
In silico identification of novel PqsD inhibitors: promising molecules for quorum sensing interference in Pseudomonas aeruginosa†
Tatiana F.
Vieira
 ab,
Nuno M. F. S. A.
Cerqueira
ab,
Manuel
Simões
c and
Sérgio F.
Sousa
*ab
ab,
Nuno M. F. S. A.
Cerqueira
ab,
Manuel
Simões
c and
Sérgio F.
Sousa
*ab
aUCIBIO/REQUIMTE, BioSIM, Departamento de Medicina, Faculdade de Medicina da Universidade do Porto, Alameda Prof. Hernâni Monteiro, 4200-319 Porto, Portugal. E-mail: sergiofsousa@med.up.pt
bAssociate Laboratory i4HB – Institute for Health and Bioeconomy, Faculdade de Medicina, Universidade do Porto, 4200-319 Porto, Portugal
cLEPABE Laboratory for Process Engineering, Environment, Biotechnology and Energy, Faculty of Engineering, University of Porto, Rua Dr. Roberto Frias, s/n, 4200-465 Porto, Portugal
First published on 25th September 2023
Abstract
PqsD is an anthraniloyl-CoA anthraniloyltransferase involved in the synthesis of the secondary metabolites essential to the formation of Pseudomonas quinolone signal (PQS) inducer molecules. Its main substrate is anthraniloyl-coenzyme A (ACoA) but it can accept malonyl-CoA as secondary substrate. Suppression of PqsD activity has been connected to the inhibition of biofilm formation and can also be a good target for dual inhibition, when combined with PqsR inhibition. Here we describe the validation and application of an in silico methodology to find new compounds to inhibit PqsD. Using molecular docking and structure-based virtual screening protocols, five databases of compounds were screened (FDA approved subset of the ZINC database, Chimiothèque Nationale, Mu.Ta.Lig. Virtual Chemotheca, Interbioscreen (IBS), and Comprehensive Marine Natural Products Database (CMNPD)), representing a total of 221![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 146 molecules. The top five compounds of each database were selected to be further analysed using molecular dynamics simulations. Binding affinity was validated using free energy calculations, enabling the selection and characterization of eight compounds for future studies aiming to develop new quorum sensing inhibitors.
146 molecules. The top five compounds of each database were selected to be further analysed using molecular dynamics simulations. Binding affinity was validated using free energy calculations, enabling the selection and characterization of eight compounds for future studies aiming to develop new quorum sensing inhibitors.
Design, System, ApplicationThis project employs a fully integrated multi-technique in silico optimization protocol that combines molecular docking (validated through re- and cross-docking experiments), development of a robust virtual screening procedure with enhanced ability in active ligand identification, application of this protocol to large libraries of chemically distinct potential molecules (a total of 221146 molecules) from various origin and, finally, a thorough molecule selection stage. For selection, molecular dynamics simulations, free energy calculations, visual inspection and similarity analysis are combined to identify the most promising molecules. The eight selected ligands can now be experimentally tested and used for rational and target-specific drug design against PqsD, an anthraniloyl-CoA anthraniloyltransferase involved in the synthesis of the secondary metabolites essential to the formation of Pseudomonas quinolone signal (PQS), an important target for quorum sensing interference in Pseudomonas aeruginosa.
|
Introduction
Pseudomonas aeruginosa is a Gram-negative opportunistic pathogen that exhibits a large genome, a highly adaptable nature and a plethora of regulatory systems.1 These characteristics make the bacteria very difficult to eradicate, particularly in immunocompromised patients. Additionally, antimicrobial therapy is often hampered due to the presence of efflux pumps, β-lactamases and the ability of P. aeruginosa to start growing as sessile communities commonly called biofilms. The thick biofilm matrix composed of extracellular polymeric substances (EPS) is highly impermeable to antimicrobial agents. P. aeruginosa controls the expression of virulence factors and biofilm formation through a complex and cell dependent communication system called quorum sensing (QS). In this bacterium, there are three main QS systems that communicate with each other and are highly hierarchical: las, rhl systems that use homoserine lactones as signal molecules and the pqs system that uses quinolones as the autoinducer. These QS inducer molecules, by accumulating in a growing bacterial population, start influencing the production and release of virulence factors.2 A simplified overview of the pqs system is displayed in Fig. 1.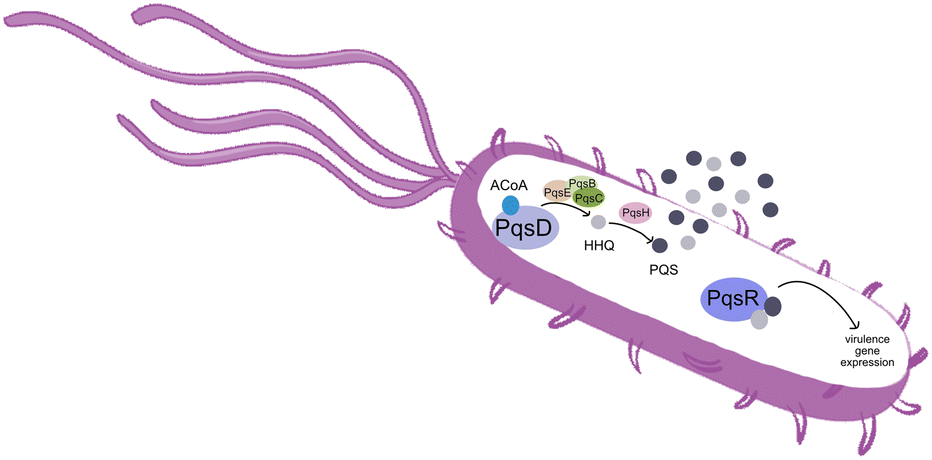 | ||
| Fig. 1 Simplified overview of the PQS system in P. aeruginosa. | ||
PqsD is an anthraniloyl-CoA anthraniloyltransferase involved in the synthesis of the secondary metabolites 2-heptyl-3-hydroxy-4(1H)-quinolone and 4-hydroxy-2(1H)-quinolone, mediators in the formation of the pqs inducer molecules 2-heptyl-4-hydroxyquinoline (HHQ) and 2-heptyl-3-hydroxy-4-quinolone (PQS).3 It belongs to the β-ketoacyl-ACP-synthase family and is similar in structure to the β-ketoacyl-ACP-synthase III (FabH) in E. coli and M. tuberculosis.4 Its main substrate is anthraniloyl-coenzyme A (ACoA) but it can accept malonyl-CoA as secondary substrate. PqsD has also been shown to be responsible for the synthesis of 2,4-dihydroxyquinolone (DHQ), an abundant extracellular metabolite of P. aeruginosa.5,6
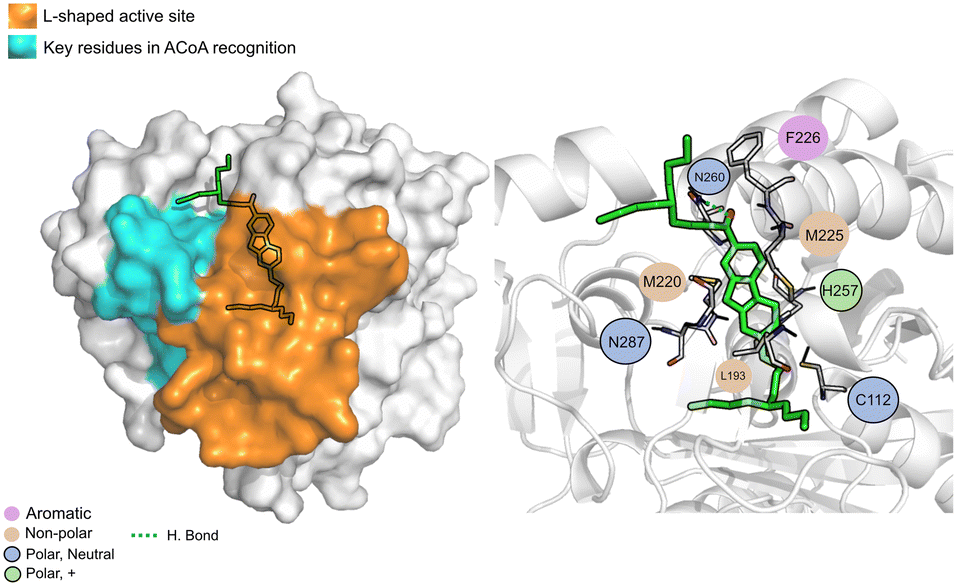
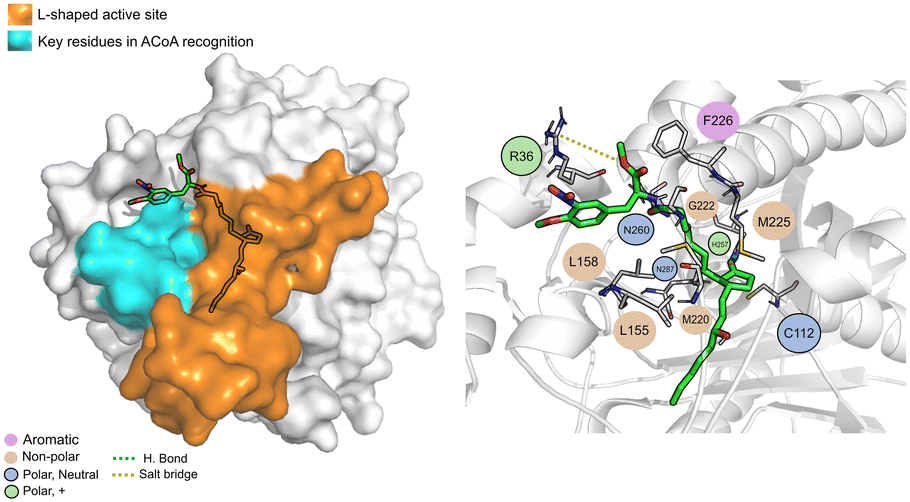
The PqsD binding pocket is long and narrow (15 Å) and can be divided into three parts: a positively charged entrance, a hydrophobic middle and a polar bottom delimited by the catalytic triad Cys112–His257–Asn287.4 Residues between Glu140 to Gly162 are key in CoA recognition and binding 6 and residues 186 to 222 form a L-shaped active side. A detailed view of the PqsD structure is presented in Fig. 2.
 | ||
| Fig. 2 Schematic representation of PqsD bound to ACoA (PDB: 3H77). Protein represented in cartoon, with key features highlighted with different colors, and ACoA represented by atom VdW radius (spheres). | ||
Suppression of PqsD activity has been shown to inhibit biofilm formation7 and when combined with MvfR/PqsR inhibition can lead to more highly efficient attenuation of P. aeruginosa pathogenicity, in a phenomenon called dual inhibition.8 Furthermore, targeting the PQS system, highly specific to P. aeruginosa, can minimize the development of side effects by not interfering with other bacteria important for the health maintenance in humans.1
Some inhibitors for β-ketoacyl-ACP-synthase III (FabH) have been described, namely cerulenin, thiolactomycin, indole derivatives, Schiff bases, benzoylaminobenzoic acid derivatives and compounds containing sulfur atoms.9 Cerulenin and thiolactomycin also target other type II FAS enzymes. Cerulenin forms an adduct with the catalytic cysteine of FabF and thiolactomycin acts like a reversible inhibitor.10 Some of these have been tested against PqsD such as benzoylaminobenzoic acid derivatives and compounds containing sulfur atoms. Benzoylaminobenzoic acid derivatives have shown promising in vitro activity against PqsD.11–13 More specifically regarding sulfonamide substituted 2-benzamidobenzoic acids, researchers realized that the presence of carboxylic acid was essential for activity and that the variation of substituents of the 2-aminobenzoic acid part could increase the potency of the possible PqsD inhibitors.12 Furthermore, it has been suggested that there are only two possible inhibition modes of action; either by direct interaction between the inhibitor molecule and the residues in the binding site or simply by blocking the entrance of the PqsD pocket.3,12,14
The use of computer aided drug design tools, namely protein–ligand docking and structured-based virtual screening (VS)15,16 has been shown to help narrow down the list of compounds to test experimentally, with promising results,17 particularly regarding molecular docking in the PqsD binding pocket.18,19 The workflow of the present study is presented in Fig. 3 and has been applied to other projects and other bacterial targets.20–24
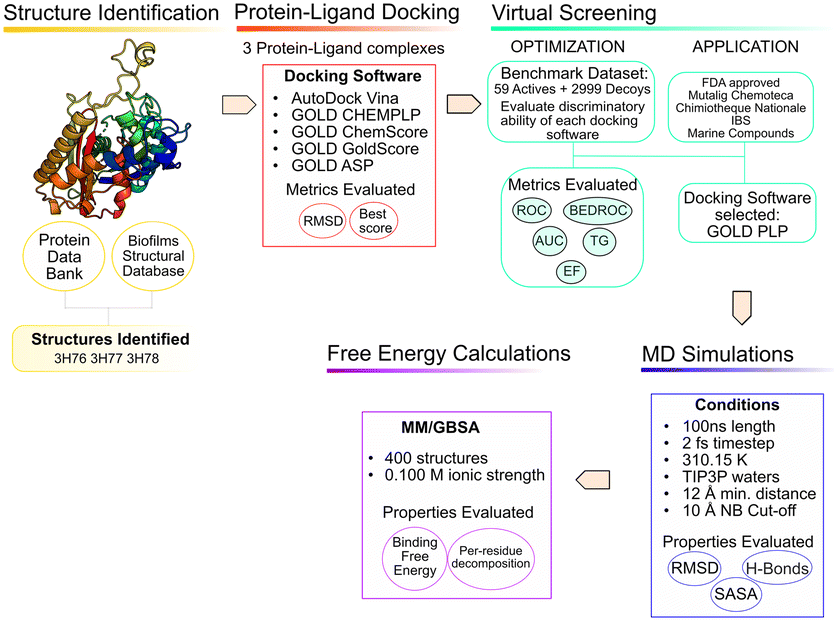 | ||
| Fig. 3 In silico workflow used in the current study. | ||
Methodology
Structure analysis and preparation
Searching the Protein Data Bank25 and the Biofilms Structural Database,26 3 X-ray structures of PqsD were identified. The PqsD model predicted by Alphafold (P20582)27,28 and available at the Alphafold Protein Structure Database was also taken into consideration, to evaluate possible structural variations but it was not used for the in silico study because the priority was given to choosing an experimental structure with a crystallographic ligand in the binding pocket.Structure 3H76 represents the structure in the apo form. Structure 3H77 presents the Cys112 residue covalently bound to anthranilate, a reaction intermediate. Structure 3H78 has one mutation (Cys112Ala) to evaluate the role of Cys112 in the catalytic mechanism of PqsD. Also, this structure has anthranilic acid in the binding pocket. The presence of alanine does not affect the binding of ACoA but affects the kinetic activity of the enzyme, since in the experimental assays with this structure, there was no product detected.6 Further details on these X-ray structures are shown in Table 1.
For target preparation, polar hydrogens were added with GOLD at physiological pH and subject to visual inspection of the amino acid residues at the binding pocket. Waters and molecules resulting from the crystallographic process, were removed. The three structures were aligned to ensure consistency in the docking coordinates. The crystallographic ligands were extracted and saved in separate files, to be used in the validation of the docking conditions. For GOLD, all the targets were saved in .mol2 file format, and for Vina in .pdbqt file format.
Protein–ligand docking protocol validation
Two docking software alternatives and five scoring functions were used for this project. Autodock Vina,29 and GOLD30 (CHEMPLP, GoldScore, ChemScore and ASP scoring functions). To ensure consistency, consistent docking conditions were applied to all the SF, in terms of binding pocket definition.Testing a variety of scoring functions (SF) is intended to evaluate which is the best for this specific target with a long, narrow pocket, since docking results have been shown to differ significantly depending on the specific target protein and ligand type.31,32
Re-docking and cross-docking were the methods used for protocol validation. Re-docking is used to evaluate the ability of each SF and protocol to correctly reproduce the geometry and orientation of the respective crystallographic ligand pose. By evaluating the binding affinity and the root mean square deviation (RMSD) between the heavy atoms of the crystallographic and docked poses, one can evaluate the quality of the protocol in correctly identifying the experimental pose. Cross-docking evaluates the universality of the protocol, evaluating the ability of each experimental structure of the target protein in accommodating the binding of the different confirmed binders. It involves the docking of all the X-ray ligand structures into all the protein structures available. In this case, only one crystallographic ligand (ACoA) was extracted using pymol,33 the protonation was corrected using OpenBabel34 and was subsequently docked into the three PDB PqsD structures. The purpose of this test is to determine if individual X-ray structures can accommodate the different X-ray ligands, which are co-crystallized in other X-ray structures. The binding affinity was evaluated to determine the quality and accuracy of the method.
Virtual screening protocol validation
For the validation of the VS conditions, an active versus decoys protocol was applied. The goal was to evaluate the performance of each scoring function (SF) and protocol in discriminating between binders and non-binders. The ideal SF would provide the true binders with better scores, being able to clearly single out binders from non-binders. However, due to the impossibility to mathematically account for all the phenomena that occurs in protein–ligand binding, that is not always the case. Since the goal of a VS is to quickly screen large databases of compounds, the accuracy is somewhat penalized.35,36 Therefore, the protocol was continuously improved to maximize the ability of each SF to rank the true binders in the top solutions.The ChEMBL37 and BindingDB38 databases were explored, and 59 ligands were found to have reported experimental activity against PqsD.39,40 The DUD-E41 database was used to generate decoy molecules. Decoys are molecules that show similar physical properties to the active molecules, to eliminate bias, but have distinct chemical properties and are assumed to be non-binders. For each active molecules, 50 decoys were generated, giving a total of 2950 decoys. The complete test set was composed of a total of 3009 molecules, prepared for docking with OpenBabel.34
The results obtained with the four scoring functions applied were evaluated through a web-based application, Screening Explorer,42 as well as using Excel. Several metrics were considered: enrichment factor at 1%, receiver-operating characteristic (ROC) curves and the respective area under the curve (AUC), Boltzmann-enhanced discrimination of ROC (BEDROC), the robust initial enhancement (RIE), and total gain (TG). A SF shows a good discriminatory ability and reproducibility if it presents a TG value over 0.25, combined with an AUC over 50%.
Virtual screening of compound databases
Five libraries of compounds were screened, to narrow the search for new inhibitors, using the docking and VS conditions that were previously optimized and validated. The selected databases were: FDA approved compounds43 for drug repurposing, the French National chemical library (Chimiothèque Nationale)44 and Mu.Ta.Lig. Virtual Chemotheca45 as databases of synthetic compounds, Interbioscreen (IBS)46 and Marine Compound Database (CMNPD)47 and as sources of natural and marine compounds, respectively. The rational of screening various compound databases is to provide a more representative and diverse test set, containing synthesizable and natural compounds. At this stage, the crystallographic structure and SF used were 3H76 and ChemPLP, respectively.The FDA approved library of compounds is subset of the ZINC database,43 a free database that contains over 230 million commercially available compounds. At the time of the VS experiments, the FDA-approved drugs dataset had 3207 compounds that were all docked against PqsD.
Chimiothèque Nationale44 contains more than 70000 compound that were synthesized or isolated in different laboratories. For this project, a total of 61640 compounds were screened against PqsD.
The Mu.Ta.Lig. Virtual Chemotheca45 was the result of the European Union COST action (CA15135) and it contains molecules that were synthesized by several research groups. For this assay, 64804 compounds were screened against PqsD.
The natural compounds database of Interbioscreen (IBS)46 containing 66278 compounds was selected to find possible inhibitors for PqsD. This database includes over 68000 natural compounds and derivatives from several ecosystems around the world.
Finally, 25217 compounds retrieved form the Comprehensive Marine Natural Products Database (CMNPD)47 were docked against PqsD. This database contains manually curated chemical, physicochemical, and pharmacokinetic properties of marine natural compounds.
Prior to the virtual screening application, all the databases were verified and “cleaned”. Duplicate molecules were eliminated, as well as compounds with molecular weight above 700 g mol−1.
A similarity search was conducted using the SkelSpheres descriptor of Datawarrior, for the best predicted compounds retrieved from the VS. This descriptor encodes circular spheres of atoms and bonds into a hashed binary fingerprint of 512 bits. From every atom in the molecule, DataWarrior constructs fragments of increasing size by including n layers of atom neighbours (n = 1 to 5). These circular fragments are canonicalized considering aromaticity and stereochemistry, counts duplicate fragments, in addition encodes hetero-atom depleted skeletons. From the canonical representation a hash code is generated, which is used to set the respective bit of the fingerprint. It has a good resolution, leading to less hash collisions and it is the most accurate descriptor for calculating similarities of chemical graphs.
Molecular dynamics and free energy calculations
To validate the docking predictions, molecular dynamics (MD) simulations were performed with PqsD and the top 5 compounds of each database. In the MD simulations PqsD was treated with the ff14SB force field48 and the 25 compounds selected were parameterized with Gaussian16 (ref. 49) using ANTECHAMBER and the general Amber force field (GAFF)50 to calculate RESP HF/6-31G(d) charges. The protein–ligand complexes were then treated with the Leap module of Amber18.51 ACoA, the natural substrate, was used as a control molecule.All the PqsD–ligand systems were neutralized with the addition of sodium ions and placed into TIP3P water molecules box; with periodic boundary conditions whose edges were at least at a 12 Å distance from each protein–ligand complex atom. A time step of 2 fs was used, and the SHAKE algorithm was applied to constrain the bonds involving hydrogen atoms. The long-range electrostatic interactions were calculated with the particle mesh Ewald summation method and the cut-off value for electrostatic and Lennard-Jones interactions was set at 10.0 Å.
Four minimization steps were applied to remove clashes, followed by two equilibration steps and a final production run. The minimization steps were applied to the following groups of atoms: first minimization-water molecules (2500 steps); second minimization-hydrogens atoms (2500 steps); third minimization-chains of all the amino acid residues (2500 steps); forth minimization-full system (10000 steps). The two 50 ps equilibration steps consisted of: 1-heating of the system to 298 K using a Langevin thermostat at constant volume (NVT ensemble) 2-equilibration of the density of the system at 298 K. Lastly, the 100 ns production run was performed in an NPT ensemble with a temperature of 298 K and 1 bar pressure. VMD52 and the cpptraj tool53 were employed to explore the resulting molecular dynamics (MD) trajectories.
The end-point free energy method MM–GBSA54 (molecular mechanics–generalized Born surface area method) was employed to assess the binding free energies of each ligand towards PqsD. The MM/PBSA.py script, available in AMBER,55 was applied to calculate the energies, with a salt concentration of 0.100 mol dm−3. The final last 90 ns of the MD simulation of every complex with an interval of 10 frames were considered for the analysis, representing a total of 1800 frames per complex. The free energy decomposition option was used to obtain information about the contribution of each residue to the total free energy. The results are presented from the strongest binder to the weakest. This method involves several approximations, and the entropic term is the one that displays the highest statistical uncertainty.56 This method is very useful to provide insight and analyze the binding affinities and trends, as well as helping in narrowing down the selection of compounds for experimental testing.
Results and discussion
Table 2 shows the results of the re-docking and cross-docking for all the PqsD structures available in the PDB database. As expected, structure 3H77 showed the lowest scores due to the presence of anthranilate bound to Cys112As previously mentioned, structure 3H78 has a mutation (Cys112Ala), that is not catalytically active in vivo, however, since alanine is a small residue, roughly the same size as a cysteine, it does not cause a steric hindrance in the binding pocket and in the cross-docking with ACoA it produces a high score, which is particularly evident when using GoldScore.Regarding the all the SF studied, ChemScore performed the worst and was unable to correctly dock the ACoA in the binding site.
Even though structure 3H78 presented good docking scores it was discarded at this stage. Since the mutation of 3H78 is in the binding pocket, and in one of the residues of the catalytic triad (leading to a form that is not catalytically active in vivo), it could influence the docking results leading to a selection of compounds, later on, in the VS stage, that could not have affinity toward wt PqsD.
After the initial study of structure 3H77, the docking scores obtained were not satisfactory because there was an anthranilate bound to Cys112. Even after removal of the anthranilate group, the results did not improve (data not shown), and it was decided to also remove this structure from the VS studies.
Ultimately, the structure chosen to move on to the VS optimization stage was 3H76, the crystallographic structure of the enzyme in the apo form. At this stage, Vina was also left out because it did not provide accurate results in terms of score and crystallographic pose reproducibility.
The actives vs. decoys protocol was performed on structure 3H76 and the metrics used to evaluate its quality are depicted in Table 3 and Fig. 4. The enrichment factor at 1% (EF 1%) measures how many actives are found at 1% of the ordered database, so, naturally, the higher the EF at 1% the better. Additionally, a good discriminatory ability of the SFs, is verified by TG and ROC values above 0.25 and 50% respectively.42 GoldScore and CHEMPLP were the SFs that exhibited the best metrics, with CHEMPLP showing a slight improvement in the ability to find true binders early on. Even though ASP and ChemScore showed higher AUC, the early recognition metrics (EF1% and TG) were not satisfactory. Eventually, GoldScore and ChemPLP provided similar results (particularly in the VS metrics), ultimately GoldScore was discarded because it is much more time-consuming and ChemPLP was able to provide good results much faster.
| EF 1% | AUC% | 3H76 | RIE | BEDROC | |
|---|---|---|---|---|---|
| TG | |||||
| ASP | 0.00 | 71 | 0.02 | 1.25 | 0.09 |
| ChemScore | 0.00 | 71 | 0.02 | 0.98 | 0.06 |
| GoldScore | 1.70 | 67 | 0.25 | 3.24 | 0.20 |
| ChemPLP | 1.73 | 68 | 0.25 | 1.65 | 0.20 |
 | ||
| Fig. 4 ROC curves for 3H76 for the several scoring functions tested. | ||
The top 1% of compounds from each database were selected based on the docking score generated by the application of the VS protocol using the PLP scoring function.
The ChemPLP score and molecular weight of the top eight compounds predicted in the FDA approved database are depicted in Table 4, along with a small description of the pharmaceutical use.
| Drug name | Description | ChemPLP score | MW (g mol−1) |
|---|---|---|---|
| Tessalon | A non-narcotic oral antitussive agent57 | 93.5 | 603.7 |
| Nonoxynol-9 (N-9) | Vaginal spermicide used for the non-hormonal contraception in conjunction with other modes of contraception58 | 94.5 | 616.8 |
| Polidocanol | Sclerosing agent used for the treatment of uncomplicated spider and reticular veins in the lower extremities59 | 84.7 | 585.8 |
| Deferoxamine (Dfo) | Chelating agent used to treat iron or aluminum toxicity and some blood transfusion dependent anemias60 | 90.5 | 560.7 |
| Phytonadione (vitamin K1 aka K-Ject) | Treat and manage vitamin K deficiency61 | 93.0 | 416.6 |
| Salmeterol | Beta-2 adrenergic agonist used to treat and manage symptoms of asthma and chronic obstructive pulmonary disease (COPD)62 | 85.3 | 416.6 |
| Nefazodone | Antidepressant used in the treatment of depression. However, it has been discontinued in some countries due to the risk of hepatotoxicity63 | 85.8 | 471.0 |
| Ketoconazole | Broad spectrum imidazole antifungal used to treat seborrheic dermatitis and fungal skin infections64 | 82.4 | 531.4 |
The chemical structure of the top compounds from the all the databases can be seen in Fig. 5. Most of the compounds selected present high molecular weights with many rotatable bonds, exhibiting long tails oriented toward the polar bottom of the binding pocket similarly to the natural inducer.
 | ||
| Fig. 5 Chemical structure of the selected compounds. Orange represents the compounds from the FDA approved database. Blue represents the molecules retrieved from Chimiothèque Nationale. Red corresponds to the ones coming from Mu.Ta.Lig Chemotheca. Represented in green are the compounds retrieved from the IBS and in black the compounds selected from CMNPD. | ||
Interestingly three classes of compounds were highlighted: isoquinoline derivatives from Chemoteca, chromone derivatives from IBS and imidazole derivatives from Marine Compounds. Interestingly, isoquinoline derivatives had already been studied as antimicrobial agents65,66 with promising activity against several species of bacteria and fungi. Chromone derivatives have shown activities against Mycobacterium tuberculosis, Staphylococcus aureus, and Bacillus subtilis67 and have also appeared as top hits in another VS study regarding MvfR/PqsR.68 As for imidazole derivatives, their structure has already been proven to be a good starting point in the design of more efficient QS inhibitors.69 Consequently, both chromone and benzimidazole derivatives seem to be particularly good candidates for dual inhibition.
None of the selected compounds contains sulfur atoms, so the hypothesis of a nucleophilic attack in a similar way than ACoA it not likely. However, the selected ligands possess oxygen and nitrogen atoms capable of interacting with the sulfur atom of Cys112 or increasing the number of hydrogen bond interactions. This data was further analyzed using MD simulations and measuring the distance between these atoms and Cys112.
The similarity analysis performed shows that there is structural similarity between molecules in the same database the following compounds presenting high chemical similarity between them: Tessalon, N-9 and Polidocanol (from the FDA approved database); AB-00069970 and AB-00069855 (from the Chimiotheque database); CMLDID26439 and CMLDID51411 (from the Mu.Ta.Lig Chemotheca database); STOCK1N-00436 and STOCK1N-45295 (from the IBS database) (Fig. S1 in the ESI†). There is, however, no chemical similarity between any of the predicted molecules and the ACoA.
To further analyze and validate the docking predictions, molecular dynamics simulations were performed in the top 25 protein–ligand complexes. The results are shown in Table 5. ACoA was used as reference and the docked pose of each ligand was used as a starting point. Several parameters were considered to evaluate the stability of the protein–ligand complexes such as: protein and ligand RMSd and average number of hydrogen bonds formed. The exposure of the ligand to the solvent when bound to the protein was also evaluated measuring the solvent-accessible surface area (SASA), percentage of potential ligand SASA buried, and number of water molecules present within a radius of 3 Å from the pocket.

|
All the molecules remained bound to the protein throughout the simulations, however, because some of them are quite big and have long tails, leading to higher protein and ligand RMSDs in comparison with the initial structures. This might be explained because their movement in the pocket induces a displacement of the amino-acid residues of the active site. Even though they are big molecules, they are buried deep into the pocket, with most compounds presenting a percentage of potential ligand SASA buried above 85%.
Calculations were also performed to determine binding energies of association and evaluate residues contributing most to ligand stabilization in binding pockets using MM/GBSA. Results are presented in Table 6. For the MM/GBSA calculation, the entropic term was neglected, due to the limitations in the entropy calculation method, a common approximation.54,70,71 Therefore, the MM/GBSA values will be looked as a ranking tool, to guide the selection of compounds to test experimentally. The overall energy of binding was decomposed, and the individual residue contribution was evaluated for each protein–ligand interaction. The three most relevant residues are also represented in Table 6. The potential inhibitors selected are highlighted in purple both in Tables 5 and 6.
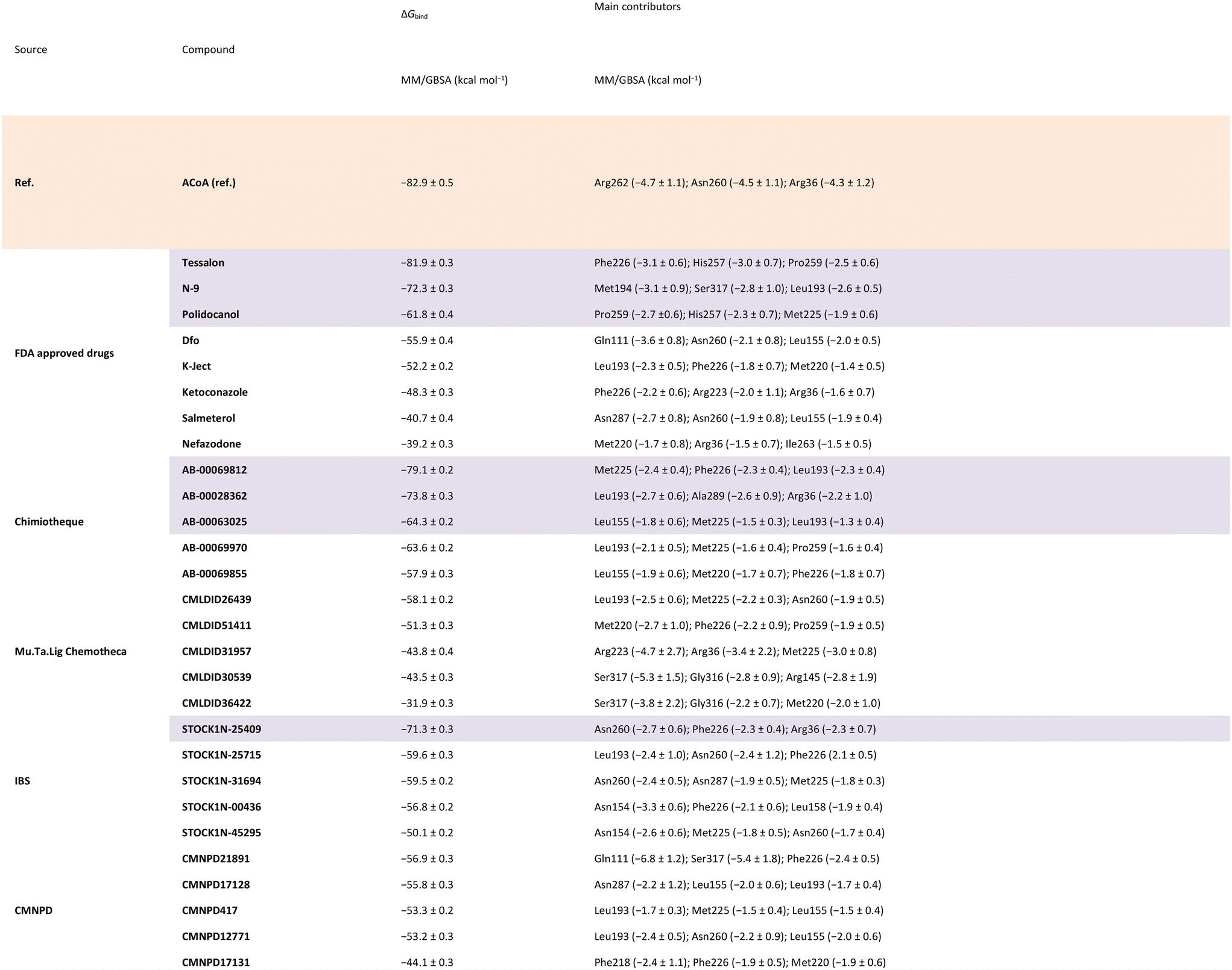
|
To narrow down the list of compounds to be studied experimentally, a graph was plotted to correlate the Docking ChemPLP score with the MM/GBSA values (kcal mol−1) (Fig. 6). This graph allows for a better visualization and confirmation of the potential of these molecules as PqsD inhibitors. Ultimately, the compounds selected were the ones that presented higher docking scores and more negative binding free energies (compounds with docking scores above 90 and MM/GBSA values below −60 kcal mol−1: Tessalon, AB-00069812, AB-00028362, N-9, STOCK1N-25409, AB-00069970, AB-00063025 and Polidocanol). A normalized score was calculated (ChemPLP score by the number of heavy atoms), to eliminate some of the docking bias (as the docking score is proportional to the number of atoms) and the results are presented in Fig. S2 in the ESI.† The selected compounds exhibit higher normalized scores than the reference (ACoA) and the tendency in terms of compound selection is preserved, because, even though the compounds present a slightly decrease normalized score, they still stand out in terms of a more favorable estimated binding free energy.
 | ||
| Fig. 6 Comparison of docking ChemPLP scores and total binding free energy (MM/GBSA). ACoA was used as reference. Blue compounds show higher logP values. | ||
It is curious to see that the molecules that were chemically similar in the FDA approved database were all selected as the most promising ones (Tessalon, N-9 and Polidocanol). While the ones from Chimiotheque, Mu.Ta.Lig Chemotheca and IBS databases were not selected.
The interactions between the selected compounds and ACoA with PqsD is depicted in detail in Fig. 7 to 15. The ligand poses characterized are the most representative structures obtained from the MD simulations.
 | ||
| Fig. 7 ACoA bound to PqsD (green licorice). Surface view emphasizing the L-shaped active site and area with key residues involved in ACoA recognition (left). Interaction map highlighting the most relevant residues (right) involved in ligand stabilization. | ||
ACoA (Fig. 7) is stabilized through electrostatic interactions but can also form hydrogen bonds with Asp150, Gly222 and Asn260. It also forms salt bridges with Arg36 and Arg262. These arginines along with Arg223 seem to be important for ACoA recognition and interact with it through the diphosphate group. The covalent bond between ACoA and Cys112 is not visible because the methodology applied is not able to study the formation/breaking of covalent bonds. However, the distance between the residue and ACoA is below 2 Å, and it was considered a good approximation.
Tessalon (Fig. 8) occupies almost the same area of ACoA, except for the interaction with Arg36 and Asp150 being in a more vertical position. It can form hydrogen bond interactions with residues His257 and Asn287, two of the residues of the catalytic triad, it is, however, very far away from Cys112 (∼7.3 Å). It can also form a salt bridge with Arg223. Tessalon caused a shift in the position of His257 and Asn287, when compared to ACoA.
 | ||
| Fig. 8 Tessalon (green licorice) bound to PqsD. Interaction map highlighting the most relevant residues (right) involved in ligand stabilization. | ||
AB00028362 (Fig. 9) can form hydrogen bonds with Cys112 (2.16 Å), Asn260 (2.07 Å) and Ala289 (1.67 Å), the other interactions are mainly hydrophobic. The key residues involved in ACoA recognition do not seem to interact with AB00028362.
 | ||
| Fig. 9 AB-00028362 (green licorice) bound to PqsD. Interaction map highlighting the most relevant residues (right) involved in ligand stabilization. | ||
AB00069812 (Fig. 10) interacts with PqsD mainly through electrostatic interactions with most of the residues and through hydrogen bond with Asn260. The aliphatic tails of AB00069812 seem to lock the compound into the L-shaped active site portion of the pocket.
 | ||
| Fig. 10 AB00069812 (green licorice) bound to PqsD. Interaction map highlighting the most relevant residues (right) involved in ligand stabilization. | ||
N-9 (Fig. 11) is stabilized mainly through electrostatic interactions and can form a hydrogen bond with Asn87. The tail of N-9 seems to trigger a shift in the position of His257, Met225 and Phe226, when compared with ACoA–PqsD complex, leading to the formation of a tunnel to accommodate the aliphatic tail. Distance to Cys112 of 6.5 ± 2.1 Å.
 | ||
| Fig. 11 N-9 (green licorice) bound to PqsD. Interaction map highlighting the most relevant residues (right) involved in ligand stabilization. | ||
STOCK1N-25409 (Fig. 12) is stabilized by non-polar interactions but can also interact via hydrogen bond with Gly222, Asn260 and His257. It can also interact with Arg36 via salt bridge. It can interact, via electrostatic interactions, with some of the key residues involved in ACoA recognition, namely Leu158 and Leu155. Distance to Cys112 is 11.5 ± 1.1 Å.
 | ||
| Fig. 12 STOCK1N-25409 (green licorice) bound to PqsD. Interaction map highlighting the most relevant residues (right) involved in ligand stabilization. | ||
AB-00063025 (Fig. 13) interacts with PqsD pocket, mainly through the establishment of non-polar interactions. The tricyclic head of this compound is in a position similar to the bicyclic head of ACoA when bound to PqsD, interacting with residues Asn154 and Leu155. However, it does not seem to interact with any of the residues of the catalytic triad (distance to Cys112 of 9.3 ± 1.7 Å).
 | ||
| Fig. 13 AB-00063025 (green licorice) bound to PqsD. Interaction map highlighting the most relevant residues (right) involved in ligand stabilization. | ||
AB-00069970 (Fig. 14), can form hydrogen bond with Asn260 but interacts mainly through non-polar interactions. The aliphatic tail sits on top of the region of the key residues involved in ACoA recognition, however, there is no interaction involved with the three arginines in the cavity entrance (Arg36, Arg223 and Arg262). Distance to Cys112 is 9.8 ± 1.2 Å.
 | ||
| Fig. 14 PqsD – AB-00069970 (green licorice) bound to PqsD. Interaction map highlighting the most relevant residues (right) involved in ligand stabilization. | ||
Polidocanol (Fig. 15) is a long chain composed of carbon and oxygen atoms, it interacts only through non-polar interactions with the L-shaped portion of the binding pocket. Distance to Cys112 is around 6.9 ± 1.4 Å.
Overall, the results suggest that these molecules could be used to block access to the active site, preventing the interaction with ACoA, and not necessarily by interacting with the catalytic triad as the natural substrate would do. The next part of the study would be to test these compounds experimentally and evaluate their ability to suppress the ACoA interaction. PqsD may not seem like an obvious target, but suppressing the formation of the autoinducer molecules could have a huge impact on the quorum sensing system. This work is a good starting point in the elucidation of mechanism of action of this enzyme and the types of interaction that it can perform with molecules other than ACoA. Furthermore, new classes of possible inhibitors have been identified, including isoquinoline and imidazole derivatives.
 | ||
| Fig. 15 Polidocanol (green licorice) bound to PqsD. Interaction map highlighting the most relevant residues (right) involved in ligand stabilization. | ||
Conclusions
An in silico methodology was created and validated to find new molecules capable of inhibiting PqsD, an anthraniloyl-CoA anthraniloyltransferase involved in the synthesis of the secondary metabolites important for the PQS quorum sensing system of Pseudomonas aeruginosa. Protein–ligand docking, structure-based VS and MD simulations were employed to select and validate a list of chemical diverse compounds subsequent experimental evaluations. Five different databases of compounds were screened in a total of 221146 molecules, and 28 compounds were selected to be further analysed with MD simulations and free energy calculations.
The MD simulations are a very good tool to consolidate the docking prediction as it allows for the search of the most stable ligand conformation eliminating the bias and improving the free energy predictions. The entropic contribution, in the free energy calculations was overlooked because is computationally time consuming and expensive to calculate. However, this work is the first that describes a complete in silico methodology (from molecular docking to molecular dynamics simulations and free energy calculations) to find possible inhibitors of PqsD and the main objective was to narrow down the list of compounds, rank them, and look for new drug design scaffold for future experimental testing. The goal was successfully achieved and, naturally, the next stage is the design of an experimental methodology to test the compounds selected and evaluate their specificity toward the pqs system of P. aeruginosa.
From the simulations it is hypothesized that these compounds may interact with the upper region of the protein tunnel by blocking the access to some of the key residues involved in ACoA recognition. Only one compound, AB00028362, seems to have the ability to directly interact with the residues in the binding site, as an inhibitor would, mainly through the establishment of a covalent bond with Cys112. Overall, this study suggests two mechanisms of action for inhibiting PqsD activity. One, by competitively binding with the natural autoinducer at the active site; a second mode, by blocking access to the protein tunnel. These modes, and the structural scaffolds identified can be further explored for the identification of new molecules with higher affinity or specificity and for experimental/theoretical optimization.
Author contributions
Conceptualization, S. F. S.; methodology, T. F. V.; software, T. F. V.; validation, T. F. V.; formal analysis, T. F. V.; investigation, T. F. V.; data curation, T. F. V.; writing – original draft preparation, T. F. V.; writing – review and editing, S. F. S., M. S.; supervision, S. F. S., M. S.; project administration, S. F. S.; funding acquisition, S. F. S., M. S.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by national funds from Fundação para a Ciência e a Tecnologia (grant numbers: SFRH/BD/137844/2018, UIDP/04378/2020, UIDB/04378/2020 and 2020.01423.CEECIND/CP1596/CT0003). Some of the calculations were produced with the support of the INCD funded by the FCT and FEDER under project 01/SAICT/2016 number 022153, and projects CPCA/A00/7140/2020 and CPCA/A00/7145/2020. This work was also financially supported by: Base Funding [LA/P/0045/2020 (ALiCE), UIDB/00511/2020 and UIDP/00511/2020 (LEPABE)], funded by national funds through [FCT/MCTES (PIDDAC)].Notes and references
- R. F. Langendonk, D. R. Neill and J. L. Fothergill, Front. Cell. Infect. Microbiol., 2021, 11, 665759 CrossRef PubMed.
- M. Prothiwa, D. Szamosvári, S. Glasmacher and T. Böttcher, Beilstein J. Org. Chem., 2016, 12, 2784–2792 CrossRef CAS PubMed.
- M. P. Storz, C. Brengel, E. Weidel, M. Hoffmann, K. Hollemeyer, A. Steinbach, R. Müller, M. Empting and R. W. Hartmann, ACS Chem. Biol., 2013, 8, 2794–2801 CrossRef CAS.
- Z. Zhou and S. Ma, ChemMedChem, 2017, 12, 420–425 CrossRef CAS.
- Y.-M. Zhang, M. W. Frank, K. Zhu, A. Mayasundari and C. O. Rock, J. Biol. Chem., 2008, 283, 28788–28794 CrossRef CAS PubMed.
- A. K. Bera, V. Atanasova, H. Robinson, E. Eisenstein, J. P. Coleman, E. C. Pesci and J. F. Parsons, Biochemistry, 2009, 48, 8644–8655 CrossRef CAS PubMed.
- M. P. Storz, C. K. Maurer, C. Zimmer, N. Wagner, C. Brengel, J. C. de Jong, S. Lucas, M. Müsken, S. Häussler, A. Steinbach and R. W. Hartmann, J. Am. Chem. Soc., 2012, 134, 16143–16146 CrossRef CAS PubMed.
- A. Thomann, C. Brengel, C. Börger, D. Kail, A. Steinbach, M. Empting and R. W. Hartmann, ChemMedChem, 2016, 11, 2522–2533 CrossRef CAS PubMed.
- Y. Luo, Y.-S. Yang, J. Fu and H.-L. Zhu, Expert Opin. Ther. Pat., 2012, 22, 1325–1336 CrossRef CAS.
- M. M. Alhamadsheh, F. Musayev, A. A. Komissarov, S. Sachdeva, H. T. Wright, N. Scarsdale, G. Florova and K. A. Reynolds, Chem. Biol., 2007, 14, 513–524 CrossRef CAS.
- D. Pistorius, A. Ullrich, S. Lucas, R. W. Hartmann, U. Kazmaier and R. Müller, ChemBioChem, 2011, 12, 850–853 CrossRef CAS PubMed.
- E. Weidel, J. C. de Jong, C. Brengel, M. P. Storz, A. Braunshausen, M. Negri, A. Plaza, A. Steinbach, R. Müller and R. W. Hartmann, J. Med. Chem., 2013, 56, 6146–6155 CrossRef CAS.
- S. Hinsberger, J. C. de Jong, M. Groh, J. Haupenthal and R. W. Hartmann, Eur. J. Med. Chem., 2014, 76, 343–351 CrossRef CAS.
- J. H. Sahner, C. Brengel, M. P. Storz, M. Groh, A. Plaza, R. Müller and R. W. Hartmann, J. Med. Chem., 2013, 56, 8656–8664 CrossRef CAS PubMed.
- S. F. Sousa, A. J. M. Ribeiro, J. T. Coimbra, R. P. Neves, S. A. Martins, N. S. H. Moorthy, P. A. Fernandes and M. J. Ramos, Curr. Med. Chem., 2013, 20, 2296–2314 CrossRef CAS PubMed.
- S. F. Sousa, P. A. Fernandes and M. J. Ramos, Proteins: Struct., Funct., Genet., 2006, 65, 15–26 CrossRef CAS.
- D. Lapaillerie, C. Charlier, V. Guyonnet-Dupérat, E. Murigneux, H. S. Fernandes, F. G. Martins, R. P. Magalhães, T. F. Vieira, C. Richetta, F. Subra, S. Lebourgeois, C. Charpentier, D. Descamps, B. Visseaux, P. Weigel, A. Favereaux, C. Beauvineau, F. Buron, M.-P. Teulade-Fichou, S. Routier, S. Gallois-Montbrun, L. Meertens, O. Delelis, S. F. Sousa and V. Parissi, Antimicrob. Agents Chemother., 2022, 66(8), e0008322 CrossRef.
- N. D. S. L. De Guzman, J. A. C. Madrono, M. A. Endriga and E. J. L. Frio, in 2021 IEEE Int. Conf. Bioinforma. Biomed., 2021, pp. 376–380 Search PubMed.
- K. D. G. Martin, K. G. V. Padilla, I. J. A. Buan, K. Dave Gloria Martin, K. Grace Vergara Padilla and I. Joyce Arenas Buan, Orient. J. Chem., 2021, 37, 143–150 CAS.
- F. G. Martins, A. Melo and S. F. Sousa, Molecules, 2021, 26, 2600 CrossRef CAS PubMed.
- T. F. Vieira, F. G. Martins, J. P. Moreira, T. Barbosa and S. F. Sousa, Molecules, 2021, 26, 6162 CrossRef CAS PubMed.
- T. F. Vieira, R. P. Magalhães, M. Simões and S. F. Sousa, Antibiotics, 2022, 11, 185 CrossRef CAS.
- R. P. Magalhães, T. F. Vieira, A. Melo and S. F. Sousa, Mol. Syst. Des. Eng., 2022, 7, 434–446 RSC.
- S. Fernandes, A. Borges, I. B. Gomes, S. F. Sousa and M. Simões, Food Res. Int., 2023, 165, 112519 CrossRef CAS PubMed.
- H. M. M. Berman, J. Westbrook, Z. Feng, G. Gilliland, T. N. Bhat, H. Weissig, I. N. Shindyalov and P. E. Bourne, Nucleic Acids Res., 2000, 28, 235–242 CrossRef CAS.
- R. P. Magalhães, T. F. Vieira, H. S. Fernandes, A. Melo, M. Simões and S. F. Sousa, Trends Biotechnol., 2020, 38, 937–940 CrossRef.
- J. Jumper, R. Evans, A. Pritzel, T. Green, M. Figurnov, O. Ronneberger, K. Tunyasuvunakool, R. Bates, A. Žídek, A. Potapenko, A. Bridgland, C. Meyer, S. A. A. Kohl, A. J. Ballard, A. Cowie, B. Romera-Paredes, S. Nikolov, R. Jain, J. Adler, T. Back, S. Petersen, D. Reiman, E. Clancy, M. Zielinski, M. Steinegger, M. Pacholska, T. Berghammer, S. Bodenstein, D. Silver, O. Vinyals, A. W. Senior, K. Kavukcuoglu, P. Kohli and D. Hassabis, Nature, 2021, 596, 583–589 CrossRef CAS.
- M. Varadi, S. Anyango, M. Deshpande, S. Nair, C. Natassia, G. Yordanova, D. Yuan, O. Stroe, G. Wood, A. Laydon, A. Žídek, T. Green, K. Tunyasuvunakool, S. Petersen, J. Jumper, E. Clancy, R. Green, A. Vora, M. Lutfi, M. Figurnov, A. Cowie, N. Hobbs, P. Kohli, G. Kleywegt, E. Birney, D. Hassabis and S. Velankar, Nucleic Acids Res., 2022, 50, D439–D444 CrossRef CAS PubMed.
- O. Trott and A. J. Olson, J. Comput. Chem., 2009, 31, 455–461 CrossRef PubMed.
- G. Jones, P. Willett, R. C. Glen, A. R. Leach and R. Taylor, J. Mol. Biol., 1997, 267, 727–748 CrossRef CAS PubMed.
- T. F. Vieira and S. F. Sousa, Appl. Sci., 2019, 9, 4538 CrossRef CAS.
- T. F. Vieira, R. P. Magalhaes and S. F. Sousa, Front. Drug Chem. Clin. Res., 2019, 2, 1–4 Search PubMed.
- The PyMOL Molecular Graphics System, Version 1.2r3pre, Schrödinger, LLC Search PubMed.
- N. M. O'Boyle, M. Banck, C. A. James, C. Morley, T. Vandermeersch and G. R. Hutchison, Aust. J. Chem., 2011, 3, 33 Search PubMed.
- S. F. Sousa, N. M. F. S. A. Cerqueira, P. A. Fernandes and M. J. Ramos, Comb. Chem. High Throughput Screening, 2010, 13, 442–453 CrossRef CAS.
- N. M. F. S. A. Cerqueira, D. Gesto, E. F. Oliveira, D. Santos-Martins, N. F. Brás, S. F. Sousa, P. A. Fernandes and M. J. Ramos, Arch. Biochem. Biophys., 2015, 582, 56–67 CrossRef CAS.
- A. Gaulton, A. Hersey, M. Nowotka, A. P. Bento, J. Chambers, D. Mendez, P. Mutowo, F. Atkinson, L. J. Bellis, E. Cibrián-Uhalte, M. Davies, N. Dedman, A. Karlsson, M. P. Magariños, J. P. Overington, G. Papadatos, I. Smit and A. R. Leach, Nucleic Acids Res., 2017, 45, D945–D954 CrossRef CAS PubMed.
- M. K. Gilson, T. Liu, M. Baitaluk, G. Nicola, L. Hwang and J. Chong, Nucleic Acids Res., 2016, 44, D1045–D1053 CrossRef CAS PubMed.
- J. H. Sahner, M. Empting, A. Kamal, E. Weidel, M. Groh, C. Börger and R. W. Hartmann, Eur. J. Med. Chem., 2015, 96, 14–21 CrossRef CAS PubMed.
- G. Allegretta, E. Weidel, M. Empting and R. W. Hartmann, Eur. J. Med. Chem., 2015, 90, 351–359 CrossRef CAS PubMed.
- M. M. Mysinger, M. Carchia, J. J. Irwin and B. K. Shoichet, J. Med. Chem., 2012, 55, 6582–6594 CrossRef CAS.
- C. Empereur-Mot, J.-F. Zagury and M. Montes, J. Chem. Inf. Model., 2016, 56, 2281–2286 CrossRef CAS PubMed.
- T. Sterling and J. J. Irwin, J. Chem. Inf. Model., 2015, 55, 2324–2337 CrossRef CAS PubMed.
- M. F. Hibert, Drug Discovery Today, 2009, 14, 723–725 CrossRef.
- F. Ortuso, D. Bagetta, A. Maruca, C. Talarico, M. L. Bolognesi, N. Haider, F. Borges, S. Bryant, T. Langer, H. Senderowitz and S. Alcaro, Front. Chem., 2018, 6, 130 CrossRef.
- IBS The Natural Compound (NC) collection, InterBioScreen ltd., can be found under https://www.ibscreen.com/natural-compounds, n.d.
- C. Lyu, T. Chen, B. Qiang, N. Liu, H. Wang, L. Zhang and Z. Liu, Nucleic Acids Res., 2021, 49, D509–D515 CrossRef CAS PubMed.
- J. A. Maier, C. Martinez, K. Kasavajhala, L. Wickstrom, K. E. Hauser and C. Simmerling, J. Chem. Theory Comput., 2015, 11, 3696–3713 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision C.01, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- J. Wang, R. M. Wolf, J. W. Caldwell, P. A. Kollman and D. A. Case, J. Comput. Chem., 2004, 25, 1157–1174 CrossRef CAS.
- D. A. Case, T. E. Cheatham, T. Darden, H. Gohlke, R. Luo, K. M. Merz, A. Onufriev, C. Simmerling, B. Wang and R. J. Woods, J. Comput. Chem., 2005, 26, 1668–1688 CrossRef CAS.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33–38 CrossRef CAS PubMed.
- D. R. Roe and T. E. Cheatham, J. Chem. Theory Comput., 2013, 9, 3084–3095 CrossRef CAS.
- E. Wang, H. Sun, J. Wang, Z. Wang, H. Liu, J. Z. H. Zhang and T. Hou, Chem. Rev., 2019, 119, 9478–9508 CrossRef CAS.
- B. R. Miller, T. D. McGee, J. M. Swails, N. Homeyer, H. Gohlke and A. E. Roitberg, J. Chem. Theory Comput., 2012, 8, 3314–3321 CrossRef CAS PubMed.
- S. Genheden and U. Ryde, Expert Opin. Drug Discovery, 2015, 10, 449–461 CrossRef CAS.
- M. Doona and D. Walsh, Palliat. Med., 1998, 12, 55–58 CrossRef CAS.
- V. Iyer and S. S. Poddar, Eur. J. Contracept. Reprod. Health Care, 2008, 13, 339–350 CrossRef CAS.
- N. Li, J. Li, M. Huang and X. Zhang, Medicine, 2021, 100, e24500 CrossRef.
- Deferoxamine, can be found under https://go.drugbank.com/drugs/DB00746, n.d.
- C. J. Ingold and S. R. Sergent, Phytonadione (Vitamin K1) [Updated 2023 Feb 11], in StatPearls, StatPearls Publishing, Treasure Island (FL), 2023, Available from: https://www.ncbi.nlm.nih.gov/books/NBK557622/ Search PubMed.
- D. I. Ball, R. T. Brittain, R. A. Coleman, L. H. Denyer, D. Jack, M. Johnson, L. H. C. Lunts, A. T. Nials, K. E. Sheldrick and I. F. Skidmore, Br. J. Pharmacol., 1991, 104, 665–671 CrossRef CAS PubMed.
- R. N. Marcus and J. Mendels, J. Clin. Psychiatry, 1996, 57, 19–23 CAS.
- C. Piérard-Franchimont, V. Goffin, J. Decroix and G. E. Piérard, Skin Pharmacol. Physiol., 2002, 15, 434–441 CrossRef PubMed.
- A. Galán, L. Moreno, J. Párraga, Á. Serrano, M. J. Sanz, D. Cortes and N. Cabedo, Bioorg. Med. Chem., 2013, 21, 3221–3230 CrossRef PubMed.
- L. Opletal, M. Ločárek, A. Fraňková, J. Chlebek, J. Šmíd, A. Hošt'álková, M. Šafratová, D. Hulcová, P. Klouček, M. Rozkot and L. Cahlíková, Nat. Prod. Commun., 2014, 9, 1709–1712 CrossRef.
- Y. Liu, L. Ding, J. He, Z. Zhang, Y. Deng, S. He and X. Yan, Fitoterapia, 2021, 154, 105004 CrossRef CAS.
- T. F. Vieira, R. P. Magalhães, N. M. F. S. A. Cerqueira, M. Simões and S. F. Sousa, Mol. Syst. Des. Eng., 2022, 7, 1294–1306 RSC.
- M. Starkey, T. Kitao, J. He, A. Tzika, S. Milot, B. Lesic, V. Righi, F. Lepine, A. Bandyopadhaya, L. Rahme, D. Maura, A. Bandyopadhaya, B. Lesic, J. He, T. Kitao, V. Righi, S. Milot, A. Tzika and L. Rahme, PLoS Pathog., 2014, 10, e1004321 CrossRef PubMed.
- B. Shaker, S. Ahmad, J. Lee, C. Jung and D. Na, Comput. Biol. Med., 2021, 137, 104851 CrossRef PubMed.
- C. Wang, D. Greene, L. Xiao, R. Qi and R. Luo, Front. Mol. Biosci., 2018, 4, 1–18 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3me00107e |
| This journal is © The Royal Society of Chemistry 2024 |