrup, T. Markussen, P. Vancraeyveld, J. Wellendorff, J. Schneider, T. Gunst, B. Verstichel, D. Stradi, P. A. Khomyakov, U. G. Vej-Hansen and M. E. Lee, QuantumATK: an integrated platform of electronic and atomic-scale modelling tools, Phys. Condens. Matter, 2019, 32, 015901 CrossRef PubMed.
- N. Bokka, V. Adepu, A. Tiwari, S. Kanungo and P. Sahatiya, A detailed comparative performance analysis of the Transition Metal Di-chalcogenides (TMDs) based strain sensors through experimental realisations and first principle calculations, FlatChem, 2022, 32, 100344 CrossRef CAS.
- N. Bokka, D. Som, S. Kanungo and P. Sahatiya, Investigation of the Transduction Mechanism of Few Layer SnS2 for Pressure and Strain Sensing: Experimental Correlation With First Principles Study, IEEE Sens. J., 2021, 21, 17254–17261 CAS.
- M. Y. Hwang and L. H. Kang, Data-driven pursuit of electrochemically stable 2D materials with basal plane activity toward oxygen electrocatalysis, J. Mech. Sci. Technol., 2017, 31, 3787–3789 CrossRef.
- E. Y. Li and N. Marzari, Improving the electrical conductivity of carbon nanotube networks: A first-principles study, ACS Nano, 2011, 5, 9726–9736 CrossRef CAS PubMed.
- L. P. Simoneau, J. Villeneuve, C. M. Aguirre, R. Martel, P. Desjardins and A. Rochefort, Influence of statistical distributions on the electrical properties of disordered and aligned carbon nanotube networks, J. Appl. Phys., 2013, 114, 114310 CrossRef.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- J. R. Miller, Intermolecular electron transfer by quantum mechanical tunneling, Science, 1975, 189, 221–222 CrossRef CAS PubMed.
- Y. Liu, L. Ornago, M. Carlotti, Y. Ai, M. El Abbassi, S. Soni, A. Asyuda, M. Zharnikov, H. S. J. Van Der Zant and R. C. Chiechi, Intermolecular Effects on Tunneling through Acenes in Large-Area and Single-Molecule Junctions, J. Phys. Chem. C, 2020, 124, 22776–22783 CrossRef CAS PubMed.
- W. Deng, S. N. Du, Z. Y. Ruan, X. J. Zhao, Y. C. Chen, J. L. Liu and M. L. Tong, Aggregation-induced suppression of quantum tunneling by manipulating intermolecular arrangements of magnetic dipoles, Aggregate, 2024, 5, e441 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ma00846d |
| This journal is © The Royal Society of Chemistry 2024 |