
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRationally modified SNX-class Hsp90 inhibitors disrupt extracellular fibronectin assembly without intracellular Hsp90 activity†
Gciniwe S.
Mathenjwa
ab,
Abir
Chakraborty
 c,
Abantika
Chakraborty
c,
Ronel
Muller
b,
Mathew P.
Akerman
b,
Moira L.
Bode
d,
Adrienne L.
Edkins
*c and
Clinton G. L.
Veale
*a
c,
Abantika
Chakraborty
c,
Ronel
Muller
b,
Mathew P.
Akerman
b,
Moira L.
Bode
d,
Adrienne L.
Edkins
*c and
Clinton G. L.
Veale
*a
aDepartment of Chemistry, University of Cape Town, Rondebosch, Cape Town 7701, South Africa. E-mail: clinton.veale@uct.ac.za
bSchool of Chemistry and Physics, University of KwaZulu-Natal, Private Bag X01, Scottsville, 3209, South Africa
cThe Biomedical Biotechnology Research Unit (BioBRU), Department of Biochemistry and Microbiology, Rhodes University, Makhanda, 6139, South Africa. E-mail: a.edkins@ru.ac.za
dMolecular Sciences Institute, School of Chemistry, University of the Witwatersrand, Private Bag 3, PO WITS, 2050, Johannesburg, South Africa
First published on 2nd September 2024
Abstract
Despite Hsp90's well documented promise as a target for developing cancer chemotherapeutics, its inhibitors have struggled to progress through clinical trials. This is, in part, attributed to the cytoprotective compensatory heat shock response (HSR) stimulated through intracellular Hsp90 inhibition. Beyond its intracellular role, secreted extracellular Hsp90 (eHsp90) interacts with numerous pro-oncogenic extracellular clients. This includes fibronectin, which in the tumour microenvironment enhances cell invasiveness and metastasis. Through the rational modification of known Hsp90 inhibitors (SNX2112 and SNX25a) we developed four Hsp90 inhibitory compounds, whose alterations restricted their interaction with intracellular Hsp90 and did not stimulate the HSR. Two of the modified cohort (compounds 10 and 11) were able to disrupt the assembly of the extracellular fibronectin network at non-cytotoxic concentrations, and thus represent promising new tool compounds for studying the druggability of eHsp90 as a target for inhibition of tumour invasiveness and metastasis.
1. Introduction
Heat shock protein 90 (Hsp90) is a highly conserved and abundant ATP-dependent chaperone protein, which promotes the formation of functional conformers of client proteins involved in critical cellular processes, including a host of signalling intermediates and transcription factors.1 This cytoprotective function extends to many forms of cancers where high growth rates and mutated oncoproteins cause major perturbations in cellular proteostasis.2,3 Because of this, the inhibition of Hsp90 function can be sufficient to induce the simultaneous degradation of pro-oncogenic factors.2,4 This has seen Hsp90 designated as a sought-after target for developing cancer chemotherapeutics, with a particular emphasis on N-terminal interacting ATP antagonists.5–8However, despite this promise and numerous candidates entering trials, there has been a disappointing lack of translation into the clinic.9 The lack of translational efficacy has been linked to detrimental effects associated with pan-Hsp90 inhibition of non-isoform selective ATP antagonists,10,11 as well as the cytoprotective heat shock response (HSR).
The HSR is a phenomenon in which Hsp90 inhibition stimulates the expression of additional chaperones, including high levels of intracellular Hsp70, to support pro-oncogenic cellular processes, thus compromising Hsp90 inhibitory efficacy.12,13 However, the observation that the compensatory expression of Hsp70 is only observed in the presence of Hsp90 N-terminal interacting ATPase inhibitors has led to the development of small molecules which inhibit Hsp90 activity, either through C-terminal engagement14–16 or disruption of co-chaperone protein–protein interaction formation.17–19
In addition to its intracellular function, secreted Hsp90 or extracellular Hsp90 (eHsp90) interacts with numerous cell surface receptors involved in cell signalling, such as toll-like receptors (TLRs) lipoprotein receptor-related protein 1 (LRP1) and the epidermal growth factor receptor (EGFR) family as well as extracellular client proteins such as matrix metalloproteinases (MMP) and fibronectin required for regulating the extracellular matrix (ECM).20 The dysregulated equilibrium of cancer cells results in consistent excretion of eHsp90, whose interaction with the extracellular environment stimulates pro-oncogenic signal transduction and remodelling of the ECM, thus enhancing cancer cell-growth, migration, invasiveness and metastasis.20–23
The stress-inducible Hsp90α is the predominantly secreted isoform and plays a particularly important role in tumour progression. Knock out of Hsp90α restricts cancer cell migration, invasiveness and capacity to metastasise, without impacting cell survival or growth.24 However, the constitutive Hsp90β isoform, whose knock down results in cancer cell death, has been shown to interact with extracellular fibronectin, and MMP-3.25–27
Accordingly, inhibition of eHsp90 provides a substantial opportunity to simultaneously disrupt several pro-oncogenic pathways without accessing the intracellular environment, thus avoiding triggering the HSR. It has further been postulated that it is the unintentional effect on eHsp90, and not the cytosolic forms, which is responsible for the promising results of pre-clinical Hsp90 inhibitors.28 Therefore, traditional medicinal chemistry strategies, which seek to promote cell-penetration, could unwittingly be hindering access to the effective target of Hsp90 inhibitors.
This phenomenon was first demonstrated through the alteration of pan-Hsp90 inhibitor 17-DMAG (1) into the cell impermeable zwitterionic DMAG-N-oxide (2), which was found to disrupt in vitro tumour cell migration and extracellular matrix-dependent cytoskeletal reorganisation, with no effect on intracellular Hsp90 function (Fig. 1). This in turn translated into a significant reduction in the in vivo tumour colonisation of mice lungs with intravenously administered melanoma cells.29
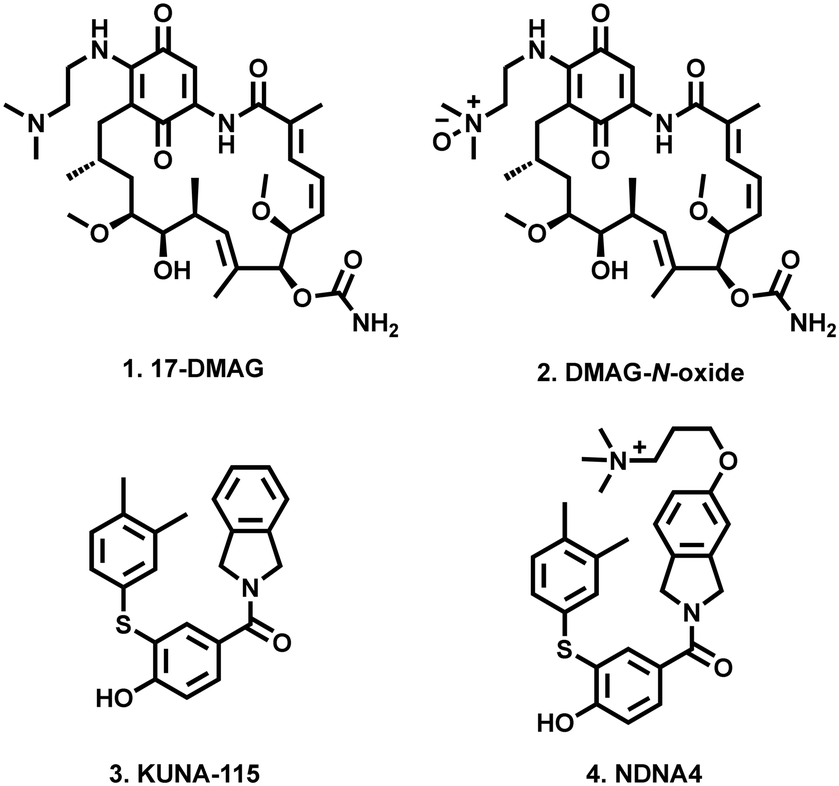 | ||
| Fig. 1 Examples of cell impermeable Hsp90 inhibitors (2 and 4), and their parent compounds (1 and 3). | ||
Most recently Blagg and co-workers reported that through modification of their previously reported Hsp90α inhibitor (KUNA-115, 3)30 with a cationic quaternary ammonium containing alkyl ether (NDNA4, 4), they were able to almost completely restrict cell permeability. These modified compounds which retained Hsp90 isoform selectivity, did not induce intracellular Hsp90 client degradation, stimulation of the HSR or disruption of hERG channel maturation.31
SNX2112 (5, Fig. 2) and SNX25a (6) are pan-Hsp90 inhibitory compounds, with on-target activity that results in the degradation of multiple pro-oncogenic Hsp90 clients and subsequent potent broad-spectrum activity against a range of cancer cell-lines.32–34 A glycine ester mesylate containing orally bioavailable pro-drug of 5 (7, SNX5422) displayed early success, entering phase 1 clinic trails. Unfortunately, while generally well tolerated, development was halted due to observations of ocular toxicity.35 However, despite this setback, their significant promise has seen clinical interest in the SNX class of Hsp90 inhibitors retained.36–38 We therefore reasoned that the development of SNX compounds, which exclusively target the extracellular environment, may be an effective means of maximising the potential of this class of pan-Hsp90 inhibitors as potent anticancer agents which do not engage with intracellular Hsp90. Accordingly, we report the design and synthesis of four new members of the SNX class (8–11, Fig. 2), which have been structurally altered to include polar alkyl tethers.
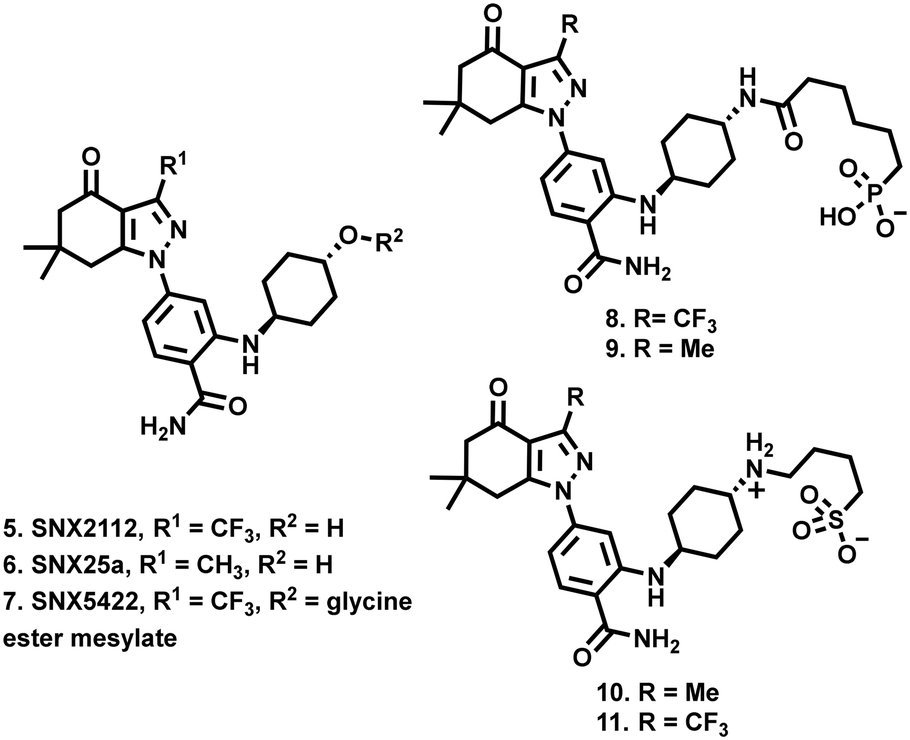 | ||
| Fig. 2 Compounds 8–11 described in this study, derived from the SNX class of HSP90 inhibitory compounds 5–7. | ||
Importantly, while these modifications did not diminish in vitro Hsp90 activity, they significantly reduced cytotoxicity. Furthermore, our data indicate that these compounds do not induce the degradation of cyclin dependent kinase 4 (CDK4) an obligate intracellular Hsp90 client, nor do they stimulate the expression of Hsp70 (a marker of the HSR), suggesting that they do not inhibit intracellular Hsp90. Finally, we demonstrate that at non-toxic concentrations, two of this cohort of compounds decreased the intensity of the extracellular fibronectin network matrix, suggesting that these compounds act as eHSP90 inhibitors.
2. Results and discussion
2.1 Compound design
Previous reports in which the SNX scaffold was modified into Hsp90 interacting probes showed that alkyl tethers substituted ortho to the benzamide did not have a significant impact on Hsp90 N-terminal binding.39–41 Taldone et al. also showed that isosteric replacement of the 4-aminocyclohexan-1-ol moiety of 5 with a 1,4-diamino cyclohexane, followed by amide coupling to an alkyl chain, had no significant impact of Hsp90 binding.42Based on this information, we reasoned that substituting polar tethers on the 1,4-diamino cyclohexane ring would be a synthetically tractable means to generate compounds with the desired alteration in physicochemical properties, without disrupting Hsp90 N-terminal binding. For the purposes of this study, we opted to utilise alkyl sulfonate and phosphonate containing tethers, both of which exist in their polar anionic form at physiological pH, and have been shown to suitably alter physicochemical properties of ligands to inhibit cell permeation.43,44 It is also likely that the alkyl amino present in compounds 10 and 11, would promote zwitterion formation at physiological pH. The choice of chain length and electrophilic functional group (alkyl halide and carboxylic acid) was based on reagents which were commercially available to us at the time. To support our structural design, we conducted an in silico analysis, which suggested that the Hsp90 inhibitory core of compounds 8–11 had the same binding pose as co-crystalised SNX2112.45 This included key electrostatic interactions with Tyr139, Asp93 (shown in white) and binding site waters (shown in red). In addition, compounds 8–11 and SNX2112 both interacted in the same fashion with the hydrophobic pocket formed by Leu103, Phe138, Val150 and Trp162 (shown in green). Importantly, the polar tethers oriented out the binding pocket into the bulk solvent without disrupting pharmacophore binding (Fig. 3).
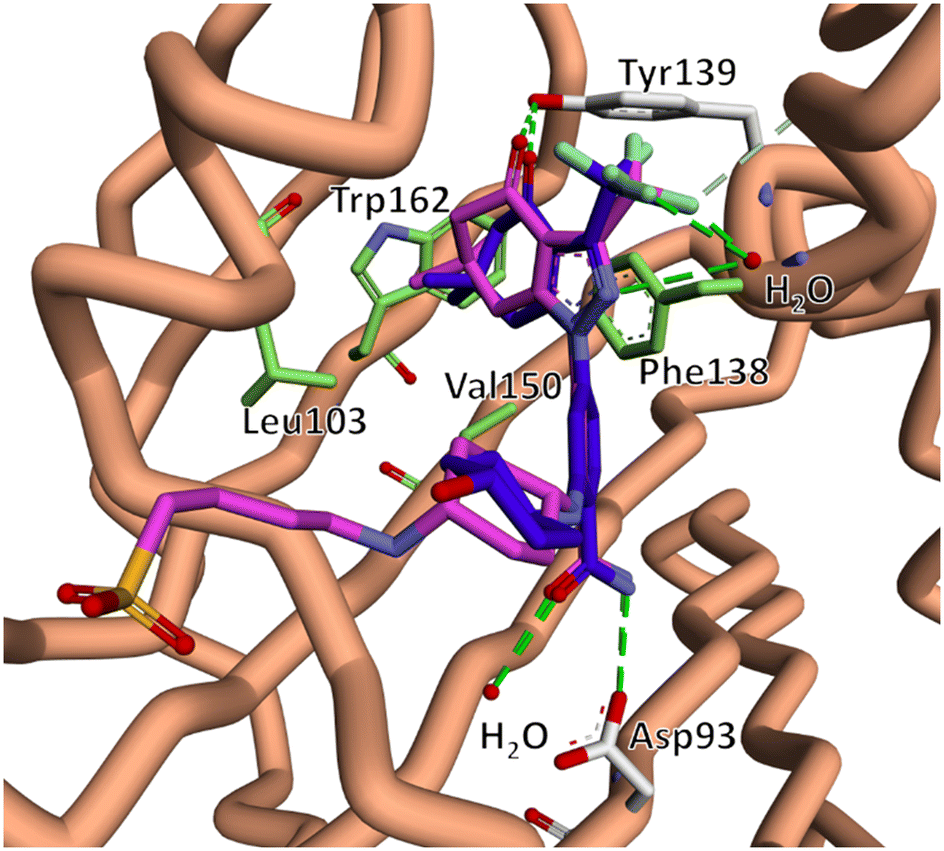 | ||
| Fig. 3 The lowest energy docked binding mode of compound 11 (pink) overlaid with x-ray co-crystal structure of SNX2112 (5, blue) in complex with the N-terminal domain of Hsp90 (PDB 6LTK).45 The predicted binding pose suggests that compounds 8–11 retained key binding site interactions including electrostatic interactions with Tyr139, Asp93 (white) and binding site waters (red), as well as hydrophobic interactions with Leu103, Phe138, Val150 and Trp162 (green). This data indicated that our proposed structural modifications would not disrupt engagement with the SNX2112 binding pocket. | ||
2.2 Chemistry
To access our desired compounds, we utilised two separate reaction pathways, both of which were adapted from the methodology reported by Huang et al. (Scheme 1).32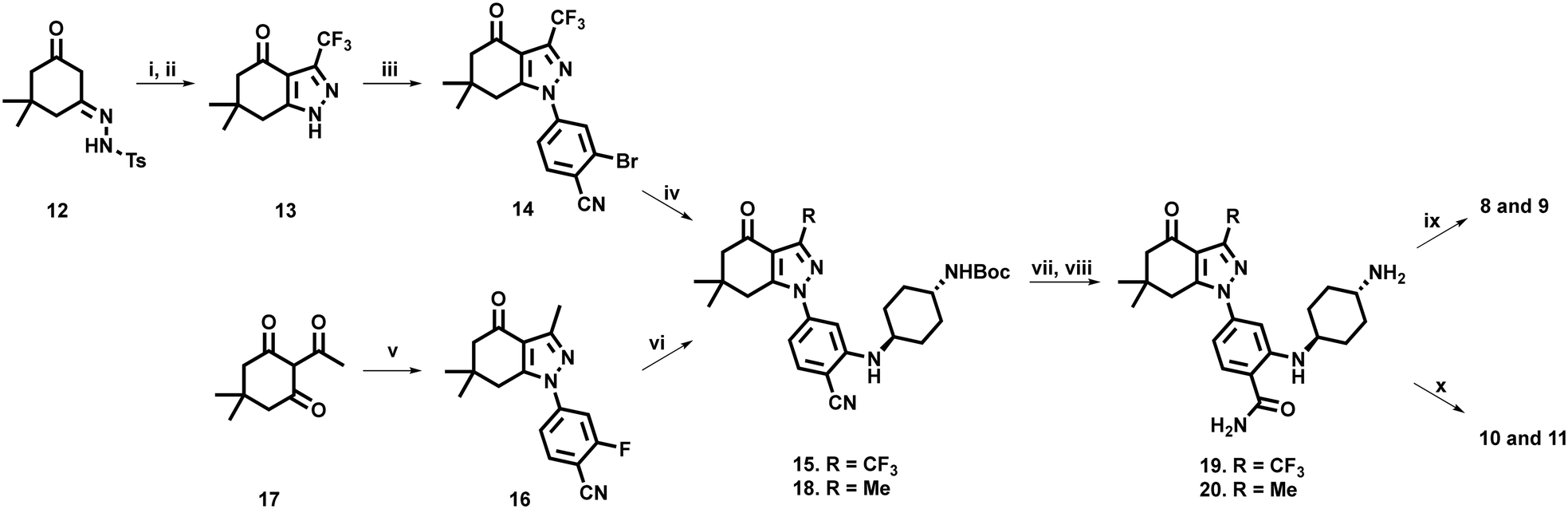 | ||
| Scheme 1 i) (CF3CO)2O, TNF, NEt3, 55 °C, 2 h; ii) NaOH, MeOH, H2O, r.t., 3 h, 45%; iii) 2-bromo-4-fluorobenzonitrile, NaH, anhy. DMSO, 45 °C, 40 h, 57%; iv) trans-N-Boc-1,4-cyclohexanediamine, DPPF[PdCl2], DPPF, NatOBu, THF, 65 °C, 65%; v) 2-fluoro-4-hydrazinylbenzonitrile, MeOH, AcOH, r.t., 3 d, 67%; vi) trans-N-Boc-1,4-cyclohexanediamine, DIPEA, DMSO, 90 °C, 60 min, 74%; vii) EtOH, NaOH, 30% H2O2, H2O, r.t., 16 h, 60%. viii) TFA, DCM, r.t., 5 h, 41–79%. ix) 6-Phosphonohexanoic acid, DIPEA, EDCI, NHS, DMF, r.t., 42 h, 53–62%; x) 4-bromo-1-butanesulfonic acid, DIPEA, DMF, 50 °C, 18 h, 39–52%. | ||
In a one-pot, two-step procedure, tosyl hydrazone dimedone derivative 12 was cyclised with trifluoromethyl acetic acid anhydride, to generate the tetrahydroindazolone core structure 13, followed by selective nucleophilic aromatic substitution on 2-bromo-4-fluorobenzonitrile, to generate intermediate 14. Standard Buchwald–Hartwig conditions,32,46 with an N-Boc protected 1,4-diamino cyclohexane, successfully yielded key intermediate 15. Unfortunately, this approach to access the tetrahydroindazolone scaffold of 14, was not applicable for the synthesis of the methyl- containing analogue 16.47 Accordingly, we pivoted to an alternative approach in which 2-acetyldimedone (17) was cyclised with a suitably functionalised phenylhydrazine to generate intermediate 16. On this occasion, the presence of an aromatic fluoride facilitated the formation of 18via simpler base-mediated nucleophilic aromatic substitution using N-Boc protected 1,4-diaminocyclohexane.
Nitrile hydrolysis of both 15 and 18, followed by N-Boc deprotection afforded the free amines 19 and 20. Preparation of phosphonate containing compounds 8 and 9 was achieved via condensation of 19 and 20 with 6-phosphonohexanoic acid, under standard EDCI mediated amide coupling conditions. Sulfonate containing analogues 10 and 11 were successfully prepared via nucleophilic substitution of 4-bromo-1-butanesulfonic acid in the presence of DIPEA at 50 °C.
2.3 Biological evaluation
We progressed onto preliminary in vitro biological assessment in an Hsp90 ATPase assay. Here compounds 8–11 were determined to inhibit ATP hydrolysis and were equipotent to SNX2112, indicating that our structural modifications had no measurable impact on Hsp90 inhibitory activity (Table 1).Furthermore, assessment against a HeLa cell line showed that compounds 8–11 were in excess of an order of magnitude less cytotoxic than SNX2112 (Table 1), which is a reduction in activity roughly in line with previous reports of extracellular pan-Hsp90 inhibitors.48
To assess whether compounds 8–11 had an impact on intracellular Hsp90 activity we used western blotting to measure changes in the intracellular abundance of the obligate Hsp90 client protein, CDK4 and Hsp70 in HeLa cells (Fig. 4). As expected, inhibition of intracellular Hsp90 by SNX2112 (5) resulted in a significant decrease in the abundance of CDK4, whilst simultaneously stimulating intracellular Hsp70 expression, presumably as a result of the HSR. Conversely, treatment of HeLa cells with compounds 8–11 did not decrease the abundance of CDK4, nor was any change in Hsp70 abundance observed. The combined observations of target level Hsp90 activity comparable to that of SNX2112, whilst being unable to stimulate the HSR, or induce degradation of Hsp90 client proteins, implies that the structural modifications made to generate 8–11, restricts access to intracellular Hsp90, possibly due to cell penetration impediment to the extent that it cannot impact Hsp90 function. This is also likely partly responsible for the reduced cytotoxicity observed in Table 1.
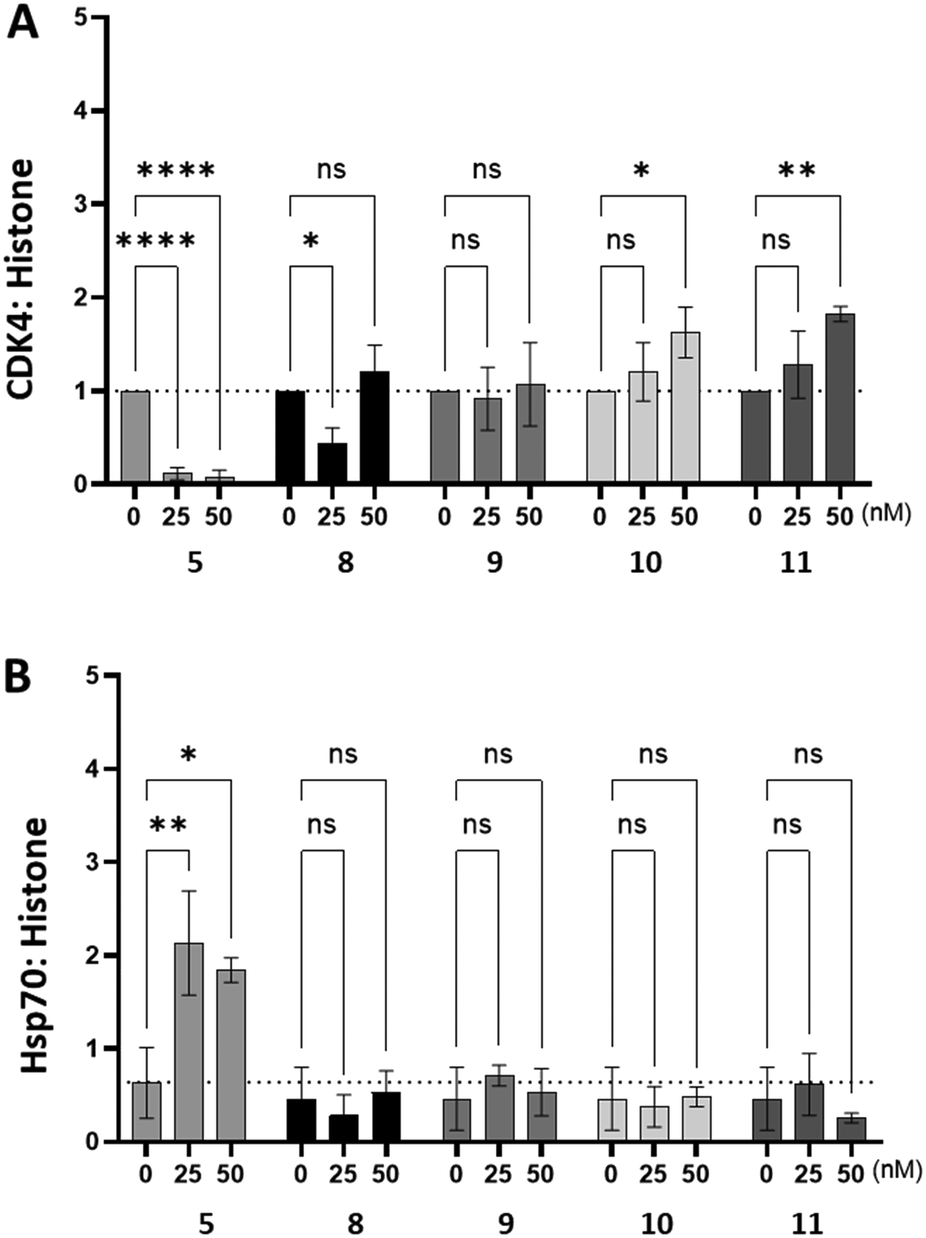 | ||
| Fig. 4 Densitometry readings following Western blot of HeLa cells treated with compounds 5, 8–11. Treatment with validated intracellular Hsp90 inhibitor SNX2112 (5) resulted in an expected reduction in levels of Hsp90 client CDK4 (A), with a concomitant increase in Hsp70 levels (B). Treatment with modified analogues 8–11 did not mirror this same effect, indicating a lack of engagement with intracellular Hsp90. | ||
We then progressed onto assessing the impact of compounds 8–11 on extracellular fibronectin matrix assembly. The Hs578T cell line endogenously produces high levels of extracellular fibronectin matrix in culture and, hence, is a convenient model to study small molecule induced changes to the extracellular environment. Hs578T cells were cultured for 6 days to establish the matrix and then treated overnight with compounds 5, 8–11 and suitable controls.
Following this, cell and soluble cell-associated proteins were removed. The remaining insoluble cell-derived extracellular matrix was stained with anti-fibronectin antibodies, visualised by fluorescence microscopy, and quantified by mean fluorescence intensity per area of matrix (MFI/area, Fig. 5).49 Treatment with wild- type functional upstream domain (WT-FUD), a known inhibitor of fibronectin matrix assembly,50 as well as high concentration (200 μM) exposure to novobiocin (Nov)25 resulted in the expected substantial reduction in MFI/area compared to the normalised DMSO control.49 This same effect was not observed in the control experiments using the mutant FUD (MT-FUD), whose reduced fibronectin affinity inhibited its ability to disrupt fibronectin fibrillogenesis.49 Hsp90α was used as a positive control, where its addition resulted in a significant increase of the MFI/area of the fibronectin matrix. Treatment of Hs578T cells at a sub IC50 concentration (5 nM) of SNX-2112, had no significant impact on the fibronectin matrix, which is likely as a result of cellular penetration, thus limiting access to eHsp90. Compounds 8 and 9, which both feature the phosphonate tether, were also inactive in this assay. In contrast, however, treatment with both sulfonate tether containing compounds 10 and 11 at concentrations substantially below their cytotoxic IC50 values resulted in a reduction of the fibronectin matrix, comparable to that of both wild-type FUD and 200 μM novobiocin treatments.
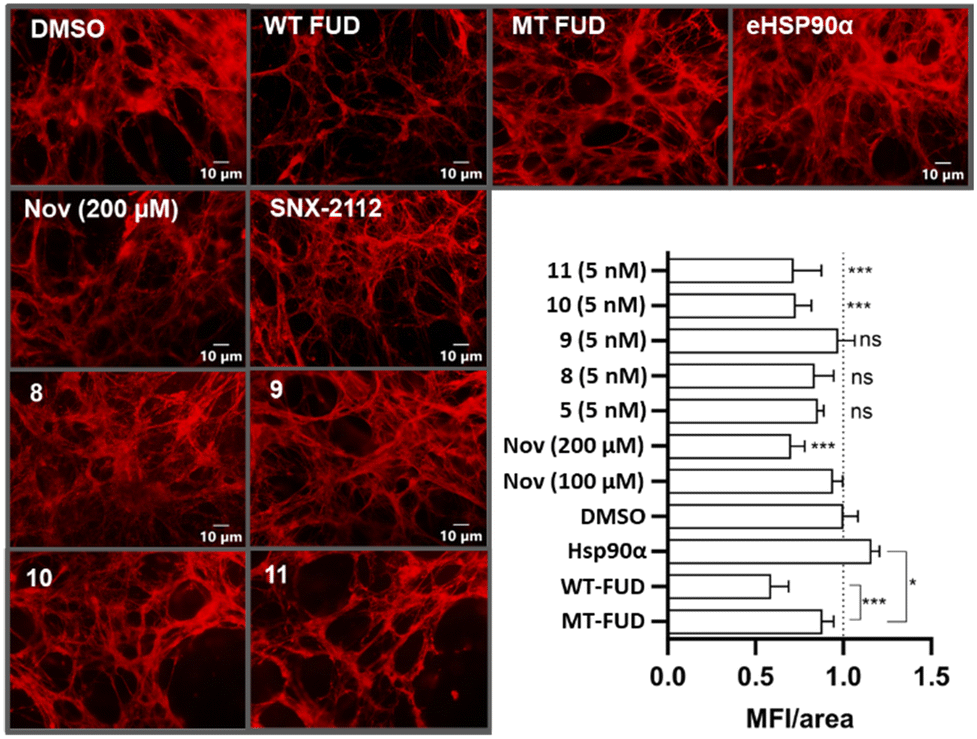 | ||
| Fig. 5 Fluorescence microscopy images and fluorescence intensity quantification of fibronectin matrix assembly. This data indicates that, at sub-cytotoxic concentrations, two modified SNX analogues 10 and 11 disrupted fibronectin matrix assembly in a similar fashion to wild-type functional upstream domain (WT-FUD), a known fibronectin inhibitor. | ||
Conclusions
In this study, we sought to develop eHsp90 inhibitors through the rational modification of N-terminal interacting ATP antagonists SNX2112 and SNX25a. The four new compounds (8–11) disclosed in this study were found to possess Hsp90 inhibitory activity equal to that of SNX2112. However, this activity was not maintained in the intracellular environment, having no impact on either the relative abundance of CDK4 or Hsp70. In addition, these modifications resulted in a substantial reduction in cytotoxicity. While the cell impermeability of 8–11 has not been explicitly proven, when taken together, these data imply a significant impediment of cell permeability. Based on this assertion, we further tested the impact that relegating Hsp90 inhibitors to the extracellular environment would have on the assembly of the fibronectin matrix. Here we showed that, while cell permeable SNX2112 had no impact matrix assembly, alkyl sulfonate containing Hsp90 inhibitors 10 and 11 both inhibited fibronectin accumulation akin to that observed for cells treated with WT-FUD, a natural fibronectin matrix assembly inhibiting peptide. It is currently unclear why this effect was not observed with the alkyl phosphonates 8 and 9. It is possible that the observed effect is not related to the phosphonate or sulfonate moieties, but rather due to the exitance of a zwitterionic form of compounds 10 and 11. We are currently expanding aspects of the SAR to attempt to elucidate this phenomenon. Significantly, however, the inhibition of fibronectin is an increasingly attractive strategy for developing non-toxic adjuvant therapies for suppressing cancer proliferation.51–53 This study, therefore, provides an important pharmacological validation of eHsp90 as a target for mimicking the phenotypic response of FUD. Furthermore, compounds 10 and 11 in particular represent useful tool compounds for advancing eHSP90 inhibition as an anticancer drug discovery strategy.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
GSM developed and conducted all chemical synthesis and helped draft the manuscript. AC and AC both conducted biological evaluations. RM conducted in silico analysis. MPA and MLB both provided student supervision. ALE and CGLV conceptualised the project and drafted the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge support from UCT, UKZN, Rhodes University, the South African Research Chairs Initiative of the DSI and NRF (Grant No. 98566), the NRF CPRR (Grant No 129262), and Future Leaders – African Independent Research (FLAIR), a partnership between the AAS and the Royal Society that is funded by the UK Government as part of the Global Challenge Research Fund (GCRF). GSM, AC, AC and RM gratefully acknowledge doctoral and post-doctoral support from the NRF (RM Grant number 120771). We also thank the Centre for High Performance Computing (CHPC) for access to Schrodinger's modelling suite.References
- F. U. Hartl, A. Bracher and M. Hayer-Hartl, Nature, 2011, 475, 324–332 CrossRef CAS PubMed.
- J. Trepel, M. Mollapour, G. Giaccone and L. Neckers, Nat. Rev. Cancer, 2010, 10, 537–549 CrossRef CAS PubMed.
- R. Freilich, T. Arhar, J. L. Abrams and J. E. Gestwicki, Acc. Chem. Res., 2018, 51, 940–949 CrossRef CAS PubMed.
- D. Mahalingam, R. Swords, J. S. Carew, S. T. Nawrocki, K. Bhalla and F. J. Giles, Br. J. Cancer, 2009, 100, 1523–1529 CrossRef CAS PubMed.
- W. B. Pratt and D. O. Toft, Exp. Biol. Med., 2003, 228, 111–133 CrossRef CAS PubMed.
- A. Donnelly and B. Blagg, Curr. Med. Chem., 2008, 15, 2702–2717 CrossRef CAS PubMed.
- D. C. DeZwaan and B. C. Freeman, Trends Biochem. Sci., 2010, 35, 384–391 CrossRef CAS PubMed.
- G. Garg, H. Zhao and B. S. J. Blagg, ACS Med. Chem. Lett., 2015, 6, 204–209 CrossRef CAS PubMed.
- L. Neckers and P. Workman, Clin. Cancer Res., 2012, 18, 64–76 CrossRef CAS PubMed.
- S. J. Mishra, T. S. Reynolds, T. Merfeld, M. Balch, S. Peng, J. Deng, R. Matts and B. S. J. Blagg, ACS Med. Chem. Lett., 2022, 13, 1870–1878 CrossRef CAS PubMed.
- S. J. Mishra, W. Liu, K. Beebe, M. Banerjee, C. N. Kent, V. Munthali, J. Koren, J. A. Taylor, L. M. Neckers, J. Holzbeierlein and B. S. J. Blagg, J. Med. Chem., 2021, 64, 1545–1557 CrossRef CAS PubMed.
- E. Schmitt, L. Maingret, P.-E. Puig, A.-L. Rerole, F. Ghiringhelli, A. Hammann, E. Solary, G. Kroemer and C. Garrido, Cancer Res., 2006, 66, 4191–4197 CrossRef CAS PubMed.
- Y. Wang and S. R. McAlpine, Org. Biomol. Chem., 2015, 13, 3691–3698 RSC.
- M. N. Rahimi, L. K. Buckton, S. S. Zaiter, J. Kho, V. Chan, A. Guo, J. Konesan, S. Kwon, L. K. O. Lam, M. F. Lawler, M. Leong, G. D. Moldovan, D. A. Neale, G. Thornton and S. R. McAlpine, ACS Med. Chem. Lett., 2018, 9, 73–77 CrossRef CAS PubMed.
- S. S. Zaiter, Y. Huo, F. Y. Tiew, J. E. Gestwicki and S. R. McAlpine, J. Med. Chem., 2019, 62, 742–761 CrossRef CAS PubMed.
- O. W. Mak, N. Sharma, J. Reynisson and I. K. H. Leung, Bioorg. Med. Chem. Lett., 2021, 38, 127857 CrossRef CAS PubMed.
- C. G. L. Veale, M. O. Okpara, M. C. Vaaltyn, A. Mahama, F. Albericio, B. de la Torre, D. J. Clarke and A. L. Edkins, ChemRxIV, 2024, preprint, DOI:10.26434/chemrxiv-2024-2q612.
- C. G. L. Veale, M. Mateos-Jiménez, M. C. Vaaltyn, R. Müller, M. P. Makhubu, M. Alhassan, B. G. de la Torre, F. Albericio, C. L. Mackay, A. L. Edkins and D. J. Clarke, Chem. Commun., 2021, 57, 10919–10922 RSC.
- G. Pimienta, K. M. Herbert and L. Regan, Mol. Pharmaceutics, 2011, 8, 2252–2261 CrossRef CAS PubMed.
- L. Seclì, F. Fusella, L. Avalle and M. Brancaccio, Cell. Mol. Life Sci., 2021, 78, 4069–4083 CrossRef PubMed.
- J. A. de la Mare, T. Jurgens and A. L. Edkins, BMC Cancer, 2017, 17, 202 CrossRef PubMed.
- A. Chakraborty and A. L. Edkins, Biochem. Soc. Trans., 2021, 49, 2611–2625 CrossRef CAS PubMed.
- R. A. Sager, F. Khan, L. Toneatto, S. B. D. Votra, S. J. Backe, M. R. Woodford, M. Mollapour and D. Bourboulia, Front. Mol. Biosci., 2022, 9, 982593 CrossRef CAS PubMed.
- M. Zou, A. Bhatia, H. Dong, P. Jayaprakash, J. Guo, D. Sahu, Y. Hou, F. Tsen, C. Tong, K. O'Brien, A. J. Situ, T. Schmidt, M. Chen, Q. Ying, T. S. Ulmer, D. T. Woodley and W. Li, Oncogene, 2024, 36, 2160–2171 CrossRef PubMed.
- M. C. Hunter, K. L. O'Hagan, A. Kenyon, K. C. H. Dhanani, E. Prinsloo and A. L. Edkins, PLoS One, 2014, 9, e86842 CrossRef PubMed.
- K. C. H. Dhanani, W. J. Samson and A. L. Edkins, Sci. Rep., 2017, 7, 17617 CrossRef PubMed.
- A. L. Correia, H. Mori, E. I. Chen, F. C. Schmitt and M. J. Bissell, Genes Dev., 2013, 27, 805–817 CrossRef CAS PubMed.
- W. Li, F. Tsen, D. Sahu, A. Bhatia, M. Chen, G. Multhoff and D. T. Woodley, Int. Rev. Cell Mol. Biol., 2013, 303, 203–235 CAS.
- S. Tsutsumi, B. Scroggins, F. Koga, M.-J. Lee, J. Trepel, S. Felts, C. Carreras and L. Neckers, Oncogene, 2008, 27, 2478–2487 CrossRef CAS PubMed.
- S. J. Mishra, A. Khandelwal, M. Banerjee, M. Balch, S. Peng, R. E. Davis, T. Merfeld, V. Munthali, J. Deng, R. L. Matts and B. S. J. Blagg, Angew. Chem., Int. Ed., 2021, 60, 10547–10551 CrossRef CAS PubMed.
- T. S. Reynolds and B. S. J. Blagg, ACS Med. Chem. Lett., 2023, 14, 1250–1256 CrossRef CAS PubMed.
- K. H. Huang, J. M. Veal, R. P. Fadden, J. W. Rice, J. Eaves, J. P. Strachan, A. F. Barabasz, B. E. Foley, T. E. Barta, W. Ma, M. A. Silinski, M. Hu, J. M. Partridge, A. Scott, L. G. DuBois, T. Freed, P. M. Steed, A. J. Ommen, E. D. Smith, P. F. Hughes, A. R. Woodward, G. J. Hanson, W. S. McCall, C. J. Markworth, L. Hinkley, M. Jenks, L. Geng, M. Lewis, J. Otto, B. Pronk, K. Verleysen and S. E. Hall, J. Med. Chem., 2009, 52, 4288–4305 CrossRef CAS PubMed.
- Y. Okawa, T. Hideshima, P. Steed, S. Vallet, S. Hall, K. Huang, J. Rice, A. Barabasz, B. Foley, H. Ikeda, N. Raje, T. Kiziltepe, H. Yasui, S. Enatsu and K. C. Anderson, Blood, 2009, 113, 846–855 CrossRef CAS PubMed.
- S. Chandarlapaty, A. Sawai, Q. Ye, A. Scott, M. Silinski, K. Huang, P. Fadden, J. Partdrige, S. Hall, P. Steed, L. Norton, N. Rosen and D. B. Solit, Clin. Cancer Res., 2008, 14, 240–248 CrossRef CAS PubMed.
- A. Rajan, R. J. Kelly, J. B. Trepel, Y. S. Kim, S. V. Alarcon, S. Kummar, M. Gutierrez, S. Crandon, W. M. Zein, L. Jain, B. Mannargudi, W. D. Figg, B. E. Houk, M. Shnaidman, N. Brega and G. Giaccone, Clin. Cancer Res., 2011, 17, 6831–6839 CrossRef CAS PubMed.
- T. L. Chen, B. Harrington, J. Truxall, R. Wasmuth, A. Prouty, S. Sloan, A. M. Lehman, D. Sampath, E. Orlemans, R. A. Baiocchi, L. Alinari, J. C. Byrd, J. A. Woyach and E. Hertlein, J. Hematol. Oncol., 2021, 14, 36 CrossRef CAS PubMed.
- J. R. Infante, G. J. Weiss, S. Jones, R. Tibes, T. M. Bauer, J. C. Bendell, J. M. Hinson, D. D. Von Hoff, H. A. Burris, E. O. Orlemans and R. K. Ramanathan, Eur. J. Cancer, 2014, 50, 2897–2904 CrossRef CAS PubMed.
- M. Gutierrez, R. Guo, G. Giaccone, S. V. Liu, Z. Hao, C. Hilton, J. M. Hinson, M. G. Kris, E. O. Orlemans and A. Drilon, Lung Cancer, 2021, 162, 23–28 CrossRef CAS PubMed.
- P. F. Hughes, J. J. Barrott, D. A. Carlson, D. R. Loiselle, B. L. Speer, K. Bodoor, L. A. Rund and T. A. J. Haystead, Bioorg. Med. Chem., 2012, 20, 3298–3305 CrossRef CAS PubMed.
- J. J. Barrott, P. F. Hughes, T. Osada, X. Yang, Z. C. Hartman, D. R. Loiselle, N. L. Spector, L. Neckers, N. Rajaram, F. Hu, N. Ramanujam, G. Vaidyanathan, M. R. Zalutsky, H. K. Lyerly and T. A. Haystead, Chem. Biol., 2013, 20, 1187–1197 CrossRef CAS PubMed.
- L. B. Crowe, P. F. Hughes, D. A. Alcorta, T. Osada, A. P. Smith, J. Totzke, D. R. Loiselle, I. D. Lutz, M. Gargesha, D. Roy, J. Roques, D. Darr, H. K. Lyerly, N. L. Spector and T. A. J. Haystead, ACS Chem. Biol., 2017, 12, 1047–1055 CrossRef CAS PubMed.
- T. Taldone, D. Zatorska, P. D. Patel, H. Zong, A. Rodina, J. H. Ahn, K. Moulick, M. L. Guzman and G. Chiosis, Bioorg. Med. Chem., 2011, 19, 2603–2614 CrossRef CAS PubMed.
- J. A. Carozza, J. A. Brown, V. Böhnert, D. Fernandez, Y. AlSaif, R. E. Mardjuki, M. Smith and L. Li, Cell Chem. Biol., 2020, 27, 1347–1358.e5 CrossRef CAS PubMed.
- J. R. Walker, M. P. Hall, C. A. Zimprich, M. B. Robers, S. J. Duellman, T. Machleidt, J. Rodriguez and W. Zhou, ACS Chem. Biol., 2017, 12, 1028–1037 CrossRef CAS PubMed.
- D. Zhao, Y. M. Xu, L. Q. Cao, F. Yu, H. Zhou, W. Qin, H. J. Li, C. X. He, L. Xing, X. Zhou, P. Q. Li, X. Jin, Y. He, J. H. He and H. L. Cao, Front. Cell Dev. Biol., 2021, 9, 650106 CrossRef PubMed.
- R. Dorel, C. P. Grugel and A. M. Haydl, Angew. Chem., Int. Ed., 2019, 58, 17118–17129 CrossRef CAS PubMed.
- G. S. Mathenjwa, M. P. Akerman, M. L. Bode and C. G. L. Veale, Synlett, 2022, 33, 1907–1912 CrossRef CAS.
- J. Mccready, D. S. Wong, J. A. Burlison, W. Ying and D. G. Jay, Cancers, 2014, 6, 1031–1046 CrossRef PubMed.
- A. Chakraborty, R. Tonui and A. L. Edkins, Cell Stress Chaperones, 2023, 28, 697–707 CrossRef CAS PubMed.
- L. M. Maurer, B. R. Tomasini-Johansson, W. Ma, D. S. Annis, N. L. Eickstaedt, M. G. Ensenberger, K. A. Satyshur and D. F. Mosher, J. Biol. Chem., 2010, 285, 41087–41099 CrossRef CAS PubMed.
- Z. Gong, M. Chen, Q. Ren, X. Yue and Z. Dai, Signal Transduction Targeted Ther., 2020, 5, 12 CrossRef CAS PubMed.
- H. J. Lee, M. K. Gari, D. R. Inman, Z. T. Rosenkrans, B. M. Burkel, A. P. Olson, J. W. Engle, R. Hernandez, S. M. Ponik and G. S. Kwon, J. Controlled Release, 2022, 350, 284–297 CrossRef CAS PubMed.
- H. Ghura, M. Keimer, A. von Au, N. Hackl, V. Klemis and I. A. Nakchbandi, Neoplasia, 2021, 23, 837–850 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4md00501e |
| This journal is © The Royal Society of Chemistry 2024 |