
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInhibition of DXR in the MEP pathway with lipophilic N-alkoxyaryl FR900098 analogs†
Darean
Bague‡
a,
Ruiqin
Wang‡
a,
Dana
Hodge
 d,
Marwa O.
Mikati
e,
Jose S.
Roma
c,
Helena I.
Boshoff
c,
Allyson L.
Dailey
b,
Misgina
Girma
b,
Robin D.
Couch
b,
Audrey R.
Odom John
de and
Cynthia S.
Dowd
*a
d,
Marwa O.
Mikati
e,
Jose S.
Roma
c,
Helena I.
Boshoff
c,
Allyson L.
Dailey
b,
Misgina
Girma
b,
Robin D.
Couch
b,
Audrey R.
Odom John
de and
Cynthia S.
Dowd
*a
aDepartment of Chemistry, George Washington University, Washington, D.C. 20052, USA. E-mail: cdowd@gwu.edu
bDepartment of Chemistry and Biochemistry, George Mason University, Fairfax, VA 22030, USA
cTuberculosis Research Section, LCIM, NIAID/NIH, Bethesda, MD 20892, USA
dDivision of Infectious Diseases, Children's Hospital of Philadelphia, Philadelphia, PA 19104, USA
eDepartment of Molecular Microbiology, Washington University School of Medicine, St. Louis, MO 63110, USA
First published on 22nd May 2024
Abstract
In Mycobacterium tuberculosis (Mtb) and Plasmodium falciparum (Pf), the methylerythritol phosphate (MEP) pathway is responsible for isoprene synthesis. This pathway and its products are vital to bacterial/parasitic metabolism and survival, and represent an attractive set of drug targets due to their essentiality in these pathogens but absence in humans. The second step in the MEP pathway is the conversion of 1-deoxy-D-xylulose-5-phosphate (DXP) to MEP and is catalyzed by 1-deoxy-D-xylulose-5-phosphate reductoisomerase (DXR). Natural products fosmidomycin and FR900098 inhibit DXR, but are too polar to reach the desired target inside some cells, such as Mtb. Synthesized FR900098 analogs with lipophilic substitution in the position α to the phosphorous atom showed promise, resulting in increased activity against Mtb and Pf. Here, an α substitution, consisting of a 3,4-dichlorophenyl substituent, in combination with various O-linked alkylaryl substituents on the hydroxamate moiety is utilized in the synthesis of a novel series of FR900098 analogs. The purpose of the O-linked alkylaryl substituents is to further enhance DXR inhibition by extending the structure into the adjacent NADPH binding pocket, blocking the binding of both DXP and NADPH. Of the initial O-linked alkylaryl substituted analogs, compound 6e showed most potent activity against Pf parasites at 3.60 μM. Additional compounds varying the phenyl ring of 6e were synthesized. The most potent phosphonic acids, 6l and 6n, display nM activity against PfDXR and low μM activity against Pf parasites. Prodrugs of these compounds were less effective against Pf parasites but showed modest activity against Mtb cells. Data from this series of compounds suggests that this combination of substituents can be advantageous in designing a new generation of antimicrobials.
Introduction
Tuberculosis (TB) and malaria still pose a significant threat to global public health despite important research efforts toward eradication. The major causative agents of TB and malaria are Mycobacterium tuberculosis (Mtb) and Plasmodium falciparum (Pf), respectively. The World Health Organization (WHO) reported an estimated combined 2.2 million deaths related to TB and malaria globally in 2021.1,2 Further exacerbating the situation, there has been a rapid increase in Mtb and Pf drug resistance, leaving limited therapeutic options.3–10 The WHO reported 1.5 million cases of drug-resistant TB in 2021.2 Similarly, in regions with a high malaria burden, such as the Greater Mekong Subregion (GMS) and Africa, resistance to artemisinin-combination therapy (ACT) is rising at an alarming rate.1,5–7,10 This threat to global public health requires the discovery and validation of novel drug targets that are not currently being used by existing antibiotics.The 2C-methyl-D-erythritol-4-phosphate (MEP, or nonmevalonate) pathway for isoprene synthesis (Fig. 1) is found in several pathogens, including Mtb and Pf.11–16 Isoprene isomers isopentenyl pyrophosphate (IPP) and dimethylallyl pyrophosphate (DMAPP) are vital as they are converted to isoprenoids which are essential to cell wall synthesis and other cellular functions.17–20 Although isoprene synthesis is also vital to humans, we synthesize isoprenes through an alternate pathway. This makes the MEP pathway an attractive set of drug targets. Interrupting synthesis of isoprenes is lethal for Mtb and Pf cells, thus providing a novel method to combat both drug-sensitive and drug-resistant strains of Mtb and Pf.
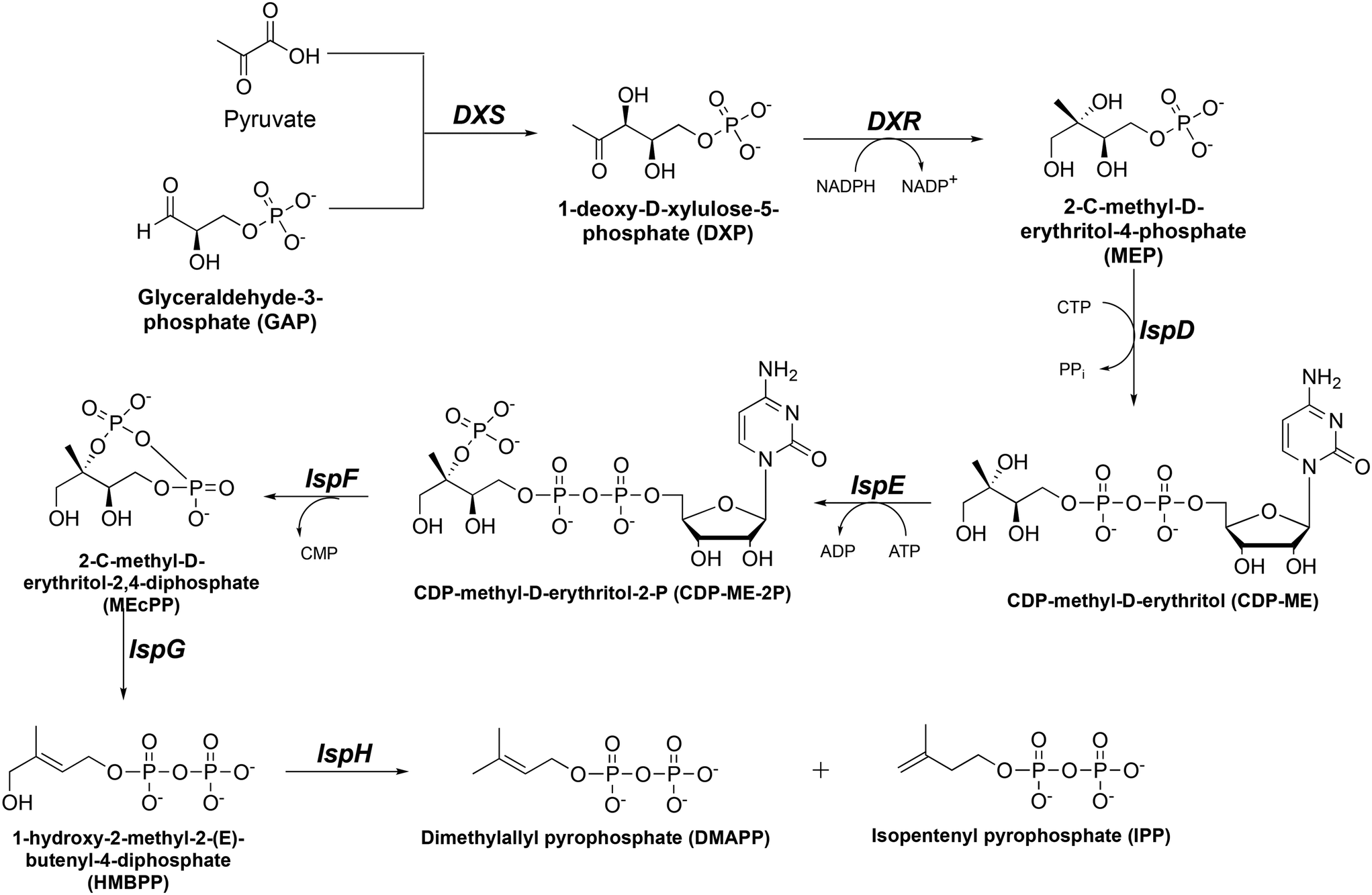 | ||
| Fig. 1 The methyl erythritol phosphate (MEP) pathway of isoprene synthesis. | ||
In the MEP pathway, 1-deoxy-D-xylulose-5-phosphate (DXP) is isomerized and reduced to MEP (Fig. 1).14–20 This step is catalyzed by 1-deoxy-D-xylulose 5-phosphate reductoisomerase (DXR), which is a validated drug target for malaria.21–24 Fosmidomycin (Fos, 1) and FR900098 (2) are natural products produced by Streptomyces lavendulae and Streptomyces rubellomurinus, respectively, which have antimicrobial activities against Gram-negative bacteria (Fig. 2).25–32 These compounds are potent inhibitors of DXR, mimic the DXP substrate, and bind to DXR at the DXP binding site. Although potent, Fos and FR900098 are very polar and can display poor cellular activity.16–28 Thus, they have been frequent starting points for inhibitor design. Fos and FR900098 have four positions where modifications could enhance Mtb and Pf activity: the phosphonate, the propyl backbone, the N-hydroxyl moiety, and the N-acyl substituent. We and others have synthesized analogs in which all of these moieties were modified, and the resulting analogs were tested for activity against Mtb and Pf.33–53 Additionally, these compounds (for example, 3 and 4) often have better cell permeability due to increased lipophilicity.29,54
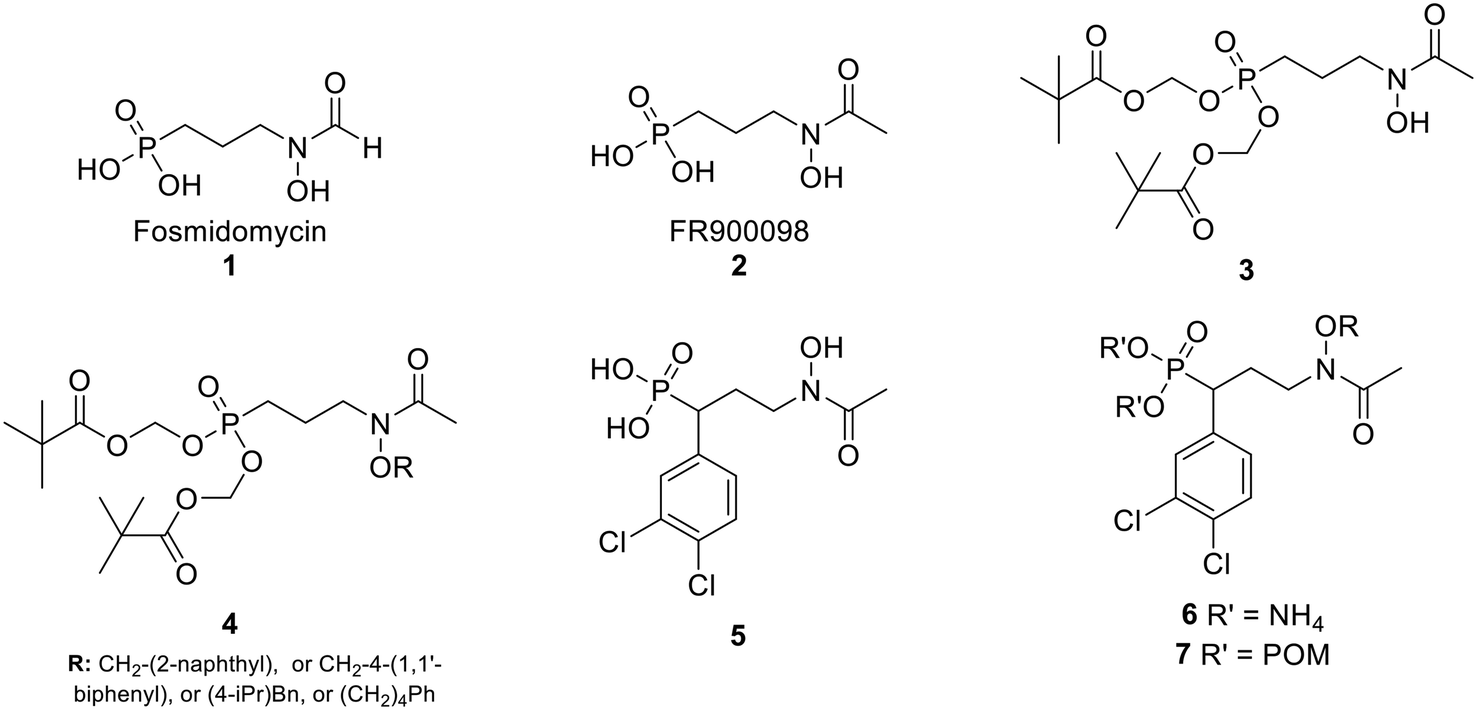 | ||
| Fig. 2 Fosmidomycin (1), FR900098 (2), previously reported compounds (3–5), and compounds in current work (6–7). | ||
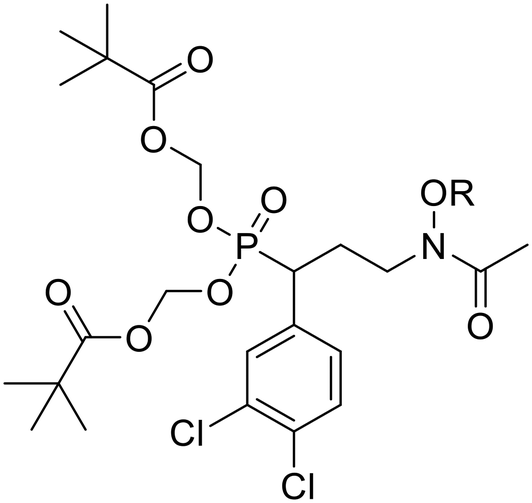
In the current work, we combine two structural features, previously shown to be advantageous to improving antimicrobial activity, in an effort to design a novel family of MEP pathway inhibitors, termed MEPicides. In 2006, the Van Calenbergh group reported a series of analogs bearing an aryl substituent alpha to the phosphonate moiety.55,56 Specifically, the analog with 3,4-dichlorophenyl substituent in this position (5) had improved activity against Pf. In our own work, we previously explored Fos/FR900098 analogs that bind to both the DXP binding site and the adjacent NADPH binding site through extension of the retrohydroxamate with either an N-acyl or N-alkoxy substituent (4).34,38,54 Here, we report a series of FR900098 analogs that combine features of both of these efforts: an α-3,4-dichlorophenyl group and an N-alkoxy substituent (6 and 7). Aromatic N-alkoxy substituents were chosen in order to explore binding to the NADPH binding site, as in earlier work.34,54 The phosphonic acid salts (6) and pivaloyloxymethylene prodrugs (7) were made to explore both enzyme inhibition and cellular activity.
Results & discussion
Synthesis
The route used to prepare the compounds is shown in Scheme 1. Starting from 3,4-dichlorobenzaldehyde, compound 8 was synthesized as previously described.55 Compound 8 was then subjected to reductive amination to yield benzyloxyamine 9. Upon acetylation, the retrohydroxamate moiety was completed to give the diethyl ester phosphonate 10. The diethyl ester of 10 was cleaved using trimethylsilylbromide (TMSBr), and the dipivaloyloxymethyl (diPOM) prodrug 11 was synthesized using pivaloyloxymethylchloride (POMCl). Compound 11 was debenzylated with boron trichloride (BCl3) to give compound 12. The hydroxylamine of compound 12 was then reacted with the appropriate alkyl bromide to undergo Williamson ether synthesis, yielding diPOM prodrug 7l. For the remaining ethers, the benzyl protecting group of the hydroxylamine in compound 10 was cleaved using BCl3 to give intermediate 13. Application of Williamson ether conditions with commercially available alkyl bromides yielded N-O-ethers 14b–h,j–n. Compound 14h was subjected to Suzuki coupling to give compound 14i. Diethyl ester phosphonate 13 and N-alkoxy compounds 14b–n were converted to diammonium salts 6a–nvia cleavage of the diethyl groups with TMSBr followed by treatment with 7 M NH3 in methanol. Reaction of salts 6a–k,m,n with POMCl yielded diPOM ester phosphonates 7a–k,m,n.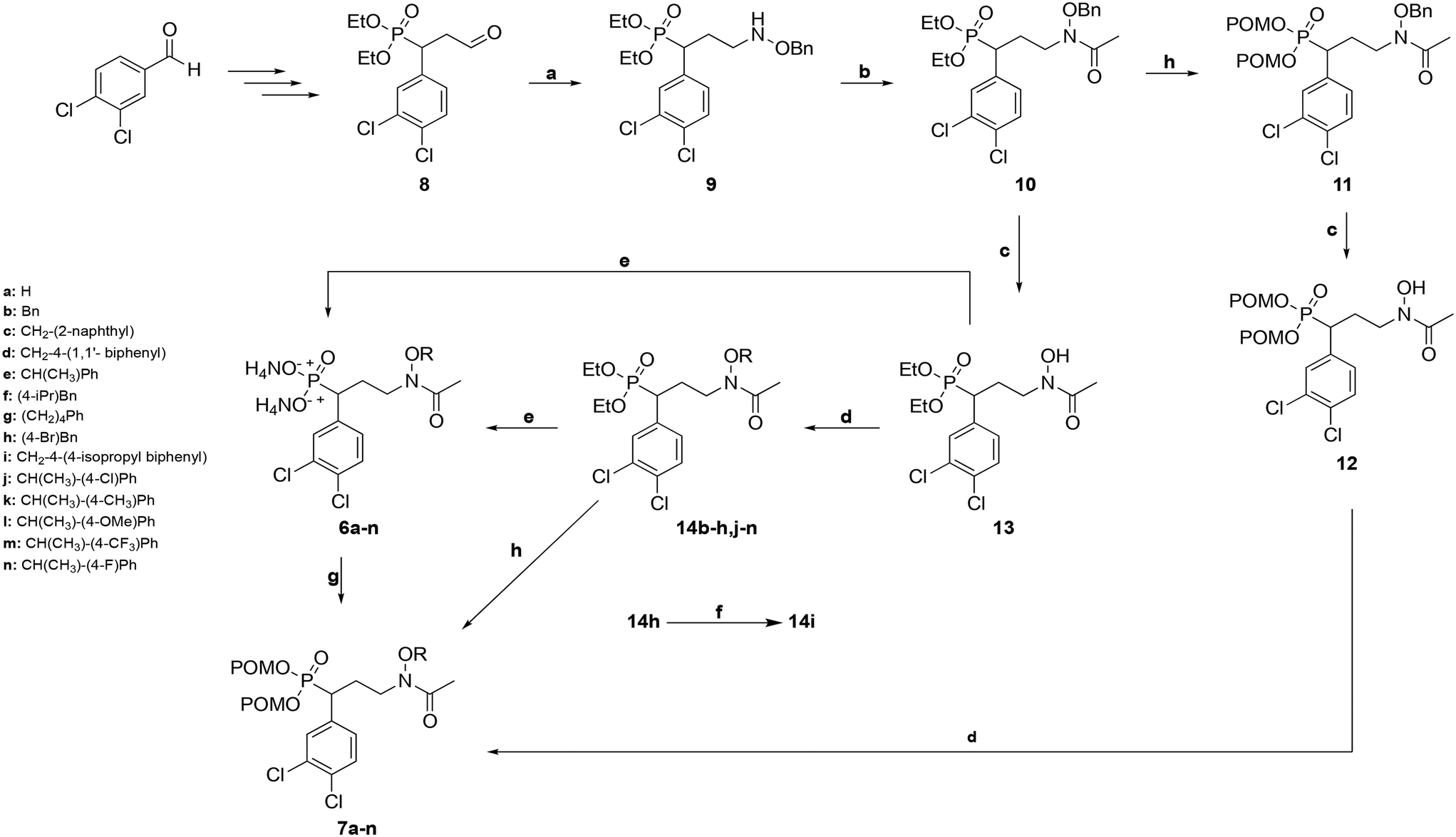 | ||
| Scheme 1 Synthesis of FR900098 Analogs. Reagents and conditions: (a) (i) OBHA, MeOH, rt, (ii) NaBH3CN, acetic acid, MeOH, rt, 41%; (b) AcCl, Et3N, DCM 0 °C to rt, 85%; (c) BCl3, DCM, −78 °C, 95–97%; (d) alkyl bromide, NaH, THF, 0 °C to rt, 4–97%; (e) (i) TMSBr, DCM, 0 °C to rt, (ii) 7M NH3 in MeOH, 57%-quantitative; (f) 4-isopropylphenyl boronic acid, Pd(PPh3)4, diethyl ether, Na2CO3, EtOH, 70 °C, 19%; (g) POMCl, DIPEA, NaI, THF, 60 °C, 13–60%; (h) (i) TMSBr, DCM, 0 °C to rt, (ii) POMCl, DIPEA, NaI, THF, 60 °C, 15%. Abbreviations: OBHA = O-benzylhydroxylamine, AcCl = acetyl chloride, RBr = alkyl bromide, TMSBr = trimethylsilylbromide, POMCl = pivaloyloxymethylchloride, DCM = dichloromethane, DIPEA = N,N-diisopropylethylamine, THF = tetrahydrofuran. | ||
Biological evaluation
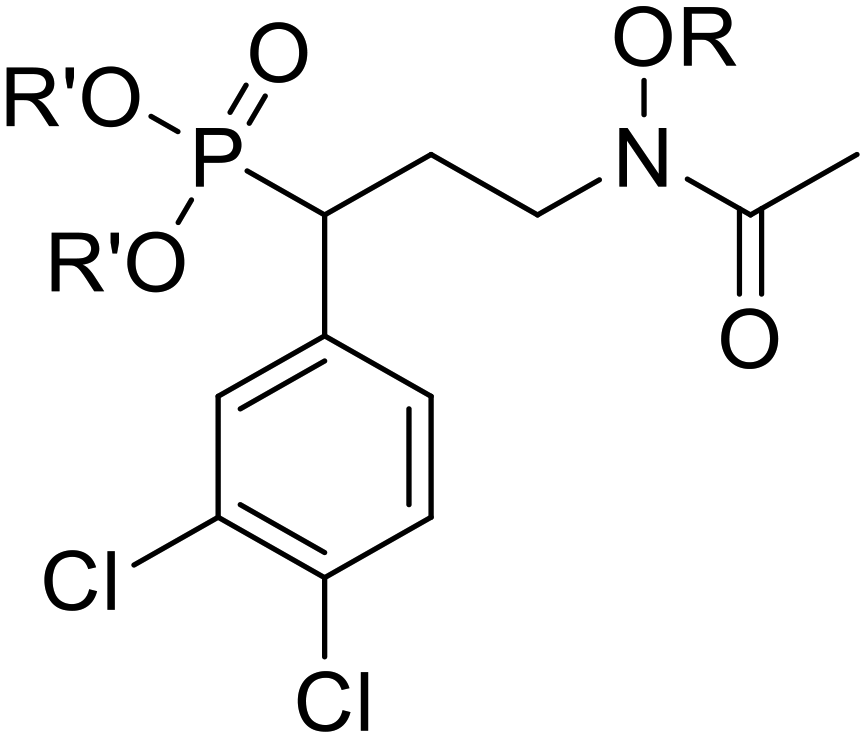
The compounds were subjected to a series of assays to evaluate their activity as inhibitors of DXR, their ability to inhibit Mtb and Pf growth, and their cytotoxicity in mammalian cells. In addition, to verify that the compounds inhibit the MEP pathway as expected, malaria parasites treated with the most active compounds were subjected to IPP rescue, as supplementation with IPP restores growth to parasites treated with well-characterized MEP pathway inhibitors.57 These compounds were also examined against Pf parasites bearing the had1 mutation, which is resistant to fosmidomycin due to increased substrate levels.58Evaluation of phosphonic acid salts against PfDXR and MtbDXR
Salts 6a–n were tested for activity against both PfDXR and MtbDXR, and this data is shown in Table 1. Enzyme activity was initially measured at a single concentration of 100 μM. For compounds inhibiting DXR more than 90%, the half-maximal inhibitory concentration (IC50) was determined (Fig. S1 and S2†). Previous docking studies showed that aromatic N-alkoxy substituents could occupy the NADPH cofactor binding site, and a series of aromatic N-O-ethers were most successful.13,14 To explore the effect of adding a 3,4-dichlorophenyl substituent (alpha to the phosphorous atom) to the previously synthesized N-O-ethers, compounds 6b–6i were synthesized.|
|
||||||
|---|---|---|---|---|---|---|
| Compound | R | Pf DXRa | Pf IC50a | Mtb DXRa | Mtb MICb | clog![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Pc Pc |
| μM | μM | μM | μg mL−1 (μM) | |||
| a Half-maximal inhibitory activity (IC50). Values in parentheses are percent remaining enzyme activity at 100 μM. b 7H9/glucose/casitone/tyloxapol media. c Calculated using MarvinSketch. | ||||||
| 1 (Fos) | H | 0.063 (ref. 59) | 1.021 (ref. 59) | 0.44 (ref. 34) | >500 | −2.64 |
| 2 (FR) | H | 0.022 (ref. 59) | 0.511 (ref. 59) | 2.91 (ref. 34) | >500 | −2.54 |
| 6a | H | 13.62 | 18.63 ± 3.81 | (15.70) | >100 (>266) | 0.31 |
| 6b | CH2Ph | (26.66) | 31.46 ± 7.85 | (34.20) | >100 (>214) | 3.06 |
| 6c | CH2-(2-Naphthyl) | (46.64) | 60.91 ± 4.97 | (82.10) | 12.5 (24.2) | 4.06 |
| 6d | CH2-4-(1,1′-Biphenyl) | (26.97) | 86.37 ± 10.12 | (93.90) | 12.5 (23.1) | 4.72 |
| 6e | CH(CH3)Ph | 16.83 | 3.60 ± 0.80 | (64.60) | >100 (>208) | 3.47 |
| 6f | CH2-(4-iPr)Ph | 26.46 | 68.73 ± 9.44 | (80.90) | 6.25 (12.3) | 4.29 |
| 6g | (CH2)4Ph | (25.64) | 63.31 ± 16.73 | (99.10) | 1.56 (3.1) | 3.86 |
| 6h | CH2-(4-Br)Ph | (53.07) | 67.82 ± 3.57 | (72.30) | >50 (>92) | 3.83 |
| 6i | CH2-4-[4′-iPr-(1,1′-biphenyl)] | (56.11) | 38.14 ± 0.43 | (63.20) | 25 (45) | 4.72 |
| 6j | CH(CH3)-(4-Cl)Ph | 20.35 | 68.59 ± 20.90 | (66.20) | 37 (72) | 3.62 |
| 6k | CH(CH3)-(4-CH3)Ph | 1.21 | 3.53 ± 0.66 | (30.70) | >50 (>101) | 3.56 |
| 6l | CH(CH3)-(4-OCH3)Ph | 0.11 | 0.30 ± 0.04 | 0.78 | >50 (>98) | 2.86 |
| 6m | CH(CH3)-(4-CF3)Ph | (74.69) | 79.24 ± 12.11 | (59.60) | >50 (>91) | 3.94 |
| 6n | CH(CH3)-(4-F)Ph | 0.47 | 2.42 ± 0.43 | 4.78 | >100 (>200) | 3.62 |
Among the first set of nine analogs (6a–i), compounds 6a and 6e were the most potent against PfDXR with IC50 values of 13.62 and 16.83 μM, respectively. Interestingly, compound 6e is the only analog of the initial set which bears a methyl branch at the benzyl position of the N-O-ether. SAR exploration of the phenyl ring of 6e yielded compounds 6j–n. Of these five 4-position analogs, three showed improved PfDXR activity relative to 6e. Compound 6k (4-OCH3) displayed an IC50 value against PfDXR of 1.21 μM. Although not as active as parent compounds fosmidomycin (1) and FR900098 (2), compound 6l (4-OCH3) and 6n (4-F) were the quite potent against PfDXR with IC50 values of 0.11 μM and 0.47 μM, respectively.
When evaluated for activity against MtbDXR, the majority of salts 6a–n were inactive. Two exceptions were compounds 6l (4-OCH3) and 6n (4-F), with IC50 values of 0.78 μM and 4.78 μM, respectively. Interestingly, the IC50 value of 6l is a significant improvement over FR900098 (2) and on par with that of fosmidomycin (1). The increase in potency for these new compounds could be due to the increased electron density of the para substituents. Additional analogs will be needed to show a clear trend.
Evaluation of phosphonic acid salts against P. falciparum and M. tuberculosis
Compounds 6a–n were also evaluated for activity against Pf3D7 malaria parasites and MtbH37Rv (Table 1, Fig. S3†). Among the first set of compounds (6a–i), compound 6e was again the most potent against Pf (IC50 value of 3.60 μM). This data is further evidence that the branch methyl group is beneficial for activity. Without the branch methyl group, potency suffers as seen in compound 6b. Among compounds 6j–6n, compounds 6k, 6l, and 6n had improved activity compared with the parent compound 6e. This trend is similar to the compounds' activities against the enzyme. Compound 6l is the most potent phosphonic acid salt of the series against Pf parasites, with an IC50 value of 0.30 μM, and the only analog with potency against Pf parasites better than that of fosmidomycin (1). Against Mtb, several compounds showed modest activity, which is an improvement over both parent compounds. The most potent salt was compound 6g. None of these salts, however, were active against MtbDXR, indicating the compounds likely inhibit Mtb growth through an alternate mechanism.Evaluation of analog prodrugs against P. falciparum and M. tuberculosis
Prodrugs 7a–n were evaluated for activity against Pf3D7 parasites and Mtb cells (Table 2 and Fig. S3†). Compound 7l, the prodrug of phosphonic acid 6l, is the most potent prodrug against Pf parasites with an IC50 value of 0.37 μM. This compound is more active against Pf parasites than either fosmidomycin (1) or FR900098 (2). Further, this data confirms the branch methyl group and the 4-OCH3 substituent are beneficial for the potency of these compounds. When evaluated for activity against Mtb, several prodrugs had moderate activity. Prodrugs 7a, 7l, and 7n had the greatest potency with MIC values of 4.60 μg mL−1 (6.5–8.1 μM). This is a significant improvement in activity compared with the parent salts which were inactive, confirming the lipophilic prodrug is important for activity, likely due to increased cell penetration.|
|
||||
|---|---|---|---|---|
| Compound | R | Pf IC50a | Mtb MICb | clogPc |
| μM | μg mL−1 (μM) | |||
| a Pf3D7 parasites. b Mtb H37Rv; 7H9/glucose/casitone/tyloxapol media. c Calculated using MarvinSketch; Pf = P. falciparum; Mtb = M. tuberculosis; IC50 = half-maximal inhibitory concentration; MIC = minimal inhibitory concentration. | ||||
| 1 (Fos) | H | 1.021 (ref. 59) | >500 (ref. 34) | −2.64 |
| 2 (FR) | H | 0.511 (ref. 59) | >500 (ref. 34) | −2.54 |
| 7a | H | 13.01 ± 0.03 | 4.60 (8.1) | 5.59 |
| 7b | CH2Ph | 41.60 ± 4.20 | 6.25 (9.5) | 7.65 |
| 7c | CH2-(2-naphthyl) | 13.96 ± 1.91 | 6.25 (8.8) | 8.65 |
| 7d | CH2-4-(1,1′-biphenyl) | 8.78 ± 0.92 | 12.50 (17.0) | 9.77 |
| 7e | CH(CH3)Ph | 5.22 ± 1.33 | 9.40 (13.9) | 8.06 |
| 7f | CH2-(4-iPr-Ph) | 13.86 ± 1.79 | 12.50 (17.8) | 8.88 |
| 7g | (CH2)4Ph | 13.94 ± 0.92 | 12.50 (17.8) | 8.77 |
| 7h | CH2-(4-Br)Ph | 34.80 ± 6.80 | 6.25 (8.5) | 8.42 |
| 7i | CH2-4-[4′-iPr-(1,1′-biphenyl)] | 14.92 ± 5.55 | >100 (>128) | 10.54 |
| 7j | CH(CH3)-(4-Cl)Ph | 30.07 ± 6.64 | 9.40 (13.3) | 9.02 |
| 7k | CH(CH3)-(4-CH3)Ph | 31.91 ± 8.09 | 12.50 (18.2) | 8.97 |
| 7l | CH(CH3)-(4-OCH3)Ph | 0.37 ± 0.06 | 4.60 (6.5) | 8.25 |
| 7m | CH(CH3)-(4-CF3)Ph | 23.84 ± 3.91 | >100 (>134) | 9.39 |
| 7n | CH(CH3)-(4-F)Ph | 21.16 ± 1.15 | 4.60 (6.6) | 8.21 |
Confirming on-target MEP pathway inhibition: IPP rescue and had1 mutant resistance
The most active inhibitors of PfDXR and Pf parasite growth were further examined to confirm inhibition of the MEP pathway. The IPP rescue assay is a useful method of determining whether inhibitors are acting through the MEP pathway. Inhibition of DXR stops production of IPP, blocking the entire MEP pathway, and killing the parasite. In the presence of media supplemented with excess IPP, parasites are rescued from inhibition and growth is restored. The most potent antiparasitic compounds were evaluated in the IPP rescue assay (Fig. 3). The addition of exogenous IPP rescues the growth of P. falciparum parasites treated with phosphonic acid salts 6k, 6l, 6n, and prodrug 7l. This data suggests these analogs inhibit Pf parasite growth via blocking the MEP pathway.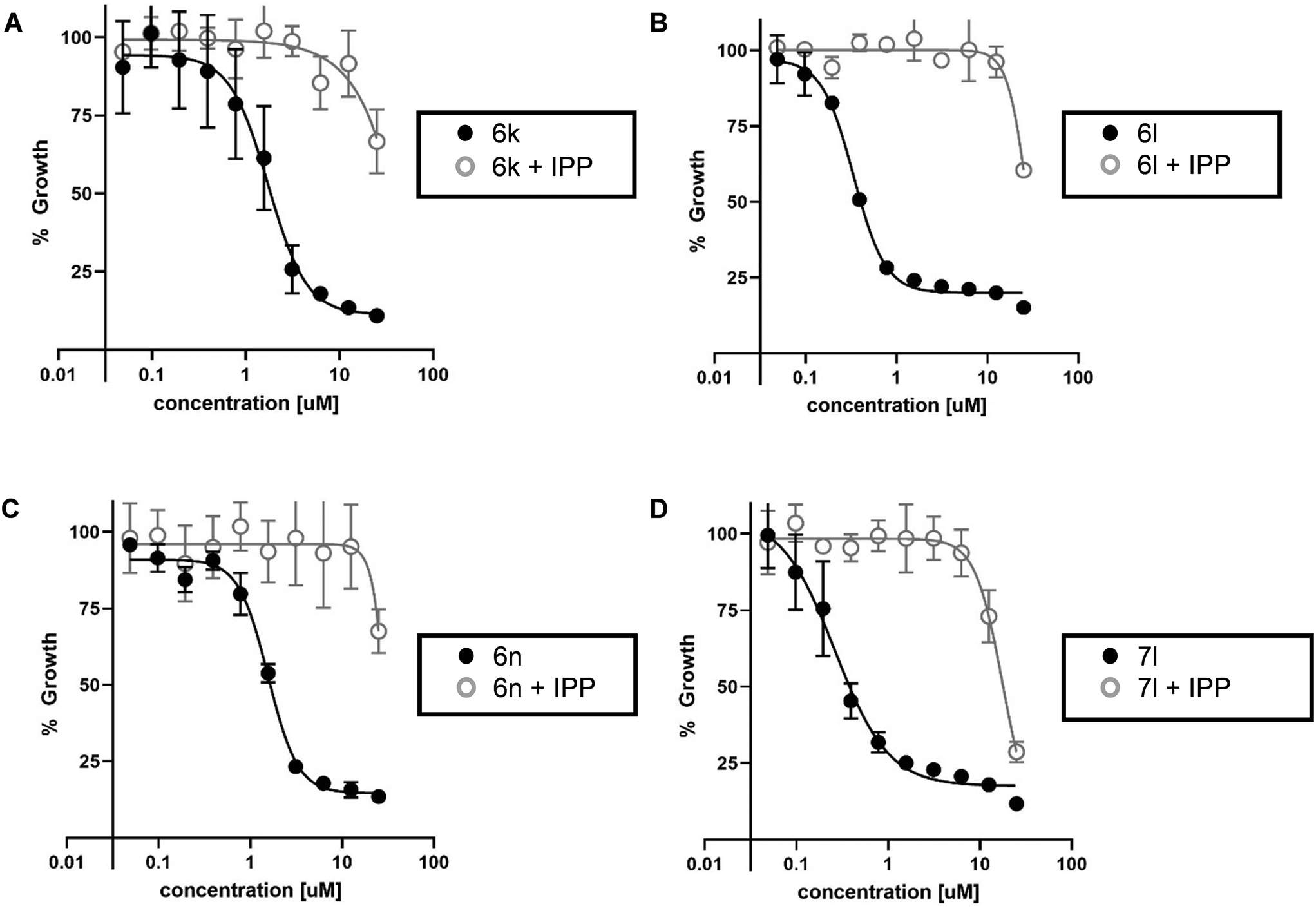 | ||
| Fig. 3 IPP rescue of inhibitor-treated P. falciparum. Drug treatment kills Pf parasites (closed circles). IPP, the product of the MEP pathway, rescues growth of drug-treated parasites (open circles). Shown are graphs from four independent experiments: A) 6k); B) 6l; C) 6n; D) 7l. | ||
IPP rescue alone does not fully confirm MEP pathway inhibition as compounds with alternate mechanisms of action, related to the apicoplast but not the MEP pathway, can also be rescued by IPP.57 To fully confirm the mechanism of action of 6k, 6l, 6n, and 7l is through inhibition of the MEP pathway, they were tested for cross resistance to the fosmidomycin-resistant strain of Pf bearing the had1 mutation. This mutation confers fosmidomycin resistance via increased levels of DXR substrate DXP. Indeed, compounds 6k, 6l, 6n, and 7l also displayed increased resistance to had1 mutant Pf strain (Fig. S3 and S4†).
Cytotoxicity
Compounds that had the most potent activity against Pf parasites (6k, 6l, 6n, and 7l) were evaluated for cytotoxic properties against human hepatocytes (HepG2, Table 3). None of the compounds were cytotoxic up to 100 μg mL−1.|
|
|||
|---|---|---|---|
| Compound | R | R′ | HepG2 IC50 μg mL−1 (μM) |
| IC50 = half-maximal inhibitory concentration. | |||
| 6k | CH(CH3)-(4-CH3)Ph | NH4 | >100 (>200) |
| 6l | CH(CH3)-(4-OCH3)Ph | NH4 | >100 (>200) |
| 6n | CH(CH3)-(4-F)Ph | NH4 | >100 (>200) |
| 7l | CH(CH3)-(4-OCH3)Ph | POM | >100 (>140) |
| Tamoxifen | — | — | 11.72 μM |
Conclusion
We set out to design a novel set of FR900098 analogs based on compounds 4 and 5, which have reported potent activities against P. falciparum and M. tuberculosis. This work complements our prior work by asking if combining the α-3,4-dichlorophenyl and N-alkoxyaryl substituents would enhance activity against PfDXR, MtbDXR, and the intact organisms. We also questioned if a prodrug approach would impact the potency of the compounds. Finally, we queried whether our most potent compounds were on-target inside the cell, while also evaluating their cytotoxicity.The data shows that the disubstituted α-3,4-dichlorophenyl, N-alkoxyaryl analogs are most potent against PfDXR and Pf parasites. The 4-OCH3 (6l) and 4-F (6n) compounds were the most potent salts with IC50 values against PfDXR of 0.11 and 0.47 μM, respectively, and with IC50 values against Pf parasites of 0.30 and 2.42 μM, respectively. We speculate that slightly lower logP values of these compounds, compared to the rest of the series, play a role in their enhanced activities. Interestingly, prodrugs of these compounds did not show a robust improvement in activity against Pf parasites. Aside from 7l, all of the prodrug compounds displayed a dramatic reduction in antimalarial potency relative to their salt parent compounds. This may be due to insufficient deprotection of the bisphosphonate ester by Plasmodium spp. Indeed, steric hindrance due to the α-3,4-dichlorophenyl substituent could prevent or slow esterase activity on these compounds. Nonetheless, compound 7l (4-OCH3, IC50Pf3D7 = 0.37 μM), was very potent against Pf parasites, exceeding the activity of parent compounds fosmidomycin (1) and FR900098 (2).
Interestingly, most of the phosphonic acid salts (6a–6n) were only weakly active against MtbDXR. The most active compounds were again 6l and 6n at 0.78 and 4.78 μM, respectively. One reason for this lower activity could be explained by an earlier observation. Andaloussi et al. stated that addition of the α-3,4-dichlorophenyl group could clash with a Trp residue near the binding site of MtbDXR, resulting in lower inhibition of the protein.33 Many of the salt compounds displayed weak activity against Mtb cells. Because most of these compounds did not inhibit MtbDXR, it is presumed that their cellular activity is through an alternate target. Prodrugging the phosphonates led to improved Mtb cellular activity. For compounds 7l and 7n (MIC value of 4.60 μg mL−1 for both analogs), this activity may be due to DXR inhibition. The activity of these compounds, as well as salt 6l, exceeds the activity of fosmidomycin (1) and FR900098 (2).
Compounds displaying the most potent activity against PfDXR and Pf parasites were examined further to determine if their mechanism of action was through the MEP pathway. Drug-treated parasites were effectively rescued by addition of exogenous IPP, the product of the MEP pathway. Similar to known MEP pathway inhibitor fosmidomycin, IPP rescued parasites from treatment with compounds 6k, 6l, 6n, and 7l, confirming the intracellular target.19,37 Additionally, these compounds displayed cross resistance to the had1 mutant strain of Pf, mimicking fosmidomycin again. Thus, these data confirm the on-target mechanism of action of these compounds as MEP pathway inhibitors. Finally, these four most active compounds were tested for cytotoxicity against human HepG2 cells and found to display negligible cytotoxicity.
Thus, taken together, the data show that FR900098 analogs bearing both the α-3,4-dichlorophenyl and N-branched-alkoxyaryl substituents are very potent compounds against PfDXR and Pf parasites. Compounds 6k, 6l, 6n, and 7l are especially effective, and the prodrug is not required for high antimalarial activity. The compounds have an on-target mechanism of action and display low human cytotoxicity. While it is clear that the activity of these compounds against MtbDXR and Mtb cells is not straightforward, future studies will focus on refinement of these structures and further biological evaluation as antimalarial compounds.
Experimental methods
General
All reagents were purchased from commercial suppliers and used without further purification. Anhydrous solvents were purified by MBRAUN MB-SPS solvent purification system before use. Air sensitive reactions were carried out under a nitrogen atmosphere. 1H and 13C NMR spectra were recorded in CDCl3 or C2D6OS on an Agilent spectrometer at 400 MHz or 101 MHz, respectively, with TMS, H2O, or solvent signal as internal standard. Chemical shifts are given in parts per million (ppm). Spin multiplicities are given with the following abbreviations: s (singlet), bs (broad singlet), d (doublet), dd (doublet of doublets), ddd (doublet of doublets of doublets), t (triplet), dt (doublet of triplets), ddt (doublet of doublet of triplets), q (quartet), qt (quintet), and m (multiplet). Mass spectra were measured in the ESI mode on an HPLC-MS (Shimadzu LCMS-2020) or in the EI mode on an GC-MS (Shimadzu GCMS-QP2010S). Thin layer chromatography (TLC) was performed on Baker-flex Silica Gel IB2-F silica plates. The purity of synthesized compounds (>95%) was determined by HPLC-MS (Shimadzu LCMS-2020) using Shimadzu Nexcol C18 5 μm particle (50 × 3.0 mm). A linear gradient of acetonitrile (50–100%) in water (containing 0.1% formic acid) was used, at a flow rate of 0.2 mL min−1 from 0.5 to 15.5 min, followed by 100% acetonitrile for an additional 0.5 min. Quantification of compound purity was determined via integration of the area under the DAD curve using the LabSolutions software. High-resolution mass spectroscopy spectra (HRMS) were recorded in positive or negative ESI mode on a Waters Q-TOF Ultima mass spectrometer (UIUC Mass Spectrometry Laboratory) or in positive FAB mode on a VG Analytical VG70SE magnetic sector mass spectrometer (JHU Mass Spectrometry Facility).:1 dichloromethane:hexanes) to afford a sightly yellow solid (4.04 g, 65%). 1H NMR (400 MHz, CDCl3) δ (ppm): 6.68 (dd, 1H), 7.37 (d, 1H), 7.45 (m, 3H), 9.71 (d, 1H).
:2 hexanes:ethyl acetate) to yield a yellow oil (6.91 g, 67%). 1H NMR (400 MHz, CDCl3) δ (ppm): 1.13 (t, 3H), 1.25 (t, 3H), 2.51 (m, 1H), 2.76 (m, 1H), 3.32–3.45 (m, 1H), 3.74–3.87 (m, 1H), 3.89–4.17 (m, 2H), 5.68 (dd, 1H), 6.82–6.93 (m, 3H), 6.95–7.03 (m, 2H), 7.17–7.26 (m, 6H), 7.36–7.47 (m, 2H).
:2 dichloromethane:ethyl acetate) to afford a colorless oil as the product (2.02 g, 78%). 1H NMR (400 MHz, CDCl3) δ (ppm): 1.11 (t, 3H), 1.23 (t, 3H), 2.99–3.26 (m, 2H), 3.59–3.74 (ddd, 1H), 3.79–3.89 (m, 1H), 3.91–3.99 (m, 2H), 4.00–4.13 (m, 2H), 7.12–7.52 (m, 3H), 9.66 (s, 1H).
:3 dichloromethane:methanol) and yielded a colorless oil (1.28 g, 42%). 1H NMR (400 MHz, CDCl3) δ (ppm): 1.15 (t, 3H), 1.28 (t, 3H, J = 7.6 Hz), 1.95–2.13 (m, 1H), 2.27–2.42 (m, 1H), 2.58–2.74 (m, 1H), 2.79–2.90 (m, 1H), 3.11–3.27 (ddd, 1H), 3.76–3.88 (m, 1H), 3.90–3.98 (m, 1H), 4.01–4.12 (m, 2H), 4.54–4.76 (s, 2H), 5.49 (bs, 1H), 7.01–7.20 (m, 1H), 7.23–7.45 (m, 7H). LCMS (ESI+): 446.25 m/z [M + H]+.
:3 dichloromethane:methanol), to afford a slightly yellow oil (0.200 g, 46%). 1H NMR (400 MHz, CDCl3) δ (ppm): 1.21 (s, 18H), 1.90–2.02 (m, 3H), 2.08–2.40 (m, 2H), 2.40–2.63 (m, 1H), 3.00–3.24 (m, 1H), 3.26–3.56 (m, 2H), 3.67–4.08 (m, 1H), 5.25–5.68 (m, 4H), 7.18 (bs, 1H), 7.37–7.41 (m, 3H). 13C NMR (100 MHz, CDCl3) δ (ppm): 20.1, 25.9, 26.5, 38.4, 41.0, 43.0, 81.4, 128.5, 128.9, 130.6, 131.6, 132.5, 134.1, 171.9, 176.1. HRMS (ESI+) calculated for C23H35Cl2NO9P 570.1426, found 570.1420 [M + H]+. Purity = 97.3%.
:3 dichloromethane:methanol), to afford an orange, viscous oil (1.15 g, 97%). 1H NMR (400 MHz, CDCl3) δ (ppm): 1.15–1.36 (m, 6H), 1.87 (m, 1H), 2.08 (s, 3H), 2.43 (m, 1H), 3.12 (m, 1H), 2.79–2.90 (m, 1H), 3.42 (m, 1H), 3.68 (m, 1H), 3.90–4.11 (m, 4H), 7.13–7.24 (m, 1H), 7.38–7.49 (m, 2H), 9.45 (bs, 1H). LCMS (ESI+): 398.25 m/z [M + H]+.
General procedure for synthesis of N-O-ethers 14b–h,j–n
Compound 13 (1 eq.) was dissolved in anhydrous tetrahydrofuran (0.1 M) under a nitrogen atmosphere and cooled to 0 °C. Sodium hydride (1.2 eq.) was added to the solution, and the reaction was stirred for 20 minutes. To the reaction mixture was added aryl chloride (1.2 eq.). The mixture was warmed to room temperature and stirred for 18–48 hours. After removing the tetrahydrofuran under reduced pressure, the residue was dissolved in dichloromethane and quenched with water. The aqueous phase was extracted with dichloromethane (5 × 20 mL). The organic phase was dried over sodium sulfate and filtered. The solvent was removed under reduced pressure. The crude product was purified via column chromatography (97:3 dichloromethane:methanol) or silica gel preparatory plate (97:3 dichloromethane:methanol) to give the pure compound.
:1 dichloromethane:ethyl acetate) to afford a yellow oil (0.065 g, 19%). 1H NMR (400 MHz, CDCl3) δ (ppm): 1.14–1.28 (m, 12H), 1.58 (t, 2H), 2.00 (s, 3H), 2.45 (m, 1H), 2.97 (m, 2H), 3.48 (m, 2H), 3.79–4.10 (m, 3H), 4.79 (t, 2H), 7.09–7.73 (m, 11H). 13C NMR (101 MHz, CDCl3) δ (ppm): 16.3, 23.8, 23.9, 33.8, 59.9, 113.9, 126.9, 127.0, 127.3, 128.4, 128.6, 129.6, 131.1, 132.0, 132.1, 133.8. LCMS (ESI+): 606.25 m/z [M + H]+. Purity = 93.9%.
General procedure for synthesis of diammonium salt compounds 6a–n
A solution of 13, 14b–n (1 eq.) in anhydrous dichloromethane (0.1 M) under nitrogen was cooled to 0 °C. Trimethylsilyl bromide (3–5 eq.) was added dropwise to the solution. The mixture was warmed to room temperature and stirred overnight. Trimethylsilyl bromide and dichloromethane were removed under reduced pressure. The crude residue was stirred at room temperature in anhydrous methanol for 1–2 hours. The methanol was removed under reduced pressure. 7 M NH3 in methanol (2 eq.) was added to the mixture, and it was allowed to stir at room temperature for 1–2 hours. The excess methanol was removed under reduced pressure to yield the pure salts.General procedure for synthesis of dipivaloyloxymethylene prodrugs 7c–g,i
Salts 6c–g,i (1 eq.) were dissolved in anhydrous N,N-dimethylformamide (0.01 M). Triethyl amine (6 eq.) and pivaloyloxymethyl chloride (6 eq.) were added to the solution at room temperature. The reaction mixture was heated to 60 °C and was stirred overnight. The reaction mixture was extracted with water (3 × 50 mL) and diethyl ether (3 × 50 mL). Excess ether was removed under reduced pressure. The crude residue was dissolved in hexanes (50 mL) and washed with water (3 × 50 mL). The organic layer was dried over sodium sulfate and filtered. The hexanes was removed under reduced pressure and the residue was purified by normal phase silica column chromatography with 60:40 hexanes:ethyl acetate.
General procedure for synthesis of dipivaloyloxymethylene prodrugs 7h,j–k
To a solution of salts 6h,j–l (1 eq.) in anhydrous tetrahydrofuran (0.1 M) in a nitrogen atmosphere were added N,N-diisopropylethylamine (6 eq.) and pivaloyloxymethyl chloride (6 eq.) at room temperature. The reaction mixture was heated to 60 °C and stirred for 48–72 hours. The tetrahydrofuran was removed under reduced pressure. The crude residue was dissolved in dichloromethane (20 mL) and washed with saturated NaHCO3 (3 × 10 mL), followed by brine (1 × 15 mL). The organic phase was dried over anhydrous sodium sulfate and filtered. The dichloromethane was removed under reduced pressure, and the residue was purified via column chromatography (5:1 dichloromethane:ethyl acetate) to yield the desired compound.
:3 dichloromethane:methanol). Slightly yellow oil (0.010 g, 4%). 1H NMR (400 MHz, CDCl3) δ (ppm): 1.18 (s, 9H), 1.22 (s, 9H), 1.85 (s, 3H), 2.09–2.19 (m, 1H), 2.65–2.39 (m, 1H), 3.14–3.28 (m, 1H), 3.44–3.70 (m, 2H), 3.65 (s, 3H), 3.77–3.89 (m, 1H), 5.46–5.66 (m, 4H), 6.94 (bs, 1H), 7.15 (d, 1H), 7.26 (s, 2H), 7.38–7.43 (m, 2H). 13C NMR (100 MHz, CDCl3) δ (ppm): 20.2, 20.7, 26.9, 27.3, 38.9, 40.2, 46.2, 55.3, 113.8, 126.3, 126.7, 129.0, 130.1, 132.0, 134.4, 135.4, 139.2, 159.1, 169.1, 175.2. HRMS (ESI+) calculated for C32H45Cl2NO10P 704.2158, found 704.1966 [M + H]+. Purity = 97.2%.
General procedure for synthesis of dipivaloyloxymethylene prodrugs 7m,n
A solution of 14m,n (1 eq.) in anhydrous dichloromethane (0.1 M) under nitrogen was cooled to 0 °C. Trimethylsilyl bromide (3 eq.) was added dropwise to the solution under nitrogen. The mixture was warmed to room temperature and stirred overnight. Trimethylsilyl bromide and dichloromethane were removed under reduced pressure. The crude residue was stirred at room temperature in anhydrous methanol for one to two hours. The methanol was removed under reduced pressure. To a solution of the crude residue in tetrahydrofuran (0.1 M) were added N,N-diisopropylethylamine (6 eq.) and pivaloyloxymethyl chloride (6 eq.) at room temperature. The reaction mixture was heated to 60 °C and stirred for 48–72 hours. The tetrahydrofuran was removed under reduced pressure. The crude residue was dissolved in dichloromethane (20 mL) and washed with saturated NaHCO3 (3 × 10 mL), followed by brine (1 × 15 mL). The organic phase was dried over anhydrous sodium sulfate and filtered. The dichloromethane was removed under reduced pressure, and the residue was purified via column chromatography (97:3 dichloromethane:methanol) to yield the desired compound.
P. falciparum and M. tuberculosis dxr cloning, expression, and purification
The P. falciparum and M. tuberculosis dxr genes were cloned, expressed, and purified as previously described.37,60 Briefly, the Pf and Mtb dxr genes were each cloned into a pET101/DTOPO vector. The recombinant plasmid was transformed into chemically competent E. coli BL21 CodonPlus (DE3)-RIL cells (Stratagene, LA Jolla, CA). For protein expression, a 10 mL overnight seed culture of E. coli BL21 CodonPlus (DE3)-RIL + MtbDXR (or PfDXR) was added to 1 L of LB media and incubated with shaking at 37 °C and 250 rpm. Once an OD600 of 1.8 was achieved, protein expression was induced using 0.5 mM isopropyl β-D-thiogalactopyranoside (IPTG), and the culture was allowed to incubate for an additional 18 h. Cells were harvested via centrifugation (4650 × g, 20 min) and stored at −80 °C. Protein was isolated and purified from the cells via chemical lysis and affinity chromatography. Cell lysis was achieved using lysis buffer A (100 mM Tris pH 8, 0.032% lysozyme, 3 mL per mg cell pellet), followed by lysis buffer B (0.1 M CaCl2, 0.1 M MgCl2, 0.1 M NaCl, 0.020% DNASE, 3 mL per mg cell pellet). Centrifugation (48000 × g, 20 min) yielded the clarified cell lysate that was passed through a TALON immobilized metal affinity column (Clontech Laboratories, Mountain View, CA). The column was washed with 20 column volumes of 1× equilibrium buffer (50 mM HEPES pH 7.5, 300 mM NaCl), 10 column volumes of 1× wash buffer (50 mM HEPES pH 7.5, 300 mM NaCl, 10 mM imidazole), and 15 column volumes of 2× wash buffer (100 mM HEPES pH 7.5, 600 mM NaCl, 20 mM imidazole). The protein was then eluted with 5 column volumes of 1× elution buffer (150 mM imidazole pH 7.0, 300 mM NaCl). Buffer was exchanged with 0.1 M Tris pH 7.5, 1 mM NaCl, 5 mM DTT during concentration by ultrafiltration. Protein concentration was determined using Advanced Protein Assay Reagent (Cytoskeleton, Denver CO) with γ-globulins (Sigma-Aldrich) as the standard. Purified protein was visualized via Coomassie stained SDS-PAGE. The yield of MtbDXR and PfDXR averaged 5 mg and 1 mg per 1 L shake flask, respectively.
DXR enzyme inhibition assay
P. falciparum DXR activity was assayed at 37 °C by spectrophotometrically monitoring the enzyme catalyzed oxidation of NADPH upon addition of 1-deoxy-D-xylulose 5-phosphate (DOXP; Echelon Biosciences, Salt Lake City, UT) to the assay mixture, as described previously.47,48 Briefly, the assay system contained 100 mM Tris pH 7.8, 25 mM MgCl2, 0.86 μM PfDXR, and 150 μM NADPH. The reaction was initiated by adding 144 μM DOXP to the complete assay mixture. For the MtbDXR assays, the assay contained 100 mM MES pH 6.5, 25 mM MgCl2, 0.89 μM MtbDXR, 150 μM NADPH, and 47 μM DOXP. The oxidation of NADPH was monitored at 340 nm using an Agilent 8453 UV-visible spectrophotometer equipped with a temperature regulated cuvette holder. All assays were performed in technical duplicates. All determined IC50 values were independently validated (by a second enzymologist) to be within 5% of the determined value. If they were not, then additional replicate assays were performed to further refine the dose–response plots (and resulting IC50 value) until validation within the 5% cutoff was achieved.P. falciparum growth inhibition assay
Asynchronous P. falciparum cultures were maintained in 2% hematocrit suspended in complete media (RPMI-1640 (Millipore-Sigma) supplemented with 27 mM sodium bicarbonate, 11 mM glucose, 5 mM HEPES, 1 mM sodium pyruvate, 0.37 mM hypoxanthine, 0.01 mM thymidine, 10 μg ml−1 gentamycin, and 0.5% Albumax (Life Technologies)) under 5% O2/5% CO2/90% N2 atmosphere at 37 °C. Wild type strain 3D7 (BEI Resources, MRA-102) and fosmidomycin resistant strain (had1) E1-C12 (ref. 58) were diluted to 0.5% parasitemia and treated with inhibitors at concentrations ranging from 1.2 nM to 492.4 μM at the start of assays. Growth inhibition assays were performed in opaque 96-well plates at 100 μL culture volume. After 3 days, parasite growth was quantified by measuring DNA content using PicoGreen (Life Technologies) as described.58 Fluorescence was measured on a FLUOstar Omega microplate reader (BMG Labtech) at 485 nm excitation and 528 nm emission. Half-maximal inhibitory concentration (IC50) values were calculated by nonlinear regression analysis using GraphPad Prism software. Data was fit using a 4-parameter dose–response curve. For isopentenyl pyrophosphate (IPP) (Isoprenoids, LC) rescue experiments, 125 μM IPP was added to the appropriate wells for the duration of the experiment. Each assay was performed as a technical duplicate. Each compound was assayed as a biological triplicate.M. tuberculosis growth inhibition assay
A stock culture of Mtb H37Rv (ATCC 27294) was grown to OD 0.5 in 7H9/glucose/casitone/tyloxapol consisting of Middlebrook 7H9 broth (Difco) supplemented with 0.08% NaCl, 0.4% glucose, 0.05% Bacto casitone and 0.05% tyloxapol. The culture was diluted 1:500 in 7H9/glucose/casitone/tyloxapol before aliquoting 50 μL into each well of a 96-well plate. The inhibitors were dissolved in DMSO to make stock solutions of 20 mg mL−1. Inhibitors were added to the first row of wells of the 96-well plate with 100 μL of 7H9/glucose/casitone/tyloxapol with remaining wells containing 50 μL of 7H9/glucose/casitone/tyloxapol. After pipet mixing and use of a multichannel pipet, 50 μL was removed from each well in the first row and added to the second row. 2-Fold dilution in this manner was carried out to give eleven dilutions of each inhibitor. The plates were incubated for 2 weeks at 37 °C, and the MIC99 values were read macroscopically using an inverted plate reader. Each measurement was made three independent times.
HepG2 cell inhibition assay
For cytotoxicity assays, HepG2 cells (ATCC HB-8065) were grown in DMEM supplemented with 4 mM L-glutamine (Gibco #11966-025) with 4.5 g L−1D-glucose as carbon source. Cells were trypsinized, resuspended in DMEM/glutamine/glucose to 4 × 105 cells per mL and 50 μL per well transferred to flat-bottom white opaque tissue culture plates (Falcon #353296) containing 50 μL per well of the respective medium with test compound. Compound concentrations were two-fold dilutions ranging from 100 to 0.1 μg mL−1 as well as the drug-free DMSO-only control. All concentrations were tested in duplicate. After 24 h incubation at 5% CO2, 37 °C, 10 μL per well of Celltiter-Glo reagent (Promega #G9241) was added and luminescence recorded after 20 min incubation in the dark.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was generously supported by the George Washington University (GWU) Department of Chemistry, the NIH (AI 123433 to C. S. D. and AI 123808 to A. R. O. J.), and the Division of Intramural Research, NIAID, NIH. We thank Furong Sun and Haijun Yao (UIUC) for assistance with HRMS analysis.References
- World Malaria Report 2022, World Health Organization, Geneva, 2022, https://www.who.int/publications/i/item/9789240064898 Search PubMed.
- Global Tuberculosis Report 2022, World Health Organization, Geneva, 2022, https://www.who.int/teams/global-tuberculosis-programme/tb-reports Search PubMed.
- A. Sharma, M. De Rosa, N. Singla, G. Singh, R. Barnwal and A. Pandey, Tuberculosis: An Overview of the Immunogenic Response, Disease Progression, and Medicinal Chemistry Efforts in the Last Decade toward the Development of Potential Drugs for Extensively Drug-Resistant Tuberculosis Strains, J. Med. Chem., 2021, 64, 4359–4395 CrossRef CAS PubMed.
- S. Wellington and D. Hung, The Expanding Diversity of Mycobacterium tuberculosis Drug Targets, ACS Infect. Dis., 2018, 4, 696–714 CrossRef CAS PubMed.
- G. Pines, E. Oh, M. Bassalso, A. Choudhury, A. Garst, R. Fankhauser, C. Eckert and R. Gill, Genomic Deoxyxylulose Phosphate Reductoisomerase (DXR) Mutations Conferring Resistance to the Antimalarial Drug Fosmidomycin in E. coli, ACS Synth. Biol., 2018, 7, 2824–2832 CrossRef CAS PubMed.
- B. Rigel and P. Roepe, Altered Drug Transport by Plasmodium falciparum Chloroquine Resistance Transporter Isoforms Harboring Mutations Associated with Piperaquine Resistance, Biochemistry, 2020, 59, 2484–2493 CrossRef PubMed.
- J. Okombo and K. Chibale, Insights into Integrated Lead Generation and Target Identification in Malaria and Tuberculosis Drug Discovery, Acc. Chem. Res., 2017, 50, 1606–1616 CrossRef CAS PubMed.
- J. McKie, K. Douglas, C. Chan, S. Roser, R. Yates, M. Read, J. Hyde, M. Dascombe, Y. Yuthavong and W. Sirawaporn, Rational Drug Design Approach for Overcoming Drug Resistance: Application to Pyrimethamine Resistance in Malaria, J. Med. Chem., 1998, 41, 1367–1370 CrossRef CAS PubMed.
- V. Singh and K. Chibale, Strategies to Combat Multi-Drug Resistance in Tuberculosis, Acc. Chem. Res., 2021, 54, 2361–2376 CrossRef CAS PubMed.
- G. Pines, E. J. Oh, M. C. Bassalo, A. Choudhury, A. D. Garst, R. G. Fankhauser, C. A. Eckert and R. T. Gill, Genomic Deoxyxylulose Phosphate Reductoisomerase (DXR) Mutations Conferring Resistance to the Antimalarial Drug Fosmidomycin in E. Coli, ACS Synth. Biol., 2018, 7, 2824–2832 CrossRef CAS PubMed.
- T. Kuzuyama, T. Shimizu, S. Takahashi and H. Seto, Fosmidomycin, a specific inhibitor of 1-deoxy-D-xylulose 5-phosphate reductoisomerase in the nonmevalonate pathway for terpenoid biosynthesis, Tetrahedron Lett., 1998, 39, 7913–7916 CrossRef CAS.
- L. Kuntz, D. Tritsch, C. Grosdemange-Billiard, A. Hemmerlin, A. Willem, T. J. Bach and M. Rohmer, Isoprenoid biosynthesis as a target for antibacterial and antiparasitic drugs: phosphonohydroxamic acids as inhibitors of deoxyxylulose phosphate reducto-isomerase, Biochem. J., 2005, 386, 127–135 CrossRef CAS PubMed.
- S. Takahashi, T. Kuzuyama, H. Watanabe and H. Seto, A 1-deoxy-d-xylulose 5-phosphate reductoisomerase catalyzing the formation of 2-C-methyl-d-erythritol 4-phosphate in an alternative nonmevalonate pathway for terpenoid biosynthesis, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 9879–9884 CrossRef CAS PubMed.
- X. Wang and C. S. Dowd, The Methylerythritol Phosphate Pathway: Promising Drug Targets in the Fight against Tuberculosis, ACS Infect. Dis., 2018, 4, 278–290 CrossRef CAS PubMed.
- A. Frank and M. Groll, The Methylerythritol Phosphate Pathway to Isoprenoids, Chem. Rev., 2017, 117, 5675–5703 CrossRef CAS PubMed.
- A. Argyrou and J. S. Blanchard, Kinetic and Chemical Mechanism of Mycobacterium Tuberculosis 1-Deoxy-D-Xylulose-5-Phosphate Isomeroreductase, Biochemistry, 2004, 43, 4375–4384 CrossRef CAS PubMed.
- T. Kuzuyama, T. Shimizu, S. Takahashi and H. Seto, Fosmidomycin, a specific inhibitor of 1-deoxy-D-xylulose 5-phosphate reductoisomerase in the nonmevalonate pathway for terpenoid biosynthesis, Tetrahedron Lett., 1998, 39, 7913–7916 CrossRef CAS.
- E. R. Jackson, G. San Jose, R. C. Brothers, E. K. Edelstein, Z. Sheldon, A. Haymond, C. Johny, H. I. Boshoff, R. D. Couch and C. S. Dowd, The effect of chain length and unsaturation on Mtb Dxr inhibition and antitubercular killing activity of FR900098 analogs, Bioorg. Med. Chem. Lett., 2014, 24, 649–653 CrossRef CAS PubMed.
- R. L. Edwards, R. C. Brothers, X. Wang, M. I. Maron, P. D. Ziniel, P. S. Tsang, T. E. Kraft, P. W. Hruz, K. C. Williamson, C. S. Dowd and A. R. O. John, MEPicides: Potent Antimalarial Prodrugs Targeting Isoprenoid Biosynthesis, Sci. Rep., 2017, 7, 8400 CrossRef PubMed.
- T. Masini and A. K. H. Hirsch, Development of Inhibitors of the 2 C-Methyl-D-Erythritol 4-Phosphate (MEP) Pathway Enzymes as Potential Anti-Infective Agents, J. Med. Chem., 2014, 57, 9740–9763 CrossRef CAS PubMed.
- N. Singh, G. Cheve, M. Avery and C. McCurdy, Comparative Protein Modeling of 1-Deoxy-D-xylulose-5-phosphate Reductoisomerase Enzyme from Plasmodium falciparum: A Potential Target for Antimalarial Drug Discovery, J. Chem. Inf. Model., 2006, 46, 1360–1370 CrossRef CAS PubMed.
- J. Munos, X. Pu, S. Mansoorabadi, H. Kim and H. Liu, A Secondary Kinetic Isotope Effect Study of the 1-Deoxy-D-xylulose-5-phosphate Reductoisomerase-Catalyzed, J. Am. Chem. Soc., 2009, 131, 2048–2049 CrossRef CAS PubMed.
- L. Deng, K. Endo, M. Kato, G. Cheng, S. Yajima and Y. Song, Structures of 1-Deoxy-D-Xylulose5-Phosphate Reductoisomerase/Lipophilic Phosphonate Complexes, ACS Med. Chem. Lett., 2011, 2, 165–170 CrossRef CAS PubMed.
- A. Wong, J. W. Munos, V. Devasthali, K. A. Johnson and H. Liu, Study of 1-Deoxy-D–Xylulose-5-Phosphate Reductoisomerase: Synthesis and Evaluation of Fluorinated Substrate Analogues, Org. Lett., 2004, 6, 3625–3628 CrossRef CAS PubMed.
- B. Lell, R. Ruangweerayut, J. Wiesner, M. A. Missinou, A. Schindler, T. Baranek, M. Hintz, D. Hutchinson, H. Jomaa and P. G. Kremsner, Fosmidomycin, a novel chemotherapeutic agent for malaria, Antimicrob. Agents Chemother., 2003, 47, 735–738 CrossRef CAS PubMed.
- T. Umeda, N. Tanaka, Y. Kusakabe, M. Nakanishi, Y. Kitade and K. T. Nakamura, Molecular basis of fosmidomycin's action on the human malaria parasite Plasmodium falciparum, Sci. Rep., 2011, 1, 1–8 CrossRef PubMed.
- A. M. Jansson, A. Wieckowska, C. Bjorkelid, S. Yahiaoui, S. Sooriyaarachchi, M. Lindh and S. L. Mowbray, DXR inhibition by potent mono- and disubstituted fosmidomycin analogues, J. Med. Chem., 2013, 56, 6190–6199 CrossRef CAS PubMed.
- B. Zhang, K. Watts, D. Hodge, L. Kemp, D. Hunstad, L. Hicks and A. Odom, A Second Target of the Antimalarial and Antibacterial Agent Fosmidomycin Revealed by Cellular Metabolic Profiling, Biochemistry, 2011, 50, 3570–3577 CrossRef CAS PubMed.
- E. Uh, E. Jackson, G. San Jose, M. Maddox, R. Lee, R. Lee, H. Boshoff and C. Dowd, Antibacterial and antitubercular activity of fosmidomycin, FR900098, and their lipophilic analogs, Bioorg. Med. Chem. Lett., 2011, 21, 6973–6976 CrossRef CAS PubMed.
- S. Kholodar, G. Tombline, J. Liu, Z. Tan, C. Allen, A. Gulick and A. Murkin, Alteration of the Flexible Loop in 1-Deoxy-D-xylulose-5-phosphate Reductoisomerase Boosts Enthalpy-Driven Inhibition by Fosmidomycin, Biochemistry, 2014, 53, 3423–3431 CrossRef CAS PubMed.
- A. Leon, L. Liu, Y. Yang, M. P. Hudock, P. Hall, F. Yin, D. Studer, K.-J. Puan, C. T. Morita and E. Oldfield, Isoprenoid Biosynthesis as a Drug Target: Bisphosphonate Inhibition of Escherichia Coli K12 Growth and Synergistic Effects of Fosmidomycin, J. Med. Chem., 2006, 49, 7331–7341 CrossRef CAS PubMed.
- C. Zinglé, L. Kuntz, D. Tritsch, C. Grosdemange-Billiard and M. Rohmer, Isoprenoid Biosynthesis via the Methylerythritol Phosphate Pathway: Structural Variations around Phosphonate Anchor and Spacer of Fosmidomycin, a Potent Inhibitor of Deoxyxylulose Phosphate Reductoisomerase, J. Org. Chem., 2010, 75, 3203–3207 CrossRef PubMed.
- M. Andaloussi, L. M. Henriksson, A. Wieckowska, M. Lindh, C. Bjorkelid, A. M. Larsson, S. Suresh, H. Iyer, B. R. Srinivasa, T. Bergfors, T. Unge, S. L. Mowbray, M. Larhed, T. A. Jones and A. Karlen, Design, synthesis, and X-ray crystallographic studies of alpha-aryl substituted fosmidomycin analogues as inhibitors of Mycobacterium tuberculosis 1-deoxy-D-xylulose 5-phosphate reductoisomerase, J. Med. Chem., 2011, 54, 4964–4976 CrossRef CAS PubMed.
- G. San Jose, E. R. Jackson, A. Haymond, C. Johny, R. L. Edwards, X. Wang, R. C. Brothers, E. K. Edelstein, A. R. Odom, H. I. Boshoff, R. D. Couch and C. S. Dowd, Structure-Activity Relationships of the MEPicides: N-Acyl and O-Linked Analogs of FR900098 as Inhibitors of Dxr from Mycobacterium tuberculosis and Yersinia pestis, ACS Infect. Dis., 2016, 2, 923–935 CrossRef CAS PubMed.
- H. S. Ball, M. Girma, M. Zainab, H. Riley, C. T. Behrendt, C. Lienau, S. Konzuch, L. A. A. Avelar, B. Lungerich, I. Soojhawon, S. M. Noble, T. Kurz and R. D. Couch, Inhibition of the Yersinia Pestis Methylerythritol Phosphate Pathway of Isoprenoid Biosynthesis by α-Phenyl-Substituted Reverse Fosmidomycin Analogues, ACS Omega, 2020, 5, 5170–5175 CrossRef CAS PubMed.
- T. Verbrugghen, P. Vandum, J. Pouyez, L. Maes, J. Wouters and S. Van Calenbergh, Alpha-Heteroatom Derivatized Analogues of 3-(Acetylhydroxyamino)propyl Phosphonic Acid (FR900098) as Antimalarials, J. Med. Chem., 2013, 56, 376–380 CrossRef CAS PubMed.
- X. Wang, R. L. Edwards, H. Ball, C. Johnson, A. Haymond, M. Girma, M. Manikkam, R. C. Brothers, K. T. McKay, S. D. Arnett, D. M. Osbourn, S. Alvarez, H. I. Boshoff, M. J. Meyers, R. D. Couch, A. R. Odom John and C. S. Dowd, MEPicides: α,β-Unsaturated Fosmidomycin Analogues as DXR Inhibitors against Malaria, J. Med. Chem., 2018, 61, 8847–8858 CrossRef CAS PubMed.
- M. B. Girma, H. S. Ball, X. Wang, R. C. Brothers, E. R. Jackson, M. J. Meyers, C. S. Dowd and R. D. Couch, Mechanism of Action of N-Acyl and N-Alkoxy Fosmidomycin Analogs: Mono- and Bisubstrate Inhibition of IspC from Plasmodium Falciparum, a Causative Agent of Malaria, ACS Omega, 2021, 6, 27630–27639 CrossRef CAS PubMed.
- S. Konzuch, T. Umeda, J. Held, S. Hähn, K. Brücher, C. Lienau, C. T. Behrendt, T. Gräwert, A. Bacher, B. Illarionov, M. Fischer, B. Mordmüller, N. Tanaka and T. Kurz, Binding Modes of Reverse Fosmidomycin Analogs toward the Antimalarial Target IspC, J. Med. Chem., 2014, 57, 8827–8838 CrossRef CAS PubMed.
- L. Deng, S. Sundriyal, V. Rubio, Z. Shi and Y. Song, Coordination Chemistry Based Approach to Lipophilic Inhibitors of 1-Deoxy-D-Xylulose-5-Phosphate Reductoisomerase, J. Med. Chem., 2009, 52, 6539–6542 CrossRef CAS PubMed.
- S. Yajima, K. Hara, J. M. Sanders, F. Yin, K. Ohsawa, J. Wiesner, H. Jomaa and E. Oldfield, Crystallographic Structures of Two Bisphosphonate:1-Deoxyxylulose-5-Phosphate Reductoisomerase Complexes, J. Am. Chem. Soc., 2004, 126, 10824–10825 CrossRef CAS PubMed.
- A. Leon, L. Liu, Y. Yang, M. P. Hudock, P. Hall, F. Yin, D. Studer, K.-J. Puan, C. T. Morita and E. Oldfield, Isoprenoid Biosynthesis as a Drug Target: Bisphosphonate Inhibition of Escherichia Coli K12 Growth and Synergistic Effects of Fosmidomycin, J. Med. Chem., 2006, 49, 7331–7341 CrossRef CAS PubMed.
- C. Zinglé, L. Kuntz, D. Tritsch, C. Grosdemange-Billiard and M. Rohmer, Isoprenoid Biosynthesis via the Methylerythritol Phosphate Pathway: Structural Variations around Phosphonate Anchor and Spacer of Fosmidomycin, a Potent Inhibitor of Deoxyxylulose Phosphate Reductoisomerase, J. Org. Chem., 2010, 75, 3203–3207 CrossRef PubMed.
- T. A. Wencewicz, B. Yang, J. R. Rudloff, A. G. Oliver and M. J. Miller, N–O Chemistry for Antibiotics: Discovery of N-Alkyl-N-(Pyridin-2-Yl)Hydroxylamine Scaffolds as Selective Antibacterial Agents Using Nitroso Diels–Alder and Ene Chemistry, J. Med. Chem., 2011, 54, 6843–6858 CrossRef CAS PubMed.
- T. Knak, M. A. Abdullaziz, S. Höfmann, L. A. Alves Avelar, S. Klein, M. Martin, M. Fischer, N. Tanaka and T. Kurz, Over 40 Years of Fosmidomycin Drug Research: A Comprehensive Review and Future Opportunities, Pharmaceuticals, 2022, 15, 1553 CrossRef CAS PubMed.
- K. Brücher, T. Gräwert, S. Konzuch, J. Held, C. Lienau, C. Behrendt, B. Illarionov, L. Maes, A. Bacher, S. Wittlin, B. Mordmüller, M. Fischer and T. Kurz, Prodrugs of Reverse Fosmidomycin Analogues, J. Med. Chem., 2015, 58, 2025–2035 CrossRef PubMed.
- C. T. Behrendt, A. Kunfermann, V. Illarionova, A. Matheeussen, M. K. Pein, T. Gräwert, J. Kaiser, A. Bacher, W. Eisenreich, B. Illarionov, M. Fischer, L. Maes, M. Groll and T. Kurz, Reverse Fosmidomycin Derivatives against the Antimalarial Drug Target IspC (Dxr), J. Med. Chem., 2011, 54, 6796–6802 CrossRef CAS PubMed.
- L. Deng, K. Endo, M. Kato, G. Cheng, S. Yajima and Y. Song, Structures of 1-Deoxy-D-Xylulose5-Phosphate Reductoisomerase/Lipophilic Phosphonate Complexes, ACS Med. Chem. Lett., 2011, 2, 165–170 CrossRef CAS PubMed.
- A. Wong, J. W. Munos, V. Devasthali, K. A. Johnson and H. Liu, Study of 1-Deoxy-D–Xylulose-5-Phosphate Reductoisomerase: Synthesis and Evaluation of Fluorinated Substrate Analogues, Org. Lett., 2004, 6, 3625–3628 CrossRef CAS PubMed.
- R. Chofor, S. Sooriyaarachchi, M. D. P. Risseeuw, T. Bergfors, J. Pouyez, C. Johny, A. Haymond, A. Everaert, C. S. Dowd, L. Maes, T. Coenye, A. Alex, R. D. Couch, T. A. Jones, J. Wouters, S. L. Mowbray and S. Van Calenbergh, Synthesis and Bioactivity of β-Substituted Fosmidomycin Analogues Targeting 1-Deoxy-D-Xylulose-5-Phosphate Reductoisomerase, J. Med. Chem., 2015, 58, 2988–3001 CrossRef CAS PubMed.
- A. Nordqvist, C. Björkelid, M. Andaloussi, A. M. Jansson, S. L. Mowbray, A. Karlén and M. Larhed, Synthesis of Functionalized Cinnamaldehyde Derivatives by an Oxidative Heck Reaction and Their Use as Starting Materials for Preparation of Mycobacterium Tuberculosis 1-Deoxy-D-Xylulose-5-Phosphate Reductoisomerase Inhibitors, J. Org. Chem., 2011, 76, 8986–8998 CrossRef CAS PubMed.
- G. Cai, L. Deng, B. G. Fryszczyn, N. G. Brown, Z. Liu, H. Jiang, T. Palzkill and Y. Song, Thermodynamic Investigation of Inhibitor Binding to 1-Deoxy-D-Xylulose-5-Phosphate Reductoisomerase, ACS Med. Chem. Lett., 2012, 3, 496–500 CrossRef CAS PubMed.
- K. Brücher, B. Illarionov, J. Held, S. Tschan, A. Kunfermann, M. K. Pein, A. Bacher, T. Gräwert, L. Maes, B. Mordmüller, M. Fischer and T. Kurz, α-Substituted β-Oxa Isosteres of Fosmidomycin: Synthesis and Biological Evaluation, J. Med. Chem., 2012, 55, 6566–6575 CrossRef PubMed.
- G. San Jose, E. R. Jackson, E. Uh, C. Johny, A. Haymond, L. Lundberg, C. Pinkham, K. Kehn-Hall, H. I. Boshoff, R. D. Couch and C. S. Dowd, Design of Potential Bisubstrate Inhibitors against Mycobacterium tuberculosis (Mtb) 1-Deoxy-D-Xylulose 5-Phosphate Reductoisomerase (Dxr)-Evidence of a Novel Binding Mode, MedChemComm, 2013, 4, 1099–1104 RSC.
- T. Haemers, J. Wiesner, S. Van Poecke, J. Goeman, D. Henschker, E. Beck, H. Jomaa and S. Van Calenbergh, Synthesis of α-substituted fosmidomycin analogues as highly potent Plasmodium falciparum growth inhibitors, Bioorg. Med. Chem. Lett., 2006, 16, 1888–1891 CrossRef CAS PubMed.
- T. Haemers, J. Wiesner, R. Busson, H. Jomaa and S. Van Calenbergh, Synthesis of α-Aryl-Substituted and Conformationally Restricted Fosmidomycin Analogues as Promising Antimalarials, Eur. J. Org. Chem., 2006, 2006, 3856–3863 CrossRef.
- E. Yeh and J. DeRisi, Chemical Rescue of Malaria Parasites Lacking an Apicoplast Defines Organelle Function in Blood-Stage Plasmodium falciparum, PLoS Biol., 2011, 9, e1001138 CrossRef CAS PubMed.
- A. M. Guggisberg, J. Park, R. L. Edwards, M. L. Kelly, D. M. Hodge, N. H. Tolia and A. R. Odom, A sugar phosphatase regulates the methylerythritol phosphate (MEP) pathway in malaria parasites, Nat. Commun., 2014, 5, 4467 CrossRef CAS PubMed.
- X. Wang, R. L. Edwards, H. S. Ball, K. M. Heidel, R. C. Brothers, C. Johnson, A. Haymond, M. Girma, A. Dailey, J. S. Roma, H. I. Boshoff, D. M. Osbourn, M. J. Meyers, R. D. Couch, A. R. Odom John and C. S. Dowd, MEPicides: α,β Unsaturated Fosmidomycin N-Acyl Analogs as Efficient Inhibitors of Plasmodium Falciparum 1-Deoxy-D-Xylulose-5-Phosphate Reductoisomerase, ACS Infect. Dis., 2023, 9(7), 1387–1395 CrossRef CAS PubMed.
- A. Haymond, C. Johny, T. Dowdy, B. Schweibenz, K. Villarroel, R. Young, C. J. Mantooth, T. Patel, J. Bases, G. S. Jose, E. R. Jackson, C. S. Dowd and R. D. Couch, Kinetic Characterization and Allosteric Inhibition of the Yersinia Pestis 1-Deoxy-D-Xylulose 5-Phosphate Reductoisomerase (MEP Synthase), PLoS One, 2014, 9(8), e106243 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3md00642e |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |