
Discovery of a dual-acting inhibitor of interleukin-1β and STATs for the treatment of inflammatory bowel disease†
Haowei
Cai‡
,
Zhuorong
Liu‡
,
Ping
Sun‡
,
Yinghua
Zhou
,
Yuyun
Yan
,
Yiming
Luo
,
Xiuxiu
Zhang
,
Ruiwen
Wu
,
Xiangting
Liang
,
Dan
Wu
,
Wenhui
Hu
* and
Zhongjin
Yang
 *
*
Guangdong Provincial Key Laboratory of Molecular Target & Clinical Pharmacology, The NMPA and State Key Laboratory of Respiratory Disease, School of Pharmaceutical Sciences and the Fifth Affiliated Hospital, Guangzhou Medical University, Guangzhou 511436, China. E-mail: huwenhui@gzhmu.edu.cn; yzj23@gzhmu.edu.cn
First published on 9th November 2023
Abstract
Currently, a significant proportion of inflammatory bowel disease (IBD) patients fail to respond to conventional drug therapy such as immunosuppressants and biologic agents. Interference with the JAK/STAT pathway and blocking of IL-1 signaling are two promising therapeutic strategies for these unresponsive IBD patients. This work describes the discovery of an inhibitor 10v that not only blocks NLRP3 and AIM-2 inflammasome-mediated IL-1β signaling, but also reduces the expression of STAT1 and STAT5 in the JAK/STAT pathway. Importantly, 10v exhibits a significant anti-IL-1β effect and decreases the levels of STAT1 and STAT5 in a mouse model of colitis. As a result, a novel small molecule is identified with a dual inhibitory capacity towards both inflammasomes/IL-1β and STAT pathways, which supports further exploration of the therapeutic potential for IBD patients that do not respond to current drug therapy.
1. Introduction
Inflammatory bowel disease (IBD) is a chronic relapsing inflammatory intestinal disease characterized by refractoriness and heterogeneity, which encompasses two forms—Crohn's disease (CD) and ulcerative colitis (UC).1 Although the cause of IBD remains largely unknown, it involves complex pathological features, such as ulcers, lymphoid aggregates, crypt abscesses, and granulomas, and clinical symptoms, including diarrhea, bleeding, and weight loss.2–4 Treatment of IBD includes general immunosuppressants (such as corticosteroids and thiopurines) or biologic agents (such as anti-TNF-α and anti-interleukin-12/23) to modulate inflammatory mediators.5,6 However, unfortunately, there is still a large proportion of patients who are still unresponsive to current treatments.7 The treatment of resistance remains a major challenge.Recently, IL-1-driven stromal–neutrophil interactions were identified to be the critical signaling in individuals with IBD that did not respond to anti-TNF and other therapeutics.8 These suggest that the IL-1 signaling blockade is a rational strategy for the treatment of IBD. As an individual form of IL-1, IL-1β plays a pivotal role for promoting inflammation.9 In IBD patients, IL-1β induces a strong chemokine response and drives neutrophil infiltrates, inflammatory fibroblast activation, and epithelial cell loss in ulcerated intestinal tissue.8 The production of IL-1β was mediated by multiple inflammasomes including NLRP3, AIM2, NLRC4, and NLRP1.10 This is consistent with the fact that the tissue from patients with IBD express high amounts of inflammasome genes.8,11 Taken together, therapies that inhibit inflammasomes/IL-1β pathway are promising for the treatment of IBD.
Besides, JAK/STAT is also a crucial pro-inflammatory signaling pathway that mediates the biological effects of cytokines involving pathological processes in IBD.12,13 Interfering with the JAK family and the STAT family is a new approach for some patients that remain unresponsive to current therapies.12,14 For example, tofacitinib, a non-specific inhibitor of JAKs, affects multiple pro-inflammatory cytokine-dependent and STAT-mediated pathways and is helpful in the treatment of patients who have failed or did not tolerate conventional or biological therapies.14 Despite this, the JAK-mediated signaling induced by different cytokines can lead to complex and unpredictable effects including adverse reactions.15 Thus, studying the effect of STAT proteins on the pathology of IBD has become an active area. However, current reported data on STATs and their roles in IBD is somewhat conflicting. For example, increased expression of STAT1, but not its phosphorylated (activated) form, has been observed in patients with Crohn's disease, while other reports have indicated higher levels of STAT1 expression and activation in both forms of IBD compared to healthy humans.16,17 Although the functional role of STAT1 is complex and remains controversial, improvements in colitis were observed in STAT1-null mice.18 This complexity is also reflected in the case of STAT5, which can be activated by many cytokines, such as IL-3, the IL-2 family, EGF, platelet-derived growth factors, and so on.15 STAT5-deficient mice exhibited perinatal lethality, but STAT5 dimers were sufficient to survive.19 STAT5 activation was found to increase the proliferation of the intestinal epithelial stem cells, which engaged in intestinal crypt regeneration.20 However, the STAT5 tetramer seems to promote autoimmune-mediated inflammation, such as regulatory T cells, CD8+ T cells, CD4+ T cells, natural killer (NK) cells, and chemokine.21,22 In addition, human monocyte-derived macrophages (MDMs) from IBD-risk variant carriers expressing high levels of STAT3 and STAT5 are more sensitive to promoting an inflammatory response relative to non-risk variant carrier MDMs after pattern-recognition receptor (PRR) stimulation.23 The threshold of STAT3 and STAT5 expression appears to determine the immune outcomes.23 Given the complex function of STATs themselves and their phosphorylation, rationally inhibiting their expression or promoting their degradation could provide potential strategies for the treatment of IBD.
In previous studies, we have shown that chalcone analogues can inhibit the NLRP3/IL-1β pathway.24 Notably, chalcones could be potential inhibitors that can target the JAK/STAT pathway.25 This work describes the identification of a new lead compound 10v, which not only inhibits the secretion of NLRP3 and AIM-2 inflammasome-mediated IL-1β, but also reduces the expression of STAT1 and STAT5. In vivo pharmacological models of colitis have shown that 10v has a significantly potent anti-IL-1β effect and partially blocks the STAT pathway function.
2. Results and discussion
2.1. Chemistry
Based on previously reported chalcone analogues that exhibited potential as NLRP3 inflammasome inhibitors,24 we designed a series of small molecules relying on strategies such as functional group reversal and combining active fragments. All derivatives were synthesized by the route described in Scheme 1. Generally, hydroxybenzaldehyde (1 or 6) as the starting material was protected by chloromethoxyethane (EOMCl) followed by the Claisen–Schmidt reaction with substituted acetophenones to give compounds 4 or 9a, 9b, 9d, 9e, 9j, and 9u. Compound 9a underwent the C–S bond metathesis with ethyl bromoacetate, providing 9c in good yield. The condensation of 9e with carboxylic acid or acid chloride produced compounds 9f and 9g. Compound 9g underwent the Michael addition reaction with dimethylamine or diethylcarbamodithioate to obtain 9h and 9i. The acid 9j was esterified with ethanol under the catalysis of concentrated H2SO4 to give 9k or condensed with the corresponding amino acid derivatives to yield 9l, 9p, 9t, 9n, and 9v. The hydrolysis of 9l or 9n produced 9m or 9o, respectively. The subsequent condensation of 9m with different amines gave the amide 9q–s. The deprotection of 4, 9a–d, 9f, and 9h–v with hydrochloric acid or trifluoroacetic acid provided the target compounds 5, 10a–d, 10f, and 10h–v.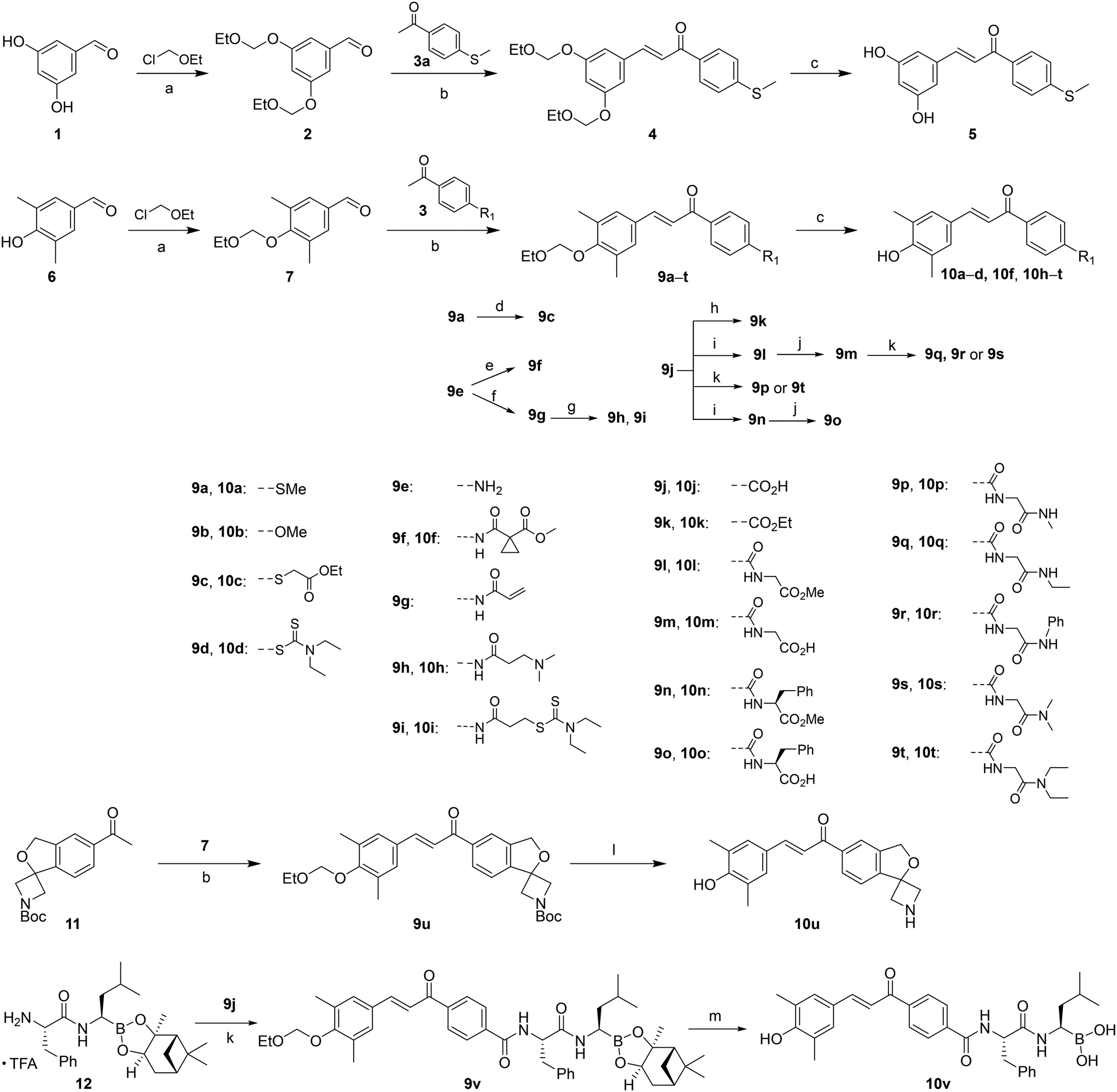 | ||
| Scheme 1 Chemical synthesis of the derivatives 5 and 10a–v. Reagents and conditions: (a) DMAP, DIPEA, and DCM, rt. (b) NaOH and MeOH, rt. (c) 1 M aqueous HCl and MeOH, 60 °C. (d) Ethyl bromoacetate and trifluoroethanol, 100 °C. (e) 1-Methoxycarbonylcyclopropane-1-carboxylic acid, HATU, and DIPEA, rt. (f) Acryloyl chloride, TEA, and DCM, rt. (g) Dimethylamine or sodium diethylcarbamodithioate, TEA, and DCM, rt. (h) Concentrated sulfuric acid and EtOH, 60 °C. (i) Various amino acid methyl esters, DIPEA, HATU, and DCM, 0 °C to rt. (j) LiOH, H2O, and MeOH, rt. (k) Various amino acetamides, EDCI-HCl, HOBt, DIPEA, and DCM, 0 °C to rt. (l) Trifluoroacetic acid and DCM, rt. (m) Isobutyl boronic acid, 1 M aqueous HCl, and heptane/MeOH. | ||
2.2. Inhibitory activity of compounds on NLRP3-dependent IL-1β
Of all the inflammasome pathways, the NLRP3 inflammasome/IL-1β pathway is the best characterized. The NLRP3 inflammasome is closely associated with various types of sterile inflammation and chronic inflammatory reactions.26 In our previously established cellular screening model, the J774A.1 cell line was firstly primed with lipopolysaccharide (LPS) to express NLRP3 and pro-IL-1β, followed by activation of NLRP3 with nigericin. Active NLRP3 assembles into NLRP3 inflammasomes, which induce pro-caspase-1 autoproteolysis into caspase-1, finally driving the secretion of proinflammatory cytokine IL-1β and pyroptosis.27 The drug treatment occurs after LPS priming NLRP3 expression and before nigericin activating NLRP3. This means that our model will screen out the inhibitors that act in the NLRP3 activation step.As shown in Table 1, our previously reported compound 10a showed a micromolar activity against NLRP3-dependent IL-1β with the IC50 value of 3.67 μM. Introducing two phenolic hydroxyl groups (5) on the left benzene ring slightly increased the inhibitory activity, but the solubility in organic solvents is greatly reduced, which is not conducive to chemical modification. With the aim of expanding the compound library, the right benzene ring was further chemically derived. The substitution of the methylthiol moiety with methoxyl (10b) exhibited a 4-fold decrease of inhibitory potency, while the substitution with 2-ethoxy-2-oxoethylthiol (10c) maintained the inhibitory activity. When the diethylcarbamodithioate moiety, a fragment from disulfiram that effectively inhibits NLRP3 inflammasome activation, was combined into the chalcone scaffold, a significantly reduced inhibitory potency (∼7-fold) was observed in 10d. In general, replacement of a sulfur atom with a nitrogen atom, while introducing different acylation groups (10f–10i), still maintained or slightly reduced NLRP3-dependent IL-1β activity. Analogue 10j with a 4-CO2H lost its inhibitory activity, and the esterified 10k showed a recovery of activity with the IC50 value of 8.36 μM, indicating that suitable lipophilicity at the position is required for activity. The inversion of the N-acylated functional groups led to the amide analogues (10p, 10q, 10s, and 10t) which showed inhibitory potency comparable to that of 10f and 10h. An exception was compound 10r with aniline replacing the methylamine moiety of 10p, which exhibited no activity. The change transforms Gly-NHMe (10p) into Gly-OMe (10l, IC50 = 1.08 μM) and Phe-OMe (10n, IC50 = 1.09 μM), which remarkably improved the inhibitory potency. Similar to 10j, when the methyl ester group in 10l changed into a carboxyl group (10m), a complete loss of activity was observed in the inhibition of IL-1β. However, compound 10o with a carboxyl group of phenylalanine exhibited relatively good inhibitory activity (IC50 = 3.37 μM), possibly due to the presence of the benzyl group complementing lipophilicity at this position. Compound 10u, bearing a spirocyclical structure containing an oxygen atom and a nitrogen atom, also exhibited moderate inhibitory activity with an IC50 value of 5.21 μM. Boronic acid-based fragments are a key pharmacophore, which inhibit NLRP3 inflammasome activation. Thus, a combination of 10o and isopentylboronic acid through an amide bond led to the new compound 10v, which exhibited excellent activity against NLRP3-dependent IL-1β with an IC50 of 0.13 μM and had an appropriate clog![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P of 4.13.
P of 4.13.
|
|
|||
|---|---|---|---|
| Compound | R1 | Inhibition of IL-1β | clogP |
| IC50 (μM) | |||
| a J774A.1 cells were primed by LPS (1 μg mL−1) for 5 h, treated with the compounds for 1 h, stimulated with nigericin (10 μM) for 1 h, and finally analyzed with ELISA for IL-1β from the supernatants (three separate determinations). | |||
| 5 | — | 1.57 ± 0.12 | 2.95 |
| 10a | - -SME | 3.67 ± 0.19 | 4.52 |
| 10b | - -OMe | 14.4 ± 1.8 | 4.07 |
| 10c |

|
4.93 ± 0.56 | 4.60 |
| 10d |
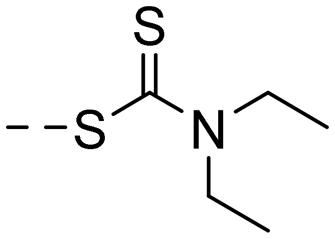
|
27.1 ± 8.2 | 4.40 |
| 10f |
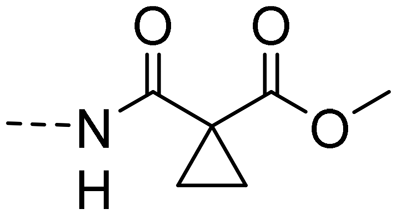
|
3.01 ± 0.43 | 3.50 |
| 10h |

|
5.97 ± 0.37 | 3.68 |
| 10i |
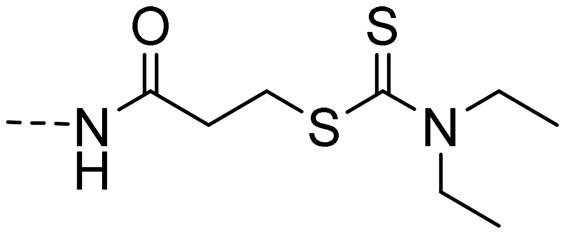
|
3.80 ± 0.73 | 5.45 |
| 10j | - -CO2H | >50.0 | 3.86 |
| 10k | - -CO2Et | 8.36 ± 1.14 | 4.49 |
| 10l |
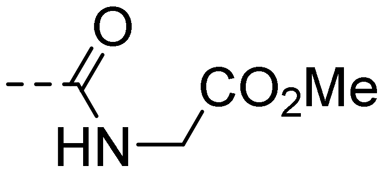
|
1.08 ± 0.08 | 3.03 |
| 10m |
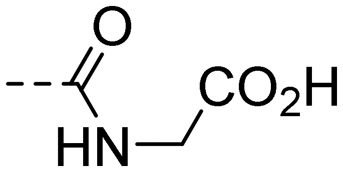
|
>50.0 | 2.82 |
| 10n |

|
1.09 ± 0.40 | 4.76 |
| 10o |

|
3.37 ± 0.98 | 4.56 |
| 10p |
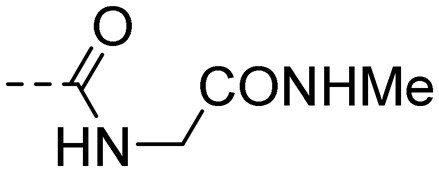
|
3.02 ± 0.62 | 2.17 |
| 10q |
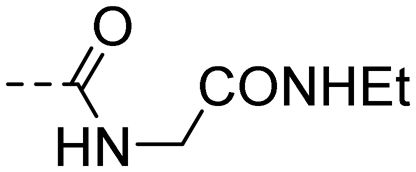
|
3.96 ± 0.23 | 2.70 |
| 10r |
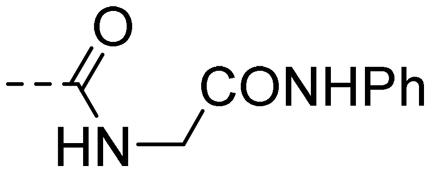
|
>50.0 | 4.03 |
| 10s |
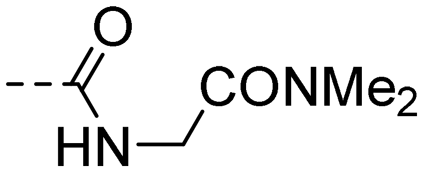
|
5.97 ± 0.36 | 2.60 |
| 10t |
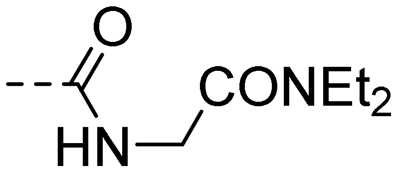
|
5.95 ± 0.49 | 3.66 |
| 10u | — | 5.21 ± 2.38 | 2.28 |
| 10v |
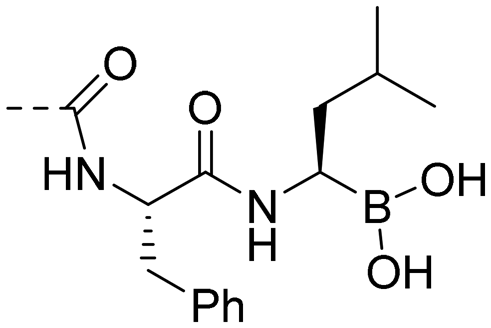
|
0.13 ± 0.02 | 4.13 |
Furthermore, we measured the cytotoxicity of 10v using J774A.1 and HEK293 cells. As shown in Fig. S1 (ESI†), 10v exhibited mild cytotoxicity against J774A.1 after 2 h of incubation and significant toxicity at 24 h (data not shown). For HEK293 cells, 10v did not show any toxicity in 2 h or 24 h of incubation.
2.3. Biological mechanism of 10v inhibiting NLRP3/IL-1β
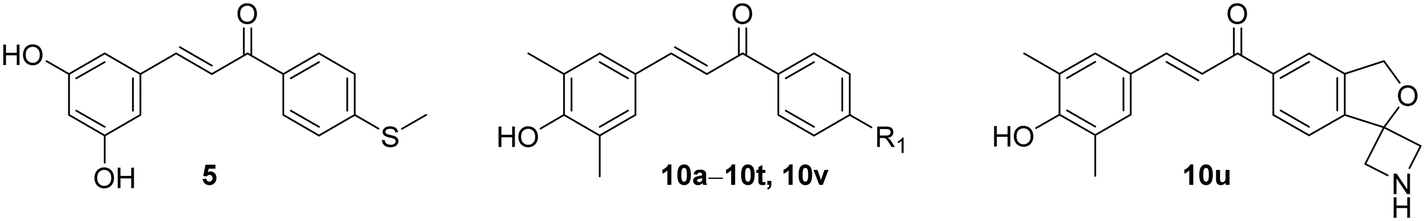
To confirm the inhibitory effect on the NLRP3-mediated pyroptotic signaling, we tested the effects of 10v on the NLRP3 pathway-associated proteins by the western blot analysis. The data showed that 10v significantly inhibited the release of mature IL-1β and caspase-1 p20 subunits and the production of cleaved GSDMD (Fig. 1A). These factors determine pyroptotic cell death, which involves the immune system and triggers inflammation. ASC specks represent the assembled NLRP3 inflammasome, and immunofluorescence analyses showed that 10v significantly inhibited the formation of ASC specks (Fig. 1B and C). During inflammasome assembly, NLRP3 binds to the adapter protein ASC via PYD–PYD interactions. Thus, we tested whether 10v could influence the NLRP3-ASC interaction via a coimmunoprecipitation (Co-IP) assay. As shown in Fig. 1D, compound 10v remarkably inhibited the interaction of NLRP3 and ASC. Notably, the structure of 10v incorporates two potent fragments, a chalcone and boric acid. Our previous studies have shown that chalcone-based analogues inhibit NLRP3 inflammasome activation through suppressing ROS production,24 while boric acid-based derivatives do so by promoting ubiquitination of NLRP3.27,28 Thus, the effect of 10v on intracellular ROS was accessed by utilizing a fluorescent probe DCFH-DA. Unexpectedly, treatment with 10v did not decrease the generation of intracellular ROS (Fig. 1E). Furthermore, we examined whether 10v could promote the NLRP3 ubiquitination, which can block NLRP3 inflammasome assembly and activation. As shown in Fig. 1F, compound 10v remarkably promoted the ubiquitination of NLRP3. These results indicate that compound 10v prevents the NLRP3 inflammasome activation possibly through promoting the ubiquitination of NLRP3.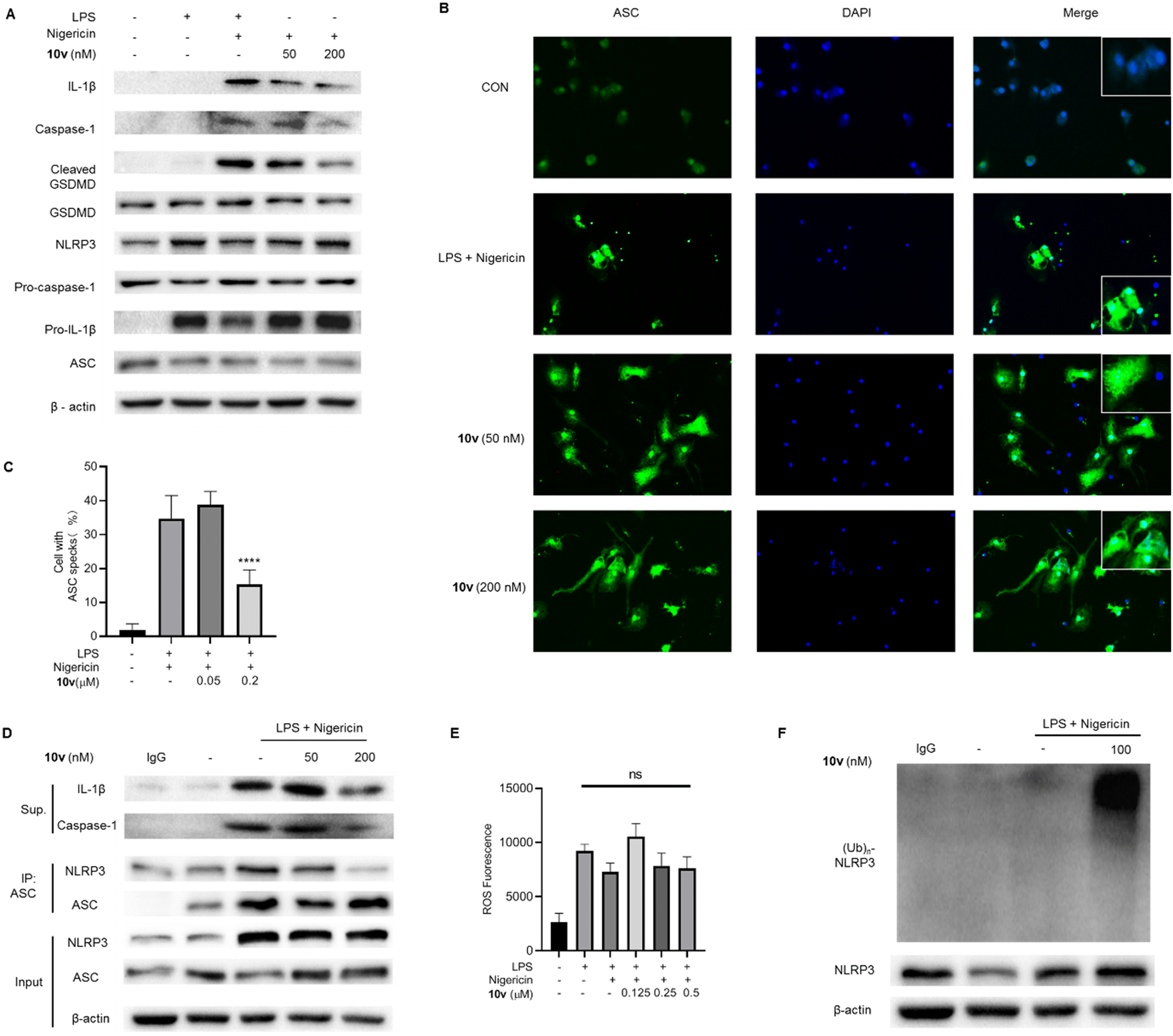 | ||
| Fig. 1 Compound 10v exhibited inhibitory effects on the NLRP3/IL-1β pathway. (A) Western blot analysis of IL-1β, caspase-1, cleaved GSDMD, GSDMD, pro-IL-1β, pro-caspase-1, NLRP3, and ASC. (B) Immunofluorescence detection of ASC specks. (C) Statistical plot of ASC specks. (D) IP analysis of the NLRP3–ASC interaction. (E) Analysis of ROS levels in LPS primed J774A.1 cells by DCFH-DA staining and fluorescence microscopy. (F) Analysis of NLRP3 ubiquitination levels in J774A.1 cells. | ||
2.4. Effect of 10v on activation of different inflammasomes
IL-1β production is dependent on multiple inflammasomes such as NLRP3, AIM2, NLRC4, and so on. Therefore, we tested the effect of 10v on different inflammasomes in mouse bone marrow-derived macrophages (BMDMs). BMDMs were primed with LPS before pretreating with compound 10v, and then stimulated with different inflammasome stimuli. As shown in Fig. 2A and B, compound 10v significantly inhibited the IL-1β release mediated by the NLRP3 inflammasome in response to stimulation by nigericin or imiquimod (IMQ). The FLA-ST Ultrapure stimulus triggered NLRC4 inflammasome-dependent IL-1β production from BMDMs, while 10v treatment did not reduce the NLRC4 pathway (Fig. 2C). In contrast, 10v moderately inhibited AIM2-dependent secretion of IL-1β stimulated by the dsDNA analog poly(dA:dT) (Fig. 2D). Taken together, 10v can block the activation of NLRP3 and AIM2 inflammasomes, but not the NLRC4 inflammasome.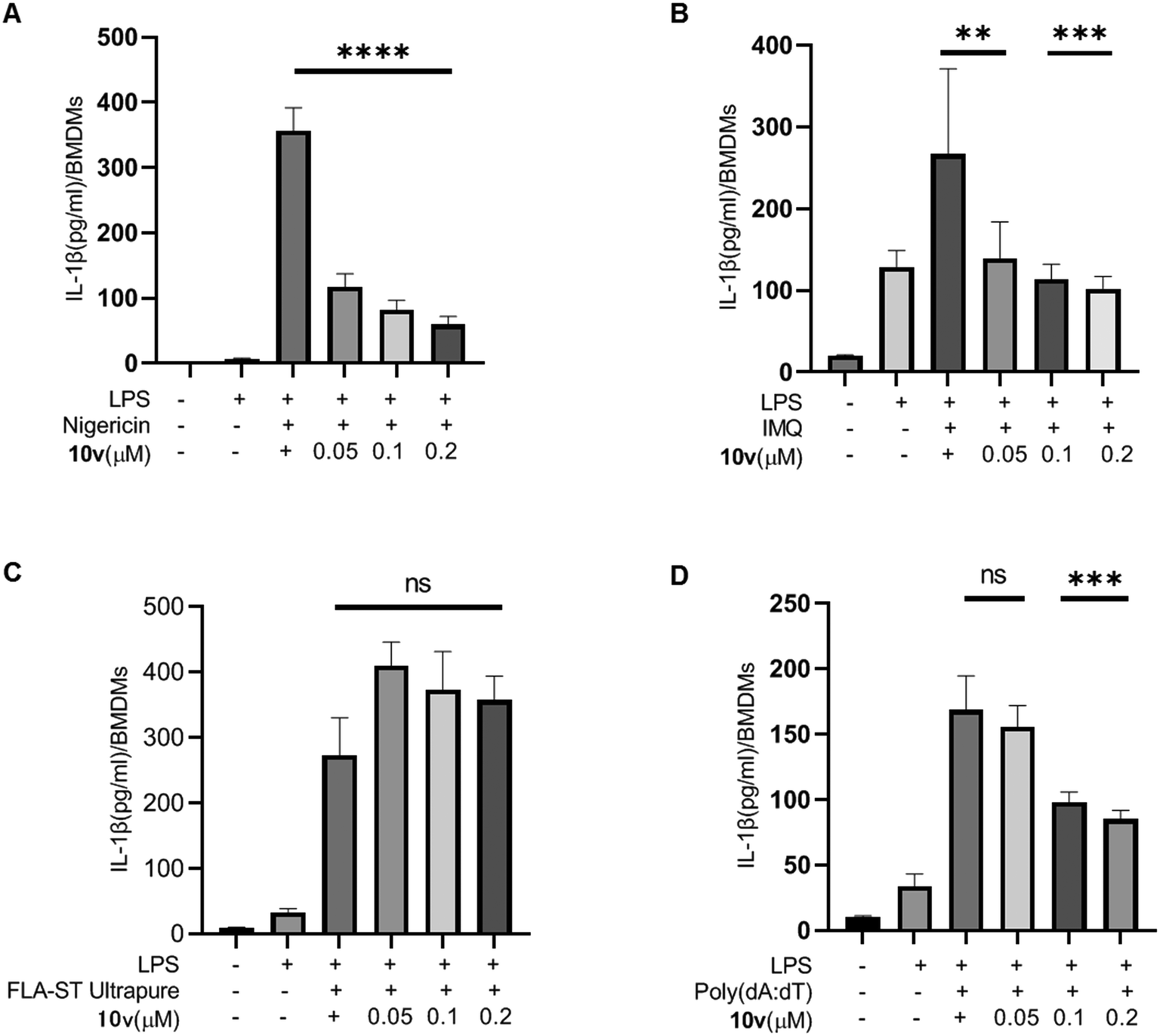 | ||
| Fig. 2 10v exhibited inhibitory effects on both NLRP3 and AIM2 inflammasomes. (A) ELISA detection of IL-1β from BMDMs with treatment of LPS, 10v, and then the NLRP3 stimulus nigericin. (B) ELISA detection of IL-1β from BMDMs with treatment of LPS, 10v, and then NLRP3 stimulus IMQ. (C) ELISA detection of IL-1β in BMDMs with treatment of LPS, 10v, and then NLRC4 stimulus FLA-ST Ultrapure. (D) ELISA detection of IL-1β in BMDMs with treatment of LPS, 10v, and then AIM2 stimulus poly(dA:dT) salt and lipofectamine 3000. | ||
2.5. Inhibitory effect of 10v on the STAT pathway
Inhibiting the JAK/STAT pathway is a promising approach for treating IBD patients that are unresponsive to current therapies. STATs are key mediators of the pro-inflammatory effects of this pathway. Thus, we examined whether 10v influenced the STAT proteins. As shown in Fig. 3, compound 10v significantly inhibited the expression of STAT1 and STAT5, but not STAT3. This effect led to the reduction of the phosphorylation of STAT1 and STAT5. It is unclear why 10v can cause a decrease in STAT protein expression, we cannot rule out the notion that it may have something to do with the inflammasome or proteasome. Anyhow, our data suggest 10v does block the STAT1 and STAT5 signaling. | ||
| Fig. 3 Compound 10v inhibited the expression of STATs. (A–C) Western blot analysis of pSTAT1, STAT1, pSTAT3, STAT3, pSTAT5, and STAT5 from J774A.1 cells pretreated with LPS and 10v. | ||
2.6. In vivo efficacy of 10v
Finally, we determine whether 10v can ameliorate ulcerative colitis in vivo. In the preliminary toxicity experiments, we found that when attempting to inject 10v intraperitoneally at a dose of 2 mg kg−1 per day (n = 2) for 7 days, the mice were in a poor state and inactive. Based on our previous research experience,27 we chose to perform intermittent treatment with 10v every 72 h for 7 days. Mice with colitis were modeled by feeding them drinking water containing 2.5% dextran sulfate sodium (DSS) for 7 days. Starting from the first day, compound 10v (0.2 and 0.6 mg kg−1 at the 2nd, 5th, and 8th day, i.p.) or the positive drug salicylazosulfapyridine (SASP, 10 mg kg−1 per day, p.o.) were administered for 9 days. As shown in Fig. 4A–D, mice in the model group were observed to lose body weight, shorten colon length, and increase fecal occult blood. Nine days after SASP administration, mice displayed no improvement in bodyweight loss and hemafecia, but a slight increase in colonic length. In contrast, treatment with 10v improved the symptoms of bodyweight loss, hemafecia, and colonic shortening, especially at the dose of 0.6 mg kg−1. Such a relief was also observed in the tissue-associated inflammatory factor IL-1β, although drug treatment did not decrease the amount of TNF-α and IL-6 (Fig. 4E–G). Next, the histological and pathological changes were observed with hematoxylin and eosin. DSS administration induced severe intestinal mucosal damage, such as infiltration of inflammatory cells, depletion of goblet cells, and loss of crypt architecture. After treatment with 10v or SASP, histological damage was significantly alleviated (Fig. 4H and I). Furthermore, we investigated the effect of 10v on the NLRP3 pathway and STAT pathway in vivo. Our results showed that 10v decreased the amount of caspase-1, STAT1, and STAT5 at a dose of 0.6 mg kg−1, but did not decrease the amount of STAT3 (Fig. 4J). These data suggest that 10v possesses a positive therapeutic effect on ulcerative colitis in vivo. | ||
| Fig. 4 10v ameliorates DSS-induced ulcerative colitis. (A) The change in bodyweight of the mice. (B) The blood stool index. (C) The colon length in each group. (D) Representative pictures of colon length. (E–G) ELISA detection of IL-1β, IL-6, and TNF-α in colon tissues. (H) Representative photos of the histopathological damage in the colons using H&E staining (×20). Scale bar: 200 μm. (I) Scores of H&E staining. (J) Western blot analysis of STAT1, STAT3, STAT5, NLRP3, and caspase-1 from intestinal tissue. | ||
3. Conclusions
Building upon previous reports of chalcone analogues with NLRP3 inflammasome inhibition potential, we further designed and optimized a chalcone scaffold by utilizing substitution, functional group reversal, and active fragment combination strategies. A preliminary SAR study suggested that an appropriate lipophilicity at the C-4′ position favors inhibitory activity. Further combination with a boric acid fragment led to a new lead compound 10v, which effectively exhibited potency at the submicromolar level. Mechanistic characterization indicated that compound 10v inhibited NLRP3 inflammasome assembly and activation though blocking the binding between NLRP3 and ASC but not via suppressing ROS production. In addition, compound 10v also inhibited AIM2 inflammasome-dependent IL-1β. Notably, 10v could inhibit STAT1 and STAT5 expression in the JAK/STAT pathway. It was confirmed that 10v can reduce the in vivo expression levels of IL-1β and STAT1 and STAT5 and has the ability to ameliorate DSS-induced colitis. In summary, our work identifies a promising inhibitor 10v with dual inhibition of inflammasomes/IL-1β and the STAT pathway, which supports further chemical development for the treatment of IBD patients who do not respond to current drug therapies.4. Experimental section
4.1. Chemistry
All reactions were carried out under a N2 atmosphere using anhydrous solvents of commercial grade. Reactions were monitored by thin layer chromatography by visualization using ultraviolet light. Flash column chromatography was used to purify the product on Tsingdao silica gel (200–300 mesh). 1HNMR and 13CNMR spectra were recorded on a JEOL 400YH spectrometer. The multiplicity was described as follows: s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet, and br = broad.Procedure a: etherification reaction. To a solution of hydroxybenzaldehyde (1.0 equiv.) in DCM (0.3 mol L−1), DIPEA (2.0 equiv.) and DAMP (0.03 equiv.) were added in an ice water bath. Then chloromethoxyethane (2.0 equiv.) was added dropwise and gradually warmed to room temperature for 3 h. The solvent of the reaction solution was evaporated under reduced pressure, and the remaining residue was purified by column chromatography [PE/EA = 6
:1] to obtain the product.
Procedure b: preparation of chalcones. Various benzaldehydes (1.0 equivalent) were dissolved in MeOH (0.2–0.5 mol L−1) and then saturated aqueous NaOH solution (2.0 equiv.) was added. After stirring for 10 min, the product from Procedure a was added dropwise to the mixture. The reaction was stirred at room temperature for 24 h and TLC detected the reaction process. MeOH in the reaction solution was removed under vacuum evaporation, and the pH value was adjusted to 4 with 1 M hydrochloric acid. Then methanol was removed and the residues were extracted with EA and water. The organic layer was dried with NaSO4, filtered, and concentrated to obtain the crude product, which was purified by flash column chromatography to provide the final product.
Procedure c: removal of the protective group. The product from Procedure b was dissolved in MeOH (0.4 mol L−1), and then 1 M HCl (2.0 equiv.) was added. The reaction solution was refluxed for 1 h. After the completion of the reaction, the solvent was removed under reduced pressure. The residues were extracted with EA and water, dried with NaSO4, filtered and concentrated to obtain the crude product, which was purified by flash column chromatography to provide the target product.
Procedure d: preparation of 9c. Compound 9a (1.0 equiv.) and ethyl bromomethylacetate (6.0 equiv.) was dissolved in trifluoroethanol (0.2–0.5 mol L−1). The reaction was stirred for 24 h at 100 °C. The reaction solvent was removed under vacuum. The residue was purified by flash column chromatography to get the target product.
Procedure e: preparation of 9f. 1-(Methoxycarbonyl)cyclopropane-carboxylic acid (1.0 equiv.) and HOBT (1.1 equiv.) were dissolved in DCM (0.4 mol L−1) at room temperature. EDCI (1.2 equiv.) was added and the mixture was stirred for 20 min. Then compound 9e (1.2 equiv.) and DIPEA (3.0 equiv.) were added to the reaction solution. After being stirred at room temperature for 4 h, the solvent was removed under vacuum. The reaction residue was re-dissolved with EA, washed three times with saturated sodium bicarbonate and 3% citric acid, dried with anhydrous sodium sulfate. The solvent is evaporated and the residue is purified by flash column chromatography.
Procedure f: preparation of 9g. Compound 9e (1.0 equiv.) and TEA (1.4 equiv.) were dissolved in anhydrous THF (0.3 mol L−1), followed by the addition of acryloyl chloride (1.1 equiv.) in an ice water bath. The mixture was stirred for 5 h at room temperature. The reaction solution is filtered and then extracted with water and EA. The organic layer is concentrated to obtain the residue, which was purified by flash column chromatography to give the product.
Procedure g: preparation of 9h and 9j. Compound 9g (1.0 equiv.) and sodium diethyldithiocarbamate (1.5 equiv.) or dimethylamine hydrochloride (1.5 equiv.) were dissolved in MeOH (0.4 mol L−1), followed by the addition of triethylamine. The mixture was stirred for 4 h at room temperature. The solvent was removed under reduced pressure and the residue was purified by flash column chromatography.
Procedure h: esterification reaction. Compound 9j (1.0 equiv.) was dissolved in ethanol (0.5 mol L−1), and then concentrated sulfuric acid (1.0 equiv.) was added. The reaction solution was refluxed for 4 h. The solvent was removed under vacuum. The reaction residue was extracted with water and EA. The organic layer was concentrated to give the residue, which was purified by flash column chromatography to obtain the product.
Procedure i: conversion of carboxylic acids to amides. To a solution of carboxylic acid (1.0 equiv.) in DCM (0.5 mol L−1), HATU (1.2 equiv.) was added. After being stirred for 30 min at room temperature, the corresponding amines (1.1 equiv.) were added, followed by the addition of DIPEA (3.0 equiv.). The reaction solution was stirred at room temperature for 4 h. The reaction solvent was removed by vacuum evaporation. The residue was re-dissolved with EA, washed three times with saturated sodium bicarbonate aqueous solution, 3% citric acid aqueous solution, and dried anhydrous sodium sulfate. The solvent was concentrated to give the residue, which was purified by flash column chromatography to obtain the target product.
Procedure j: hydrolysis reaction. The corresponding methyl ester (1.0 equiv.) was dissolved in methanol (0.2–0.4 mol L−1). Aqueous LiOH solution (2.0 equiv.) was added. The reaction was stirred for 4 h at room temperature. The pH was adjusted to 5–6 with 1 M HCl, the solvent was removed by vacuum evaporation, and extraction was carried out three times with EA. The combined organic phase was dried with anhydrous sodium sulfate, concentrated and purified by column chromatography to give the acid product.
Procedure k: condensation of acids with amines. To a solution of carboxylic acid (1.0 equiv.) and HOBT (1.1 equiv.) in DCM (0.3 mol L−1), EDCI (1.2 equiv.) was added at −4 °C. The mixture was stirred for 20 min. Then compound 9j (1.2 equiv.) and DIPEA (3.0 equiv.) were added and stirred continuously at room temperature for 4 h. The reaction mixture was extracted with DCM and washed three times with saturated aqueous sodium bicarbonate and 3% citric acid. The organic phase was dried with anhydrous sodium sulfate, concentrated and purified by flash column chromatography to give the target compound.
Procedure l: removal of the Boc protecting group. To compound 9u (1.0 equiv.) in DCM (0.2 mol L−1), trifluoroacetic acid (10 equiv.) was added at room temperature. The solution was stirred for 3 h. The reaction solvent was removed by vacuum evaporation, and the residue was extracted with EA and saturated aqueous sodium bicarbonate. The organic phase was dried with anhydrous sodium sulfate, concentrated, and purified by flash column chromatography to give compound 10u.
Procedure m: removal of protecting groups. To a mixture of compound 9v (1.0 equiv.) in methanol and n-hexane (1
:1) solution (0.2 mol L−1), 1 M HCl (2.0 equiv.) and isobutylboronic acid (3.0 equiv.) were added. The reaction was stirred for 6 h at room temperature. After the completion of the reaction, the methanol phase was separated and adjusted to pH ∼ 7 with saturated aqueous sodium bicarbonate. The reaction solution was washed three times with n-hexane and extracted with EA and water. The organic phase was dried with anhydrous sodium sulfate, concentrated, and purified by flash column chromatography to give compound 10v.
(E)-3-(3,5-Dihydroxyphenyl)-1-(4-(methylthio)phenyl) prop-2-en-1-one (5). An amorphous yellow solid; yield: 76% for 3 steps; 1HNMR (400 MHz, DMSO-d6) δ 9.47 (s, 2H), 8.01 (d, J = 8.6 Hz, 2H), 7.65 (d, J = 15.6 Hz, 1H), 7.46 (d, J = 15.5 Hz, 1H), 7.35 (d, J = 8.4 Hz, 2H), 6.63 (d, J = 2.2 Hz, 2H), 6.29 (t, J = 2.1 Hz, 1H), 2.51 (s, 3H). 13CNMR (101 MHz, DMSO-d6) δ 188.53, 159.21, 146.01, 144.82, 136.91, 134.32, 129.55, 125.43 (d, J = 13.0 Hz), 122.05, 107.40, 105.62, 14.45; HRMS (ESI) calcd for C16H14O3S [M + H]+ 287.0697, found 287.0738.
(E)-3-(4-Hydroxy-3,5-dimethylphenyl)-1-(4-methoxyphenyl)prop-2-en-1-one (10b). An amorphous yellow solid; yield: 62% for 3 steps; 1HNMR (400 MHz, DMSO-d6) δ 8.08 (dd, J = 9.2, 2.5 Hz, 2H), 7.66 (d, J = 15.5 Hz, 1H), 7.53 (d, J = 15.4 Hz, 1H), 7.43 (s, 2H), 7.02 (dd, J = 9.2, 2.5 Hz, 2H), 3.81 (d, J = 2.2 Hz, 3H), 2.16 (s, 6H). 13CNMR (101 MHz, DMSO-d6) δ 187.82, 163.51, 156.60, 144.53, 131.33, 131.21, 130.01, 126.33, 125.15, 118.77, 114.46, 56.04, 17.03; HRMS (ESI) calcd for C18H18O3 [M + H]+ 283.1289, found 283.1326.
(E)-4-(3-(4-Hydroxy-3,5-dimethylphenyl)acryloyl)phenyl diethylcarbamodithioate (10d). An amorphous black-brown solid; yield: 51% for 3 steps; 1HNMR (400 MHz, DMSO-d6) δ 8.94 (d, 1H), 8.10 (dd, J = 8.5, 2.1 Hz, 2H), 7.68 (d, J = 15.6 Hz, 1H), 7.60 (d, J = 15.4 Hz, 1H), 7.55 (dd, J = 8.5, 2.1 Hz, 2H), 7.46 (s, 2H), 3.91 (q, J = 7.1 Hz, 2H), 3.83 (q, J = 7.2 Hz, 2H), 2.16 (s, 6H), 1.30 (t, J = 7.1 Hz, 3H), 1.15 (t, J = 7.1 Hz, 3H). 13CNMR (101 MHz, DMSO-d6) δ 193.04, 188.96, 157.04, 145.95, 139.32, 137.55, 136.45, 130.38, 129.26, 126.15, 125.20, 118.64, 49.91, 47.91, 17.08, 13.23, 11.87; HRMS (ESI) calcd for C22H25NO2S2 [M + H]+ 400.1360, found 400.1395.
Ethyl (E)-2-((4-(3-(4-hydroxy-3,5-dimethylphenyl)acryloyl)phenyl)thio)acetate (10c). An amorphous yellow solid; yield: 73% for 3 steps; 1HNMR (400 MHz, DMSO-d6) 1H NMR (400 MHz, DMSO-d6) δ 8.88 (s, 1H), 8.02 (dd, J = 8.6, 1.9 Hz, 2H), 7.66 (d, J = 15.5 Hz, 1H), 7.56 (d, J = 15.4 Hz, 1H), 7.44 (s, 2H), 7.40 (dd, J = 8.6, 1.9 Hz, 2H), 4.07 (q, J = 7.1 Hz, 2H), 4.02 (s, 2H), 2.16 (s, 6H), 1.12 (t, J = 7.1 Hz, 3H). 13CNMR (101 MHz, CDCl3) δ 169.37, 155.12, 145.55, 136.13, 129.54, 129.15, 127.27, 127.00, 123.85, 118.81, 62.08, 35.09, 16.09, 14.22; HRMS (ESI) calcd for C21H22O4S3 [M + H]+ 371.1272, found 371.1310.
Methyl (E)-1-((4-(3-(4-hydroxy-3,5-dimethylphenyl)acryloyl)phenyl)carbamoyl)cyclopropane-1-carboxylate (10f). An amorphous yellow solid; yield: 85% for 4 steps; 1HNMR (400 MHz, DMSO-d6) δ 10.63 (s, 1H), 8.07 (dd, J = 8.8, 2.0 Hz, 2H), 7.73 (dd, J = 8.8, 2.0 Hz, 2H), 7.67 (d, J = 15.5 Hz, 1H), 7.54 (d, J = 15.4 Hz, 1H), 7.43 (s, 2H), 3.64 (s, 3H), 2.16 (s, 6H), 1.44–1.39 (m, 2H), 1.39–1.33 (m, 2H). 13CNMR (101 MHz, DMSO-d6) δ 187.81, 172.21, 167.00, 156.71, 144.79, 143.64, 133.39, 130.20, 130.11, 126.32, 125.14, 119.14, 118.68, 53.05, 30.79, 17.08, 16.85; HRMS (ESI) calcd for C23H23NO5 [M + H]+ 394.1610, found 394.1647.
(E)-3-(Dimethylamino)-N-(4-(3-(4-hydroxy-3,5-dimethylphenyl)acryloyl)phenyl)propanamide (10h). An amorphous yellow solid; yield: 65% for 4 steps; 1HNMR (400 MHz, DMSO-d6) δ 10.38 (s, 1H), 8.07 (d, J = 8.8 Hz, 2H), 7.71 (d, J = 8.8 Hz, 2H), 7.66 (d, J = 15.5 Hz, 1H), 7.54 (d, J = 15.4 Hz, 1H), 7.43 (s, 2H), 2.62 (t, J = 7.0 Hz, 2H), 2.50 (t, J = 7.0 Hz, 2H), 2.20 (s, 6H), 2.16 (s, 6H). 13CNMR (101 MHz, DMSO-d6) δ 187.82, 171.13, 156.67, 144.69, 143.84, 133.12, 130.19, 130.06, 126.31, 125.14, 118.87, 118.71, 55.17, 45.15, 34.89, 17.05; HRMS (ESI) calcd for C22H26N2O3 [M + H]+ 367.1977, found 367.2013.
(E)-3-((4-(3-(4-Hydroxy-3,5-dimethylphenyl)acryloyl)phenyl) amino)-3-oxopropyl diethyl-carbamodithioate (10i). An amorphous yellow solid; yield: 83% for 4 steps; 1HNMR (400 MHz, DMSO-d6) δ 10.31 (s, 1H), 8.87 (s, 1H), 8.07 (d, J = 8.7 Hz, 2H), 7.71 (d, J = 8.7 Hz, 2H), 7.67 (d, J = 15.5 Hz, 1H), 7.54 (d, J = 15.4 Hz, 1H), 7.44 (s, 3H), 3.92 (q, J = 7.1 Hz, 2H), 3.67 (q, J = 7.1 Hz, 2H), 3.46 (t, J = 6.8 Hz, 2H), 2.78 (t, J = 6.7 Hz, 2H), 2.16 (s, 6H), 1.14 (q, J = 7.1 Hz, 6H). 13CNMR (101 MHz, DMSO-d6) δ 194.20, 187.82, 170.49, 156.67, 144.70, 143.68, 133.20, 130.21, 130.08, 126.32, 125.14, 118.88, 118.71, 49.55, 47.02, 36.29, 32.05, 17.05, 12.84, 11.90; HRMS (ESI) calcd for C25H30N2O3S2 [M + H]+ 471.1737, found 471.1769.
(E)-4-(3-(4-Hydroxy-3,5-dimethylphenyl)acryloyl)benzoic acid (10j). A light yellow solid; yield: 58% for 3 steps; mp 205.6 °C; 1HNMR (400 MHz, methanol-d4) δ 8.20–8.13 (m, 2H), 8.08–8.02 (m, 2H), 7.69 (d, J = 15.5 Hz, 1H), 7.61 (d, J = 15.5 Hz, 1H), 7.48 (s, 2H), 2.17 (s, 6H). 13CNMR (101 MHz, DMSO-d6) δ 189.18, 167.34, 157.12, 146.29, 141.77, 134.64, 130.43, 130.13, 129.03, 126.05, 125.21, 118.67, 17.08; HRMS (ESI) calcd for C18H16O4 [M + H]+ 297.1082, found 297.1122.
Ethyl (E)-4-(3-(4-hydroxy-3,5-dimethylphenyl)acryloyl)benzoate (10k). An amorphous yellow solid; yield: 98% for 3 steps; 1HNMR (400 MHz, methanol-d4) δ 8.12 (d, J = 8.0 Hz, 2H), 8.08 (d, J = 8.0 Hz, 2H), 7.68 (d, J = 15.7 Hz, 1H), 7.50 (d, J = 15.6, 3.0 Hz, 1H), 7.34 (s, 2H), 4.37 (q, J = 7.2 Hz, 2H), 2.21 (s, 6H), 1.38 (t, J = 7.1 Hz, 3H). 13CNMR (101 MHz, DMSO-d6) δ 189.08, 165.71, 157.15, 146.38, 142.06, 133.59, 130.43, 129.92, 129.11, 126.06, 125.20, 118.67, 61.72, 17.07, 14.65; HRMS (ESI) calcd for C20H20O4 [M + H]+ 325.1395, found 325.1432.
Methyl (E)-(4-(3-(4-hydroxy-3,5-dimethylphenyl)acryloyl)benzoyl)glycinate (10l). An amorphous yellow solid; yield: 65% for 4 steps; 1HNMR (400 MHz, CD3OD) δ 9.17 (t, J = 5.9 Hz, 1H), 8.24–8.18 (m, 2H), 8.04–7.98 (m, 2H), 7.73 (d, J = 15.4 Hz, 1H), 7.65 (d, J = 15.4 Hz, 1H), 7.51 (s, 2H), 4.06 (d, J = 5.7 Hz, 2H), 3.67 (s, 3H), 2.21 (s, 6H). 13CNMR (101 MHz, DMSO-d6) δ 188.97, 170.84, 166.49, 157.28, 146.12, 140.77, 137.41, 130.42, 128.97, 128.17, 126.01, 125.22, 118.59, 52.37, 41.83, 17.10. HRMS (ESI) calcd for C28H27NO5 [M + H]+ 368.1453, found 368.1493.
Methyl (E)-(4-(3-(4-hydroxy-3,5-dimethylphenyl)acryloyl)benzoyl)-L-phenylalaninate (10n). An amorphous yellow solid; yield: 86% for 4 steps; [α]D25 +5.3° (c 0.2, MeOH); 1HNMR (400 MHz, DMSO-d6) δ 9.04 (d, J = 7.8 Hz, 1H), 8.94 (s, 1H), 8.14 (d, J = 8.0 Hz, 2H), 7.89 (d, J = 7.9 Hz, 2H), 7.68 (J = 15.4 Hz, 1H), 7.60 (J = 15.4 Hz, 1H), 7.47 (s, 2H), 7.31–7.20 (m, 5H), 7.19–7.13 (m, 1H), 4.70–4.60 (m, 1H), 3.61 (s, 3H), 2.17 (s, 6H). 13CNMR (101 MHz, DMSO-D6) δ 188.99, 172.64, 166.33, 157.07, 146.08, 140.72, 138.17, 137.48, 130.42, 129.65, 128.88, 128.83, 128.28, 127.10, 126.11, 125.18, 118.69, 54.93, 52.61, 36.69, 17.10; HRMS (ESI) calcd for C28H27NO5 [M + H]+ 458.1923, found 458.1959.
(E)-(4-(3-(4-Hydroxy-3,5-dimethylphenyl)acryloyl)benzoyl)glycine (10m). A foamed yellow solid; yield: 52% for 5 steps; 1HNMR (400 MHz, DMSO-d6) δ 9.00 (t, J = 5.9 Hz, 1H), 8.16 (d, J = 8.2 Hz, 2H), 7.97 (d, J = 8.2 Hz, 2H), 7.69 (d, J = 15.5 Hz, 1H), 7.60 (d, J = 15.4 Hz, 1H), 7.46 (s, 2H), 3.91 (d, J = 4.9 Hz, 2H), 2.16 (s, 6H). 13CNMR (101 MHz, DMSO-d6) δ 189.15, 171.69, 166.45, 157.00, 146.09, 140.65, 137.69, 130.35, 128.91, 128.12, 126.11, 125.21, 118.74, 41.83, 17.00; HRMS (ESI) calcd for C20H19NO5 [M + H]+ 354.1297, found 354.1334.
(E)-(4-(3-(4-Hydroxy-3,5-dimethylphenyl)acryloyl)benzoyl)-L-phenylalanine (10o). An amorphous yellow solid; yield: 75% for 5 steps; [α]D25 +28.0° (c 0.1, MeOH); 1HNMR (400 MHz, DMSO-d6) δ 8.99 (s, 1H), 8.92 (d, J = 8.2 Hz, 1H), 8.18–8.11 (m, 2H), 7.93–7.86 (m, 2H), 7.69 (d, J = 15.4 Hz, 1H), 7.60 (d, J = 15.4 Hz, 1H), 7.48 (s, 2H), 7.33–7.22 (m, 4H), 7.20–7.11 (m, 1H), 4.66–4.56 (m, 1H), 3.23–3.14 (m, 1H), 3.10–3.00 (m, 1H), 2.18 (s, 6H). 13CNMR (101 MHz, DMSO-d6) δ 189.06, 173.62, 166.25, 157.05, 146.05, 140.62, 138.66, 137.80, 130.40, 129.63, 128.80 (d, J = 6.5 Hz), 128.23, 126.97, 126.12, 125.20, 118.74, 54.87, 36.76, 17.08; HRMS (ESI) calcd for C27H25NO5 [M + H]+ 444.1766, found 444.1805.
(E)-N-(2-(Ethylamino)-2-oxoethyl)-4-(3-(4-hydroxy-3,5-dimethylphenyl)acryloyl)benzamide (10q). An amorphous yellow solid; yield: 26% for 6 steps; 1HNMR (400 MHz, DMSO-d6) δ 8.97 (s, 1H), 8.91 (t, J = 6.0 Hz, 1H), 8.22–8.15 (m, 2H), 8.04–7.98 (m, 2H), 7.95 (t, J = 5.6 Hz, 1H), 7.73 (d, J = 15.5 Hz, 1H), 7.62 (d, J = 15.4 Hz, 1H), 7.50 (s, 2H), 3.83 (d, J = 5.9 Hz, 2H), 3.15–3.02 (m, 2H), 2.18 (s, 6H), 1.00 (t, J = 7.2 Hz, 3H). 13CNMR (101 MHz, DMSO-d6) δ 188.98, 168.92, 166.25, 157.04, 146.02, 140.51, 137.98, 130.41, 128.82, 128.28, 126.14, 125.19, 118.70, 43.22, 33.97, 17.09, 15.33; HRMS (ESI) calcd for C22H24N2O4 [M + Na]+ 403.1667, found 403.1624.
(E)-4-(3-(4-Hydroxy-3,5-dimethylphenyl)acryloyl)-N-(2-oxo-2-(phenylamino)ethyl)benzamide (10r). An amorphous yellow solid; yield: 38% for 6 steps; 1HNMR (400 MHz, DMSO-d6) δ 10.10 (s, 1H), 9.06 (t, J = 5.9 Hz, 1H), 9.01 (s, 1H), 8.22–8.17 (m, 2H), 8.05–8.01 (m, 2H), 7.73 (d, J = 15.4 Hz, 1H), 7.63 (d, J = 15.4 Hz, 1H), 7.60–7.56 (m, 2H), 7.50 (s, 2H), 7.32–7.25 (m, 2H), 7.06–6.99 (m, 1H), 4.07 (d, J = 5.8 Hz, 2H), 2.18 (s, 6H). 13CNMR (101 MHz, DMSO-d6) δ 189.04, 168.19, 166.53, 157.06, 146.08, 140.59, 139.44, 137.84, 130.42, 129.34, 128.92, 128.25, 126.12, 125.20, 119.67, 118.69, 43.82, 17.08; HRMS (ESI) calcd for C26H24N2O4 [M + H]+ 429.1770, found 429.1809.
(E)-N-(2-(Dimethylamino)-2-oxoethyl)-4-(3-(4-hydroxy-3,5-dimethylphenyl)acryloyl)benzamide (10s). An amorphous yellow solid; yield: 23% for 6 steps; 1HNMR (400 MHz, DMSO-d6) δ 8.93 (s, 1H), 8.72 (t, J = 5.6 Hz, 1H), 8.16 (d, J = 8.1 Hz, 2H), 7.98 (d, J = 8.2 Hz, 2H), 7.70 (d, J = 15.5 Hz, 1H), 7.60 (d, J = 15.4 Hz, 1H), 7.48 (s, 2H), 4.09 (d, J = 5.6 Hz, 2H), 2.99 (s, 3H), 2.82 (s, 3H), 2.17 (s, 6H). 13CNMR (101 MHz, DMSO-d6) δ 188.97, 168.63, 166.22, 157.39, 146.07, 140.57, 138.06, 130.42, 128.89, 128.15, 125.95, 125.23, 118.54, 41.51, 36.31, 35.67, 17.11; HRMS (ESI) calcd for C22H24N2O4 [M + Na]+ 403.1634, found 403.1628.
(E)-4-(3-(4-Hydroxy-3,5-dimethylphenyl)acryloyl)-N-(2-(methylamino)-2-oxoethyl)benzamide (10p). A yellow solid; yield: 63% for 4 steps; mp 179.3°C; 1HNMR (400 MHz, DMSO-d6) δ 8.97–8.86 (m, 2H), 8.20–8.13 (m, 2H), 8.03–7.96 (m, 2H), 7.83 (d, J = 4.6 Hz, 1H), 7.71 (d, J = 15.5 Hz, 1H), 7.60 (d, J = 15.5 Hz, 1H), 7.48 (s, 2H), 3.81 (d, J = 5.9 Hz, 2H), 2.56 (d, J = 4.5 Hz, 3H), 2.17 (s, 6H). 13CNMR (101 MHz, DMSO-d6) δ 188.98, 168.92, 166.25, 157.04, 146.02, 140.51, 137.98, 130.41, 128.82, 128.28, 126.14, 125.19, 118.70, 43.22, 33.97, 17.09, 15.33; HRMS (ESI) calcd for C21H22N2O4 [M + Na]+ 389.1477, found 389.1471.
(E)-N-(2-(Diethylamino)-2-oxoethyl)-4-(3-(4-hydroxy-3,5-dimethylphenyl)acryloyl)benzamide (10t). An amorphous yellow solid; yield: 76% for 4 steps; 1HNMR (400 MHz, CDCl3) δ 7.95 (d, J = 8.4 Hz, 2H), 7.86 (d, J = 8.4 Hz, 2H), 7.63 (d, J = 15.5 Hz, 1H), 7.26 (d, J = 15.6 Hz, 1H), 7.21 (s, 2H), 7.16 (d, J = 2.2 Hz, 1H), 4.17 (d, J = 3.8 Hz, 2H), 3.37 (q, J = 7.2 Hz, 2H), 3.25 (q, J = 7.2 Hz, 2H), 2.19 (s, 6H), 1.16 (t, 3H), 1.09 (t, J = 7.2 Hz, 3H); 13CNMR (101 MHz, DMSO-d6) δ 188.97, 167.75, 166.15, 157.07, 146.02, 140.51, 138.03, 130.42, 128.92, 128.14, 126.12, 125.17, 118.66, 41.32, 41.07, 40.30, 17.10, 14.60, 13.59. HRMS (ESI) calcd for C24H28N2O4 [M + H]+ 409.2083, found 409.2118.
(E)-3-(4-Hydroxy-3,5-dimethylphenyl)-1-(3′H-spiro[azetidine-3,1′-isobenzofuran]-5′-yl)prop-2-en-1-one (10u). An amorphous yellow solid; yield: 63% for 3 steps; 1HNMR (400 MHz, CD3OD) δ 8.13 (d, J = 8.0 Hz, 1H), 7.98 (s, 1H), 7.83 (d, J = 7.9 Hz, 1H), 7.69 (d, J = 15.5 Hz, 1H), 7.52 (d, J = 15.5 Hz, 1H), 7.35 (s, 2H), 5.21 (s, 2H), 4.50 (d, J = 11.8 Hz, 2H), 4.44 (d, J = 11.9 Hz, 2H), 2.22 (s, 6H). 13CNMR (101 MHz, DMSO-d6) δ 188.86, 157.09, 145.93, 143.99, 140.33, 139.42, 130.37, 129.40, 126.16, 125.24, 122.43, 121.81, 118.71, 84.71, 72.89, 60.04, 17.16; HRMS (ESI) calcd for C21H21NO3 [M + H]+ 336.1555, found 336.1593.
((R)-1-((S)-2-(4-((E)-3-(4-hydroxy-3,5-dimethylphenyl) acryloyl)benzamido)-3-phenylpropanamido)-3-methylbutyl)boronic acid (10v). An amorphous light yellow solid; purity: 100%; yield: 68% for 4 steps; [α]D25 −59.0° (c 0.1, MeOH); 1HNMR (400 MHz, methanol-d4) δ 8.08 (d, J = 8.1 Hz, 2H), 7.89 (d, J = 7.9 Hz, 2H), 7.68 (d, J = 15.7, 3.1 Hz, 1H), 7.51 (d, J = 15.5, 3.1 Hz, 1H), 7.34 (s, 2H), 7.29 (d, J = 4.4 Hz, 4H), 7.25–7.20 (m, 1H), 4.96 (t, J = 8.1 Hz, 1H), 3.21 (dd, J = 7.9, 2.6 Hz, 2H), 2.64 (t, J = 7.7 Hz, 1H), 2.22 (s, 6H), 1.36–1.23 (m, 2H), 1.12 (t, J = 7.5 Hz, 2H), 0.84–0.78 (m, 6H). 13CNMR (101 MHz, CD3OD) δ 190.48, 176.22, 168.12, 156.85, 146.91, 141.14, 137.03, 135.88, 129.57, 129.20, 128.43, 128.22, 127.62, 126.95, 126.08, 124.78, 117.81, 52.04, 48.51, 39.52, 37.00, 25.31, 22.59, 20.59, 15.28; HRMS (ESI) calcd for C32H37BN2O6 [M + Na]+ 579.2642, found 579.2639.
4.2. Biological screening
Abbreviations
| AIM2 | Absent in melanoma 2 |
| ASC | Apoptosis-associated speck-like protein containing a CARD protein |
| BCA | Bicinchoninic acid |
| DCM | Dichloromethane |
| DMF | N,N-Dimethylformamide |
| DSS | Dextran sulfate sodium |
| Gly | Glycine |
| HEPES | N-2-Hydroxyethylpiperazine-N-ethane-sulphonic acid |
| NLRC4 | NOD-like receptor family CARD-containing 4 |
| NLRP3 | NOD-, LRR-, and pyrin domain-containing protein 3 |
| SASP | Salicylazosulfapyridine |
| SDS-PAGE | Sodium dodecyl sulfate polyacrylamide gel electrophoresis |
Author contributions
Zhongjin Yang and Wenhui Hu designed this project. Haowei Cai and Zhuorong Liu performed the chemical synthesis. Yuyun Yan assisted in the chemical synthesis. Ping Sun and Yinghua Zhou performed the in vitro mechanism assays. All authors participated in the animal experiments of DSS-induced ulcerative colitis. Dan Wu carried out the identification experiment of the compounds. Zhongjin Yang and Ping Sun contributed to the writing, review and editing of the manuscript. All authors have given their approval to the final version of the manuscript.Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
This work was supported by the National Natural Science Foundation of China (NSFC) (no. 81803364 to Z. Yang and no. 81872743 to W. Hu), Guangzhou Municipal Science and Technology Project (CN) (no. 202102020546 to Z. Yang) and High-level University Construction Fund of Guangdong Province (no. 06-410-2107213 to Z. Yang).References
- Z. Liu, R. Liu, H. Gao, S. Jung, X. Gao, R. Sun, X. Liu, Y. Kim, H.-S. Lee, Y. Kawai, M. Nagasaki, J. Umeno, K. Tokunaga, Y. Kinouchi, A. Masamune, W. Shi, C. Shen, Z. Guo, K. Yuan, M. Abreu, J.-P. Achkar, V. Andersen, C. Bernstein, S. R. Brant, L. Bujanda, S. C. Ng, L. A. Denson, R. H. Duerr, L. R. Ferguson, D. Franchimont, A. Franke, R. Gearry, H. Hakonarson, J. Halfvarson, C. Heller, A. Julià, J. Kelsen, H. Khalili, S. Kugathasan, J. Kupcinskas, A. Latiano, E. Louis, R. Malekzadeh, J. L. McCauley, C. Moran, D. Okou, T. Orchard, A. Palotie, M. Parkes, J. Pekow, U. Potočnik, G. Radford-Smith, J. D. Rioux, G. Rogler, B. Sands, M. Silverberg, H. Sokol, S. Vermeire, R. K. Weersma, R. J. Xavier, N. Hu, Q. Cao, Y. Wang, Y. Miao, H. Zhang, X. Lv, X. Gao, H. Zhang, J. Su, B. Feng, Y. Zhao, L. Zhu, Y. Chen, L. Zhu, C. Chen, Y. Wang, Y. Wang, Z. Pang, Y. Chen, X. Zhang, H. Li, Q. Yu, M. Ye, S. Zhang, W. Tang, M. Wang, X. Cao, R. Zhu, G. Zhou, Z. Bian, X. Guo, X. Wu, J. Liu, W. Xu, Y. Li, Q. Guo, Z. Guo, S. Zhu, D. Li, J. Liu, T. Ge, J. Cho, M. J. Daly, D. P. B. McGovern, B. D. Ye, K. Song, Y. Kakuta, M. Li and H. Huang, FinnGen, C. International Inflammatory Bowel Disease Genetics, C. Chinese Inflammatory Bowel Disease Genetics, Genetic architecture of the inflammatory bowel diseases across East Asian and European ancestries, Nat. Genet., 2023, 55, 796–806 CrossRef CAS PubMed
.
- R. Ungaro, S. Mehandru, P. B. Allen, L. Peyrin-Biroulet and J. F. Colombel, Ulcerative colitis, Lancet, 2017, 389, 1756–1770 CrossRef
- J. Torres, S. Mehandru, J. F. Colombel and L. Peyrin-Biroulet, Crohn's disease, Lancet, 2017, 389, 1741–1755 CrossRef
- V. Sinopoulou, M. Gordon, A. K. Akobeng, M. Gasparetto, M. Sammaan, J. Vasiliou and T. M. Dovey, Interventions for the management of abdominal pain in Crohn's disease and inflammatory bowel disease, Cochrane Database Syst. Rev., 2021, 11, Cd013531 Search PubMed
- M. Agrawal, E. A. Spencer, J. F. Colombel and R. C. Ungaro, Approach to the Management of Recently Diagnosed Inflammatory Bowel Disease Patients: A User's Guide for Adult and Pediatric Gastroenterologists, Gastroenterology, 2021, 161, 47–65 CrossRef
- B. Verstockt, A. Salas, B. E. Sands, C. Abraham, H. Leibovitzh, M. F. Neurath, N. Vande Casteele, S. Danese, G. D'Haens, L. Eckmann, W. A. Faubion, B. G. Feagan, V. Jairath, C. Ma, S. Mehandru, J. Panes, F. Rieder, W. J. Sandborn, M. S. Silverberg, M. Veny, S. Vermeire and S. Vetrano, ATRC, IL-12 and IL-23 pathway inhibition in inflammatory bowel disease, Nat. Rev. Gastroenterol. Hepatol., 2023, 20, 433–446 CrossRef CAS
- K. Papamichael, W. Afif, D. Drobne, M. C. Dubinsky, M. Ferrante, P. M. Irving, N. Kamperidis, T. Kobayashi, P. G. Kotze, J. Lambert, N. M. Noor, X. Roblin, G. Roda, N. Vande Casteele, A. J. Yarur, N. Arebi, S. Danese, S. Paul, W. J. Sandborn, S. Vermeire, A. S. Cheifetz and L. Peyrin-Biroulet, Therapeutic drug monitoring of biologics in inflammatory bowel disease: unmet needs and future perspectives, Lancet Gastroenterol. Hepatol., 2022, 7, 171–185 CrossRef
- M. Friedrich, M. Pohin, M. A. Jackson, I. Korsunsky, S. J. Bullers, K. Rue-Albrecht, Z. Christoforidou, D. Sathananthan, T. Thomas, R. Ravindran, R. Tandon, R. S. Peres, H. Sharpe, K. Wei, G. F. M. Watts, E. H. Mann, A. Geremia, M. Attar, F. Barone, M. Brenner, C. D. Buckley, M. Coles, A. P. Frei, K. G. Lassen, F. M. Powrie, S. McCuaig, L. Thomas, E. Collantes, H. H. Uhlig, S. N. Sansom, A. Easton, S. Raychaudhuri, S. P. Travis and F. M. Powrie, IL-1-driven stromal–neutrophil interactions define a subset of patients with inflammatory bowel disease that does not respond to therapies, Nat. Med., 2021, 27, 1970–1981 CrossRef CAS
- C. A. Dinarello, The IL-1 family of cytokines and receptors in rheumatic diseases, Nat. Rev. Rheumatol., 2019, 15, 612–632 CrossRef CAS PubMed
- Y. Li, H. Huang, B. Liu, Y. Zhang, X. Pan, X.-Y. Yu, Z. Shen and Y.-H. Song, Inflammasomes as therapeutic targets in human diseases, Signal Transduction Targeted Ther., 2021, 6, 247 CrossRef CAS
- V. Khatri and R. Kalyanasundaram, Therapeutic implications of inflammasome in inflammatory bowel disease, FASEB J., 2021, 35, e21439 CrossRef CAS PubMed
- A. Salas, C. Hernandez-Rocha, M. Duijvestein, W. Faubion, D. McGovern, S. Vermeire, S. Vetrano and N. Vande Casteele, JAK-STAT pathway targeting for the treatment of inflammatory bowel disease, Nat. Rev. Gastroenterol. Hepatol., 2020, 17, 323–337 CrossRef
- S. Banerjee, A. Biehl, M. Gadina, S. Hasni and D. M. Schwartz, JAK-STAT Signaling as a Target for Inflammatory and Autoimmune Diseases: Current and Future Prospects, Drugs, 2017, 77, 521–546 CrossRef CAS PubMed
- S. Mishra, A. Jena, R. Kakadiya, V. Sharma and V. Ahuja, Positioning of tofacitinib in treatment of ulcerative colitis: a global perspective, Expert Rev. Gastroenterol. Hepatol., 2022, 16, 737–752 CrossRef CAS PubMed
- X. Hu, J. Li, M. Fu, X. Zhao and W. Wang, The JAK/STAT signaling pathway: from bench to clinic, Signal Transduction Targeted Ther., 2021, 6, 402 CrossRef
- S. Schreiber, P. Rosenstiel, J. Hampe, S. Nikolaus, B. Groessner, A. Schottelius, T. Kühbacher, J. Hämling, U. R. Fölsch and D. Seegert, Activation of signal transducer and activator of transcription
(STAT) 1 in human chronic inflammatory bowel disease, Gut, 2002, 51, 379–385 CrossRef CAS PubMed
- J. Mudter, B. Weigmann, B. Bartsch, R. Kiesslich, D. Strand, P. R. Galle, H. A. Lehr, J. Schmidt and M. F. Neurath, Activation pattern of signal transducers and activators of transcription (STAT) factors in inflammatory bowel diseases, Am. J. Gastroenterol., 2005, 100, 64–72 CrossRef CAS PubMed
- S. K. Bandyopadhyay, C. A. de la Motte, S. P. Kessler, V. C. Hascall, D. R. Hill and S. A. Strong, Hyaluronan-mediated leukocyte adhesion and dextran sulfate sodium-induced colitis are attenuated in the absence of signal transducer and activator of transcription 1, Am. J. Pathol., 2008, 173, 1361–1368 CrossRef CAS PubMed
- J. X. Lin, P. Li, D. Liu, H. T. Jin, J. He, M. A. U. Rasheed, Y. Rochman, L. Wang, K. Cui, C. Liu, B. L. Kelsall, R. Ahmed and W. J. Leonard, Critical Role of STAT5 transcription factor tetramerization for cytokine responses and normal immune function, Immunity, 2012, 36, 586–599 CrossRef CAS PubMed
- S. Gilbert, H. Nivarthi, C. N. Mayhew, Y.-H. Lo, T. K. Noah, J. Vallance, T. Rülicke, M. Müller, A. G. Jegga, W. Tang, D. Zhang, M. Helmrath, N. Shroyer, R. Moriggl and X. Han, Activated STAT5 Confers Resistance to Intestinal Injury by Increasing Intestinal Stem Cell Proliferation and Regeneration, Stem Cell Rep., 2015, 4, 209–225 CrossRef CAS PubMed
- K. L. Monaghan, D. Aesoph, A. G. Ammer, W. Zheng, S. Rahimpour, B. Y. Farris, C. A. Spinner, P. Li, J. X. Lin, Z. X. Yu, V. Lazarevic, G. Hu, W. J. Leonard and E. C. K. Wan, Tetramerization of STAT5 promotes autoimmune-mediated neuroinflammation, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2116256118 CrossRef CAS
- J.-X. Lin, P. Li, D. Liu, H. T. Jin, J. He, M. A. U. Rasheed, Y. Rochman, L. Wang, K. Cui, C. Liu, B. L. Kelsall, R. Ahmed and W. J. Leonard, Critical Role of STAT5 Transcription Factor Tetramerization for Cytokine Responses and Normal Immune Function, Immunity, 2012, 36, 586–599 CrossRef CAS PubMed
- M. Hedl, R. Sun, C. Huang and C. Abraham, STAT3 and STAT5 Signaling Thresholds Determine Distinct Regulation for Innate Receptor-Induced Inflammatory Cytokines, and STAT3/STAT5 Disease Variants Modulate These Outcomes, J. Immunol., 2019, 203, 3325–3338 CrossRef
- C. Zhang, H. Yue, P. Sun, L. Hua, S. Liang, Y. Ou, D. Wu, X. Wu, H. Chen, Y. Hao, W. Hu and Z. Yang, Discovery of chalcone analogues as novel NLRP3 inflammasome inhibitors with potent anti-inflammation activities, Eur. J. Med. Chem., 2021, 219, 113417 CrossRef CAS
- A. A. WalyEldeen, S. Sabet, H. M. El-Shorbagy, I. A. Abdelhamid and S. A. Ibrahim, Chalcones: Promising therapeutic agents targeting key players and signaling pathways regulating the hallmarks of cancer, Chem.-Biol. Interact., 2023, 369, 110297 CrossRef CAS PubMed
- M. S. J. Mangan, E. J. Olhava, W. R. Roush, H. M. Seidel, G. D. Glick and E. Latz, Targeting the NLRP3 inflammasome in inflammatory diseases, Nat. Rev. Drug Discovery, 2018, 17, 588–606 CrossRef CAS PubMed
- X. Wu, P. Sun, X. Chen, L. Hua, H. Cai, Z. Liu, C. Zhang, S. Liang, Y. Chen, D. Wu, Y. Ou, W. Hu and Z. Yang, Discovery of a Novel Oral Proteasome Inhibitor to Block NLRP3 Inflammasome Activation with Anti-inflammation Activity, J. Med. Chem., 2022, 65, 11985–12001 CrossRef CAS PubMed
- J. Tang, S. Tu, G. Lin, H. Guo, C. Yan, Q. Liu, L. Huang, N. Tang, Y. Xiao, R. M. Pope, M. V. S. Rajaram, A. O. Amer, B. M. Ahmer, J. S. Gunn, D. J. Wozniak, L. Tao, V. Coppola, L. Zhang, W. Y. Langdon, J. B. Torrelles, S. Lipkowitz and J. Zhang, Sequential ubiquitination of NLRP3 by RNF125 and Cbl-b limits inflammasome activation and endotoxemia, J. Exp. Med., 2020, 217, e20182091 CrossRef PubMed
Footnotes |
| † Electronic supplementary information (ESI) available: The cytotoxicity profile for 10v; the 1H NMR, 13C NMR, and HRMS of all target compounds; and the HPLC purity of 10v are available free of charge. See DOI: https://doi.org/10.1039/d3md00451a |
| ‡ All these authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |