
Guggulsterone – a potent bioactive phytosteroid: synthesis, structural modification, and its improved bioactivities
T. P.
Adarsh Krishna†
 *a,
T. P.
Ajeesh Krishna†
bc,
Baldev
Edachery
a and
S.
Antony Ceasar
bc
*a,
T. P.
Ajeesh Krishna†
bc,
Baldev
Edachery
a and
S.
Antony Ceasar
bc
aR & D Division, Sreedhareeyam Farmherbs India Pvt. Ltd, Kerala 686 662, India. E-mail: adarshkrishnatp@gmail.com
bDivision of Plant Molecular Biology and Biotechnology, Department of Bioscience, Rajagiri College of Social Sciences, Kochi, Kerala 683 104, India
cDivision of Phytochemistry and Drug-Design, Department of Bioscience, Rajagiri College of Social Sciences, Kochi, Kerala 683 104, India
First published on 2nd November 2023
Abstract
Guggulsterone is a phytosteroid derived from the oleo-gum resin of the critically endangered plant Commiphora wightii. This molecule has attracted increasing attention due to its excellent biochemistry potential and the compound has consequently been evaluated in clinical trials. With a low concentration in natural resources but wide medicinal and therapeutic value, chemists have developed several synthetic routes for guggulsterone starting from various steroid precursors. Moreover, numerous studies have attempted to modify its structure to improve the biological properties. Nowadays, green and sustainable chemistry has also attracted more attention for advanced chemical processes and reactions in steroid chemistry. The present review aimed to summarize the literature and provide an update about the improvements in the chemical synthesis and structural modification of guggulsterone from the view of green chemistry. Moreover, this review encompasses the improved activities of structurally modified guggulsterone derivatives. We expect that the information provided here will be useful to researchers working in this field and on this molecule.
1. Introduction
Steroids are important biodynamically active agents that have cyclopentanoperhydrophenanthrene (CPPP) nuclei with different chemical functionalities. Currently, steroid molecules are still essential in the field of drug discovery, due to their binding ability with different nuclear receptors, especially for receptor-mediated diseases.1 This class of molecules is the broadest group of therapeutics next to antibiotics.2 In recent years, researchers have paid increasing attention to the structural modification of steroids, and several structurally functionalized/modified steroids are considered as important medicinal lead molecules, particularly as chemoprotective and synergistic agents in cancer chemotherapy.3–5 Structurally functionalized steroids have emerged as interesting molecules with potentially improved pharmacological and pharmaceutical properties.6 Unluckily, despite the therapeutic potency of steroids in treating different syndromes, they can also cause many side effects.7 Fascinatingly, several compounds of plant origin (Fig. 1) and their synthetic derivatives have been suggested to have several potent biological activities, especially as anticancer agents.8–13 These compounds are easily available and inexpensive. The various chemical entities in their structures make them attractive lead compounds for medicinal chemists to synthesize a large number of analogues.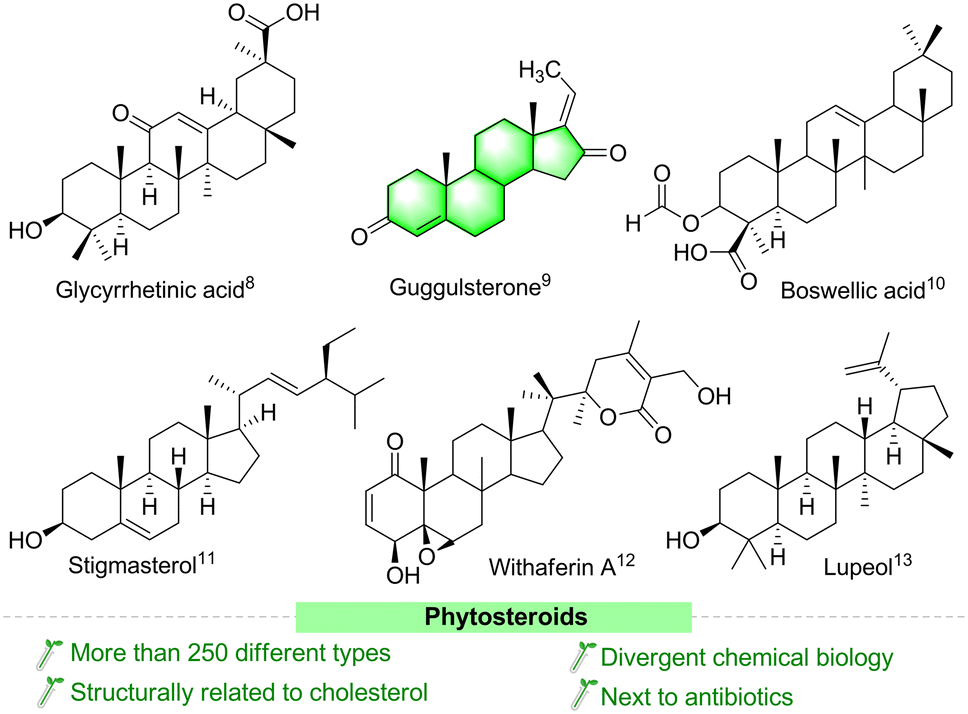 | ||
| Fig. 1 Representative examples of biologically active phytosteroids isolated from various plants. | ||
Guggulsterone (GS) is a phytosteroid derived from the oleo-gum resin of Commiphora wightii.14 This resin has been widely used for centuries to treat various ailments in the Ayurveda tradition,15 and many commercial herbal combinations contain guggulu as the main ingredient.14,16 From chemical investigations, it has been reported that the isomers (E/Z)-guggulsterone is the main active ingredient in this gum resin. These isomers exhibit several potent biological effects, such as thyroid stimulatory activity,17 platelet aggregation,18 anticancer,19 antioxidant,20 anti-inflammatory,21 antiatherosclerotic,22 cardioprotective,23 and cytotoxic,24 antihyperglycemic.25 Moreover, this molecule shows a high-affinity binding to the active site of the SARS-CoV-2 major protease.26 In addition, its chemically modified analogues have shown promise as good anticancer agents.27–29 Due to their different biochemistry, these molecules have gained increasing importance in recent years. Based on the above considerations, there is a strong demand for GS and its analogues for further research and development, and application. However, the biologically active lipids from C. wightii are not readily available, and many trees are needed to collect the resin. Moreover, the resin has a low concentration of GS.30 In this context, organic chemists have developed several synthetic routes for GS and its derivatives starting from various steroid precursors. Efficient protocols for complex molecules with divergent functional groups have great importance in the pharmaceutical field and are needed in drug research and development.31,32 Therefore, in this review, we give an insight into the bioactivity, chemical synthesis, and structural modification of GS. Moreover, this analysis encompasses the improved activities of structurally modified GS derivatives. We hope that the information provided here will be helpful to researchers working in this field, especially on this molecule.
2. Therapeutic potential of guggulsterone for cancer
The uncontrolled division of abnormal cells is called cancer, which is one of the most serious problems in the world, being the leading cause of death worldwide.33 Nowadays, multiple strategies are practiced for cancer therapy, but they are generally not potent or effective enough for cancer. For example, conventional treatments, such as surgery, radiation, and chemotherapy, are widely used for cancer, but they have some side effects.34 Many effective drug molecules are derived from plant origins,35 and they can be used to improve human health and prevent lifestyle diseases.36–38 Therefore, researchers are trying to find novel plant-based bioactive molecules with therapeutic potential for cancer therapy. Moreover, they are also trying to understand the therapeutic effects at the gene levels of these bioactive molecules for the better management of cancer. Recent studies have shown that some genes play differential roles in various cancers.39–42 Therefore, better cancer therapy could be possible by regulating the expression of the desirable genes with the help of bioactive molecules, which would represent a promising approach to control the proliferation of cancerous cells.In cancer therapy, the inhibition of cancerous cell multiplication and apoptosis (cell death) is essential for achieving anticancer effects. Currently, more than 60% of bioactive molecules show some anticancer properties, and these may be found from various sources, such as plants, and microorganisms.43 GS is a promising phytosterol that can activate survival pathways and regulate/activate anticancer genes/transcription factors/enzymes/proteins, etc. It can help to regulate the proliferation of cancerous cells. In in vitro studies, GS has been shown to target constitutively activated cancer survival pathways, such as phosphatidylinositol 3-kinase (PI3K)/protein kinase B (AKT), Janus kinase (JAK)/signal transducer and activator of transcription protein (STAT), and nuclear factor-kappaB (NF-κB). These signaling pathways regulate the growth and inflammatory responses of cancerous cells via regulation of anti-apoptotic and inflammatory genes.44–46 For example, GS treatment was reported to decrease mucin4 (MUC4) expression in pancreatic cancer cells (Capan1 and CD18/HPAF) through transcriptional regulation by inhibiting the JAK/STAT pathway.47 This study revealed that GS-induced apoptosis and cell cycle arrest in pancreatic cancer, as assessed via annexin-V assay and FACS analysis.47 Furthermore, Yamada et al.48 revealed that GS suppressed the deoxycholic acid (DCA)-induced activation of caudal type homeobox 2 (CDX2) and cyclooxygenase-2 (COX-2) expression through the inhibition of NF-κB activity. The transcription of CDX2 and COX-2 through NF-κB activation plays an important role in the development of Barrett's esophagus (BE) and esophageal adenocarcinoma (EAC). Therefore, the NF-κB inhibition activity of GS showed that it is a promising bioactive molecule to prevent and treat BE and EAC.48 Furthermore, many scientific studies have revealed that GS regulates the expression of cancer-related genes to control cell growth or induce apoptosis to prevent cancer.49–54 However, it is essential to understand the effect of GS on the upregulation of pro-apoptotic genes or the downregulation of anti-apoptotic genes, as well as the role of their transcription factors. Recently, many researchers have proven the potential therapeutic targets of GS in cancer,55,56 highlighting the promise of natural molecules for cancer therapy.
Interestingly, GS has the ability to induce apoptosis in various cancers, such as liver cancer,57 colon cancer,58 head and neck cancer,59 prostate cancer,60 bladder cancer,61 and gastric cancer.62 Therefore, GS is a potentially useful bioactive molecule for all types of cancers. GS has two isomeric forms, namely trans (E-guggulsterone) and cis (Z-guggulsterone) isomers,63 and it has also been shown to have potent anticancer properties.64a The Z-guggulsterone treatment showed the inhibition of angiogenesis in both in vitro and in vivo studies.64b For example, Xiao and Singh64b revealed that the Z-guggulsterone treatment of DU145 (human prostate cancer cells) implanted cells in mice inhibited angiogenesis via the vascular endothelial growth factor (VEGF), and by downregulation of the VEGF receptor 2 (VEGF-R2) and the inactivation of Akt. Furthermore, the in vivo studies showed a significant decrease in tumor burden, microvessel area, and VEGF-R2 protein expression in mice.64b Therefore, Z-guggulsterone has shown promising therapeutic potential as a molecule against human prostate cancer. Moreover, GS derivatives have also shown cancer chemo-preventive and therapeutic potential in both in vitro and in vivo studies.28,29,65 Recently, Kohyama et al.64c synthesized nine GS derivatives (GSDs) and evaluated them for their activity against human PANC-1 pancreatic cancer cells. Among the GSDs, GSD1 (30a) and GSD7 (30) showed better cytotoxicity against human PANC-1 cells and inhibited PANC-1 cancerous cell colony formation.64c Comparatively, the various derivatives/analogues have demonstrated higher efficiency than their naturally derived molecules. Therefore, developing new synthetic routes for producing GS and its derivatives/analogs is an important area in current research, and could lead to utilizing derivatives/analogs of GS for the development of new drugs for cancer. Therefore, we need to understand the chemistry, biology, and various synthetic routes and modifications of GS and its derivatives.
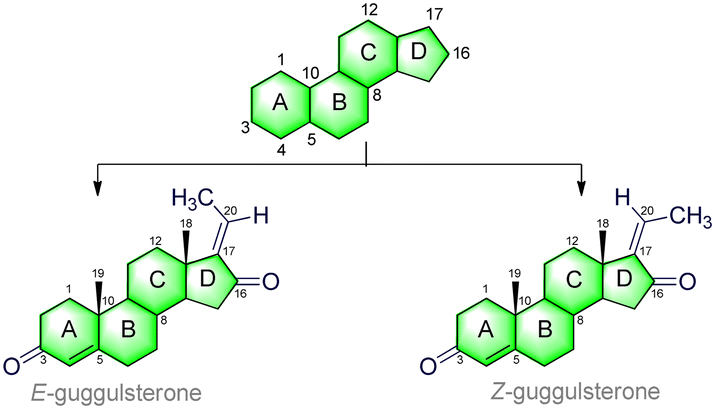
3. Structure of guggulsterone
GS (C21H28O2) is a phytosteroid family of natural products, chemically classed as pregna-4,17-diene-3-16-dione.66 It is a tetracyclic ring-fused molecule consisting of three numbers of six-membered cyclohexane rings (A, B, and C) and one five-membered cyclopentane ring (D); the basic structure is represented in Table 1. These rings (A–D) are constructed in a highly specific tetracyclic 17-carbon core that has a perhydrocyclopentano[α]phenanthrene orientation. The 17 carbons are numbered in ascending order starting with the ‘A’ ring of the GS molecule, continuing onto the ‘B’ ring, onto the ‘C’ ring, and ending in the ‘D’ ring. GS has carbonyl (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) groups in positions C-3 and C-16, and also possesses an unsaturated (CC) at the junction of the C(4) and C(5) carbons. The ethylidene (CH–CH3) group is attached to the C(17) position, which gives its two isomeric forms: cis (Z) and trans (E). Moreover, a total of five numbers of stereocenters (at C*(8), C*(9), C*(10), C*(13), and C*(14) positions) can be found in the skeleton. This molecule follows Pfizer's rule of five, including a MW < 500 Da (here, MW = 312.45 g mol−1), number of H-bond donors < 5 (here, HBDs = 0), and number of H-bond acceptors < 10 (here, HBAs = 2). Also, there is no violation of Veber and co-workers rules, including number of rotatable bonds < 10 (here, NBRs = 0), and polar surface area < 140 Å2 (here, TPSA = 34.14 Å2).67 More details are presented in Table 1.
O) groups in positions C-3 and C-16, and also possesses an unsaturated (CC) at the junction of the C(4) and C(5) carbons. The ethylidene (CH–CH3) group is attached to the C(17) position, which gives its two isomeric forms: cis (Z) and trans (E). Moreover, a total of five numbers of stereocenters (at C*(8), C*(9), C*(10), C*(13), and C*(14) positions) can be found in the skeleton. This molecule follows Pfizer's rule of five, including a MW < 500 Da (here, MW = 312.45 g mol−1), number of H-bond donors < 5 (here, HBDs = 0), and number of H-bond acceptors < 10 (here, HBAs = 2). Also, there is no violation of Veber and co-workers rules, including number of rotatable bonds < 10 (here, NBRs = 0), and polar surface area < 140 Å2 (here, TPSA = 34.14 Å2).67 More details are presented in Table 1.
|
|
||
|---|---|---|
| No | Structural details | |
| 1 | Molecule class | Steroid |
| 2 | Molecular formula | C21H28O2 |
| 3 | Molecular weight | 312.45 g mol−1 |
| 4 | Num. heavy atoms | 23 |
| 5 | Num. arom. heavy atoms | 0 |
| 6 | Fraction C-sp3 | 0.71 |
| 7 | Num. rotatable bonds | 0 |
| 8 | Num. H-bond acceptors | 2 |
| 9 | Num. H-bond donors | 0 |
| 10 | Complexity | 640 |
| 11 | Stereocenter count | 5 |
| 12 | Molar refractivity | 93.54 |
| 13 | TPSA | 34.14 Å2 |
In 2001, Sarkhel and co-workers68 reported the crystal structure of the cis-isomer, which is represented in Fig. 2. The bond distances and angles of the trans-isomer were in good agreement with those of the cis-isomer.69 The trans-isomers, ‘A’ ring exists in the distorted sofa conformation, while the ‘B’ and ‘C’ rings adopt the distorted chair conformation. The cyclopentane ‘D’ ring is intermediate between the half-chair and envelope conformations. The ‘A/B’ ring junctions is quasi-trans, while the ‘B/C’ and ‘C/D’ ring systems are trans-fused about the C(8)–C(9) and C(13)–C(14) bonds, respectively. The steroid CO has a small twist, as shown by the C(19)–C(10)⋯C(13)–C(18) carbon and pseudo-torsion angle being found to be 7.2°. Moreover, the intramolecular and intermolecular hydrogen bonding (C–H⋯O) in GS stabilizes the crystal structure.
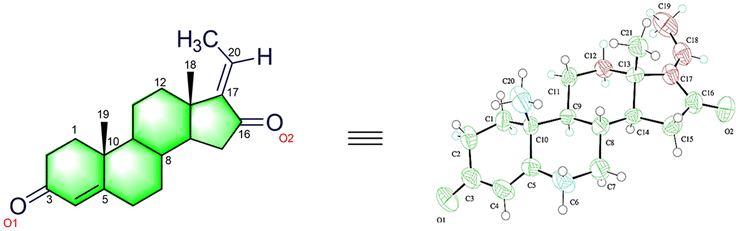 | ||
| Fig. 2 Crystal diagram of (E)-guggulsterone reported by Sarkhel and co-workers. | ||
4. Chemical synthesis of guggulsterone
The molecules that make up living systems have always fascinated and inspired synthetic organic chemists. The synthesis and spatial arrangements of isomeric GS were reported long before its isolation.70 The isolation was done by Patil and co-workers71 from the oleo-resin or gum of the tree C. wightii. The isolated compounds of GS are similar to the synthesized compounds in all aspects. The therapeutic value, usage, and demand for these compounds is increasing. However, the biologically active lipids from C. wightii are not readily available, and many trees are needed to collect the resin. Moreover, the resin has a low concentration of GS. In this context, organic chemists have developed several synthetic routes for GS starting from various steroid precursors. Ever since the discovery and first characterization of steroids in the 1920s and 1930s. Scientists have faced several challenges in their synthesis,72 including the establishment of all-carbon quaternary stereocenters, multiple redox manipulations, and the assembly of polycyclic ring systems with defined stereo-chemistries at ring junctions.Continuous improvements in chemical synthesis and transformation over the past two centuries have been of great importance in improving the chemical processing efficiency and in introducing some aspects of green chemistry using cost-effective synthetic precursors and catalysts. Steroidal precursors are important substrates for producing several bioactive synthetic and natural steroids, including GS. Generally, 16-DPA (16-dehydropregnenolone acetate), 4-AD (4-androstene-3,17-dione), ADD (1,4-androstadiene-3,17-dione), and 9-OH-AD (9α-hydroxyandrost-4- ene-3,17-dione) are the most widely used precursors for the synthesis of several bioactive steroid molecules. Consequently, these molecules are highly in demand in the pharma industry.73,74 Several commercially available steroids have been used as starting materials for GS synthesis (Fig. 3).
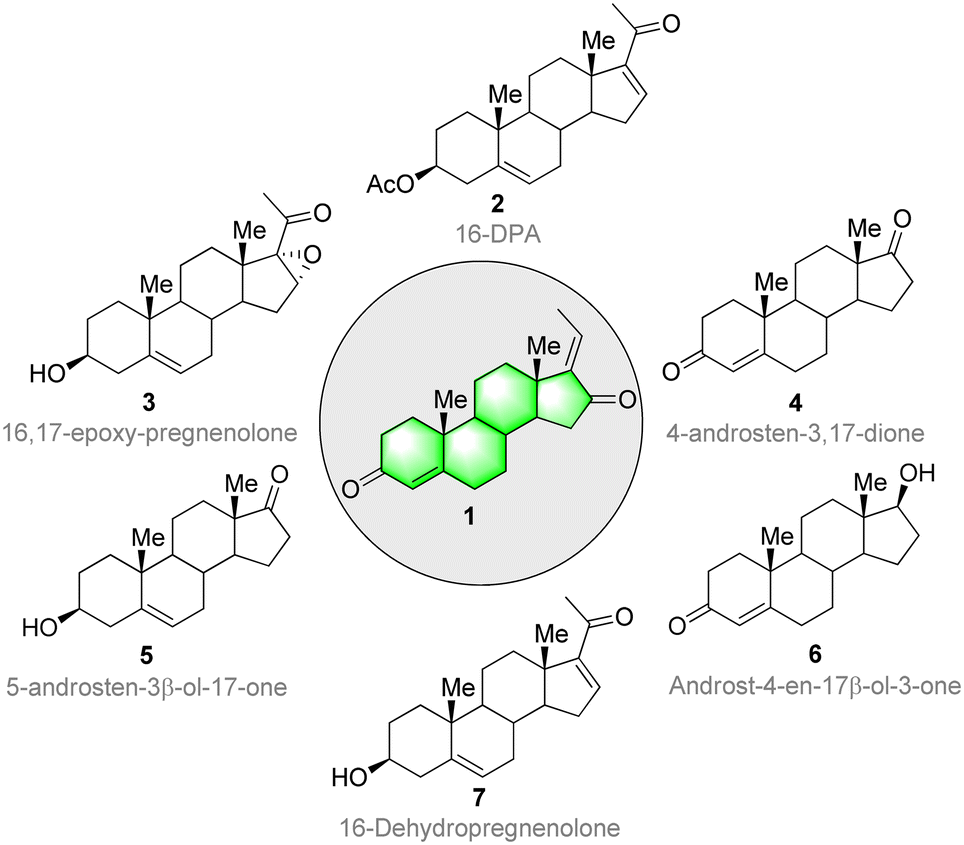 | ||
| Fig. 3 Structures of starting materials for guggulsterone synthesis. | ||
The first synthetic strategy for isomeric GS started from 16-DPA (2). The isomeric allylic alcohol 5,17(20)-pregnadiene-3β,16-diol (9) was the key intermediate in the process, and was generated through a reduction of the α,β-unsaturated fraction of 16-DPA (2) followed by an allylic rearrangement (Fig. 4). Also, the Oppenauer oxidation of compound 10 afforded a mixture of trans–cis diones (Z/E GS) in the ratio of about 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, respectively.70 However, the process had several drawbacks. Since the yield of the diol compound 9 was only obtained in a poor yield (21.7%), it was impossible to prepare the final product on a large scale. Moreover, in the reduction process, a by-product through 1,4-hydride addition is always inevitable, so a purification step was necessary, while the use of pyrophoric and flammable reagents, such as lithium aluminum hydride (LiAlH4), and highly volatile solvents at the industrial scale is another limiting factor. In another alternative process, the α,β-unsaturated fraction of 16-DPA (2) was converted to 16,17-epoxy-carbonyl (3) instead of allylic alcohols (9). Furthermore, conversion of the carbonyl fraction to the methylene group by the hydrazone derivative under Huang–Minlon conditions afforded the key intermediate. Then, a further Oppenauer oxidation of compound 10 led to the formation of GS. In this method, the formation of a by-product, pyrazone, in appreciably high yields lowered the yield of the final product drastically. Moreover, the conversion of CC into oxiranes with H2O2 was not reproducible and instead a conjugate addition product was obtained in the reported conditions.
:1, respectively.70 However, the process had several drawbacks. Since the yield of the diol compound 9 was only obtained in a poor yield (21.7%), it was impossible to prepare the final product on a large scale. Moreover, in the reduction process, a by-product through 1,4-hydride addition is always inevitable, so a purification step was necessary, while the use of pyrophoric and flammable reagents, such as lithium aluminum hydride (LiAlH4), and highly volatile solvents at the industrial scale is another limiting factor. In another alternative process, the α,β-unsaturated fraction of 16-DPA (2) was converted to 16,17-epoxy-carbonyl (3) instead of allylic alcohols (9). Furthermore, conversion of the carbonyl fraction to the methylene group by the hydrazone derivative under Huang–Minlon conditions afforded the key intermediate. Then, a further Oppenauer oxidation of compound 10 led to the formation of GS. In this method, the formation of a by-product, pyrazone, in appreciably high yields lowered the yield of the final product drastically. Moreover, the conversion of CC into oxiranes with H2O2 was not reproducible and instead a conjugate addition product was obtained in the reported conditions.
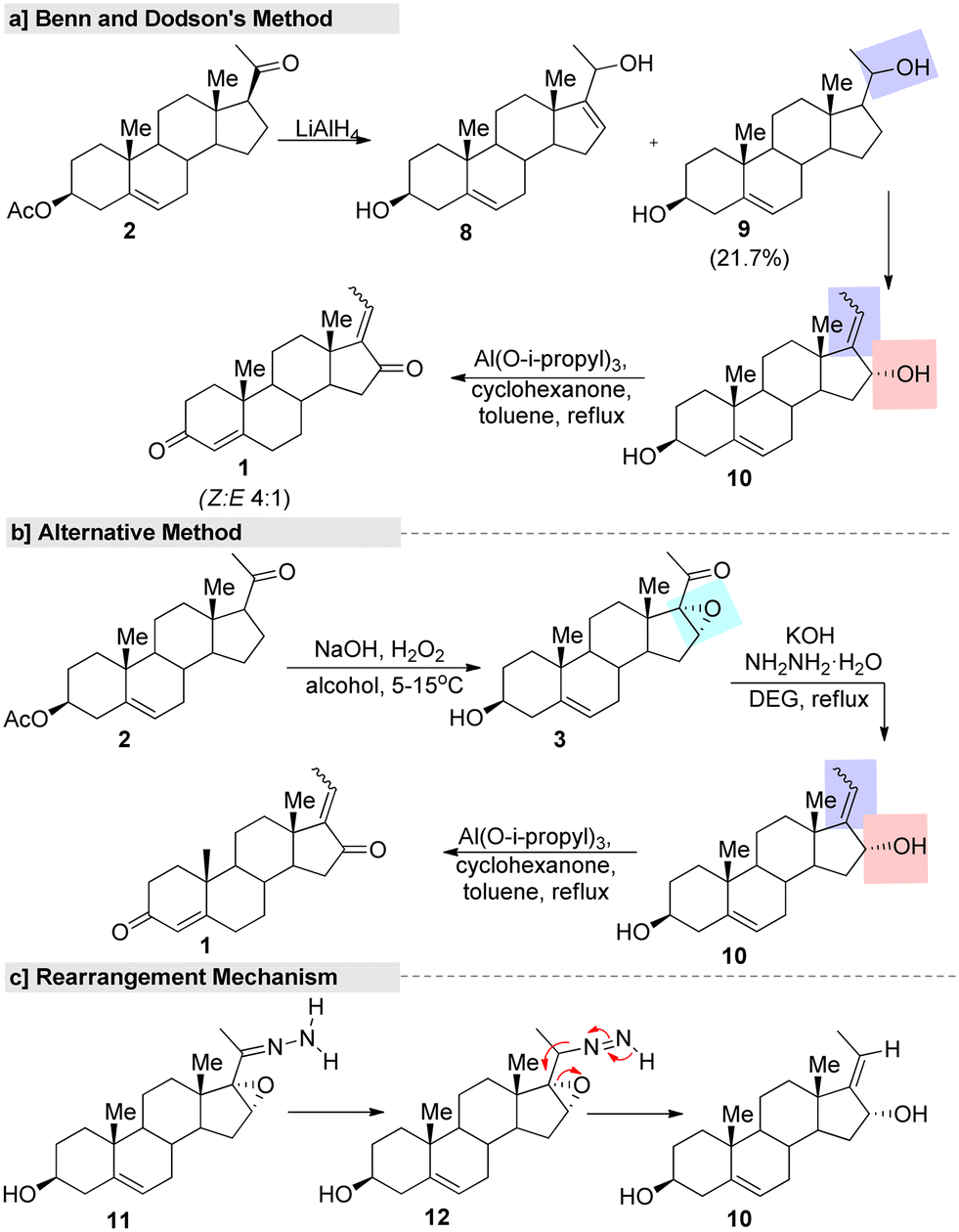 | ||
| Fig. 4 First synthetic route for (E/Z)-guggulsterone, (a) Benn and Dodson's method, (b) alternative method and (c) rearrangement mechanism. | ||
In this context, several improved and efficient methods for the preparation of GS have been developed by various research groups. Notably, Pratap's research group75 reported an upgraded method for the synthesis of GS in which they converted 16-DPA (2) into 3-β-hydroxy-16-α and 17-oxido-5 pregnen-20-one (3) under a hydrogen peroxide-urea (UHP) adduct system. Further, the hydroxylated product 10 was obtained from a key intermediate 3, with this reaction promoted by hydrazine. Later, the Oppenauer oxidation of compound 10 led to the formation of the desired GS in a good yield (Fig. 5a). Additionally, they developed another improved method in which the unsaturated carbonyl fraction of compound 2 was converted into 16,17-epozy carbonyls followed by a Kishner reduction–elimination under Huang–Minlon conditions to produce the key intermediate.75 In the same year, Gokaraju and co-workers76 invented a three-step method that involved the epoxidation of 2 by H2O2 oxidant with the assistance of a co-base. Further, compound 3 was subjected to a Wolff–Kishner reduction and elimination under hydrazine and a strong base. Finally, isomeric GS was obtained through an Oppenauer oxidation (Fig. 5b). The ration of Z and E in the final product was found to be in the range of 10 to 0.1 and the purity of GS was >99% according to HPLC.
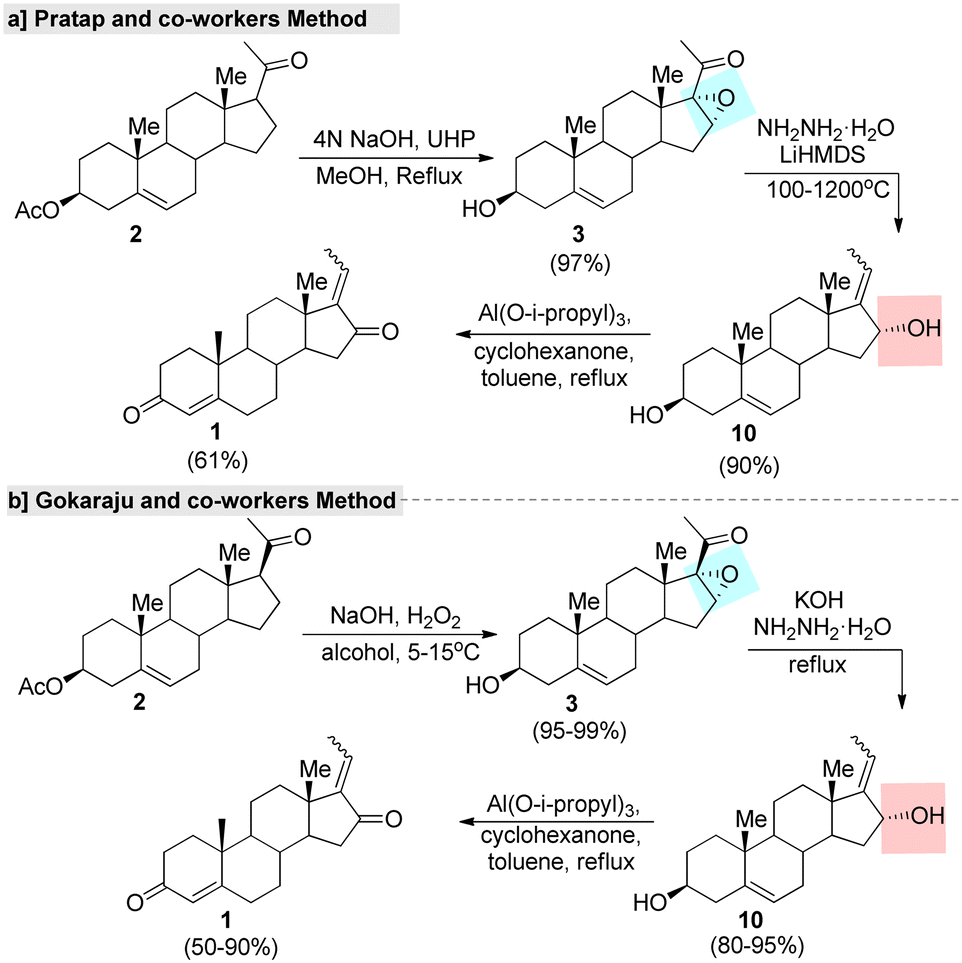 | ||
| Fig. 5 Improved methods for guggulsterone, (a) Pratap and co-worker's method and (b) Gokaraju and co-worker's method. | ||
Ham and co-workers77 described a regioselective synthesis of GS using 98% pure hydrazine monohydrate (Fig. 6). In this two-step synthesis, initially the cis-diol was prepared from a steroid 16,17-epoxy-pregnenolone (3) via hydrazine reduction. The stereoselectivity and reaction outcome of this method differed from those reported in the literature. For example, Benn and Dodson's method gave cis- and trans-diol as a mixture with an overall reaction yield of 22%, under specific conditions. In another report, a total yield of 47% as a mixture of cis- and trans-diols was prepared by Kessar and Rampal78 using 80% pure hydrazine. These reports, clearly showed that the regio-selectivity and reaction yield of the cis-diol depends on the purity of the hydrazine monohydrate and the course of the reaction and the reaction conditions.
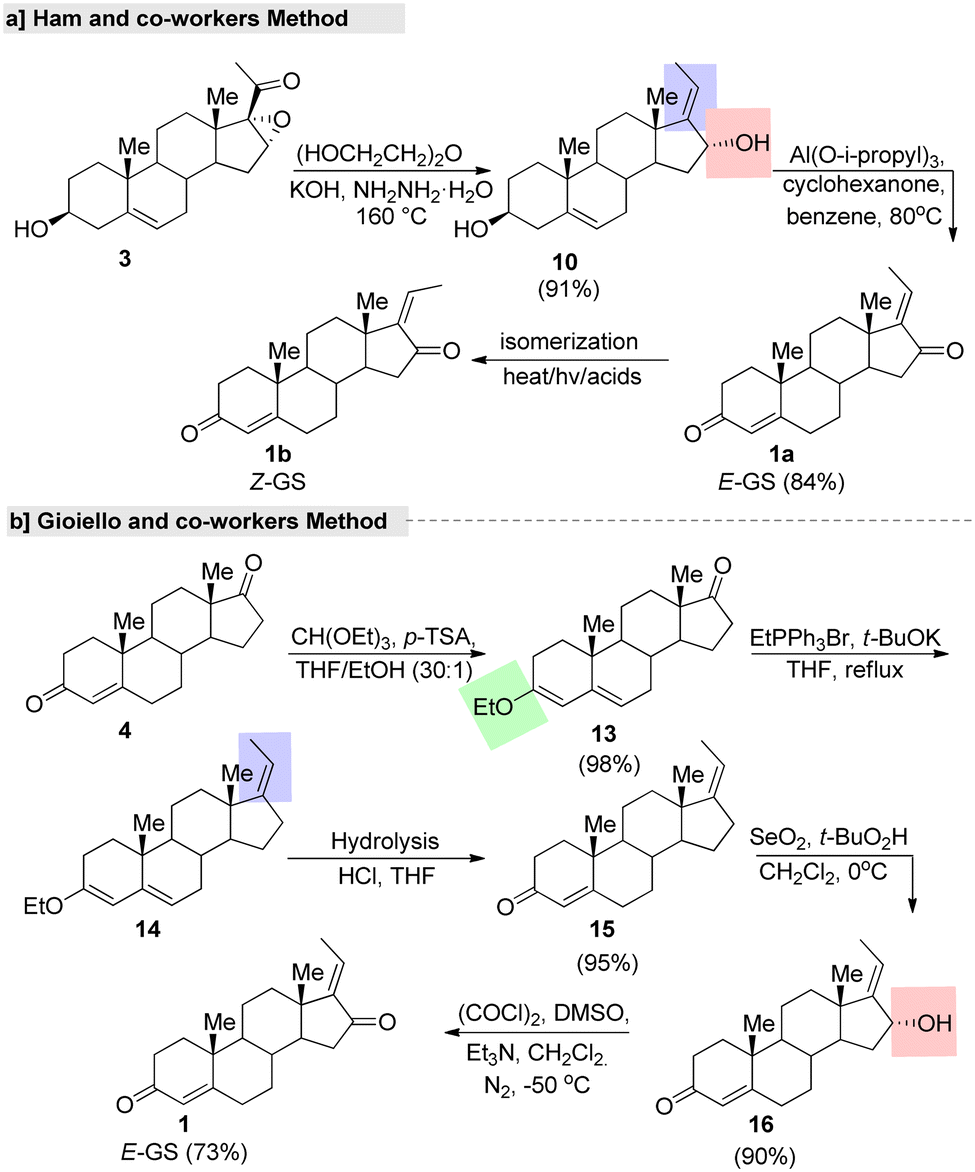 | ||
| Fig. 6 Shortest fabrication routes for guggulsterone, (a) Ham and co-worker's method and (b) Gioiello and co-worker's method. | ||
In another study, an efficient and gram-scale regioselective production of GS was reported by Gioiello and co-workers.79 This total synthesis started with protection of the keto group of androsten-3,17-dione (4), followed by regioselective alkylation (C-17 side chain generation) by the commercially available EtPPh3Br under t-BuOK as a base in THF. Further, C-16 oxidation afforded (E)-GS in good yields with a high stereoselective manner (Fig. 6b). Further, the same group explored the synthesis of (Z)-GS by an isomerization reaction under a strongly acidic cation exchange resin (Amberlyst 15®) in a benzene system through sonication. After silica gel purification of the isomer (E:Z = 39:61), (Z)-GS was isolated in a 58% yield. We can adopt this straightforward synthetic route for the gram-scale synthesis of isomeric pure (E)-GS. The main advantages of this strategy are the use of low-cost easily available starting materials and the highest rate of reaction. In addition, they established the relevant isomeric purity (equal to 98.1% for the (E)-GS isomer) via normal-phase HPLC analysis.
Chin et al.80 utilized natural steroids, such as androst-4-ene-3,17-dione (androstenedione, 4), 5-androsten-3-β-ol-17-one (androstenolone, 5), and 17-β-hydroxyandrost-4-en-3-one (testosterone, 6) for assembling GS. The synthetic methods using these three precursors are outlined in Fig. 8. Androstenedione (4) is generally used as a precursor of testosterone and other androgens, as well as of estrogens like estrone. In this method, first ketone protection was carried out with ethyane-1,2-dithiol (EDT) under a BF3 system. Further, the Wittig reaction provided the C-17 olefine steroid. This was followed by deprotection, sterioselective hydroxylation, and oxidation, resulting in 16α-(E)-GS (16). Finally, the treatment of compound 16 with activated MnO2 led to the formation of the single enantiomer (E)-GS in 94% purity with an overall yield of 68% (Fig. 7a). The second method started with androstenolone 5 (Fig. 8b). Initially, olefination on this compound 5 was achieved by a Wittig reaction without the C-3 hydroxyl (–OH) group protection. Further, compound 19 was treated under a SeO2-TBHP system, followed by the Oppenauer oxidation of compound 10 to give the formation of the desired product.
 | ||
| Fig. 7 Chin and co-workers' synthesis of guggulsterone from natural steroids, (a) androstenedione and (b) androstenolone. | ||
 | ||
| Fig. 8 Chin and co-workers' synthesis of guggulsterone from natural precursors, (a) androstenolone and (b) testosterone. | ||
In another method,80 the oxidation of compound 19 with pyridinium chlorochromate (PCC) gave the desired oxidized product 20, which was treated under the strong base t-BuOK to give the formation of compound 15 in a good yield. Further, compound 15 was oxidized under a SeO2–TBHP system to form the intermediate 16. Further, the MnO2 system afforded the single enantiomer of GS from 16α-(E)-GS (16) with a 94% yield (Fig. 8a). Finally, GS was synthesized from testosteron (6). Protection of the allylic ketone of 6 by ketals in the presence of TsOH (p-toluenesulfonic acid) afforded compound 21. Next, a keto group was introduced at the C-(17) position of compound 21 under a PCC system gave another intermediate, 22, which underwent a Wittig reaction with ethyltriphenyl phosphonium ylide to give the desired C-17 olefinated compound 23. Further, deprotection of the ketal group on 23 followed by sterioselective hydroxylative and oxidation under the SeO2–TBHP system afforded the final product (Fig. 8b).
Very recently, Reddy and co-workers81 developed an efficient method for enone transposition with the help of a silyl group. Regioselective enantio switching, substituent shuffling, and Z-selectivity are the highlights of the method. This approach was successfully applied to the formal synthesis of (E)-GS, which was accomplished in a short sequence (Fig. 9). Initially, 16-dehydropregnenolone (7) was treated with PhMe2SiLi, which gave the silyl addition product 24. Further, compound 24 was treated with a catalytic amount of TFA in a CH3CN/H2O system. The reaction furnished 25 as a major product, and then proto-desilylation of 25 afforded the desired alcohol 10. Finally, this intermediate 10 was transformed into the corresponding (E)-GS.
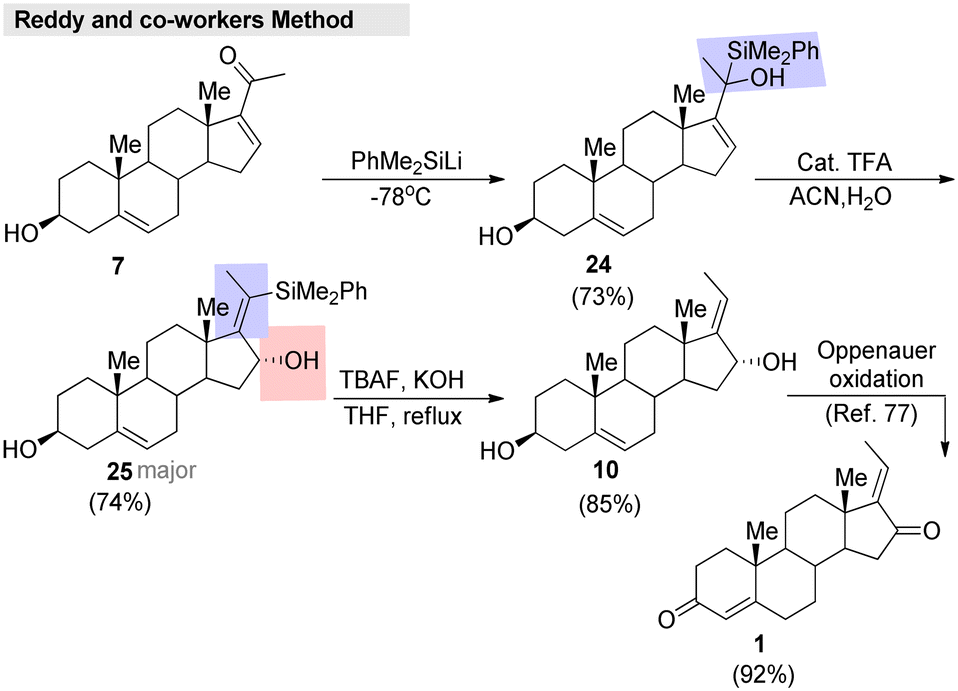 | ||
| Fig. 9 Reddy and co-workers' formal synthesis of guggulsterone. | ||
In conclusion, GS synthesis remains a topic of interest to synthetic organic chemists and the pharmaceutical industry today due to its distinct chemical biology, as clearly showed by Athawale and co-workers' efforts, among others.81 Its production can be achieved by modifying readily available steroidal compounds. In 1964, Benn and Dodson70 made the first chemical synthetic attempt from 16-DPA,which involved mainly C(16)-carbonylation and C(17)-carbonyl olefination reaction. For the carbonylation reaction, typically Oppenauer oxidation is used; this reaction can be performed with a Lewis acid derived from aluminum in a stoichiometric way. Moreover, this process is well known for oxidizing valuable alcohols into their desired ketones. In particular, it has been verified to be extremely successful in the oxidation of natural sterols. For allylation, mostly the Wittig reaction is used. This reaction has several advantages over other olefination strategies; in particular, the alkene (–CH2CH2–R) group always directly substitutes with a carbonyl group (CO). In comparison, many other carbonyl olefination reactions often happen via double-bond rearrangements. Furthermore, the factors influencing the (E)- and (Z)-stereo-selectivity are well understood and can be easily controlled by a careful choice of the reaction conditions and phosphorus reagent.
The use of steroid precursors, such as 16-DPA, has long served as a starting point for the synthesis of many steroidal molecules, including GS. As reasonable and versatile starting materials, such molecules have influenced modern synthetic chemistry. Moreover, the use of a stable polycyclic ring core with defined quaternary stereocenters at the ring junctions avoids major challenges in steroid synthesis. Although there are several conventional methods that are well known for GS synthesis, these reactions often suffer from several drawbacks, such as the need to handle toxic, sensitive, and expensive reagents and chemicals, poor selectivity, and low yields. The advanced synthetic strategies for GS are limited. Therefore, novel strategies and improved protocols are needed for the established methods to meet modern synthesis criteria, such as short, sustainable, safe, and cost-effective methods, or using a framework of 12 green principles.82 Globally, green chemistry demonstrates high atom economy, and promotes the use of green-reaction conditions over conventional batch synthesis (Fig. 10). Ultrasound-mediated synthesis,83,84 microwave irradiation technology,85,86 photo-catalysis,87,88 and microbial/enzymatic transformations89,90 all belong to this category. Many researchers have developed several eco-friendly methods/approaches for different steroid molecules.2 However, organic chemists must learn and adopt green synthetic approaches for the synthesis of steroid molecules, especially GS-like bioactive molecules.
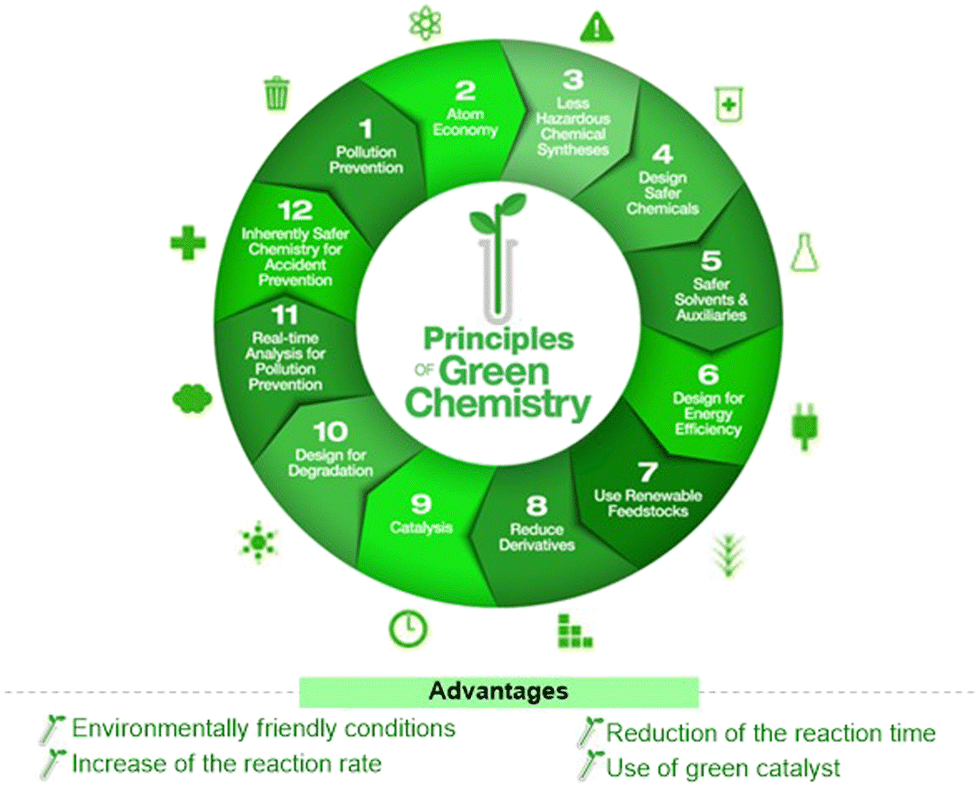 | ||
| Fig. 10 Advantages of green chemistry processes over conventional chemistry synthesis. | ||
5. Structural modification and improved activities
Steroids are a major focus of research not only because of their unique structural complexity and diversity but also because of their excellent ability to bind nuclear and membrane receptors and as they display diverse biochemistries.91 Making even a small structural modification to one of their key parts is one of the most suitable ways to increase their potency and selectivity toward various targets.92a The basic underlying principle is that similar structures will present similar biological activities. This approach is used in many drug-development programs worldwide. There are several clinical anticancer drug molecules optimized from natural lead compounds. For example, calusterone92b and fluoxymesterone92c are derived from the chemical manipulation of testosterone.As discussed in the above section, GS is a potent biologically active molecule with a multi-step chemical synthetic route. Structural modifications of the GS molecule can alter its selectivity, efficacy, and potency. In general, structurally modified analogues are prepared either from its moiety or other steroid precursors. Herein we sequentially discussed the possible modification sites (Fig. 11) and reported synthetic approaches for GS derivatives and their modified activities.
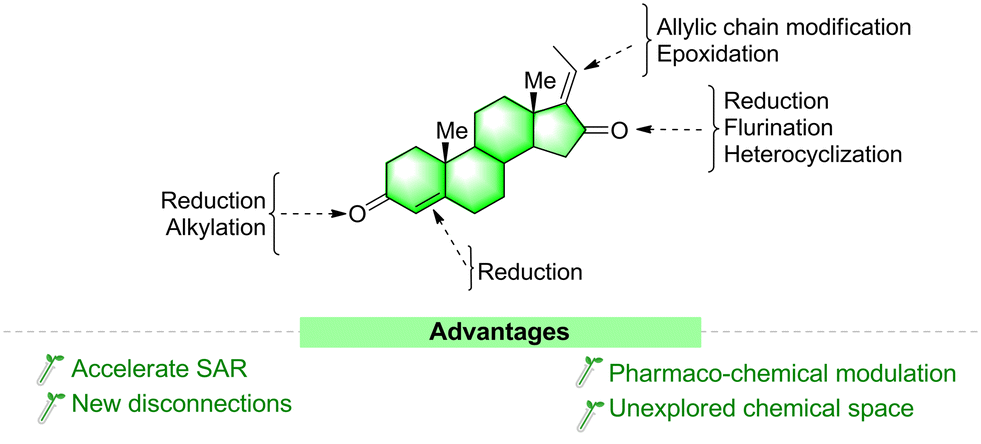 | ||
| Fig. 11 Structural modification sites of guggulsterone. | ||
Hybrid compounds have captured an important position in cancer therapy, and the hybridization of triazole scaffolds with other anticancer pharmacophores may furnish valuable therapeutic interventions for cancer treatment. A study by Lee and colleagues27 reported on triazole-guggulsterone hybrids as novel kidney cell protective agents. Based on a preliminary investigation, they focused on modifying the structure of the (E) isomer because it is less efficient than the (Z) form. To improve the efficacy of the (E) isomer, they modified the ketone (C-16) group by introducing various alkyl and phenyl functionalities with a triazole moiety through a Cu(II)-catalyzed [3 + 2] click reaction. The multi-step synthetic routes are summarized in Fig. 12. The reaction was started from the steroidal precursor's 5-androsten-3β-ol-17-one (5), and the regular three-step process (alkylation, oxidation, and hydroxylation) afforded 16-α-(E)-ol (16) in a good yield. Further, mesylation followed by azide anion (N3−) addition on compound 16 afforded a key intermediate 26. For various triazole-guggulsterone derivatives, they treated various aliphatic and aromatic alkynes 27 with 26 under a CuSO4/sodium ascrobate (Cu-SA) system. After the successful synthesis of triazole-guggulsterone hybrids, they tested their cellular protective effects through a cisplatin-treated cultured LLCPK1 kidney epithelial cell line model. Among the tested derivatives, (4-(1H-1,2,3-triazol-4-yl)phenyl) methanol-linked compound 28b was found to be the most potent.27 The protection effect of this compound was stronger than that of the positive control compound N-acetylcysteine. The cell viability of compound 28b at a concentration of 25 μM was found to be >90% and it was more potent than (Z)-GS. The other derivative with the 4-fluorophenyl (28c) group also showed a similar potency, and due to the steric hindrance of the 4-methyl phenyl group, compound 28a exhibited lower potency. Further investigation into 28b showed significant reductions in p-JNK, p-p38, and cleaved caspase-3 protein levels. These results indicate that compound 28b inhibited the activation of the MAPK signaling pathway, which is involved in apoptosis.
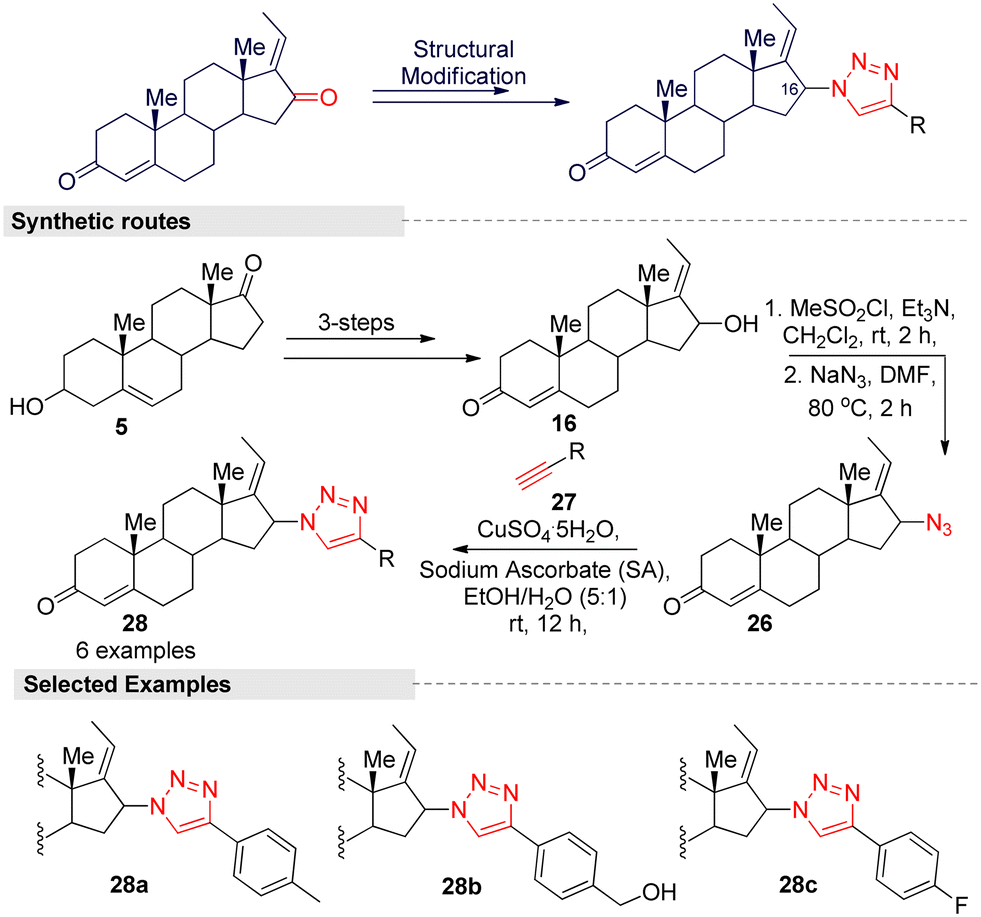 | ||
| Fig. 12 Lee and co-workers' method for the synthesis of (E)-guggulsterone-triazole derivatives. | ||
It is well known that the triazole skeletal moiety is an elite building block in the discovery of potent anticancer agents, and some of its analogues have already been made available to healthcare and clinics or are under clinical trials for fighting against various cancers.93,94 Moreover, GS is known to inhibit cancer cell proliferation and also to sensitize drug resistance in cancer cells. From this interesting result, we also hope that this type of triazole-guggulsterone hybrid molecules, especially 28b, may lead to the development of kidney protective agents, and that the co-treatment of compound 28b cisplatins may alleviate nephrotoxicity without compromising the therapeutic efficacy of cisplatin.
Kohyama, et al.28 designed GS derivatives (30–31) by ‘A’ and ‘D’ ring modification, for which they adopted the Kang and co-worker methods. This multi-step synthesis started from an endogenous steroid hormone precursor 5. The different triphenyl phosphonium ylides reacted with compound 5 to afford ‘D’ ring functionalized derivatives (29a–d). Here higher regioselective products (E)-alkenes were formed. Further, the synthesized compounds 29a–d were subjected to allylic oxidation followed by Oppenauer reaction to provide (E)-isomers (30a–c) and natural GS (1a). Later, they converted 30a–b into its corresponding (Z)-isomers 31a–b under Kang's isomerization conditions (Fig. 13).
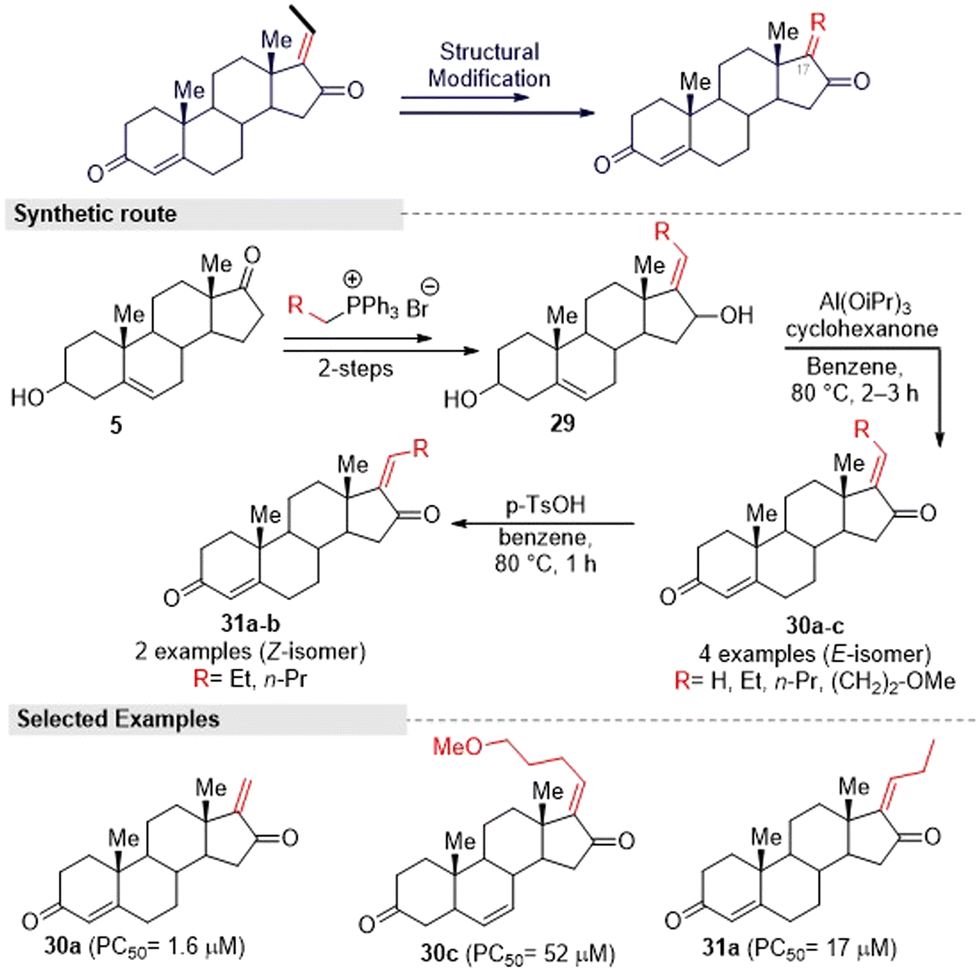 | ||
| Fig. 13 Kohyama and co-workers' designed method for obtaining guggulsterone derivatives via A- and D-ring modification. | ||
Also, for the synthesis of an enone-deficient derivative (32), they initiated a reaction with compound 19, which was obtained via the Wittig reaction. The selective Oppenauer reaction produced an inseparable mixture of (Z:E) isomers of 32 in a 9:1 ratio (Fig. 14, synthetic route I). Furthermore, the synthesis of compound 34 was achieved from the known compound 5 through a three-step sequence. The hydrogenation of compound 34 proceeded in a stereoselective manner to afford a single diastereomeric compound 35 (Fig. 14, synthetic route II). After that, they evaluated the anti-austerity activity of (E/Z) GS and the other nine synthesized compounds against the PANC-1 tumor cell line in nutrient nutrient-deprived medium (NDM). Among the tested compounds, 30a and 34 showed the most potent preferential cytotoxicity against the tested cells with PC50 values of 1.6 μM and 3.2 μM, respectively. The selectivity index of 30a and 34 in NDM was 6.5 times and 34 times compared to under nutrient-rich conditions (DMEM), which showed the structure–activity relationship. The unsubstituted exocyclic double bond within the enone moiety in the D-ring of GS derivatives seems essential for the potent activity, and GS derivatives with substituents at the exocyclic double bond displayed weak activity. Further investigation, it was found that both of these compounds altered PANC-1 cell morphology, leading to cell death at the sub-micromolar concentration range, which shows their potential as lead structures for anti-austerity drug development.
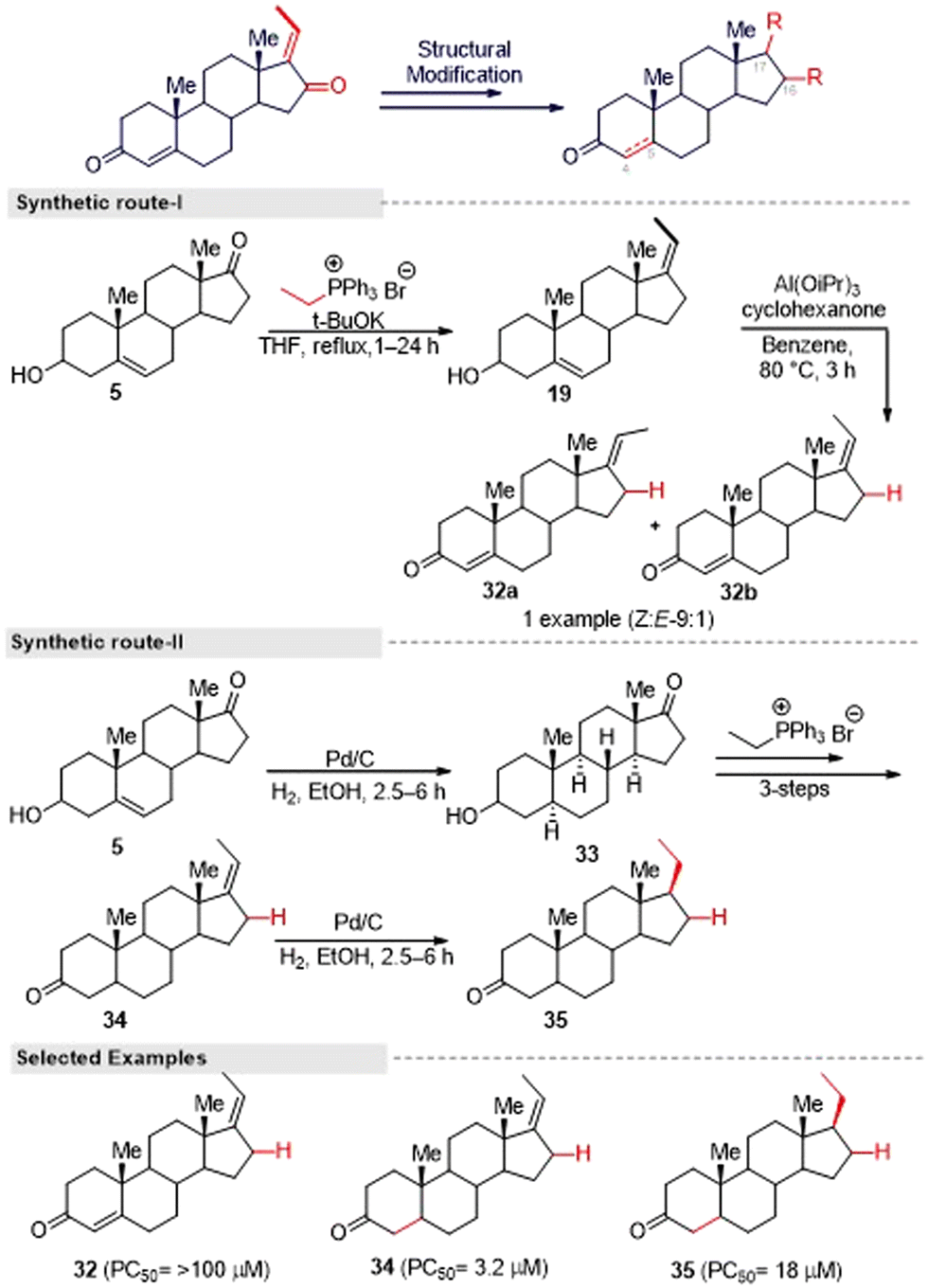 | ||
| Fig. 14 Lee and co-workers' method for the synthesis of (E)-guggulsterone-triazole derivatives. | ||
Interestingly, Abdellatef and colleagues29 adopted a multi-step synthetic approach described by Kohyama's group28 and prepared a series of GS derivatives (30a–f) via alkyl chain modification. Further, they successfully evaluated their IKK and NF-κB signaling strength and metastatic potential for breast cancer cells. Compound 30a was identified as the most potent molecule and strongly inhibited NF-κB activity and nuclear translocation in conjunction with displaying potent toxicity in NF-κB-dependent breast cancer cell lines. In addition, it could inhibit breast cancer cell invasion and migration, and 30a treatment blocked the IKK/NF-κB pathways and the expression of the target genes of NF-κB, such as MMP-2 and MMP9, in triple-negative breast cancer. Therefore, 30a is an attractive candidate to target NF-κB activation in breast cancer and has the potential for treating breast cancer growth and metastasis.
Inspired by the above hit GS analogues, Kohyama, et al.65 synthesized another two sets of GS derivatives via enone-deficient and electrophilic derivatives and successfully evaluated their cytotoxicity against the pancreas ductal adenocarcinoma cell line (PANC-1). For enone-deficient derivatives, dehydroepiandrosterone (5) was used as a synthetic precursor. The key intermediate 37 was synthesized via a three-step process, which involved alkylation, oxidation, and hydroxylation reactions. Further, it was treated with MOM and converted into the corresponding ether 38. Allylic oxidation of compound 30 to compound 37 proceeded in a diastereo-selective manner. Similarly, compounds 40 and 41 were synthesized from 5 and 39 in three steps (Fig. 15). Next, lower electrophilic derivatives (47–49) were synthesized via the Horner–Wadsworth–Emmons (HWE) reaction. The reaction of compound 42 and 43 with HWE reagent 44 led to the formation of the difluoroalkene silyl ethers 45 and 46 in moderate yields. The deprotection of these silyl ethers and subsequent oxidation gave the GS derivatives 47 and 48. Moreover, the MOM deprotection of 48 provided another derivative 49.
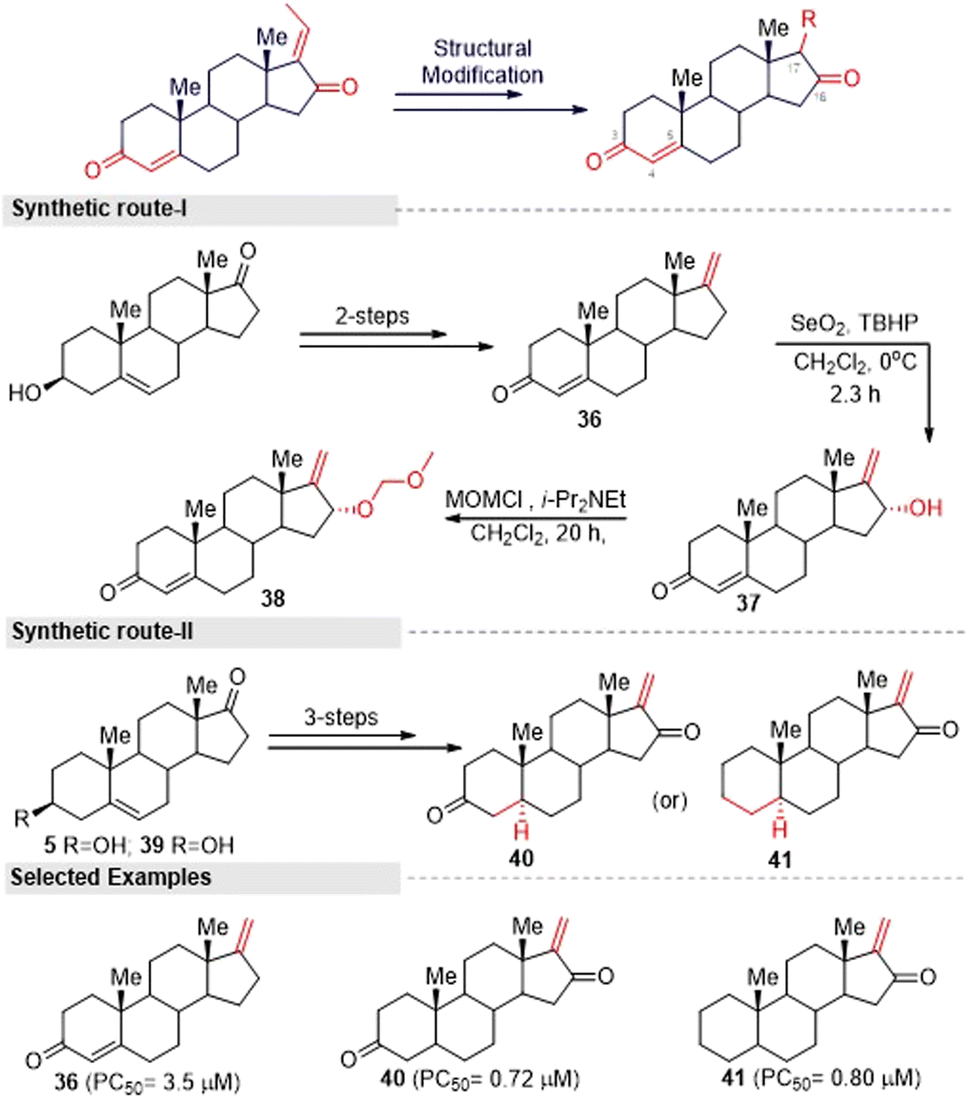 | ||
| Fig. 15 Kohyama, and co-workers' method for synthesizing enone-deficient guggulsterone derivatives. | ||
Furthermore, they synthesized compound 51 as an advanced enone-deficient derivative without an allylic H on the D-ring. This two-step sequence was effective for the conversion of 42 to the α-difluorinated alkene 50 (Fig. 16, synthetic route I). Finally, compound 51 was obtained through the TBS deprotection of 50 and subsequent oxidation (Fig. 16, synthetic route II). After the successful synthesis of GS derivatives, they evaluated their anticancer activity against human pancreatic cells (PANC-1). Among the tested derivatives, compounds 40 and 41 were found to be the most active against PANC-1 cells in NDM, exhibiting PC50 values of 0.72 μM and 0.80 μM, respectively. Their common structure was α-methylene cyclopentenone, a highly reactive Michael acceptor. Moreover, the derivatives that contained a β-di-substituted enone on the A-ring and lacked other enones or oxygen on the D-ring derivatives (for example, 36 and 47) displayed only moderate preferential cytotoxicities under NDM conditions and were inactive under DMEM conditions. Thus, compound 40 could be a promising lead compound for anticancer drug discovery. This compound was able to preferentially kill PANC-1 cells under NDM conditions. It also demonstrated antimetastatic potential by inhibiting the cell migratory and colony formation potentials of PANC-1 cells under DMEM conditions. Mechanistically, compound 40 was found to target the Akt/mTOR signaling pathway.
 | ||
| Fig. 16 Kohyama and co-workers' method for synthesizing gem-difluoroalkene guggulsterone derivatives. | ||
Additionally, Feng and Zhang developed a novel one-pot system for 18-norsteroid compound under peracetic acid (PAA)/acetic acid condition.95 The reaction consisted of three steps: epoxidation, stereoselective 1,2-migration, and a chemo-selective epoxidation reaction (Fig. 17). In general, the Wagner–Meerwein-type rearrangement is simpler than other methods for obtaining 18-norsteroids. However, the starting materials are limited to C(17)-oxygenated, such as 17-hydroxy or 16,17-epoxy steroids, and there is a tendency to crop just Δ13-18-norsteroids. Even worse, large amounts of toxic and corrosive acid reagents (H2SO4, HF, TsOH, etc.) are needed, as well as harsh reaction conditions and tedious work-up procedures. Therefore, this method is a modest and efficient method for the further screening of more potent 18-norsteroid actives.
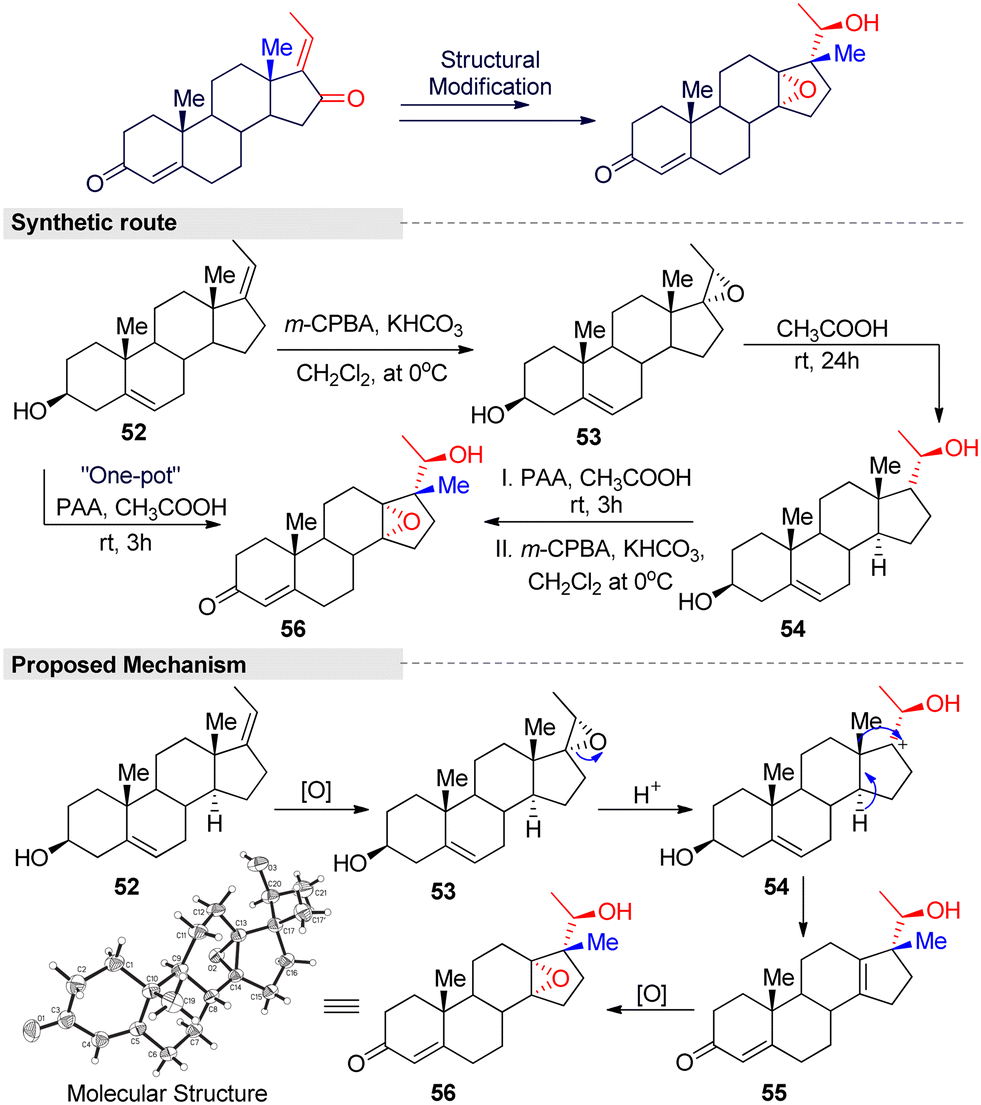 | ||
| Fig. 17 Feng and Zhang's method for obtaining the 18-norsteroid derivative. | ||
In conclusion, steroids differ from each other in the nature of the functional groups attached, their location, and the configuration of the steroid moieties. A small modification in the molecular skeleton of steroids can produce remarkable differences in their biological activities.96 Looking at the synthetic routes for structurally modified GS analogs, androstenolone is the main starting material for GS derivatives. The functionalization sites and upcoming groups were already described in the above sections and shown in Fig. 11. The improved activity of GS derivatives has been reported by several groups27–29,94,95 and compound 30a is one of the hit structurally modified GS molecules showing potent anticancer activity. Therefore, those currently working on this molecule should pay more attention to the area of structural modification and to the aspects of green chemistry and late-stage diversification, especially through C–H bond activation. C–H transformations have many advantages over conventional methods, such as new disconnections, step economy, sustainability, and efficiency.97–100 So this approach is an efficient way to quickly generate new chemical entities on a lead molecule without resorting to de novo synthesis. There are several late-stage diversification approaches in many natural products, such as estrone,101 resveratrol,102 artemisinin,103 juglone,104 bakuchiol,105 vitamin K3,106 and curcumin,107 were reported in the literature.
In addition, hetero-functionalized or fused steroids are considered important and effective compounds that are widely applied in medicinal and pharmaceutical fields. Thus, several research groups have developed novel steroidal molecules containing a heterocyclic linkage, either annulated or spiro-coupled to steroid A and D rings.108–110 For example, Kovács et al.111 developed a green catalytic system for androstene-oxadiazole hybrid molecules (Fig. 18).
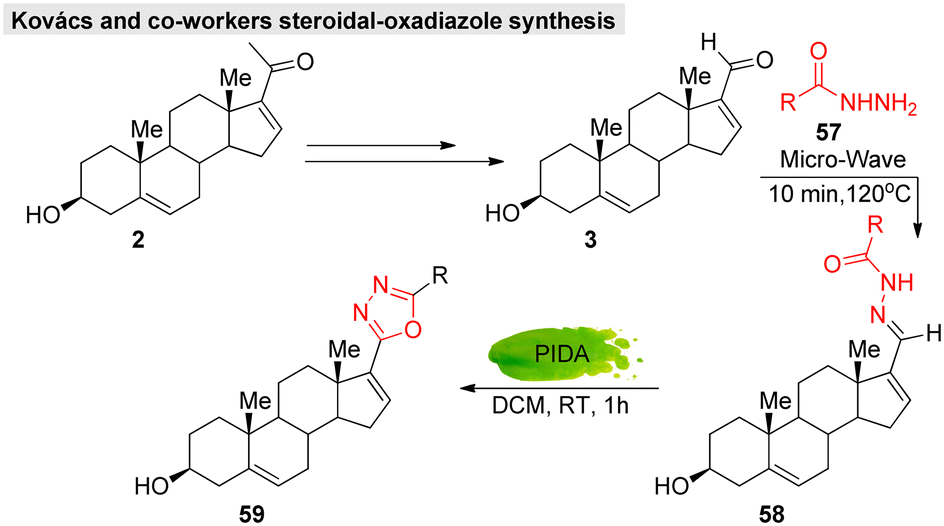 | ||
| Fig. 18 Kovacs and co-workers' green catalytic system for obtaining steroidal-oxadiazole derivatives. | ||
Moreover, they successfully evaluated their anticancer activity against various malignant cell lines, such as HeLa, MCF7, A2780, and A43. Likewise, several approaches have been reported for steroidal-thiazole,112 imidazolidine,113,114 isoxazoline,115 pyrazole,116 oxazole,117 tetrazole,118 pyrazoline,119 pyridine,120 and pyrimidine121 derivative syntheses. In GS chemistry, hetero-functionalized derivatives have so far been limited. Lee's group was the only one to work on the synthesis of guggulsterone-triazole hybrid molecules that showed potent cytotoxicity. Therefore, we can adopt these existing methodologies (with or without modifications) on GS, which may provide a new opportunity to explore its functionalities.
6. Conclusion and future perspectives
After 1990, more than half of the drugs introduced into the market are derivatives of natural products. Moreover, in the last decade, the steroidal market has experienced a significant increase in activity and attention due to the SARS-CoV2 pandemic. The development of advanced chemical technologies/catalysis systems have greatly influenced the field of natural product synthesis, including the synthesis of steroids.122b,c Since the discovery of the active principle “GS” from the oleo-resin or gum of the C. wightii tree in 1972, it has been identified as effective against several chronic diseases, and this molecule is now one of the highlights in medicinal chemistry. Day by day, the number of publications appearing in the scientific community is increasing, highlighting the ever-growing research interests, importance, and applications of this molecule (Fig. 19).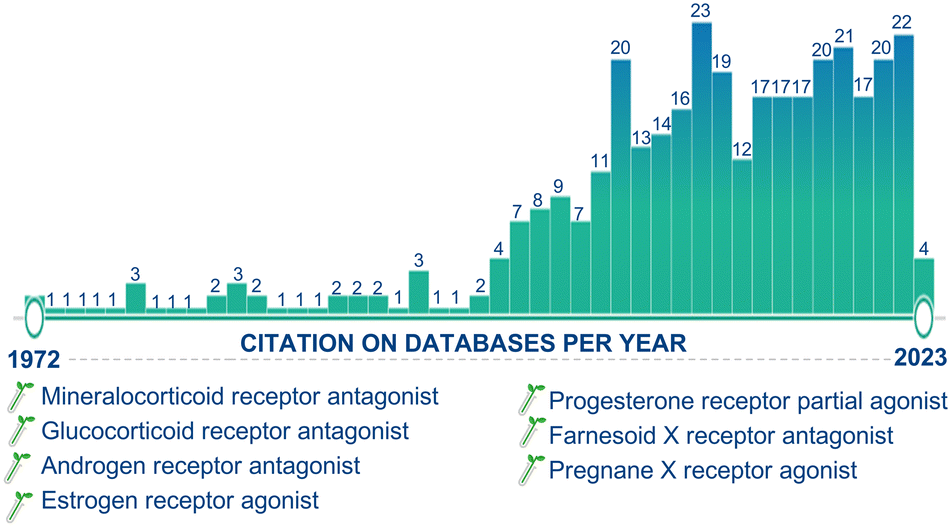 | ||
| Fig. 19 Guggulsterone citations that appear along the time line from 1972 till 2023. The Pubmed database was searched by the use of “guggulsterone” as a keyword. The plot shows the cumulative number of hits identified for each year after the first report of guggulsterone. | ||
Notably, GS has exhibited diverse biological activities by binding to nuclear receptors and modulating the expression of proteins involved in carcinogenesis. It has also been reported to regulate gene expression by exhibiting the regulation of other molecular targets, including transcription factors, such as nuclear factor (NF)-κB, signal transducer, and activator of transcription, and steroid receptors. The synthesis and spatial arrangements of isomeric GS have been reported long before their isolation. The first synthetic strategy for isomeric GS started from 16-DPA, and involved mainly C(16)-carbonylation and C(17)-carbonyl olefination reactions. After that, several conventional methods have been reported for GS synthesis, but these reactions often suffer from several drawbacks, such as the use of toxic and expensive chemicals, poor selectivity, and low yields. Advanced synthetic strategies for GS are limited. Therefore, novel strategies and improved/modified protocols are needed for the established methods to meet modern synthesis criteria, such as short, sustainable, safe, and cost-effective processes. We have discussed several synthetic methods with the concept of energy-efficient and environmentally friendly synthetic transformation for various bioactive molecules, including steroids. From this point of view, these methods can be adopted or modified for the production of GS and its derivatives for further development, application, and research.
As is well known, all biological activity depends on events occurring at the molecular level, and bioavailability is crucial for effective drugs. In general, phytosterols have a hydrophobic steroid skeleton, poor chemical stability, and limited bioavailability, which limit their utilization and efficacy. The bioavailability of phytosterols depends on several factors, including certain intestinal transporters, their different molecules, and genetic factors. Phytosterols and cholesterol have similar chemical structures, but the intestinal absorption rate of phytosterols is much lower than that of cholesterol; whereby less than 5% of phytosterols are absorbed, while 50–60% of cholesterol is absorbed.123a GS has been found in animal research to be orally active, displaying an absolute bioavailability of 42.9% after oral administration in rats, with a half-life of around 10 h in this species, indicating its good pharmacokinetic profile.123b Moreover, GS is characterized by a physiological solubility up to 50 μM, high plasma protein binding, and limited bioavailability.123c
To expand the application of GS-like phytosterols, their solubility, dispersibility, and bioavailability need to be increased by chemical or physical modifications.124 The structural modifications of phytosterols are performed in the following aspects: (i) chemical modification by esterification and transesterification with hydrophobic fatty acids and triacylglycerols to improve their oil solubility, (ii) chemical manipulation by coupling with various hydrophilic moieties to enhance their water solubility. These can be done by chemical,125 enzyme-catalyzed,126 or ionic liquid-catalyzed127 synthesis. Moreover, colloidal delivery systems,128 such as emulsions, oleogels, liposomes, and nanoparticles, have been shown to be effective in improving the water distribution, stability, and bioavailability of phytosterols via promoting their dissolution in the gastrointestinal tract, enhancing their uptake in targeted tissues and providing sustained release. From this perspective, these studies may provide further insights into the modulation of the pharmacodynamics and pharmacokinetics properties of GS and its derivatives in the future.
Author contributions
AKTP designed the work and down overall supervision. The authors contributed to the data preparation and drafted and revised the manuscript. All authors have read and approved the final manuscript.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
We gratefully acknowledge the support from Sreedhareeyam Farmherbs India Pvt. Ltd, Kerala India for carrying out this work.References
- W.-S. He, H. Zhu and Z.-Y. Chen, J. Agric. Food Chem., 2018, 66, 3047–3062 CrossRef CAS PubMed.
- P. Gupta and A. Mahajan, Environ. Chem. Lett., 2019, 17, 879–895 CrossRef CAS.
- A. Gupta, B. S. Kumar and A. S. Negi, J. Steroid Biochem. Mol. Biol., 2013, 137, 242–270 CrossRef CAS.
- J. A. R. Salvador, J. F. S. Carvalho, M. A. C. Neves, S. M. Silvestre, A. J. Leitao, M. M. C. Silva and M. L. S. E. Melo, Nat. Prod. Rep., 2013, 30, 324–374 RSC.
- N. Abbas, G. S. P. Matada, P. S. Dhiwar, S. Patel and G. Devasahayam, Anti-Cancer Agents Med. Chem., 2021, 21, 861–893 CAS.
- M. M. D. S. Cepa, E. J. Tavares da Silva, G. Correia-da-Silva, F. M. F. Roleira and N. A. A. Teixeira, J. Med. Chem., 2005, 48, 6379–6385 CrossRef CAS PubMed.
- S. C. Manson, R. E. Brown, A. Cerulli and C. F. Vidaurre, Respir. Med., 2009, 103, 975–994 CrossRef.
- A. Roohbakhsh, M. Iranshahy and M. Iranshahi, Curr. Med. Chem., 2016, 23, 498–517 CrossRef CAS PubMed.
- J. F. Yang, T. M. Chen, H. H. Chang, Y. L. Tsai, W. C. Tsai, W. Y. Huang, C. H. Lo, C. S. Lin, P. C. Shen and Y. Chen, Eur. J. Pharmacol., 2023, 938, 175411 CrossRef CAS PubMed.
- S. K. A. N. U. Rehman, F. Khan, O. Ullah, S. A. Halim, A. Khan, M. U. Anwar, S. M. Rahman, R. Csuk and A. Al-Harrasi, J. Mol. Struct., 2013, 1282, 135181 Search PubMed.
- Y. S. Kim, X. F. Li, K. H. Kang, B. Ryu and S. K. Kim, BMB Rep., 2014, 47, 433 CrossRef.
- S. K. Yousuf, R. Majeed, M. Ahmad, P. L. Sangwan, B. Purnima, A. K. Saxsena, K. A. Suri, D. Mukherjee and S. C. Taneja, Steroids, 2011, 76, 1213–1222 CrossRef CAS PubMed.
- N. Sharma, P. Palia, A. Chaudhary, K. Verma and I. Kumar, J. Drug Delivery Ther., 2020, 15, 325–332 CrossRef.
- P. Sarup, S. Bala and S. Kamboj, Scientifica, 2015, 2015, 138039 CrossRef.
- S. Sabarathinam, S. K. Rajappan Chandra and V. Thangavel Mahalingam, Sci. Rep., 2021, 11, 715 CrossRef CAS.
- P. Agarwal, R. R. M. S. Vaishnav and M. S. Ranawat, Biomed. J. Sci. Technol. Res., 2018, 6, 5355–5361 Search PubMed.
- Y. B. Tripathi, O. P. Malhotra and S. N. Tripathi, Planta Med., 1984, 50, 78–80 CrossRef CAS PubMed.
- L. Mester, M. Mester and S. Nityanand, Planta Med., 1979, 37, 367–369 CrossRef CAS.
- R. J. Leeman-Neill, S. E. Wheeler, S. V. Singh, S. M. Thomas, R. R. Seethala, D. B. Neill, M. C. Panahandeh, E.-R. Hahm, S. C. Joyce and M. Sen, Carcinogenesis, 2009, 30, 1848–1856 CrossRef CAS PubMed.
- R. Chander, A. K. Khanna and R. Pratap, J. Med. Aromat. Plant Sci., 2002, 24, 371–375 CAS.
- J. A. Francis, S. N. Raja and M. G. Nair, Chem. Biodiversity, 2004, 1, 1842–1853 CrossRef CAS PubMed.
- X. Wang, J. Greilberger, G. Ledinski, G. Kager, B. Paigen and G. Jürgens, Atherosclerosis, 2004, 172, 239–246 CrossRef CAS PubMed.
- R. Chander, F. Rizvi, A. K. Khanna and R. Pratap, Indian J. Clin. Biochem., 2003, 18, 71–79 CrossRef CAS PubMed.
- D. Xiao, Y. Zeng, L. Prakash, V. Badmaev, M. Majeed and S. V. Singh, Mol. Pharmacol., 2011, 79, 499–507 CrossRef CAS.
- B. Sharma, R. Salunke, S. Srivastava, C. Majumder and P. Roy, Food Chem. Toxicol., 2009, 47, 2631–2639 CrossRef CAS.
- A. Mishra, Y. Pathak, G. Choudhir, A. Kumar, S. K. Mishra and V. Tripathi, Cancer Res., 2021, 81, 712 CrossRef.
- D. Lee, T. Kim, K. H. Kim, J. Ham, T. S. Jang, K. S. Kang and J. W. Lee, Bioorg. Med. Chem. Lett., 2017, 27, 3156–3161 CrossRef CAS.
- A. Kohyama, R. Yokoyama, D. F. Dibwe, S. El-Mekkawy, M. R. Meselhy, S. Awale and Y. Matsuya, Bioorg. Med. Chem. Lett., 2020, 30, 126964 CrossRef CAS PubMed.
- A. A. Abdellatef, Y. Zhou, A. Yamada, S. A. Elmekkawy, A. Kohyama, S. Yokoyama, M. R. Meselhy, Y. Matsuya, H. Sakurai and Y. Hayakawa, Biomed. Pharmacother., 2021, 140, 111737 CrossRef CAS.
- N. L. Urizar and D. D. Moore, Annu. Rev. Nutr., 2003, 23, 303 CrossRef CAS.
- S. Rajasekar, T. P. A. Krishna, N. Tharmalingam, I. Andivelu and E. Mylonakis, ChemistrySelect, 2019, 4, 2281–2287 CrossRef CAS.
- T. P. A. Krishna, S. Pandaram and A. Ilangovan, Org. Chem. Front., 2019, 6, 3244–3251 RSC.
- H. Sung, J. Ferlay, R. L. Siegel, M. Laversanne, I. Soerjomataram, A. Jemal and F. Bray, Ca-Cancer J. Clin., 2021, 71, 209–249 CrossRef PubMed.
- A. Sartaj, S. Baboota and J. Ali, Curr. Pharm. Des., 2021, 27, 4630–4648 CrossRef CAS.
- T. P. Adarsh Krishna, T. P. Ajeesh Krishna, V. Sanyo Raj, S. Juliet, S. Nair, R. Ravindran and S. Sujith, Res. J. Pharm. Sci., 2013, 2, 1–6 Search PubMed.
- T. P. Ajeesh Krishna, T. P. A. Krishna, R. Kumuthakallavalli, V. N. S. Raj, S. Juliet, T. S. Rani, U. Darsana, S. N. Nair and R. Ravindran, J. Pharmacogn. Phytochem., 2014, 3, 121–124 Search PubMed.
- D. Albano, M. Benenati, A. Bruno, F. Bruno, M. Calandri, D. Caruso, D. Cozzi, R. De Robertis, F. Gentili and I. Grazzini, Insights Imaging, 2021, 12, 1–28 CrossRef PubMed.
- S. R. Ahmad and P. Ghosh, J. Indian Chem. Soc., 2022, 99, 100293 CrossRef CAS.
- J. Yan, X. Yang, X. Jiao, X. Yang, M. Guo, Y. Chen, L. Zhan and W. Chen, J. Cell. Mol. Med., 2020, 24, 814–829 CrossRef CAS PubMed.
- T. Gao, L. Zhao, F. Zhang, C. Cao, S. Fan and X. Shi, World J. Surg. Oncol., 2022, 20, 1–19 CrossRef.
- N. Kurowska, B. Strzalka-Mrozik, M. Madej, K. Pająk, C. Kruszniewska-Rajs, W. Kaspera and J. M. Gola, Cancers, 2022, 14, 1876 CrossRef CAS.
- G. Jain and P. Das, Int. J. Pharma Sci. Res., 2020, 11, 1527–1536 CAS.
- W. Kooti, K. Servatyari, M. Behzadifar, M. Asadi-Samani, F. Sadeghi, B. Nouri and H. Zare Marzouni, J. Evidence-Based Complementary Altern. Med., 2017, 22, 982–995 CrossRef CAS.
- J. H. Cheon, J. S. Kim, J. M. Kim, N. Kim, H. C. Jung and I. S. Song, Inflammatory Bowel Dis., 2006, 12, 1152–1161 CrossRef PubMed.
- S. Shishodia, G. Sethi, K. S. Ahn and B. B. Aggarwal, Biochem. Pharmacol., 2007, 74, 118–130 CrossRef CAS PubMed.
- H.-B. Xu, X.-Z. Chen, Z.-L. Yu and F. Xue, J. Ethnopharmacol., 2022, 115855 Search PubMed.
- M. A. Macha, S. Rachagani, S. Gupta, P. Pai, M. P. Ponnusamy, S. K. Batra and M. Jain, Cancer Lett., 2013, 341, 166–177 CrossRef CAS PubMed.
- T. Yamada, S. Osawa, M. Ikuma, M. Kajimura, M. Sugimoto, T. Furuta, M. Iwaizumi and K. Sugimoto, Digestion, 2014, 90, 208–217 CAS.
- S. V. Singh, Y. Zeng, D. Xiao, V. G. Vogel, J. B. Nelson, R. Dhir and Y. B. Tripathi, Mol. Cancer Ther., 2005, 4, 1747–1754 CrossRef CAS PubMed.
- K. S. Ahn, G. Sethi, B. Sung, A. Goel, R. Ralhan and B. B. Aggarwal, Cancer Res., 2008, 68, 4406–4415 CrossRef CAS.
- E. S. Kim, S. Y. Hong, H. K. Lee, S. W. Kim, M. J. An, T. Il Kim, K. R. Lee, W. H. Kim and J. H. Cheon, Oncol. Rep., 2008, 20, 1321–1327 CAS.
- M. J. An, J. H. Cheon, S. W. Kim, E. S. Kim, T. Il Kim and W. H. Kim, Cancer Lett., 2009, 279, 93–100 CrossRef CAS PubMed.
- E.-M. Noh, E. Y. Chung, H. J. Youn, S. H. Jung, H. Hur, Y.-R. Lee and J.-S. Kim, Int. J. Mol. Med., 2013, 31, 393–399 CrossRef CAS PubMed.
- H. Tian, Y. Gui, Y. Wei, B. Shang, J. Sun, S. Ma, W. You and S. Jiang, Int. Immunopharmacol., 2021, 93, 107395 CrossRef CAS.
- (a) S. Shishodia, G. Sethi, K. S. Ahn and B. B. Aggarwal, Biochem. Pharmacol., 2007, 74, 118–130 CrossRef CAS; (b) S. V. Singh, Y. Zeng, D. Xiao, V. G. Vogel, J. B. Nelson, R. Dhir and Y. B. Tripathi, Mol. Cancer Ther., 2005, 4, 1747–1754 CrossRef CAS PubMed; (c) M. A. Macha, S. Rachagani, S. Gupta, P. Pai, M. P. Ponnusamy, S. K. Batra and M. Jain, Cancer Lett., 2013, 341, 166–177 CrossRef CAS PubMed; (d) G. Jiang, X. Xiao, Y. Zeng, K. Nagabhushanam, M. Majeed and D. Xiao, BMC Complementary Altern. Med., 2013, 13, 203 CrossRef; (e) M. A. Macha, A. Matta, S. S. Chauhan, K. W. Siu and R. Ralhan, Carcinogenesis, 2011, 32, 368–380 CrossRef CAS PubMed; (f) R. J. Leeman-Neill, S. E. Wheeler, S. V. Singh, S. M. Thomas, R. R. Seethala, D. B. Neill, M. C. Panahandeh, E. R. Hahm, S. C. Joyce and M. Sen, et al. , Carcinogenesis, 2009, 30, 1848–1856 CrossRef CAS PubMed.
- S. V. Singh, S. Choi, Y. Zeng, E. R. Hahm and D. Xiao, Cancer Res., 2007, 67(15), 7439–7449 CrossRef CAS.
- J.-J. Shi, X.-L. Jia, M. Li, N. Yang, Y.-P. Li, X. Zhang, N. Gao and S.-S. Dang, World J. Gastroenterol., 2015, 21, 13277 CrossRef CAS PubMed.
- R. Leo, L. Therachiyil, S. K. Siveen, S. Uddin, M. Kulinski, J. Buddenkotte, M. Steinhoff and R. Krishnankutty, Cancers, 2019, 11, 1478 CrossRef CAS PubMed.
- M. A. Macha, A. Matta, S. S. Chauhan, K. W. M. Siu and R. Ralhan, BMC Cancer, 2010, 10, 655 CrossRef CAS PubMed.
- S. V. Singh, S. Choi, Y. Zeng, E.-R. Hahm and D. Xiao, Cancer Res., 2007, 67, 7439–7449 CrossRef CAS PubMed.
- Y. Chen, H.-H. Wang, H.-H. Chang, Y.-H. Huang, J. R. Wang, C.-Y. Changchien and S.-T. Wu, Phytomedicine, 2021, 87, 153587 CrossRef CAS PubMed.
- R. Lv, M. Zhu, K. Chen, H. Xie, H. Bai and Q. Chen, Sci. World J., 2021, 1–6 Search PubMed.
- P. Patil, S. Nipanikar, H. Bhusnar and D. Nagore, Int. J. Pharm. Sci. Drug Res., 2015, 7, 89–95 CAS.
- (a) M. Gupta, S. M. Ghufran, T. Kausar, R. Ali, S. Biswas, S. M. Nayeem, R. Ishrat, S. Ali, A. Ahmad and I. A. Rather, Molecules, 2022, 27, 5104 CrossRef CAS PubMed; (b) D. Xiao and S. V. Singh, Mol. Cancer Ther., 2008, 7(1), 171–180 CrossRef CAS PubMed; (c) A. Kohyama, R. Yokoyama, D. F. Dibwe, S. El-Mekkawy, M. R. Meselhy, S. Awale and Y. Matsuya, Bioorg. Med. Chem. Lett., 2020, 30, 126964 CrossRef CAS PubMed.
- A. Kohyama, M. J. Kim, R. Yokoyama, S. Sun, A. M. Omar, N. D. Phan, M. R. Meselhy, K. Tsuge, S. Awale and Y. Matsuya, Bioorg. Med. Chem., 2022, 54, 116563 CrossRef CAS PubMed.
- M. H. Bhat, M. Fayaz, A. Kumar and A. K. Jain, in Plant and Human Health, Springer, 2019, vol. 3, pp. 301–319 Search PubMed.
- J. A. Abdulhakim, J. King Saud Univ., Sci., 2022, 102140 CrossRef.
- P. P. S. Sarkhel and U. Yadava, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2001, 28, 285 CrossRef.
- V. K. Gupta, P. Bandhoria, B. D. Gupta and K. K. Gupta, Crystallogr. Rep., 2006, 51, 265–270 CrossRef CAS.
- W. R. Benn and R. M. Dodson, J. Org. Chem., 1964, 29, 1142–1148 CrossRef CAS.
- V. D. Patil, U. R. Nayak and S. Dev, Tetrahedron, 1972, 28, 2341–2352 CrossRef CAS.
- E. G. Mackay and M. S. Sherburn, Synthesis, 2015, 47, 1–21 CrossRef CAS.
- X. Li, T. Chen, F. Peng, S. Song, J. Yu, D. N. Sidoine, X. Cheng, Y. Huang, Y. He and Z. Su, Microb. Cell Fact., 2021, 20, 1–15 CrossRef.
- V. Mancino, B. Cerra, A. Piccinno and A. Gioiello, Org. Process Res. Dev., 2018, 22, 600–607 CrossRef CAS.
- R. Pratap, D. P. Singh, R. Pal and S. Singh, Council of Scientific and Industrial Research, Process for preparing guggulsterones, US Pat., 7365218, 2008 Search PubMed.
- G. Gokaraju, R. Gokaraju, V. Gottumukkala and V. Somepalli, Process for the synthesis of pharmacologically active (z/e)-guggulsterones, US Pat., US10498316, 2005 Search PubMed.
- J. Ham, J. Chin and H. Kang, Molecules, 2011, 16, 4165–4171 CrossRef CAS.
- S. V. Kessar and A. L. Rampal, Tetrahedron, 1968, 24, 887–892 CrossRef CAS PubMed.
- A. Gioiello, R. Sardella, E. Rosatelli, B. M. Sadeghpour, B. Natalini and R. Pellicciari, Steroids, 2012, 77, 250–254 CrossRef CAS PubMed.
- J. Chin, D. Hahn, D. H. Won, S. J. Cho, K. Kim, J. Ham and H. Kang, Bull. Korean Chem. Soc., 2017, 38, 525–529 CrossRef CAS.
- P. R. Athawale, V. M. Zade, G. Rama Krishna and D. S. Reddy, Org. Lett., 2021, 23, 6642–6647 CrossRef CAS PubMed.
- O. V. Kharissova, B. I. Kharisov, C. M. Oliva González, Y. P. Méndez and I. López, R. Soc. Open Sci., 2019, 6, 191378 CrossRef PubMed.
- A. Barge, S. Tagliapietra, A. Binello and G. Cravotto, Curr. Org. Chem., 2011, 15, 189–203 CrossRef CAS.
- B. Banerjee, Ultrason. Sonochem., 2017, 35, 1–14 CrossRef CAS PubMed.
- M. B. Gawande, S. N. Shelke, R. Zboril and R. S. Varma, Acc. Chem. Res., 2014, 47, 1338–1348 CrossRef CAS PubMed.
- R. Nishanth Rao, S. Jena, M. Mukherjee, B. Maiti and K. Chanda, Environ. Chem. Lett., 2021, 19, 3315–3358 CrossRef CAS.
- B. König, Eur. J. Org. Chem., 2017, 2017, 1979–1981 CrossRef.
- S. Reischauer and B. Pieber, iScience, 2021, 24, 102209 CrossRef CAS PubMed.
- M.-E. F. Hegazy, T. A. Mohamed, A. I. ElShamy, H. M. Abou-El-Hamd, U. A. Mahalel, E. H. Reda, A. M. Shaheen, W. A. Tawfik, A. A. Shahat and K. A. Shams, J. Adv. Res., 2015, 6, 17–33 CrossRef CAS PubMed.
- R. A. Sheldon, D. Brady and M. L. Bode, Chem. Sci., 2020, 11, 2587–2605 RSC.
- K. Atkovska, J. Klingler, J. Oberwinkler, S. Keller and J. S. Hub, ACS Cent. Sci., 2018, 4, 1155–1165 CrossRef CAS PubMed.
- (a) V. Brito, G. Alves, P. Almeida and S. Silvestre, Molecules, 2021, 26, 2032 CrossRef CAS PubMed; (b) Y. Horn, A. Halden and G. S. Gordan, Blood, 1972, 40(684–687), 46 Search PubMed; (c) B. J. Kennedy, Cancer, 1957, 10, 813–818 CrossRef CAS.
- S. Kumar, S. L. Khokra and A. Yadav, Future J. Pharm. Sci., 2021, 7, 1–22 CrossRef.
- G. S. Kumari, B. Siva, S. D. Reddy, V. L. Nayak, A. K. Tiwari, B. G. Rao and K. S. Babu, Nat. Prod. Res., 2021, 1–7 Search PubMed.
- S.-X. Feng and Y.-D. Zhang, Steroids, 2013, 78, 1332–1338 CrossRef CAS PubMed.
- Y. Peng, C. Gao, Z. Zhang, S. Wu, J. Zhao and A. Li, ACS Catal., 2022, 12, 2907–2914 CrossRef CAS.
- L. J. Durak, J. T. Payne and J. C. Lewis, ACS Catal., 2016, 6, 1451–1454 CrossRef CAS PubMed.
- D. J. Abrams, P. A. Provencher and E. J. Sorensen, Chem. Soc. Rev., 2018, 47, 8925–8967 RSC.
- T. P. A. Krishna, S. Pandaram, S. Chinnasamy and A. Ilangovan, RSC Adv., 2020, 10, 19454–19462 RSC.
- L. Guillemard, N. Kaplaneris, L. Ackermann and M. J. Johansson, Nat. Rev. Chem., 2021, 5, 522–545 CrossRef CAS PubMed.
- Y. Fujiwara, V. Domingo, I. B. Seiple, R. Gianatassio, M. Del Bel and P. S. Baran, J. Am. Chem. Soc., 2011, 133, 3292–3295 CrossRef CAS PubMed.
- M. Yanagi, N. Uchida and H. Hamada, Nat. Prod. Commun., 2019, 14, 1934578X19876210 CrossRef CAS.
- B. Hong, T. Luo and X. Lei, ACS Cent. Sci., 2020, 6, 622–635 CrossRef CAS PubMed.
- S. Pandaram, T. P. A. Krishna and A. Ilangovan, Org. Biomol. Chem., 2020, 18, 3027–3031 RSC.
- T. P. Adarsh Krishna, B. Edachery and S. Athalathil, RSC Adv., 2022, 12, 8815–8832 RSC.
- P. Sakthivel, T. P. Adarsh Krishna and A. Ilangovan, ChemistrySelect, 2021, 6, 2094–2100 CrossRef CAS.
- M. Aggarwal, R. Singh, P. Ahlawat and K. Singh, Curr. Bioact. Compd., 2022, 18, 30–49 CrossRef.
- M. Monier, A. El-Mekabaty, D. Abdel-Latif, B. D. Mert and K. M. Elattar, Steroids, 2020, 154, 108548 CrossRef CAS PubMed.
- A. M. Farhan, Q. K. Alshamusi and M. H. Jebur, J. Phys.: Conf. Ser., 2021, 1853, 12057 CrossRef CAS.
- L.-H. Shan, H.-M. Liu, K.-X. Huang, G.-F. Dai, C. Cao and R.-J. Dong, Bioorg. Med. Chem. Lett., 2009, 19, 6637–6639 CrossRef CAS PubMed.
- D. Kovács, G. Mótyán, J. Wölfling, I. Kovács, I. Zupkó and É. Frank, Bioorg. Med. Chem. Lett., 2014, 24, 1265–1268 CrossRef PubMed.
- M. M. Abdelhalim, M. M. T. El-Saidi, S. T. Rabie and G. A. Elmegeed, Steroids, 2007, 72, 459–465 CrossRef CAS PubMed.
- A. M. Dar and S. Khan, J. Fluoresc., 2016, 26, 639–649 CrossRef CAS PubMed.
- A. M. Dar, B. Rah, S. Mir, R. Nabi, M. A. Gatoo, A. Mashrai and Y. Khan, Int. J. Biol. Macromol., 2018, 111, 52–61 CrossRef CAS PubMed.
- G. Mótyán, Á. Baji, M. A. Marć, M. K. Gopisetty, D. I. Adamecz, M. Kiricsi, É. A. Enyedy and É. Frank, Appl. Sci., 2019, 10, 229 CrossRef.
- Á. Baji, F. Kovács, G. Mótyán, G. Schneider, J. Wölfling, I. Sinka, I. Zupkó, I. Ocsovszki and É. Frank, Steroids, 2017, 126, 35–49 CrossRef PubMed.
- A. Ali, M. Asif, H. Khanam, A. Mashrai, M. A. Sherwani and M. Owais, RSC Adv., 2015, 5, 75964–75984 RSC.
- M. A. Shamsuzzaman, A. Ali, A. Mashrai, H. Khanam, A. Sherwani and M. Owais, Eur. Chem. Bull., 2014, 3, 1075–1080 Search PubMed.
- A. E.-G. E. Amr, M. El-Naggar, M. A. Al-Omar, E. A. Elsayed and M. M. Abdalla, Molecules, 2018, 23, 1572 CrossRef PubMed.
- G. S. Nongthombam, K. Borah, T. Muinao, Y. Silla, M. Pal, H. P. Deka Boruah and R. C. Boruah, ACS Comb. Sci., 2018, 21, 11–27 CrossRef.
- S. M. M. Yahya, A. O. Abdelhamid, M. M. Abd-Elhalim, G. H. Elsayed and E. F. Eskander, Steroids, 2017, 126, 15–23 CrossRef CAS PubMed.
- (a) D. E. V. Pires, T. L. Blundell and D. B. Ascher, J. Med. Chem., 2015, 58, 4066–4072 CrossRef CAS PubMed; (b) H. R. Khatri, N. Carney, R. Rutkoski, B. Bhattarai and P. Nagorny, Eur. J. Org. Chem., 2020, 7, 755–776 CrossRef; (c) E. Tomarelli, B. Cerra, F. G. Mutti and A. Gioiello, Adv. Synth. Catal., 2023, 365, 1–26 CrossRef.
- (a) B. Salehi, C. Quispe, J. Sharifi-Rad, N. Cruz-Martins, M. Nigam, A. P. Mishra, D. A. Konovalov, V. Orobinskaya, I. M. Abu-Reidah, W. Zam, F. Sharopov, T. Venneri, R. Capasso, W. Kukula-Koch, A. Wawruszak and W. Koch, Front. Pharmacol., 2020, 11, 599959 CrossRef CAS PubMed; (b) N. Verma, S. K. Singh and R. C. Gupta, Pharm. Pharmacol. Commun., 1999, 5, 349–354 CrossRef CAS; (c) Y. S. Chhonker, H. Chandasana, R. Mukkavilli, Y. D. Prasad, T. S. Laxman and S. Vangala, Drug Test. Anal., 2016, 8, 966–975 CrossRef CAS PubMed.
- (a) H. Wen-Sen, Z. Hanyue and C. Zhen-Yu, J. Agric. Food Chem., 2018, 66(12), 3047–3062 CrossRef PubMed; (b) Y. Hu, C. Ma, X. Chen, G. Bai and S. Guo, Grain Oil Sci. Technol., 2022, 5, 146–155 CrossRef CAS; A. Agbay, L. De La Vega, G. Nixon and S. Willerth, Biomed. Mater., 2018, 13, 034104 Search PubMed; H. Lamba, L. Sabikhi, D. Aggarwal, U. Choudhary, S. Reddi, S. Kapila and R. Kapila, Int. J. Food Sci., 2018, 53, 626–633 Search PubMed; (c) R. Sharma, C. Benwood and S. M. Willerth, Curr. Protoc., 2021, 12, 331 CrossRef PubMed; (d) N. Shahzad, W. Khan, M. D. Shadab, A. Ali, S. S. Saluja, S. Sharma, F. A. Al-Allaf, Z. Abduljaleel, I. A. Ibrahim, A. F. Abdel-Wahab and M. A. Afify, Biomed. Pharmacol. J., 2017, 88, 786–794 CrossRef CAS PubMed.
- Chemical synthesis (a) H. S. Ewart, L. K. Cole, J. Kralovec, H. Layton, J. M. Curtis, J. L. C. Wright and M. G. Murphy, J. Nutr., 2002, 132(6), 1149–1152 CrossRef CAS PubMed; (b) Y. Pouilloux, G. Courtois, M. Boisseau, A. Piccirilli and J. Barrault, Green Chem., 2003, 5(1), 89–91 RSC; (c) S. Valange, A. Beauchaud, J. Barrault, Z. Gabelica, M. Daturi and F. Can, J. Catal., 2007, 251, 113–122 CrossRef CAS; (d) X. Meng, P. Sun, Q. Pan, Z. Shi, K. Yang and R. He, Eur. J. Lipid Sci. Technol., 2006, 108, 13–18 CrossRef CAS; (e) C. Jia, X. Xia, H. Wang, M. Bertrand, G. Chen and X. Zhang, Food Chem., 2020, 33, 127200 CrossRef PubMed; (f) H. C. Nguyen, K.-C. Huang and C. H. Su, Chem. Eng. J., 2020, 382, 122796 CrossRef CAS.
- Enzyme-catalyzed synthesis (a) N. L. S. Hakalin, M. Molina Gutierrez, A. Prieto and M. J. Martinez, Food Chem., 2018, 261, 139–148 CrossRef CAS PubMed; (b) W. He, D. Hu, Y. Wang, X. Chen, C. Jia, H. Ma and B. Feng, Food Chem., 2016, 192, 557–565 CrossRef CAS PubMed; (c) W. He, J. Li, X. Pan, Y. Zhou, C. Jia, X. Zhang and B. Feng, Bioresour. Technol., 2012, 114, 1–5 CrossRef CAS PubMed; (d) L. Hu, S. Llibin, J. Li, L. Qi, X. Zhang, D. Yu, E. Walid and L. Jiang, Bioprocess Biosyst. Eng., 2015, 38, 2343–2347 CrossRef CAS; (e) B. H. Kim and C. C. Akoh, Food Chem., 2007, 102, 336–342 CrossRef CAS; (f) M. Miao, H. Liu, B. Jiang, C. Yang, X. Xia and T. Zhang, J. Funct. Foods, 2014, 7, 452–461 CrossRef CAS; (g) M. Molina-Gutierrez, N. L. S. Hakalin, L. Rodrıguez-Sanchez, A. Prieto and M. J. Martınez, Food Chem., 2017, 221, 1458–1465 CrossRef CAS PubMed; (h) Z. Wang, S. H. Hwang and S. S. Lim, Eur. J. Lipid Sci. Technol., 2015, 117, 1037–1048 CrossRef CAS.
- Ionic liquid-catalyzed synthesis (a) W. He, L. Li, Q. Huang, J. Yin and X. Cao, Food Chem., 2018, 263, 1–7 CrossRef CAS PubMed; (b) W. He, Q. Liu, H. Yu, X. Si and J. Zhang, J. Am. Oil Chem. Soc., 2016, 93, 509–517 CrossRef CAS; (c) Z. Bin, F. Ting, Y. Yan, L. Feng, O. A. Idowu and S. Hongbo, Catal. Sci. Technol., 2022, 12, 6405–6415 RSC; (d) W. S. He, H. H. Wang, Z. M. Jing, D. D. Cui, J. Q. Zhu, Z. I. Li and H. L. Ma, J. Am. Oil Chem. Soc., 2018, 95, 89–100 CrossRef CAS.
- Physical change (a) B. Mansoori, B. A. Mohammadi, F. Abedi-Gaballu, S. Abbaspour, M. Ghasabi, R. Yekta, S. Shirjang, G. Dehghan, M. R. Hamblin and B. Baradaran, J. Cell. Physiol., 2020, 235, 6817–6830 CrossRef CAS PubMed; (b) M. Mohammadi, S. M. Jafari, H. Hamishehkar and B. Ghanbarzadeh, Trends Food Sci. Technol., 2020, 101, 73–88 CrossRef CAS; (c) N. Mosallaei, A. Mahmoudi, H. Ghandehari, V. K. Yellepeddi, M. R. Jaafari and B. Malaekeh-Nikouei, Eur. J. Pharm. Biopharm., 2016, 104, 42–50 CrossRef CAS PubMed; (d) U. Paaver, I. Laidmae, H. A. Santos, J. Yliruusi, J. Aruvali, K. Kogermann and J. Heinamaki, Asian J. Pharm. Sci., 2016, 11, 500–506 CrossRef; (e) S. H. Park and H. K. Kim, Enzyme Microb. Technol., 2020, 139, 109581 CrossRef CAS PubMed; (f) H. S. Ribeiro, R. Gupta, K. W. Smith, K. F. van Malssen, A. K. Popp and K. P. Velikov, Soft Matter, 2016, 12, 5835–5846 RSC; (g) S. Rozner, A. Kogan, S. Mehta, P. Somasundaran, A. Aserin, N. Garti and M. F. Ottaviani, J. Phys. Chem. B, 2009, 113, 700–707 CrossRef CAS PubMed; (h) H. Sasako, F. Kihara, K. Koyama, K. Higashi, K. Yamamoto and K. Moribe, Food Chem., 2016, 210, 269–275 CrossRef CAS PubMed; (i) K. Tai, M. Rappolt, X. He, Y. Wei, S. Zhu, J. Zhang, L. Mao, Y. Gao and F. Yuan, Food Chem., 2019, 293, 92–102 CrossRef CAS PubMed; (j) F. C. Wang, N. Acevedo and A. G. Marangoni, Food Funct., 2017, 8, 3964–3969 RSC.
Footnote |
| † Contributed equally. |
| This journal is © The Royal Society of Chemistry 2024 |