
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe role of silicon in drug discovery: a review
Jenny-Lee
Panayides
 *a,
Darren Lyall
Riley
*b,
Felix
Hasenmaile
c and
Willem A. L.
van Otterlo
c
*a,
Darren Lyall
Riley
*b,
Felix
Hasenmaile
c and
Willem A. L.
van Otterlo
c
aPharmaceutical Technologies, Future Production: Chemicals, Council for Scientific and Industrial Research (CSIR), Meiring Naude Road, Brummeria, Pretoria, South Africa. E-mail: jpanayides@csir.co.za
bDepartment of Chemistry, Faculty of Natural and Agricultural Sciences, University of Pretoria, Lynnwood Road, Pretoria, South Africa. E-mail: darren.riley@up.ac.za
cDepartment of Chemistry and Polymer Science, Stellenbosch University, Matieland, Stellenbosch, 7600, South Africa
First published on 1st July 2024
Abstract
This review aims to highlight the role of silicon in drug discovery. Silicon and carbon are often regarded as being similar with silicon located directly beneath carbon in the same group in the periodic table. That being noted, in many instances a clear dichotomy also exists between silicon and carbon, and these differences often lead to vastly different physiochemical and biological properties. As a result, the utility of silicon in drug discovery has attracted significant attention and has grown rapidly over the past decade. This review showcases some recent advances in synthetic organosilicon chemistry and examples of the ways in which silicon has been employed in the drug-discovery field.
 Jenny-Lee Panayides | Dr Jenny-Lee Panayides is a Principal Researcher and Research Group Leader (Pharmaceutical Technologies) at the Council for Scientific and Industrial Research, South Africa. She holds a PhD in synthetic chemistry and microbiology (University of the Witwatersrand), and certification in advanced project management (University of Pretoria). Her early research supported malaria transmission-blocking drug discovery efforts through high-throughput screening cascades, while she has shifted her research focus in recent years to supporting the self-sufficiency of African pharmaceutical manufacturing. She now leads an applied research program focusing on the local production of active pharmaceutical ingredients, integrating continuous flow chemistry with intelligent process transformation, and provides thought leadership on various working groups in the pharmaceutical and 4IR technologies space. |
 Darren Lyall Riley | Prof. Riley obtained his doctorate in organic chemistry at the University of the Witwatersrand, Johannesburg, South Africa in 2007. This was followed by a post-doctoral fellowship at the same institution. In 2010 he was appointed as a principal scientist at iThemba Pharmaceuticals and thereafter returned to academia joining the University of Pretoria in 2013. His research interests include small molecule-based drug discovery and the application of flow chemistry for the development of more sustainable approaches for manufacturing pharmaceuticals. |
 Felix Hasenmaile | Felix studied Chemistry at the University of Freiburg and visited the Université Paris-Sud for a research stay working on Ca(II)-catalysed hydroaminations with Prof. Gandon and Prof. LeBoeuf. He obtained his Ph.D. in the group of Prof. Brückner, where he investigated the atropisomerism of macrocyclic biaryls and developed catalysts for asymmetric synthesis. He joined the group of Prof. van Otterlo as a post-doctoral fellow pursuing the atroposelective synthesis of naphthylisoquinoline alkaloids. He now works at the research department of Boehringer Ingelheim. |
 Willem A. L. van Otterlo | Willem van Otterlo received his PhD in 1999 from the University of the Witwatersrand (WITS), Johannesburg, South Africa, followed by a postdoctoral fellowship at the University of Montreal, Quebec, Canada. In 2001, he returned to WITS and progressed to the position of Associate Professor in 2008. In 2008/2009 he took up a von Humboldt Georg-Forster Research Fellowship at the Max Planck Institute, Dortmund, and in 2010 was appointed to the Chair of Organic Chemistry in the Department of Chemistry and Polymer Science at the University of Stellenbosch. His research interests are focused on the synthesis of small molecules with potential bioactivity. |
Introduction
“Silicon and carbon are alike in so many ways, and yet, they are so different. This dichotomy was behind a good portion of the interest in silicon chemistry in the 20th century... In recent decades, silicon has thoroughly penetrated areas as diverse as organic synthesis and microelectronics” – Josef Michl, Editor, Chemical Reviews, 1995.1Silicon, the 14th element in the periodic table, accounts for about a quarter of all the material in the Earth's crust.2 It is however never found naturally in its elemental form, but always in combinations with other elements, especially oxygen. Despite its abundance, silicon plays only a minor role in living organisms. While inorganic forms, such as silica (SiO2) or silicic acid (Si(OH)4) are of importance in the biochemistry of algae and plants,3 it has generally been accepted that there are no naturally occurring organosilicon compounds, i.e. compounds containing at least one silicon–carbon bond.4
Silicon in drug discovery
“Some of the fundamental differences between carbon and silicon can lead to marked alterations in the physiochemical and biological properties of the silicon-containing analogues… and the resulting benefits can be exploited in the drug design process” – Graham A. Showell, Amedis Pharmaceuticals Ltd.5The design of new and improved drug-like compounds for the treatment of disease requires not only good activity and selectivity but also low toxicity and physiologically acceptable pharmacokinetics. Traditionally the medicinal chemist tackles the task of developing lead active pharmaceutical ingredients (API's) in an iterative design approach weighing up activity against parameters such as selectivity, solubility, absorption, distribution, metabolism, excretion, and toxicology (ADMET) – see the following recent example for a study which included determining the ADMET properties of silicon–nitrogen heterocycles.6
Currently, drug developers often look to known families of drugs to draw inspiration for the design of new biologically active compounds. Recent data would suggest that only about 3 dozen scaffolds exist which are routinely utilized and that they account for approximately 50% of all compounds which have been FDA approved.7 These same studies have also demonstrated that most of the small bioactive molecule side-chains observed in modern drug structures have been designed by using a rather small number of different functional groups.8 These observations has also led to the application of bioisosteres as a modern strategy in drug design which aims to utilize bioisosteres to improve the ADMET properties of small molecule drug-like entities or to obtain new subclasses of compounds with modified properties.5,9
During the 1970's the notion of using a “silicon switch”, or effectively a silicon–carbon substitution to design novel analogues of bioactive molecules was developed by Tacke and colleagues. In this strategy, an existing molecule could be modified by replacing one or more carbon-atoms by silicon atoms (or silicon atom-containing functional/structural groups). It should be appreciated that a “silicon switch” of this type could be treated conceptually as a classical tetravalent bioisostere.5 However, in our opinion, application of this approach still remains undervalued and therefore underutilized. For example, in 2001 it was estimated that less than 1% of all patent cooperation treaty (PCT) applications in the field of drug discovery referred to compounds that contained silicon or other less frequently used elements.10 Recently, however, in addition to the aforementioned rapid growth in organosilicon chemistry there has been increased interest in the incorporation of silicon as a bioisostere of carbon into drug-like scaffolds in an effort to manipulate pharmacokinetically important parameters.
Impact of silicon on medicinal chemistry
Both carbon and silicon atoms possess four valence electrons and as such, similarities with respect to the chemistry of silicon and carbon are apparent. However, in terms of several chemical aspects the two elements differ substantially from one another.5 A 2013 review by Franz and Wilson highlighted the fact that the different chemical properties of organosilicon-based compounds are of particular relevance to medicinal chemistry.11 Some of these fundamental differences can result in marked variations in the physico- and biochemical properties of silicon-containing analogues with respect to their carbon parent compounds. These differences can of course be rationally exploited in the drug design process, of which some concepts are described in Table 1.| Property | Difference | Potential benefit |
|---|---|---|
| Bonding | Altered bond strength and disfavoured multiple bonds | • Silane diol peptidomimetics |
| • Access to bioisosteres not structurally available to carbon | ||
| Atomic size | Altered bond lengths and bond angles | • Altered in vitro potency |
| • Modified selectivity | ||
| • Altered rate of metabolism | ||
| Electronegativity | Increased H-bond strength and acidity of silanols | • Improved potency in pharmacophores where H-bonding is important |
| Lipophilicity | Increased lipophilicity of silicon-containing compounds | • Improved in vivo half-life |
| • Enhanced tissue distribution | ||
| • Enhanced cell penetration |
Silicon has a high affinity towards oxygen and forms stronger bonds to oxygen and the halogens than carbon. On the other hand, bonds between silicon and carbon or hydrogen are weaker than the respective carbon–carbon or carbon–hydrogen bonds. The crux of modern organosilicon synthetic chemistry hinges on the generation of exceptionally stable bonds of silicon to oxygen or fluoride whilst at the same time cleaving weaker silicon bonds.12
The chemistry of Si–C and Si–O single bonds dominates the overall chemistry of silicon, and as such, its application as a tetrahedral bioisostere of carbon is therefore not unexpected. In contrast to the strength of some of the σ Si bonds, π Si bonds are generally weak.12 For this reason, generally no stable sila-equivalents exist for alkenes and carbonyl functional groups even though compounds with sterically or electronically stabilised silicon–carbon13 and silicon–oxygen14 double bonds can be isolated. It should also be noted that whilst the carbonyl double bond is favoured over its hydrated form, the formation of a Si![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O double bond is disfavoured over its hydrate (the silicon diol or silanediol).15 It has also been observed that silicon cannot form physiologically stable Si–H-containing compounds. Not only is the Si–H bond weaker than its C–H comparator, it also experiences reversed polarity.5 As such, the Si–H bond behaves quite differently to the C–H bond. For an example, the Si–H bond is readily cleaved with water under non-acidic conditions to afford the corresponding silanol featuring a Si–OH bond, conditions under which the C–H bond remains quite stable.5 These crucial bond differences can therefore be used to synthesise sila-analogues where carbon counterparts are much more difficult to obtain, if not impossible to access under normal experimental conditions (see brief discussions of novel synthetic methodology concerning the synthesis of Si-containing molecules elsewhere in this review).
O double bond is disfavoured over its hydrate (the silicon diol or silanediol).15 It has also been observed that silicon cannot form physiologically stable Si–H-containing compounds. Not only is the Si–H bond weaker than its C–H comparator, it also experiences reversed polarity.5 As such, the Si–H bond behaves quite differently to the C–H bond. For an example, the Si–H bond is readily cleaved with water under non-acidic conditions to afford the corresponding silanol featuring a Si–OH bond, conditions under which the C–H bond remains quite stable.5 These crucial bond differences can therefore be used to synthesise sila-analogues where carbon counterparts are much more difficult to obtain, if not impossible to access under normal experimental conditions (see brief discussions of novel synthetic methodology concerning the synthesis of Si-containing molecules elsewhere in this review).
Silicon containing bonds are always longer than the corresponding carbon analogues – the C–C bond length is ∼1.54 Å versus the C–Si bond length of ∼1.87 Å.16 A popular example demonstrating this is seen in that the trimethylsilyl (TMS) group is less sterically demanding than its carbon analogue, the tert-butyl arrangement – at least when measured in terms of A-values. The longer bond to the SiMe3-group decreases steric interactions, even though the size of the whole group is indeed larger.17 This bond length divergence leads to subtle but important changes in terms of the size and shape of silicon-containing compounds, when compared with their carbon-only parent compounds.15 This in turn can result in changes in terms of how the silicon analogues interact with their biological targets to ultimately have a marked effect on the overall pharmacological profiles of the silyl-containing molecules.5,15 Silicon is also more electropositive than carbon, and because of this carbon-silicon bonds are considerably polarized.12 For the same reason, polarisations of bonds between and other elements will also be different when compared to the respective bonds to silicon.
A research area which has potential benefits for the “silicon switch” approach involves investigations into hydrogen-bond strengths and acidities.5 It is important to note that the hydrogen-bond strength of a silanol makes it more favourable as a donor when compared to the corresponding carbinol.15,18 As a result, in molecules where a carbon-bonded OH functions as a hydrogen-bond donor, the application of a silanol functional group can result in improved biochemical potency.15 It should thus be stressed that due to the importance of hydrogen bond donors in drug discovery,19 investigations into the physical properties of molecules like the silanols, including that of hydrogen bond acidity, are of significant value.20
Generally, silicon-containing compounds are more lipophilic (and in turn hydrophobic) than their corresponding carbon counterparts. When the lipophilicity of a molecule is modulated, this in turn can be expected to alter the in vivo effects due to the small molecule. For example, small increases in lipophilicity have been found to significantly increase molecular tissue penetration abilities.15,21,22 As a result, silicon-containing molecules have proven to be less prone to hepatic metabolism resulting in subsequent higher molecular plasma half-lives (in comparison to the carbon-based parent molecules).15 In addition, lipophilicity can play a crucial role in the ability of small molecule drug candidates to cross the blood–brain barrier (BBB).15 Research has shown that many computational models predicting BBB permeation contain a lipophilicity criterion.23
The area of computational research involving small silicon-containing molecules has also seen recent attention, and in silico studies have regularly been utilized to support observed bond characteristics of silicon atoms in small molecules. Advances in this field include new Amber-compatible organosilane force fields24 and other computational approaches to investigating molecules of this type.25,26
Another exciting area of research that will certainly impact the field of silicon-containing bioactive compounds is the development of enzymes capable of performing biocatalytic transformations of the silicon fragments contained in these compounds, as exemplified by the following reference.27 Findings in this field may also be relevant in terms of evaluating the advantages of incorporating silicon into bioactive molecules. Care should also be taken to establish the broader implications of this switch in terms of biodegradability (or alternatively recyclability) of the new compounds, all with a view on responsible sustainability of utilizing the novel silicon-incorporated molecules (see for example the following reference).28 Also of importance, in terms of the interaction of environmental microbes with silicon-containing molecules, is the recent research into the biodegradative formation29 or cleavage30 of Si–C bonds. These processes, which seem to occur under at least very mild environmental (natural) conditions, would appear yet to be proven in a satisfactory scientifically rigorous manner.
Silyl ethers as protecting groups
The major use of silicon in synthetic organic chemistry has to date been in the application of organosilyl protecting groups as parts of an essential synthetic strategy. Due to this use, there has been significant growth of Si-protecting group technology, to the point where they are now very often applied in any organic synthesis requiring the protection of an OH group.31 The first group, that found larger application was the trimethylsilyl group (TMS) 1. Since then sterically bulkier, and hence more stable groups, such as triethylsilyl (TES) 2, triisopropyl (TIPS) 3, tert-butyldimethylsilyl (TBDMS) 4, tert-butyldiphenyl (TBDPS) 5 and tert-hexyldimethyl (TDS) 6, have come into more regular use, as well as disiloxane protecting groups such as tetraisopropyldisoloxanediyl (TIPD) 7 (Fig. 1).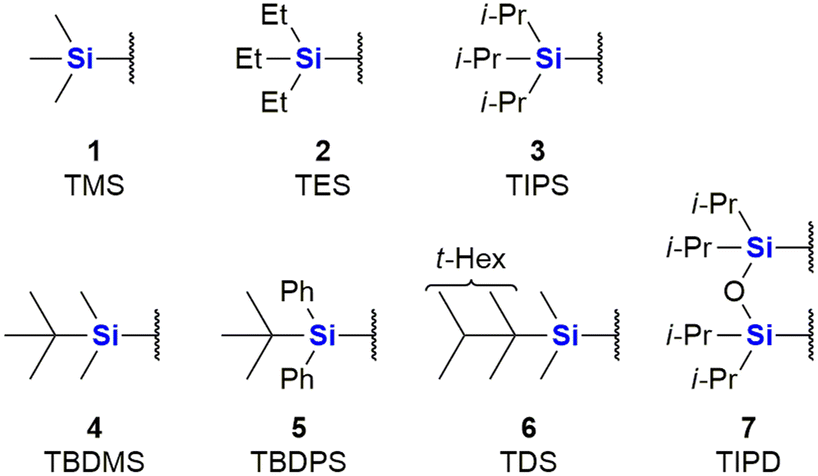 | ||
| Fig. 1 Common organosilyl O-protecting groups. | ||
Compounds containing organosilyl protecting groups are routinely deprotected under either acidic or basic conditions or by the application of reagents providing fluoride ions (as for example, by using the reagent, tetrabutylammonium fluoride which often is applied as the hydrate).32 As a result of the deprotection, silanols (Si–OH), or, in the case of deprotection with fluoride ions, mixtures of silanols and silyl fluorides are obtained as side-products, which are usually considered waste.31 The discarding of silicon protecting group waste occurs even on industrial scale, and is obviously still a significant issue in terms of the principles of Green Chemistry and reaction atom economy (AE). It is, however, possible to recycle the silanols by recovery from the aqueous waste and subsequently converting them back into their respective chlorosilanes, which can be used again for O-protection.31
Recent progress in organosilicon chemistry
The field of organosilicon chemistry has grown rapidly. Although not the focus of this review, the reader is referred to some recent highlights of the last decade in the area showcasing the large number of synthetic tools available for the synthesis of organosilicon compounds. A special emphasis is put on methods which are relevant for the synthesis of silicon containing scaffolds presented in this review (Fig. 2–6). In addition to the transformations shown in Fig. 2–6, the reader is further referred to several new reviews and perspectives (2020–2023) highlighting recent developments in electrochemistry,33 C–H-activation,34–37 cross-coupling reactions,38–41 hydrosilylations,42–46 carbosilylations,47 construction of silicon stereogenic compounds,48–50 silicon-containing small rings,51,52 synthesis of silanols53 and various other synthetic aspects.54–60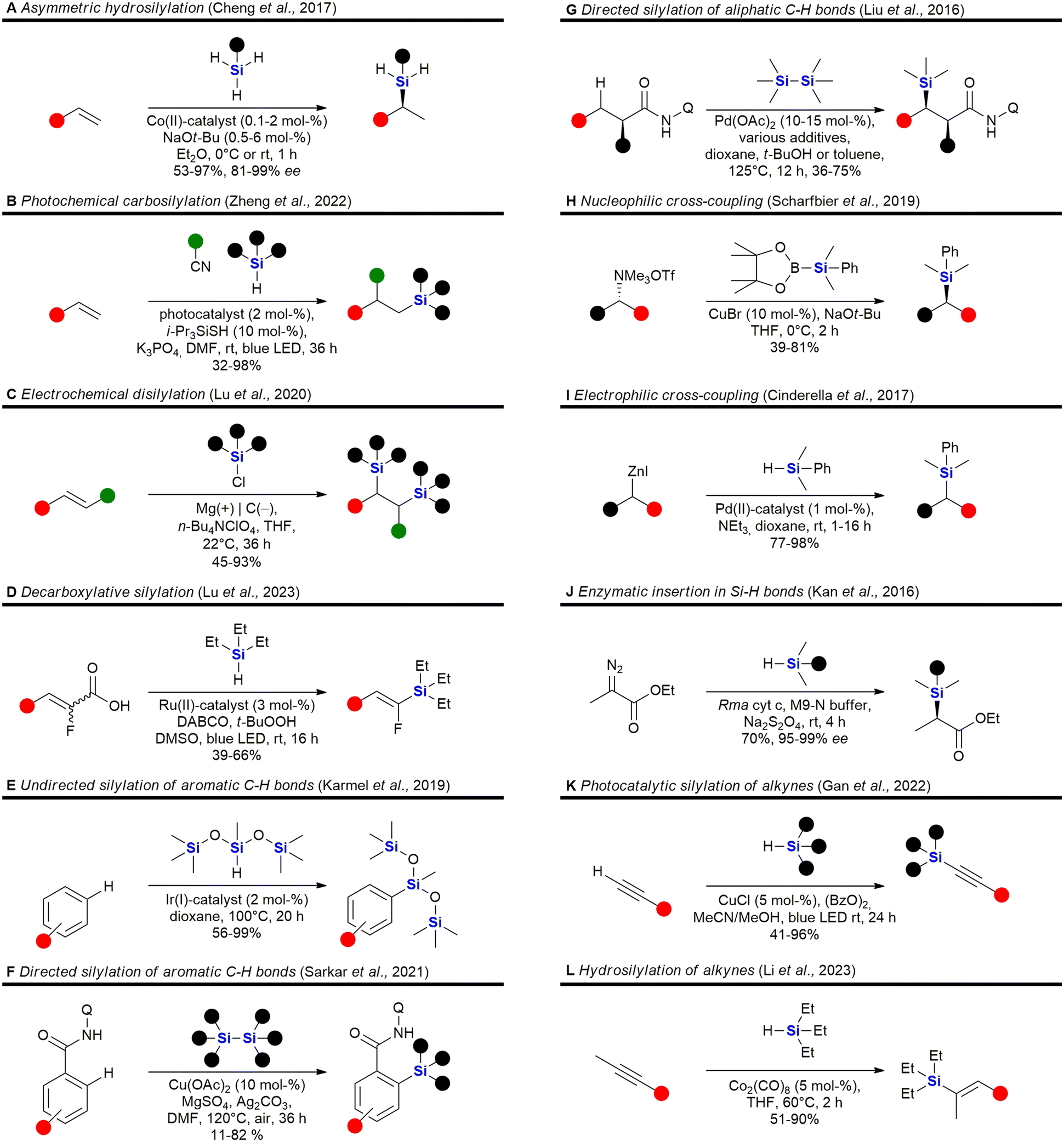 | ||
| Fig. 2 Recent advances in the field of C–Si bond formation – examples A,61 B,62 C,63 D,64 E,65 F,66 G,67 H,68 I,69 J,70 K71 and L.72 Q = 8-aminoquinoline. | ||
 | ||
| Fig. 4 Recent advances in the synthesis of sila-heterocycles – examples A,79 B,80 C,81 D,82 E,83 F,84 G85 and H.86 | ||
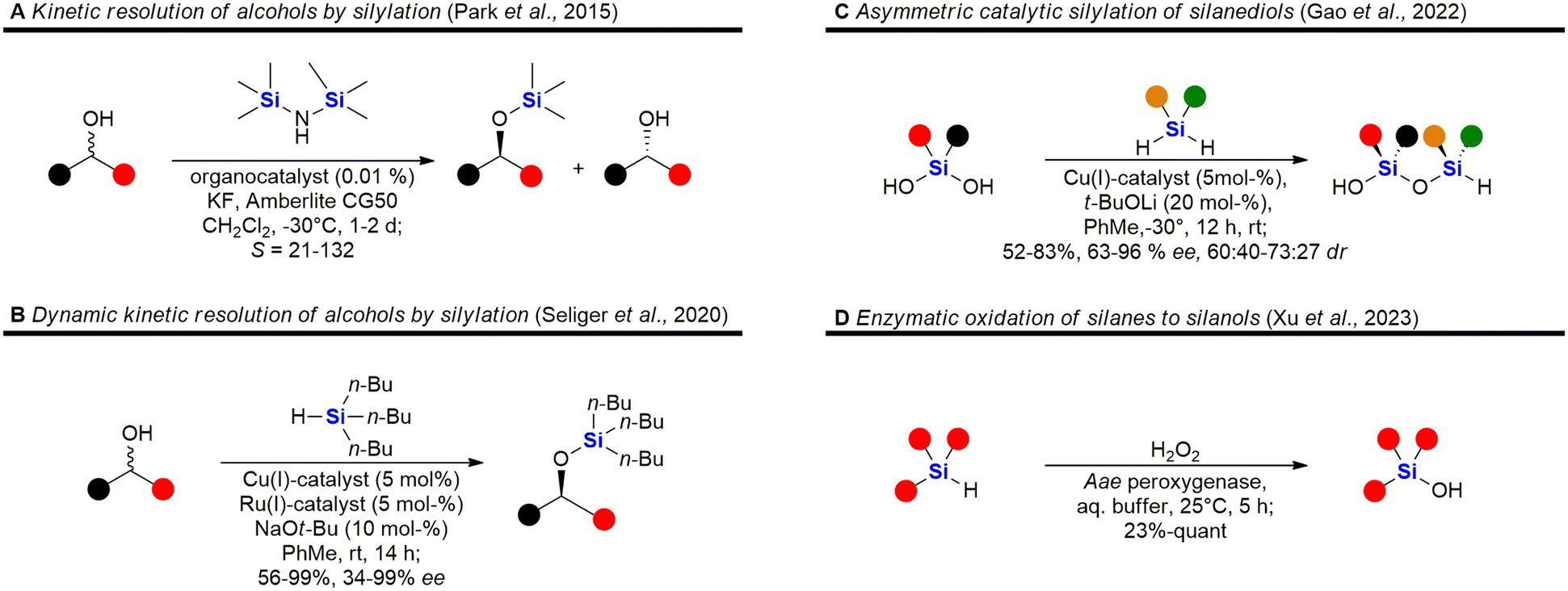 | ||
| Fig. 5 Recent advances in the synthesis of silyl ethers, siloxanes and silanols – examples A,87 B,88 C89 and D.90 | ||
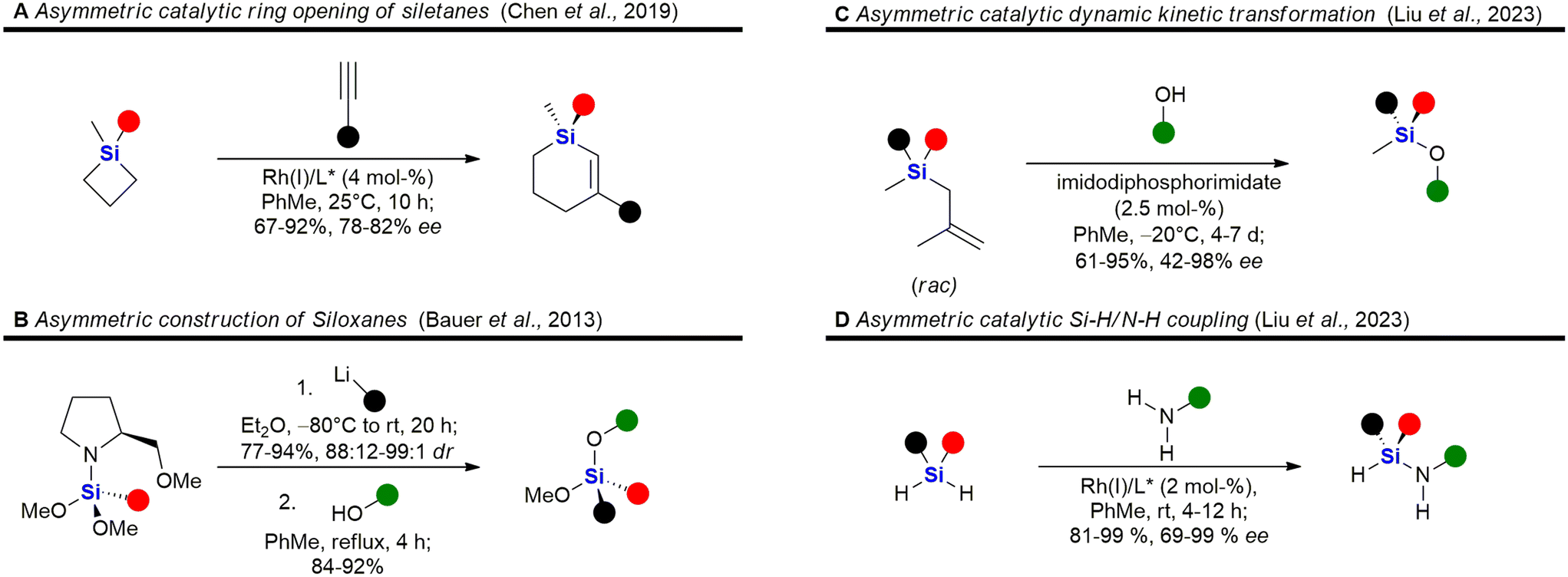 | ||
| Fig. 6 Recent advances in the asymmetric synthesis of chiral silanes – examples A,91 B,92 C93 and D.94 | ||
Incorporation of silicon in the scaffold of biologically active molecules necessarily requires the formation of Si–C-bonds. Significant advances have been made in this field and Fig. 2 presents some recent examples. Hydrosilylation of olefins,61,79,95–113 strained rings114,115 or Michael-acceptors116–119 are popular methods for the construction of C–Si-bonds as a large variety of hydrosilanes are commercially available. Cheng et al. recently reported the asymmetric hydrosilylation of both styrenes and unactivated olefins by an earth-abundant Co-catalyst which proceeds under very mild conditions and with high stereoselectivity Fig. 2A).61 Carbosilylations120–130 of olefins are less common, yet Zheng et al. reported a photochemical procedure using arylnitriles and silanes as reaction partners (Fig. 2B).62 Disilylations of olefins can be accomplished by an electrochemical process developed by Lu et al. using chlorosilanes as the silyl source (Fig. 2C).63 Recently, also an electrochemical silyl-oxygenation131 and a Ni-catalysed silyl-acylation132 were reported. Among the numerous reports for the synthesis of vinylsilanes133–137 or arylsilanes.138–150 by cross-coupling, Lu et al. reported a particularly interesting method using photocatalysis and α-fluoroacrylic acids as coupling partners (Fig. 2D).64 Incorporation of silyl groups by C(sp2)–H-activation has seen tremendous progress,66,139,142,151–169 exemplified here by the undirected Ir-catalysed silylation reported by Karmel et al.65 (Fig. 2E) or the directed silylation by Sarkar et al.66 who employed an inexpensive Cu-catalyst (Fig. 2F). Silylation of aliphatic C–H bonds80,157,170–178 is also rapidly developing. Recently Liu et al. reported the diastereoselective silylation of amides using Pd-catalysis and an aminoquinoline directing group (Fig. 2G).67 Other means of C(sp3)–Si bond formation179 include processes in which the silyl group is introduced as a nucleophile180–186 – see for example the work of Scharfbier et al. (Fig. 2H)68 – or as an electrophile187 – see for example Cinderella et al. (Fig. 2I).69 A recent milestone in the field was the discovery of Kan et al. that enzymes are able to catalyse C–Si bond formation by the reaction of α-diazo esters with organsilanes (Fig. 2J)70 – even though silicon is not naturally encountered in biomolecules. Terminal alkynes can be silylated91,188–191 with organosilanes by a dehydrogenative photochemical reaction using a Cu-catalyst (Fig. 2K).71 Recently, a regioselective hydrosilylation of methylsubstituted internal alkynes was reported by Li et al. using Co-catalysis (Fig. 2L).72 There has also been progress in the field of carbosilylation of alkynes:192 and silylation of arynes.193
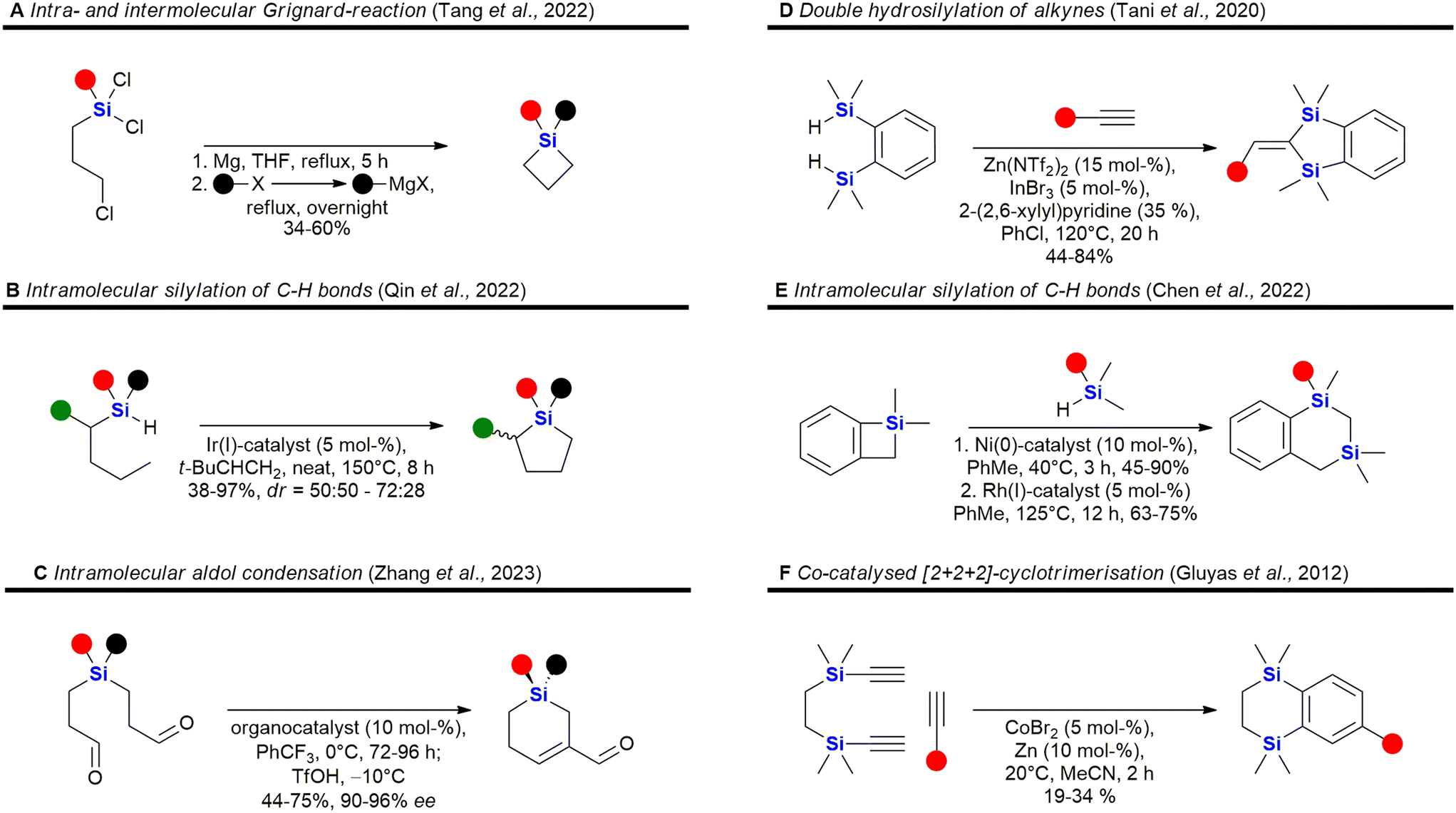
Some recent reports for the synthesis of 4–6 membered silacycles with one or two silicon atoms are shown in Fig. 3. The synthesis of silacyclobutanes is almost exclusively achieved by the intramolecular Grignard reaction of γ-haloalkylchlorosilanes (Fig. 3A). In this work, Tang et al. reported a convenient one-pot procedure in which a second substituent was also introduced (see ESI in ref. 73). Qin et al. reported the synthesis of silacyclopentanes186,194–204 (Fig. 3B) by an iridium-catalysed C–H-activation followed by an intramolecular silylation.74 Among the recent methods to construct 6-membered silacycles82,198,200,205–212 the application of an organocatalysed intramolecular aldol condensation (Fig. 3C) stands out, as it provides a silicon centred stereocentre with high enantiomeric control.75 See ref. 211, 213 and 214 for the synthesis of larger rings with one silicon atom. Five membered rings with two silicon atoms215 may be obtained by a double hydrosilylation of terminal alkynes (Fig. 3D) by a Zn/In co-catalytic system.76 Chen et al. reported the synthesis of benzannulated 1,3-disilacyclohexanes from silacyclobutanes by a nickel-catalysed silylative ring-opening, followed by a Rh-catalysed intramolecular C–H-silylation (Fig. 3E).77 Synthetic methods to synthesise 1,4-disilycyclohexanes, which are incorporated in retinoid X receptor agonists (Fig. 15), seem to be less developed78 and mostly rely on the Co-catalysed [2 + 2 + 2]-cyclotrimerisation reaction (Fig. 3F).216
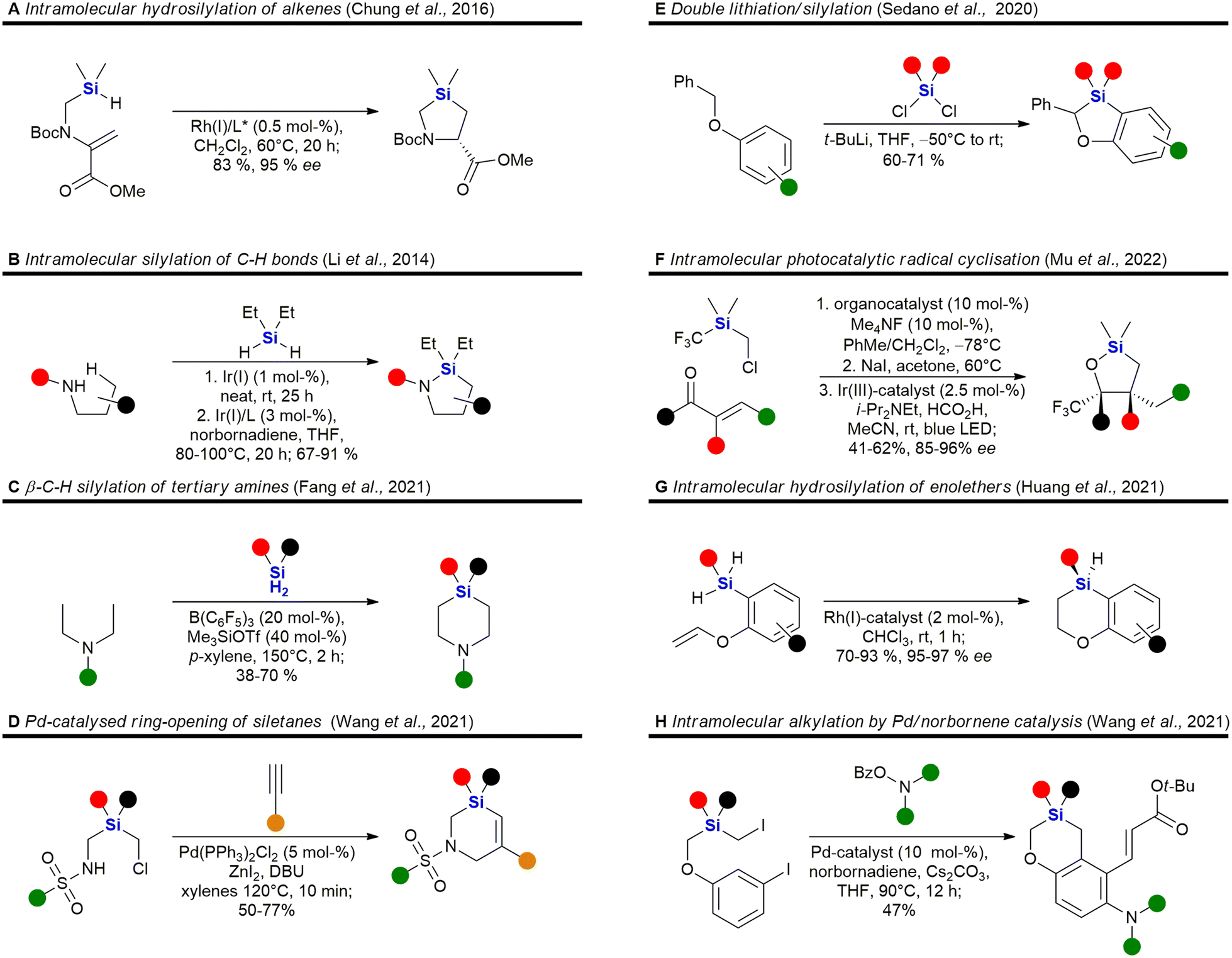
Heterocycles, especially those containing nitrogen, are privileged structural motives in small molecule drugs.217,218 Therefore, there is considerable interest in the synthesis of silicon-containing heterocycles (Fig. 4).219 A recent highlight in the synthesis of 3-sila-pyrrolidines was the development of a Rh(I)-catalysed asymmetric intramolecular hydrosilylation for the synthesis of silaproline (Fig. 4A).79 This unnatural amino acid [see ref. 220 and 221 for the synthesis of other unnatural silicon-containing amino acids] has been incorporated in several biologically active peptides (Fig. 59, 98, 117 and 119). 2-Sila-pyrrolidines are accessible by consecutive iridium-catalysed N–H and C–H silylations (Fig. 4B), but have not been reported as part of biologically active molecules so far.80 4-Sila-piperidines (Fig. 4C) are rather commonly used in small molecule drugs (Fig. 60 and 64–66). While traditionally prepared by double alkylation of a nitrogen-nucleophile,222 Fang et al. recently reported their synthesis through a double dehydrogenative silylation of dialkylamines.81 3-Sila-piperidines are less frequently encountered (Fig. 108). They are accessible by the palladium-catalysed ring-opening of in situ prepared siletanes, followed by carbosilylation of terminal alkynes (Fig. 4D).82 See also ref. 223 for the synthesis of silazepanes. There is also some interest in the synthesis of heterocycles containing both oxygen and silicon, yet these have not been incorporated into small molecule drugs so far. Benzannulated 3-silaoxolanes can be prepared by double lithiation of phenylbenzyl ethers, followed by addition of a dichlorosilane as an example (Fig. 4E).83
Recently, an interesting two-step procedure for the synthesis of 2-silafuranes was reported. It involved an organocatalytic 1,2-addition of a trifluoromethylsilane to an enone and a subsequent photocatalytic radical addition (Fig. 4F).84 An asymmetric rhodium-catalysed intramolecular hydrosilylation of phenylvinyl ethers gave access to benzannulated 4-silatetrahydropyrans and also established a silicon stereocentre (Fig. 4G).85 3-Silatetrahydropyrans are less explored, but recently a palladium/norbornene cooperative catalysis approach provided one example of a complex compound with such a structural motif (Fig. 4H).86 In addition, see the following ref. 224 for the synthesis of siloxepanes by a Pd-catalysed ring-expansion of siletanes.
Fig. 5 shows some recent examples in the chemistry of silyl ethers, silanols and siloxanes. Derivatisation of natural products and known small molecule drugs by formation of silyl ethers is the most common approach to include silicon in biologically active molecules. Even though largely considered a standard procedure, new methods have been investigated.195,196,225 A special focus has been laid on the asymmetric synthesis of silyl ethers. Park et al. reported the kinetic resolution of secondary alcohols by an organocatalytic silylation using hexamethylsilazane as a silyl source (Fig. 5A).87 Later, a dynamic kinetic resolution was reported by Seliger et al. who employed a ruthenium-catalyst for the racemisation of secondary alcohols and a copper-catalyst for the asymmetric silylation (Fig. 5B).88 Besides alcohols, silanediols can also be silylated in an asymmetric manner, as exemplified by Gao et al. who reported the desymmetrisation of silanediols with dihydrosilanes under Cu-catalysis (Fig. 5C).89 This report also includes a few examples of a double desymmetrisation of both the silanediol and the dihydrosilane, resulting in disiloxanes with two silicon stereocentres. Disiloxanes are present in several biologically active molecules (e.g.Fig. 13 and 55). Numerous new methods for the oxidation of hydrosilanes to silanols, which possess interesting biological activities (e.g.Fig. 55, 63, 101, 104, 107 and 111), have been reported,226–229 among these the enzymatic oxidation by Xu et al. under physiological conditions (Fig. 5D).90
A selection of recently developed methods for the asymmetric construction of silicon stereocentres85,230–246 are shown in Fig. 6, yet only one example of a silicon-containing drug with a silicon stereocentre has been reported so far. Desymmetrisation of siletanes by a rhodium-catalysed sequence of ring-opening and carbosilylation of alkynes provides enantioenriched sila-cyclohexenes (Fig. 6A).91 This methodology was later applied to the asymmetric synthesis of sila-mesembralon (Fig. 116). Prolinolmethyl ether was used as a chiral auxiliary in the desymmetrisation of dimethoxysilanes by nucleophilic attack of organolithium compounds (Fig. 6B). The auxiliary was cleaved by alcoholysis, which occurred under retention of configuration.92 Very recently, a dynamic kinetic transformation of racemic methallylsilanes by an organocatalytic alcoholysis was disclosed (Fig. 6C).93 Dihydrosilanes can also be desymmetrised by rhodium-catalysed dehydrosilylation of primary amines (Fig. 6D).94
The rest of this review summarises findings linked to the incorporation of silicon as a means of modifying both activity and pharmacokinetic parameters in drug-like molecules. These bioactive silicon-containing molecules will be briefly described in the specific fields of medicinal chemistry in which they have been applied, including cancer, anti-bacterial, anti-fungal, anti-viral, anti-inflammatory, cardiovascular and neurological diseases. Where appropriate, the presented organosilicon API's are contrasted against that of the non-silicon containing parent compounds which formed the basis for the silyl-derivation or carbon–silicon-switch, to allow the reader an understanding of how incorporation of silicon atom(s) has resulted in a modified molecule with new (and potentially improved) properties and/or bioactivity. The reader is further referred to earlier reviews and perspectives describing both the synthesis and biological evaluation of organosilicon compounds,5,11,15,247–262 as well as some popular online science discussions that have highlighted the rather slow journey of silicon within the broader medicinal chemistry environment.263–266 It should also be mentioned at this point that that focus of the rest of this review will be on medicinally-relevant “small molecules” and not on the broader area “silicon-based materials” which has also demonstrated significant value in modern medical therapeutics and devices.
Anti-cancer agents
The use of organosilicon compounds as anti-cancer agents can be traced back to the 1980's when Chiu et al. reported the use of silylated pro-drugs as potential anti-cancer agents.267 These researchers examined the use of N- and O-silylated compounds, that were related to various “mustard-like” anti-cancer drugs, as potential prodrugs (Fig. 7). In this research, several O-silylated carbamate derivatives 8 showed very good activity against P-388 lymphocytic leukaemia in mice.267 Since then the carbon/silicon switch has received much attention as a means of designing compounds with high anti-tumour activity and low toxicity towards non-cancerous cells.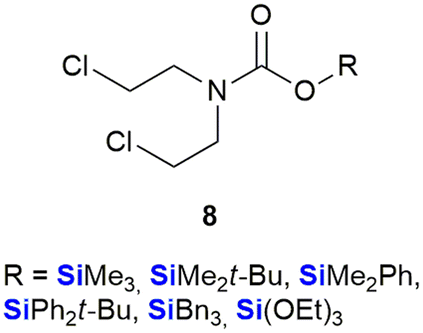 | ||
| Fig. 7 N- and O-triorganosilylated compounds as potential pro-drugs for the treatment of cancer. | ||
Bexarotene 9 is a retinoid X receptor (RXR)-selective retinoid agonist that is retailed under the brand name Targretin (Fig. 8). It is currently applied in therapies targeting cutaneous T-cell lymphoma and underwent phase III clinical trials for the treatment of non-small cell lung cancer,268–270 albeit without broader success.271 Retinoids are small molecule mimics of retinoic acid, a vitamin A metabolite. Retinoids have been demonstrated to function as retinoid X receptor agonists impacting the regulation of gene expression and thereby also cell differentiation and proliferation.272 In comparison to carbon-based structural groups, the hydrophobic nature of silyl functional groups made them particularly useful for probing the chemical space associated with the hydrophobic retinoid pharmacophore.
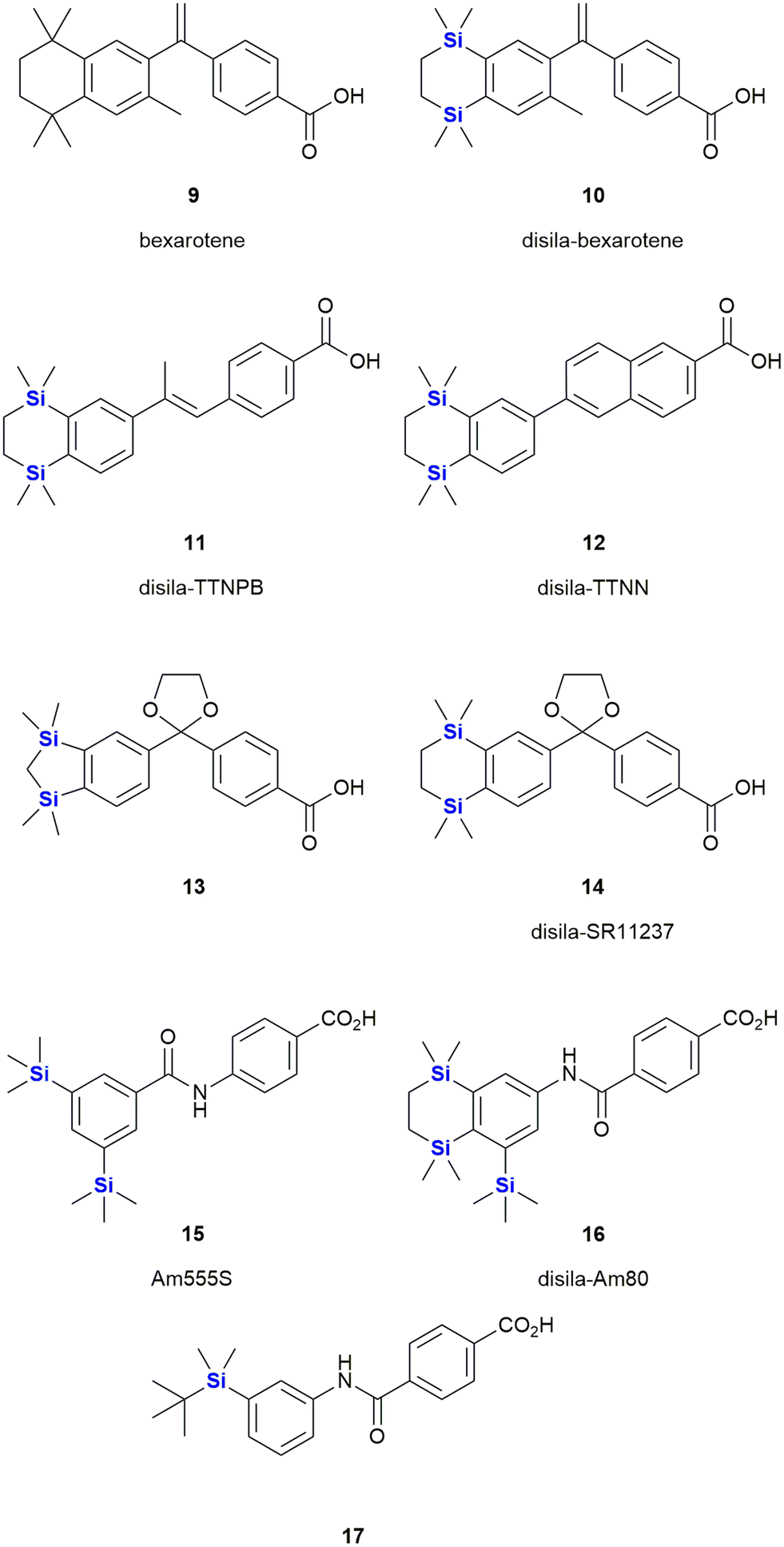 | ||
| Fig. 8 Retinoid X receptor agonist bexarotene 9 and silicon-containing analogues. | ||
In terms of an example, in 2005 Tacke and co-workers reported the agonist activity screening of a synthesized disila-substituted derivative 10, in which a pair of carbon atoms of bexarotene 9 had been substituted with silicon.273 Upon screening of 10 for agonist activity (involving a hRXR β cell-based luciferase assay), the researchers found that compound 10 had a comparable response when compared to bexarotene 9. This research group have since also reported the synthesis of a structurally related disila-TTNPB analogue 11, which was shown to act as a strong pan-RAR agonist. Compound 11 was also shown to display the same strong differentiation and apoptosis-inducing activity in NB4 promyelocytic leukaemia cells as its non-silyl source compound (Fig. 8)274 (and other research involved the testing of these types of compounds against human pluripotent stem cells275). Interestingly, through in silico assessment of the electron densities of bexarotene 9 and disila-bexarotene 10, Tacke and co-workers further showed in 2013 that the replacement of the carbon atoms with silicon atoms did not significantly influence the electronic structures of the molecules, or the RXR receptor activation.26 Furthermore, the group also reported the synthesis and biological evaluation of disila-TTNN 12, a powerful RARBγ-selective retinoid, where again, the carbon silicon exchange afforded a novel compound with a significant increase in activity.78
In another report, the Tacke group reported the RXR agonists 13 and 14. In both cases, there were slight, but reproducible improvements in the activity of these disila-compounds when compared to their respective carba-analogues.276
The Kagechika and Tacke groups further reported research on the retinoids Am555S 15 and disila-Am80 16 respectively.277,278 Am555S 15 had improved α-subunit activity and was evaluated in phase II clinical trials,279 whilst disila-Am80 16 had increased potency towards β- and γ-RAR relative to the analogous non-sila parent compound. Most recently, Kagechika reported the synthesis and screening of several structurally related analogues. During this work the researchers also identified compound 17 which had an EC50 of 7.2 nM against the human acute promyelocytic leukaemia cell line HL-60 and which was 120 times more active than bexarotene 9 and showed strong selectivity towards RARγ.280
The Zablotskaya group have extensively studied the effect of modifying biologically active molecules through the incorporation of silicon groups.281–292 Their research efforts have primarily focused on the development of neurological agents which are discussed later in this review; however, they have also reported the synthesis and biological evaluation of several potent anti-cancer agents. The Zablotskaya group's research has been driven by the hypothesis that the incorporation of silicon would potentially improve the biological properties of existing silicon-free active pharmaceutical ingredients (APIs) by increasing drug lipophilicity and therapeutic index, prolonging action, lowering toxicity and lowering therapeutic doses.
The group reported the positive influence of oxygen and nitrogen silylation, silyl alkylation, silyloxyalkylation and sila-substitution with regards to psychotropic and anti-tumour activity.281 A review of the literature by the authors indicated that many synthetic anti-microbial APIs contain the structural N–C–C–O– sequence. The researchers thus undertook to study the structure activity relationships (SARs) of a range of trialkylsilyl substituents at the oxygen atom of various β-aminoethanol derivatives. The in vitro anti-tumour and anti-microbial properties of the novel compounds were studied and it was found that the silylmethiodide 18 showed high anti-tumour activity (IC50 0.3 μg mL−1) against HT-1080 (human fibrosarcoma) and NO-generation activity (IC50 1 μg mL−1) against MG-22A (mouse hepatoma) (Fig. 9) and also exhibited some anti-bacterial and anti-fungal activity.
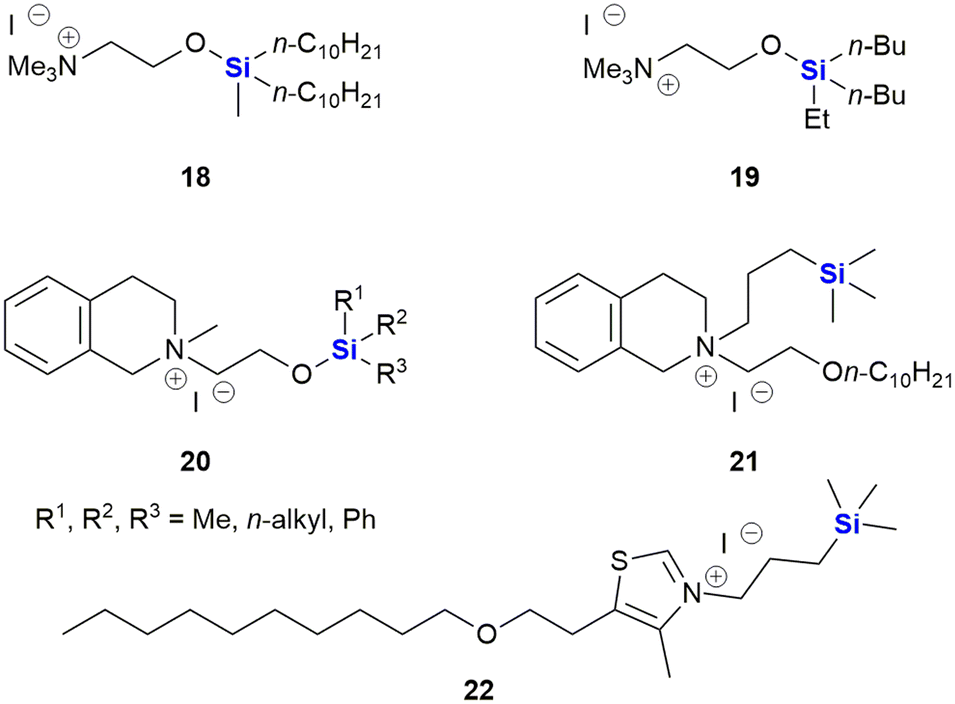 | ||
| Fig. 9 Anti-cancer silyl ether derivatives of β-aminoethanols. | ||
In the same family, the silylmethiodid 19 showed comparable IC50 values of 3 μg mL−1 and 1 μg mL−1 against HT-1080 and MG22-A cell lines respectively (Fig. 9). In general, a more potent cytotoxic effect was observed with increasing bulk surrounding the silicon atom. In a subsequent report, these compounds were incorporated into novel water-soluble iron-oxide based magnetic nanoparticles which exhibited low to moderate cytotoxicity effects on tumour cell lines.293
In subsequent publications, the Zablotskaya group reported on the preparation and biological evaluation of a range of N-methyl-N-(2-triorganylsiloxyethyl)1,2,3,4-tetrahydro(iso)quinolinium iodide derivatives. The investigation of quinolone derivatives was driven by the fact that they commonly display a wide range of biological activities and are regarded as being “privileged” structures (Fig. 9).282 Silylated derivatives showed improved cytotoxicity activity across the board when contrasted against the non-silylated parent analogues. The study also showed that the HT-1080 and MG-22A cell lines were highly responsive to the organosilicon substituents. Compound 20 displayed significant inhibition of HT-1080 and MG-22A cells when R1, R2, R3 = C2H5, C2H5, C2H5 or CH3, C7H15, C7H15 with LC50 values of <1 and 1 μg mL−1 respectively and selective inhibition against MG-22A (LC50 2 μg mL−1) when R1, R2, R3 = CH3, CH3, C11H23.
Zablotskaya and co-workers have continued with the development of silylated tetrahydroisoquinoline derivatives and most recently have reported the synthesis and biological evaluation of O-decyl substituted tetrahydroisoquinoline 21 and thiazolium organosilicon iodo salt 22, which displayed LC50 values ranging from 0.1–0.2 μg mL−1 when screened against HT-1080, MG-22A and NIH 3T3 cell lines.292 The silylated analogues possessed high NO generation abilities, which was absent in the unsilylated parent compounds; furthermore, the compounds were determined to be non-toxic, with LD50 values ranging from 200–1600 mg kg−1.
Ignatovich and co-workers reported the synthesis of novel silicon-containing bisfuranes 23, and their work included investigating the effect of the amine structure and the silicon atom's alkyl substituent with respect to the compounds' cytotoxicities (Fig. 10).294 Cytotoxicity (IC50) and toxicity (LC50) were determined against two cancer cell lines, namely HT-1080 (human fibrosarcoma) and MG-22A (murine hepatocellular carcinoma), as well as normal NIH 3T3 (murine embryonic fibroblasts) cells, respectively. The researchers found that regardless of the amines utilized, the compounds exhibited moderate toxicity (174–360 mg kg−1) and all derivatives with TMS substituents were more active than those with the corresponding TES substituents. The presence of an amine-containing group at position R1 resulted in higher toxicity towards the normal NIH-3T3 cells, except for the morpholine group which showed low cytotoxicity across all three cell lines. Further studies on the morpholine-substituted derivative indicated that the introduction of the TMS group onto the furan ring greatly increased the cytotoxicity (3 μg mL−1 for HT-1080 and MG-22A cells). In contrast, examples with the TES group (no cytotoxic effect) or unsubstituted analogues were less active (100 μg mL−1 and 38 μg mL−1 for HT-1080 and MG-22A cell lines respectively).
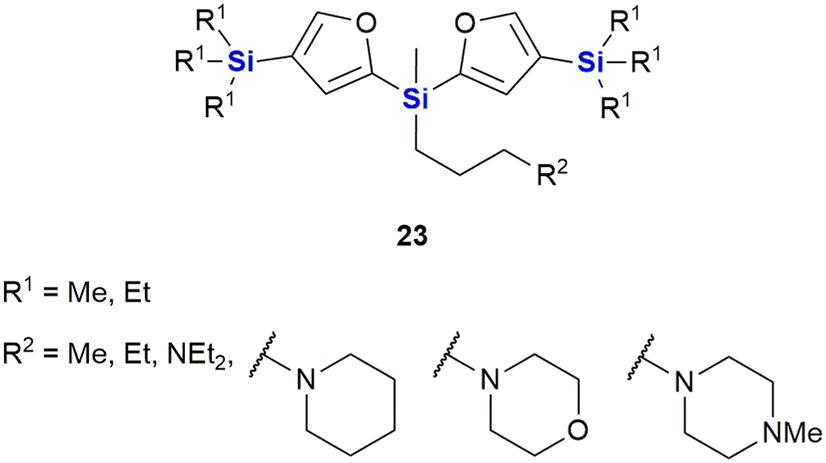 | ||
| Fig. 10 Silicon containing bisfuranes 23 with anti-cancer properties. | ||
In 2007, Lo and co-workers reported the synthesis of two novel glucoconjugated silicon(IV) phthalocyanines 24 and 25 (Fig. 11). Both compounds showed highly photocytotoxic activity against HT-29 (human colorectal carcinoma) and HepG2 (human hepatocarcinoma) cell lines, the most active being the non-chlorinated phthalocyanine 24 which demonstrated IC50 values as low as 6 nM.295 It was proposed by the researchers that the chlorinated analogue 25 had reduced activity (IC50 17–21 nM) due to a higher aggregation tendency in biological media, which in turn led to a decrease in the ability to form reactive oxygen species inside the cells. The non-chlorinated analogue 24 was also shown to have significant and selective affinity to lysosomes, but not mitochondria, of the HT-29 cells. The application of various thiophenic silicon phthalocyanines has received considerable interest as drug delivery systems for anti-cancer photodynamic therapy (PDT).296–298 PDT allows the non-invasive treatment of malignant tumours through the employment of photochemical transformations which themselves are mediated by photosensitizers. Phthalocyanine derivatives are excellent photosensitizers; however, their planar geometry typically leads to molecular aggregation and often their hydrophobic nature prevents them from existing as monomeric dispersions in aqueous media.299 In the latter case, strong collisional quenching of the molecules excited states occurs.299 The use of silicon as a central atom helps overcome these issues as silicon can accommodate axial substituents preventing the formation of aggregates. Notably, Jaung, Jang and co-workers recently reported the development of non-aggregated photosensitisers for use as PDT agents.299 In this instance the silicon-based photosensitisers were housed in carbohydrate-based block glycopolymer polymeric micelles, the use of polymeric micelles having been shown to enhance tumour localisation, permeation, and retention. For other representative examples of silicon phthalocyanines with biological application from the last decade, see the following references300–312 and reviews.313,314
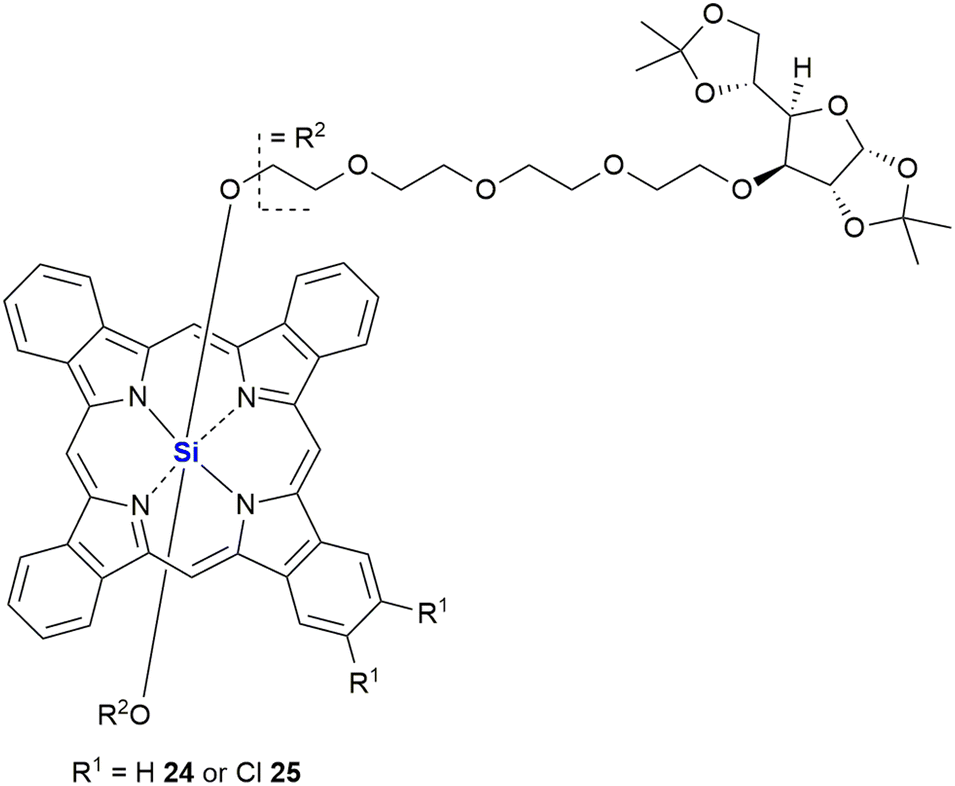 | ||
| Fig. 11 Novel glucoconjugated silicon(IV) phthalocyanines for photodynamic cancer therapy. | ||
Following on from the clinical efficacy of cis-dichlorodiamineplatinum(II) (cisplatin), Anderson et al. prepared and assessed the anti-tumour activity of platinum(II) and platinum(IV) complexes with silicon containing diamines (Fig. 12).315 Silaplatin 26 and the related platinum(IV) analogue 27 increased the life span of mice bearing intraperitoneally implanted murine L1210 leukaemia by 109–248% (26) and 115–214% (27) at doses of 10–40 mg kg−1. Furthermore, the platinum(II) complex 26 showed high activity against a cisplatin-resistant L1210 cellular subline (35–98% at doses of 5–30 mg kg−1) which compared favourably to the activity of cisplatin (8% at 10 mg kg−1). A related cyclobutanedicarboxylic acid complex 28 showed activity against both cisplatin-sensitive (22–77% at doses of 10–40 mg kg−1) and -resistant (7–41% at doses of 10–40 mg kg−1) L1210, but was less potent than the dichloro analogue 26.
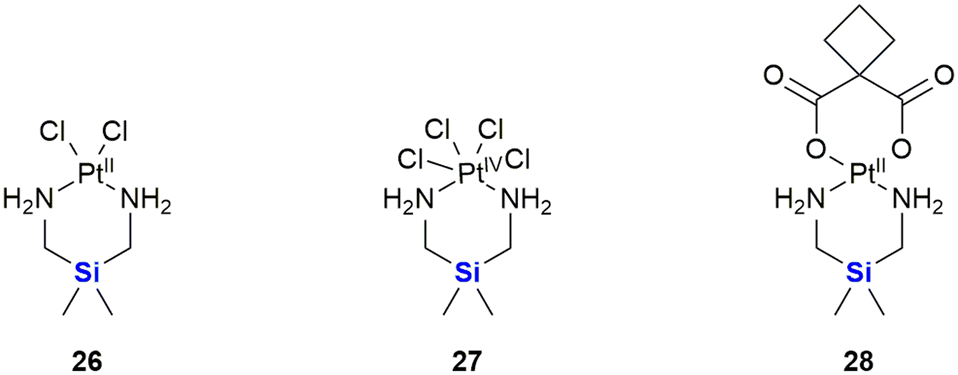 | ||
| Fig. 12 Silicon-containing-platin complexes of platinum(II) and platinum(IV) with anti-cancer activities. | ||
SILA compounds 409 29 and 421 30 were patented in 1999 by Hegyes and co-workers and were shown to act as resistance modifiers against multidrug-resistant cancer cells through the inhibition of cellular efflux pumps (Fig. 13).316,317 These compounds were demonstrated to have anti-proliferative effects without the induction of apoptosis (although other researchers saw increased apoptotic activity)318 and initial results indicated that these SILA derivatives specifically acted on the MDR1 p-glycoprotein 170 and subsequent investigations suggested further effects on cell-proliferation and viability.319 Later, it was shown that the same compounds had moderate potential to prevent the formation of tumours caused by carcinogenic chemicals or the Epstein–Barr-virus320 and increase the sensitivity of adenocarcinoma cells LoVo/Dx cells to doxorubicin.321
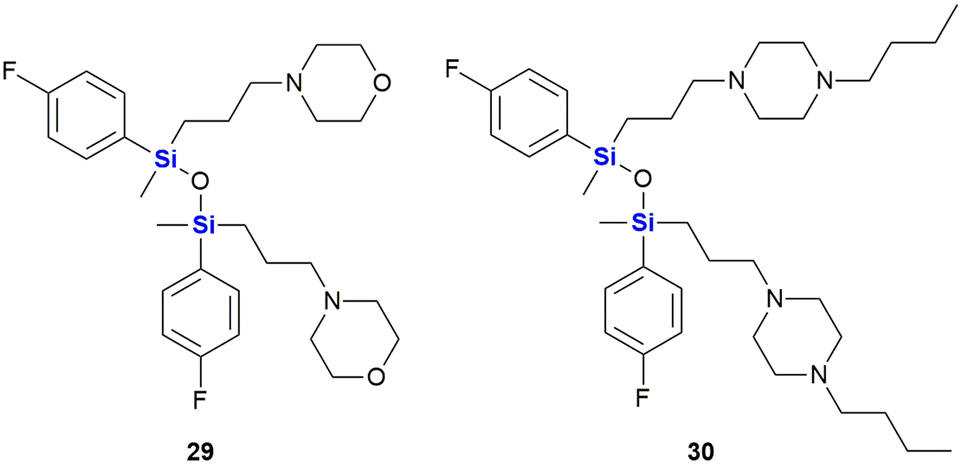 | ||
| Fig. 13 Anti-proliferative SILA compounds 409 29 and 421 30. | ||
Thompson et al. described the isolation and structures of two potent anti-viral and anti-tumour compounds, mycalamides A 31 and B 32, which were isolated from the New Zealand sponge Mycale sp.322 Apart from acyl and alkyl derivatives, several silyl derivatives 33 were prepared to further explore the biological properties of these compounds (Fig. 14).
 | ||
| Fig. 14 Mycalamide A 29 and B 30 and silyl ether derivatives 31 with anti-cancer properties. | ||
The TMS ethers were found to all be active against the murine leukaemia P388 cell line (IC50 = 0.1 to 1.3 ng mL−1), though it is postulated that the TMS ethers were simply hydrolysing back to the active parent compounds (IC50 0.07 to 0.5 ng mL−1). The bulkier, less labile, TBDMS ethers showed a marked decrease in activity (IC50 = 23 to >12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500 ng mL−1) relative to the parent compounds.
500 ng mL−1) relative to the parent compounds.
Members of the combretastatin family have been demonstrated to potently inhibit microtubule activity through tubulin-binding at the colchicine site, and in doing so interrupting cell growth and proliferation.323 Combretastatin A-4 34 showed the strongest anti-tumour cell growth-inhibition (IC50 = 0.004 μM) in an MCF-7 (breast cancer) cell proliferation assay. It also acted as a vascular disrupting agent (VDA) reducing blood flow and leading to tumour cell death (Fig. 15). Nakamura et al. reported the synthesis of silicon-containing combretastatin analogues 35 in which the ethene linker was replaced with a silicon bridge, thereby affording analogues in which the distance between the two ring systems remained almost unchanged (3.00 Å vs. 3.04 Å for the silyl-analogues). One of the compounds, 35, which contains a Si(CH3)H linker, exhibited potent inhibition of tubulin polymerization, as well as significant cancer cell growth inhibition (IC50 = 0.007 μM) in an MCF-7 cell proliferation assay.324
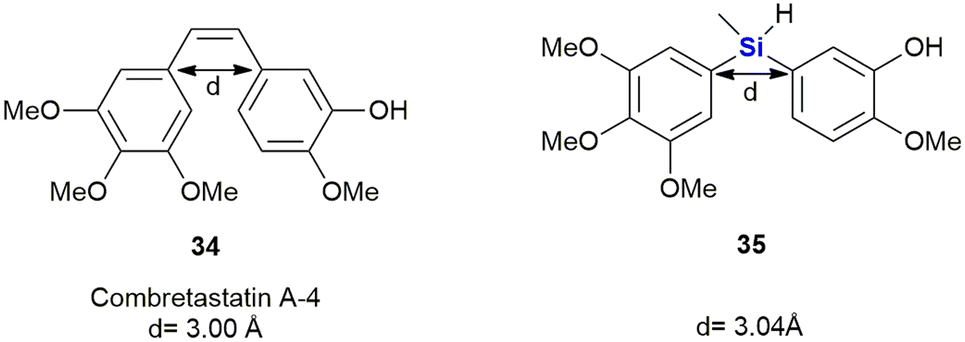 | ||
| Fig. 15 Combretastatin A-4 34 and silicon analogues 35. | ||
In addition, this compound also prevented [3H]colchicine binding (∼91% inhibition at 3 μM) and the researchers surmised that these biochemical characteristics were comparable to that of combretastatin A-4 34. Furthermore, the silicon-containing analogue 35 showed improved physio-chemical stability. The authors therefore proposed that the silicon linker could thus act as a bioisostere of a cis alkene bond.
In a continuation of their work looking at diphenylsilanes as a cis-stilbene mimetics, Fujii and co-workers also turned their attention to investigating whether silyl groups could also mimic aliphatic cis-olefins (Fig. 16).325 During this research, series of silylated oleoylethanolamide derivatives were prepared (which included compounds 37 and 38), based upon a known endogenous cis-olefin-containing peroxisome proliferator-activated receptor PPARα agonist 36 (ref. 326) and evaluated in terms of their PPAR α/β/γ agonistic activities, as PPARγ agonists not only do they act as insulin sensitizers but they also possess anti-proliferative activity.327 To this end, the design rationale revolved around the fact that the silicon-containing compound would mimic the cis-alkene functional arrangement. From molecular orbital calculations it was demonstrated that the distance between the carbons with a silicon bridge was similar to that of the two carbons in a cis-relationship (3.132 vs. 3.214 Å). In comparison, two carbons linked by a carbon bridging atom would according to the modelling have a significantly smaller distance between them of 2.579 Å. In terms of interesting outcomes, the diethylsilyl derivative 37 was found to exhibit PPAR α/δ agonistic activity while the related compound 38 was demonstrated to be a PPARδ-selective agonist.
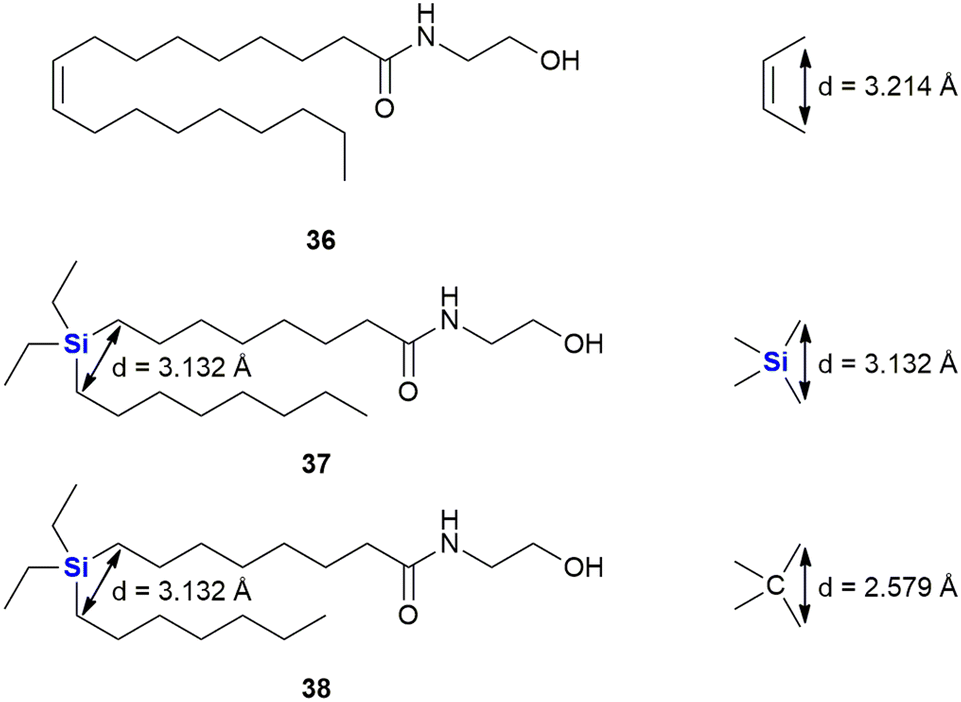 | ||
| Fig. 16 The use of silyl groups as mimics of aliphatic cis-olefins. | ||
The same researchers further reported the synthesis of bisphenol analogues possessing vitamin D receptor (VDR) agonistic activity and androgen receptor (AR) antagonistic activity (Fig. 17).328 The researchers observed that the replacement of the quaternary carbon linker in the known non-secosteroidal vitamin D mimic LG190178 39 (ref. 329) with silicon, increased the compound's AR antagonistic activity, but reduced its VDR-agonistic ability. The results thus represented the first example of a nuclear receptor (NR) selectivity switch by carbon–silicon substitution. Of note was that the most potent AR-selective analogue 40 inhibited the testosterone-induced growth of murine mammary carcinoma cell line SC-3 cells more efficiently (IC50 = 0.072 μM) than the well-known androgen antagonist hydroxyflutamide (IC50 = 1.4 μM).328,330
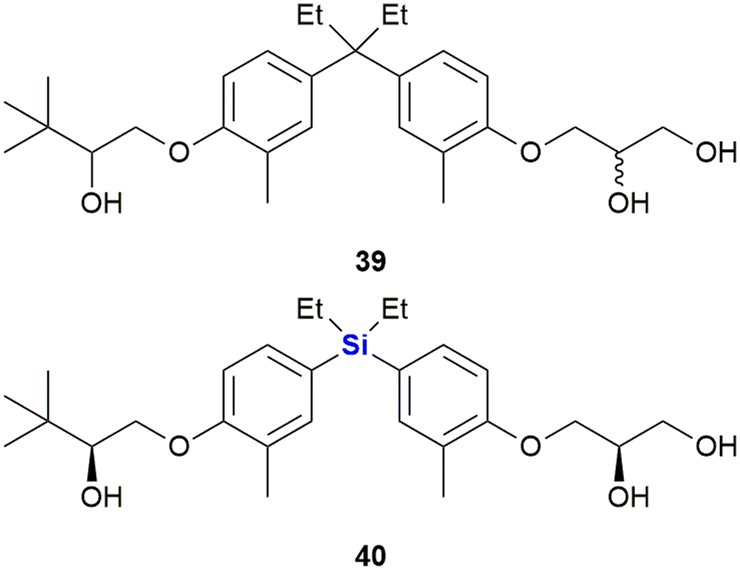 | ||
| Fig. 17 Non-secosteroidal vitamin D mimic LG190178 39 and sila-analogue 40. | ||
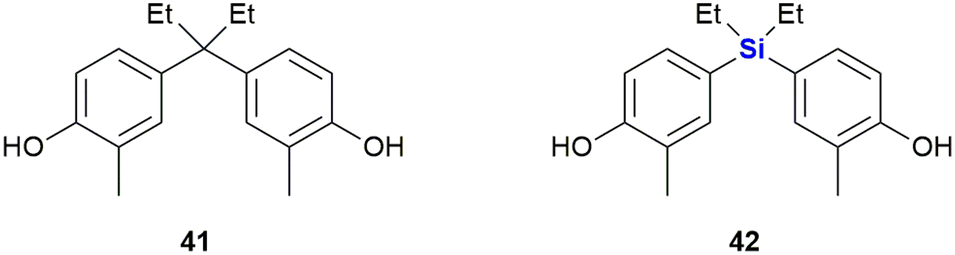
In another study, Maruyama et al. investigated the SARs of bisphenol analogues as oestrogen receptor antagonists, potential drug candidates for the treatment of oestrogen-sensitive breast cancer (Fig. 18). The bisphenol 41 was identified as a potent antagonist of oestrogen receptor ERα (IC50 = 25 nM) and less so of ERβ (IC50 = 260 nM). Its sila-analogue 42 however was less potent against both ERα (IC50 = 72 nM) and ERβ (IC50 = 800 nM).328
 | ||
| Fig. 18 Bisphenol 41 and sila-analogue 42 as oestrogen receptor antagonists. | ||
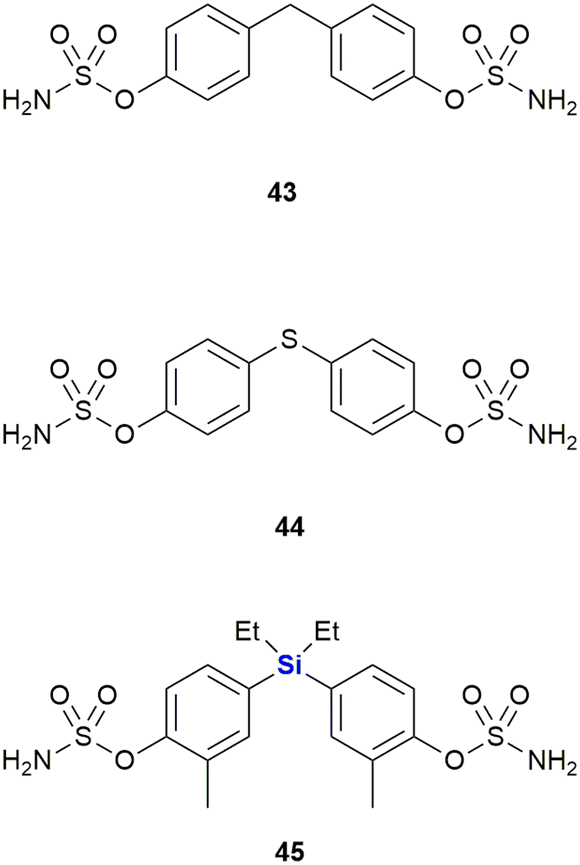
Steroidsulphatase (STS) plays a crucial role in the oestrogen synthesis of postmenopausal women and is therefore a possible target for postmenopausal hormone-dependent breast cancer therapy. Although several steroid-based STS inhibitors are now known, these compounds have been found difficult to optimize and have the potential to interact with other targets. While investigating this challenge, Kajita et al. postulated that diphenylmethanebisphenol derivatives would potentially bind to the ligand-binding domain (LBD) of oestrogen receptors like attributed to oestradiol, thereby acting as a steroid proxy molecule (Fig. 19). The silicon linked bisphenol 45 showed an inhibitory activity of STS (IC50 = 170 nM) between that of the carbon analogue 43 (IC50 = 1024 nM) and the sulphur analogue 44 (IC50 = 88 nM). Upon hydrolysis of the sulphonamide moiety, these inhibitors would liberate bisphenol metabolites which might possess oestrogen receptor antagonistic properties (vide supra). Hydrolysis of the sulphonamide groups of 45 by STS generates the metabolite 42 (Fig. 18) which possess antagonistic properties against oestrogen receptor ERα, thus adding to the anti-proliferative properties of 45 against breast cancer cells. In contrast, the metabolites of carbon analogue 43 and sulphur analogue 44 possessed weak agonistic properties against ERα, which is unwanted as it might promote the growth of breast cancer cells.331
 | ||
| Fig. 19 Bis-phenol sulphonamides 43, 44 and 45 as STS inhibitors. | ||
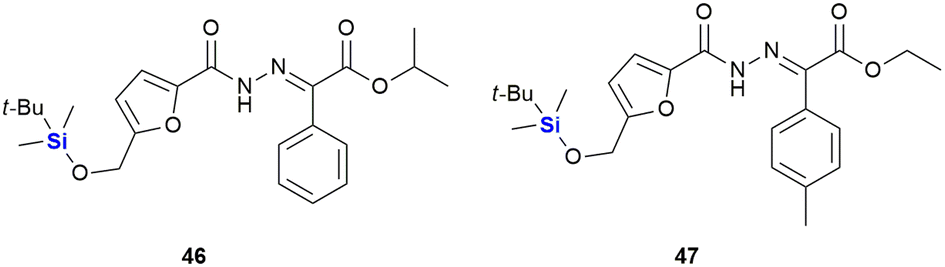
In 2014, Trindade and co-workers reported an NHC-catalysed umpolung reaction between 5-hydroxymethyl furfural (HMF) derivatives and diazo compounds affording a set of HMF-substituted acylhydrazones which exhibit anti-tumour activity (Fig. 20).332 Upon screening of various HMF derivatives to determine their anti-proliferative activities against HT-29 (colon), MCF-7 (breast), NCI-H460 (lung) cancer cell lines, it was observed that the hydroxyl and O-benzyl derivatives displayed insignificant activity. However, the respective TBDMS derivatives were shown to be active against MCF-7 cell lines with IC50 values as low as 3.5–7.29 μM. It was postulated that the TBDMS group increased the lipophilicity of the compound, and that this aided in the biological activity. Interestingly, the two most active compounds, 46 (IC50 3.60–6.37 μM) and 47 (IC50 3.50–7.29 μM), displayed the lowest toxicities towards differentiated CaCo-2 (colon cancer) monolayer cells at IC50 values of 40.12 and 27.90 μM respectively.
 | ||
| Fig. 20 Silicon-containing acylhydrazones displaying anti-tumour activity. | ||
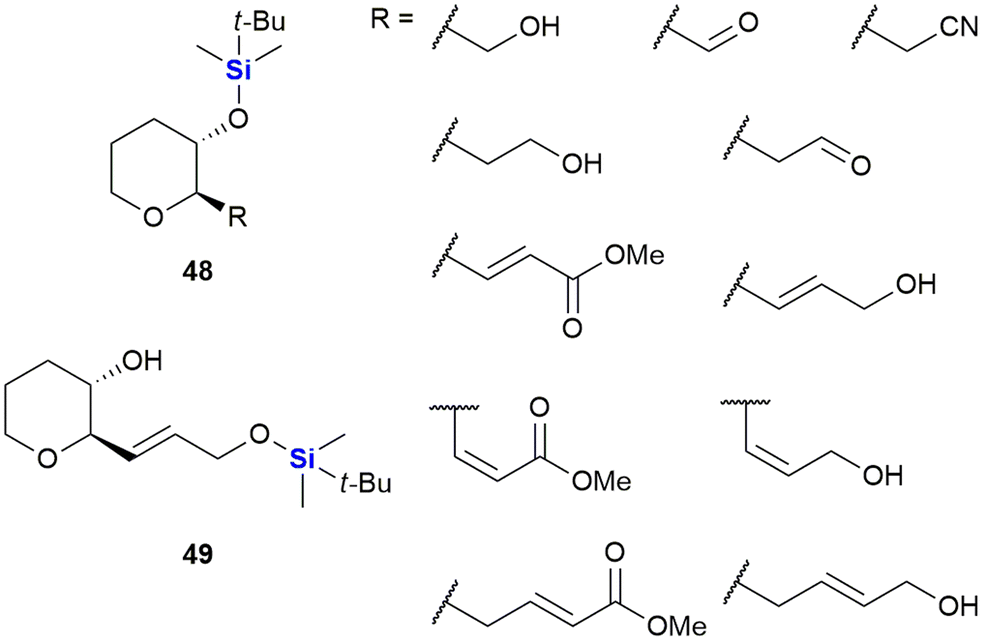
In 2005, Padrón and co-workers described research in which it was demonstrated that incorporation of the TBDMS group onto small molecules could positively modulate their cytotoxicity against human-based tumour cells.333 In this work, the researchers synthesized a set of enantiomerically pure (2R,3S)-disubstituted tetrahydropyrans 48 and 49 bearing a variety of diverse functionalities (Fig. 21). In addition to the TBDMS group, other common hydroxyl functionality protecting groups were utilized (including allyl, acetate and benzoate groups – not shown). The compounds were subsequently screened in vitro against HL-60 human leukaemia cells and MCF-7 human breast cancer cells. Significant observations included that the synthetic derivatives bearing the TBDMS group at tetrahydropyran ring position 3 elicited considerable cancer cell cytotoxicity.333 When the SAR of the set of molecules was evaluated, the results indicated that the TBDMS group should not only be seen as a functional protecting group for organic synthesis, but that it could also be utilized as part of a plausible strategy to introduce lipophilicity in drug-like molecules.
 | ||
| Fig. 21 Silyl-(2R,3S)-disubstituted tetrahydropyrans with anti-cancer properties. | ||
This increased lipophilicity was anticipated to aid in the cellular uptake of the TBDMS-containing molecules and thus to potentially modulate their cytotoxic activity. The Padrón group furthermore investigated this enhanced drug cytotoxicity by further in vitro screening of the aforementioned compounds and some other derivatives against a range of solid tumour cell lines, namely: T-47D (breast cancer), HeLa (cervix cancer), SW-1573 (non-small cell lung cancer), WiDr (colon cancer) and A-2780 (ovarian cancer) cells. The results obtained were published the following year (2006) and supported the conclusions from the original paper.334
In 2007, the same authors extended the tetrahydropyran scaffolds to structurally similar chlorovinyl-TMS containing dihydropyrans which were prepared and assessed for anti-proliferative activity against human solid tumour cell lines A-2780 (ovarian cancer), SW-1573 (non-small cell lung cancer) and WiDr (colon cancer).335 The non-sila parent compound 50 showed no growth inhibition against the human solid tumour cell lines; however, in general the silylated analogues 51 showed an enhancement in cytotoxicity (Fig. 22). The introduction of the TMS group at position 3 of the oxacyclic ring system in general improved activity against all the cell lines screened. When compared against previously reported cytotoxic non-sila alkyl chloro dihydropyrans (52, 53 and 54) the sila analogues showed improved activity when R3 was a cyclohexyl or but-3-ynyl group.
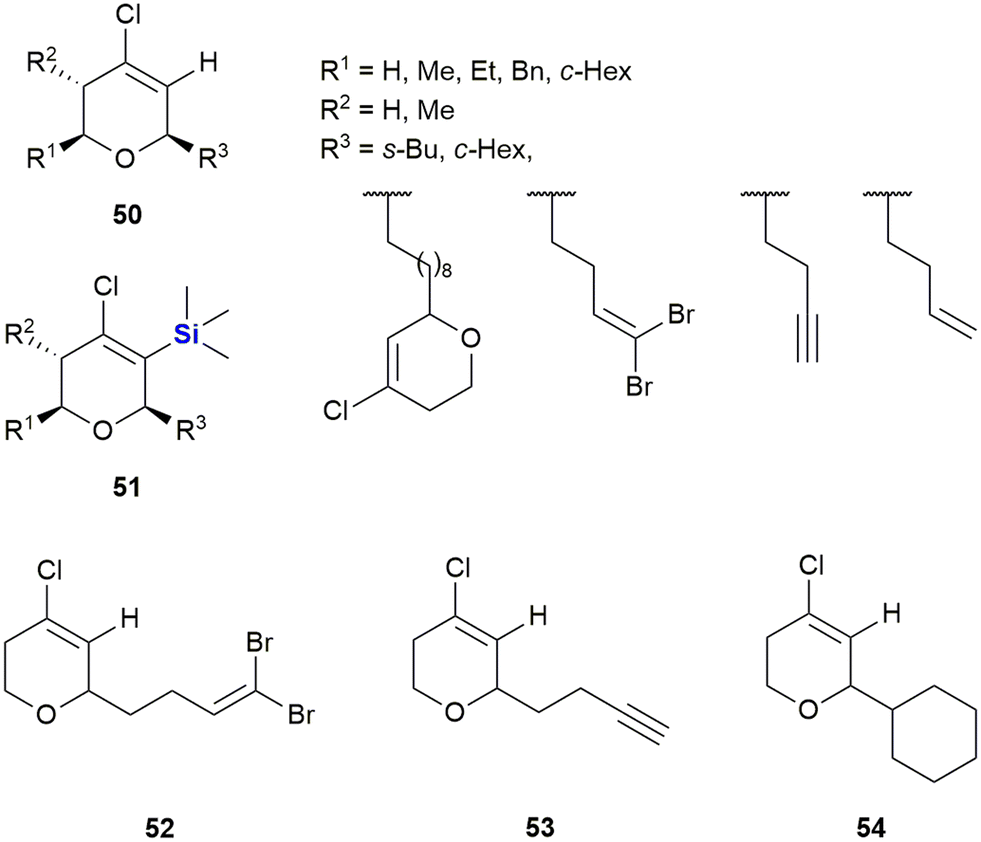 | ||
| Fig. 22 Chlorovinyl-TMS dihydropyrans displaying anti-proliferative activity against human solid tumour cell lines. | ||
Growth inhibition against A-2780 and SW-1573 cell lines was generally better (17–34 μM) than that of WiDr cells (15–67 μM). Notably, in the case of the non-sila analogues 53 and 54 no inhibition was observed against the WiDr cells.
Saeeng and co-workers have reported the synthesis of novel C-19-analogues of andrographolide 55, a major diterpenoid from Andrographis paniculata with potent cytotoxic activities.336 SAR studies showed that the incorporation of a silyl ether or triphenylmethyl ether group at the C-19 position increased toxicity against various cancer cells such as P-388 (leukaemia), KB (oral), COL-2 (colon), MCF-7 (breast), LU-1 (lung) and ASK (rat glioma) (Fig. 23). Seven of the ten sila-analogues showed improved activity across all cells lines relative to the parent compound 55. The most potent sila-analogues 56 and 57 displayed effective inhibitory doses ranging from 0.34–3.62 μM and 0.88–3.01 μM respectively. The activities against P-388 (0.34–0.88 μM) and ASK (1.22–2.85 μM) cells were better than that of the potent anti-cancer drug ellipticine which gave values of 2.44 and 3.56 μM. Against the remaining cell lines activities were comparable. The authors hypothesized that the improved activity of the silylated compounds was potentially due to the bulky silyl groups at the C-19 position aiding in transport across cellular membranes.
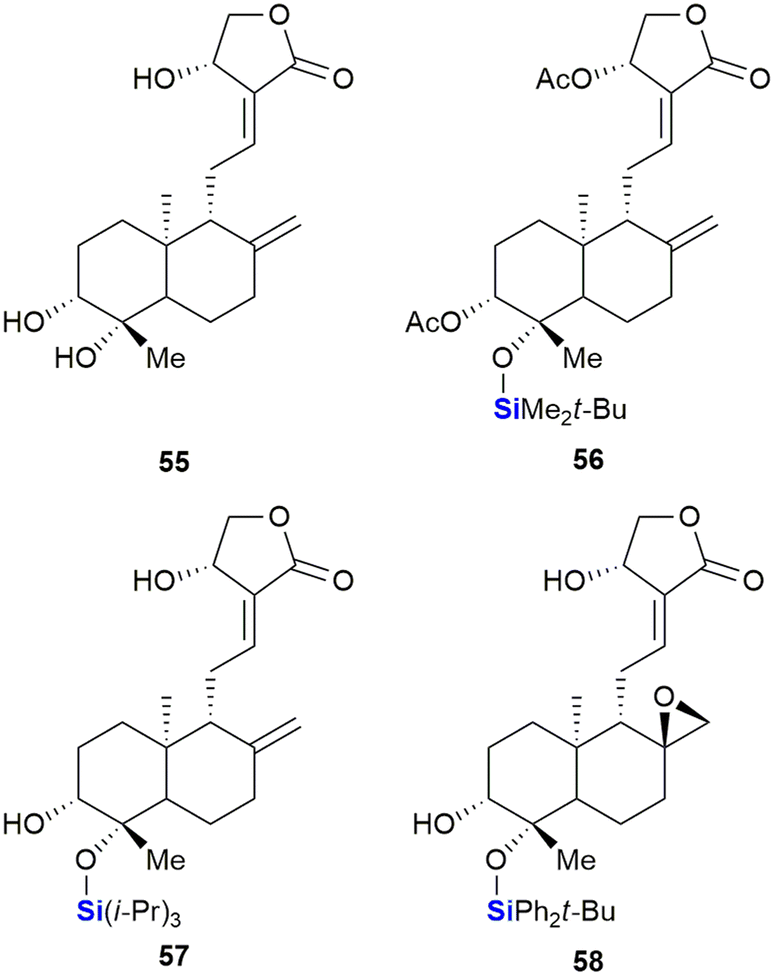 | ||
| Fig. 23 Andrographolide 51 and silyl ether derivatives with potent cytotoxic activities. | ||
Thereafter, the group reported the identification of exo-epoxide-containing 58, which was shown to be highly cytotoxic against CHO (Chinese hamster ovary), HepG2 (liver), UISO-BCA1 (breast) and HeLa (cervix) cancer cells,337 by inhibiting the activity of Topo-IIα.336,338 In further studies, compound 58 was shown to function by targeting the Wnt/β-catenin/GSK-3β pathway in HT-29 colorectal cancer cells.339
The Saeeng group also reported the isolation and derivatisation of durantoside I 59 from Citharexylum spinosum L. (Fig. 24).340 During their studies it was noted that removal of the cinnamate group at position C-7 and silylation of the sugar group afforded the most active compounds. The TBDPS ether 60 proved most active of the compounds synthesized, with an ED50 of 8.2–9.19 μM against the MCF-7 (breast), A-549 (lung) and ASK (rat glioma) cancer cell lines. In contrast, durantoside I 59 showed no appreciable activity against the cancer cell lines.
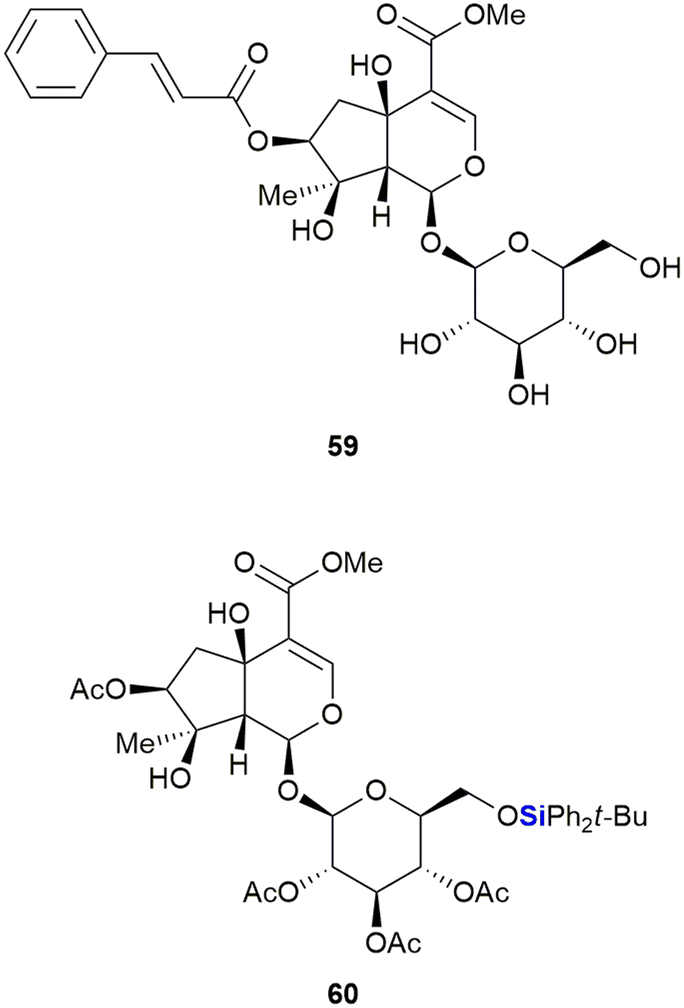 | ||
| Fig. 24 Durantoside I 59 and silyl ether derivative 60. | ||
The iridoids, catalpol 61 and 6-epi-catalpol 66 (Fig. 25), showed significant inhibition of Taq DNA polymerase, with IC50 values of 48 μM and 22 μM, respectively. Padrón et al. subsequently synthesised catalpol silyl ether derivatives 62–65 and investigated their anti-proliferative activity against several solid tumour cell lines.341 The bioactivity results indicated that all the silyl ethers were significantly more active than the parent compound or related ester derivatives. As an example, compound 65 inhibited the growth of tumours by 50% at concentrations of 1.8–2.7 μM. Later, Garro, Pungitore et al. investigated the silyl ether derivatives 67–70 of 6-epi-catalpol.342 In terms of this set of compounds, 67–68 showed no inhibition of Taq DNA polymerase, while 69 was slightly active. Of interest was that only TBDMS-derivative 70 showed promising activity (IC50 = 5.57 μM).
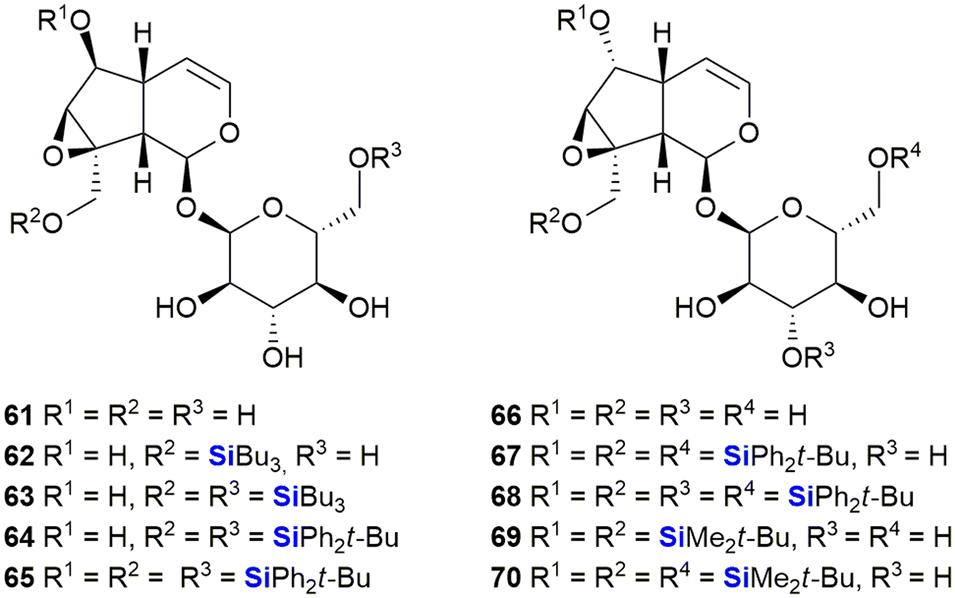 | ||
| Fig. 25 Catalpol and 6-epi-catalpol silyl ethers. | ||
Camptothecin (20S)-71 is the forerunner of a set of well-known anti-tumour therapeutics that has been the focus of new cancer treatments. The closely related topotecan 72 was approved for curative use in the USA whilst the prodrug irinotecan 73 was similarly approved as parts of anti-cancer therapy in the USA, France and Japan.343,344 In addition, a number of other camptothecin-related drugs have provided promising in vivo results and have been in various stages of clinical trials (Fig. 26).345,346
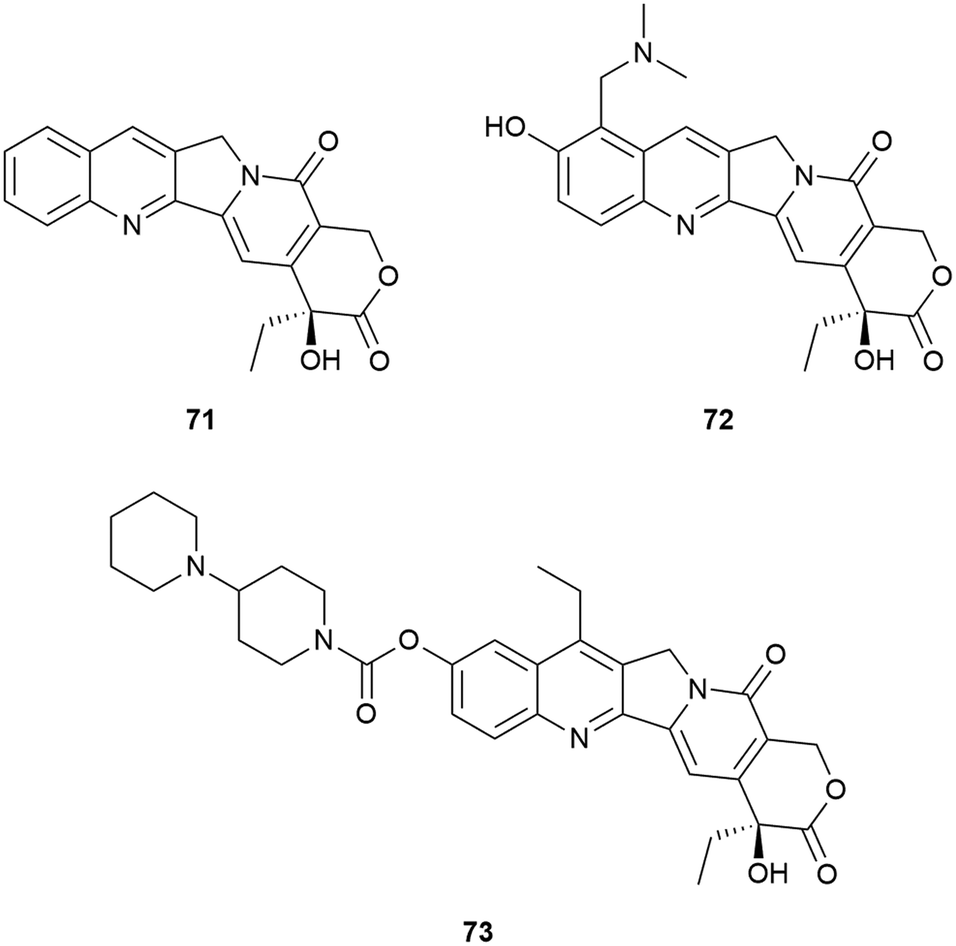 | ||
| Fig. 26 Camptothecin 71, topotecan 72 and irinotecan 73. | ||
With respect to the synthesis of camptothecin derivatives and analogues, the Curran research group developed an innovative cascade radical annulation route, in order to obtain a range of novel substituted (20S)-7-silylcamptothecins (which were given the general name of the “silatecans”).347 During the same research, a series of unsubstituted 7-silylcamptothecins were also prepared using established Friedlander synthetic methods. A selection of these derivatives (74–78), all containing one or more TMS substituents on the camptothecin skeleton, are depicted in Fig. 27. The synthetic camptothecin derivatives were all tested in terms of their abilities to inhibit the growth of HL-60 (human promyelocyticleukaemia), 833 K (human teratorcarcinoma) and DC-3F (hamster lung) cells in vitro. The researchers observed that the newly synthesized compounds were at least equipotent, with some eliciting even more impressive growth inhibitory activity than camptothecin 71. This outcome was particularly impressive because even by current standards, the anti-cancer activity of camptothecin 71 is still regarded as a challenging benchmark, not easy to eclipse.
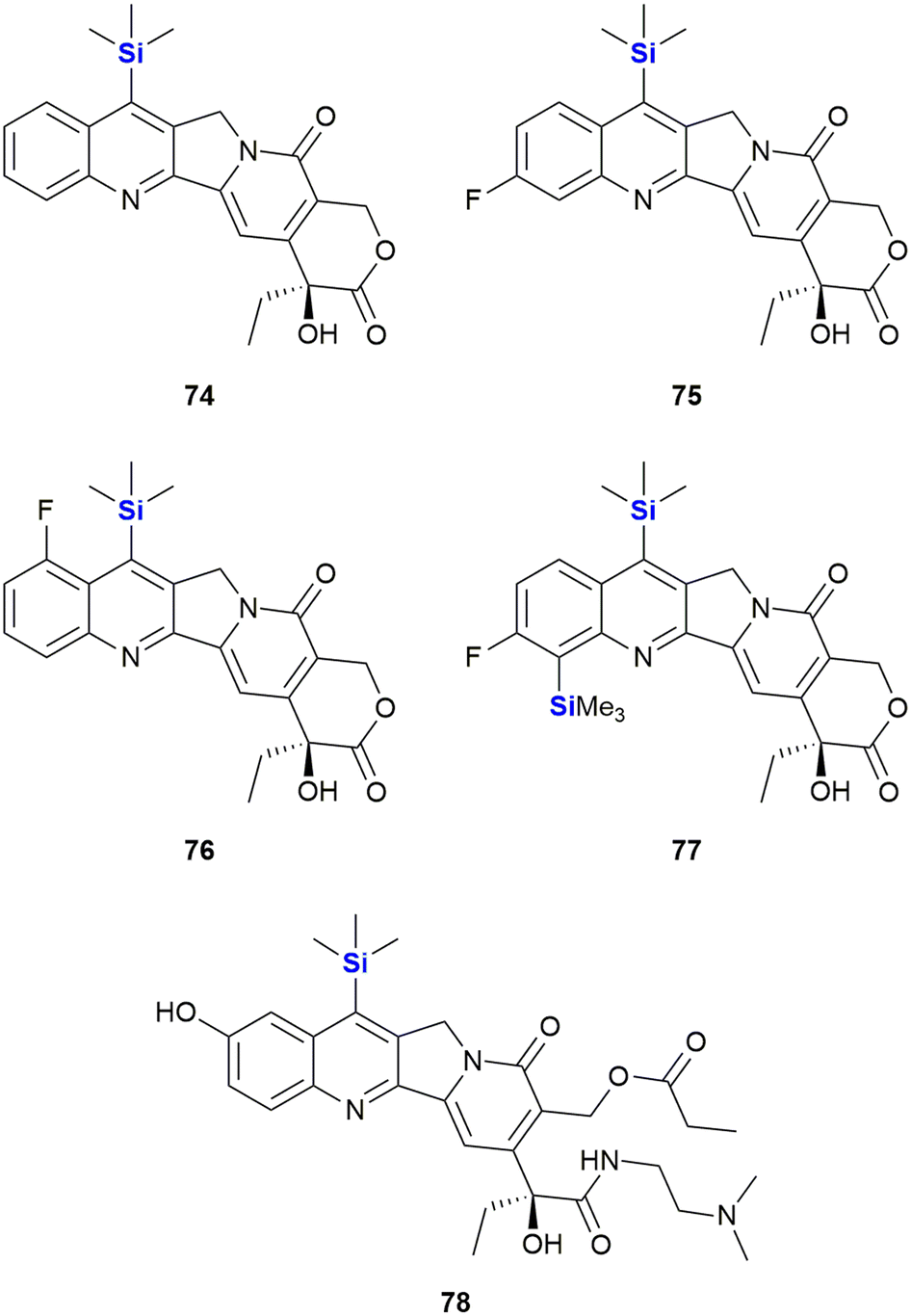 | ||
| Fig. 27 A selection of further silylcamptothecins. | ||
Of interest was that the cellular results demonstrated that the TMS-camptothecin derivatives are also highly active. TMS-bearing 74 showed surprising in vivo chemotherapeutic outcomes (demonstrated by way of tumour volume reduction against sarcoma-180 in B6D2F1 mice) compared to camptothecin 71.347 Compound 75 showed a comparable anti-tumour effect to camptothecin 71, when evaluated on Lewis lung carcinoma, albeit at a four-fold lower dose. These in vitro results indicated the significant promise this class of compounds was expected to have in terms of in vivo activity, though only limited in vivo data has been published. Compounds 74 and 75 were superior to camptothecin 71 when tested against Lewis lung carcinoma and sarcoma-180 tumour models in murine xenografts.348
The Burke group introduced the camptothecin derivative silatecan 79, which showed potent activity in the National Cancer Institute (NCI) cytotoxicity screen with an average tumour growth inhibition of GI50 = 21 nM (Fig. 28).349 While the activity of camptothecin against MDA-MB-435 S human breast cancer cells dropped significantly in the presence of humane serum albumin (from IC50 = 15 nM to IC50 = 375 nM), 79 largely retained much of its activity (with a change from IC50 = 14 nM to IC50 = 90 nM). The in vivo potency of 79 was subsequently proven by the successful treatment of U87 subcutaneous glioma and intracranial glioma in murine xenografts at a dose of 30 mg kg−1.
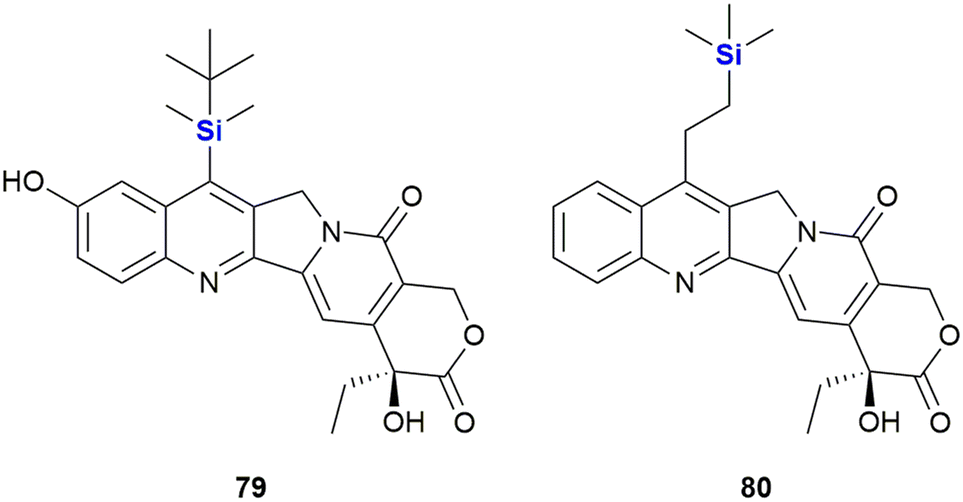 | ||
| Fig. 28 Silatecan 79 and karenitecin 80. | ||
During the investigation of more effective camptothecin derivatives, van Hattum et al. synthesized and investigated karenitecin 80 (Fig. 28).350,351 It was determined that the lipophilic silylated side chain increased oral bioavailability and stability of the active lactone form, probably due to stabilisation by alpha-1 acid glycoprotein,.352 In addition, the structural modification provided resistance to metabolic conversion. Karenitecin was shown to be less susceptible to tumour-mediated drug resistance mechanisms and showed potent activity against ovarian (IC50 = 1.8–2.2 nM) and colon cancer cell lines (IC50 = 1.5–3.2 nM). It also showed promising results in phase I and II clinical trials for the treatment of refractory melanoma.353,354 Finally, for a review on the important topic of silylated analogues of camptothecin and their value as anti-cancer agents. See the following ref. 355.
Schreiber and co-workers performed the synthesis of a diverse range of small organosilicon molecules and then evaluated their cellular activity profiles. As part of this research the authors developed a general method for the annulation of isatins allowing for the preparation of a range of spiro-oxindoles (Fig. 29).356 As an outcome, the results of ninety representative compounds from 41 diverse assays were profiled, all with the goal of evaluating the potential of organosilicon-containing compounds as cellular process modulators. A wide range of activities for organosilicon-containing small molecules was demonstrated and representative examples were subjected to secondary screens for dose-dependent activity and EC50 determination. Activities of selected examples against lung adenocarcinoma (A-549) and hepatocellular carcinoma (HepG2) included EC50 inhibition of 42.8 μM and 32.5 μM for silyl-containing triazole 81 and 16.6 μM for the HepG2 assay in the case of pyrrolidine 82.
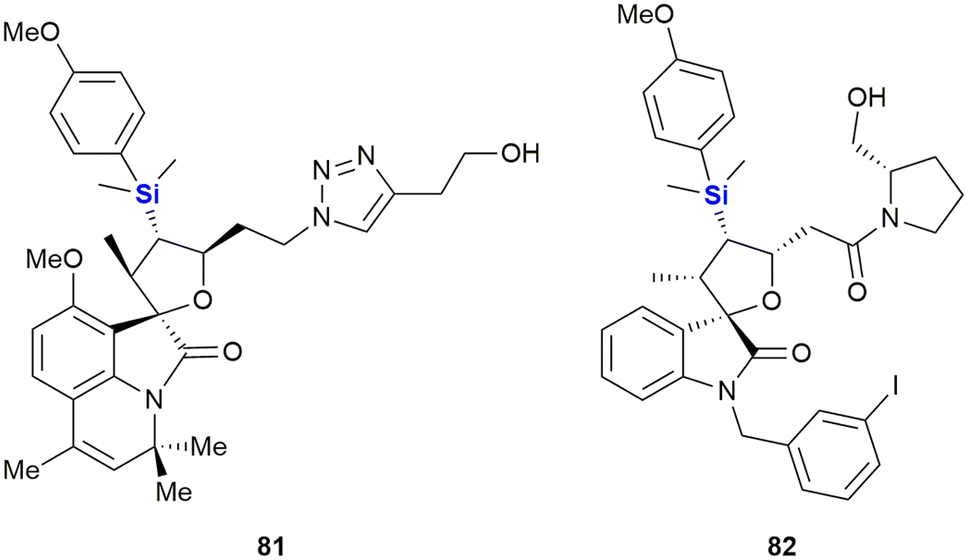 | ||
| Fig. 29 Anti-cancer spiro-oxindoles with silyl groups. | ||
Indomethacin 83, a non-steroidal anti-inflammatory drug (NSAID) has been utilized in treatments for rheumatoid arthritis, ankylosing spondylitis and osteoarthritis (Fig. 30). Indomethacin 83 has also shown promise in the treatment and prevention of human cancers;357–360 however, the observance of severe side-effects unfortunately limited its application in the treatment of cancer patients. In research performed by West and co-workers, aimed at improving the safety profile and biocellular activity of indomethacin 83,361 the design, synthesis and testing of a series of sila-substituted indomethacin derivatives 84–86 was performed.
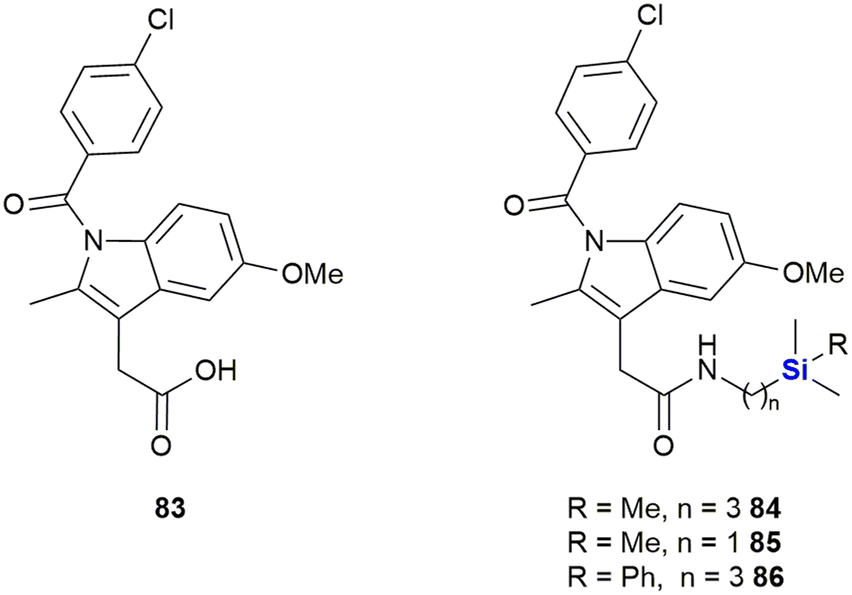 | ||
| Fig. 30 Indomethacin 83 and silicon containing derivatives. | ||
In terms of cellular testing against the human MiaPaCa-2 pancreatic cell line, it was found that the cell growth inhibitory activity for the sila-indomethacin derivatives 84–86 was indeed significant (indomethacin 83 was previously shown to have an IC50 value of 170 μM against the same cell line). The West research group demonstrated that their novel sila-indomethacin derivatives had IC50 values of 4.78–20.0 μM in the in vitro assays.361 Further in vitro pharmacology enzyme assays determined that the sila-indomethacin derivatives 84 and 86 were selective COX-2 inhibitors that were in essence superior to indomethacin 83 in this regard.
In a paper by Lukevits and co-workers the biological implications of in vitro silyl-removal processes involving triorganosilyl derivatives of some biologically active heterocyclic bases (including 5-fluorouracil 87, barbituric acid 88 and xanthine 89 and uridine 90 derivatives), was investigated.362 The researchers found that temporarily masking hydrophilic functional groups on small molecules appeared to have a significant effect on their biological activities. For example, in contrast to the pyrimidine nucleoside uridine, TBDMS-protected uridine 90 (R = SiMe2t-Bu) inhibited the development and proliferation of HT-1080 (fibrosarcoma in human lungs – 96% inhibition at 100 μg mL−1) and NiH 3T3 cells (fibroblasts in mice – 95% inhibition at 100 μg mL−1) in cell culture experiments (Fig. 31).
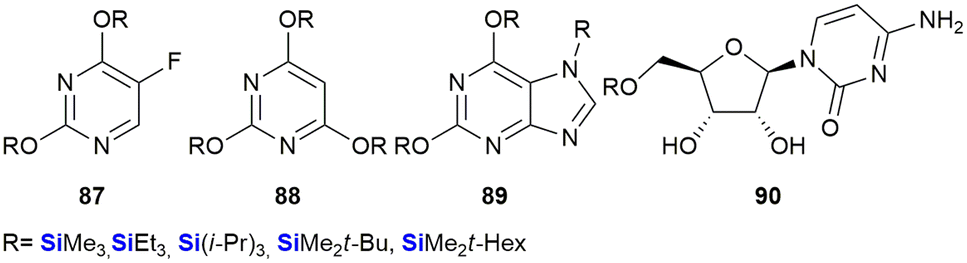 | ||
| Fig. 31 Anti-cancer silyl ether derivatives of heterocyclic bases and uridine. | ||
This type of impact was also recognized in research performed by Herczegh and co-workers who synthesised a series of leinamycin mimetics (92–95) by introducing the biologically active 1,2-dithiolan-3-one-1-oxide moiety of leinamycin 91 onto an aldehyde-D-arabinose coupled to a uridine or methyluridine fragment (Fig. 32).363 In doing so the researchers noted that the lipophilic silyl group derivatives 92 and 94 demonstrated improved cytotoxic activities against HeLa3 cervix tumour cells (IC50 = 3.68–10.04 μM) when compared to the analogues without the silyl groups (IC50 = 50.71–62.23 μM). The desoxythymidine derivatives 93 (no IC50 determined) and 95 (IC50 = 43.12 μM) were less active.
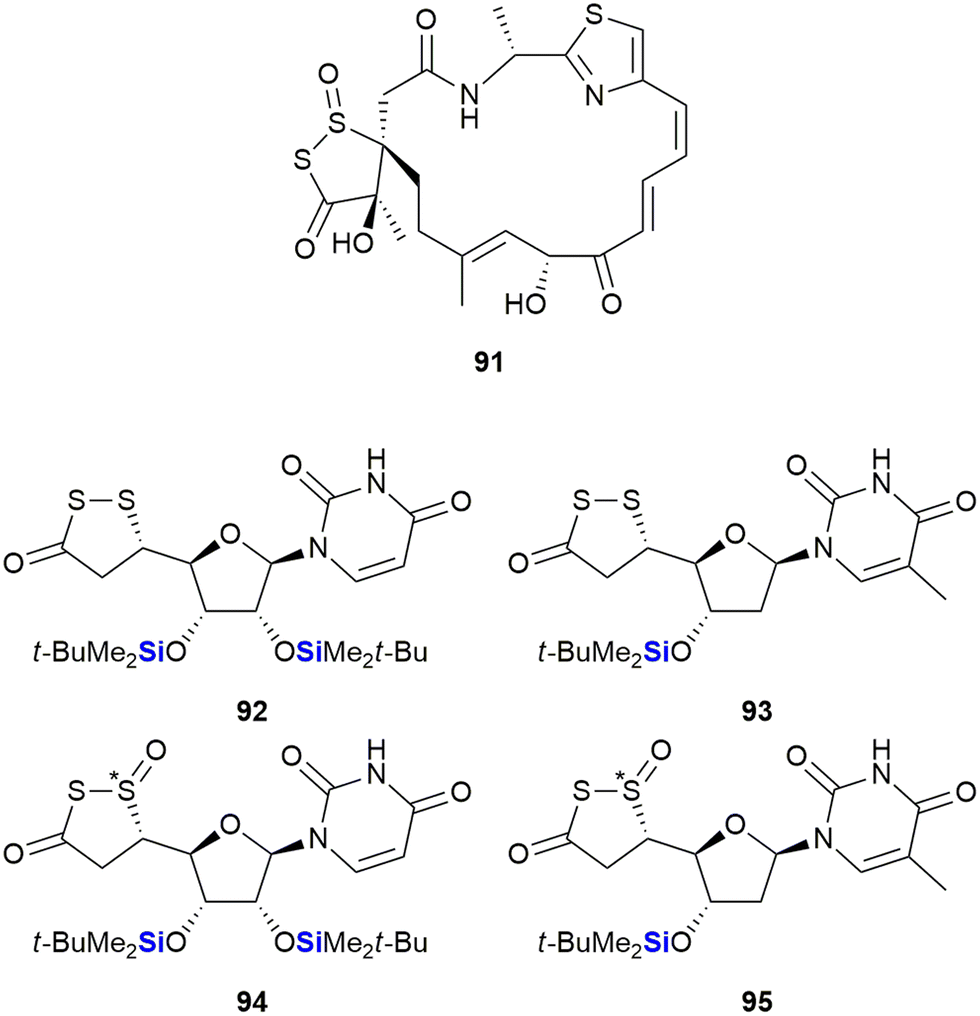 | ||
| Fig. 32 Leinamycin 91 and sila-leinamycin mimetics (note that the stereochemistry of the silatrane unit was not given in the original publication). | ||
A further example of a silyl nucleoside derivative demonstrating anti-cancer activity was reported by Fenlon and colleagues in 2002.364 This research involved the synthesis and anti-cancer screening of a set of 3′-O-silatranylthymidines (Fig. 33). The diastereomeric mixture of 3′-O-(trimethylsilatranyl)thymidine 96 was evaluated versus human breast (MDA-MB-435) and central nervous system (CNS) (SNB-19 and U251) cancer cell lines at a concentration of 0.1 mM over 48 hours. The researchers found that the growth percentages of cells treated with compound 96 were as follows: (i) breast cells 54% and (ii) CNS cells 83%, when compared to untreated control cells (where growth was taken to equal 100%). When compared to the closely related silatranylthymidine 97, activity was absent in the CNS and lung cells, while 81% growth was observed in the breast cell line (correlating to 19% inhibition). It should be noted that 96 is a mixture of four diastereomers, and it would thus be expected that each diastereoisomer would have a unique activity, given that stereochemistry influences pharmacological activity.364 The researchers thus reported that work on the synthesis and screening of each of the individual diastereomers was ongoing. More recently, Ye and co-workers reported structurally similar analogues wherein the sugar moiety was replaced with a γ-amino group.365 The 5-fluorouracil derivative 98 was the most active silatrane derivative of a nucleobase identified in this study and was assayed against human breast (MDA-MB-435, IC50 = 17.1 μM), cervix (HeLa, IC50 = 18.3 μM) and intestinal (HT-29, IC50 = 11.6 μM)) cancer cell lines.
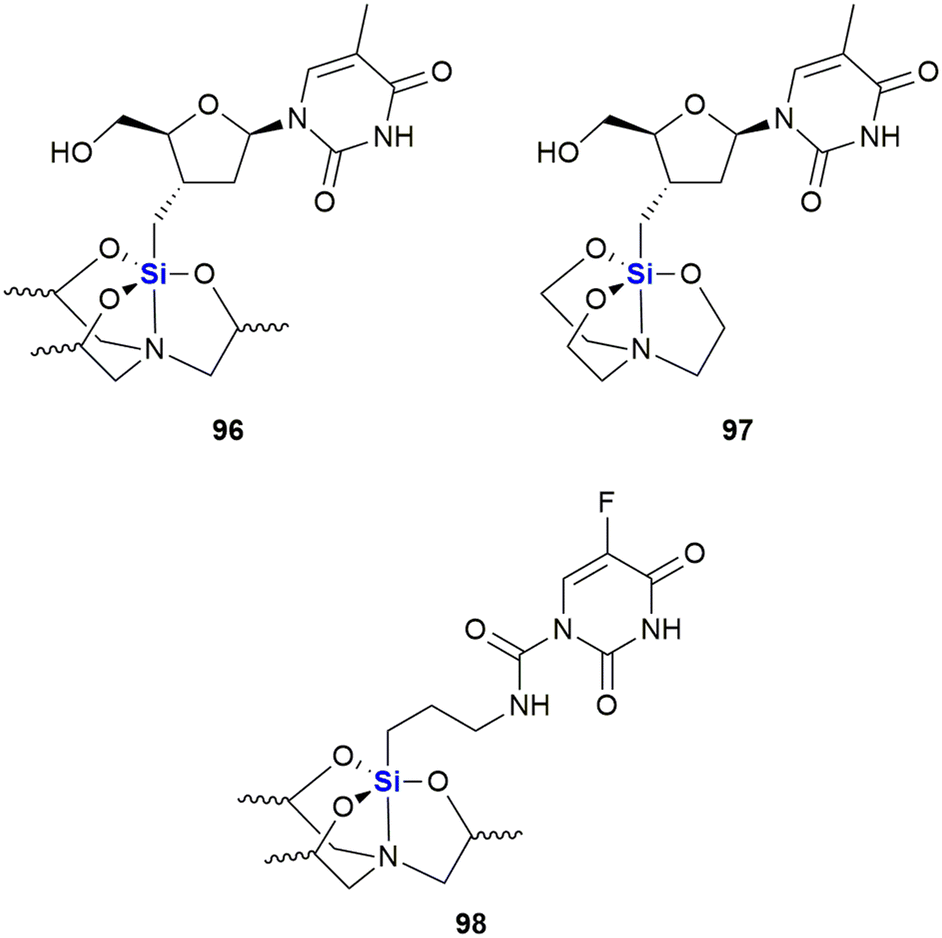 | ||
| Fig. 33 Silatrane modified thymidines which possess anti-cancer activity (note that the stereochemistry of the silatrane unit was not given in the original publication). | ||
In 2016, van Otterlo and co-workers evaluated the application of novel nucleoside derivatives to induce apoptosis and cell death in two colorectal adeno-carcinoma cell lines, namely Caco-2 and HT-29.366 As part of this research, three novel nucleosides 99–101 were prepared and shown to have cytotoxic activities against both cell lines with IC50 values ranging from 3–37 μM, with the Caco-2 cells being more sensitive to the compounds (Fig. 34). The nucleoside analogues proved to be significantly less toxic to normal, unstimulated leukocytes when compared to camptothecin 71. In addition, it was demonstrated that the induction of apoptosis was promoted by an increase of caspase 8 and caspase 3 activities. The group also reported the synthesis of novel silyl- and trityl-modified nucleosides 102–109, followed by biological screening of the compounds against several tumour lines: HL-60 (leukaemia), K562 (leukaemia), Jurkat (T-cell leukaemia), Caco-2 and HT-29. In addition, the small library of compounds was tested at varying concentrations against two human glioma lines (U373 and Hs683) to obtain GI50 values (Fig. 35).366
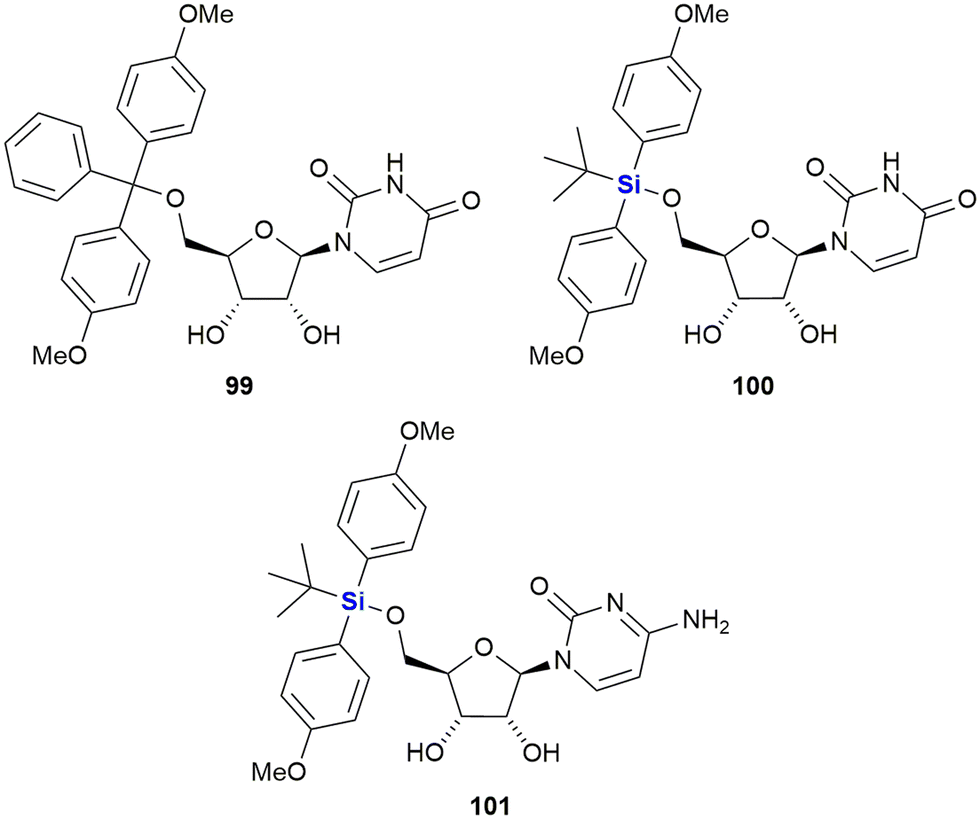 | ||
| Fig. 34 Nucleoside silyl ether derivatives displaying activity against Caco-2 and HT-29 cell lines. | ||
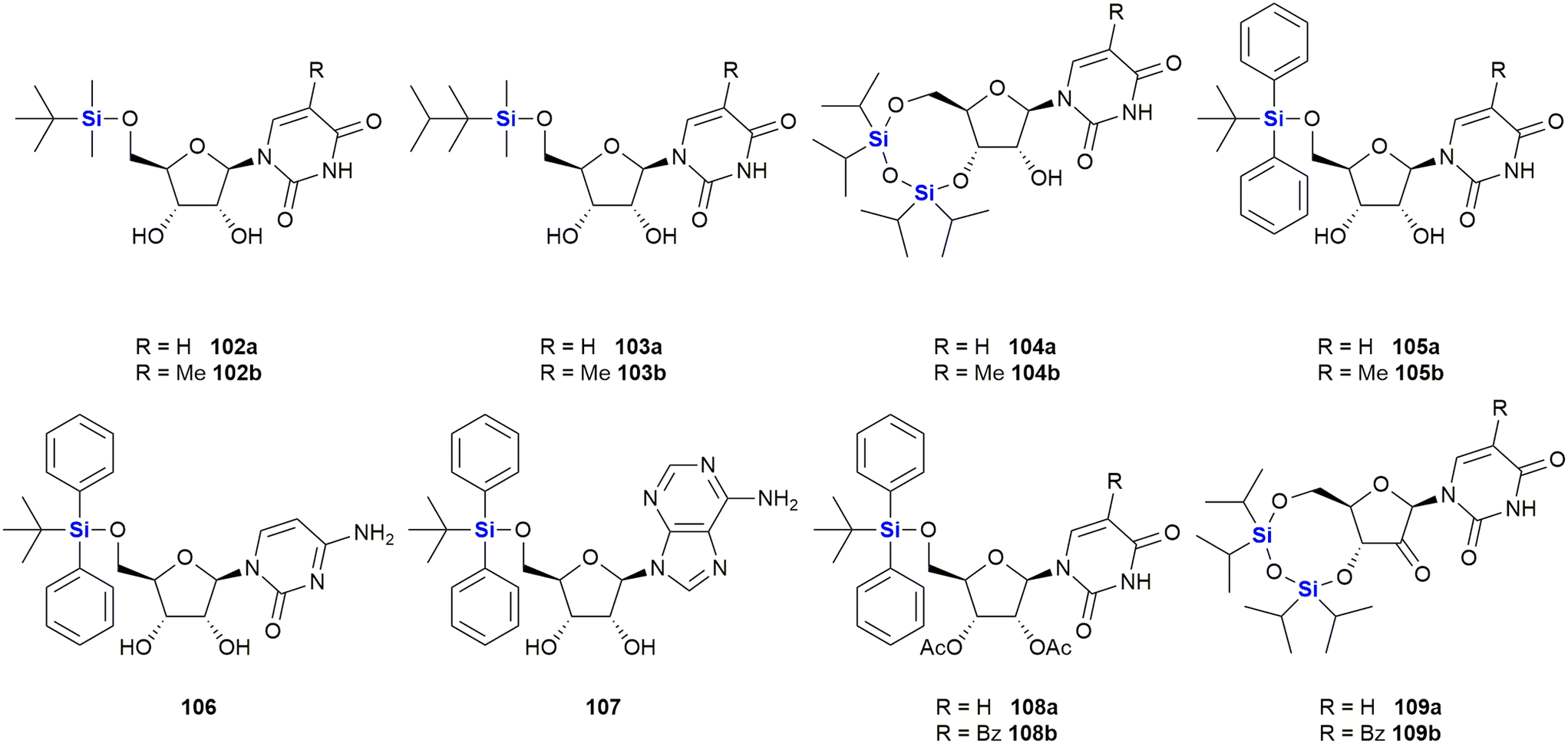 | ||
| Fig. 35 Nucleoside silyl ether derivatives displaying activity against Caco-2 and HT-29 cell lines. | ||
The effect of a selection of the compounds on the growth of cancer cell lines and white blood cells is shown in Table 2. TBDPS-containing compounds showed activity with the best being the uridine derivative 105a which demonstrated slight inhibition of the leukaemia suspension cell line (<10%), but was better against the adherent cell lines as the colorectal Caco-2 and HT-29 cell proliferation were inhibited to below 40 and 35% respectively.366 Interestingly, a non-silyl trityl-nucleoside 99 also displayed comparable activity to the TBDMPS-containing systems and acetylation of the carbohydrate rings in both the silyl and non-silyl cases resulted in a loss of activity. When cell viability was determined against freshly isolated leucocytes it was noted that all the synthetic derivatives showed reduced in vitro growth inhibition when compared to the 100 μM camptothecin control 71, suggesting that they may be less impactful on non-cancerous cells.
| Compound | Cell viability (% surviving at 100 μM as compared to control (100%)) | |||||
|---|---|---|---|---|---|---|
| HL-60 | Jurkat | K-562 | Caco-2 | HT-29 | WBC | |
| NA = not active, NI = no inhibition (cell viability >50% for cell lines, cell viability 100% for WBC). | ||||||
| 105a | 5.6 ± 0.3 | 6.3 ± 0.6 | 9.4 ± 0.1 | 37.4 ± 1.0 | 29.4 ± 0.6 | 61.5 ± 4.5 |
| 106 | 26.0 ± 0.6 | 16.0 ± 1.3 | 35.6 ± 0.9 | 37.2 ± 0.4 | 25.6 ± 0.7 | NI |
| 107 | 27.9 ± 0.5 | 18.3 ± 1.2 | 45.5 ± 0.7 | NA | NA | 59.3 ± 2.0 |
| 108b | 11.2 ± 0.5 | NA | 35.5 ± 2.4 | NA | NA | NI |
| 71 | Camptothecin | 8.9 ± 0.7 | 39.6 ± 0.9 | 12.8 ± 0.2 | 11.4 ± 0.2 | 34.6 ± 2.0 |
Finally, 105a and 107 were also shown to induce apoptosis (in combination with extensive cytoplasmic vacuole formation) in both HT-29 and Caco-2 cell lines, demonstrating comparable or better activity than the camptothecin 71 reference.367
Further investigations into the effect of silyl-decorated nucleoside derivatives on cancer cell population growth revealed 8 compounds showing GI50 values in the range of 25–40 μM. Five of these compounds contained TBDPS-groups (namely 105b, 106, 8107, 108a and 108b), one contained the TIPD-protecting group (88a) and the remainder were the non-silyl trityl derivatives (see Fig. 35).366 AlogP calculations suggested that there was a trend wherein the more lipophilic molecules showed improved GI50 values, thus suggesting that bulkier non-polar silyl groups could be improving cell membrane permeation, or could be causing activity by disrupting cell membranes.366
Cutler and co-workers also reported on the anti-proliferative activity of nucleoside derivatives in the form of N6,5′-bis-ureidoadenosine analogues (Fig. 36).368 In this research, the prepared compounds were screened against the NCI60 panel of human cancers. The presence of a 2′-OTBS group proved necessary for activity; however, acceptable anti-proliferative effects also required the presence of a N6,5′-bis-ureido group. In a follow-up manuscript, Peterson and co-workers investigated if the 3′C-carboxymethyl moiety in 110, which was synthetically challenging to install, was in fact necessary for activity.369 The studies showed that replacement of this moiety with a second-OTBS group afforded a lead compound 111 with improved potency. Notably, the new analogue afforded IC50's in the range of 6–10 μM for five of the six human colon cancer lines, with three of the renal cancer lines displaying similar activities.369 The researchers attempted thereafter to improve the activity of 111 by preparing a library of analogues wherein the TBDPS groups were replaced by other silyl or acyl groups; however, biological assessment showed no improvement over lead compound 111. Interestingly, the desilylated derivative of 111 was screened for binding affinity to the bone morphogenetic protein receptor, together with representative examples from the silyl and acyl derivatives, and it was only the desilylated compound that showed appreciable activity (Kd = 11.7 ± 0.5 μM). These results, together with computational docking studies, suggested that 111 was in fact a prodrug of the active (desilylated) derivative.370,371 Finally, modification of the 5′-bis-ureido group to give 112, afforded a compound which demonstrated potent inhibition of the NCl-H522 lung adenocarcinoma cell line at nanomolar concentrations (GI50 = 9.7 nM).372
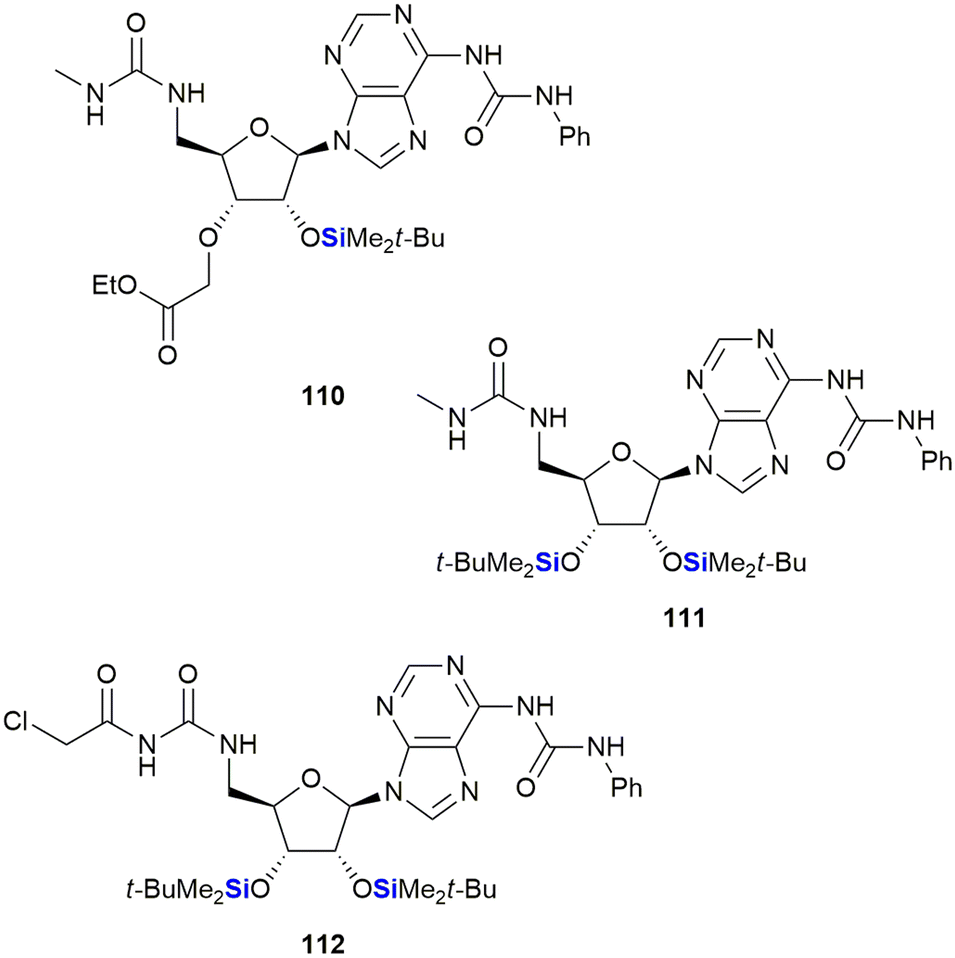 | ||
| Fig. 36 Anti-proliferative nucleoside silyl ether derivatives. | ||
Staying with the theme of silylated nucleosides, Lanver and Schmalz reported the synthesis of silyl ethers of carbocyclic nucleoside analogues (CNAs) involving a Pauson–Khand reaction. The-CNA analogues were synthesised based upon a CNA scaffold known to be apoptosis-inducing (Fig. 37).373 In a subsequent study by the same team, an enantioselective synthesis of further CNA analogues was developed and the compounds were assessed in terms of their anti-tumour activity against a Burkitt-like lymphoma cell line model. Activities for the compounds were observed to be in the low micro-molar range. Generally, TBDMS ethers were more active than TDS ethers, with the silyl ether CNA 114 being the most active compound of the whole series (LD50 = 46 μM 113, LD50 = 9 μM 114).374
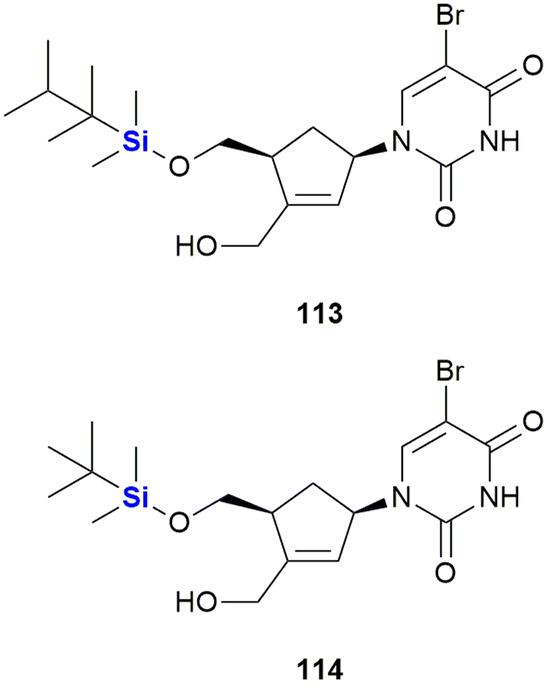 | ||
| Fig. 37 Carbocyclic nucleoside analogues shown to induce apoptosis. | ||
Schmalz and co-workers also reported the synthesis and biological evaluation of iron-containing nucleoside analogues (115–120) characterised by the presence of a 1,3-butadiene-Fe(CO)3 sub-scaffold (Fig. 38).375,376 A lead silyl-containing compound 115 was identified displaying potent apoptosis with an LD50 of 10 μM when screened against Burkitt-like lymphoma BJAB cancer cells. A further three silyl-compounds (106–108) displayed LD50 values of 18 μM (106), 30 μM (107) and 14 μM (108). In a more recent paper, silylated metal-containing nucleosides (119–120) were reported to show selective cytotoxicity against CLL leukaemia cells. Equal activity was also observed in cells from high-risk disease groups (e.g., del11q/del17p cytogenetic and fludarabine resistant cells).377
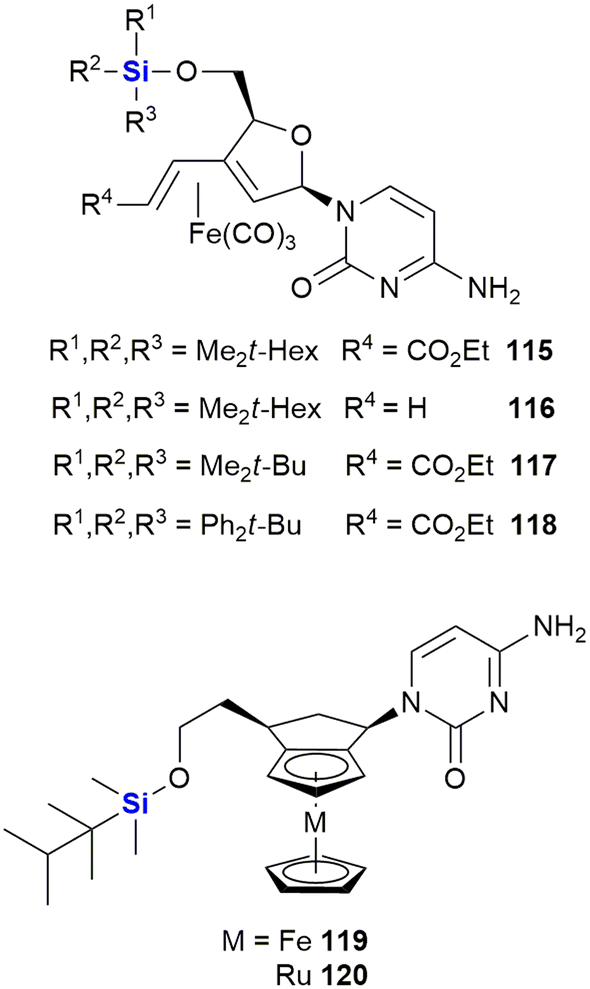 | ||
| Fig. 38 Iron-containing nucleoside analogues which show activity against BJAB and CLL cell lines. | ||
In their synthesis of C1,C2 functional group derivatives of the Amaryllidaceae alkaloid lycorine, Kornienko and co-workers demonstrated that there was a strong correlation between compound lipophilicity and anti-cancer activity. These results thus suggested that their compound design needed to take cell permeability into account (Fig. 39).378 To this end, the use of silyl groups afforded increased lipophilicity. This was showcased by the TIPS-silyl ether derivative 121 which was shown to be equipotent to lycorine when tested against A-549 (lung), MCF-7 (breast), T98-G (glioblastoma), HS-683 (human glioma), SK-MEL (melanoma) and B16 (melanoma) cancer cell lines.
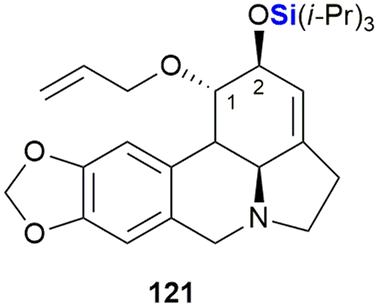 | ||
| Fig. 39 Silyl ether lycorine derivative with anti-cancer properties. | ||
Recently, Nelson et al. reported the development of novel silyl cyanocinnamic acid derivatives as metabolic plasticity inhibitors for potential anti-cancer treatment (Fig. 40).379 The researchers rationalised that insertion of a bulky, acid stable TBDPS ether on the phenolic hydroxy of the monocarboxylate transporter cyanohydroxycinnamic acid 122 would increase lipophilicity, metabolic stability and the ability of the small molecule to influence the mitochondrial function, all the while retaining the activity of 122. With this objective in mind, silylated analogues 123 and 124 were synthesized and demonstrated to show enhanced inhibition of proliferation in cancer cells through glycolysis and mitochondrial dysfunction. When applied in in vivo studies against a colorectal cancer cell WiDr (colon cancer) tumour xenograft, the silylated derivatives were also well tolerated.
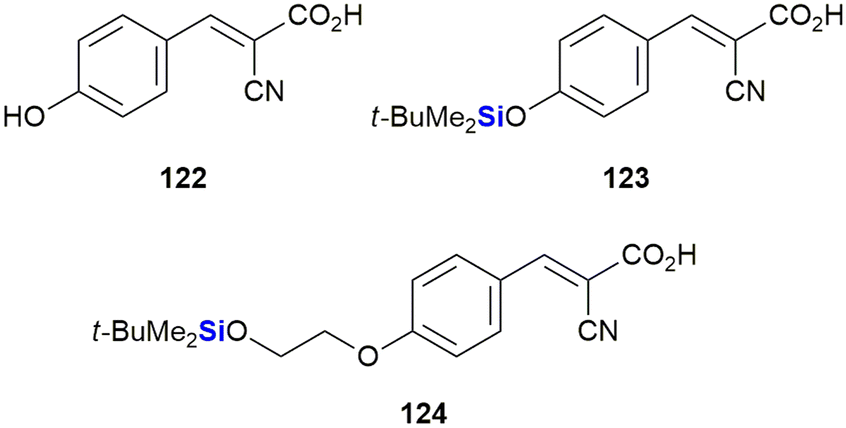 | ||
| Fig. 40 Anti-proliferative cyanohydroxycinnamic acid 122 and related sila-analogues. | ||
In 2021, Walczak and co-workers reported the outcomes of an investigation into the effects of adding silicon groups to mucobromic acid 125, a molecule containing the furan-2(5H)-one core (Fig. 41).380 In previous studies, the authors had shown that the incorporation of aliphatic groups at the C-5 position of mucobromic acid increased the compound's anti-proliferative activity and improved its selectivity towards the non-small cell lung cancer A-549 cell line.381 The researchers then investigated the effect of introducing silyl ethers at the same position, thus allowing them to control both the hydrophobicity and selectivity of the new compounds. Three of the four silyl derivatives screened showed increased activity relative to the parent compound 125, with compounds 126 and 127 showing selective anti-proliferative activity for the HCT-116 colon cancer cell line (IC50 = 1.3 and 1.6 μM respectively). Critically, the compounds were shown to be most active against colorectal cancer cell lines.
 | ||
| Fig. 41 Anti-proliferative sila-analogues of mucobromic acid. | ||
In the last decade, Bazzocchi and co-workers have explored the chemical space surrounding withaferin A 128 which has included the synthesis of silyl ether-containing withaferin A derivatives (Fig. 42). These investigations resulted in derivatives with increased cytotoxicity against HeLa (cervical carcinoma), A-549 (lung carcinoma) and MCF-7 (breast adenocarcinoma) cell lines.382–384 In an effort to improve on their initial findings, a library of thirty analogues was prepared leading to the identification of eight analogues with activities ranging from 1–32 nM when screened against human epithelial ovarian carcinoma cisplatin-sensitive and -resistant cell lines. From these experiments, silyl-containing compounds 129–131 were identified by the researchers as potential candidates for clinical translation in individuals suffering from relapsed ovarian cancer.
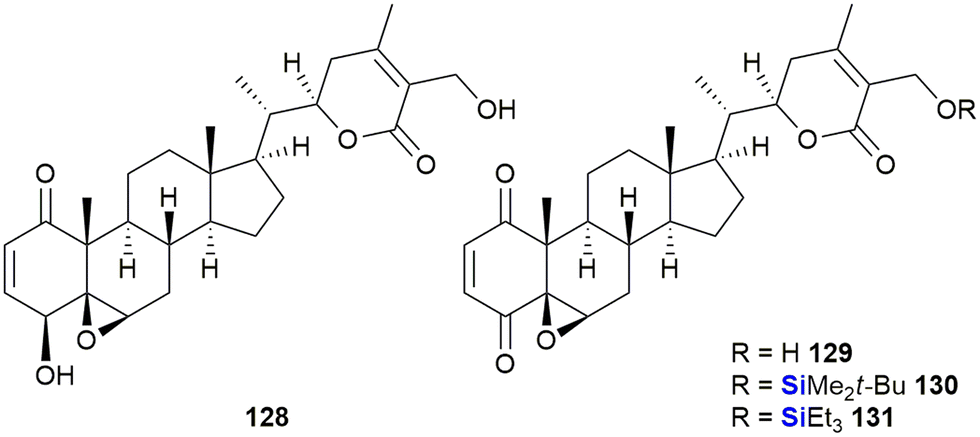 | ||
| Fig. 42 Withaferin A-based silyl ether analogues with anti-cancer properties. | ||
Zhao et al. designed dipeptides with a silicon-containing alkyl chain for the treatment of prostate cancer resistant to androgen-deprivation therapy and conventional chemotherapeutics (Fig. 43).385 The dipeptides 132 exhibited potent cytotoxicity against multiple cancer cell lines from the NCI-60 cell line panel at low micromolar or even nanomolar concentrations. They were further evaluated against resistant prostate cancer cell lines and showed an average IC50 value of 1.50 μM. The authors determined the mode of action of these compounds to be comprised of the promotion of androgen receptor AR protein degradation, its variant Ar-v7 and survivin, a protein essential for cell division and which also inhibits cell death.
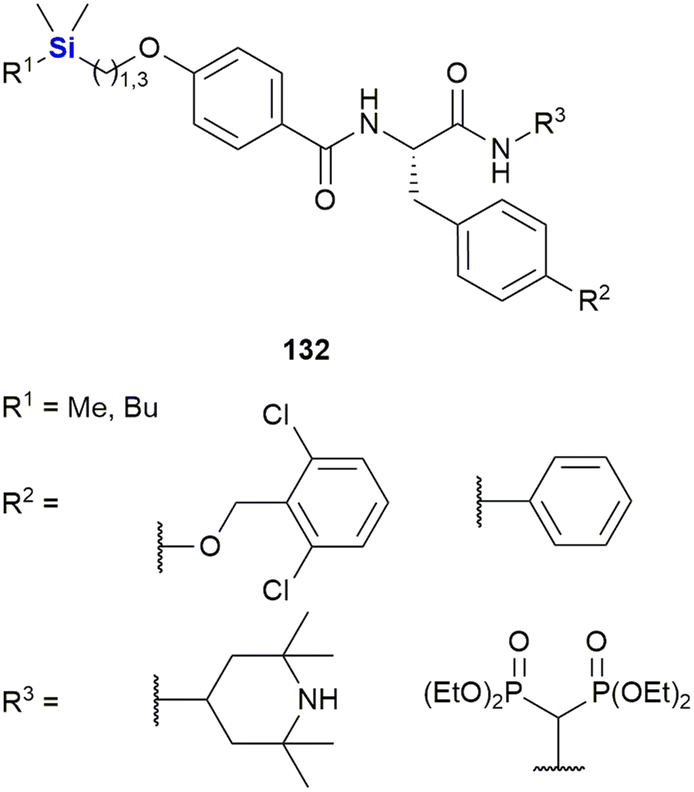 | ||
| Fig. 43 Silyl modified dipeptides against prostate cancer. | ||
Garro et al. investigated the anti-proliferative activity of the silyl ether derivatives 133 and 134 (Fig. 44) against human solid tumour cell lines.386 Experiments determined that while 134 was inactive, 133 showed moderate activity against cervix, breast and colon tumour cell lines (HeLa: GI50 24,1 μL mL−1, T-47D: GI50 26.6 μL mL−1, WiDr: GI50 32.2 μL mL−1).
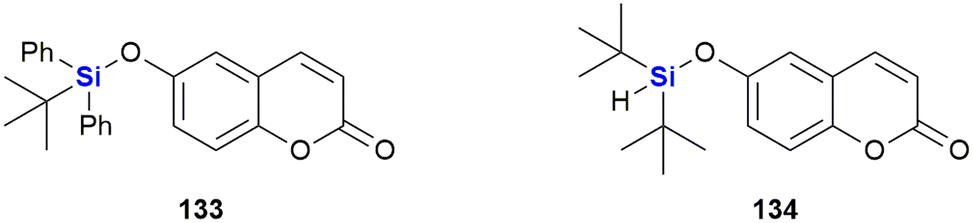 | ||
| Fig. 44 Anti-proliferative silyl ether derivatives of 6-hydroxycoumarins. | ||
While silicon-containing drugs are well investigated, reports of biologically active germanium compounds are less common. Kagechika et al. synthesised a series of para-substituted phenols with either a carbon-, silicon- or germanium-containing substituent (Fig. 45).387 While the hydrophobicity of the compounds increased significantly by the incorporation of a silicon instead of a carbon atom (clogP = 3.50 (135) and 4.12 (136)), switching from silicon to germanium did not to have a profound impact (clogP = 4.11 (137)). This effect, however, was less pronounced in the case of ethyl instead of methyl substituents. The authors also determined that these synthetic phenols promoted the proliferation of the breast cancer cell line MCF-7 by binding to the oestrogen receptor. Both the silicon (EC50 = 63 nM (136) and 3.4 nM (139)) and germanium (EC50 = 100 nM (137) and 2.1 nM (140)) compounds were significantly more active than their carbon counterparts (EC50 = 870 nM (135) and 23 nM (138)). For a computational study on Si/Ge isosterism in the fungicide flusilazole see the following ref. 388.
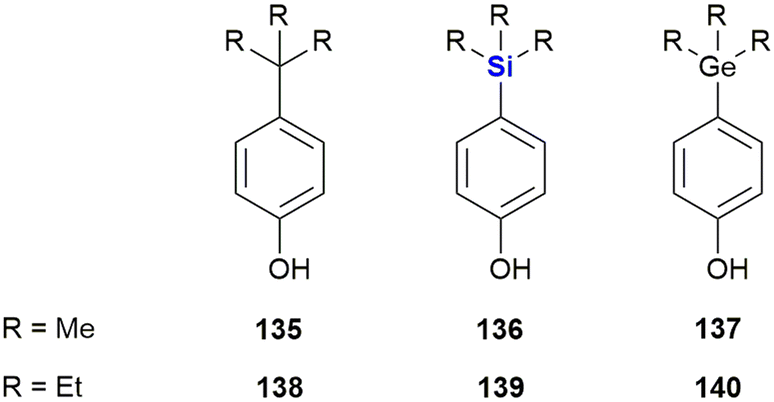 | ||
| Fig. 45 Carbon, silicon and germanium containing phenols with oestrogenic activity. | ||
Mousazadeh et al. reported the triazoles 141 and 142 with a unique combination of sulphur- and silicon-containing functional groups (Fig. 46). These compounds possessed potent cytotoxic activity against the human breast cancer cell line MCF-7 with an IC50 = 30.3 μg mL−1 for the most potent derivative, which is close to the activity of doxorubicin (IC50 = 28.89 μg mL−1). The silylalkynes 141 also showed promising activities against both Gram-positive and Gram-negative bacteria, while the disilyl derivatives 142 were only active against Gram-positive strains.389 See the following references for related synthetic work from the same group.390,391
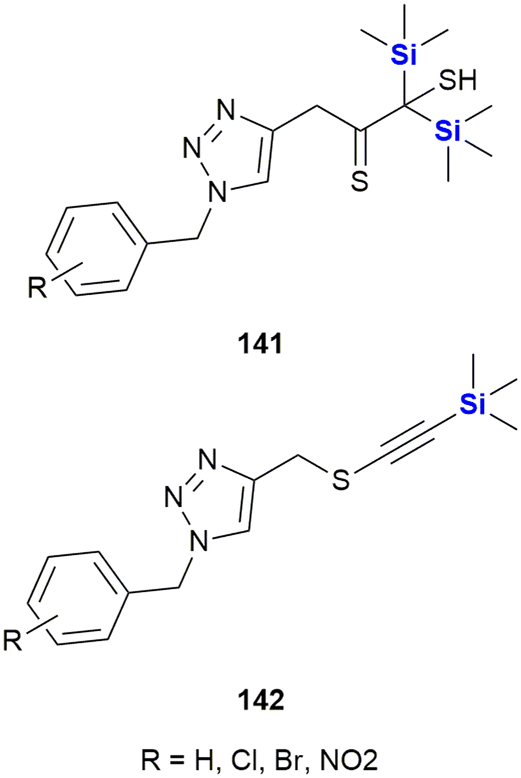 | ||
| Fig. 46 Sulphur- and silicon-containing cytotoxic triazoles. | ||
In a computational study, Eryilmaz investigated the pharmacological properties of all FDA approved drugs for the treatment of acute lymphoblastic leukaemia.392 Among these, nelarabine 143 (Fig. 47) was predicted to exhibit the most promising activity against four out of six possible target proteins. Further modelling investigations involving the nelarabine scaffold suggested that the disilylated analogue 144 might not only possess improved molecular properties, but also more potent bioactivity against even five out of six target proteins. These predictions must still be borne out by the actual synthesis and testing of the disylated nelarabine compound.
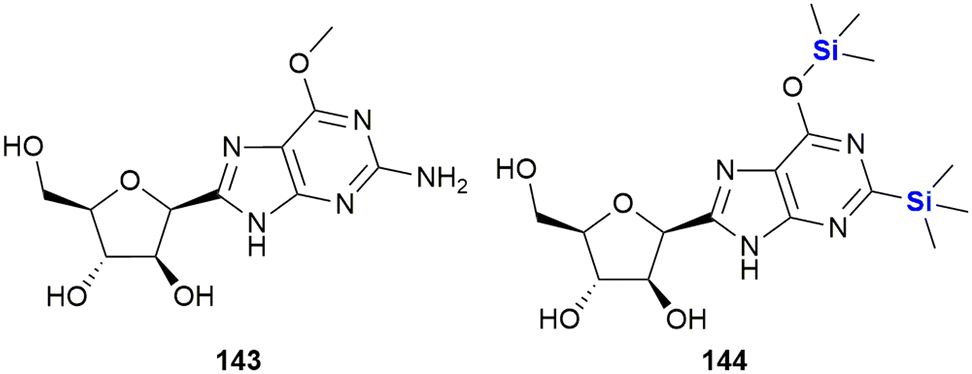 | ||
| Fig. 47 Nelarabine 143 and siliyl-containing analogue 144. | ||
The neurotensin receptor NTS1 is over-expressed in several cancer cell lines and therefore is an attractive target for tumour diagnostics and therapy.393 In 2020, Morgat, Cavelier and co-workers investigated neurotensin derivatives for tumour imaging (Fig. 48).394 In this research, the shortest functional pentapeptide fragment of neurotensin was connected to a 68Ga-labeled complex. The researchers found that the exchange of the C-terminal leucine found in natural neurotensin by the non-natural amino acid TMS-alanine shown in 145 improved binding affinity towards NTS1 (KD = 6.29 nM), increased stability in human plasma (t1/2 = 7.17–24.63 min) and lowered efflux.
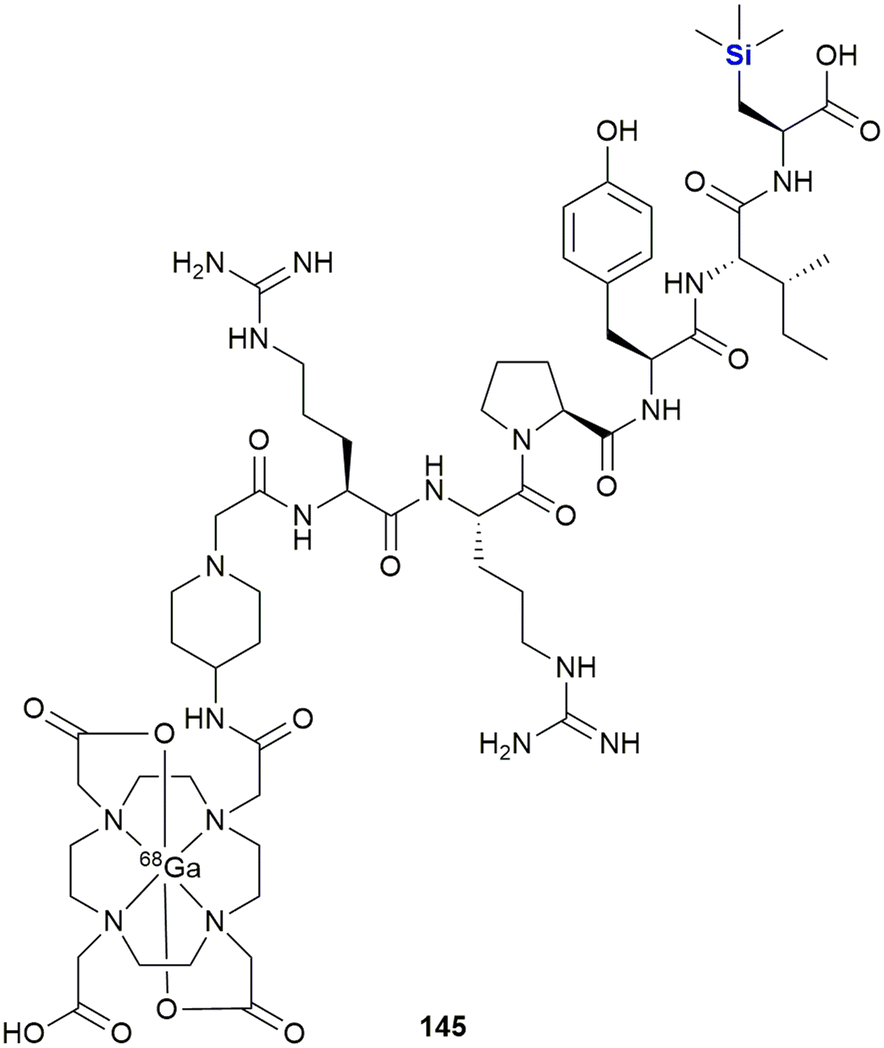 | ||
| Fig. 48 68Ga-radiolabeled silicon-containing neurotensin analogue. | ||
Vorinostat 146 (Fig. 49) is an inhibitor of Zn(II) dependent histone deacetylases (HDACs) and is approved by the FDA for the treatment of cutaneous T-cell lymphoma.395,396 Olsen et al. synthesised and investigated 147, in which the Zn(II) chelating hydroxamic acid group of 146 was exchanged for a silanediol group,397 as it was proposed that silanediols should possess a similar affinity for Zn(II) ions as hydroxamic acids.398 When tested, 147 showed inhibitory activity against 6 out of 10 tested HDACs at micromolar concentrations (IC50 = 0.6–28 μM), but was generally less active than vorinostat 146 (IC50 = 0.021–70 μM). For a patent detailing the description of Si-containing HDACS, see the following ref. 399.
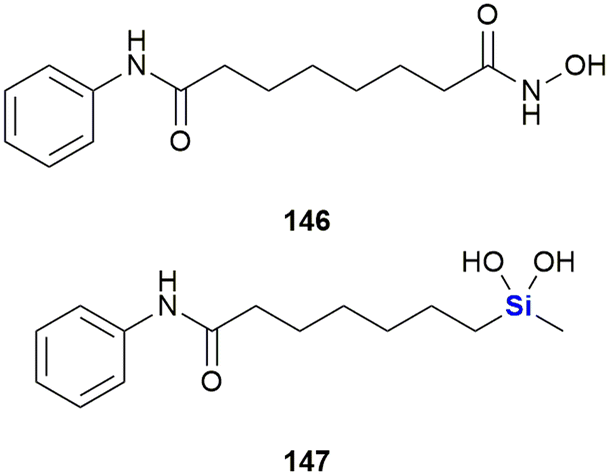 | ||
| Fig. 49 Vorinostat (146) and silanediol analogue 147. | ||
Very recently (2022), Zaltrariov et al. investigated the activity of the silatrane 148 and its imine derivative 149 (Fig. 50) against hepatocarcinoma (HepG2) and breast adenocarcinoma (MCF-7) cell lines.400 The silatrane 148, with a free amine functional group, was determined to be inactive even at a high concentration of 300 μg mL−1. Its imine derivative 149, however, showed dose dependant activity against both cancer cell lines with IC50 values of 150 μg mL−1 (HepG2) and 65 μg mL−1 (MCF-7) respectively. The cytotoxic properties were therefore linked to the aldimine moiety and not to the silatrane structure. On the other hand, the free amine group enhanced the activity of 148 against several fungal and bacterial strains, where 149 showed MICs in the range of 1.20–2.90 μg mL−1.
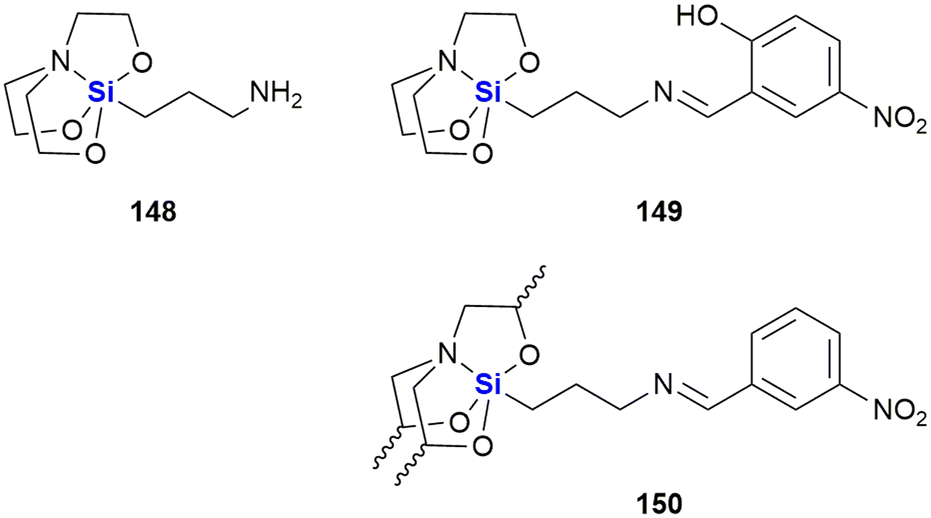 | ||
| Fig. 50 Anti-cancer silatrane containing amine 148 and its imine derivatives 149 and 150 (note that the stereochemistry of the silatrane unit was not given in the original publication). | ||
Within the broader silatrane topic, Ping et al. investigated amide, carbamate, imine and 5-fluoruracil derivatives of γ-aminopropylsilatrane. Among the tested compounds, 150 showed the highest activity and inhibited the growth of melanoma (MDA-MB-435, IC50 = 12.5 μg mL−1), cervix (HeLa, IC50 = 11.8 μg mL−1) and colon cancer cell lines (HT-29, IC50 = 12.3 μg mL−1).401
Staying with silatranes, Shi et al. reported the synthesis of a silatrane modified 5-fluoro uracil and its bioactivity (Fig. 51). The compound 151 showed some inhibition effect against colon (HCT-8, 29.33%) and liver (Bel7402, 29.26%) cancer cell lines at 5 μg mL−1.402
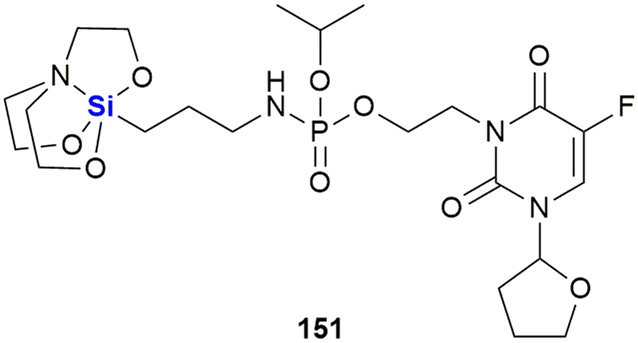 | ||
| Fig. 51 Silatrane modified 5-fluorouracil with anti-cancer properties. | ||
El-Hussieny et al. presented the synthesis of a range of silicon-containing heterocycles 152–158 by condensation of silylated isocyanates or isothiocyanates with phosphorous ylids (Fig. 52).403 The resulting compounds were tested as inhibitors of the Zn(II) dependent matrix metalloprotease MMP-2, a prospective target for the prevention of tumour growth.404 In the biochemical evaluations, the compounds showed significant activity at low concentrations (IC50 = ∼41–429 nM) with the thiolactam 157 being the most potent. Molecular docking studies revealed that 157 could be inhibiting MMP-2 by binding of the sulphur atom to the Zn(II) cofactor in the active centre.
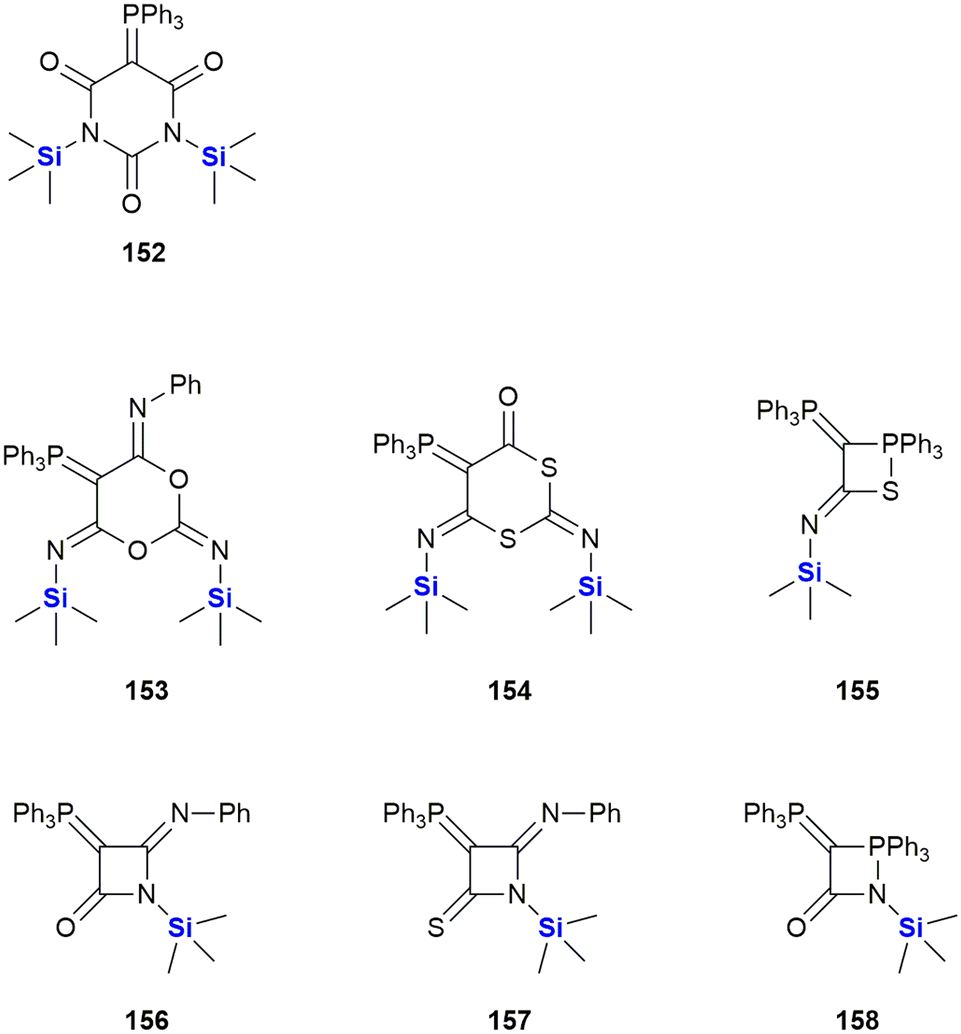 | ||
| Fig. 52 Silicon-containing MMP2 inhibitors. | ||
Anti-viral agents
Interest in the use of organosilicon regents as anti-viral agents can be traced back to the 1990's when Camarasa and co-workers reported the synthesis and testing of a new class of 3′-spiro nucleosides as HIV inhibitors (Fig. 53).405 In this particular research, a number of xylo- and ribo-nucleoside analogues (159–161) decorated with TBDMS groups were evaluated for their anti-HIV-1 activity. It was found that compounds 160a–c with a 3′-spiro moiety in the R configuration and which contained silyl groups at the C-2′ and C-5′ positions showed strong anti-HIV activity while the S-configured spirocycles 159a–c were significantly less active.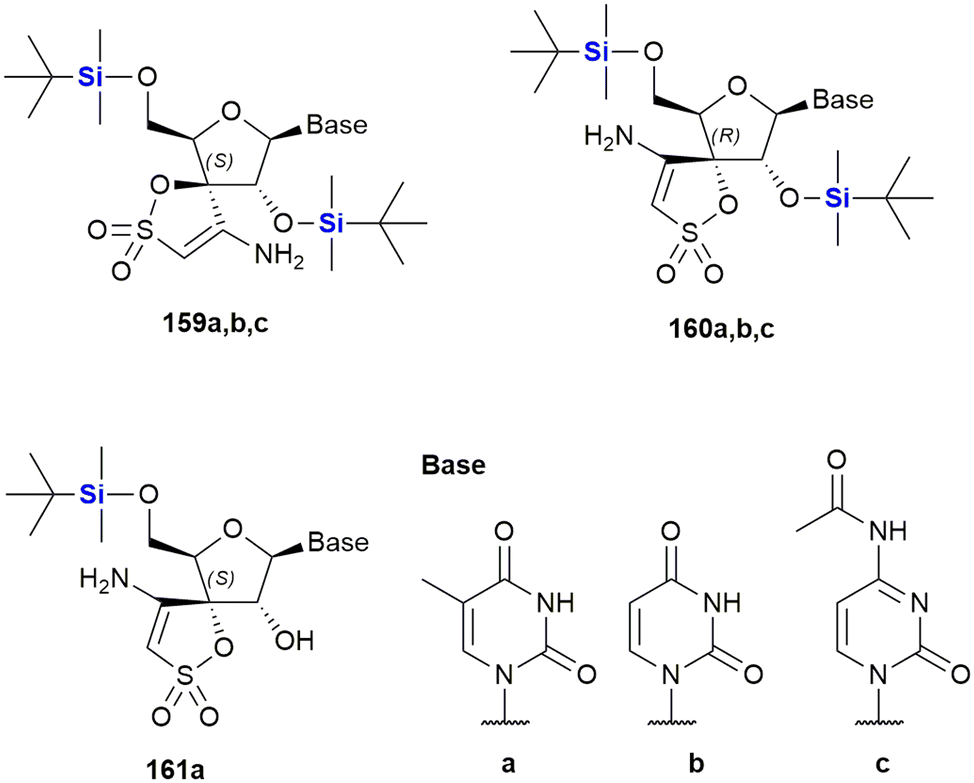 | ||
| Fig. 53 Silicon containing 3′-spiro nucleosides displaying strong anti-HIV activity. | ||
The thymine derivative (160a) showed an EC50 of 0.034 μg mL−1 against HIV-1 induced cytopathicity in MT-4 cells, whilst the analogous uracil 160b (EC50 = 0.114 μg mL−1) and 4-N-cytosine 160c (EC50 = 0.097 μg mL−1) compounds being three-fold less active. Removal of one or both silyl groups at either position, like in 161a (EC50 > 100 μg mL−1) caused a complete loss of activity, suggesting a critical role of the O-trialkylsilicon moieties. Eventually, the pharmacological profile of these molecules found them to be unfavourable for further clinical development. These compounds have however been shown to be highly useful for studying certain biological aspects of RT in HIV-1 and they have thus been used to study the importance of the p51 subunit in RT inhibition, showing non-peptide molecular interaction at the p66/p51 subunits and proving that the β7–β8 loop of p51 is required for RT dimerization.406
During the same time period, Sieburth and co-workers reported the development of silanediol peptidomimetics wherein the tetrahedral diol core 163, normally formed as result of the hydrolysis of a peptide bond 162 by a protease enzyme, was replaced with the nonhydrolyzable silanediol 164 (Fig. 54).407 These derivatives are neutral and cell-permeable and were shown to be active within an order of magnitude of the HIV protease inhibitor indinavir. In addition, for a recent manuscript disclosing approaches to late-stage site-selective modifications of activated synthetic peptides in complex structures through the application of photoredox catalysis, see the following ref. 408, and for a book chapter review covering silylated amino acids, peptides and peptidomimetics see ref. 409.
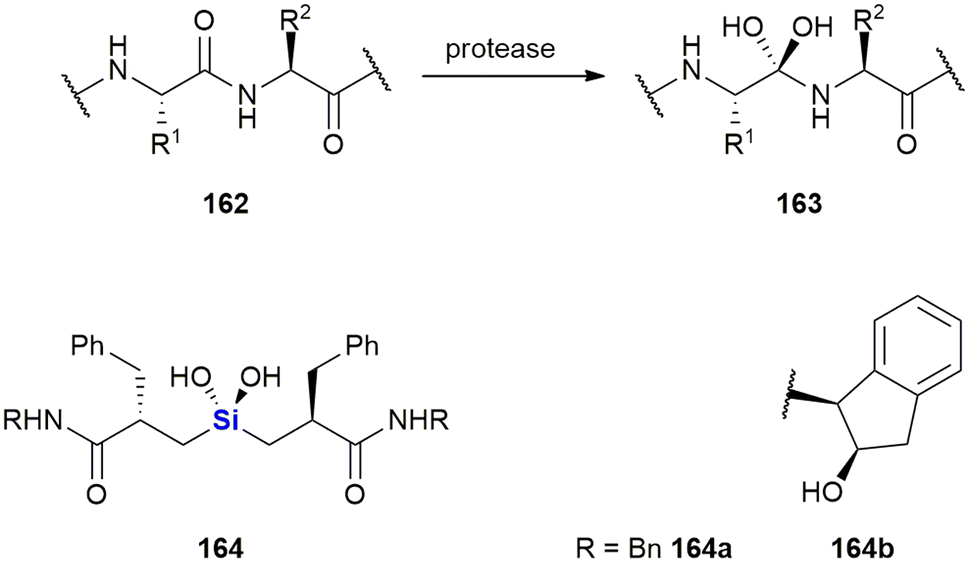 | ||
| Fig. 54 Silanediol peptidomimetics targeting HIV protease. | ||
The Déléris group synthesised novel C-alkylated heterocyclic derivatives (165–166) as leads for reverse-transcriptase-mediated anti-HIV-1 activity (Fig. 55).410 The synthesised nucleobases were designed to specifically interact with the catalytic site of HIV-1 reverse transcriptase (RT). Compound 165 was identified as being both a good inhibitor of HIV-1 replication (IC50 10–15 μM) and of recombinant HIV-1 RT (IC50 6–14 μM) with insignificant cytotoxicity. The compound was shown to be a non-competitive inhibitor selective for HIV-1 with no activity against other DNA and RNA viruses being observed. The degree of inhibition was dependent on the template primer used and the compound's selectivity for HIV-1 over HIV-2 was also established.
 | ||
| Fig. 55 Silanols as HIV-1 reverse transcriptase inhibitors. | ||
More recently, Miller and co-workers reported on the ω-functionalisation of prodrugs of the gold standard HIV nucleotide reverse transcriptase inhibitor, tenofovir 167 (Fig. 56).411 Tenovofir 167 unfortunately suffers from poor cell permeability and oral bioavailability and must be administered in a pro-drug form to be therapeutically effective. The pro-drug forms in turn are facilely and prematurely metabolized in the liver and as a result significant research has focussed on the development of derivatives with better pharmacokinetic properties. Of specific interest to this review was the phospholipid-based pro-drug TXL 168 which was developed to address this issue; however, the molecule unfortunately suffered from substantial hepatic extraction (t1/2 = 42 min). In response, the research team involved in this work, reported the synthesis of two pro-drugs, 169 and 170, which had increased half-lives of 8.66 h and 2.49 h respectively in human liver microsomes, potent anti-HIV activity (49 nM and 69 nM, respectively) and enhanced pharmacokinetic properties.
 | ||
| Fig. 56 HIV reverse transcribase inhibitor tenofovir 167 and prodrug forms 168–170. | ||
Last year (2023), Chen et al. published an investigation of HEPTe derivatives for the inhibition of HIV-1 reverse transcriptase (Fig. 57).412 These researchers exchanged the thioether linker for either a CHOH or a CHOTMS group. While generally compounds with a free alcohol showed higher activity against wild type HIV-1 than their silylated counterparts, the silyl ether 173 (EC50 = 0.20 μM) showed the second highest activity of all compounds tested and even slightly surpassed its unsilylated analogue 172 (EC50 = 0.20 μM). Compound 173 also showed moderate activity against clinically relevant mutant strains and exhibited nanomolar inhibition of HIV-1 reverse transcriptase (IC50 = 0.65 μmol L−1).
 | ||
| Fig. 57 HEPT (171) derived non-nucleosidic HIV-1 reverse transcriptase inhibitors. | ||
Ye et al. investigated a series of acyclovir (174) derivatives in which the drug was attached to a silatrane by a linker chain (Fig. 58). The silatrane unit was expected to stimulate the immune response, while the acyclovir part was supposed to inhibit virus replication. Indeed, 175 increased proliferation of splenic lymphocytes and upregulated the expression of interferone IF-γ, interleukine IL-2 and T-cell CD markers, while it maintained the anti-viral activity of acyclovir 174 against herpes simplex viruses HSV-1 and HSV-2.413 In another study, 176 showed similar immunomodulatory effects and decreased secretion of hepatitis B virus antigens HBsAg and HBeAg with IC50 = 34.5 μM and 89.8 μM, respectively.414
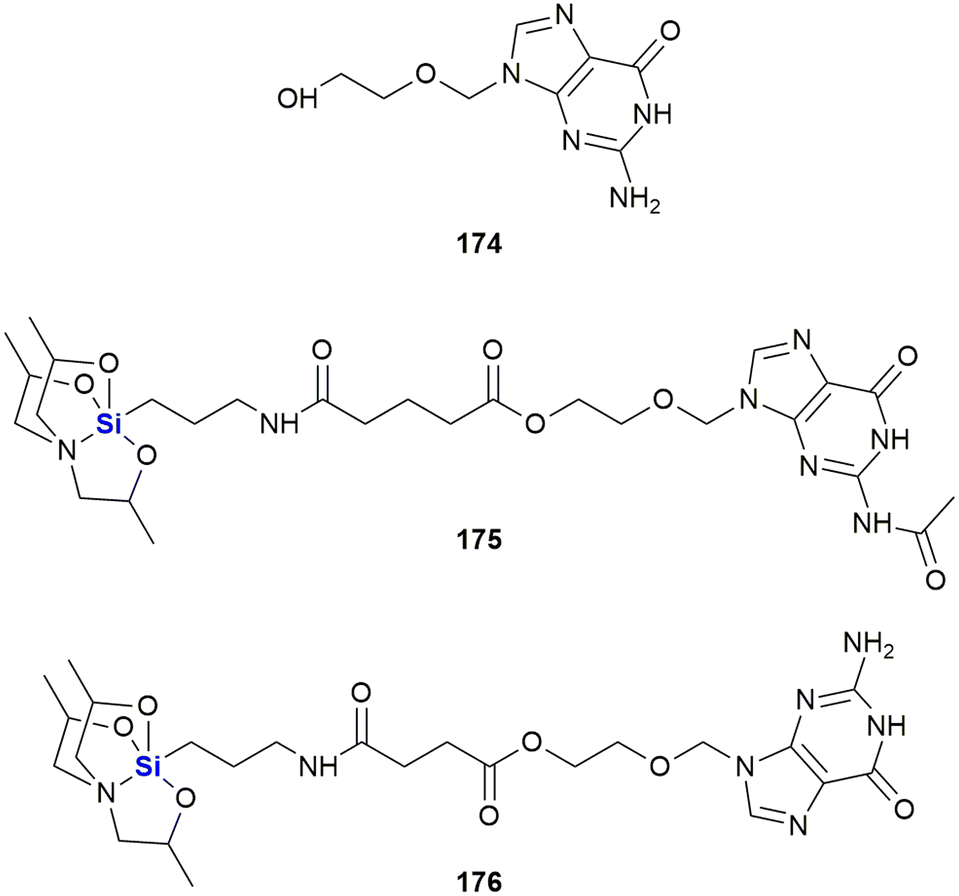 | ||
| Fig. 58 Acyclovir 174 and silatrane modified acyclovir derivatives 175 and 176 (note that the stereochemistry of the silatrane unit was not given in the original publication). | ||
In 2012, the Merck Sharpe and Dohme Corporation filed a patent detailing the preparation of silyl-containing compounds for the treatment of viral diseases, and in particular the hepatitis C virus (HCV) (Fig. 59).415–417 NS5A RCI 177, which contains a silaproline moiety,418 inhibited GT-1a and GT-1b replicons (EC50 16 and 3 pM respectively) and inhibited chimeric GT-2a (JFH), GT-3 and GT-4a (EC50 27, 350 and 20 pM respectively).
 | ||
| Fig. 59 Anti-viral silyl-containing compounds against HCV. | ||
In the related compound 178 the utility of a bridging silicon group was investigated; potent anti-viral activity was observed in the GT-1a and GT-1b (EC90 76 and 43 pM respectively). The compound was however found to be sensitive to the Y93H mutation in the GT-1a replicon with anEC50 of 811 nM.
In 2018, Zhang and co-workers reported the synthesis and biological evaluation of a series of silicon-containing HCV NS5A inhibitors based upon ombitasvir 179, a compound which displays pan-genotype activity (Fig. 60).419 The compounds were prepared by the bioisosteric replacement of a quaternary carbon with a silicon atom. The most promising analogue 180 displayed improved pan genotype NS5A inhibition with picomolar potency against GT1a (7 pM), GT1b (3 pM), GT2a (0.8 pM), GT3a (10 pM), GT4a (0.1 pM), GT5a (1 pM) and GT6a (445 pM). In all cases, the sila-analogues were more potent than ombitasvir 179. Pharmokinetic evaluations revealed that 180 had similar plasma protein binding and liver distribution patterns to ombitasvir 170, and no cytotoxicity or inhibition of hERG and CYP 450. More recently (2020), the same researchers reported the identification of compound 181 based on the same core, this time incorporating a 4-silapiperidine group in place of the triethylsilyl group. Again, this compound had excellent potency against genotype 1 subtype a, GT1a, GT1b, GT2a, GT3a, GT4a, GT5a and GT6a, with EC50 values in the range of 0.33–17 pM.420 Compound 181 also had good pharmacokinetic parameters and high liver distribution in preclinical animal models.
 | ||
| Fig. 60 Ombitasvir 179 and sila-analogues 180 and 181. | ||
Wang and co-workers prepared 5′-silyated 3′-1,2,3,3-triazolyl thymidine analogues 182 and 183 which act as inhibitors of the tropical diseases West Nile virus (WNV) and Dengue virus (DENV) (Fig. 61).421 5′-Silylated analogues of 3′aziothymidine (AZT) were prepared which were able to selectively inhibit WNV and DENV. SAR studies indicated that this activity was heavily dependent on the 5′-silyl group and the 3′-triazole. The activity appeared highly selective, with compound 182 showing potent anti-HIV-1 activity (EC50 = 67 nM) with no appreciable WNV or DENV activity (7.4% and 0% inhibition respectively at 10 μM concentration). In contrast, the TBDMS ether analogue 183 showed potent WNV and DENV activity (97% and 85% inhibition respectively at 10 μM concentration), but insignificant anti-HIV-1 activity. Assessment of the SAR studies performed indicated that large bulky silyl groups (e.g., TBS, TIPS and TBDMS) all inhibited both WNV and DENV with 85–100% inhibition at 10 μM; conversely, small non-silyl groups, such as acetyl, methyl, ethoxymethyl and tetrahydropyran, elicited poor activity. When large non-silyl groups were used (such as DMTr), only moderate activity was observed, with 23% and 46% inhibition at 10 μM being measured for WNV and DENV respectively. The SAR also highlighted that in addition to a bulky silylated substituent at the 5′ position, the triazole at the 3′ position needed a bulky aromatic ring system(s), although bulky alkyl groups also conferred some activity. Within the libraries of silylated analogues there was also some selectivity observed between the viruses WNV and DENV.
 | ||
| Fig. 61 Anti-viral silyated thymidine analogues against West Nile virus (WNV) and Dengue virus (DENV). | ||
Wang and co-workers further reported on the development of organosilanes as potent anti-virals targeting multi-drug resistant influenza-A viruses (Fig. 62).422,423 The research was conducted in response to the growing resistance to the only oral FDA-approved treatment oseltamivir (Tamiflu) and targeted the AM2-S31N mutant proton channel present in 95% of the current circulating influenza virus. In this work, organosilanes 184–185 were designed as potent channel blockers and silane 185 showed improved activity in inhibiting the A/WSN/33 (H1N1) virus over the related carbon analogue (EC50 0.4 μM vs. 2.5 μM). The compound was also highly active against oseltamivir-sensitive (EC50 0.6–1.2 μM) and -resistant (EC50 0.6–3.0 μM) influenza A viruses.
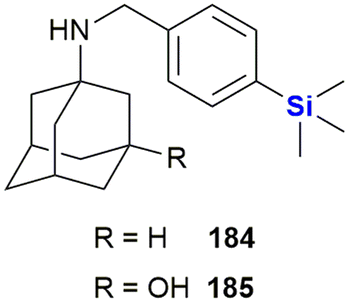 | ||
| Fig. 62 Organosilanes against multi-drug resistant influenza-A. | ||
Anti-bacterial agents
In 2006 (ref. 424) and 2007,425 Kim, Baney and co-workers published articles outlining research involving the possible application of trialkylsilanols (186–194) as a novel class of anti-microbials. These research outputs described the discovery of an unexpected class of powerful biocides based on fairly straightforward-to-synthesize silanols.424 Anti-microbial screens with the synthetic silanols, tert-butanol and siloxanes were subsequently conducted against Gram-negative (Escherichia coli) and Gram-positive (Staphylococcus aureus) bacteria. The researchers found that when the silanol treatments were used, the number of viable bacteria were reduced by more than eight orders of magnitude when compared to the application of the corresponding alcohols. Of note was that in particular, triethylsilanol 187 exhibited a potent biocidal effect at a very low concentration and within ten minutes of application. A subsequent study of the SAR for these silanols revealed that the increased O–H bond acidity, as well as the increased lipophilicity, were the primary factors governing the anti-bacterial properties of the compounds.426 Examples of the compounds tested are provided in Fig. 63.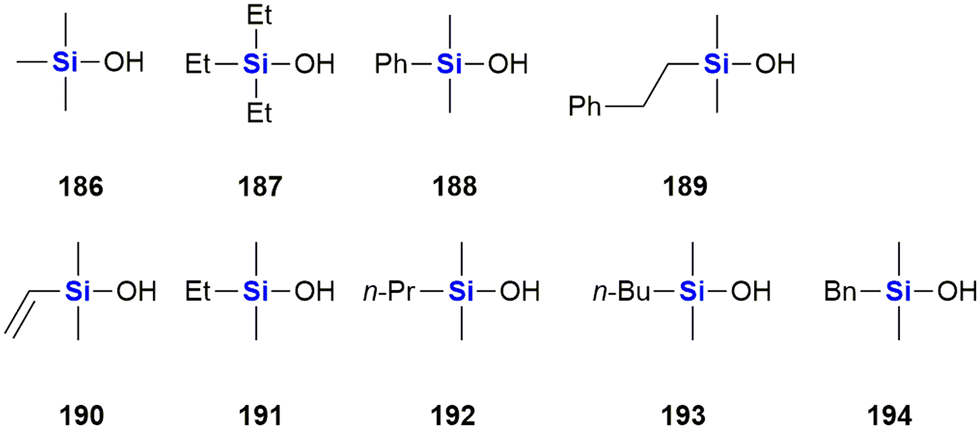 | ||
| Fig. 63 Representative examples of silanols showing anti-bacterial activity. | ||
In 2014, Reddy and co-workers patented a number of silicon analogues of piperine 195, an alkaloid originally isolated from black pepper as this compound possessed respectable anti-tubercular activity (Fig. 64).427,428 Two of the compounds developed, 196 and 197, displayed MIC values of 50 μg L−1 when screened against Mycobacterium tuberculosis (H37Rv). In comparison, piperine 195 showed no observable activity under the same conditions.
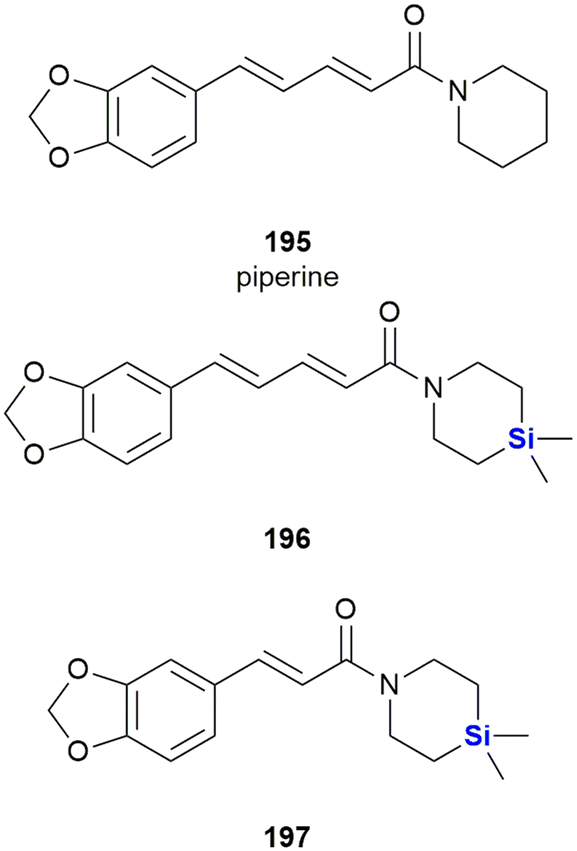 | ||
| Fig. 64 Silicon analogues of a piperine alkaloid originally isolated from black pepper possessing anti-tubercular activity. | ||
The researchers further patented silicon analogues of the diarylpyrrole compound BM212 198.428 Compounds 199 and 200 showed improved activity when screened against H37Rv, with MIC values ranging from 0.2–1.5 μM in the case of scaffold 199, while 198 showed a MIC of 1.7 μM. Interestingly, when the position of the silyl containing group was moved to the adjacent position on the pyrrole ring system, as in compound 199 (MIC = 3.6–23.1 μM), a significant decrease in activity was noted (Fig. 65). More recently, the mechanism of action of these compounds was investigated by docking studies.429 The authors suggest that diarylpyrroles 199 bind to mycolic acid transporter MmpL3. As hydrophobic interactions between the ligand and MmpL3 are dominant, it is easily understood why the incorporation of silicon increases the binding affinity compared to the parent compounds 198. The position of the amino group is crucial for the interaction with Asp256 and Tyr257, thus explaining the lower affinity of arylpyrroles 200.
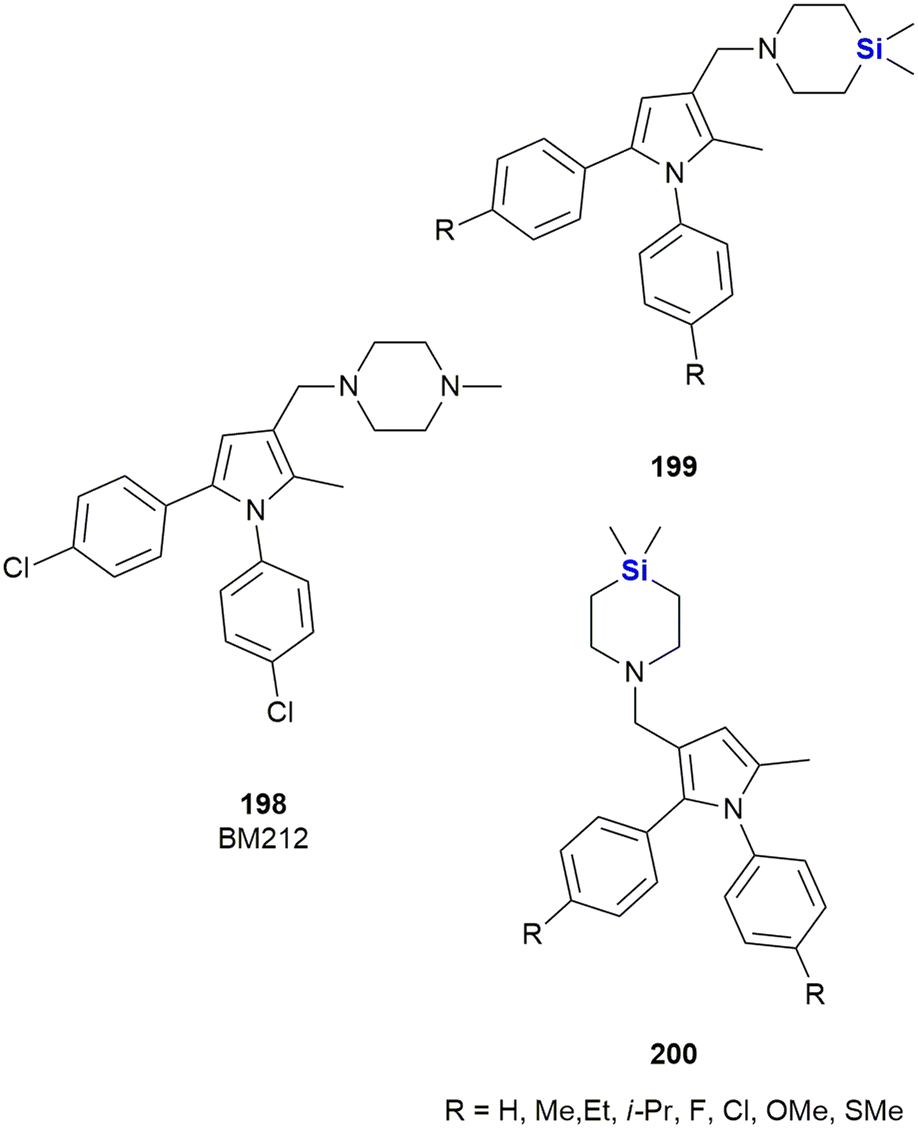 | ||
| Fig. 65 Anti-tubercular silicon containing pyrroles. | ||
More recently (2016), the Reddy group has reported the identification of further novel sila analogues of rimonabant 201 as anti-tubercular agents. The most potent of these was compound 202, which displayed an MIC of 31 ng mL−1 when screened against M. tuberculosis (H37Rv).430 The researchers found that inclusion of the silicon atom showed greatly improved potency over the non-sila derivatives (Fig. 66).
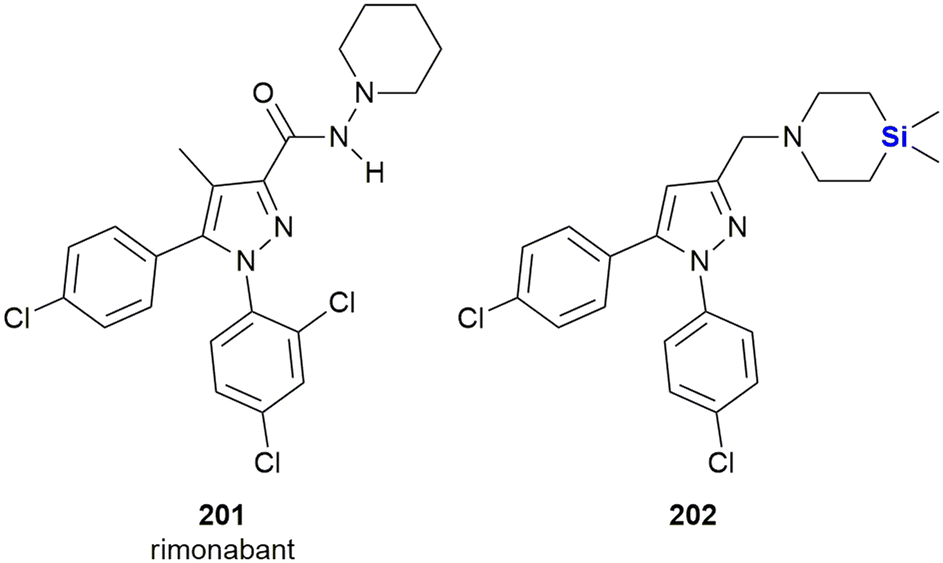 | ||
| Fig. 66 Rimobant 201 and sila-analogue 202. | ||
The potency of the disiloxan 30 (Fig. 13) to inhibit efflux pumps in cancer cells (vide supra) raised attention to its use in the treatment of multidrug resistant tuberculosis. Initial investigations by Martins et al. showed that compound 30 (but not the related compound SILA 409 29, Fig. 13) enhanced the intracellular killing activity of human macrophages infected with extensively drug-resistant tuberculosis XDR-TB and possessed in vitro activity against XDR-TB (MIC = 3.125 mg L−1).43130 thus transformed non-killing macrophages into killers of phagocytosed bacteria with no observable cytotoxicity. Furthermore, Simons et al. showed that 30 possessed inhibitory activity against 21 clinical Mycobacterium tuberculosis isolates,432 some of them drug and multidrug resistant, with MIC50 values in the range of 2–16 mg L−1.431,433 Later, Knegt et al. demonstrated in vitro that 30 significantly enhanced the susceptibility of M. tuberculosis against isoniazid and especially against rifampicin. While the therapy of tuberculosis-infected mice with a combination of 30 and isoniazid, rifampicin and pyrazinamide proved to be beneficial in the short term, it did not prevent relapse of TB after treatment was curtailed after 13 weeks.432
The development of neurologically active agents is often hampered by poor brain penetration across the BBB, Reddy and co-workers reported that silicon analogues of the marketed oxazolidinone broad spectrum anti-biotic linezolid afforded dramatic improvements in the ability to cross the BBB, brain/plasma ratio and brain exposure (Fig. 67).434 As part of their investigations, the researchers replaced the morpholine or thiomorpholine groups of linezolid 203 or sutezolid 204 (phase II clinical candidate for treating TB) giving silicon analogue 203, a compound for which solubility clogP increased from 0.17 (203) and 1.00 (204) to 4.15 (205). Silicon analogue 205 also demonstrated up to a 30-fold improvement in the brain/plasma ratio when compared to linezolid 203. However, anti-bacterial activity against several bacteria, including Streptococcus pneumoniae (4 to 16 μg L−1) and Neisseria meningitidis (32 μg L−1) was less than that of linezolid 203 which had anti-bacterial activities of 0.5 to 8 μg L−1. This would indicate that although there was increased ability of 205 to cross the blood–brain barrier, the incorporation of the silicon had an adverse effect on the ability of the compound to induce anti-bacterial activity.
 | ||
| Fig. 67 Oxazolidinone anti-biotics linezolid 203 and sutezolid 204 and silicon analogue 205. | ||
The choline and colamine derivatives 18 and 19 developed by the Zablotskaya group shown previously in this review (Fig. 9) also displayed good anti-bacterial activity when screened against two Gram-positive (Bacillus cereus ATCC 11778 and Staphylococcus aureus ATCC 25923) and two Gram-negative (Proteus mirabilis NCIM 2241 and Escherichia coli ATCC 25922) bacteria using the agar dish diffusion method.281 The results showed that the non-silylated parent compound showed no activity, whereas the silylated analogues displayed comparable or improved activities when compared to the standard anti-microbials, gentamicin and piperacillin. Furthermore, O-decyl substituted derivatives of tetrahydroisoquinoline and thiazolium organosilicon salts (Fig. 9) have been reported by the same group. These, in addition to having good anti-tumour activity, displayed good anti-bacterial and anti-fungal properties, suggesting their potential as monotherapeutic agents for the treatment of bacterial and/or fungal infections in cancer patients.
In 2023, Bishai, Yu and co-workers, investigated triazole derivatives as inhibitors of mycobacterial membrane protein large 3 (MmpL3) for the treatment of Mycobacterium tuberculosis.435 The silicon-containing 206 (Fig. 68) emerged as the most promising drug candidate from 37 compounds synthesized and showed >90% inhibition at concentrations as low as 0.03125 μg mL−1, while showing negligible cytotoxicity. Furthermore, compound 206 was eight times more potent than its unsilylated analogue 207, an observation which the researchers explained was probably due to increased hydrophobic interactions of 206 with the target protein.
 | ||
| Fig. 68 Anti-tuberculosis 1,2,4-triazoles with (206) and without (207) silicon. | ||
In 2022, Cebrián, Kuipers, Morales et al. investigated modifications of the well-known natural product resveratrol by converting this molecule into its respective silyl ethers (Fig. 69).436 In terms of activity, the resveratrol silyl ethers 208 showed inhibitory activity against several strains of Gram-positive bacteria at micromolar concentrations (MIC 4–128 μM). Their mode of action involved membrane permeability and ionophore-related activities. While some of the silyl ethers were also quite cytotoxic, with IC50 values against MRC5 cells of up to 1.8 μM, the silyl ether 209 showed low cytotoxicity, all the while maintaining high bactericidal activity. Furthermore, some synergistic effects with traditional antibiotics were observed. In 2022, Choudary and co-workers reported the design of novel zinc ionophores 210 (Fig. 70).437 The researchers determined that introduction of a lipophilic silyl group ortho to the phenol shielded the hydrophilic part of the complex and thus increased cellular membrane permeability.
 | ||
| Fig. 69 Anti-bacterial silyl ether derivatives of resveratrol. | ||
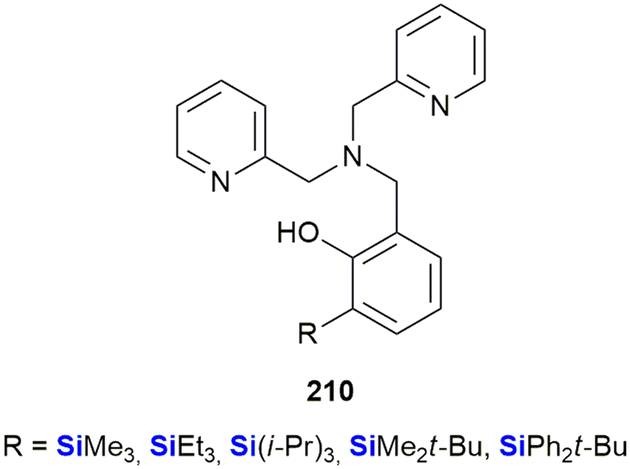 | ||
| Fig. 70 Silylated zinc ionophores with anti-bacterial properties. | ||
The silyl group also lowered the binding affinity to Zn(II) ions which improved the compounds' ability to release Zn(II) ions after transporting them across a cell membrane. In bioactivity assays, the silylated zinc ionophores 210 showed anti-bacterial activity against various Gram-positive bacteria strains, for example Straphylococus epidermidis, showed growth inhibition at concentrations as low as 1.2–8.4 μg mL−1. In terms of other characteristics, while commercial pyrithione showed comparable activity, the silylated zinc ionophores 210 showed much lower cytotoxicity against human cells.
Very recently (2023), the silicon-containing triazoles 211 and 212 (Fig. 71) were evaluated for their anti-bacterial properties.438 In this research, it was demonstrated by Singh et al. that the triazoles showed weak activity against Escherichia coli and Pseudomonas aeruginosa (211: 6.3 mg mL−1; 212: 15 mg L−1), yet moderate activity against Straphylococcus aureus (211: 0.195 mg mL−1; 212: 1.87 mg mL−1). Finally, 211 was indicated to be a potential inhibitor of the DNA gyrase from S. aureus by molecular docking predictions.
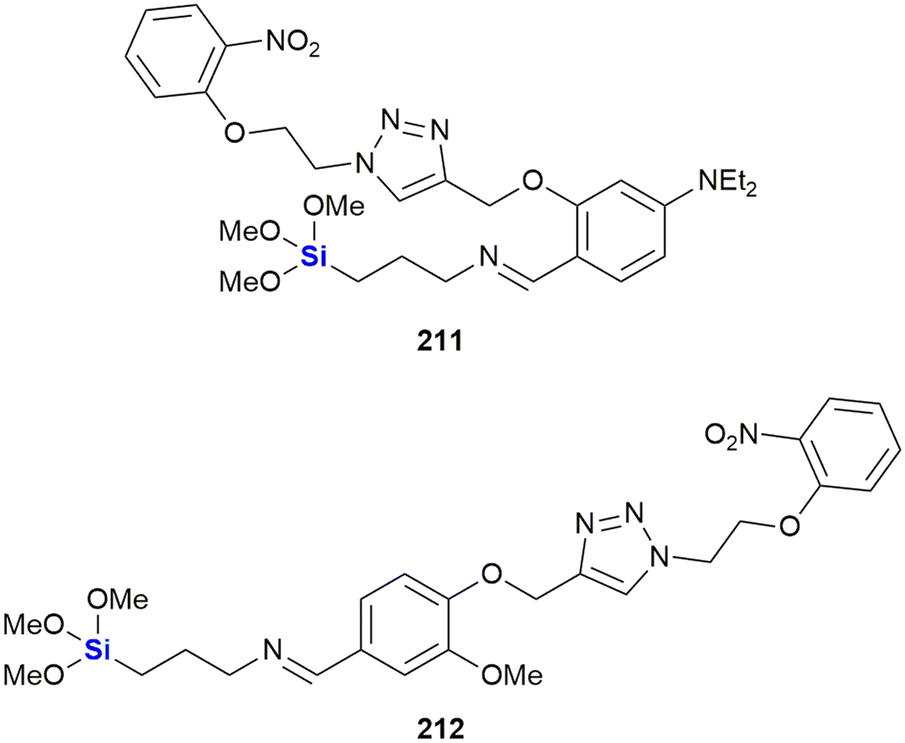 | ||
| Fig. 71 Anti-bacterial silicon-containing 1,2,3-trizoles. | ||
Dhingra et al. studied the anti-bacterial properties of silicon complexes with imine ligands (Fig. 72).439 While both complexes 213 and 214 inhibited the growth of several Gram-positive and Gram-negative bacteria strains, 214 was generally more active and even surpassed the activity of streptomycin against Staphylococcus aureus (MIC 1.56 μL mL−1). The researchers also determined that complex 214 possessed anti-fungal activity comparable to fluconazole. In another study, Singh et al. investigated the anti-bacterial effects of silicon complexes with amino acid-derived imine ligands. The zone of inhibition against Gram-positive and Gram-negative bacteria were measured and quantitative SARs were investigated. Generally, complexes 215 were more active than 216 and the serine derived ligands were more potent than ligands derived from other amino acids.440
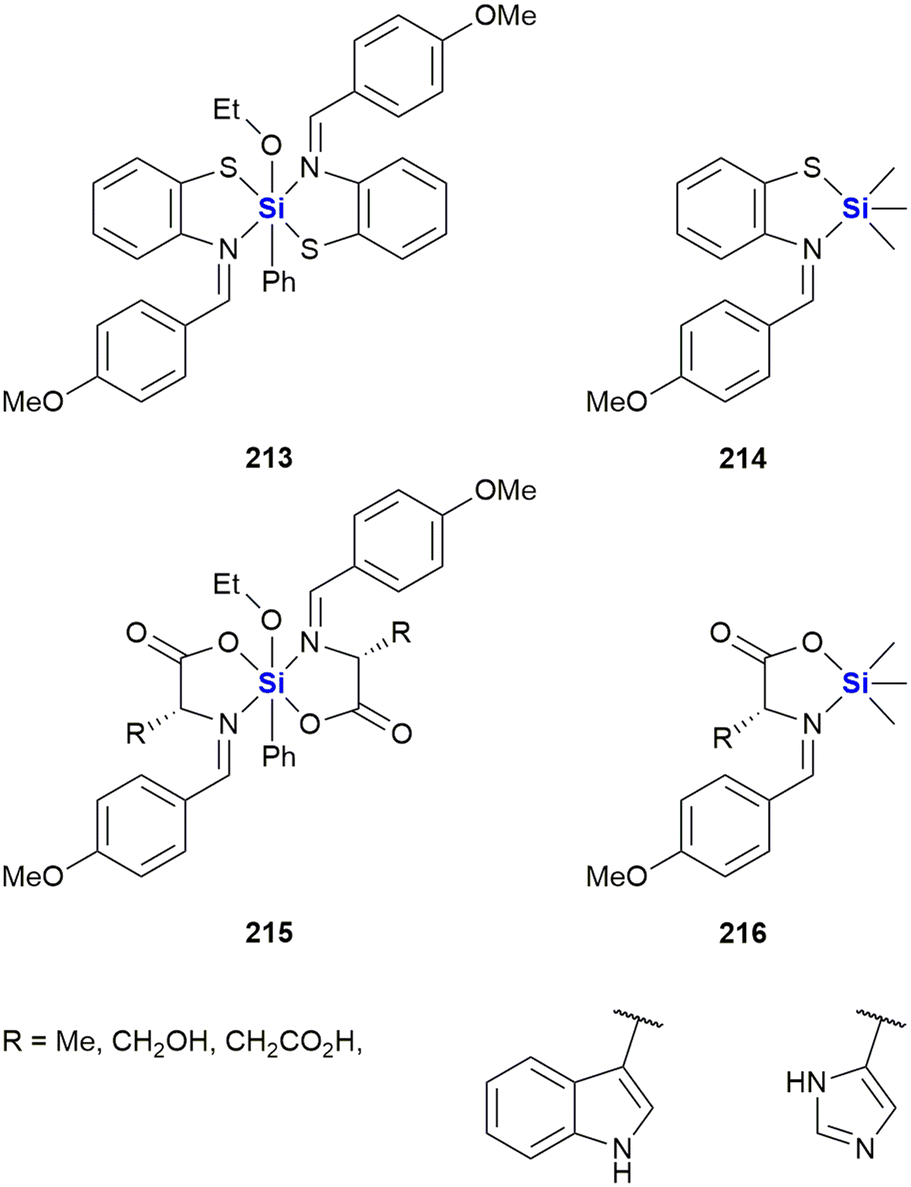 | ||
| Fig. 72 Anti-bacterial silicon complexes with imine ligands. | ||
Adamovich and coworkers reported the synthesis and anti-bacterial properties of silatrane pyrrole-2-carboxamide hybrids which were linked by an amide bond (Fig. 73). Generally, these compounds were more active against Gram-positive bacteria than Gram-negative bacteria. Compound 217 showed high activity against Gram-positive Enterococcus durans (MIC = 3.1 μg mL−1) and Bacillus subtilis (MIC = 6.2 μg mL−1), whereas 218 showed the highest activity against Gram-negative Escherichia coli (MIC = 62.5 μg mL−1).441
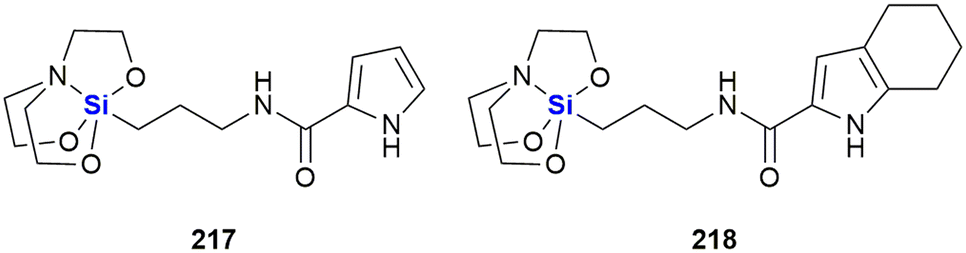 | ||
| Fig. 73 Anti-bacterial silatrane pyrrole-2-carboxamide hybrids. | ||
Adamovich and co-workers furthermore investigated the anti-bacterial properties of a series of isoxazole derivatives of γ-aminopropylsilatranes (Fig. 74). Among these compounds, 219 showed high activity against Enterococcus durans (MIC = 12.5 μg mL−1) and Bacillus subtilis (MIC = 6.2 μg mL−1).442
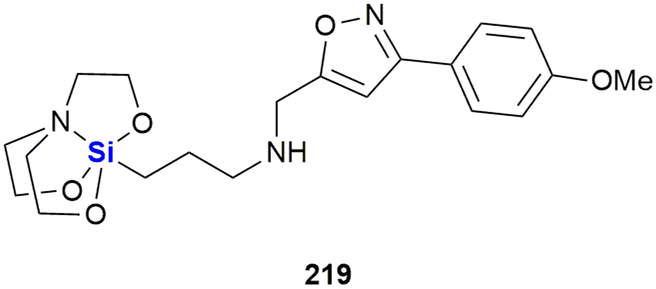 | ||
| Fig. 74 Anti-bacterial isooxazol derivatives of γ-aminopropylsilatrane. | ||
Singh et al. reported the synthesis of silatrane dithiocarbamate 220 and investigated its anti-bacterial properties (Fig. 75). Compound 220 weakly inhibited the growth of Bacillus subtillus (MIC 0.8–3.94 mg mL−1) and Escherichia coli (MIC = 0.765–3.62 mg mL−1).443
 | ||
| Fig. 75 Anti-bacterial silatrane dithiocarbamate 220. | ||
In 2018, Singh et al. connected a series of imines to a silatrane group by forming a triazole linker using click-chemistry (Fig. 76). Compound 221 showed the most potent anti-bacterial activity and inhibited the growth of both Gram-negative (MIC = 13.25–26.5 μg mL−1) and Gram-positive (MIC = 3.25 μg mL−1) bacteria.444
 | ||
| Fig. 76 Anti-bacterial silatrane-containing imines. | ||
Singh et al. also investigated the anti-bacterial properties of silatrane-containing phthalimides (Fig. 77). Compounds 222 and 223 showed similar but moderate activity against Staphylococcus aureus, Acinetobacter baumannii, Pseudomonas aeruginosa and Escherichia coli with MICs in the range of 0.20–0.25 mg mL−1.445 For the anti-cancer activity of structurally related amino derivatives of trimethylsilyl-containing 4-azatricyclo-[5.2.1.02,6]dec-8-ene-3,5-diones see the research of Napiórkowska et al.446
 | ||
| Fig. 77 Anti-bacterial silatrane containing phthalimides (note that the stereochemistry of the silatrane unit was not given in the original publication). | ||
In another study, Singh et al. synthesised derivatives of 5-halo uracils (224–226) and thymine (227) containing two silatrane moieties (Fig. 78). These compounds were tested against Escherichia coli, Bacillus subtillus, Staphylococcus aureus and Vibrio cholera using a microbroth dilution assay and MIC50 values were in the range of 0.18–0.33 mg mL−1.447
 | ||
| Fig. 78 Anti-bacterial silatrane containing thymine and 5-halo uracil derivatives. | ||
In 2023, Adamovich et al. reported the Michael addition of 3-aminopropyl-silatrane to several unsaturated nitriles, esters and amides yielding the silatranes 228–230 (Fig. 79).448 While these compounds inhibited the growth of several Gram-positive and Gram-negative bacterial strains at high concentrations of >100 μg mL−1, they stimulated bacterial growth at lower concentrations of <12 μg mL−1. Further in silico investigations predicted anti-neoplastic and macrophage colony stimulating activity of these silatranes.
 | ||
| Fig. 79 Bioactive Michael adducts of 3-aminopropylsilatranes. | ||
Baghershiroudi et al. reported the synthesis of the sulphur- and silyl-containing tetrazoles 231–233 and the evaluation of their anti-bacterial properties against several Gram-positive and Gram-negative strains (Fig. 80). Thiones 231 showed promising activities with MICs in the range of 3.91–31.25 μg mL−1 for the most potent derivative. The thioalkynes 232 showed lower activity, whereas the tris(trimethylsilyl) compounds 233 showed no anti-bacterial activity at all.449
 | ||
| Fig. 80 Sulphur- and silicon-containing anti-bacterial tetrazoles. | ||
Anti-fungal agents
The Zablotskaya's group's choline and colamine derivatives 18 and 19 also displayed anti-fungal activity when screened against two fungi strains, Candida tropicalis ATCC 4563 and Candida albicans ATCC 2091 (Fig. 9).281 When compared to the standard anti-fungals, nystatin and fluconazole, both the silylated and non-silylated parent compounds displayed comparable activity to nystatin, but in all but one case were not as potent as fluconazole.In 2015, Reddy and co-workers reported the development and anti-fungal screening of a series of silicon analogues of the known anti-fungals, fenpropimorph 234, amarolfine 235 and fenpropodin 236 (Fig. 81).450 A small library of 15 sila-analogues were prepared of which 12 (237–248) showed potent activity against C. albicans, Candida glabrata, C. tropicalis, Cryptococcus neoformans, and Aspergillus niger. The most potent candidate of the new silicon-containing molecules was compound 245, which displayed better anti-fungal activity than fluconazole, fenpropimorph and fenpropodin across all five pathogen lines. In addition, the MIC values for 245 were comparable or better than those of amorolfine, as it elicited the most potent fungicidal effect against all of the tested strains.
 | ||
| Fig. 81 Anti-fungal fenpropimorph 234, amarolfine 235 and fenpropodin 236 and silicon-containing analogues 237–248. | ||
Fu, Liao and co-workers have reported the development of an octahedral silicon complex as a potent anti-fungal agent. Their study involved an effort to prepare silicon complexes as template-analogues of ruthenium complexes which had previously been shown to display a range of biological activities.451
The use of silicon was anticipated to allow the formation of more stable and less toxic complexes. To this end, complex 249 was prepared and shown to exhibit significant inhibition of the growth of the fungus, C. neoformans with MIC and MFC of 4.5 and 11.3 μM respectively. Compound 249 was also shown to act in a fungicidal manner against both the proliferative and quiescent Cryptococcus cells (Fig. 82).
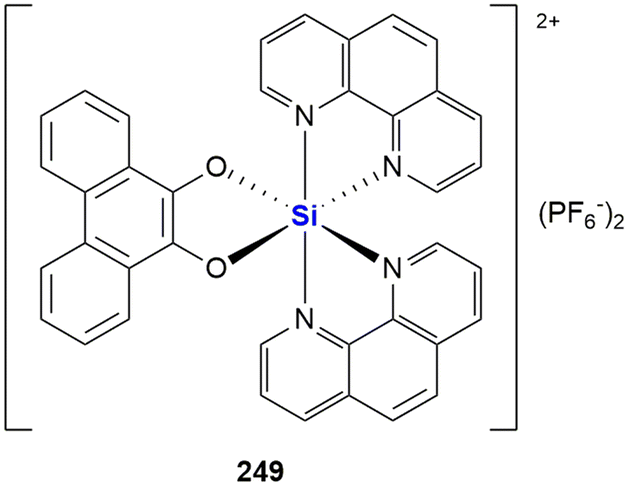 | ||
| Fig. 82 Anti-fungal silicon arenediolate complex 249. | ||
Bargan et al. reported the synthesis the anti-bacterial and anti-fungal properties of imine derivatives of γ-aminopropylsilatranes 250–252 (Fig. 83). 250 showed high anti-fungal activity against Aspergillus fumigatus, Penicillium chrysogenum and Fusarium (MIC = 0.08 μg mL−1) while no anti-bacterial activity was reported. Compounds 251 and 252 were inactive.452
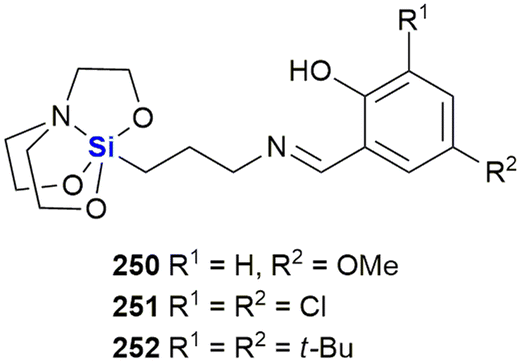 | ||
| Fig. 83 Anti-fungal γ-aminopropylsilatrane-coupled imines. | ||
Anti-parasitic agents
Smith and co-workers described the preparation and anti-parasitic activity of ferrocenyl- and aryl-functionalised organosilanethiosemicarbazones which were converted into half-sandwich ruthenium(II) and rhodium(III) complexes (253–262) (Table 3).453 The complexes were assessed for activity against chloroquine-sensitive (NF54) and resistant (Dd2) Plasmodium falciparum strains and found to have activities in the range of 2.92–7.81 μM and 1.01–6.66 μM respectively. Although marginally less active than the non-silicon analogues (2.57–3.41 μM and 1.01–2.29 μM for NF54 and Dd2 respectively), the inclusion of the silicon atoms reduced cytotoxicity towards a Chinese hamster ovarian cell line. Limited additional screening against the G3 strain of Trichomonas vaginalis suggested that the silicon-containing compounds were more effective than the non-silicon ones.|
|
|||||
|---|---|---|---|---|---|
| 253 | 254 | 255 | 256 | 257 | |
| IC50 (μM) NF54 | — | 7.81 | 2.92 | 4.19 | 2.57 |
| IC50 (μM) Dd2 | — | — | 4.28 | 6.66 | 2.29 |
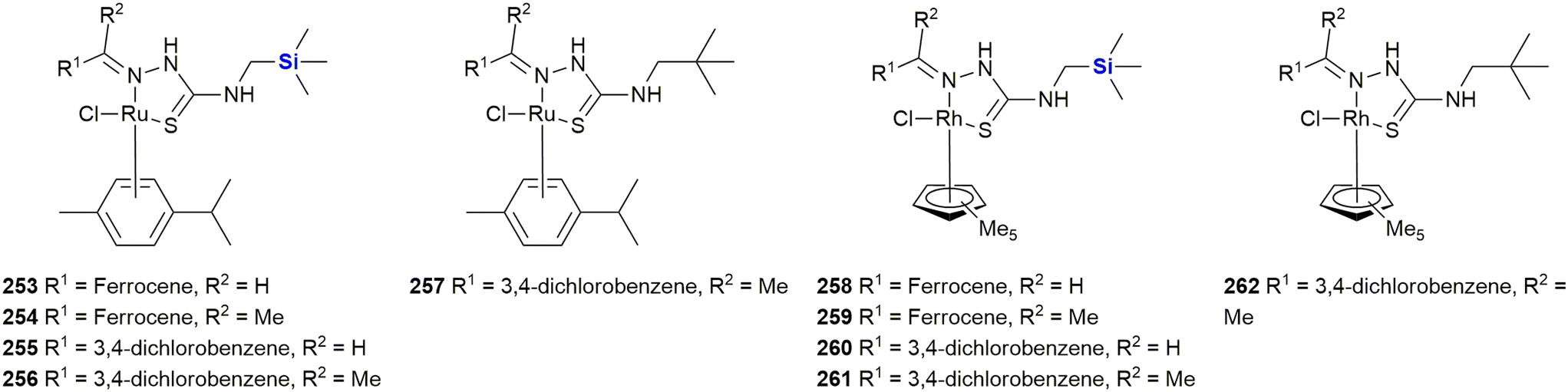
| 258 | 259 | 260 | 261 | 262 | |
|---|---|---|---|---|---|
| IC50 (μM) NF54 | 1.80 | — | 3.73 | 1.31 | 3.41 |
| IC50 (μM) Dd2 | 2.27 | — | 3.11 | 1.18 | 1.01 |
Smith and co-workers also synthesised two silicon-containing chloroquine analogues 263 and 264 and derived a series of transition metal complexes 269–278 of these scaffolds (Fig. 84).454 Both the free quinolines and their ruthenium and rhodium complexes showed potent activity against a chloroquine-sensitive strain of Plasmodium falciparum (NF54), but only lower activity against the chloroquine-resistant strain Dd2. Generally, it was found that a longer alkyl chain increased anti-plasmodial activity. Of additional interest was that although the compounds showed weak anti-mycobacterial activity against Mycobacterium tuberculosis H37Rv they demonstrated fairly potent anti-tumour activity against the oesophageal WHCO1 cancer cell line (IC50 = 4.41–51.92 μM).
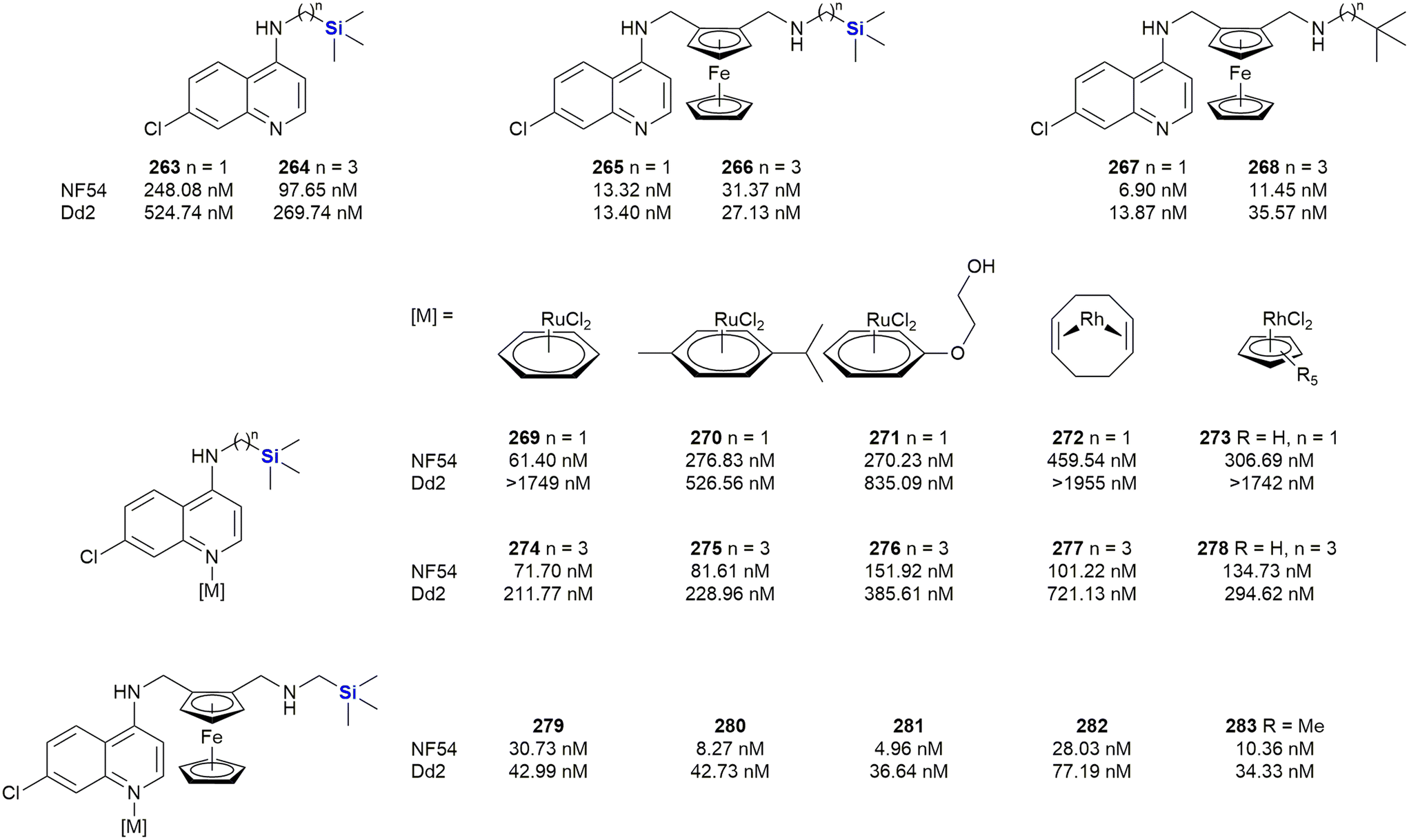 | ||
| Fig. 84 Anti-plasmodial quinolines with an organosilane side chain. | ||
The same group reported the synthesis of ferrrocenyl-functionalised quinolines 265 and 266 with a silylated side chain (Fig. 84). These compounds were evaluated for their anti-plasmodial properties.455 The compounds were found to be less active than their carbon analogues 267 and 268 against the chloroquine-sensitive strain of Plasmodium falciparum NF54 (IC50 = 13.32–31.37 nM), but, in contrast to the carbon analogues, did not lose activity when tested against the chloroquine-resistant strain Dd2 (IC50 = 13.4–27.1 nM). Furthermore, compounds 265 and 266 also inhibited the growth of Trichomonas vaginalis by 24% and 77%, respectively, at a concentration of 50 μM.
In a subsequent study, ruthenium and rhodium complexes 279–283 were investigated, some of which displayed even higher activity than the parent quinoline 265.456
Adams et al. reported the synthesis and anti-plasmodial properties of palladacycles 284–289 with thiosemicarbazone ligands (Fig. 85). The compounds were again tested against a chloroquine sensitive (NF54) and a chloroquine resistant strain (NFDd2) of Plasmodium falciparum and showed similar activity against both strains at micromolar concentrations. The free ligands were also tested, but showed much lower activity than their corresponding palladacyclic complexes, especially against the chloroquine resistant strain.457
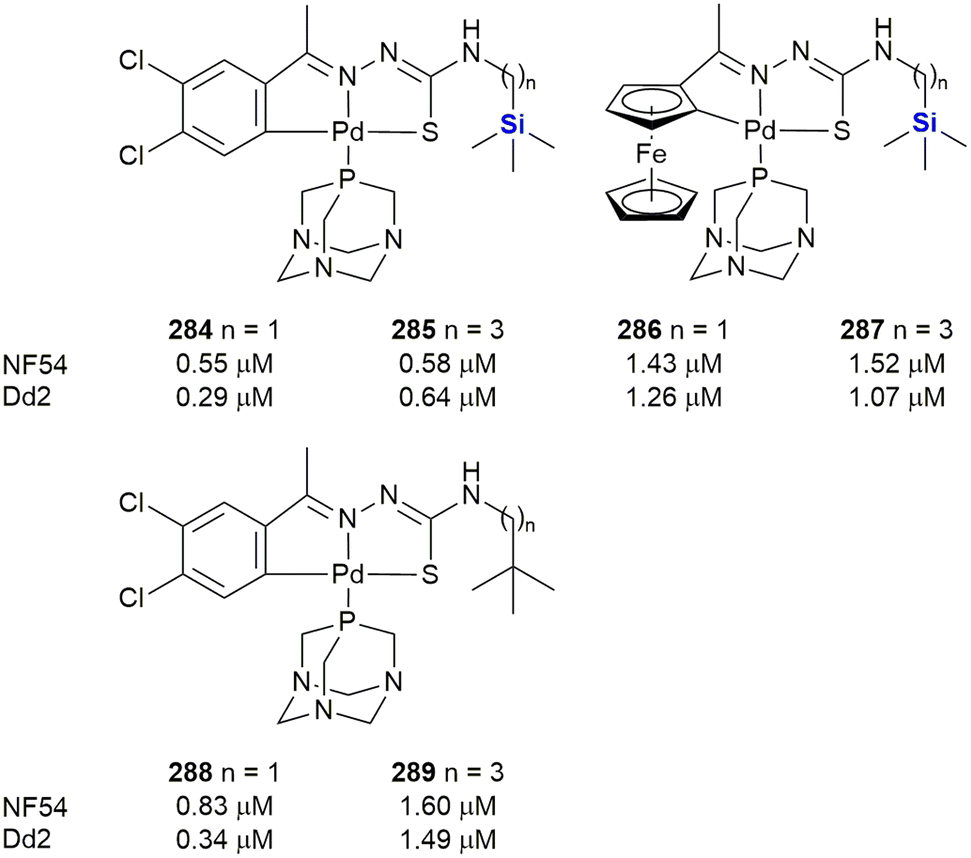 | ||
| Fig. 85 Thiosemicarbazone palladium complexes with an organosilane side chain. | ||
In 2016, Singh and co-workers published their efforts in the design and synthesis of molecules based on chalcones connected via triazole linker to silatranes, such as generalized example 290, as anti-parasitics targeting giardicidal and trichomonacidal unicellular paraites (Table 4).458 The compound libraries prepared all showed potent activity against Giardia lamblia and Trichomonas vaginalis with IC50 value ranges from 19.58–131.2 μM and 18.24–101.26 μM respectively. Almost the entire library displayed better activity than the current therapeutic treatment involving the drug metronidazole. The group further published data on related anti-parasitic and anti-oxidative pyrene-functionalised silatranes showing activity against Giardia lamblia and Entamoeba histolytica.459,460
|
|
||||||
|---|---|---|---|---|---|---|
| ortho | meta | para | ||||
| R | G. lamblia | T. vaginalis | G. lamblia | T. vaginalis | G. lamblia | T. vaginalis |
| Ph | 59.695 | 27.24 | 28.08 | 23.26 | 80.96 | 65.595 |
| 4-MeOPh | 54.855 | 21.605 | 32.16 | 26.69 | NS | 30.275 |
| 2-MeOPh | 40.94 | 22.69 | NS | 23.81 | 43.73 | 43.125 |
| 2,4-MeOPh | 31.945 | 22.855 | 35.395 | 22.125 | 43.885 | 43.805 |
| 2,3,4-MeOPh | 30.175 | 18.245 | NS | 21.265 | 19.58 | 42.095 |
| 4-MePh | 33.245 | 24.82 | 47.995 | 50.025 | 131.2 | 101.265 |
| 4-ClPh | 33.08 | 21.925 | 33.935 | 22.27 | 78.45 | 55.68 |
| Metronidazole | 62.48 μM (for G. lamblia) 55.85 μM (for T. vaginalis) | |||||
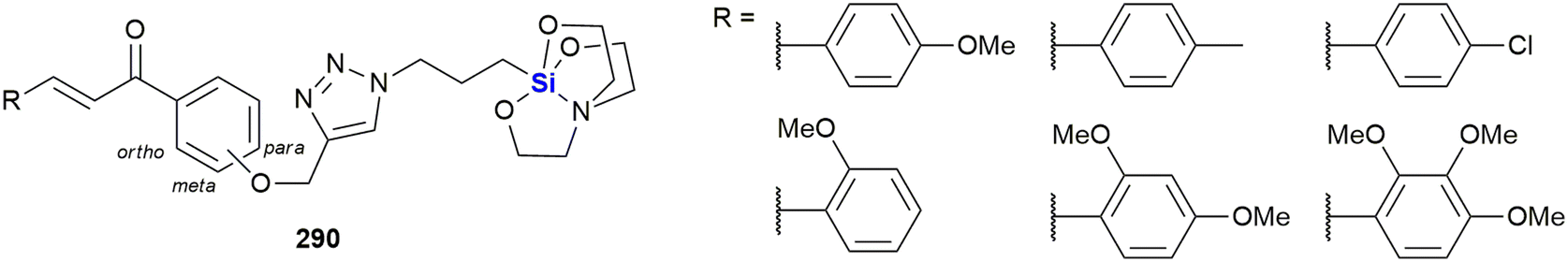
Singh et al. reported the synthesis of a series of hybrid molecules 291 and 292 with a chalcone and a silatrane group (Fig. 86). While the anti-bacterial and anti-fungal activity was low, they showed potent activity against Giardia lamblia (up to IC50 = 0.57 μM after 24 h) and Trichomonas vaginalis (up to IC50 = 57.72 μM after 24 h).461
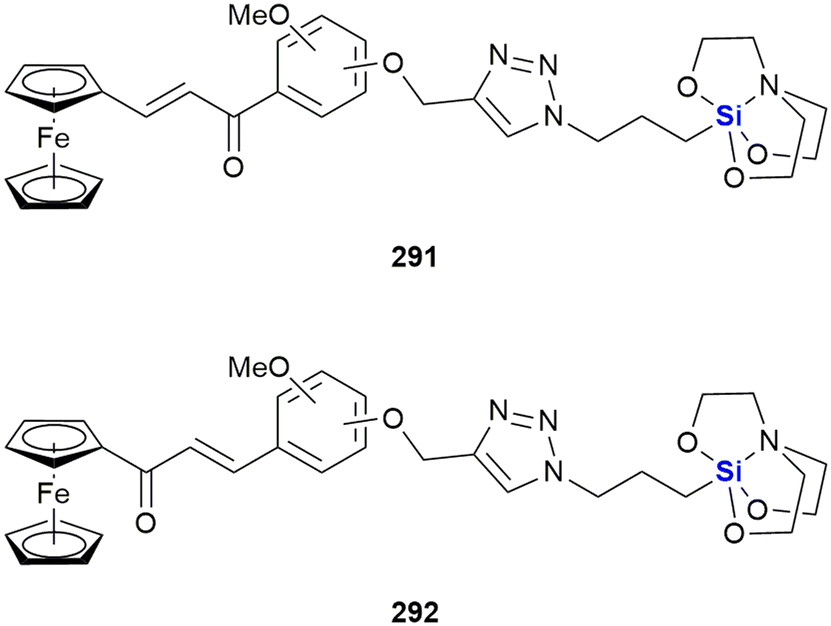 | ||
| Fig. 86 Anti-parasitic silatranes. | ||
Singh et al. also reported the synthesis of pyrene functionalised silatranes 293 and 294 (Fig. 87). Compound 293 showed slightly lower anti-parasitic activity than the methylated silatrane 294 against Giardia lamblia (IC50 = 53.27–63.06 μM) and Tryposomas vaginalis (IC50 = 1.75–3.40 μM).459
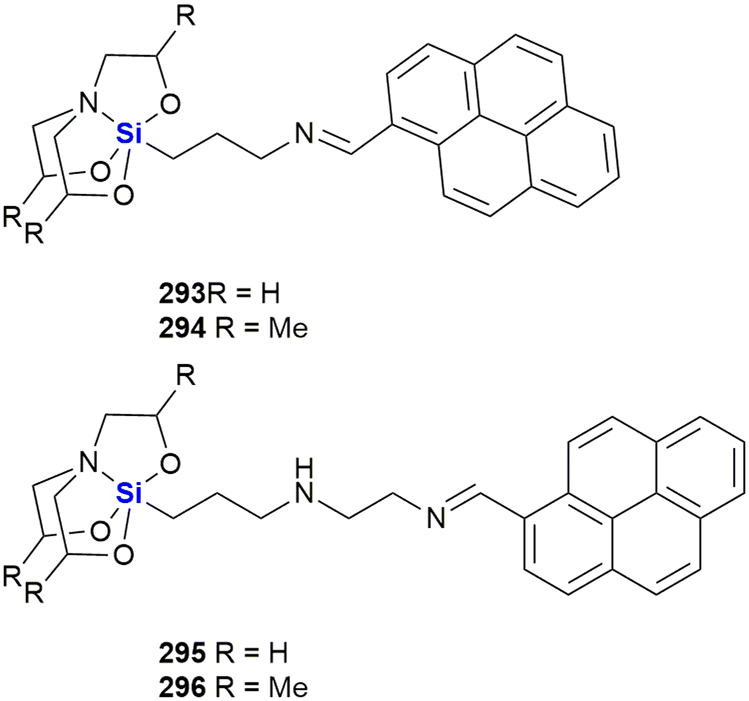 | ||
| Fig. 87 Anti-parasitic pyrene functionalised silatranes. | ||
Later, the same group reported the synthesis of the closely related pyrene functionalised silatranes 295 and 296 with an additional ethylenediamine linker (Fig. 87). Interestingly, the 295 showed higher activity than its methylated counterpart 296 against Giardia lamblia (IC50 = 0.56 μM and 44.55 μM, respectively) but lower activity against Entamoeba histolytica (IC50 = 56.91 μM and 1.14 μM, respectively).460
In 2023, San Nicolás-Hernández et al. investigated 24 silylated derivatives of the known leishmanicidal and trypanocidal drug. Withaferin A 297 and its 4-dehydro congener 298, in order to optimize their respective ADMET profiles (Fig. 88).462 Most of the silyl ethers showed higher anti-parasitic activity than the established reference drugs, miltefosine and benznidazole.
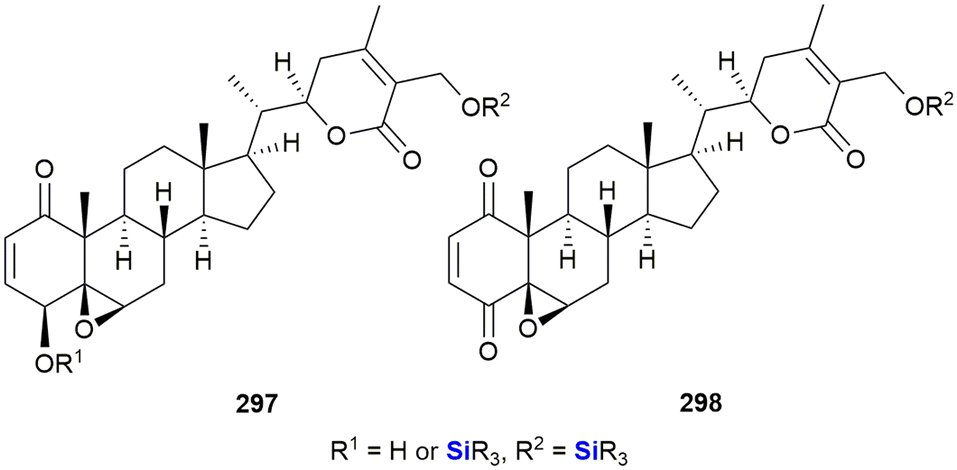 | ||
| Fig. 88 Silyl ether derivatives of withaferin A (297) and 4-dehydrowithaferin A (298). | ||
Anti-inflammatory agents
Geronikaki et al. have reported the synthesis of silicon-containing thiazole derivatives 299 as both lipoxygenase and anti-inflammatory agents (Fig. 89).463 The class of compounds was found to possess good anti-inflammatory action (55% CPE inhibition at a dose of 0.01 mmol kg−1 for 299), mild protection against edema formation and inhibition against soybean lipoxygenase. The substituent at the 4-position of the thiazole ring was linked to lipoxygenase inhibition and cytotoxicity, with the most potent inhibitor (IC50 LOX 0.01 mM) containing a bulky p-MeOPh-group at this position. It was further shown that increasing the distance between the thiazole ring and the piperidyl ring in both the silyl- and non-silyl compounds was linked to an increase in cytotoxicity. | ||
| Fig. 89 Silicon-containing thiazole derivatives with anti-inflammatory activity. | ||
Ibuprofen 300 is the gold standard NSAID used for pain management (Fig. 90),464,465 and recently Beckmann, Grabowsky and co-workers reported the synthesis and bioevaluation of sila-ibuprofen 301. In this research the sila-analogue was designed to contain a SiHMe2 group as a bioisostere for the isobutyl group in ibuprofen. Furthermore, the sila-analogue 301 displayed a similar inhibitory profile to ibuprofen when screen against COX-I and COX-II, but with improved solubility characteristics. Pérez et al. prepared esters and amides 302 from ibuprofen with a silicon containing alkyl chain.466 These derivatives were tested for their anti-inflammatory properties by testing them against nuclear factor κβ, a transcription factor which is involved in inflammation response and chronic inflammatory diseases. While the esters did not inhibit NF-κβ, the amides showed potent activity (IC50 = 45.5–92.0 μM) and performed even better than ibuprofen (IC50 = 241.5 μM). Further work performed by these researchers included in vivo and in silico studies on the novel silicon-containing ibuprofen derivatives synthesized.467
 | ||
| Fig. 90 Ibuprofen 300 and sila-analogues 301 and 302. | ||
In 2007, Barnes et al. reported the synthesis of two silylated derivatives (304 and 305) of the p38 mitogen-activated protein (MAP) kinase inhibitor BIRP-796 (303) and investigated their anti-inflammatory activity (Fig. 91).468 The researchers demonstrated that 304 (IC50 = 64 nM) showed similar activity to 303 (IC50 = 55 nM), but was more stable in human liver microsomes. Compound 305 was only briefly investigated and showed low nanomolar activity.
 | ||
| Fig. 91 p38 MAP kinase inhibitors BIRP-796 303 and sila-analogues 304 and 305. | ||
In 2021, Sen, Reddy and co-workers identified the sila-piperidine amide 306 (Fig. 92) as a potent mosquito repellent.469 The investigators demonstrated that 306 provided protection from Anopheles aegypti for 756 ± 5 min at a dose of 0.5 mg cm−2 and was more effective than its carba-analogue 307 (520 ± 4 min) and the common insect repellent, N,N-diethyl-3-methyl benzamide (DEET) 308 (616 ± 5 min).
 | ||
| Fig. 92 Sila-piperidine amide mosquito repellent. | ||
Shete et al. reported the synthesis of a series of cannabigerol (309, CBG) and cannabidiol (310, CBD) analogues, among these the silylated chinones 311 and 312 (Fig. 93). They found that 311 and especially 312 possessed potent anti-inflammatory activity against heme-induced NLRP3 inflammasome by inhibiting several cytokines (IL-1β, TNF-α, IL-6) at concentrations of 10 μM. The compounds showed no cytotoxicity in vitro against J774A.1 cells.
 | ||
| Fig. 93 Anti-inflammatory silicon-containing CBD-analogues. | ||
The anti-inflammatory activity of CDB-analogue 312 was further investigated in vivo and significantly decreased heme-induced IL-1β levels in mice. This suggests that 312 could be a potential drug candidate against diseases such as sickle cell anemia and thalassemia which involve activation of the NLRP3 inflammatory response by hemolysis.470
Cardiovascular diseases
Sieburth and co-workers have reported the synthesis and biological evaluation of a range of silanediol peptidomimetics as transition state analogue inhibitors of several hydrolases. That the silanediol group is indeed essential for the activity of such compounds was proven by Sieburth and co-workers by the synthesis of the pair of silanol 313 and silanediol 314 (Fig. 94). While the mixture of diastereomers of 314 inhibited ACE with an IC50 of 14 nM, replacing one of the OH groups by a methyl group caused a significant drop of activity of the diastereomeric mixture of 313 (IC50 = 3040 nM).471 | ||
| Fig. 94 Silanol and silanediol peptidomimetic 313 and 314 targeting ACE. | ||
In a subsequent study, Sieburth and co-workers reported the synthesis and biological evaluation of a silanediol peptidomimetic 316 as an angiotensin-converting enzyme (ACE) inhibitor,472 which was based on the ketone inhibitor 315 reported by Almquist and co-workers.(Fig. 95).473–475
 | ||
| Fig. 95 Ketone 318 and silanediol 319 targeting ACE. The asterisks indicate variable stereocentres. | ||
Inhibition was shown to be highly stereo-dependent. The R,S,S configuration was most active in both the parent ketone 315 (IC50 = 1 nM) and the silanediol 316 (IC50 = 3.8 nM). In comparison, the other ketone (IC50 = 8.2, 46 and 3200 nM) and silanediol diastereomers (IC50 = 19, 207 and 72 nM) were significantly less active. Interestingly, in the case of the least active ketone diastereomer (IC50 = 3200 nM), the corresponding sila-analogue displayed an unusually low IC50 value of 72 nM suggesting that the silanediol may be better able to adapt its conformation for the required biochemical interactions. The Sieburth group also published research based on further efforts towards the development of improved stereoselective syntheses of a variety of silanol peptidomimetics, this time as protease inhibitors.476 The reader is further referred to a micro review on the subject of silandiols as protease inhibitors by the same authors.477
More recently, in 2018, Sieburth and Duong reported on the asymmetric synthesis of silanediol peptidomimetics 317 targeting the serine protease coagulation cascade enzyme FXIa (Fig. 96). These efforts were aimed at the development of a novel thrombosis treatment.478 The prepared Arg-[Si]-Ala analogues represented the first silanediol dipeptide to carry a guanidine group, and it should be noted that inhibition of Factor Xia catalysis is still to be reported.
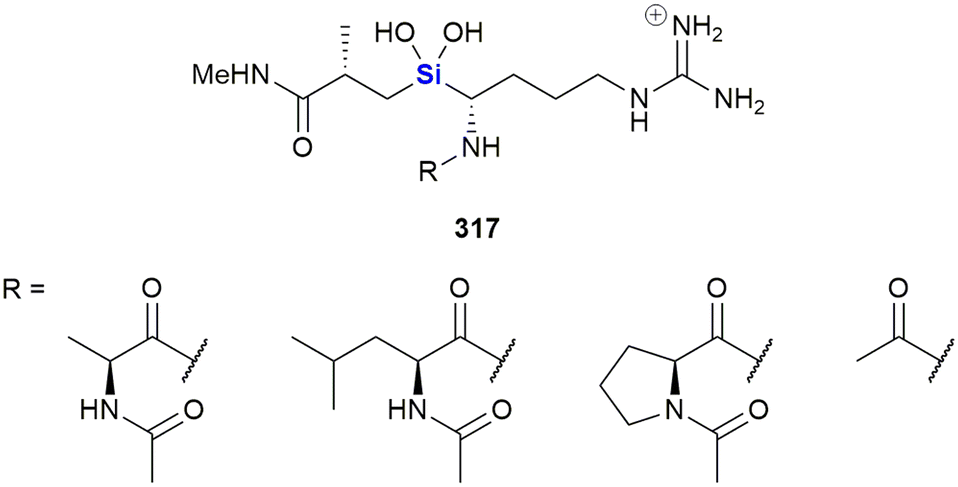 | ||
| Fig. 96 Silanediol peptidomimetics targeting FXIa. | ||
The authors further reported on the preparation and structural analysis of silanediols as transition-state-analogue inhibitors of the metalloprotease thermolysin (Fig. 97).479–481 Notably, the replacement of the phosphinic acid (318) with a silanediol afforded an organosilane which was determined to bind to a natural receptor with the silicon atom interacting with the binding site.
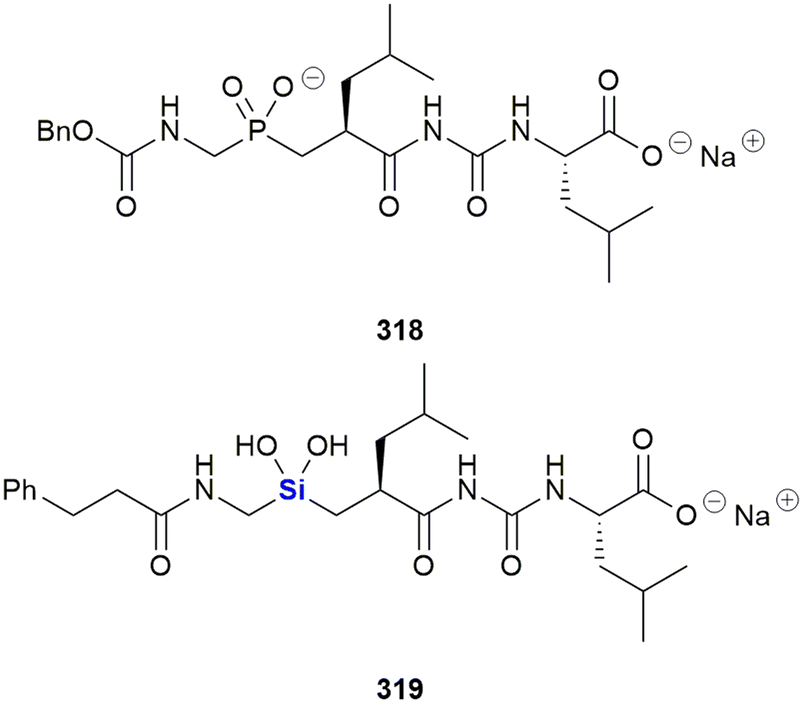 | ||
| Fig. 97 Silanediol peptidomimetic targeting thermolysin. | ||
The activities reported (Ki = 40 nM) were comparable to previously reported phosphorus analogues like 319 (Ki = 10 nM), this despite the fact that the silanediols are electronically neutral whilst the phosphorus groups are anionic in nature.
Captopril 320 (Fig. 98) is also a competitive inhibitor of the angiotensin converting enzyme (ACE) and used to treat hypertension. Dalkas et al. reported the design and synthesis of its silicon-analogue 321, which was synthesised as a diastereomeric mixture and used as such in in vitro studies.482 So called silacaptopril 321 (IC50 = 43 nM) showed lower inhibitory activity against ACE than its carbon analogue 320 (IC50 = 6.3 nM). In addition, docking studies revealed that captopril S,S-320 and silacaptopril, although modelled in silico as pure S,R-321, adopted similar orientations into the ACE ligand binding site. In addition, the modelling software predicted a higher docking score for R,S-321.
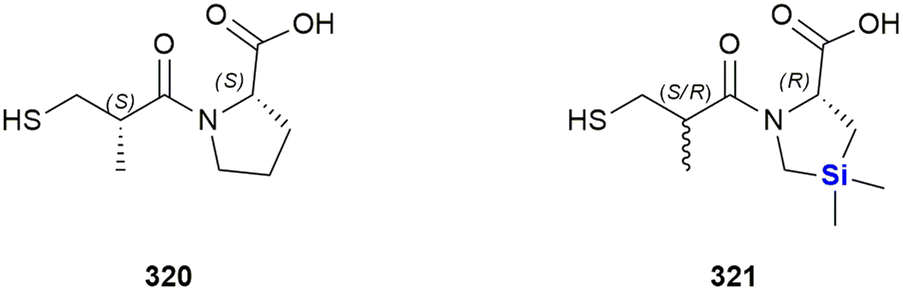 | ||
| Fig. 98 Captopril 320 and silacaptopril 321. | ||
Anti-histamine agents
The Reddy group has demonstrated how the incorporation of silicon into oxadiazoles afforded histaminic suppressors 322–327 that were more effective than the typically prescribed anti-histamine, diphenylhydramine (DPH) (Fig. 99).483 The efficacy of 327 was evaluated using RBL-2H3 as an in vitro model and an in vivo anaphylactic mouse model. It was observed that the silyl compound suppressed DNP-HAS-induced mast cell degranulation, as well as the expression of THF-α mRNA and Akt phosphorylation in antigen-stimulated RBL-2H3 cells.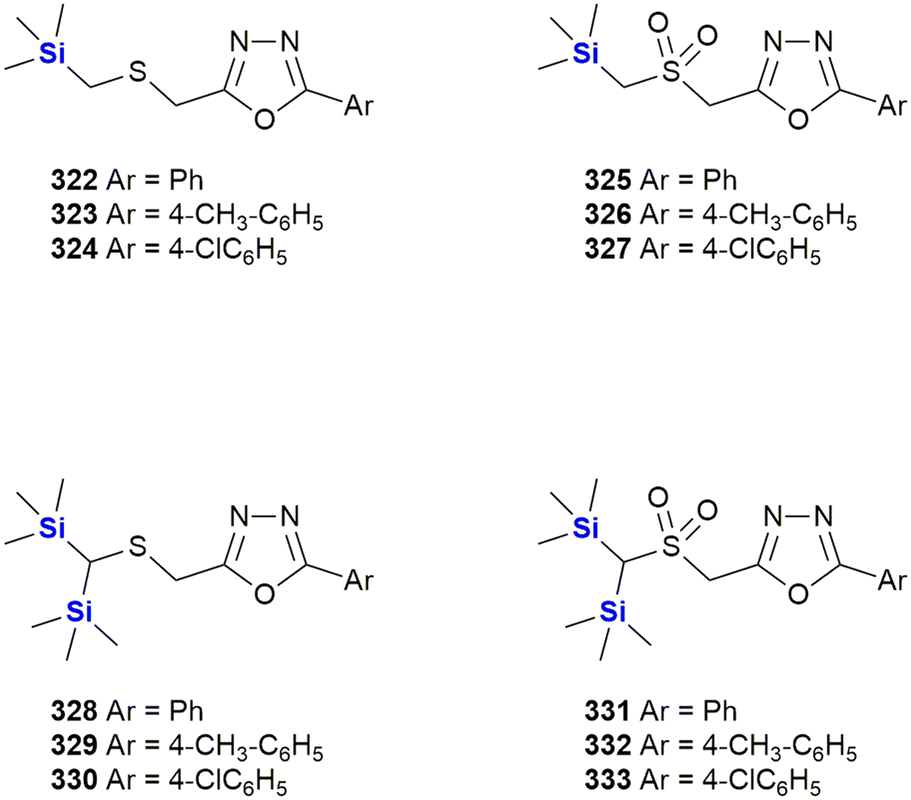 | ||
| Fig. 99 Anti-allergic silicon-containing oxadiazoles. | ||
The same group later introduced the derivatives 328–333 with two instead of one TMS groups (Fig. 99).484 Among this set of compounds, the sulphones 331–333 showed higher anti-allergic activity than the thioethers 328–330 and seemed to surpass slightly the activity of their mono-silylated counterparts. While 331 was most active, its cytotoxicity made the other sulphones more promising as potential therapeutic leads.
Diabetes
Tacke and co-workers reported the synthesis and biological evaluation of silicon-containing GPR81 and GPR109A agonists which are potential targets for certain metabolic diseases and type 2 diabetes (Fig. 100).485 During the study, several 6-sila-4,5,6,7-tetrahydrobenzo[d]thiazoles were synthesised, and the role of the quaternary silicon atom was studied by contrasting the activity of the new compounds against their respective non-sila variants 334 and 336. Sila-analogues 335 and 337 showed high GPR81 and GPR109A potency. The EC50's of 335 were in the range of 1.0–4.4 μM (GPR81) and 1.2–4.8 μM (GPR109A), while the pyrazole analogues 337 were generally more active with EC50's in the range of 0.21–2.2 μM (GPR81) and 0.47–1.8 μM (GPR109A). In contrast, the non-silylated parent compounds 334 (EC50 = 0.24 μM against GPPR81 and 2.1 μM against GPR109A) and 336 (EC50 = 0.018 μM against GPPR81 and 0.25 μM against GPR109A) generally showed higher activity. In terms of metabolic stability, the sila-analogues displayed comparable or slightly improved stabilities relative to 334 and 336.485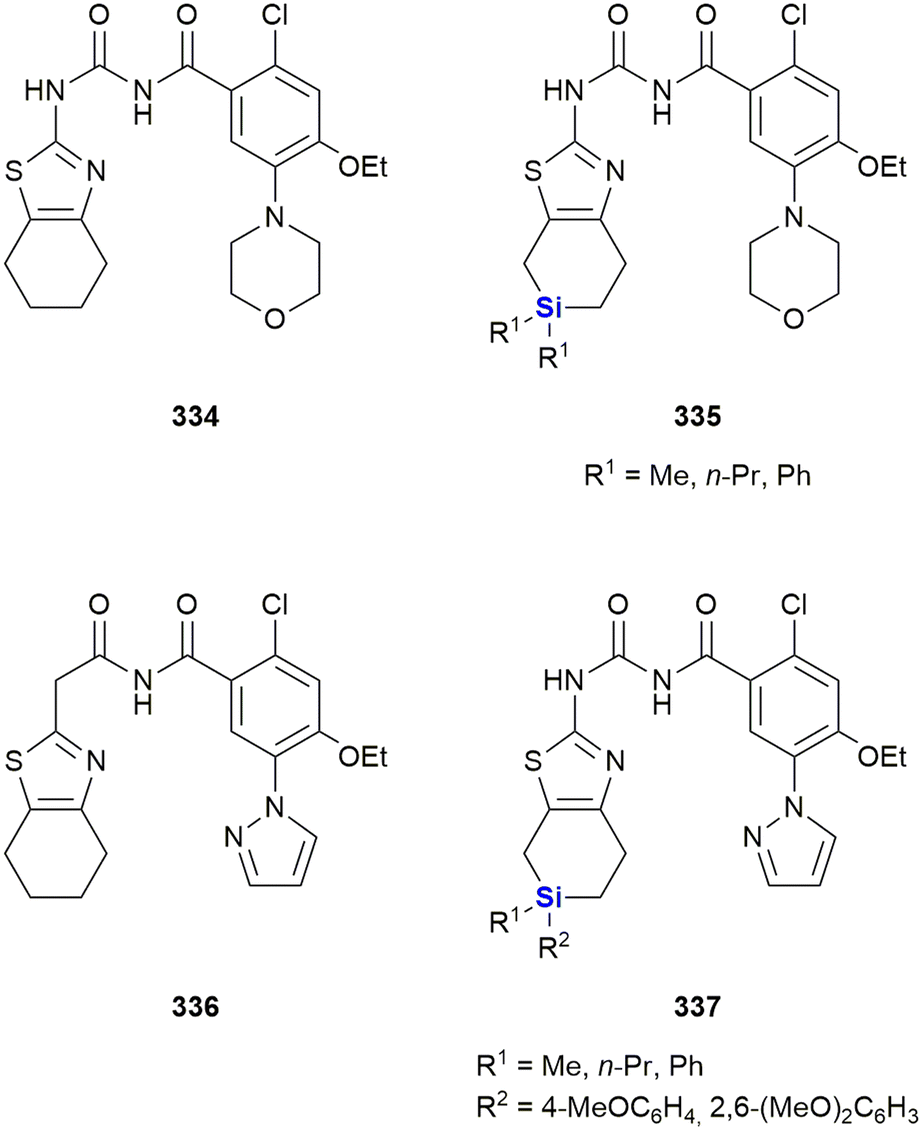 | ||
| Fig. 100 Silicon-containing GPR81 and GPR109A agonists. | ||
Anti-diarrheal agents
In 2015, Tacke and co-workers also developed a silicon analogue of loperamide 338, an opioid receptor agonist which is used clinically as an anti-diarrheal agent. Of note was that the sila-analogue 339 featured a structural carbon–silicon exchange at position 4 of the piperidine ring (Fig. 101). The physiochemical properties of 338 and 339 were found to be very similar and in both instances sub-nanomolar receptor binding affinities and agonist affinity was noted for the μ1 opioid receptor, the main target of loperamide 338. The compounds differed in their ability to cross Caco-2 cell membranes with the sila-analogue 339 having lower permeability and increased efflux abilities; however, both compounds were strong P-gp substrates and readily oxidised by human liver microsomes.486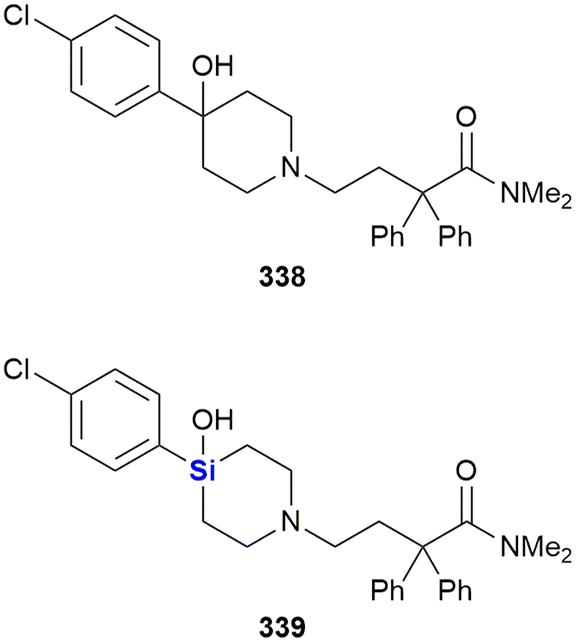 | ||
| Fig. 101 Loperamide 341 and sila-loperamide 342. | ||
Neurotropic agents
Some of the of the earliest research involving carbon–silicon switches was reported by Fessenden and Coon in 1965 (some of the initial research by this group also involved potentially interesting Si-containing pharmacological scaffolds, but without significant biological data being supplied – see for example the following ref. 487 and 488). In an example of interest to this review, these researchers systematically investigated mono- and dicarbamates, such as anxiolytic meprobamate 340 and its sila-analogue 341 (Fig. 102).489 During this bioactivity investigation it was determined that the sila-analogue 341 (LD50 > 1000 mg kg−1) was slightly less toxic than 340 (LD50 = 700 mg kg−1), but that the duration of its effects were significantly reduced from 56 min to 13 min.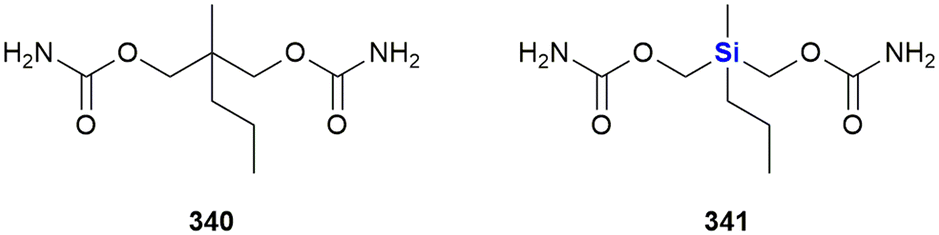 | ||
| Fig. 102 Meprobamate 340 and sila-meprobamate 341. | ||
The use of silicon atom “swaps” as a strategy in the search for and development of neurologically active compounds has attracted attention in light of the ability of silicon to dramatically alter a compounds lipophilicity. As previously mentioned in this review, in their development of anti-bacterials for the treatment of meningitis, Reddy and co-workers showed that increasing the lipophilicity of drugs can lead to improved brain exposure.434 The use of silicon bioisosteres therefore offers an attractive means of improving efficacy through improved membrane penetration of the blood brain barrier.
One of the earlier examples of utilising this approach was performed by preparing a sila-analogue of budipine 342, a monoamine uptake inhibitor used as part of a therapy for Parkinson's disease. In the late 1980s, Stasch and colleagues disseminated research focussed on the replacement of the C-atom within the 4,4-diphenyl core of budipine by a Si-atom, thereby providing compound 343 (Fig. 103).490 This sila-analogue had an increased lipophilicity (relative to the non-Si parent compound), an established advantage for therapeutics acting on the CNS (as in Parkinson's disease therapy) because this compound characteristic has been identified as being very important in improving BBB penetration.490 In terms of the neuropsychiatric disorder schizophrenia, it is widely accepted that changes in complex neurotransmitter systems (such as the excessive stimulation of dopamine D2 receptors) are impacted, even though the exact pathogenesis mechanism are still under investigation. As such, dopamine over-activity is associated with the psychotic symptoms of schizophrenia, such as delusions, hallucinations and thought disorders. In terms of current treatments, the anti-psychotic agent haloperidol 344 acts by blocking the dopamine D2 receptor, as well as other serotonin receptors (Fig. 104).491 With this in mind, in 2004, Tacke and co-workers reported research involving the synthesis of the silicon-containing derivative, sila-haloperidol 346.492 Upon testing the binding affinities of haloperidol 344 and its silicon-analogue 346 against various recombinant dopamine receptors, the researchers were able to show that sila-substitution of haloperidol had quite clearly modulated the receptor selectivity profile. These effects suggested that the biochemical impact of haloperidol 344 and sila-haloperidol 346 would differ substantially in vivo. Additional research by the group, involving radio-ligand binding studies, revealed that sila-haloperidol 346 had comparable receptor binding affinities to haloperidol 344 for the dopamine hD1, hD2, hD3, hD4 and hD5 receptors.493 The same research group also investigated trifluperidol 345 and its sila analogue 347.494 In this case, the sila-analogue was a less effective inhibitor of hD1 and hD2 and showed lower selectivity for the hD2 receptor. In contrast, in the pair of 345 and 347, the sila-substitution lead to an increase of both inhibitory potency and selectivity for the hD2 receptor.
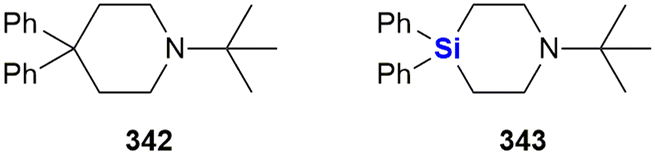 | ||
| Fig. 103 Budipine 342 and sila-budipine 343. | ||
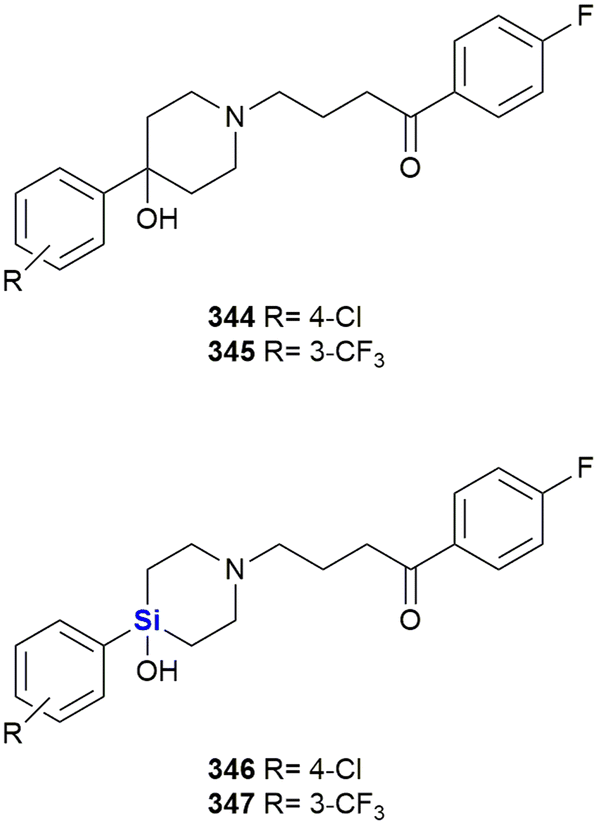 | ||
| Fig. 104 Haloperidol 344, Trifluperidol 345 and sila-analogues. | ||
Tacke et al. reported the synthesis of spirocyclic ligands for σ1 receptors (Fig. 105). The σ1 receptor is an attractive target for the treatment of several diseases of the CNS such as depression, schizophrenia, anxiety, Alzheimer's and Parkinson's diseases. Compared to their respective carba-analogues 348, introduction of silicon in the spirocyclic scaffold of 349 increased both the affinity (Ki = 0.3–2.9 mM) and the selectivity for σ1 ligands.495
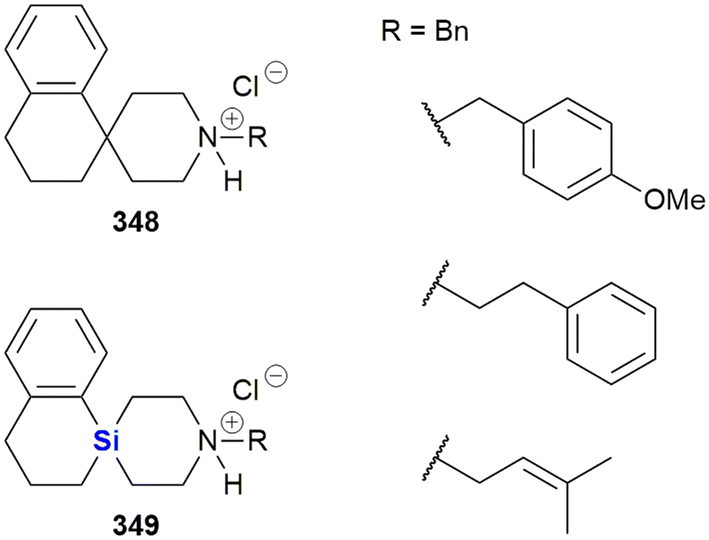 | ||
| Fig. 105 Spirocyclic σ receptor ligands 348 and sila-analogues 349. | ||
Tacke and co-workers also reported on the synthesis of silicon-based W84-type allosteric modulators for ligand binding to muscarinic M2 receptors (Fig. 106).496 The non-sila parent compound W84 350 is known to inhibit the dissociation of the orthosteric ligand [3H] N-methylscopolamine, whereas the researchers developed a silicon-containing analogue wherein one ammonium nitrogen atom was exchanged with a silicon atom affording enhanced binding. The silylated compound 351 was further shown to be an M2-selective allosteric enhancer and therefore did not affect binding at the M1, M3, M4 and M5 receptors. Interestingly, compound 352 with two silicon atoms instead of nitrogen was completely inactive.497
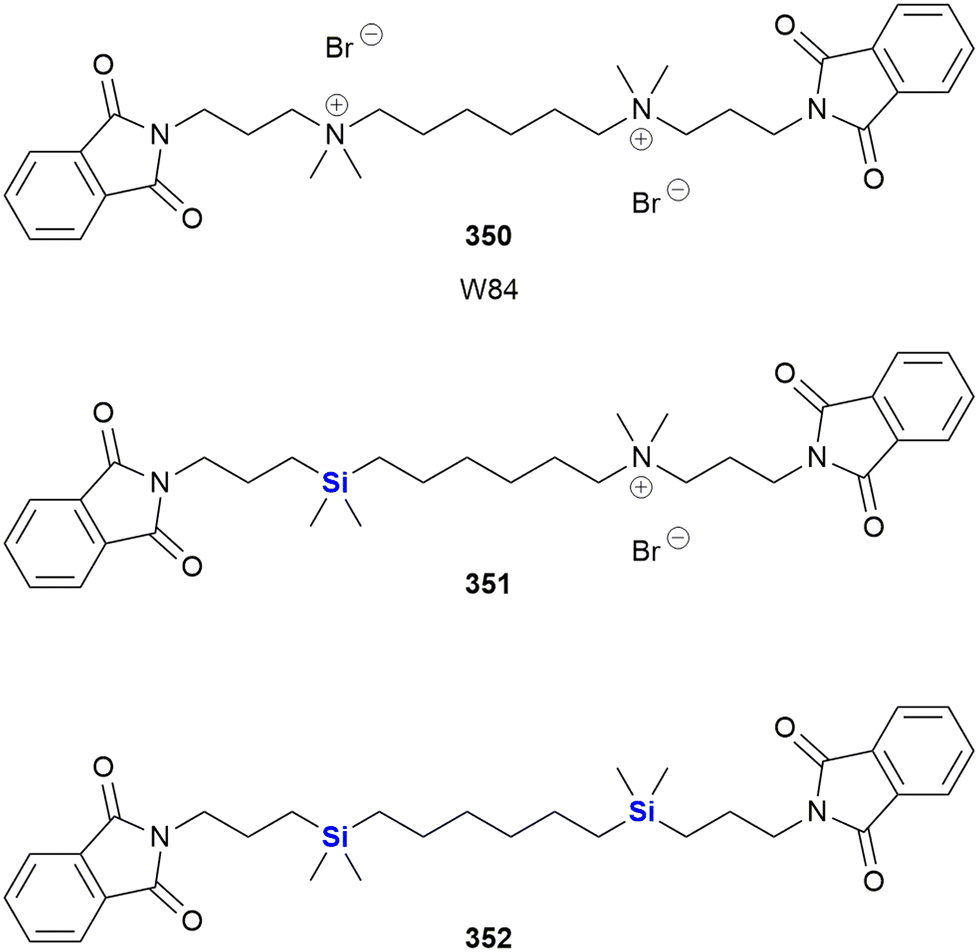 | ||
| Fig. 106 Silicon-based w84-type allosteric modulators. | ||
The Tacke research group also reported the synthesis and biological evaluation of sila-venlafaxine 354, a silicon analogue of the serotonin/noradrenaline reuptake inhibitor venlafaxine 353 (Fig. 107).498–500 Furthermore, studies suggested that sila-substitution dramatically influenced the pharmacological selectivity profiles of compound 353 with respect to serotonin, noradrenaline and dopamine reuptake inhibition. (R)-Sila-venlafaxine 354 showed 10-fold more potent inhibition of noradrenaline transporters than serotonin and dopamine transporters, which might be beneficial for the selective treatment of diseases of the CNS. The venlafaxine analogue 355 with a sila-cyclopentanol instead of a sila-cyclohexanol ring showed lower inhibition of the serotonine transporter than venlafaxine 353 and showed similar activity regarding the reuptake inhibition of serotonin, noradrenaline or dopamine.
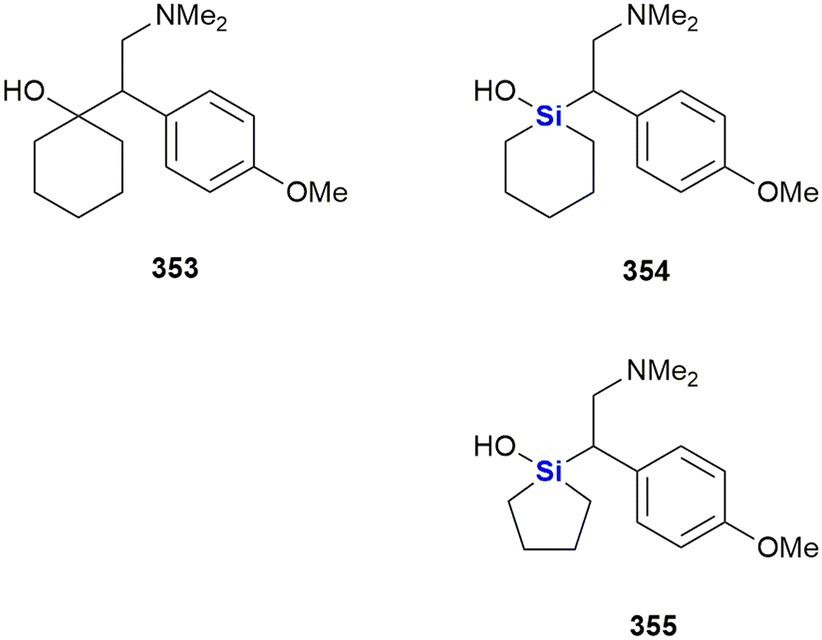 | ||
| Fig. 107 Venlafaxine 353, sila-venlafaxine 354 and a related sila-compound 355 as selective noradrenaline reuptake inhibitor. | ||
Zablotskaya and co-workers have extensively studied the effect of incorporating silicon into neurologically active compounds since the early 1990's, during which time several potent psychotropic agents were identified by these researchers (Fig. 108).289 The group demonstrated that the silylation of aminoalcohols (a strategy previously utilized for pharmacokinetic modulation of this family of compounds)501 afforded compounds which were agonists or antagonists of choline, and caused an increase in psychotropic activity. It was thereby proposed that the mechanism of action might be linked to the passage of the more hydrophobic silyl derivatives through lipophilic membranes.289 As part of this research, tetrahydroquinolines 356 and 357 as well as tetrahydroisoquinoline salts 358 and 359 were synthesised and screened for neurotropic activity. Subsequent studies showed that the substituent on the 2-hydroxyethyl side chain influenced the tone of skeletal musculature and movement co-ordination. This activity was found to decrease when moving from the unsubstituted parent compound to bulkier silyl ethers. Furthermore, weak hypothermic activity was noted for derivatives of N-(2′-hydroxyethyl)tetrahydroquinolines and almost all the novel compounds showed anti-hypoxic activity (30–55%) and anti-convulsive action on Corazol-induced convulsions, with TMS ether 356 showing the best activity.
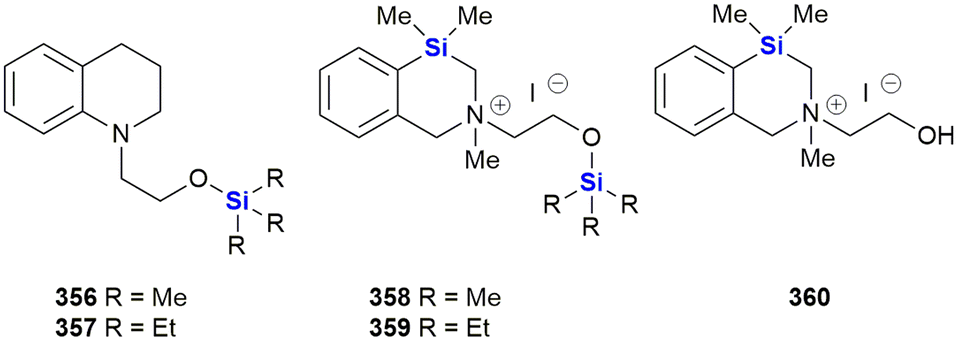 | ||
| Fig. 108 Silicon-containing compounds displaying psychotropic activity. | ||
Further investigations by Zablotskaya and co-workers, into the hydrolytic stability of trialkylsilyl derivatives of heterocyclic bases 87–90 (Fig. 31) as potentially active compounds, was undertaken to see if the rate of hydrolysis could be controlled by changing the substituents at the silicon atom.290 The compounds were attractive as the by-products of hydrolysis were determined to be particularly non-toxic. Biological assessment of the effect of the compounds on locomotor activity, muscular tone and memory processes showed that the TBDMS protected barbituric acid 88 (R = SiMe2t-Bu) had a higher sedative action and a stronger effect on memory processes, fully suppressing retrograde amnesia and higher anti-stress activity than the unsilylated barbituric acid.
In 1999, rhenium(V)-containing organosilicon complexes 361 were prepared by Lukevics and co-workers, some of which showed anti-convulsive activity and did not affect the skeletal muscle tone and movement co-ordination of mice (Fig. 109). In addition, some of the synthetic derivatives displayed tranquilizing effects increasing the length of barbiturate narcosis by 50%, strengthened the effect of amphetamines, increased motor activity and lowered murine body temperature.286
 | ||
| Fig. 109 Anti-convulsive silylated rhenium complexes. | ||
The Zablotskaya group further showed that TMS ethers of hydroxyl-containing thiazole derivatives 362 and 363, which acted as amphetamine antagonists, possessed sedative effects on mice, and that both the silylated and unsilylated ethers displayed similar activities (Fig. 110).284
 | ||
| Fig. 110 Silylated thiazole 365 and thiazolium 366 amphetamine-antagonists. | ||
In 2011, Pietschnig and co-workers reported the synthesis of three silanetriols 364–366, designed to act as reversible acetylcholinesterase inhibitors, in their search for potential anti-Alzheimer's agents (Fig. 111).502 The compounds were found to display moderate activities and negligible cytoxicities in this regard. The most active compound, cyclohexylsilanetriol 365, exhibited 45% inhibition of acetylcholinesterase relative to galantamine hydrobromide, its activity corresponding to an IC50 value of 121 μM.
 | ||
| Fig. 111 Stable silanetriols displaying moderate AChE activities. | ||
Furthermore, more recently in 2021, Morales, González-Rey and co-workers designed several silyl resveratrol derivatives and potential neuroprotective agents.503 Resveratrol 367 has been shown to exhibit neuroprotective properties; however, the low bioavailability of this natural product has limited its utility (Fig. 112). In an attempt to ameliorate this issue, the researchers designed several silyl resveratrol analogues and accessed them synthetically. Notably, compound 368 displayed improved neuroprotective activity relative to resveratrol 367, when it was screened for toxicity and neuroprotection using a zebra fish embryo model system. In addition, compound 368 also reduced motor-coordination loss in a murine model of Huntington's disease and diminished the progression of autoimmune encephalomyelitis in a multiple sclerosis murine model.
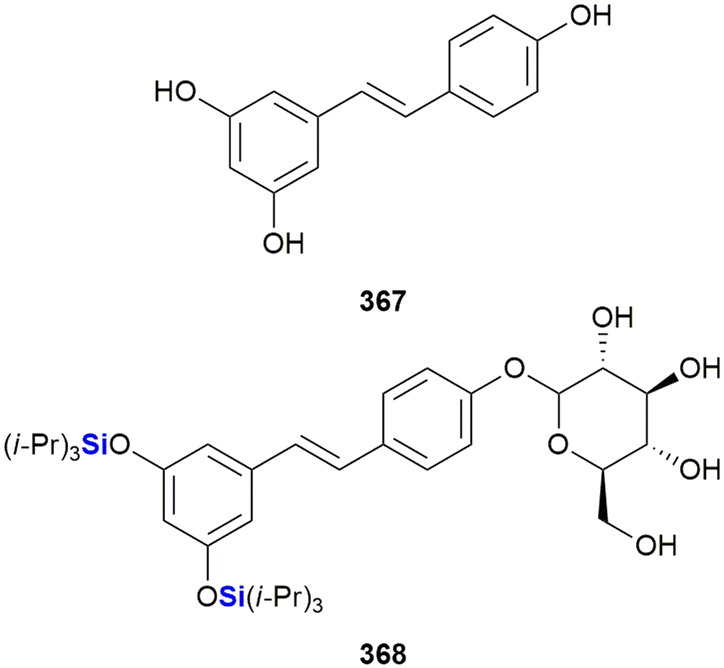 | ||
| Fig. 112 Resveratrol 367 and neuroprotective sila-resveratrol 368. | ||
In 2021, Duan, Sun, Wu et al. reported synthetic cannabinoids incorporating a carbon-silicon switch in the alkyl chain attached to the benzene ring (Fig. 113).504 The CBD-derived 369 showed mixed agonistic activity at both the CB1- and CB2-receptor, while 370 was selective for CB2. The novel cannabinoids reduced the proportion of infarcted brain tissue in experimental autoimmune encephalomyelitis (EAE) mice by 50% (369) and 90% (370) at a dose of 10 mg kg−1 or 30 mg kg−1, respectively. The CB2-selective agonist 370 was the more promising therapeutic agent for the treatment of multiple sclerosis, as 369 induced ataxia and tremors due to its CB1 agonist activity.
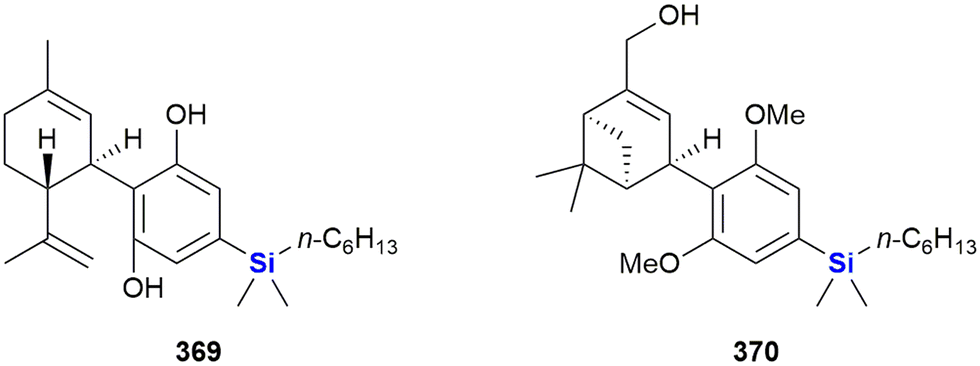 | ||
| Fig. 113 Synthetic sila-cannabinoids. | ||
Last year, El-Hussieny et al. reported the synthesis of the two sila-cycles 371 and 372 as well as the disilylated butenone 373 (Fig. 114).505 These compounds inhibited acetylcholinesterase AChE (IC50 = 113–345 nM) but were not investigated further as potential treatments of Alzheimer's disease as in this work a non-silylated compound proved to be more potent.
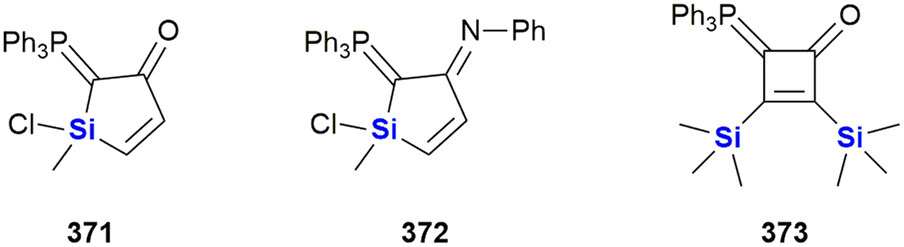 | ||
| Fig. 114 Silicon-containing AChE inhibitors. | ||
Hornsperger et al. investigated silylated trifluormethylketones as AChE inhibitors for the treatment of Alzheimer's disease with the intention to increase the lipophilicity and duration of action by the incorporation of silicon (Fig. 115). Compound 374 was a very effective inhibitor of AChE, with an even lower dissociation constant Ki = 47 pM than the ammonium ion 375 (Ki = 80 pM). It showed long-lasting, but reversible inhibition of AChE in vitro while 375 formed a stable complex with AChE and was found to be an almost irreversible inhibitor. Compound 374 showed dose-dependent inhibition of rat brain AChE with an ED50 of 2.5 mg kg−1. Due to its high lipophilicity, 374 could be dermally administered to dogs, thus avoiding a significant first-pass clearance after oral administration. Furthermore, 374 showed a significantly lower toxicity (LD50 = 180–250 mg kg−1 in mice and rats) than 375.506
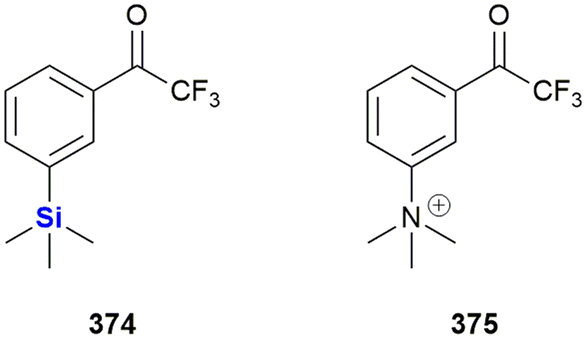 | ||
| Fig. 115 Silylated trifluormethylketones as potential Alzheimer drug candidates. | ||
In 2021, Song, Jiang and co-workers disclosed the synthesis of the anti-depressant (−)-sila-mesembranol, 376, a silicon analogue of (−)-mesembranol 377.507 Furthermore, the researchers went on to demonstrate that the sila-analogue 376 showed improved activity in two murine depression models, thereby suggesting clinical potential of this compound (Fig. 116).
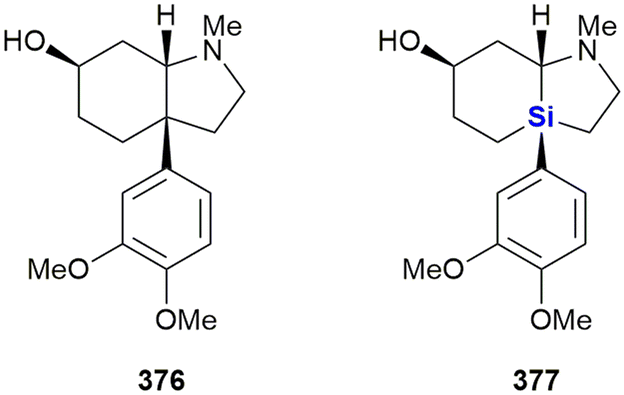 | ||
| Fig. 116 Anti-depressant (−)-mesembranol 376 and (−)-sila-mesembranol 377. | ||
Madica et al. investigated analogues of the anti-depressant tetrapeptide rapastinel 378, in which one (379, 380) or both (381) proline residues were exchanged for a silaproline (Fig. 117).508 According to predictions, the proline-silaproline switch does not influence the conformational properties of the compounds, but was supposed to increase lipophilicity and stability towards enzymatic degradation. So far, the biological activity of these compounds has not been reported.
 | ||
| Fig. 117 Silaproline derivatives of the anti-depressant rapastinel. | ||
Aminoalkylsilanes have been investigated as irreversible inhibitors of monoamine oxidase, an enzyme with several isoforms some which are linked to the metabolism of monoamine neurotransmitters. Inhibitors of these monoamine oxidases, such as 382 (Fig. 118), might therefore exhibit anti-depressant activity.509 The physiological function of another isoform, benzylamine oxidase, is however not fully understood, but its preponderance in vascular tissues might indicate a cardiovascular function. Of interest was that the aminoalkylsilane 383 caused irreversible inactivation of this isoform.510
 | ||
| Fig. 118 Silicon-containing amines as irreversible inhibitors of monoamine oxidase. | ||
Miscellaneous
Cavelier, Sarret and co-workers reported the synthesis of the silaproline analogue 384 of the neurotensin[8-13] pentapeptide fragment (Fig. 119).511 The presence of the bulky dimethylsilyl-group significantly increased the compound's resistance to proteolytic degradation, a notorious problem in the development and use of peptide therapeutics, and thus increased its bioavailability. Of interest is that 384 showed promising analgesic properties in a range of experimental pain models and did not show the common side-effects associated with opioid therapies. | ||
| Fig. 119 Analgesic silaproline analogue of the neurotensin[8-13] fragment. | ||
Silperisone 386 is a centrally acting muscle relaxant and suppresses monosynaptic and polysynaptic spinal reflexes and is used for the treatment of muscle spasms (Fig. 120). It was developed by Farkas and colleagues as a tolperisone 385 analogue in order to increase duration, oral bioavailability and reduced side effects.512
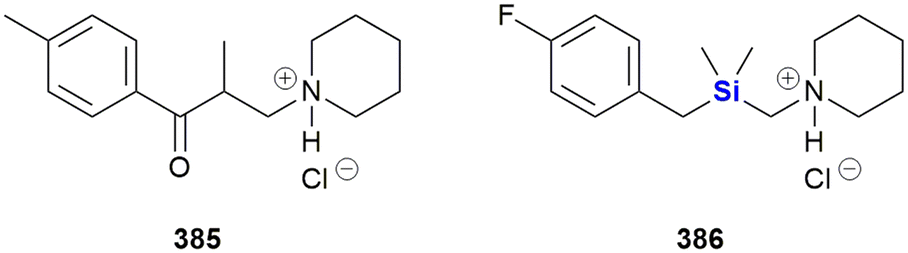 | ||
| Fig. 120 Muscle relaxants tolperisone 385 and silperisone 386. | ||
Santos and co-workers introduced silicon into 99mTc complexes with the aim of increasing the lipophilicity and bioavailability of these radiopharmaceuticals (Fig. 121). Attempts to prepare trimethylsilyl-containing complexes failed, as hydrolysis occurred during the formation of the 99mTc complex in aqueous phosphate buffer.513 Ligands with more bulky silyl ether groups were more stable and the resulting radiolabelled complexes 387 (ref. 513) and 388 (ref. 514) were obtained in high yields. Furthermore, the biodistribution of the complexes 388 were investigated in mice. Unexpectedly, they showed lower brain uptake than the non-silylated and less lipophilic reference complex 389, presumably due to their larger size.
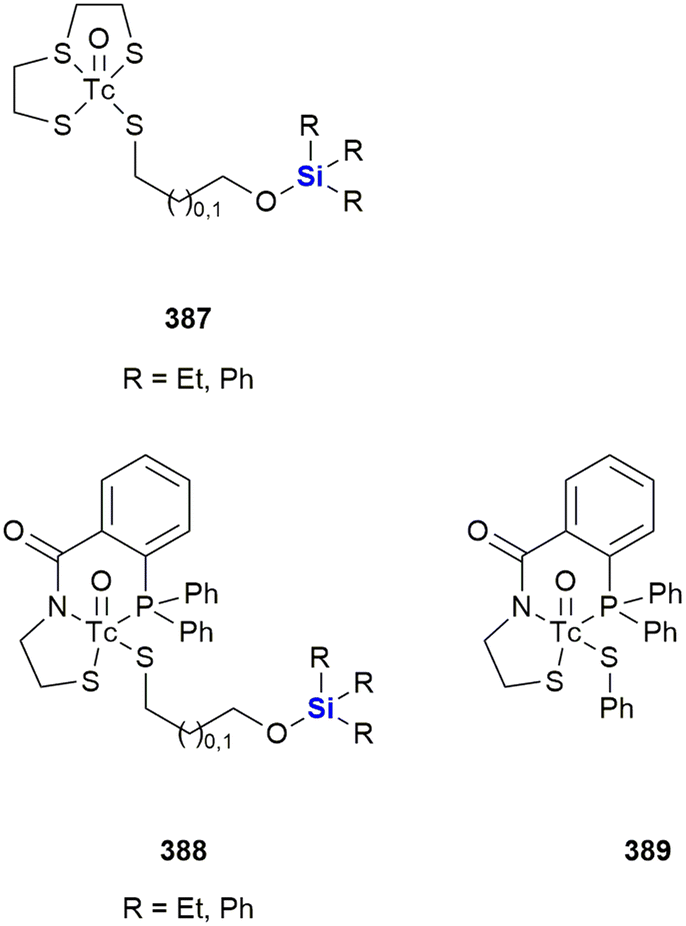 | ||
| Fig. 121 99mTc complexes with a silylated thiopropanol ligand. | ||
Van Dorsselaer investigated bis(aminoalkyl)silanes 390–394 as irreversible inhibitors of diamine oxidase (Fig. 122).515 This copper-containing enzyme which plays a key role in histamine metabolism and is linked to a range of diseases while its precise role is not clear in any case.516 Among all tested compounds, only 390 and 392 irreversibly inhibited diamine oxidase while the other compounds were completely inactive and showed neither reversible nor irreversible inhibition. It should also be recognized that the value of N-(silylmethyl)amines, -amides, and -amino acids as bioactive molecules has already been reviewed.256
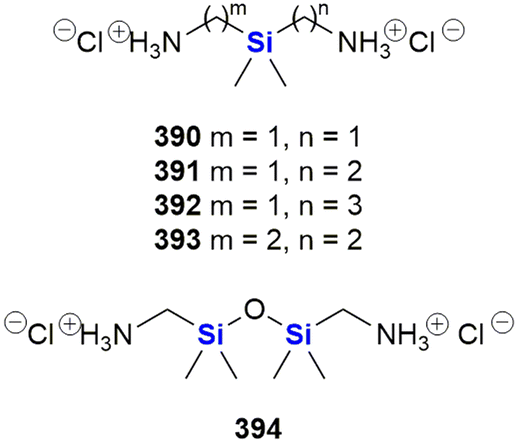 | ||
| Fig. 122 Silicon-containing diamines 390–394 as irreversible inhibitors of diamine oxidase. | ||
In 2022, Singh, Sushma and Priyanka reported the synthesis of organotriethoxysilanes 395 containing a thiosemicarbazone and a triazole group (Fig. 123).517 The compounds were used for the selective complexation and detection of Hg(II) ions by fluorescence spectroscopy with a detection limit of ∼40 nM. The compounds were only weakly cytotoxic and could therefore be used as chemosensors for Hg(II) ions in living cells. The same group also reported a series of silatranes as fluorescence sensors for a variety of heavy metal ions and investigated their potential biological activity in silico.518–527
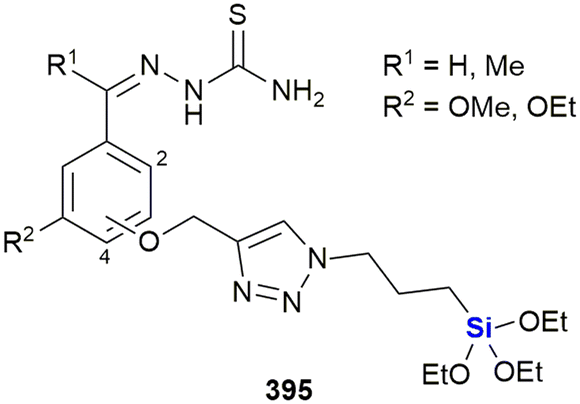 | ||
| Fig. 123 Thiosemicarbazone-triazole linked organotriethoxysilanes as Hg(II) chemosensors. | ||
Retinoic acid-related orphan receptors (RORs) regulate DNA transcription, yet their natural ligands are unknown. They exist in the form of three subtypes which are related to a variety of biological functions. RORα is related to neural functions, RORβ is thought to be involved in the processing of sensory information while RORγ is linked to insulin resistance and the immune system. RORs are therefore interesting targets for new therapeutic applications. Toyama et al. investigated a library of structurally similar dibenzosiloles which include general structures 397 and 399 as novel inverse agonists (Table 5).528 In these structures, a dialkylsilane group replaced the amide groups in the parent structures 396 and 398, which were investigated earlier as RORγ selective inverse agonists.529
|
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Inhibition at 10 μM/% | IC50/μM | |||||||||
| Compound | R | RORα | RORβ | RORγ | Compd | R1 | R2 | RORα | RORβ | RORγ |
| a Inhibition at 10 μM. | ||||||||||
| 396 | ||||||||||
| 397a | — | 37 | 29 | 37 | 399a | — | — | 7.2 | 5.7 | 4.7 |
| 397b | CH(OH)CH3 | 17 | <5.0 | <5.0 | 399b | n-Bu | n-Bu | 6.2 | 6.7 | 5.5 |
| 397c | H | 22 | <5.0 | 13 | 399c | Me | Me | 9.4 | (40%)a | 5.4 |
| 397d | Et | 7.0 | <5.0 | 18 | 399d | Et | Et | (34%)a | (28%)a | 4.2 |
| 397e | COCH3 | 13 | <5.0 | <5.0 | 399e | n-Bu | Me | 7.7 | 7.0 | 4.4 |
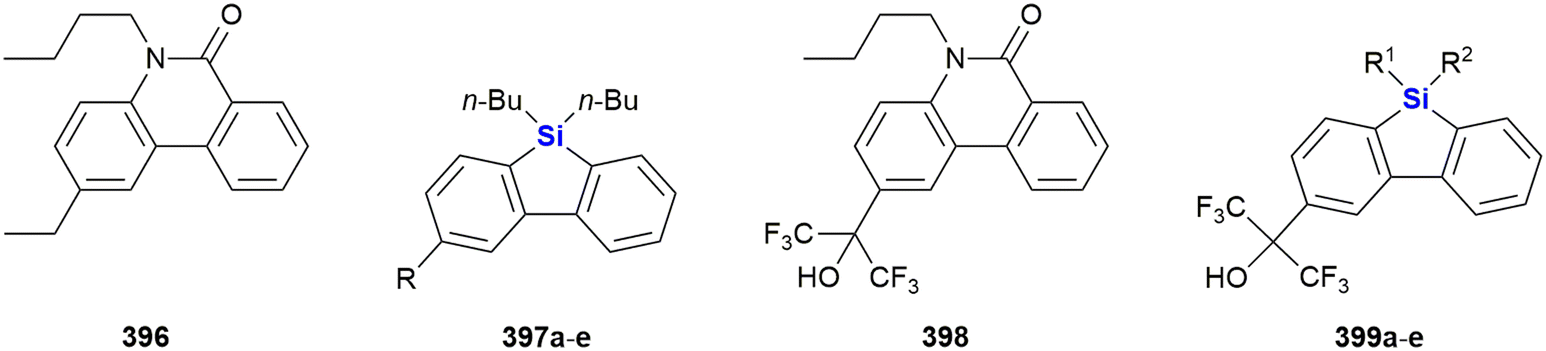
Very recently, Fujii et al. investigated silicon-containing analogues of the multi-target nuclear receptor modulator T0901317 (400), in which the sulfonamide functional group was exchanged by a silyl group (Fig. 124).530 The resulting silicon analogues were investigated as human liver X receptor (hLXR) agonists. While both 401 (EC50 = 0.43 μM against hLXRα and EC50 = 0.59 μM against hLXRβ) and 402 (EC50 = 2.15 μM against hLXRα and EC50 = 0.74 μM against hLXRβ) generally showed lower activity than T090131 (EC50 = 0.26 μM against hLXRα and hLXRβ) in a luciferase reporter gene assay using HEK293 cells. However, while T0901317 showed no selectivity, by proper choice of the silyl group it was possible to increase the selectivity of the silanes for either of the liver X receptors LXRα or LXRβ.
 | ||
| Fig. 124 Liver X receptor agonist T0901317 400 and silicon containing analogues 401, 402 and 403. | ||
The same group also reported the synthesis of the silanol derivative 403 as a multi-target nuclear receptor modulator (Fig. 124). The non-silyl parent compound 400 displays potent, but unselective, activities toward liver X receptor α (hLXRα) and β (hLXRβ), farnesoid X receptor (FXR), pregnane X receptor (PXR) and retinoic acid receptor-related orphan receptor (ROR)γ (EC50 = 0.23–3.4 μM). In contrast, the silanol derivative 403 displayed selectivity with significant activity shown only against PXR (EC50 = 0.88 μM).531,532
Agrichemicals
Silicon has not only found its way into drug discovery related to human diseases but has also found its way into the agrichemical sector. Though not the focus of this review, a few very recent examples will be presented in this section. Of importance here is to appreciate that many of the silicon-switch or Si-derivation strategies applied in medicinal chemistry are also of value in the agrichemical sector, and of course vice versa.In 2020, Maienfisch and co-workers employed a carbon–silicon switch during their development of the anti-fungal sila-cyflumetofen 405, based on the beta-ketonitrile acaricide cyflumetofen 404 (Fig. 125).533 In the development of the sila-analogue the critical for activity tert-butyl group was replaced by a TMS group. The researchers found that the silicon analogue retained good activity and had favourable pharmokinetic properties when compared to the parent compound. Sila-cyflumetofen 405 and cyflumetofen 404 both displayed complete inhibition of acaricidal activities at 200 and 50 ppm, respectively. At 12.5 ppm 404 displayed good activity (70–100% inhibition) while the sila-analogue 405 was completely inactive. That being noted, 404 and derivative 405 are prodrug forms which require conversion into the active forms 406 and 407 with carboxylesterase enzymes. In the case of the “active” forms, the sila-analogue 407 was marginally more active than 406 (IC50 for oxidoreductase activity was 0.0016 vs. 0.0025 μg mL−1).
 | ||
| Fig. 125 Acaricidal cyflumetofen 404 and sila-derivatives. | ||
In 2022, Maienfisch and co-workers investigated the effects of bioisosteric replacement by a carbon–silicon switch on cyflumetofen 404,533,534 a compound which is used as an acaricidal succinate dehydrogenase inhibitor (Fig. 126). The researchers found that simply converting the tert-butyl group into a trimethylsilyl group led to a complete loss of activity at low concentrations (12.5 mg L−1). However, if at the same time the 2-methoxyethyl ester was replaced with an isopropyl ester (408), it was found that the activity had been restored (90% activity at 12.5 mg L−1), even slightly exceeding the activity of 404 (80% activity at 12.5 mg L−1).
 | ||
| Fig. 126 Acaricidal cyflumetofen 404 and silylated derivative 408. | ||
Zhou et al. synthesised a series of silicon containing analogues of cyenopyrafen 409, a prodrug of the succinate dehydrogenase inhibitor 410 (Fig. 127).535 Among the synthesised compounds, 410 emerged as the most potent inhibitor with an LC50 of 0.062 mg L−1 against Tetranychus cinnabarinus (carmine spider mite) and slightly surpassed the activity of cyenopyrafen (LC50 of 0.141 mg L−1). As the incorporation of silicon in 410 decreased acute toxicity against fish, it might represent a promising lead for the development of more environmentally benign acaricides.
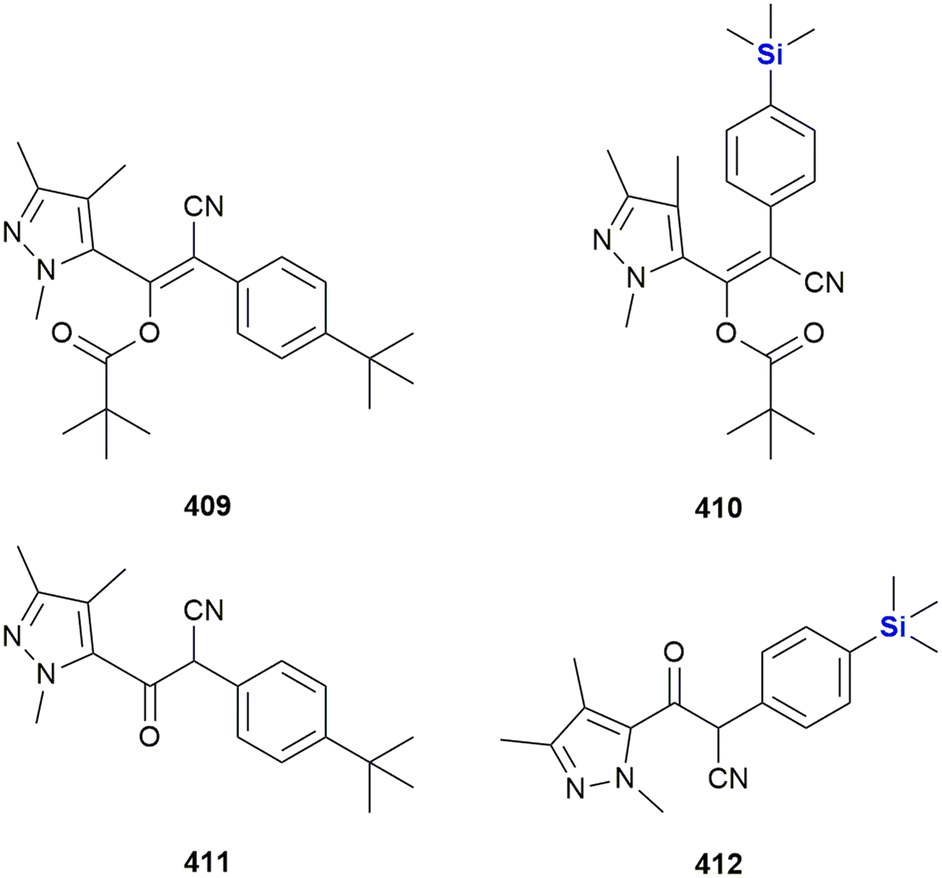 | ||
| Fig. 127 Acaricidal cyenopyrafen 409 and sila-cyenopyrafen 410 and their respective active forms 411 and 412. | ||
The same group also investigated silicon containing insecticides (Fig. 128).536 The diamide insecticide 414 was tested against the moth Mythimna separata and exhibited a low LC50 = 2.00 mg L−1, but unfortunately was less active than its carba-analogue 413 (0.27 mg L−1). In the pair of 415 and 416, the addition of a trimethylsilyl unit slightly decreased the activity against the moths Mythimna separata (LC50 = 4.68 mg L−1415, LC50 = 4.80 mg L−1416) and Plutella xylostella (LC50 = 0.21 mg L−1415, LC50 = 0.30 mg L−1416).537
 | ||
| Fig. 128 Insecticides 413 and 415 and their sila-analogues 414 and 416. | ||
The Maienfisch group also developed a series of succinate dehydrogenase inhibitors by bioisosteric replacement of the amide moiety of difluoromethyl-pyrazol amides by a β-ketonitrile moiety (Fig. 129).538 It was found that increasing hydrophobic interactions with the target protein increased the potency of the tested compounds. Indeed, the silicon-containing compounds 417–418 were among the most potent fungicides. In particular, 419 showed 91.2% inhibition against Rhizoctonia solani at low concentrations of 1 μg mL−1 and could be considered a promising agent to prevent Rice Sheath Blight.
 | ||
| Fig. 129 Silicon-containing succinate dehydrogenase inhibitors 417–419. | ||
Wei et al. also reported on the use of a carbon–silicon switch approach in the development of succinate dehydrogenase inhibitors based upon the chemical space surrounding flubeneteram 420, a recently reported anti-fungal that targets this enzyme (Fig. 130).539 Compound 421 was synthesized and found to display improved anti-fungal activities at low concentrations (0.19 mg L−1) against soybean rust control. The chlorine–silicon swap was specifically performed to improve van der Waals interactions with succinate dehydrogenase.
 | ||
| Fig. 130 Flubeneteram 420 and sila-analogue 421. | ||
Conclusions
As research on silicon containing drug molecules now spans more than six decades and currently sees many research groups actively contributing to the field, it is somewhat perplexing that even though several compounds have entered advanced clinical trials, no silicon-containing drug molecule for the treatment of human diseases and conditions has, to the best of our knowledge, ever proceeded beyond the clinical trial stage. This issue has been repeatedly raised and has generally been answered by the troubles involving the synthesis of silicon containing scaffolds on the one hand and the limited benefits of incorporating silicon on the other. However, we are of the opinion that the rich field of silicon-containing drug molecules deserves a more distinguishing and occasionally a more optimistic perspective, especially given the recent progress in synthetic methods focused on the incorporation of silicon into important drug-like scaffolds.Most examples covered in this review represent derivatives of natural products and known synthetic small molecule drugs. In these cases, the silyl moiety is introduced to optimise the pharmacological and pharmacokinetic profile but is generally not crucial for the biological activity of the molecule itself. Most derivatisations rely on the formation of silyl ethers, as many alkylchlorosilanes are commercially available at reasonable costs and cover a broad range from small, easily hydrolysable groups which cause a small increase of lipophilicity, to large, bulky groups which are both more stable and more lipophilic. Current advances in late-stage small molecule functionalisations now also include the introduction of silicon-containing groups, which should allow new derivatisations beyond the classical use of silyl ethers.540 Incorporation of silicon into biologically active scaffolds which are already identified and accessed by simple derivatisation is arguably most likely to yield commercialised silicon-containing drugs in the nearer future.
On the other hand, the use of silicon containing analogues of pharmacologically active compounds, in which silicon atoms replace carbons, or other elements, as a bioisostere has been less explored, mainly due to this being a more challenging synthetic issue. This area is however open to much opportunity, as it has been repeatedly shown that the incorporation of silicon in the actual scaffold of the drug not only can markedly improve the ADME profile, but also biological activity and selectivity. Given the significant advances in recent Si–C connective chemistry and increased possibilities of asymmetric bond formation to silicon, we will surely see this area advance swiftly, even though chances for commercialized drugs of this kind are currently admittedly low.
Last, we would like to point out an underrepresented opportunity in the field. While the different chemical behaviour and availability of silicon- or carbon-based building blocks is often an obstacle in accessing silicon-containing analogues, it might as well be seen as a chance to access new and unique biologically active scaffolds with no direct carba-analogue by creatively exploiting the unique chemistry of silicon and the broad range of readily available silicon-containing building blocks. This approach is still in its infancy, and so far, the focus has been almost exclusively on silanediols, silanetriols and silatranes. Beyond that, a huge and possibly rewarding chemical space remains to be explored.
References added in proof
During the review of this manuscript a number of papers related to the topic of this review were published. For completeness, they are listed as follows: a) an organocatalytic asymmetric synthetic approach to silicon-centered stereogenic siloxanols541 or a synthetic approach to silacyclohexanones,542 both approaches of potential importance to the synthesis of bioactive Si-containing molecules; b) the disclosure of novel silicon ethers as components of ionizable lipids as part of mRNA lipid nanoparticle complexes;543 the development of silicon-containing subtype-selective estrogen receptor modulators based on the bis(4-hydroxyphenyl)silanol structure;544 the evaluation of Si-containing β-hydroxyphenyl phosphine borane derivatives as nuclear estrogen receptor ligands;545 as well as an overview of the innovative studies involving the physical properties of organosilanes generated by way of hydrosilane–organoiodine couplings.546 Finally, the application of directed evolution to develop enzymes able to cleave the silicon–carbon bonds found in siloxane substances is also of topical interest with respect to this review.547Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported through the CSIR Parliamentary Grant. DR further acknowledges the National Research Foundation (NRF, South Africa) for funding, Thuthuka grant number 106959 and Competitive Support for Unrated Researchers grant number 116303. FH and WvO gratefully acknowledge Stellenbosch University for postdoctoral funding for FH. WvO also acknowledges Stellenbosch University (research support) and National Research Foundation, South Africa, for funding. Opinions expressed in this publication and the conclusions arrived at are those of the authors and are not necessarily attributed to the CSIR or NRF.Notes and references
- J. Michl, Chem. Rev., 1995, 95, 1135 CrossRef CAS.
- https://www.rsc.org/periodic-table/element/14/silicon (Accessed 3 May 2024).
- Z. Souri, K. Khanna, N. Karimi and P. Ahmad, J. Plant Growth Regul., 2021, 40, 906–925 CrossRef CAS.
- R. Tacke, Angew. Chem., Int. Ed., 1999, 38, 3015–3018 CrossRef CAS PubMed.
- G. A. Showell and J. S. Mills, Drug Discovery Today, 2003, 8, 551–556 CrossRef CAS PubMed.
- S. J. Barraza and S. E. Denmark, J. Am. Chem. Soc., 2018, 140, 6668–6684 CrossRef CAS PubMed.
- G. W. Bemis and M. A. Murcko, J. Med. Chem., 1996, 39, 2887–2893 CrossRef CAS PubMed.
- G. W. Bemis and M. A. Murcko, J. Med. Chem., 1999, 42, 5095–5099 CrossRef CAS PubMed.
- N. A. Meanwell, J. Agric. Food Chem., 2023, 71, 18087–18122 CrossRef CAS PubMed.
- P. Steele, Expert Opin. Ther. Pat., 2002, 12, 3–10 CrossRef CAS.
- A. K. Franz and S. O. Wilson, J. Med. Chem., 2013, 56, 388–405 CrossRef CAS PubMed.
- S. E. Thomas, Oxford Chemistry Primers, Organic Synthesis – The Roles of Boron and Silicon, Oxford University Press, USA, 1995 Search PubMed.
- A. G. Brook, F. Abdesaken, B. Gutekunst, G. Gutekunst and R. K. Kallury, J. Chem. Soc., Chem. Commun., 1981, 191–192 RSC.
- S. S. Sen, Angew. Chem., Int. Ed., 2014, 53, 8820–8822 CrossRef CAS PubMed.
- J. S. Mills and G. A. Showell, Expert Opin. Invest. Drugs, 2004, 13, 1149–1157 CrossRef CAS PubMed.
- J. Ackermann, R. Tacke and U. Wannagat, Liebigs Ann. Chem., 1979, 1915–1924 CrossRef CAS.
- W. Kitching, H. A. Olszowy, G. M. Drew and W. Adcock, J. Org. Chem., 1982, 47, 5153–5156 CrossRef CAS.
- M. M. Reichstat, U. B. Mioc, L. J. Bogunovic and S. V. Ribnikar, J. Mol. Struct., 1991, 244, 283–290 CrossRef.
- P. W. Kenny, J. Med. Chem., 2022, 65, 14261–14275 CrossRef CAS PubMed.
- J. Graton, F. Besseau, A. Goupille and J. Y. Le Questel, J. Mol. Struct., 2022, 1266, 133505 CrossRef CAS.
- D. V. Woo, J. E. Christian and R. C. Schnell, Can. J. Pharm. Sci., 1979, 14, 12–14 CAS.
- D. V. Woo, J. E. Christian and R. C. Schnell, Can. J. Pharm. Sci., 1979, 14, 65–67 CAS.
- D. E. Clark, Drug Discovery Today, 2003, 8, 927–933 CrossRef CAS PubMed.
- X. Dong, X. Yuan, Z. Song and Q. Wang, Phys. Chem. Chem. Phys., 2021, 23, 12582–12591 RSC.
- P. Luger, B. Dittrich and R. Tacke, Org. Biomol. Chem., 2015, 13, 9093–9106 RSC.
- P. Luger, M. Weber, C. Hubschle and R. Tacke, Org. Biomol. Chem., 2013, 11, 2348–2354 RSC.
- N. S. Sarai, B. J. Levin, J. M. Roberts, D. E. Katsoulis and F. H. Arnold, ACS Cent. Sci., 2021, 7, 944–953 CrossRef CAS PubMed.
- E. Grabitz, M. Reich, O. Olsson and K. Kümmerer, Sustainable Chem. Pharm., 2020, 18, 100331 CrossRef.
- C. Rücker, M. Winkelmann and K. Kümmerer, Environ. Sci. Pollut. Res., 2023, 30, 91492–91500 CrossRef PubMed.
- C. Rücker, E. Grabitz and K. Kümmerer, Chemosphere, 2023, 321, 137858 CrossRef PubMed.
- P. D. Lickiss and K. M. Stubbs, J. Organomet. Chem., 1991, 421, 171–174 CrossRef CAS.
- R. D. Crouch, Tetrahedron, 2013, 69, 2383–2417 CrossRef CAS.
- W. Zhang, W. Guan, J. I. Martinez Alvarado, L. F. T. Novaes and S. Lin, ACS Catal., 2023, 13, 8038–8048 CrossRef CAS PubMed.
- L. Zheng, X.-X. Nie, Y. Wu and P. Wang, Eur. J. Org. Chem., 2021, 2021, 6006–6014 CrossRef CAS.
- Y. Kong and D. Mu, Chem. – Asian J., 2022, 17, e202200104 CrossRef CAS PubMed.
- W. Yuan and C. He, Synthesis, 2022, 54, 1939–1950 CrossRef CAS.
- M. Zhang, S. Gao, J. Tang, L. Chen, A. Liu, S. Sheng and A. Q. Zhang, Chem. Commun., 2021, 57, 8250–8263 RSC.
- X. Pang and X.-Z. Shu, Chem. – Eur. J., 2023, 29, e202203362 CrossRef CAS PubMed.
- N. Kranidiotis-Hisatomi and M. Oestreich, Synthesis, 2023, 55, 1497–1506 CrossRef CAS.
- W. C. Xue and M. Oestreich, ACS Cent. Sci., 2020, 6, 1070–1081 CrossRef CAS PubMed.
- K. M. Korch and D. A. Watson, Chem. Rev., 2019, 119, 8192–8228 CrossRef CAS PubMed.
- J.-W. Park, Chem. Commun., 2022, 58, 491–504 RSC.
- F. Ye and L.-W. Xu, Synlett, 2021, 32, 1281–1288 CrossRef CAS.
- X. Zhang, J. Fang, C. Cai and G. Lu, Chin. Chem. Lett., 2021, 32, 1280–1292 CrossRef CAS.
- G. L. Larson and R. J. Liberatore, Org. Process Res. Dev., 2021, 25, 1719–1787 CrossRef CAS.
- S. Shamna, J. Fairoosa, C. M. A. Afsina and G. Anilkumar, J. Organomet. Chem., 2022, 960, 122236 CrossRef CAS.
- P. P. Pal, S. Ghosh and A. Hajra, Org. Biomol. Chem., 2023, 21, 2272–2294 RSC.
- J. Gao and C. He, Chem. – Eur. J., 2023, 29, e202203475 CrossRef CAS PubMed.
- Y. Wu and P. Wang, Angew. Chem., Int. Ed., 2022, 61, e202205382 CrossRef CAS PubMed.
- Y. M. Cui, Y. Lin and L. W. Xu, Coord. Chem. Rev., 2017, 330, 37–52 CrossRef CAS.
- G. C. Nandi, Eur. J. Org. Chem., 2021, 2021, 587–606 CrossRef CAS.
- W.-S. Huang, Q. Wang, H. Yang and L.-W. Xu, Synthesis, 2022, 54, 5400–5408 CrossRef CAS.
- V. Chandrasekhar, R. Boomishankar and S. Nagendran, Chem. Rev., 2004, 104, 5847–5910 CrossRef CAS PubMed.
- S. Curpanen, G. Poli, J. Oble and A. Perez-Luna, Eur. J. Org. Chem., 2021, 2021, 1055–1071 CrossRef CAS.
- M. C. D'Amaral, K. G. Andrews, R. Denton and M. J. Adler, Synthesis, 2023, 55, 3209–3238 CrossRef.
- S. K. Ghosh, Asian J. Org. Chem., 2022, 11, e202200227 CrossRef CAS.
- C. Moberg, Synthesis, 2020, 52, 3129–3139 CrossRef CAS.
- D. L. Priebbenow, Adv. Synth. Catal., 2020, 362, 1927–1946 CrossRef CAS.
- X. Wang, F. Liu, Y. Li, Z. Yan, Q. Qiang and Z.-Q. Rong, ChemCatChem, 2020, 12, 5022–5033 CrossRef CAS.
- A. Sivaramakrishna, S. Pete, C. Mandar Mhaskar, H. Ramann, D. Venkata Ramanaiah, M. Arbaaz, M. Niyaz, S. Janardan and P. Suman, Coord. Chem. Rev., 2023, 485, 215140 CrossRef CAS.
- B. Cheng, P. Lu, H. Zhang, X. Cheng and Z. Lu, J. Am. Chem. Soc., 2017, 139, 9439–9442 CrossRef CAS PubMed.
- W. Zheng, Y. Xu, H. Luo, Y. Feng, J. Zhang and L. Lin, Org. Lett., 2022, 24, 7145–7150 CrossRef CAS PubMed.
- L. X. Lu, J. C. Siu, Y. H. Lai and S. Lin, J. Am. Chem. Soc., 2020, 142, 21272–21278 CrossRef CAS PubMed.
- X.-Y. Lu, H.-Y. Pan, R. Huang, K. Yang, X. Zhang, Z.-Z. Wang, Q.-Q. Tao, G.-X. Yang, X.-J. Wang and H.-P. Zhou, Org. Lett., 2023, 25, 2476–2481 CrossRef CAS PubMed.
- C. Karmel, Z. Chen and J. F. Hartwig, J. Am. Chem. Soc., 2019, 141, 7063–7072 CrossRef CAS PubMed.
- W. Sarkar, A. Mishra, A. Bhowmik and I. Deb, Org. Lett., 2021, 23, 4521–4526 CrossRef CAS PubMed.
- Y.-J. Liu, Y.-H. Liu, Z.-Z. Zhang, S.-Y. Yan, K. Chen and B.-F. Shi, Angew. Chem., Int. Ed., 2016, 55, 13859–13862 CrossRef CAS PubMed.
- J. Scharfbier, B. M. Gross and M. Oestreich, Angew. Chem., Int. Ed., 2020, 59, 1577–1580 CrossRef CAS PubMed.
- A. P. Cinderella, B. Vulovic and D. A. Watson, J. Am. Chem. Soc., 2017, 139, 7741–7744 CrossRef CAS PubMed.
- S. B. J. Kan, R. D. Lewis, K. Chen and F. H. Arnold, Science, 2016, 354, 1048–1051 CrossRef CAS PubMed.
- Q.-C. Gan, Z.-Q. Song, C.-H. Tung and L.-Z. Wu, Org. Lett., 2022, 24, 5192–5196 CrossRef CAS PubMed.
- H. Li, C. Yang, D. Wang and L. Deng, Organometallics, 2023, 42, 1693–1698 CrossRef CAS.
- X. Tang, Y. Zhang, Y. Tang, Y. Li, J. Zhou, D. Wang, L. Gao, Z. Su and Z. Song, ACS Catal., 2022, 12, 5185–5196 CrossRef CAS.
- C. Qin, Z. Huang, S.-B. Wu, Z. Li, Y. Yang, S. Xu, X. Zhang, G. Liu, Y.-D. Wu, L. W. Chung and Z. Huang, J. Am. Chem. Soc., 2022, 144, 20903–20914 CrossRef CAS PubMed.
- X.-X. Zhang, Y. Gao, Y.-X. Zhang, J. Zhou and J.-S. Yu, Angew. Chem., Int. Ed., 2023, 62, e202217724 CrossRef CAS PubMed.
- T. Tani, Y. Sohma and T. Tsuchimoto, Adv. Synth. Catal., 2020, 362, 4098–4108 CrossRef CAS.
- S. Chen, X. He, C. Jin, W. Zhang, Y. Yang, S. Liu, Y. Lan, K. N. Houk and X. Shen, Angew. Chem., Int. Ed., 2022, 61, e202213431 CrossRef CAS PubMed.
- J. B. Gluyas, C. Burschka, S. Dorrich, J. Vallet, H. Gronemeyer and R. Tacke, Org. Biomol. Chem., 2012, 10, 6914–6929 RSC.
- J. Y. L. Chung, M. Shevlin, A. Klapars and M. Journet, Org. Lett., 2016, 18, 1812–1815 CrossRef CAS PubMed.
- Q. Li, M. Driess and J. F. Hartwig, Angew. Chem., Int. Ed., 2014, 53, 8471–8474 CrossRef CAS PubMed.
- H. Fang, K. Xie, S. Kemper and M. Oestreich, Angew. Chem., Int. Ed., 2021, 60, 8542–8546 CrossRef CAS PubMed.
- W. S. Wang, S. Zhou, L. J. Li, Y. H. He, X. Dong, L. Gao, Q. T. Wang and Z. L. Song, J. Am. Chem. Soc., 2021, 143, 11141–11151 CrossRef CAS PubMed.
- C. Sedano, R. Velasco, C. Feberero, S. Suárez-Pantiga and R. Sanz, Org. Lett., 2020, 22, 6365–6369 CrossRef CAS PubMed.
- B.-S. Mu, Y. Gao, F.-M. Yang, W.-B. Wu, Y. Zhang, X. Wang, J.-S. Yu and J. Zhou, Angew. Chem., Int. Ed., 2022, 61, e202208861 CrossRef CAS PubMed.
- Y. H. Huang, Y. C. Wu, Z. L. Zhu, S. J. Zheng, Z. H. Ye, Q. Peng and P. Wang, Angew. Chem., Int. Ed., 2022, 61, e202113052 CrossRef CAS PubMed.
- J. Wang, H. Wang, Z. Wang, L. Li, C. Qin and X. Luan, Chin. J. Chem., 2021, 39, 2659–2667 CrossRef CAS.
- S. Y. Park, J.-W. Lee and C. E. Song, Nat. Commun., 2015, 6, 7512 CrossRef PubMed.
- J. Seliger and M. Oestreich, Angew. Chem., Int. Ed., 2021, 60, 247–251 CrossRef CAS PubMed.
- J. Gao, P.-L. Mai, Y. Ge, W. Yuan, Y. Li and C. He, ACS Catal., 2022, 12, 8476–8483 CrossRef CAS.
- X. Xu, J. M. A. van Hengst, Y. Mao, M. Martinez, S. Roda, M. Floor, V. Guallar, C. E. Paul, M. Alcalde and F. Hollmann, Angew. Chem., Int. Ed., 2023, 62, e202302844 CrossRef CAS PubMed.
- H. Chen, Y. Chen, X. X. Tang, S. F. Liu, R. P. Wang, T. B. Hu, L. Gao and Z. L. Song, Angew. Chem., Int. Ed., 2019, 58, 4695–4699 CrossRef CAS PubMed.
- J. O. Bauer and C. Strohmann, Angew. Chem., Int. Ed., 2014, 53, 720–724 CrossRef CAS PubMed.
- H. Zhou, R. Properzi, M. Leutzsch, P. Belanzoni, G. Bistoni, N. Tsuji, J. T. Han, C. Zhu and B. List, J. Am. Chem. Soc., 2023, 145, 4994–5000 CrossRef CAS PubMed.
- M.-M. Liu, Y. Xu and C. He, J. Am. Chem. Soc., 2023, 145, 11727–11734 CrossRef CAS PubMed.
- J. H. Chen, B. Cheng, M. Y. Cao and Z. Lu, Angew. Chem., Int. Ed., 2015, 54, 4661–4664 CrossRef CAS PubMed.
- M. W. Gribble, M. T. Pirnot, J. S. Bandar, R. Y. Liu and S. L. Buchwald, J. Am. Chem. Soc., 2017, 139, 2192–2195 CrossRef CAS PubMed.
- M. A. Rivero-Crespo, A. Leyva-Perez and A. Corma, Chem. – Eur. J., 2017, 23, 1702–1708 CrossRef CAS PubMed.
- C. Wang, W. J. Teo and S. Z. Ge, ACS Catal., 2017, 7, 855–863 CrossRef CAS.
- J. Guo, X. Z. Shen and Z. Lu, Angew. Chem., Int. Ed., 2017, 56, 615–618 CrossRef CAS PubMed.
- H. Fang, W. Hou, G. Liu and Z. Huang, J. Am. Chem. Soc., 2017, 139, 11601–11609 CrossRef CAS PubMed.
- J. Guo, H. L. Wang, S. P. Xing, X. Hong and Z. Lu, Chem, 2019, 5, 881–895 CAS.
- Z. Y. Cheng, S. P. Xing, J. Guo, B. Cheng, L. F. Hu, X. H. Zhang and Z. Lu, Chin. J. Chem., 2019, 37, 457–461 CrossRef CAS.
- Z. Y. Zhao, Y. X. Nie, R. H. Tang, G. W. Yin, J. Cao, Z. Xu, Y. M. Cui, Z. J. Zheng and L. W. Xu, ACS Catal., 2019, 9, 9110–9116 CrossRef CAS.
- X. W. Gu, Y. L. Sun, J. L. Xie, X. B. Wang, Z. Xu, G. W. Yin, L. Li, K. F. Yang and L. W. Xu, Nat. Commun., 2020, 11, 2904 CrossRef CAS PubMed.
- S. Park, Chin. J. Chem., 2019, 37, 1057–1071 CrossRef CAS.
- S. Nishino, K. Hirano and M. Miura, Chem. – Eur. J., 2020, 26, 8725–8728 CrossRef CAS PubMed.
- Y. Wan, J. J. Zhu, Q. Y. Yuan, W. Wang and Y. Q. Zhang, Org. Lett., 2021, 23, 1406–1410 CrossRef CAS PubMed.
- Y. E. You and S. Z. Ge, Angew. Chem., Int. Ed., 2021, 60, 12046–12052 CrossRef CAS PubMed.
- S. Neogi, A. K. Ghosh, S. Mandal, D. Ghosh, S. Ghosh and A. Hajra, Org. Lett., 2021, 23, 6510–6514 CrossRef CAS PubMed.
- M. B. Zhong, X. Pannecoucke, P. Jubault and T. Poisson, Chem. – Eur. J., 2021, 27, 11818–11822 CrossRef CAS PubMed.
- A. Vivien, L. Veyre, R. Mirgalet, C. Camp and C. Thieuleux, Chem. Commun., 2022, 58, 4091–4094 RSC.
- C.-R. Jiang, C.-L. Zhao, H.-F. Guo and W. He, Chem. Commun., 2016, 52, 7862–7865 RSC.
- A. Bokka, Y. D. Hua, A. S. Berlin and J. Jeon, ACS Catal., 2015, 5, 3189–3195 CrossRef CAS.
- Y. Takeda, K. Shibuta, S. Aoki, N. Tohnai and S. Minakata, Chem. Sci., 2019, 10, 8642–8647 RSC.
- H. Kondo, K. Itami and J. Yamaguchi, Chem. Sci., 2017, 8, 3799–3803 RSC.
- N. Gandhamsetty, J. Park, J. Jeong, S. W. Park, S. Park and S. Chang, Angew. Chem., Int. Ed., 2015, 54, 6832–6836 CrossRef CAS PubMed.
- X. D. Qiu, L. Zhou, H. R. Wang, L. Y. Lu, Y. Ling and Y. A. Zhang, RSC Adv., 2021, 11, 37083–37088 RSC.
- T. Ahmad, Q. Li, S. Q. Qiu, J. L. Xu, Y. H. Xu and T. P. Loh, Org. Biomol. Chem., 2019, 17, 6122–6126 RSC.
- J. J. Shen, Q. Gao, G. Z. Wang, M. Tong, L. L. Chen and S. M. Xu, ChemistrySelect, 2019, 4, 11358–11361 CrossRef CAS.
- J. Zhou, B. Y. Jiang, Y. Fujihira, Z. Y. Zhao, T. Imai and N. Shibata, Nat. Commun., 2021, 12, 3749 CrossRef CAS PubMed.
- M. Zheng, J. Hou, L. L. Hua, W. Y. Tang, L. W. Zhan and B. D. Li, Org. Lett., 2021, 23, 5128–5132 CrossRef CAS PubMed.
- Y. Y. Xue, Z. Z. Guo, X. Y. Chen, J. Y. Li, D. P. Zou, Y. J. Wu and Y. S. Wu, Org. Biomol. Chem., 2022, 20, 989–994 RSC.
- T. Inagaki, S. Sakurai, M. Yamanaka and M. Tobisu, Angew. Chem., Int. Ed., 2022, 61, e202202387 CrossRef CAS PubMed.
- Z. K. Zhang and X. L. Hu, ACS Catal., 2020, 10, 777–782 CrossRef CAS.
- B. Z. Zhao, Y. Q. Li, H. Y. Li, M. Belal, L. Zhu and G. Y. Yin, Sci. Bull., 2021, 66, 570–577 CrossRef CAS PubMed.
- Z. T. He, X. Q. Tang, L. B. Xie, M. Cheng, P. Tian and G. Q. Lin, Angew. Chem., Int. Ed., 2015, 54, 14815–14818 CrossRef CAS PubMed.
- C. Chen, W. Sun, L. Y. Liu, J. H. Zhao, Y. J. Huang, X. N. Shi, J. Ding, D. Q. Jiao and B. L. Zhu, Org. Chem. Front., 2021, 8, 2250–2255 RSC.
- H. Li, W. S. Huang, K. F. Yang, F. Ye, G. W. Yin, Z. Xu and L. W. Xu, Asian J. Org. Chem., 2021, 10, 2883–2887 CrossRef CAS.
- S. B. Krause, J. R. McAtee, G. P. A. Yap and D. A. Watson, Org. Lett., 2017, 19, 5641–5644 CrossRef CAS PubMed.
- K. Matsumoto, J. Huang, Y. Naganawa, H. Guo, T. Beppu, K. Sato, S. Shimada and Y. Nakajima, Org. Lett., 2018, 20, 2481–2484 CrossRef CAS PubMed.
- J. Ke, W. T. Liu, X. J. Zhu, X. F. Tan and C. He, Angew. Chem., Int. Ed., 2021, 60, 8744–8749 CrossRef CAS PubMed.
- D. S. Ni and M. K. Brown, ACS Catal., 2021, 11, 1858–1862 CrossRef CAS PubMed.
- H. Zhang, E. Wang, S. Geng, Z. Liu, Y. He, Q. Peng and Z. Feng, Angew. Chem., Int. Ed., 2021, 60, 10211–10218 CrossRef CAS PubMed.
- J. Dong, X.-A. Yuan, Z. Yan, L. Mu, J. Ma, C. Zhu and J. Xie, Nat. Chem., 2021, 13, 182–190 CrossRef CAS PubMed.
- K. Kikushima, Y. Etou, R. Kamura, I. Takeda, H. Ito, M. Ohashi and S. Ogoshi, Org. Lett., 2020, 22, 8167–8172 CrossRef CAS PubMed.
- L. Zhang and M. Oestreich, Org. Lett., 2018, 20, 8061–8063 CrossRef CAS PubMed.
- W. Yuan, P. Orecchia and M. Oestreich, Chem. – Eur. J., 2018, 24, 19175–19178 CrossRef CAS PubMed.
- Z.-Z. Zhao, X. Pang, X.-X. Wei, X.-Y. Liu and X.-Z. Shu, Angew. Chem., Int. Ed., 2022, 61, e202200215 CrossRef CAS PubMed.
- H. Yamagishi, H. Saito, J. Shimokawa and H. Yorimitsu, ACS Catal., 2021, 11, 10095–10103 CrossRef CAS.
- J. Duan, K. Wang, G.-L. Xu, S. Kang, L. Qi, X.-Y. Liu and X.-Z. Shu, Angew. Chem., Int. Ed., 2020, 59, 23083–23088 CrossRef CAS PubMed.
- J. Zhang, Y. Zhang, S. Geng, S. Chen, Z. Liu, X. Zeng, Y. He and Z. Feng, Org. Lett., 2020, 22, 2669–2674 CrossRef CAS PubMed.
- V. Murugesan, V. Balakrishnan and R. Rasappan, J. Catal., 2019, 377, 293–298 CrossRef CAS.
- T. Yoshida, L. Ilies and E. Nakamura, Org. Lett., 2018, 20, 2844–2847 CrossRef CAS PubMed.
- K. Hitoshio, H. Yamagishi, J. Shimokawa and H. Yorimitsu, Chem. Commun., 2021, 57, 6867–6870 RSC.
- H. J. Lee, C. Kwak, D. P. Kim and H. Kim, Green Chem., 2021, 23, 1193–1199 RSC.
- T. Nagata, T. Inoue, X. J. Lin, S. Ishimoto, S. Nakamichi, H. Oka, R. Kondo, T. Suzuki and Y. Obora, RSC Adv., 2019, 9, 17425–17431 RSC.
- D. Y. Wang, X. Wen, C. D. Xiong, J. N. Zhao, C. Y. Ding, Q. Meng, H. Zhou, C. Wang, M. Uchiyama, X. J. Lu and A. Zhang, iScience, 2019, 15, 307–315 CrossRef CAS PubMed.
- S. H. Zheng, T. Y. Zhang and H. Maekawa, J. Org. Chem., 2020, 85, 13965–13972 CrossRef CAS PubMed.
- L. J. You, W. Yuan and C. A. He, Eur. J. Org. Chem., 2021, 2021, 3079–3082 CrossRef CAS.
- J. N. Zhao, M. Kayumov, D. Y. Wang and A. Zhang, Org. Lett., 2019, 21, 7303–7306 CrossRef CAS PubMed.
- B. Su, T. G. Zhou, X. W. Li, X. R. Shao, P. L. Xu, W. L. Wu, J. F. Hartwig and Z. J. Shi, Angew. Chem., Int. Ed., 2017, 56, 1092–1096 CrossRef CAS PubMed.
- Y. Ma, B. Wang, L. Zhang and Z. Hou, J. Am. Chem. Soc., 2016, 138, 3663–3666 CrossRef CAS PubMed.
- Y. D. Hua, P. Asgar, T. Avullala and J. Jeon, J. Am. Chem. Soc., 2016, 138, 7982–7991 CrossRef CAS PubMed.
- H. Fang, L. Guo, Y. Zhang, W. Yao and Z. Huang, Org. Lett., 2016, 18, 5624–5627 CrossRef CAS PubMed.
- W. B. Liu, D. P. Schuman, Y. F. Yang, A. A. Toutov, Y. Liang, H. F. T. Klare, N. Nesnas, M. Oestreich, D. G. Blackmond, S. C. Virgil, S. Banerjee, R. N. Zare, R. H. Grubbs, K. N. Houk and B. M. Stoltz, J. Am. Chem. Soc., 2017, 139, 6867–6879 CrossRef CAS PubMed.
- S. H. Liu, P. Pan, H. Q. Fan, H. Li, W. Wang and Y. Q. Zhang, Chem. Sci., 2019, 10, 3817–3825 RSC.
- J. Wen, B. Dong, J. J. Zhu, Y. Zhao and Z. Z. Shi, Angew. Chem., Int. Ed., 2020, 59, 10909–10912 CrossRef CAS PubMed.
- C. H. Dai, Y. L. Zhan, P. Liu and P. P. Sun, Green Chem., 2021, 23, 314–319 RSC.
- S. Liu, S. L. Zhang, Q. Lin, Y. Q. Huang and B. Li, Org. Lett., 2019, 21, 1134–1138 CrossRef CAS PubMed.
- W. X. Xu, H. L. Teng, Y. Luo, S. J. Lou, M. Nishiura and Z. M. Hou, Chem. – Asian J., 2020, 15, 753–756 CrossRef CAS PubMed.
- W. Z. Li and Z. X. Wang, Org. Biomol. Chem., 2021, 19, 5082–5086 RSC.
- J. H. Li, M. Y. Ding and C. Jiang, Org. Lett., 2021, 23, 9036–9040 CrossRef CAS PubMed.
- Y. F. Li, K. C. Shu, P. Liu and P. P. Sun, Org. Lett., 2020, 22, 6304–6307 CrossRef CAS PubMed.
- X. Gong, P. Deng and J. H. Cheng, ChemCatChem, 2022, 14, e202200060 CrossRef CAS.
- G. Pandey, S. K. Tiwari, P. Singh and P. K. Mondal, Org. Lett., 2021, 23, 7730–7734 CrossRef CAS PubMed.
- T. Thorwart and L. Greb, Chem. Sci., 2023, 14, 11237–11242 RSC.
- Y. C. Wu, Y. H. Huang, X. Y. Chen and P. Wang, Org. Lett., 2020, 22, 6657–6661 CrossRef CAS PubMed.
- S. Som, J. Choi, D. Katsoulis and K. L. Lee, Chem. Sci., 2022, 13, 10759–10764 RSC.
- Z.-B. Yan, M. Peng, Q.-L. Chen, K. Lu, Y.-Q. Tu, K.-L. Dai, F.-M. Zhang and X.-M. Zhang, Chem. Sci., 2021, 12, 9748–9753 RSC.
- M. D. Visco, J. M. Wieting and A. E. Mattson, Org. Lett., 2016, 18, 2883–2885 CrossRef CAS PubMed.
- D. Chen, D. X. Zhu and M. H. Xu, J. Am. Chem. Soc., 2016, 138, 1498–1501 CrossRef CAS PubMed.
- V. Balakrishnan, V. Murugesan, B. Chindan and R. Rasappan, Org. Lett., 2021, 23, 1333–1338 CrossRef CAS PubMed.
- W. Y. Qi, J. S. Zhen, X. H. Xu, X. Du, Y. H. Li, H. Yuan, Y. S. Guan, X. Wei, Z. Y. Wang, G. H. Liang and Y. Luo, Org. Lett., 2021, 23, 5988–5992 CrossRef CAS PubMed.
- X. Zhang, P. Y. Geng, G. X. Liu and Z. Huang, Organometallics, 2021, 40, 2365–2370 CrossRef CAS.
- B. B. Zhan, J. Fan, L. Jin and B. F. Shi, ACS Catal., 2019, 9, 3298–3303 CrossRef CAS.
- J. L. Pan, Q. Z. Li, T. Y. Zhang, S. H. Hou, J. C. Kang and S. Y. Zhang, Chem. Commun., 2016, 52, 13151–13154 RSC.
- P. Liu, J. H. Tang and X. M. Zeng, Org. Lett., 2016, 18, 5536–5539 CrossRef CAS PubMed.
- J. H. Li and C. Jiang, Org. Lett., 2021, 23, 5359–5362 CrossRef CAS PubMed.
- S. Bähr, W. Xue and M. Oestreich, ACS Catal., 2019, 9, 16–24 CrossRef.
- X. W. Liu, C. Zarate and R. Martin, Angew. Chem., Int. Ed., 2019, 58, 2064–2068 CrossRef CAS PubMed.
- C. K. Chu, Y. Liang and G. C. Fu, J. Am. Chem. Soc., 2016, 138, 6404–6407 CrossRef CAS PubMed.
- S. Mallick, E. U. Wurthwein and A. Studer, Org. Lett., 2020, 22, 6568–6572 CrossRef CAS PubMed.
- T. Y. Zhang, S. H. Zheng, T. Kobayashi and H. Maekawa, Org. Lett., 2021, 23, 7129–7133 CrossRef CAS PubMed.
- H. F. Guo, X. Chen, C. L. Zhao and W. He, Chem. Commun., 2015, 51, 17410–17412 RSC.
- M. M. Xing, H. H. Cui and C. Zhang, Org. Lett., 2021, 23, 7645–7649 CrossRef CAS PubMed.
- B. Yang and Z. X. Wang, Org. Lett., 2019, 21, 7965–7969 CrossRef CAS PubMed.
- B. Vulovic, A. P. Cinderella and D. A. Watson, ACS Catal., 2017, 7, 8113–8117 CrossRef CAS PubMed.
- A. A. Toutov, K. N. Betz, D. P. Schuman, W. B. Liu, A. Fedorov, B. M. Stoltz and R. H. Grubbs, J. Am. Chem. Soc., 2017, 139, 1668–1674 CrossRef CAS PubMed.
- M. Y. Huang, J. M. Yang, Y. T. Zhao and S. F. Zhu, ACS Catal., 2019, 9, 5353–5357 CrossRef CAS.
- C. Y. He, L. B. Xie, R. Ding, P. Tian and G. Q. Lin, Tetrahedron, 2019, 75, 1682–1688 CrossRef CAS.
- T. He, B. Li, L. C. Liu, W. P. Ma and W. He, Chem. – Eur. J., 2021, 27, 5648–5652 CrossRef CAS PubMed.
- M. F. Wisthoff, S. B. Pawley, A. P. Cinderella and D. A. Watson, J. Am. Chem. Soc., 2020, 142, 12051–12055 CrossRef CAS PubMed.
- Y. Nakamura, S. Ozawa, S. Yoshida and T. Hosoya, Chem. Lett., 2019, 48, 1296–1299 CrossRef CAS.
- C. Bellini, V. Dorcet, J. F. Carpentier, S. Tobisch and Y. Sarazin, Chem. – Eur. J., 2016, 22, 4564–4583 CrossRef CAS PubMed.
- A. A. Toutov, K. N. Betz, M. C. Haibach, A. M. Romine and R. H. Grubbs, Org. Lett., 2016, 18, 5776–5779 CrossRef CAS PubMed.
- D. D. Xu, J. C. Chen, J. Y. Li, Y. B. Zhu, S. Y. Wu, J. Q. Zhang, X. Y. Liu and B. M. Fan, Asian J. Org. Chem., 2021, 10, 1402–1405 CrossRef CAS.
- Z. C. Wang, H. Q. Fang, G. X. Liu and Z. Huang, Org. Lett., 2021, 23, 7603–7607 CrossRef CAS PubMed.
- R. Ortega, J. Sanchez-Quesada, C. Lorenz, G. Dolega, A. Karawajczyk, M. Sanz, G. Showell and F. Giordanetto, Bioorg. Med. Chem., 2015, 23, 2716–2720 CrossRef CAS PubMed.
- T. Ohmura, I. Sasaki and M. Suginome, Org. Lett., 2019, 21, 1649–1653 CrossRef CAS PubMed.
- W. Reid, J. R. McAtee and D. A. Watson, Organometallics, 2019, 38, 3796–3803 CrossRef CAS PubMed.
- M. Murai, Asian J. Org. Chem., 2022, 11, e202100651 CrossRef CAS.
- Y. K. Xu, W. W. Xu, X. Y. Chen, X. A. Luo, H. Y. Lu, M. H. Zhang, X. M. Yang, G. B. Deng, Y. Liang and Y. Yang, Chem. Sci., 2021, 12, 11756–11761 RSC.
- J. F. Huo, K. B. Zhong, Y. Z. Xue, M. M. Lyu, Y. F. Ping, W. B. Ouyang, Z. X. Liu, Y. Lan and J. B. Wang, Chem. – Eur. J., 2022, 28, e202200191 CrossRef CAS PubMed.
- J. R. Vale, A. Valkonen, C. A. M. Afonso and N. R. Candeias, Org. Chem. Front., 2019, 6, 3793–3798 RSC.
- S. Yoshioka, Y. Fujii, H. Tsujino, T. Uno, H. Fujioka and M. Arisawa, Chem. Commun., 2017, 53, 5970–5973 RSC.
- M. Fischer, C. Burschka and R. Tacke, Organometallics, 2014, 33, 1020–1029 CrossRef CAS.
- T. He, G. Q. Wang, V. Bonetti, H. F. T. Klare and M. Oestreich, Angew. Chem., Int. Ed., 2020, 59, 12186–12191 CrossRef CAS PubMed.
- M. Czyzewski, J. D. Sellars, T. Guliashvili, J. Tibbelin, L. Johnstone, J. Bower, M. Box, R. D. M. Davies, H. Ottosson and P. G. Steel, Chem. Commun., 2014, 50, 2919–2921 RSC.
- H. Arii, Y. Yano, K. Nakabayashi, S. Yamaguchi, M. Yamamura, K. Mochida and T. Kawashima, J. Org. Chem., 2016, 81, 6314–6319 CrossRef CAS PubMed.
- Y. Yang, R. J. Song, Y. Li, X. H. Ouyang, J. H. Li and D. L. He, Chem. Commun., 2018, 54, 1441–1444 RSC.
- Y. Qin, J. L. Han, C. W. Ju and D. B. Zhao, Angew. Chem., Int. Ed., 2020, 59, 8481–8485 CrossRef CAS PubMed.
- J. W. Guo, S. Liu, Q. J. Pang, H. Y. Zhang, L. Gao, L. Chen and Z. L. Song, Org. Lett., 2022, 24, 726–730 CrossRef CAS PubMed.
- M. H. Zhu, X. W. Zhang, M. Usman, H. J. Cong and W. B. Liu, ACS Catal., 2021, 11, 5703–5708 CrossRef CAS.
- X. C. Wang, H. R. Wang, X. F. Xu and D. B. Zhao, Eur. J. Org. Chem., 2021, 2021, 3039–3042 CrossRef CAS.
- D. Y. Wang, M. J. Li, X. Y. Chen, M. Y. Wang, Y. Liang, Y. Zhao, K. N. Houk and Z. Z. Shi, Angew. Chem., Int. Ed., 2021, 60, 7066–7071 CrossRef CAS PubMed.
- K. P. C. Vollhardt, Angew. Chem., Int. Ed. Engl., 1984, 23, 539–556 CrossRef.
- A. Gomtsyan, Chem. Heterocycl. Compd., 2012, 48, 7–10 CrossRef CAS.
- E. Kabir and M. Uzzaman, Results Chem., 2022, 4, 100606 CrossRef CAS.
- W. Wang, L. Gao and Z. Song, Synthesis, 2022, 54, 2749–2764 CrossRef CAS.
- C. M. Reid, K. N. Fanning, L. S. Fowler and A. Sutherland, Tetrahedron, 2015, 71, 245–251 CrossRef CAS.
- A. Rene, N. Vanthuyne, J. Martinez and F. Cavelier, Amino Acids, 2013, 45, 301–307 CrossRef CAS PubMed.
- M. Fischer and R. Tacke, Organometallics, 2013, 32, 7181–7185 CrossRef CAS.
- L. H. Luhning, J. Strehl, M. Schmidtmann and S. Doye, Chem. – Eur. J., 2017, 23, 4197–4202 CrossRef PubMed.
- Y. Qin, L. H. Li, J. Y. Liang, K. L. Li and D. B. Zhao, Chem. Sci., 2021, 12, 14224–14229 RSC.
- H. P. Lv, R. D. Laishram, J. C. Chen, R. Khan, Y. B. Zhu, S. Y. Wu, J. Q. Zhang, X. Y. Liu and B. M. Fan, Chem. Commun., 2021, 57, 3660–3663 RSC.
- D. Limnios and C. G. Kokotos, ACS Catal., 2013, 3, 2239–2243 CrossRef CAS.
- K. K. Wang, J. M. Zhou, Y. T. Jiang, M. M. Zhang, C. Wang, D. Xue, W. J. Tang, H. M. Sun, J. L. Xiao and C. Q. Li, Angew. Chem., Int. Ed., 2019, 58, 6380–6384 CrossRef CAS PubMed.
- K. F. Zhang, L. Yang, Y. F. Hu, C. H. Fan, Y. R. Zhao, L. Bai, Y. L. Li, F. X. Shi, J. Liu and W. Xie, Angew. Chem., Int. Ed., 2020, 59, 18003–18009 CrossRef CAS PubMed.
- H. Liang, L. J. Wang, Y. X. Ji, H. Wang and B. Zhang, Angew. Chem., Int. Ed., 2021, 60, 1839–1844 CrossRef CAS PubMed.
- Z. J. Garlets and H. M. L. Davies, Org. Lett., 2018, 20, 2168–2171 CrossRef CAS PubMed.
- M. P. Wiesenfeldt, T. Knecht, C. Schlepphorst and F. Glorius, Angew. Chem., Int. Ed., 2018, 57, 8297–8300 CrossRef CAS PubMed.
- Z. Wu, S. B. J. Kan, R. D. Lewis, B. J. Wittmann and F. H. Arnold, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 8852–8858 CrossRef CAS PubMed.
- Z. Wu, S. B. J. Kan, R. D. Lewis, B. J. Wittmann and F. H. Arnold, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 788–789 CrossRef PubMed.
- J. J. Feng and M. Oestreich, Angew. Chem., Int. Ed., 2019, 58, 8211–8215 CrossRef CAS PubMed.
- L. Wei, C. Shen, Y. Z. Hu, H. Y. Tao and C. J. Wang, Chem. Commun., 2019, 55, 6672–6684 RSC.
- D. L. Mu, W. Yuan, S. Y. Chen, N. Wang, B. Yang, L. J. You, B. Zu, P. Y. Yu and C. He, J. Am. Chem. Soc., 2020, 142, 13459–13468 CrossRef CAS PubMed.
- B. Yang, W. Yang, Y. H. Guo, L. J. You and C. He, Angew. Chem., Int. Ed., 2020, 59, 22217–22222 CrossRef CAS PubMed.
- L. L. Yang, J. Ouyang, H. N. Zou, S. F. Zhu and Q. L. Zhou, J. Am. Chem. Soc., 2021, 143, 6401–6406 CrossRef CAS PubMed.
- V. Carreras, C. Besnard, V. Gandon and T. Ollevier, Org. Lett., 2019, 21, 9094–9098 CrossRef CAS PubMed.
- H. P. Zhang and D. B. Zhao, ACS Catal., 2021, 11, 10748–10753 CrossRef CAS.
- Y. J. Zhang, M. Tong, Q. Gao, P. K. Zhang and S. M. Xu, Tetrahedron Lett., 2019, 60, 1210–1212 CrossRef CAS.
- Y. Zhang, J. C. Guo, J. N. Han, X. G. Zhou, W. Cao and Z. Q. Fu, Org. Biomol. Chem., 2021, 19, 6412–6416 RSC.
- R. Chowdhury, A. K. Dubey and S. K. Ghosh, Asian J. Org. Chem., 2021, 10, 1173–1183 CrossRef CAS.
- T. Kobayashi, S. Nishino, M. Miura and K. Hirano, Org. Lett., 2022, 24, 1418–1422 CrossRef CAS PubMed.
- D. C. Bai, F. Wu, L. N. Chang, M. M. Wang, H. Wu and J. B. Chang, Angew. Chem., Int. Ed., 2022, 61, e202114918 CrossRef CAS PubMed.
- Y. Liu, M. Zhan and P. F. Li, Chin. J. Chem., 2022, 40, 1028–1032 CrossRef CAS.
- J. Fotie, C. M. Matherne and J. E. Wroblewski, Chem. Biol. Drug Des., 2023, 102, 235–254 CrossRef CAS PubMed.
- S. N. Adamovich, E. N. Oborina, A. M. Nalibayeva and I. B. Rozentsveig, Molecules, 2022, 27, 3549 CrossRef CAS PubMed.
- T. Marzo and D. La Mendola, Inorganics, 2021, 9, 46 CrossRef CAS.
- C. C. Perez, F. R. Benatti, D. P. Martins Jr and A. A. Silva, Rev. Virtual Quim., 2021, 13, 981–992 CrossRef CAS.
- R. Ramesh and D. S. Reddy, J. Med. Chem., 2018, 61, 3779–3798 CrossRef CAS PubMed.
- S. Fujii and Y. Hashimoto, Future Med. Chem., 2017, 9, 485–505 CAS.
- E. Remond, C. Martin, J. Martinez and F. Cavelier, Chem. Rev., 2016, 116, 11654–11684 CrossRef CAS PubMed.
- N. F. Lazareva and I. M. Lazarev, Russ. Chem. Bull., 2015, 64, 1221–1232 CrossRef CAS.
- B. S. Sekhon, Res. Pharm. Sci., 2013, 8, 145–158 CAS.
- N. F. Lazareva, Russ. Chem. Bull., 2011, 60, 615–632 CrossRef CAS.
- S. Gately and R. West, Drug Dev. Res., 2007, 68, 156–163 CrossRef CAS.
- P. K. Pooni and G. A. Showell, Mini-Rev. Med. Chem., 2006, 6, 1169–1177 CrossRef CAS PubMed.
- W. Bains and R. Tacke, Curr. Opin. Drug Discovery Dev., 2003, 6, 526–543 CAS.
- R. J. Fessenden and J. S. Fessenden, in Advances in Organometallic Chemistry, ed. F. G. A. Stone and R. West, Academic Press, 1980, vol. 18, pp. 275–299 Search PubMed.
- L. R. Garson and L. K. Kirchner, J. Pharm. Sci., 1971, 60, 1113–1127 CrossRef CAS PubMed.
- S. Fujii, Med. Chem. Commun., 2016, 7, 1082–1092 RSC.
- D. Lowe, Silicon In Drug Molecules: Not Quite There, in In the Pipeline, 2012, https://www.science.org/content/blog-post/silicon-drug-molecules-not-quite-there Search PubMed.
- D. Lowe, Silicon In Drug Molecules, Revisited, in In the Pipeline, 2018, https://www.science.org/content/blog-post/silicon-drug-molecules-revisited Search PubMed.
- D. Lowe, Silicon Stays in the Shadows, in In the Pipeline, 2017, https://www.science.org/content/blog-post/silicon-stays-shadows Search PubMed.
- D. Lowe, Odd Elements in Drugs: Silicon, in In the Pipeline, 2004, https://www.science.org/content/blog-post/silicon-drug-molecules-not-quite-there Search PubMed.
- F. T. Chiu, Y. H. Chang, G. Ozkan, G. Zon, K. C. Fichter and L. R. Phillips, J. Pharm. Sci., 1982, 71, 542–551 CrossRef CAS PubMed.
- L. T. Farol and K. B. Hymes, Expert Rev. Anticancer Ther., 2004, 4, 180–188 CrossRef CAS PubMed.
- R. Gniadecki, C. Assaf, M. Bagot, R. Dummer, M. Duvic, R. Knobler, A. Ranki, P. Schwandt and S. Whittaker, Br. J. Dermatol., 2007, 157, 433–440 CrossRef CAS PubMed.
- K. H. Dragnev, W. J. Petty, S. J. Shah, L. D. Lewis, C. C. Black, V. Memoli, W. C. Nugent, T. Hermann, A. Negro-Vilar, J. R. Rigas and E. Dmitrovsky, Clin. Cancer Res., 2007, 13, 1794–1800 CrossRef CAS PubMed.
- R. Ramlau, P. Zatloukal, J. Jassem, P. Schwarzenberger, S. V. Orlov, M. Gottfried, J. R. Pereira, G. Temperley, R. Negro-Vilar, S. Rahal, J. K. Zhang, A. Negro-Vilar and Z. E. Dziewanowska, J. Clin. Oncol., 2008, 26, 1886–1892 CrossRef CAS PubMed.
- M. I. Dawson and Z. Xia, Biochim. Biophys. Acta, Mol. Cell Biol. Lipids, 2012, 1821, 21–56 CrossRef CAS PubMed.
- J. O. Daiss, C. Burschka, J. S. Mills, J. G. Montana, G. A. Showell, I. Fleming, C. Gaudon, D. Ivanova, H. Gronemeyer and R. Tacke, Organometallics, 2005, 24, 3192–3199 CrossRef CAS.
- M. W. Buttner, C. Burschka, J. O. Daiss, D. Ivanova, N. Rochel, S. Kammerer, C. Peluso-Iltis, A. Bindler, C. Gaudon, P. Germain, D. Moras, H. Gronemeyer and R. Tacke, ChemBioChem, 2007, 8, 1688–1699 CrossRef PubMed.
- J. B. Bauer, W. P. Lippert, S. Dörrich, D. Tebbe, C. Burschka, V. B. Christie, D. M. Tams, A. P. Henderson, B. A. Murray, T. B. Marder, S. A. Przyborski and R. Tacke, ChemMedChem, 2011, 6, 1509–1517 CrossRef CAS PubMed.
- W. P. Lippert, C. Burschka, K. Götz, M. Kaupp, D. Ivanova, C. Gaudon, Y. Sato, P. Antony, N. Rochel, D. Moras, H. Gronemeyer and R. Tacke, ChemMedChem, 2009, 4, 1143–1152 CrossRef CAS PubMed.
- T. Yamakawa, H. Kagechika, E. Kawachi, Y. Hashimoto and K. Shudo, J. Med. Chem., 1990, 33, 1430–1437 CrossRef CAS PubMed.
- R. Tacke, V. Muller, M. W. Buttner, W. P. Lippert, R. Bertermann, J. O. Daiss, H. Khanwalkar, A. Furst, C. Gaudon and H. Gronemeyer, ChemMedChem, 2009, 4, 1797–1802 CrossRef CAS PubMed.
- K. B. Higginbotham, R. Lozano, T. Brown, Y. Z. Patt, T. Arima, J. L. Abbruzzese and M. B. Thomas, J. Cancer Res. Clin. Oncol., 2008, 134, 1325–1335 CrossRef CAS PubMed.
- T. Oikawa, S. Fujii, S. Mori, H. Masuno, E. Kawachi and H. Kagechika, ChemMedChem, 2022, 17, e202200176 CrossRef CAS PubMed.
- A. Zablotskaya, I. Segal, Y. Popelis, E. Lukevics, S. Baluja, I. Shestakova and I. Domracheva, Appl. Organomet. Chem., 2006, 20, 721–728 CrossRef CAS.
- A. Zablotskaya, I. Segal, Y. Popelis, S. Grinberga, I. Shestakova, V. Nikolajeva and D. Eze, Appl. Organomet. Chem., 2013, 27, 114–124 CrossRef CAS.
- A. Zablotskaya, I. Segal, A. Kemme, S. Germane, J. Popelis, E. Lukevics, R. Berger and H. Spies, Chem. Heterocycl. Compd., 2002, 543–555 Search PubMed.
- A. Zablotskaya, I. Segal, S. Germane, I. Shestakova, I. Domracheva, A. Nesterova, A. Geronikaki and E. Lukevics, Chem. Heterocycl. Compd., 2002, 38, 859–866 CrossRef CAS.
- A. Zablotskaya, I. Segal, S. Belyakov and E. Lukevics, Appl. Organomet. Chem., 2006, 20, 149–154 CrossRef CAS.
- H. Spies, T. Fietz, A. Zablotskaya, S. Belyakov and E. Lukevics, Chem. Heterocycl. Compd., 1999, 116–125 Search PubMed.
- I. Segal, A. Zablotskaya and E. Lukevits, J. Heterocyclic Chem., 2005, 713–725 Search PubMed.
- E. Lukevics, A. Zablotskaya, I. Segal, S. Germane and J. Popelis, Chem. Heterocycl. Compd., 2003, 941–947 Search PubMed.
- E. Lukevics, I. Segal, A. Zablotskaya and S. Germane, Chem. Heterocycl. Compd., 1996, 793–799 Search PubMed.
- E. Lukevics, I. Segal, I. Birgele and A. Zablotskaya, Chem. Heterocycl. Compd., 1998, 1253–1258 Search PubMed.
- E. Lukevics, S. Germane, I. Segal and A. Zablotskaya, Chem. Heterocycl. Compd., 1997, 270–274 Search PubMed.
- A. Zablotskaya, I. Segal, G. Kazachonokh, Y. Popelis, I. Shestakova and V. Nikolajeva, Silicon, 2018, 10, 1129–1138 CrossRef CAS.
- A. Zablotskaya, I. Segal and E. Lukevics, Appl. Organomet. Chem., 2010, 24, 150–157 CrossRef CAS.
- L. Ignatovich, V. Romanov, J. Spura, J. Popelis, I. Domracheva and I. Shestakova, Chem. Heterocycl. Compd., 2012, 47, 1502–1508 CrossRef CAS.
- P. C. Lo, C. M. H. Chan, J. Y. Liu, W. P. Fong and D. K. P. Ng, J. Med. Chem., 2007, 50, 2100–2107 CrossRef CAS PubMed.
- J. T. F. Lau, P. C. Lo, Y. M. Tsang, W. P. Fong and D. K. P. Ng, Chem. Commun., 2011, 47, 9657–9659 RSC.
- M. Bispo, P. M. R. Pereira, F. Setaro, M. S. Rodriguez-Morgade, R. Fernandes, T. Torres and J. P. C. Tome, ChemPlusChem, 2018, 83, 855–860 CrossRef CAS PubMed.
- C. M. H. Chan, P. C. Lo, S. L. Yeung, D. K. P. Ng and W. P. Fong, Cancer Biol. Ther., 2010, 10, 126–134 CrossRef CAS PubMed.
- J. M. Park, C. Y. Jung, W. D. Jang and J. Y. Jaung, ACS Appl. Bio Mater., 2021, 4, 1988–2000 CrossRef CAS PubMed.
- H. Y. Yenilmez, N. Farajzadeh, N. Güler Kuşçulu, D. Bahar, S. Özdemir, G. Tollu, M. Güllü and Z. Altuntaş Bayır, Chem. Biodiversity, 2023, 20, e202201167 CrossRef CAS PubMed.
- H. Y. Yenilmez, N. Farajzadeh, G. Tollu, N. G. Kuşçulu, D. Bahar, S. Özdemir and Z. A. Bayır, ChemistrySelect, 2023, 8, e202300856 CrossRef CAS.
- G. Y. Atmaca, M. Aksel, M. D. Bilgin and A. Erdoğmuş, Photodiagn. Photodyn. Ther., 2023, 42, 103339 CrossRef CAS PubMed.
- H. Yalazan, D. Akkaya, G. Seyhan, B. Barut and H. Kantekin, Appl. Organomet. Chem., 2023, 37, e7040 CrossRef CAS.
- E. Georgiopoulou, E. Kavetsou, E. Alexandratou, A. Detsi and K. Politopoulos, Comparative characterization of SiCl2Pc and its cyclodextrin complexes as photosensitizers in photodynamic therapy, in Translational Biophotonics: Diagnostics and Therapeutics III, Proc. SPIE, 2023, vol. 12627, p. 1262718, DOI:10.1117/12.2670864.
- N. Farajzadeh, N. Güler Kuşçulu, H. Y. Yenilmez, D. Bahar and Z. Altuntaş Bayır, Dalton Trans., 2022, 51, 7539–7550 RSC.
- N. Farajzadeh, N. Güler Kuşçulu, H. Y. Yenilmez, D. Bahar and Z. Altuntaş Bayır, New J. Chem., 2022, 46, 19863–19873 RSC.
- G. Y. Atmaca, M. Aksel, B. Keskin, M. D. Bilgin and A. Erdoğmuş, Dyes Pigm., 2021, 184, 108760 CrossRef CAS.
- D. D. Ma, X. Q. Chen, Y. H. Wang, Q. M. Guo, Q. H. Ye, R. T. Guo, S. H. Xiao, Q. Ye, Y. D. Huang and Y. R. Peng, J. Lumin., 2019, 207, 597–601 CrossRef CAS.
- Q. Liu, M. Pang, S. Tan, J. Wang, Q. Chen, K. Wang, W. Wu and Z. Hong, J. Cancer, 2018, 9, 310–320 CrossRef PubMed.
- C. Jing, R. L. Wang, H. L. Ou, A. Li, Y. L. An, S. T. Guo and L. Q. Shi, Chem. Commun., 2018, 54, 3985–3988 RSC.
- K. Z. Chen, S. J. Pan, X. M. Zhuang, H. F. Lv, S. L. Que, S. S. Xie, H. Q. Yang and Y. R. Peng, J. Nanopart. Res., 2016, 18, 197 CrossRef.
- A. R. Simioni, F. L. Primo and A. C. Tedesco, J. Laser Appl., 2012, 24, 012004 CrossRef.
- K. Mitra and M. C. T. Hartman, Org. Biomol. Chem., 2021, 19, 1168–1190 RSC.
- C. Uslan and B. Şebnem Sesalan, Inorg. Chim. Acta, 2013, 394, 353–362 CrossRef CAS.
- W. K. Anderson, R. Kasliwal, D. M. Houston, Y. S. Wang, V. L. Narayanan, R. D. Haugwitz and J. Plowman, J. Med. Chem., 1995, 38, 3789–3797 CrossRef PubMed.
- A. Varga, P. Hegyes, J. Molnar, I. Mucsi, A. Hever, D. Szabo, S. Kiesig, H. Lage, D. Gaal and J. Nacsa, Substituted disiloxanes, method for the production thereof and the use thereof for reversal of multidrug resistance (MDR), EP Pat., 1432717B1, 1999 Search PubMed.
- J. Molnar, I. Mucsi, J. Nacsa, A. Hever, N. Gyemant, K. Ugocsai, P. Hegyes, S. Kiessig, D. Gaal, H. Lage and A. Varga, Anticancer Res., 2004, 24, 865–871 CAS.
- A. Zalatnai and J. Molnár, In Vivo, 2006, 20, 137–140 CAS.
- U. Olszewski, R. Zeillinger, M. Demirel Kars, A. Zalatnai, J. Molnar and G. Hamilton, Anti-Cancer Agents Med. Chem., 2012, 12, 663–671 CrossRef CAS PubMed.
- T. Harukuni, M. Takashi, S. Nobutaka, H. Judit, V. Andrea, E. Helga, M. Ilona, O. Ulrike, H. Gerhard, A. Leonard and M. Joseph, Anticancer Res., 2013, 33, 2021–2027 Search PubMed.
- O. Wesołowska, K. Michalak, M. Błaszczyk, J. Molnár and K. Środa-Pomianek, Molecules, 2020, 25, 1654 CrossRef PubMed.
- A. M. Thompson, J. W. Blunt, M. H. G. Munro, N. B. Perry and L. K. Pannell, J. Chem. Soc., Perkin Trans. 1, 1992, 1335–1342 RSC.
- G. Ş. Karatoprak, E. Küpeli Akkol, Y. Genç, H. Bardakcı, Ç. Yücel and E. Sobarzo-Sánchez, Molecules, 2020, 25, 2560 CrossRef CAS PubMed.
- M. Nakamura, D. Kajita, Y. Matsumoto and Y. Hashimoto, Bioorg. Med. Chem., 2013, 21, 7381–7391 CrossRef CAS PubMed.
- D. Kajita, M. Nakamura, Y. Matsumoto, M. Ishikawa, Y. Hashimoto and S. Fujii, Bioorg. Med. Chem. Lett., 2015, 25, 3350–3354 CrossRef CAS PubMed.
- G. Astarita, B. Di Giacomo, S. Gaetani, F. Oveisi, T. R. Compton, S. Rivara, G. Tarzia, M. Mor and D. Piomelli, J. Pharmacol. Exp. Ther., 2006, 318, 563–570 CrossRef CAS PubMed.
- A. Belfiore, M. Genua and R. Malaguarnera, PPAR Res., 2009, 2009, 830501 CrossRef CAS PubMed.
- K. Maruyama, M. Nakamura, S. Tomoshige, K. Sugita, M. Makishima, Y. Hashimoto and M. Ishikawa, Bioorg. Med. Chem. Lett., 2013, 23, 4031–4036 CrossRef CAS PubMed.
- M. F. Boehm, P. Fitzgerald, A. Zou, M. G. Elgort, E. D. Bischoff, L. Mere, D. E. Mais, R. P. Bissonnette, R. A. Heyman, A. M. Nadzan, M. Reichman and E. A. Allegretto, Chem. Biol., 1999, 6, 265–275 CrossRef CAS PubMed.
- M. Nakamura, M. Makishima and Y. Hashimoto, Bioorg. Med. Chem., 2013, 21, 1643–1651 CrossRef CAS PubMed.
- D. Kajita, M. Nakamura, Y. Matsumoto, M. Makishima and Y. Hashimoto, Bioorg. Med. Chem. Lett., 2014, 22, 2244–2252 CrossRef CAS PubMed.
- J. P. M. Antonio, R. F. M. Frade, F. M. F. Santos, J. A. S. Coelho, C. A. M. Afonso, P. M. P. Gois and A. F. Trindade, RSC Adv., 2014, 4, 29352–29356 RSC.
- O. J. Donadel, T. Martin, V. S. Martin, J. Villar and J. M. Padrón, Bioorg. Med. Chem. Lett., 2005, 15, 3536–3539 CrossRef CAS PubMed.
- J. M. Padrón, O. J. Donadel, L. G. Leon, T. Martin and V. S. Martin, Lett. Drug Des. Discovery, 2006, 3, 29–34 CrossRef.
- L. G. Leon, P. O. Miranda, V. S. Martin, J. I. Padron and J. M. Padron, Bioorg. Med. Chem. Lett., 2007, 17, 3087–3090 CrossRef CAS PubMed.
- U. Sirion, S. Kasemsook, K. Suksen, P. Piyachaturawat, A. Suksamrarn and R. Saeeng, Bioorg. Med. Chem. Lett., 2012, 22, 49–52 CrossRef CAS PubMed.
- J. Nateewattana, R. Saeeng, S. Kasemsook, K. Suksen, S. Dutta, S. Jariyawat, A. Chairoungdua, A. Suksamrarn and P. Piyachaturawat, Invest. New Drugs, 2013, 31, 320–332 CrossRef CAS PubMed.
- J. Nateewattana, S. Dutta, S. Reabroi, R. Saeeng, S. Kasemsook, A. Chairoungdua, J. Weerachayaphorn, S. Wongkham and P. Piyachaturawat, Eur. J. Pharmacol., 2014, 723, 148–155 CrossRef CAS PubMed.
- S. Reabroi, A. Chairoungdua, R. Saeeng, T. Kasemsuk, W. Saengsawang, W. M. Zhu and P. Piyachaturawat, Biomed. Pharmacother., 2018, 101, 414–421 CrossRef CAS PubMed.
- J. Apisornopas, P. Silalai, T. Kasemsuk, A. Athipornchai, U. Sirion, K. Suksen, P. Piyachaturawat, A. Suksamrarn and R. Saeeng, Bioorg. Med. Chem. Lett., 2018, 28, 1558–1561 CrossRef CAS PubMed.
- C. R. Pungitore, L. G. León, C. García, V. S. Martín, C. E. Tonn and J. M. Padrón, Bioorg. Med. Chem. Lett., 2007, 17, 1332–1335 CrossRef CAS PubMed.
- H. A. Garro, C. García, V. S. Martín, C. E. Tonn and C. R. Pungitore, Bioorg. Med. Chem. Lett., 2015, 25, 914–918 CrossRef CAS PubMed.
- N. Khaiwa, N. R. Maarouf, M. H. Darwish, D. W. M. Alhamad, A. Sebastian, M. Hamad, H. A. Omar, G. Orive and T. H. Al-Tel, Eur. J. Med. Chem., 2021, 223, 113639 CrossRef CAS PubMed.
- E. Martino, S. Della Volpe, E. Terribile, E. Benetti, M. Sakaj, A. Centamore, A. Sala and S. Collina, Bioorg. Med. Chem. Lett., 2017, 27, 701–707 CrossRef CAS PubMed.
- V. J. Venditto and E. E. Simanek, Mol. Pharmaceutics, 2010, 7, 307–349 CrossRef CAS PubMed.
- D. Sriram, P. Yogeeswari, R. Thirumurugan and T. Ratan Bal, Nat. Prod. Res., 2005, 19, 393–412 CrossRef CAS PubMed.
- H. Josien, D. Bom, D. P. Curran, Y. H. Zheng and T. C. Chou, Bioorg. Med. Chem. Lett., 1997, 7, 3189–3194 CrossRef CAS.
- R. W. Versace, Expert Opin. Ther. Pat., 2003, 13, 751–760 CrossRef CAS.
- D. Bom, D. P. Curran, S. Kruszewski, S. G. Zimmer, J. Thompson Strode, G. Kohlhagen, W. Du, A. J. Chavan, K. A. Fraley, A. L. Bingcang, L. J. Latus, Y. Pommier and T. G. Burke, J. Med. Chem., 2000, 43, 3970–3980 CrossRef CAS PubMed.
- A. H. Van Hattum, H. M. Pinedo, H. M. M. Schlüper, F. H. Hausheer and E. Boven, Int. J. Cancer, 2000, 88, 260–266 CrossRef CAS PubMed.
- A. H. van Hattum, H. M. M. Schlüper, F. H. Hausheer, H. M. Pinedo and E. Boven, Int. J. Cancer, 2002, 100, 22–29 CrossRef CAS PubMed.
- S. Yao, P. Petluru, A. Parker, D. Ding, X. Chen, Q. Huang, H. Kochat and F. Hausheer, Cancer Chemother. Pharmacol., 2015, 75, 719–728 CrossRef CAS PubMed.
- A. Daud, N. Valkov, B. Centeno, J. Derderian, P. Sullivan, P. Munster, P. Urbas, R. C. DeConti, E. Berghorn, Z. Liu, F. Hausheer and D. Sullivan, Clin. Cancer Res., 2005, 11, 3009–3016 CrossRef CAS PubMed.
- P. N. Munster and A. I. Daud, Expert Opin. Invest. Drugs, 2011, 20, 1565–1574 CrossRef CAS PubMed.
- N. F. Lazareva, V. P. Baryshok and I. M. Lazarev, Arch. Pharm., 2018, 351, 1700297 CrossRef PubMed.
- A. K. Franz, P. D. Dreyfuss and S. L. Schreiber, J. Am. Chem. Soc., 2007, 129, 1020–1021 CrossRef CAS PubMed.
- K. J. Mccormick and W. R. Panje, Cancer Immunol., Immunother., 1986, 21, 226–232 CrossRef CAS PubMed.
- R. D. Maca and W. R. Panje, Cancer, 1982, 50, 483–489 CrossRef CAS PubMed.
- W. R. Panje, Arch. Otolaryngol., 1981, 107, 658–663 CrossRef CAS PubMed.
- C. M. Ulrich, J. Bigler and J. D. Potter, Nat. Rev. Cancer, 2006, 6, 130–140 CrossRef CAS PubMed.
- G. A. Bikzhanova, I. S. Toulokhonova, S. Gately and R. West, Silicon Chem., 2007, 3, 209–217 CrossRef.
- E. Lukevits, I. Segal, I. Birgele and A. Zablotskaya, Chem. Heterocycl. Compd., 1998, 34, 1076–1080 CrossRef CAS.
- A. Szilagyi, F. Fenyvesi, O. Majercsik, I. F. Pelyvas, I. Bacskay, P. Feher, J. Varadi, M. Vecsernyes and P. Herczegh, J. Med. Chem., 2006, 49, 5626–5630 CrossRef CAS PubMed.
- C. A. Black, J. W. Ucci, J. S. Vorpagel, M. C. Mauck and E. E. Fenlon, Bioorg. Med. Chem. Lett., 2002, 12, 3521–3523 CrossRef CAS PubMed.
- P. Guo, Y. W. Wang, X. T. Luo, X. L. Qi, L. P. Hou, Z. X. Xie and F. Q. Ye, Phosphorus, Sulfur Silicon Relat. Elem., 2014, 189, 511–518 CrossRef.
- J. L. Panayides, V. Mathieu, L. M. Y. Banuls, H. Apostolellis, N. Dahan-Farkas, H. Davids, L. Harmse, M. E. C. Rey, I. R. Green, S. C. Pelly, R. Kiss, A. Kornienko and W. A. L. van Otterlo, Bioorg. Med. Chem., 2016, 24, 2716–2724 CrossRef CAS PubMed.
- L. Harmse, N. Dahan-Farkas, J. L. Panayides, W. A. L. van Otterlo and C. Penny, PLoS One, 2015, 10, e0138607 CrossRef PubMed.
- M. A. Peterson, M. Oliveira, M. A. Christiansen and C. E. Cutler, Bioorg. Med. Chem. Lett., 2009, 19, 6775–6779 CrossRef CAS PubMed.
- J. R. Shelton, S. R. Burt and M. A. Peterson, Bioorg. Med. Chem. Lett., 2011, 21, 1484–1487 CrossRef CAS PubMed.
- J. R. Shelton, C. E. Cutler, M. S. Browning, J. Balzarini and M. A. Peterson, Bioorg. Med. Chem. Lett., 2012, 22, 6067–6071 CrossRef CAS PubMed.
- J. R. Shelton, C. E. Cutler, M. Oliveira, J. Balzarini and M. A. Peterson, Bioorg. Med. Chem., 2012, 20, 1008–1019 CrossRef CAS PubMed.
- J. R. Shelton, J. Balzarini and M. A. Peterson, Bioorg. Med. Chem. Lett., 2014, 24, 5107–5110 CrossRef CAS PubMed.
- A. Lanver and H. G. Schmalz, Eur. J. Org. Chem., 2005, 2005, 1444–1458 CrossRef.
- J. Velcicky, A. Lanver, J. Lex, A. Prokop, T. Wieder and H. G. Schmalz, Chem. – Eur. J., 2004, 10, 5087–5110 CrossRef CAS PubMed.
- C. Hirschhauser, J. Velcicky, D. Schlawe, E. Hessler, A. Majdalani, J. M. Neudorfl, A. Prokop, T. Wieder and H. G. Schmalz, Chem. – Eur. J., 2013, 19, 13017–13029 CrossRef PubMed.
- D. Schlawe, A. Majdalani, J. Velcicky, E. Hessler, T. Wieder, A. Prokop and H. G. Schmalz, Angew. Chem., Int. Ed., 2004, 43, 1731–1734 CrossRef CAS PubMed.
- C. Prinz, E. Vasyutina, G. Lohmann, A. Schrader, S. Romanski, C. Hirschhauser, P. Mayer, C. Frias, C. D. Herling, M. Hallek, H. G. Schmalz, A. Prokop, D. Mougiakakos and M. Herling, Mol. Cancer, 2015, 14, 114 CrossRef PubMed.
- R. Dasari, L. M. Banuls, M. Masi, S. C. Pelly, V. Mathieu, I. R. Green, W. A. L. van Otterlo, A. Evidente, R. Kiss and A. Kornienko, Bioorg. Med. Chem. Lett., 2014, 24, 923–927 CrossRef CAS PubMed.
- G. L. Nelson, C. T. Ronayne, L. N. Solano, S. K. Jonnalagadda, S. Jonnalagadda, J. Rumbley, J. Holy, T. Rose-Hellekant, L. R. Drewes and V. R. Mereddy, Sci. Rep., 2019, 9, 18266 CrossRef CAS PubMed.
- R. Kitel, A. Byczek-Wyrostek, K. Hopko, A. Kasprzycka and K. Walczak, Pharmaceuticals, 2021, 14, 1079 CrossRef CAS PubMed.
- A. Byczek-Wyrostek, R. Kitel, K. Rumak, M. Skonieczna, A. Kasprzycka and K. Walczak, Eur. J. Med. Chem., 2018, 150, 687–697 CrossRef CAS PubMed.
- G. G. Llanos, L. M. Araujo, I. A. Jimenez, L. M. Moujir and I. L. Bazzocchi, Eur. J. Med. Chem., 2012, 54, 499–511 CrossRef CAS PubMed.
- G. G. Llanos, L. M. Araujo, I. A. Jimenez, L. M. Moujir, J. Rodriguez, C. Jimenez and I. L. Bazzocchi, Eur. J. Med. Chem., 2017, 140, 52–64 CrossRef CAS PubMed.
- N. R. Perestelo, G. G. Llanos, C. P. Reyes, A. Amesty, K. Sooda, S. Afshinjavid, I. A. Jimenez, F. Jayid and I. L. Bazzocchi, J. Med. Chem., 2019, 62, 4571–4585 CrossRef CAS PubMed.
- R. Zhao, X. Ma, L. Bai, X. Li, K. Mamouni, Y. Yang, H. Liu, A. Danaher, N. Cook, O. Kucuk, R. S. Hodges, L. Gera and D. Wu, Neoplasia, 2021, 23, 1261–1274 CrossRef CAS PubMed.
- H. A. Garro, G. F. Reta, O. J. Donadel and C. R. Pungitore, Nat. Prod. Commun., 2016, 11, 1289–1292 CrossRef PubMed.
- S. Fujii, Y. Miyajima, H. Masuno and H. Kagechika, J. Med. Chem., 2013, 56, 160–166 CrossRef CAS PubMed.
- S. Ser
![[i with combining dot above]](https://www.rsc.org/images/entities/char_0069_0307.gif) n, J. Indian Chem. Soc., 2023, 100, 100939 CrossRef.
n, J. Indian Chem. Soc., 2023, 100, 100939 CrossRef. - H. Mousazadeh, M. Milani, N. Zarghami, E. Alizadeh and K. D. Safa, Basic Clin. Pharmacol. Toxicol., 2017, 121, 390–399 CrossRef CAS PubMed.
- K. D. Safa and H. Mousazadeh, Monatsh. Chem., 2016, 147, 1951–1961 CrossRef CAS.
- K. D. Safa and H. Mousazadeh, Synth. Commun., 2016, 46, 1595–1604 CrossRef CAS.
- E. Eryilmaz, Eur. J. Pharm. Sci., 2019, 134, 266–273 CrossRef CAS PubMed.
- M. Deluigi, A. Klipp, C. Klenk, L. Merklinger, S. A. Eberle, L. Morstein, P. Heine, P. R. E. Mittl, P. Ernst, T. M. Kamenecka, Y. He, S. Vacca, P. Egloff, A. Honegger and A. Plückthun, Sci. Adv., 2021, 7, eabe5504 CrossRef CAS PubMed.
- R. Fanelli, A. Chastel, S. Previti, E. Hindié, D. Vimont, P. Zanotti-Fregonara, P. Fernandez, P. Garrigue, F. Lamare, R. Schollhammer, L. Balasse, B. Guillet, E. Rémond, C. Morgat and F. Cavelier, Bioconjugate Chem., 2020, 31, 2339–2349 CrossRef CAS PubMed.
- S. Geurs, D. Clarisse, K. De Bosscher and M. D'hooghe, J. Med. Chem., 2023, 66, 7698–7729 CrossRef CAS PubMed.
- P. A. Marks and R. Breslow, Nat. Biotechnol., 2007, 25, 84–90 CrossRef CAS PubMed.
- A. S. Madsen, H. M. E. Kristensen, G. Lanz and C. A. Olsen, ChemMedChem, 2014, 9, 614–626 CrossRef CAS PubMed.
- I. S. Ignatyev, M. Montejo, P. G. Rodríguez Ortega and J. J. L. González, J. Mol. Model., 2013, 19, 1819–1834 CrossRef CAS PubMed.
- D. Davis, H. M. Kim, J. Y. Ramphal, J. R. Spencer, V. W. F. Tai and E. J. Verner, Silanol derivatives as inhibitors of histone deacetylase, WO-2006069096-A2006069091, 2006.
- M.-F. Zaltariov, M. Turtoi, D. Peptanariu, A.-M. Macsim, L. Clima, C. Cojocaru, N. Vornicu, B.-I. Ciubotaru, A. Bargan, M. Calin and M. Cazacu, Pharmaceutics, 2022, 14, 2838 CrossRef CAS PubMed.
- G. Ping, W. Yue-Wu, L. Xin-Tong, Q. Xiao-Lu, H. Le-Ping, X. Zi-Xin and Y. Fa-Qing, Phosphorus, Sulfur Silicon Relat. Elem., 2014, 189, 511–518 CrossRef CAS.
- D. Q. Shi, Q. Chen, Z. H. Li and X. P. Liu, Phosphorus, Sulfur Silicon Relat. Elem., 2005, 180, 1621–1627 CrossRef CAS.
- M. El-Hussieny, S. T. Mansour, A. I. Hashem, M. A. Fouad and M. A. Abd-El-Maksoud, J. Chin. Chem. Soc., 2022, 69, 1908–1923 CrossRef CAS.
- A. Winer, S. Adams and P. Mignatti, Mol. Cancer Ther., 2018, 17, 1147–1155 CrossRef CAS PubMed.
- M. J. Camarasa, M. J. Perezperez, A. Sanfelix, J. Balzarini and E. Declercq, J. Med. Chem., 1992, 35, 2721–2727 CrossRef CAS PubMed.
- M. J. Camarasa, S. Velazquez, A. San-Felix and M. J. Perez-Perez, Antiviral Chem. Chemother., 2005, 16, 147–153 CrossRef CAS PubMed.
- C. A. Chen, S. M. Sieburth, A. Glekas, G. W. Hewitt, G. L. Trainor, S. Erickson-Viitanen, S. S. Garber, B. Cordova, S. Jeffry and R. M. Klabe, Chem. Biol., 2001, 8, 1161–1166 CrossRef CAS PubMed.
- C. E. Flitcroft, K. A. Jolliffe and C. S. P. McErlean, Chem. – Eur. J., 2023, 29, e202301083 CrossRef CAS PubMed.
- S. McN. Sieburth, in Bio-Inspired Silicon-Based Materials, ed. P. M. Zelisko, Springer Dordrecht, 2014, vol. 5, pp. 103–123 Search PubMed.
- B. Delord, M. C. Guillorit, J. Lafay, M. L. Andreola, D. Tharaud, L. TarragoLitvak, H. J. A. Fleury and G. Déléris, Eur. J. Med. Chem., 1996, 31, 111–122 CrossRef CAS.
- N. Pribut, M. D'Erasmo, M. Dasari, K. E. Giesler, S. Iskandar, S. K. Sharma, P. W. Bartsch, A. Raghuram, A. Bushnev, S. S. Hwang, S. L. Burton, C. A. Derdeyn, A. E. Basson, D. C. Liotta and E. J. Miller, J. Med. Chem., 2021, 64, 12917–12937 CrossRef CAS PubMed.
- Q. Hao, X. Ling, C. Pannecouque, E. De Clercq and F. Chen, Chin. Chem. Lett., 2023, 34, 107663 CrossRef CAS.
- F. Ye, X. Song, J. Liu, X. Xu, Y. Wang, L. Hu, Y. Wang, G. Liang, P. Guo and Z. Xie, Chem. Biol. Drug Des., 2015, 86, 905–910 CrossRef CAS PubMed.
- A. Han, L. Li, K. Qing, X. Qi, L. Hou, X. Luo, S. Shi and F. Ye, Bioorg. Med. Chem. Lett., 2013, 23, 1310–1314 CrossRef CAS PubMed.
- B. D. Lesur, J.-B. Danzin and Charles, N-derivatives of 1-deoxy nojirimycin, Merrell Pharmaceuticals Inc., Cincinnati, OH, United States, 1996, p. 5536732 Search PubMed.
- A. G. Nair, Q. B. Zeng, O. Selyutin, S. B. Rosenblum, Y. H. Jiang, D. Y. Yang, K. Keertikar, G. W. Zhou, M. P. Dwyer, S. H. Kim, B. Shankar, W. S. Yu, L. Tong, L. Chen, R. Mazzola, J. Caldwell, H. Q. Tang, M. L. Allard, R. N. Buckle, P. J. F. Gauuan, C. L. Holst, G. S. Martin, K. P. Naicker, S. Vellekoop, S. Agrawal, R. Liu, R. Kong, P. Ingravallo, E. Xia, Y. Zhai, A. Nomeir and J. A. Kozlowski, Bioorg. Med. Chem. Lett., 2016, 26, 1475–1479 CrossRef CAS PubMed.
- A. G. Nair, Q. B. Zeng, O. Selyutin, S. B. Rosenblum, Y. H. Jiang, D. Y. Yang, K. Keertikar, G. W. Zhou, M. Dwyer, S. H. Kim, B. Shankar, W. S. Yu, L. Tong, L. Chen, R. Mazzola, J. Caldwell, H. Q. Tang, S. Agrawal, R. Liu, R. Kong, P. Ingravallo, E. Xia, Y. Zhai, A. Nomeir, E. Asante-Appiah and J. A. Kozlowski, Bioorg. Med. Chem. Lett., 2018, 28, 1954–1957 CrossRef CAS PubMed.
- E. Rémond, C. Martin, J. Martinez and F. Cavelier, in Peptidomimetics I, ed. W. D. Lubell, Springer International Publishing, Cham, 2017, pp. 27–50 Search PubMed.
- B. M. Liu, K. Gai, H. Qin, X. S. Liu, Y. Cao, Q. Lu, D. D. Lu, D. Y. Chen, H. Q. Shen, W. Song, Y. Zhang, X. J. Wang, H. J. Xu and Y. S. Zhang, Eur. J. Med. Chem., 2018, 148, 95–105 CrossRef CAS PubMed.
- B. M. Liu, K. Gai, H. Qin, J. Wang, X. S. Liu, Y. Cao, Q. Lu, D. D. Lu, D. Y. Chen, H. Q. Shen, W. Song, J. Mei, X. J. Wang, H. J. Xu and Y. S. Zhang, J. Med. Chem., 2020, 63, 5312–5323 CrossRef CAS PubMed.
- S. K. V. Vernekar, L. Qiu, J. Zhang, J. Kankanala, H. M. Li, R. J. Geraghty and Z. Q. Wang, J. Med. Chem., 2015, 58, 4016–4028 CrossRef CAS PubMed.
- Y. M. Hu, Y. X. Wang, F. Li, C. L. Ma and J. Wang, Eur. J. Med. Chem., 2017, 135, 70–76 CrossRef CAS PubMed.
- J. Wang, C. L. Ma, Y. B. Wu, R. A. Lamb, L. H. Pinto and W. F. DeGrado, J. Am. Chem. Soc., 2011, 133, 13844–13847 CrossRef CAS PubMed.
- Y. M. Kim, S. Farrah and R. H. Baney, Electron. J. Biotechnol., 2006, 9, 176–180 CrossRef CAS.
- Y. M. Kim, S. Farrah and R. H. Baney, Electron. J. Biotechnol., 2007, 10, 252–259 CAS.
- Y.-m. Kim, S. Farrah and R. H. Baney, Int. J. Antimicrob. Agents, 2007, 29, 217–222 CrossRef CAS PubMed.
- D. S. Reddy and R. Ramesh, Antitubercular compounds and process for the preparation thereof, Council of Scientific & Industrial Research India, 2014 Search PubMed.
- D. S. Reddy, N. Vasudevan, S. Wagh and R. Ramesh, Novel pyrrole compounds with silicon incorporation, Council of Scientific & Industrial Research India, 2014.
- N. Vasudevan, Z. Motiwala, R. Ramesh, S. B. Wagh, R. D. Shingare, R. Katte, A. Anand, S. Choudhary, A. Kumar, R. S. Gokhale, K. A. Kulkarni and D. S. Reddy, Eur. J. Med. Chem., 2023, 259, 115633 CrossRef CAS PubMed.
- R. Ramesh, R. D. Shingare, V. Kumar, A. Anand, B. Swetha, S. Veeraraghavan, S. Viswanadha, R. Ummanni, R. Gokhale and D. S. Reddy, Eur. J. Med. Chem., 2016, 122, 723–730 CrossRef CAS PubMed.
- M. Martins, M. Viveiros, J. Ramos, I. Couto, J. Molnar, M. Boeree and L. Amaral, Int. J. Antimicrob. Agents, 2009, 33, 479–482 CrossRef CAS PubMed.
- G. J. de Knegt, I. A. J. M. Bakker-Woudenberg, D. van Soolingen, R. Aarnoutse, M. J. Boeree and J. E. M. de Steenwinkel, Int. J. Antimicrob. Agents, 2015, 46, 66–72 CrossRef CAS PubMed.
- S. O. Simons, J. E. Kristiansen, G. Hajos, T. van der Laan, J. Molnár, M. J. Boeree, J. van Ingen, J. B. Christensen, M. Viveiros, Z. Riedl, L. Amaral and D. van Soolingen, Int. J. Antimicrob. Agents, 2013, 41, 488–489 CrossRef CAS PubMed.
- B. Seetharamsingh, R. Ramesh, S. S. Dange, P. V. Khairnar, S. Singhal, D. Upadhyay, S. Veeraraghavan, S. Viswanadha, S. Vakkalanka and D. S. Reddy, ACS Med. Chem. Lett., 2015, 6, 1105–1110 CrossRef CAS PubMed.
- Y. Wen, S. Lun, Y. Jiao, W. Zhang, T. Hu, T. Liu, F. Yang, J. Tang, B. Zhang, W. R. Bishai and L.-F. Yu, Chin. Chem. Lett., 2023, 108464 Search PubMed.
- R. Cebrián, Q. Li, P. Peñalver, E. Belmonte-Reche, M. Andrés-Bilbao, R. Lucas, M. V. de Paz, O. P. Kuipers and J. C. Morales, J. Nat. Prod., 2022, 85, 1459–1473 CrossRef PubMed.
- K. Yamada, A. Deb, V. M. Shoba, D. Lim, B. Maji, A. E. Modell and A. Choudhary, Angew. Chem., Int. Ed., 2022, 61, e202201698 CrossRef CAS PubMed.
- G. Singh, A. Devi, Diksha, Priyanka, N. George, J. Singh, Vikas, R. Yadav and R. Sehgal, Inorg. Chem. Commun., 2023, 153, 110742 CrossRef CAS.
- N. Dhingra, J. B. Singh and H. L. Singh, Dalton Trans., 2022, 51, 8821–8831 RSC.
- H. L. Singh, N. Dhingra and S. Bhanuka, J. Mol. Struct., 2023, 1287, 135670 CrossRef CAS.
- S. N. Adamovich, E. K. Sadykov, I. A. Ushakov, E. N. Oborina and L. A. Belovezhets, Mendeleev Commun., 2021, 31, 204–206 CrossRef CAS.
- S. N. Adamovich, E. V. Kondrashov, I. A. Ushakov, N. S. Shatokhina, E. N. Oborina, A. V. Vashchenko, L. A. Belovezhets, I. B. Rozentsveig and F. Verpoort, Appl. Organomet. Chem., 2020, 34, e5976 CrossRef CAS.
- R. Singh, J. K. Puri, V. K. Chahal, R. P. Sharma, S. Kohli, R. Kant and B. S. Gill, Synth. React. Inorg., Met.-Org., Nano-Met. Chem., 2012, 42, 823–832 CrossRef CAS.
- G. Singh, J. Singh, A. Singh, J. Singh, M. Kumar, K. Gupta and S. Chhibber, J. Organomet. Chem., 2018, 871, 21–27 CrossRef CAS.
- G. Singh, A. Saroa, S. Girdhar, S. Rani, S. Sahoo and D. Choquesillo-Lazarte, Inorg. Chim. Acta, 2015, 427, 232–239 CrossRef CAS.
- M. Napiórkowska, M. Cieślak, J. Kaźmierczak-Barańska, K. Królewska, A. Czapla and B. Kuran, Heterocycl. Commun., 2015, 21, 19–24 CrossRef.
- G. Singh, G. Sharma, Sanchita, P. Kalra, P. Satija, Pawan, B. Singh, D. Aulakh and M. Wreidt, ChemistrySelect, 2020, 5, 284–292 CrossRef CAS.
- S. N. Adamovich, I. A. Ushakov, E. N. Oborina, S. V. Lukyanova and V. Y. Komarov, Int. J. Mol. Sci., 2023, 24, 9965 CrossRef CAS PubMed.
- M. Baghershiroudi, K. D. Safa, K. Adibkia and F. Lotfipour, J. Iran. Chem. Soc., 2018, 15, 1279–1286 CrossRef CAS.
- G. R. Jachak, R. Ramesh, D. G. Sant, S. U. Jorwekar, M. R. Jadhav, S. G. Tupe, M. V. Deshpande and D. S. Reddy, ACS Med. Chem. Lett., 2015, 6, 1111–1116 CrossRef CAS PubMed.
- C. Fu, B. Fu, X. X. Peng and G. J. Liao, Molecules, 2017, 22, 637 CrossRef PubMed.
- A. Bargan, M. F. Zaltariov, A. Vlad, A.-M.-C. Dumitriu, A. Soroceanu, A.-M. Macsim, M. Dascalu, C. D. Varganici, M. Cazacu and S. Shova, Arabian J. Chem., 2020, 13, 3100–3111 CrossRef CAS.
- M. Adams, C. de Kock, P. J. Smith, K. M. Land, N. Liu, M. Hopper, A. Hsiao, A. R. Burgoyne, T. Stringer, M. Meyer, L. Wiesner, K. Chibale and G. S. Smith, Dalton Trans., 2015, 44, 2456–2468 RSC.
- Y. Li, C. de Kock, P. J. Smith, H. Guzgay, D. T. Hendricks, K. Naran, V. Mizrahi, D. F. Warner, K. Chibale and G. S. Smith, Organometallics, 2013, 32, 141–150 CrossRef CAS.
- M. Adams, T. Stringer, C. de Kock, P. J. Smith, K. M. Land, N. Liu, C. Tam, L. W. Cheng, M. Njoroge, K. Chibale and G. S. Smith, 2016, 45, 19086–19095.
- Y. Li, C. de Kock, P. J. Smith, K. Chibale and G. S. Smith, Organometallics, 2014, 33, 4345–4348 CrossRef CAS.
- M. Adams, L. Barnard, C. de Kock, P. J. Smith, L. Wiesner, K. Chibale and G. S. Smith, Dalton Trans., 2016, 45, 5514–5520 RSC.
- G. Singh, A. Arora, S. S. Mangat, S. Rani, H. Kaur, K. Goya, R. Sehgal, I. K. Maurya, R. Tewari, D. Choquesillo-Lazarte, S. Sahoo and N. Kaur, Eur. J. Med. Chem., 2016, 108, 287–300 CrossRef CAS PubMed.
- G. Singh, A. Singh, J. Singh, D. Aulakh, M. Wriedt, C. Espinosa, M. A. Esteban, R. Sehgal, K. Goyal and S. Sinha, New J. Chem., 2017, 41, 15165–15172 RSC.
- G. Singh, A. Singh, K. Chowdhary, P. Satija, Sanchita, P. Kalra, G. Sharma, S. Sinha and R. Sehgal, Polycyclic Aromat. Compd., 2021, 41, 173–183 CrossRef CAS.
- G. Singh, A. Arora, P. Kalra, I. K. Maurya, C. E. Ruizc, M. A. Estebanc, S. Sinha, K. Goyal and R. Sehgal, Bioorg. Med. Chem., 2019, 27, 188–195 CrossRef CAS PubMed.
- D. San Nicolás-Hernández, C. J. Bethencourt-Estrella, A. López-Arencibia, E. Hernández-Álvarez, I. Sifaoui, I. L. Bazzocchi, J. Lorenzo-Morales, I. A. Jiménez and J. E. Piñero, Biomed. Pharmacother., 2023, 157, 114012 CrossRef PubMed.
- A. Geronikaki, D. Hadjipavlou-Litina, A. Zablotskaya and I. Segal, Bioinorg. Chem. Appl., 2007, 2007, 92145 Search PubMed.
- F. Kleemiss, P. Puylaert, D. Duvinage, M. Fugel, K. Sugimoto, J. Beckmann and S. Grabowsky, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2021, 77, 892–905 CrossRef CAS.
- F. Kleemiss, A. Justies, D. Duvinage, P. Watermann, E. Ehrke, K. Sugimoto, M. Fugel, L. A. Malaspina, A. Dittmer, T. Kleemiss, P. Puylaert, N. R. King, A. Staubitz, T. M. Tzschentke, R. Dringen, S. Grabowsky and J. Beckmann, J. Med. Chem., 2020, 63, 12614–12622 CrossRef CAS PubMed.
- D. J. Pérez, U. I. Zakai, S. Guo, I. A. Guzei, Z. Gómez-Sandoval, R. S. Razo-Hernández, R. West and Á. Ramos-Organillo, Aust. J. Chem., 2016, 69, 662–671 CrossRef.
- D. J. Pérez, M. I. Díaz-Reval, F. Obledo-Benicio, U. I. Zakai, Z. Gómez-Sandoval, R. S. Razo-Hernández, R. West, M. T. Sumaya-Martínez, K. Pineda-Urbina and Á. Ramos-Organillo, Eur. J. Pharmacol., 2017, 814, 18–27 CrossRef PubMed.
- M. J. Barnes, R. Conroy, D. J. Miller, J. S. Mills, J. G. Montana, P. K. Pooni, G. A. Showell, L. M. Walsh and J. B. H. Warneck, Bioorg. Med. Chem. Lett., 2007, 17, 354–357 CrossRef CAS PubMed.
- A. S. Kulkarni, R. Ramesh, S. Walia, S. I. Sayyad, G. B. Gathalkar, S. Balamkundu, M. Joshi, A. Sen and D. S. Reddy, ACS Omega, 2021, 6, 31236–31243 CrossRef CAS PubMed.
- S. S. Shete, F. Iqbal, M. Bhardwaj, U. Nandi, A. Kumar and D. S. Reddy, ACS Med. Chem. Lett., 2023, 14, 1716–1723 CrossRef CAS PubMed.
- M. W. Mutahi, T. Nittoli, L. Guo and S. M. Sieburth, J. Am. Chem. Soc., 2002, 124, 7363–7375 CrossRef CAS PubMed.
- J. Kim, G. Hewitt, P. Carroll and S. M. Sieburth, J. Org. Chem., 2005, 70, 5781–5789 CrossRef CAS PubMed.
- R. G. Almquist, W.-R. Chao, M. E. Ellis and H. L. Johnson, J. Med. Chem., 1980, 23, 1392–1398 CrossRef CAS PubMed.
- R. F. Meyer, E. D. Nicolaides, F. G. Tinney, E. A. Lunney, A. Holmes, M. L. Hoefle, R. D. Smith, A. D. Essenburg, H. R. Kaplan and R. G. Almquist, J. Med. Chem., 1981, 24, 964–969 CrossRef CAS PubMed.
- R. G. Almquist, J. Crase, C. Jennings-White, R. F. Meyer, M. L. Hoefle, R. D. Smith, A. D. Essenburg and H. R. Kaplan, J. Med. Chem., 1982, 25, 1292–1299 CrossRef CAS PubMed.
- Y. Bo, S. Singh, H. Q. Duong, C. Cao and S. M. Sieburth, Org. Lett., 2011, 13, 1787–1789 CrossRef CAS PubMed.
- S. M. Sieburth and C. A. Chen, Eur. J. Org. Chem., 2006, 2006, 311–322 CrossRef.
- H. Q. Duong and S. M. Sieburth, J. Org. Chem., 2018, 83, 5398–5409 CrossRef CAS PubMed.
- J. Kim, A. Glekas and S. M. Sieburth, Bioorg. Med. Chem. Lett., 2002, 12, 3625–3627 CrossRef CAS PubMed.
- J. Kim and S. M. Sieburth, J. Org. Chem., 2004, 69, 3008–3014 CrossRef CAS PubMed.
- D. H. Juers, J. Kim, B. W. Matthews and S. M. Sieburth, Biochemistry, 2005, 44, 16524–16528 CrossRef CAS PubMed.
- G. A. Dalkas, D. Marchand, J.-C. Galleyrand, J. Martinez, G. A. Spyroulias, P. Cordopatis and F. Cavelier, J. Pept. Sci., 2010, 16, 91–97 CrossRef CAS PubMed.
- G. D. Reddy, S. J. Park, H. M. Cho, T. J. Kim and M. E. Lee, J. Med. Chem., 2012, 55, 6438–6444 CrossRef PubMed.
- D. R. Guda, S.-J. Park, M.-W. Lee, T.-J. Kim and M. E. Lee, Eur. J. Med. Chem., 2013, 62, 84–88 CrossRef CAS PubMed.
- M. Geyer, J. A. Baus, O. Fjellström, E. Wellner, L. Gustafsson and R. Tacke, ChemMedChem, 2015, 10, 2063–2070 CrossRef CAS PubMed.
- M. Geyer, E. Wellner, U. Jurva, S. Saloman, D. Armstrong and R. Tacke, ChemMedChem, 2015, 10, 911–924 CrossRef CAS PubMed.
- R. J. Fessenden, J. G. Larsen, M. D. Coon and J. S. Fessenden, J. Med. Chem., 1964, 7, 695–698 CrossRef CAS PubMed.
- R. J. Fessenden and M. D. Coon, J. Med. Chem., 1964, 7, 561–562 CrossRef CAS PubMed.
- R. J. Fessenden and M. D. Coon, J. Med. Chem., 1965, 8, 604–608 CrossRef CAS PubMed.
- J. P. Stasch, H. Russ, U. Schacht, M. Witteler, D. Neuser, M. Gerlach, M. Leven, W. Kuhn, P. Jutzi and H. Przuntek, Arzneim. Forsch., 1988, 38-2, 1075–1078 Search PubMed.
- A. Schotte, P. F. M. Janssen, A. A. H. P. Megens and J. E. Leysen, Brain Res., 1993, 631, 191–202 CrossRef CAS PubMed.
- R. Tacke, T. Heinrich, R. Bertermann, C. Burschka, A. Hamacher and M. U. Kassack, Organometallics, 2004, 23, 4468–4477 CrossRef CAS.
- R. Tacke, F. Popp, B. Muller, B. Theis, C. Burschka, A. Hamacher, M. U. Kassack, D. Schepmann, B. Wunsch, U. Jurva and E. Wellner, ChemMedChem, 2008, 3, 152–164 CrossRef CAS PubMed.
- R. Tacke, B. Nguyen, C. Burschka, W. P. Lippert, A. Hamacher, C. Urban and M. U. Kassack, Organometallics, 2010, 29, 1652–1660 CrossRef CAS.
- R. Tacke, R. Bertermann, C. Burschka, S. Dörrich, M. Fischer, B. Müller, G. Meyerhans, D. Schepmann, B. Wünsch, I. Arnason and R. Bjornsson, ChemMedChem, 2012, 7, 523–532 CrossRef CAS PubMed.
- S. Duda-Johner, J. O. Daiß, K. Mohr and R. Tacke, J. Organomet. Chem., 2003, 686, 75–83 CrossRef CAS.
- J. O. Daiss, S. Duda-Johner, C. Burschka, U. Holzgrabe, K. Mohr and R. Tacke, Organometallics, 2002, 21, 803–811 CrossRef CAS.
- J. O. Daiss, C. Burschka, J. S. Mills, J. G. Montana, G. A. Showell, J. B. H. Warneck and R. Tacke, J. Organomet. Chem., 2006, 691, 3589–3595 CrossRef CAS.
- G. A. Showell, M. J. Barnes, J. O. Daiss, J. S. Mills, J. G. Montana, R. Tacke and J. B. H. Warneck, Bioorg. Med. Chem. Lett., 2006, 16, 2555–2558 CrossRef CAS PubMed.
- J. O. Daiss, C. Burschka, J. S. Mills, J. G. Montana, G. A. Showell, J. B. H. Warneck and R. Tacke, Organometallics, 2006, 25, 1188–1198 CrossRef CAS.
- A. H. Beckett, D. C. Taylor and J. W. Gorrod, J. Pharm. Pharmacol., 1975, 27, 588–593 CrossRef CAS PubMed.
- M. Blunder, N. Hurkes, S. Spirk, M. List and R. Pietschnig, Bioorg. Med. Chem. Lett., 2011, 21, 363–365 CrossRef CAS PubMed.
- E. Belmonte-Reche, P. Penalver, M. Caro-Moreno, M. L. Mateos-Martin, N. Adan, M. Delgado, E. González-Rey and J. C. Morales, Eur. J. Med. Chem., 2021, 223, 113655 CrossRef CAS PubMed.
- W. Duan, Y. Sun, M. Wu, Z. Zhang, T. Zhang, H. Wang, F. Li, L. Yang, Y. Xu, Z.-J. Liu, T. Hua, H. Nie and J. Cheng, Eur. J. Med. Chem., 2021, 226, 113878 CrossRef CAS PubMed.
- M. El-Hussieny, M. A. Abd-El-Maksoud, F. M. Soliman, M. A. Fouad and M. K. El-Ashrey, J. Enzyme Inhib. Med. Chem., 2023, 38, 2166040 CrossRef PubMed.
- J.-M. Hornsperger, J.-N. Collard, J.-G. Heydt, E. Giacobini, S. Funes, J. Dow and D. Schirlin, Biochem. Soc. Trans., 1994, 22, 758–763 CrossRef CAS PubMed.
- G. Luo, L. M. Chen, Y. Li, Y. Fan, D. Y. Wang, Y. F. Yang, L. Gao, R. T. Jiang and Z. L. Song, Org. Chem. Front., 2021, 8, 5941–5947 RSC.
- K. Madica, K. C. Nadimpally and G. J. Sanjayan, Tetrahedron Lett., 2017, 58, 1568–1571 CrossRef CAS.
- R. B. Silverman and G. M. Banik, J. Am. Chem. Soc., 1987, 109, 2219–2220 CrossRef CAS.
- C. Danzin, J.-N. Collard, P. Marchal and D. Schirlin, Bioorg. Med. Chem. Lett., 1995, 5, 2363–2366 CrossRef CAS.
- P. Tétreault, É. Besserer-Offroy, R. L. Brouillette, A. René, A. Murza, R. Fanelli, K. Kirby, A. J. Parent, I. Dubuc, N. Beaudet, J. Côté, J.-M. Longpré, J. Martinez, F. Cavelier and P. Sarret, Eur. J. Pharmacol., 2020, 882, 173174 CrossRef PubMed.
- S. Farkas, CNS Drug Rev., 2006, 12, 218–235 CrossRef CAS PubMed.
- T. Kniess, H. Spies, I. Santos and A. Zablotska, J. Labelled Compd. Radiopharm., 2002, 45, 629–636 CrossRef CAS.
- C. Fernandes, T. Kniess, L. Gano, S. Seifert, H. Spies and I. Santos, Nucl. Med. Biol., 2004, 31, 785–793 CrossRef CAS PubMed.
- V. V. Dorsselaer, D. Schirlin, P. Marchal, F. Weber and C. Danzin, Bioorg. Chem., 1996, 24, 178–193 CrossRef.
- M. C. J. Wolvekamp and R. W. F. de Bruin, Dig. Dis., 1994, 12, 2–14 CrossRef CAS PubMed.
- G. Singh, Sushma, Priyanka, Diksha, Mohit, S. Gupta, M. Angeles Esteban, C. Espinosa-Ruíz and D. González-Silvera, J. Mol. Struct., 2022, 1255, 132446 CrossRef CAS.
- G. Singh, Tamana, A. Devi, A. Saini, Y. Thakur, S. Khurana, S. Sharma, T. Diskit and K. N. Singh, Dyes Pigm., 2023, 219, 111537 CrossRef CAS.
- G. Singh, A. Devi, Tamana, P. Malik, S. Khurana, J. Stanzin, D. Sharma, Mithun and Heena, Spectrochim. Acta, Part A, 2023, 299, 122854 CrossRef CAS PubMed.
- G. Singh, Mohit, Diksha, Pawan, P. Satija, Sushma, S. Sharma, S. Gupta and K. N. Singh, Inorg. Chim. Acta, 2023, 545, 121263 CrossRef CAS.
- G. Singh, S. Gupta, S. Sharma, A. Devi, S. Khurana, P. Malik, S. Devi, Heena and Vikas, Environ. Nanotechnol., Monit. Manage., 2023, 20, 100831 CAS.
- G. Singh, Diksha, Mohit, Priyanka, A. Devi, S. Devi, H. Kaur, J. Singh and G. Singh, New J. Chem., 2023, 47, 12608–12619 RSC.
- G. Singh, Diksha, Mohit, Suman, Sushma, A. Devi, S. Gupta, C. Espinosa-Ruíz and M. Angeles Esteban, Inorg. Chim. Acta, 2022, 537, 120926 CrossRef CAS.
- G. Singh, A. Devi, Mohit, P. Satija, Sushma, Vikas, D. Gonzalez-Silvera, C. Espinosa-Ruiz and M. Angeles Esteban, Inorg. Chim. Acta, 2022, 542, 121126 CrossRef CAS.
- G. Singh, A. Devi, Mohit, Diksha, Suman, A. Saini, J. D. Kaur, S. Gupta and Vikas, New J. Chem., 2022, 46, 21717–21729 RSC.
- G. Singh, A. Saini, Mohit, Pawan, Diksha, Sushma, Suman, Priyanka and P. Satija, Inorg. Chem. Commun., 2022, 146, 110090 CrossRef CAS.
- G. Singh, G. Sharma, P. Satija, A. Singh, Pawan, C. E. Ruiz, D. G. Silvera, M. A. Esteban and S. Soni, ChemistrySelect, 2020, 5, 15055–15060 CrossRef CAS.
- H. Toyama, M. Nakamura, Y. Hashimoto and S. Fujii, Bioorg. Med. Chem., 2015, 23, 2982–2988 CrossRef CAS PubMed.
- Y. Nishiyama, M. Nakamura, T. Misawa, M. Nakagomi, M. Makishima, M. Ishikawa and Y. Hashimoto, Bioorg. Med. Chem., 2014, 22, 2799–2808 CrossRef CAS PubMed.
- N. Namba, T. Noguchi-Yachide, Y. Matsumoto, Y. Hashimoto and S. Fujii, Bioorg. Med. Chem., 2022, 66, 116792 CrossRef CAS PubMed.
- H. Toyama, H. Shirakawa, M. Komai, Y. Hashimoto and S. Fujii, Bioorg. Med. Chem., 2018, 26, 4493–4501 CrossRef CAS PubMed.
- H. Toyama, S. Sato, H. Shirakawa, M. Komai, Y. Hashimoto and S. Fujii, Bioorg. Med. Chem. Lett., 2016, 26, 1817–1820 CrossRef CAS PubMed.
- C. Zhou, J. G. Cheng, R. Beadle, F. G. Earley, Z. Li and P. Maienfisch, Bioorg. Med. Chem., 2020, 28, 115509 CrossRef CAS PubMed.
- C. Zhou, X. Wang, X. Quan, J. Cheng, Z. Li and P. Maienfisch, Nongye Yu Shipin Kexue, 2022, 70, 11063–11074 CAS.
- C. Zhou, Z. Li, X. Qian, J. Cheng and P. Maienfisch, Nongye Yu Shipin Kexue, 2023, 71, 18239–18249 CAS.
- X. Quan, L. Xu, Z. Li and P. Maienfisch, Nongye Yu Shipin Kexue, 2023, 71, 18188–18196 CAS.
- L. Xu, X. Quan, Z. Li and P. Maienfisch, Nongye Yu Shipin Kexue, 2023, 71, 18250–18259 CAS.
- C. Zhou, X. Sun, W. Fu, Z. Li, J. Cheng and P. Maienfisch, Nongye Yu Shipin Kexue, 2023, 71, 5483–5495 CAS.
- G. Wei, M.-W. Huang, W.-J. Wang, Y. Wu, S.-F. Mei, L.-M. Zhou, L.-C. Mei, X.-L. Zhu and G.-F. Yang, J. Agric. Food Chem., 2021, 69(13), 3965–3971 CrossRef CAS PubMed.
- I. F. Yu, J. W. Wilson and J. F. Hartwig, Chem. Rev., 2023, 123, 11619–11663 CrossRef CAS PubMed.
- J. J. Dalton, A. Bernal Sánchez, A. T. Kelly, J. C. Fettinger and A. K. Franz, ACS Catal., 2024, 14, 1005–1012 CrossRef CAS PubMed.
- E. Ramesh, L. D. Nandawadekar, R. S. Rao and D. S. Reddy, Org. Lett., 2023, 25, 6881–6885 CrossRef CAS PubMed.
- R. Holland, K. Lam, S. Jeng, K. McClintock, L. Palmer, P. Schreiner, M. Wood, W. Zhao and J. Heyes, ACS Nano, 2024, 18, 10374–10387 CrossRef CAS PubMed.
- Y. Matsumoto, Y. Hashimoto and S. Fujii, RSC Adv., 2023, 13, 27359–27362 RSC.
- Y. Miyajima, T. Noguchi-Yachide, K. Ochiai and S. Fujii, RSC Med. Chem., 2024, 15, 119–126 RSC.
- Y. Yamanoi, Acc. Chem. Res., 2023, 56, 3325–3341 CrossRef CAS PubMed.
- N. S. Sarai, T. J. Fulton, R. L. O'Meara, K. E. Johnston, S. Brinkmann-Chen, R. R. Maar, R. E. Tecklenburg, J. M. Roberts, J. C. T. Reddel, D. E. Katsoulis and F. H. Arnold, Science, 2024, 383, 438–443 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2024 |