
Rational design of water-harvesting hydrogels†
Moki K.
Thanusing
 ,
Peidong
Shen
,
Brett L.
Pollard
and
Luke A.
Connal
*
,
Peidong
Shen
,
Brett L.
Pollard
and
Luke A.
Connal
*
Research School of Chemistry, The Australian National University, Australia. E-mail: luke.connal@anu.edu.au
First published on 20th October 2023
Abstract
Water-harvesting polymer materials have the potential to create new sources of potable water. However, a holistic understanding of the relationship between polymer structure and water-harvesting properties is lacking compared to studies on specific materials. In this work, we synthesised a library of methacrylic acid-co-poly(ethylene glycol) methyl ether methacrylate)-based hydrogels (poly(MAA-co-PEGMA)) with directed modifications, including composition, crosslinker lengths, crosslinking density and preparation of the hydrogels. MAA serves as a hygroscopic monomer while PEGMA provides hydrophilicity and thermoresponsive properties. The water uptake and release capabilities of all materials was also assessed. The optimised composition of the copolymer (75![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5:20 MAA:EGDMA:PEGMA, mole%) has a water uptake of 98 mg g−1 polymer at 60% RH after 24 hours. The poly(MAA-co-PEGMA) materials also show a capability for water release, showing no significant decrease in water uptake capacity after repeated uptake-release cycles. Minimum temperatures for water release could easily be adjusted with polymer composition, ranging from 50–70 °C. The data presented in this body of work serves as a foundation for future efforts in creating thermoresponsive, water-harvesting polymers with real-world applications.
:5:20 MAA:EGDMA:PEGMA, mole%) has a water uptake of 98 mg g−1 polymer at 60% RH after 24 hours. The poly(MAA-co-PEGMA) materials also show a capability for water release, showing no significant decrease in water uptake capacity after repeated uptake-release cycles. Minimum temperatures for water release could easily be adjusted with polymer composition, ranging from 50–70 °C. The data presented in this body of work serves as a foundation for future efforts in creating thermoresponsive, water-harvesting polymers with real-world applications.
Design, System, ApplicationAtmospheric water harvesting has garnered interest as a way to combat water scarcity and make use of a currently untapped water source. This work outlines the structure–property relationships in the synthesis of water-harvesting polymers. By creating a library of hydrogels with different compositions of a copolymer systems based on methacrylic acid-co-poly(ethylene glycol) methyl ether methacrylate), we demonstrate in-depth relationships on the water-harvesting hydrogel structure and the complexity of rationally designing such systems. Most notably, the trade-off between water uptake and release performance is demonstrated. The understanding of structure–property relationships allows for the streamlining of rational design for water-harvesting polymers, with a faster development process accelerating the directed synthesis and optimisation of a material that can have commercial viability. |
Introduction
Water scarcity is one of the perennial problems impacting the human population. The hydrological cycle (which interconverts fresh and saline water) redistributes 455000 billion metric tonnes (bmt) of water globally per annum.1 Humans, in comparison, consume around 4000 bmt in the same period, with groundwater, rivers and lakes being the most used water sources. The uneven distribution of the hydrological cycle gives rise to local water scarcity.1,2 Atmospheric water, while also considered easily accessible, has been largely neglected. Representing around 10000 bmt of water, it would be more than sufficient for global fresh water needs while also having several major advantages. Atmospheric water is ubiquitous and can be captured locally, eliminating the need for transportation. Unlike groundwater, atmospheric water is clean and can be used immediately upon collection. The hydrological cycle replenishes atmospheric water quickly, which makes it an almost inexhaustible source of water.1,3,4 Hence, using atmospheric water as a resource is an attractive solution to location-specific water shortages.
Desiccants have significant potential as a water-harvesting technology.5,6 These can be solid (zeolites, MOFs, silica gel) or liquid (salt solutions like LiCl).4,7–9 Existing materials show impressive water harvesting performance. Xu et al. used hygroscopic LiCl salt in a MOF matrix with 0.77 g g−1 sorption in 12 hours in low humidity conditions.10 Comparable performance is seen in LiCl-impregnated macroporous polyacrylamide, made by Lyu et al. This material absorbs water rapidly, reaching 0.32 g g−1 in the first hour in high humidity conditions.11 While desiccants can easily absorb water from the air, their high affinity for water in turn makes the water release phase more energy-intensive. Some materials, like zeolites, require temperatures far above the vaporisation temperature of water in order to extract the harvested water.7,12 For a desiccant to be viable as a water-harvesting material, therefore, water uptake needs to be sufficiently rapid without requiring a prohibitively energy-intensive water release. Desiccants based off thermoresponsive polymers fit these criteria; their switchable affinity for water reduces the energy required for water release without negative effects on water uptake. These polymers exhibit drastic and discontinuous changes in properties with a change in temperature and hence can liberate water more easily.13–15 Some prominent examples include Lee et al.'s cactus-inspired solar still, which includes PNIPAAm as the thermoresponsive component, and Guo et al.'s porous films made with sustainable raw materials, which use hydroxypropyl cellulose.16,17 These materials can yield 209 mg cm−2 h−1 at 80% RH and up to 0.96 g g−1 at 30% RH respectively.
When considering the thermoresponsive behaviour of hydrogels (or crosslinked networks), the point at which the polymer switches from hydrophilic to hydrophobic (and vice versa) is known as the volume phase transition temperature (VPTT). The mechanism of this phase transition is similar to that of a non-crosslinked polymer. In the context of water harvesting, thermoresponsive polymers absorb atmospheric water by way of their hygroscopicity, before subsequent physical contraction to release liquid water upon heating above the VPTT. This is a renewable process as water capture and release can theoretically be cycled indefinitely. The release of liquid water gives thermoresponsive polymers an advantage over other desiccants, which must release water as a vapour.16,18,19 The water-harvesting thermoresponsive polymer is therefore a material of great interest in this space (Fig. 1).
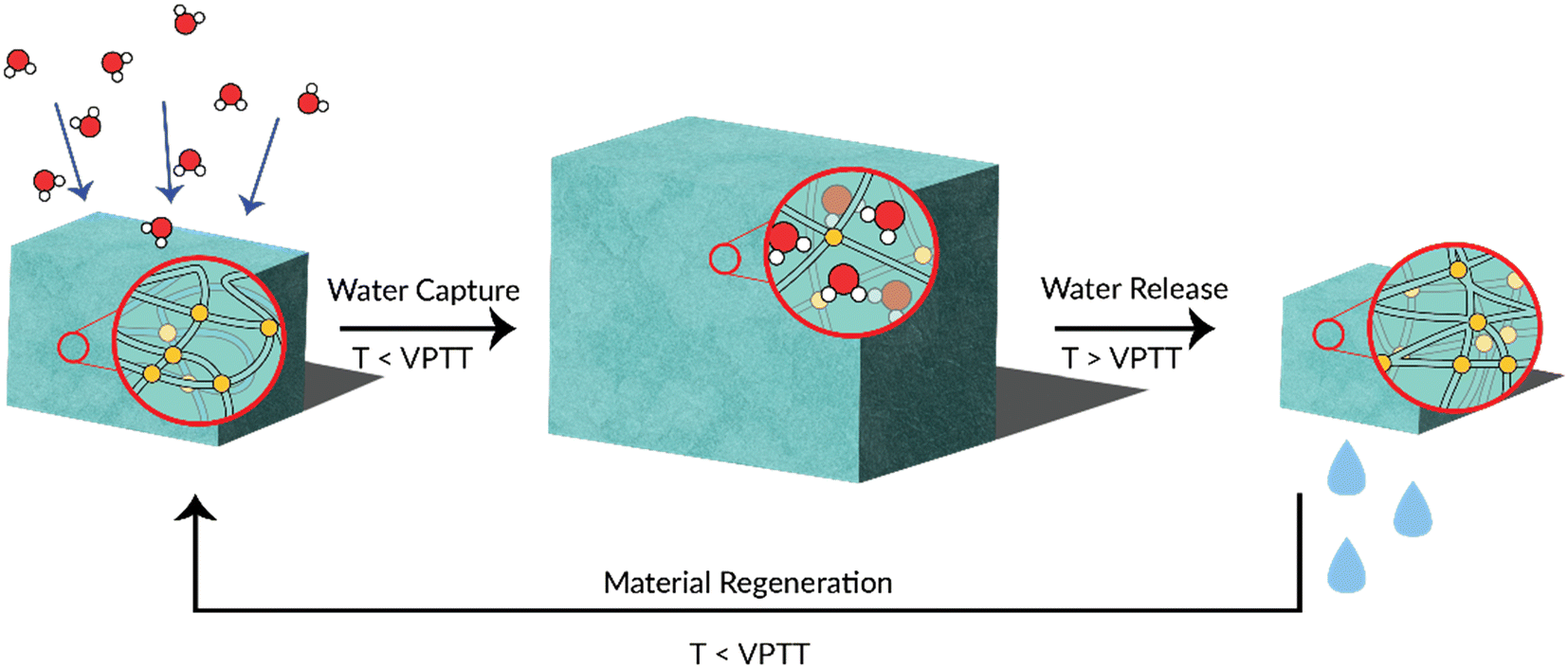 | ||
| Fig. 1 A graphical depiction of a thermoresponsive water-harvesting polymer. Water is absorbed into the hydrogel (left, middle). The system is heated above the hydrogel's volume phase transition temperature (VPTT), triggering a hydrophilic–hydrophobic transformation. As the hydrogel becomes hydrophobic, water molecules within the network aggregate and are expelled as liquid water (right). The material is renewed as the temperature is brought back below the VPTT, rendering the hydrogel hydrophilic and ready for another water-harvesting cycle. | ||
Herein, we developed a new candidate polymer with water-harvesting potential: methacrylic acid-co-poly(ethylene glycol) methyl ether methacrylate) (poly(MAA-co-PEGMA)) and its related hydrogels. We chose this material due to its hydrophilic nature and its impressive thermoresponsive, mechanical and self-healing properties.20 Currently, poly(N-isopropylacrylamide) (PNIPAAm) polymers see the most use for atmospheric water harvesting polymers in current research.16,18,19,21,22 PEGMA exhibits appreciable thermoresponsive behaviour and hydrophilicity.23 MAA is also natively hygroscopic; this is contrasted with current literature where deliquescent salts are used as the primary source of hygroscopic properties.19,22,24–26 MAA can also be easily functionalised like NIPAAm which allows for customisation of physical and chemical properties.27,28 The use of a copolymer allows for all desired functionality to be incorporated into a single polymer network. Properties such as thermoresponsive behaviour can be tuned easily by modifying the composition of the polymer. The structure–property relationships with respect to water uptake and release are investigated, which we anticipate will be critical in directing future efforts to design water-harvesting polymers.
Results and discussion
The development of polymer systems that could reversibly capture and release water at a critical temperature switch could drastically reduce the energy to harvest water from desiccant systems (Fig. 1). We initially developed a library of soluble poly(MAA-co-PEGMA) copolymers to understand the impact of composition on the resulting copolymer water uptake potential and thermoresponsitivity. MAA was chosen as the hygroscopic component of the copolymer, while PEGMA imparted thermoresponsive properties. These findings were then used to design a series of copolymer gels. Reversible addition-fragmentation transfer (RAFT) polymerisation was conducted to afford a series of copolymers (Scheme S1†). 1H NMR spectroscopy was used to characterise the poly(MAA-co-PEGMA) system (Fig. S1†). The following compositions were prepared and analysed further: poly(MAA27-co-PEGMA20), poly(MAA38-co-PEGMA16), poly(MAA40-co-PEGMA15), and poly(MAA203-co-PEGMA50).We initially screened the library in terms of ease of handling as we wanted to make consistent films to test the material's water uptake. Incremental changes in feed ratio of 10% showed marked differences in the resulting polymers's physical properties. The copolymer became physically softer with increasing PEGMA content, owing to the reduced Tg, reduced packing density between the long ethylene glycol chains of the parent monomer, as well as its strong solvation effects.20 Near-proportional changes to Tg with increasing PEGMA content were observed by Jones et al. in a similar MAA-co-PEGMA material, which corresponds with the appreciably different physical properties of our own materials when varying PEGMA content.29 We cast films from N,N-dimethylformamide (DMF) and then oven dried these films at 50 °C over 24 hours. Films cast from poly(MAA27-co-PEGMA20) were essentially low viscosity liquids and could not be easily handled. Hence, this composition was considered impractical for further investigation in this case. Gratifyingly, poly(MAA38-co-PEGMA16) could be readily shaped in a mould after casting and drying. Films cast from poly(MAA40-co-PEGMA15) were glassy but became flexible after exposure to humidity. All the aforementioned compositions were soluble in water. Finally, attempts to cast films from poly(MAA203-co-PEGMA50) were unsuccessful as the material only became viscous on attempts to dissolve. As a result, this material was not studied further.
Water uptake experiments were then conducted on the most promising compositions, namely poly(MAA38-co-PEGMA16) and poly(MAA40-co-PEGMA15). Dried samples were stored under humid conditions (70% relative humidity) in a closed environment for one day, with water uptake determined by recording their change in mass over time. There was a considerable difference in the rate of water uptake between the samples, with poly(MAA38-co-PEGMA16) showing faster water uptake in the first 400 minutes. Over 24 hours both copolymer compositions approached a similar water uptake (Fig. 2). The reduced packing caused by the long sidechains of PEGMA likely allowed the atmospheric water easier access to the polymer backbone and hydrophilic side chains.
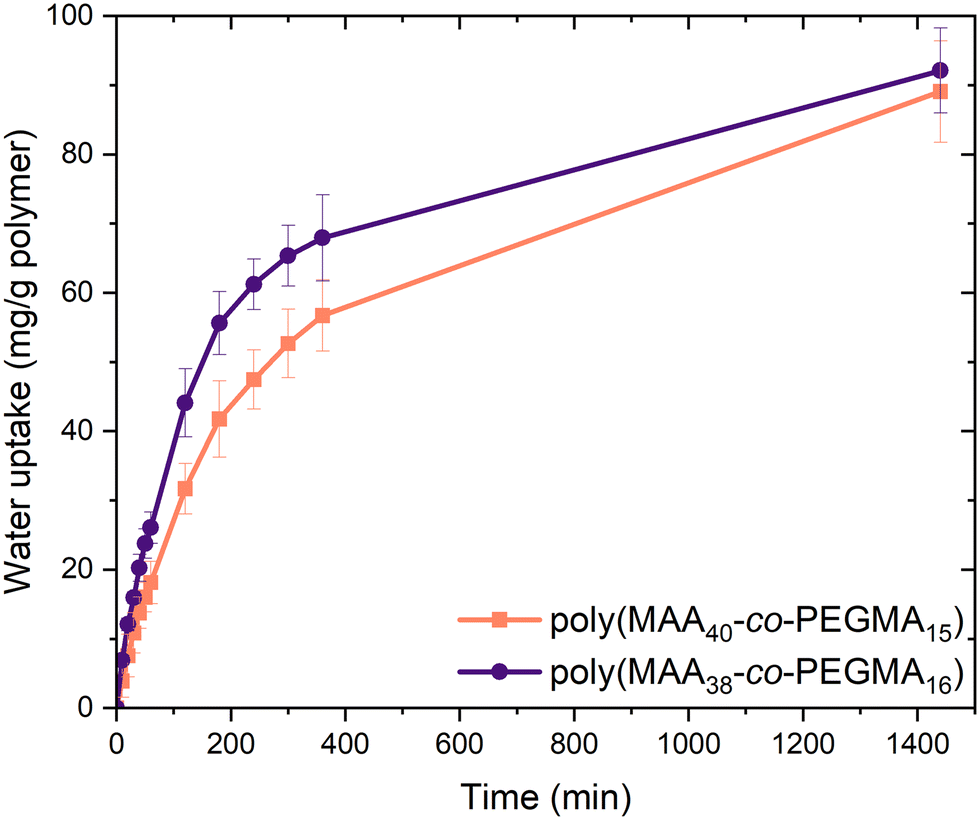 | ||
| Fig. 2 Water uptake measured over 24 hours for poly(MAA40-co-PEGMA15) and poly(MAA38-co-PEGMA16), illustrating the effects of MAA:PEGMA feed ratio. Poly(MAA40-co-PEGMA15) reached 89 mg water per g polymer after 24 hours, while poly(MAA38-co-PEGMA16) reached 92 mg g−1. Rate of uptake was also affected, evidenced by poly(MAA38-co-PEGMA16) absorbing 68 mg g−1 by 360 minutes while poly(MAA40-co-PEGMA15) absorbed 57 mg g−1 in comparison. | ||
With the above copolymer systems, we have confirmed the potential of the copolymers to absorb water. However, in a practical water harvesting system, crosslinking is required, thereby increasing the stability and applicability to water uptake and release.26,30 Thus, we next designed a covalently crosslinked hydrogel system that drew on our knowledge of the water-soluble poly(MAA-co-PEGMA) copolymer system. The general scheme for hydrogel synthesis is given below (Scheme 1). In our synthetic strategy, we utilised RAFT polymerisation to create gels with varying crosslinking density (G1–G3), PEGMA composition (G1 and G4) and crosslinker length (G1, G5, G6, G8, G9) (Table 1). We used various crosslinkers, including ethylene glycol dimethacrylate (EGDMA) and multiple lengths of poly(ethylene glycol) dimethacrylate (PEGDMA). Methacrylate crosslinkers were chosen as to keep the inter- and intramolecular interactions within the hydrogel consistent with those within the copolymer (Fig. 3).
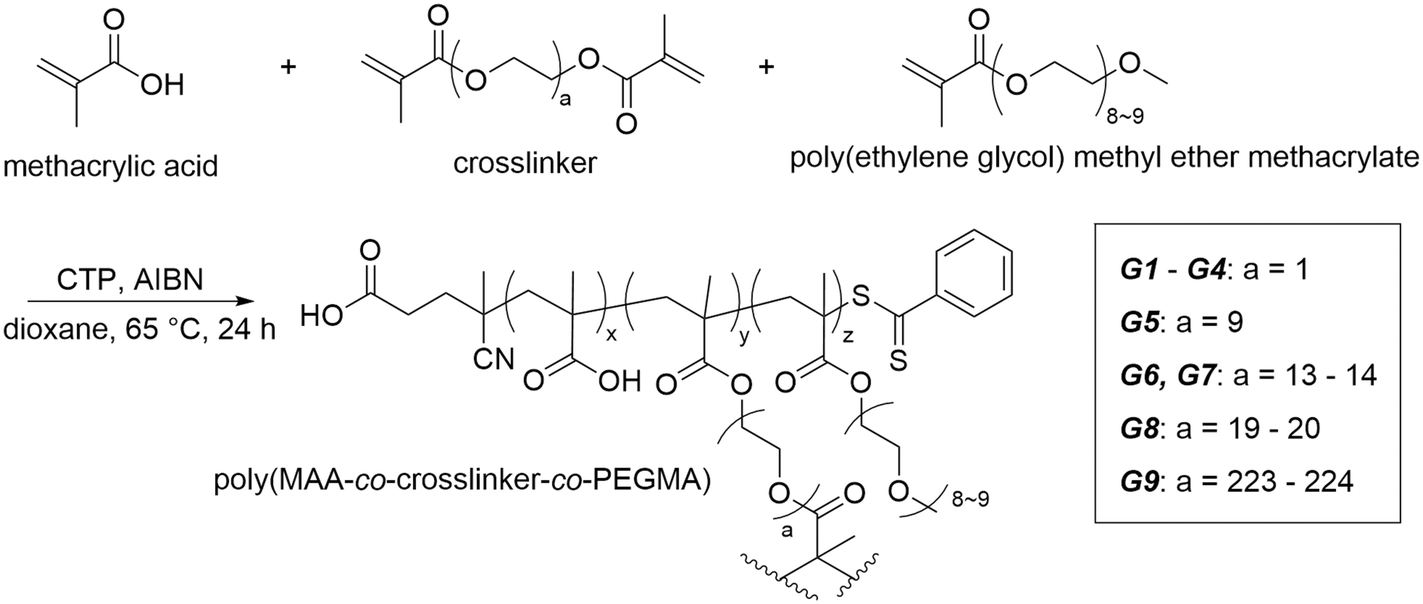 | ||
| Scheme 1 Generalised scheme for the poly(MAA-co-PEGMA) hydrogel. | ||
:crosslinker:PEGMA molar ratio
| Label | Composition (MAA:crosslinker:PEGMA) |
Crosslinker | VPTT (°C) |
|---|---|---|---|
| G1 | 75:5:20 |
EGDMA | 65.3 |
| G2 | 74:6:20 |
54.7 | |
| G3 | 73:7:20 |
52.3 | |
| G4 | 65:5:30 |
65.1 | |
| G5 | 75:5:20 |
PEGDMA 550 | 63.5 |
| G6 | 75:5:20 |
PEGDMA 750 | 57.2 |
| G7 | 75:5:20 (pristine) |
59.7 | |
| G8 | 75:5:20 |
PEGDMA 1000 | 70.6 |
| G9 | 75:5:20 |
PEGDMA 10000 | 71.3 |
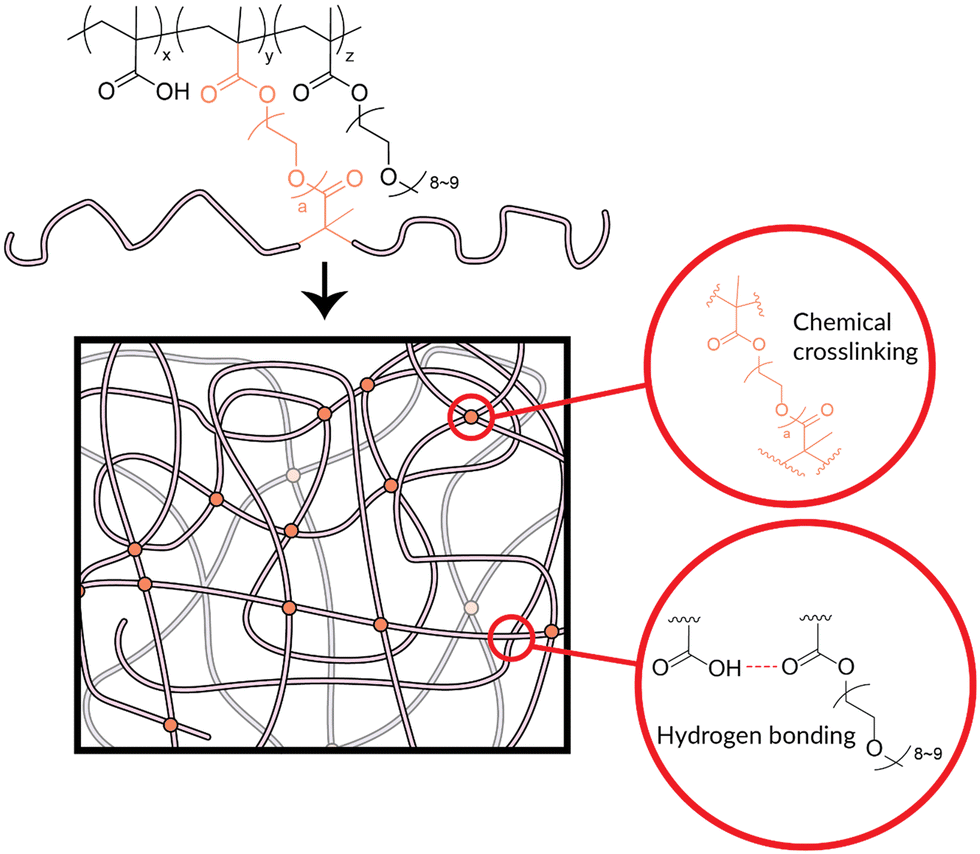 | ||
| Fig. 3 Illustration of the design of the poly(MAA-co-PEGMA)-based hydrogel. The network is held together by both chemical and physical crosslinking. Chemical crosslinking comes from the use of the dimethacrylate crosslinkers (Scheme 1, Table 1) within the hydrogel. Physical crosslinking comes principally from hydrogen bonding that occurs between the hydrogen-bond donors on the MAA units and various possible hydrogen-bond acceptors on the PEGMA or crosslinker chains. | ||
The poly(MAA-co-PEGMA)-based hydrogel was designed for stability on exposure to water using chemical crosslinking. Crosslinking already exists within the copolymer in the form of physical entanglement and hydrogen bonding. The desired application, atmospheric water-harvesting, involves consistent contact with water in vapour or liquid form. Hydrogen bonding between polymer and water is favourable during the water uptake phase, which reduces the hydrogen bonding between polymer chains and therefore the physical crosslinking. This can be observed as with increased exposure to humid conditions, copolymer samples visibly “melted”. Additionally, as water enters the copolymer system, individual polymer chains have more freedom of movement which loosens physical entanglement in the water uptake phase. Upon water release, the observed deformities in the samples were shown to be permanent. It is not practical for the desired application to have a material which readily deforms with use. The introduction of a chemical crosslinker is the simplest remedy. Chemical crosslinking is stable to water, which allows for water harvesting without deformation of the polymer.
We initially studied the effect of crosslinking density on water uptake. Dried crosslinked films (G1–G3) were held in a humid environment (70% RH) for 48 h and the weight of the gels were measured periodically. Water uptake was rapid until around the 24 hour mark, at which point water capacity was reached and the water content entered an equilibrium with the relative humidity of the environment (∼70% relative humidity (RH)). This behaviour was observed with all gels. All data was taken up to 24 hours for this reason. The gel with the highest water absorbing capacity was G1, with the lowest crosslinking density. Increasing the crosslinking density decreased the water absorption capacity (G2–G3) (Fig. 4a). The trend was expected: as crosslinking density increases, there is less network mobility and hence water permeation and absorption are decreased. Despite this, the difference in uptake for this series was more marginal than expected.
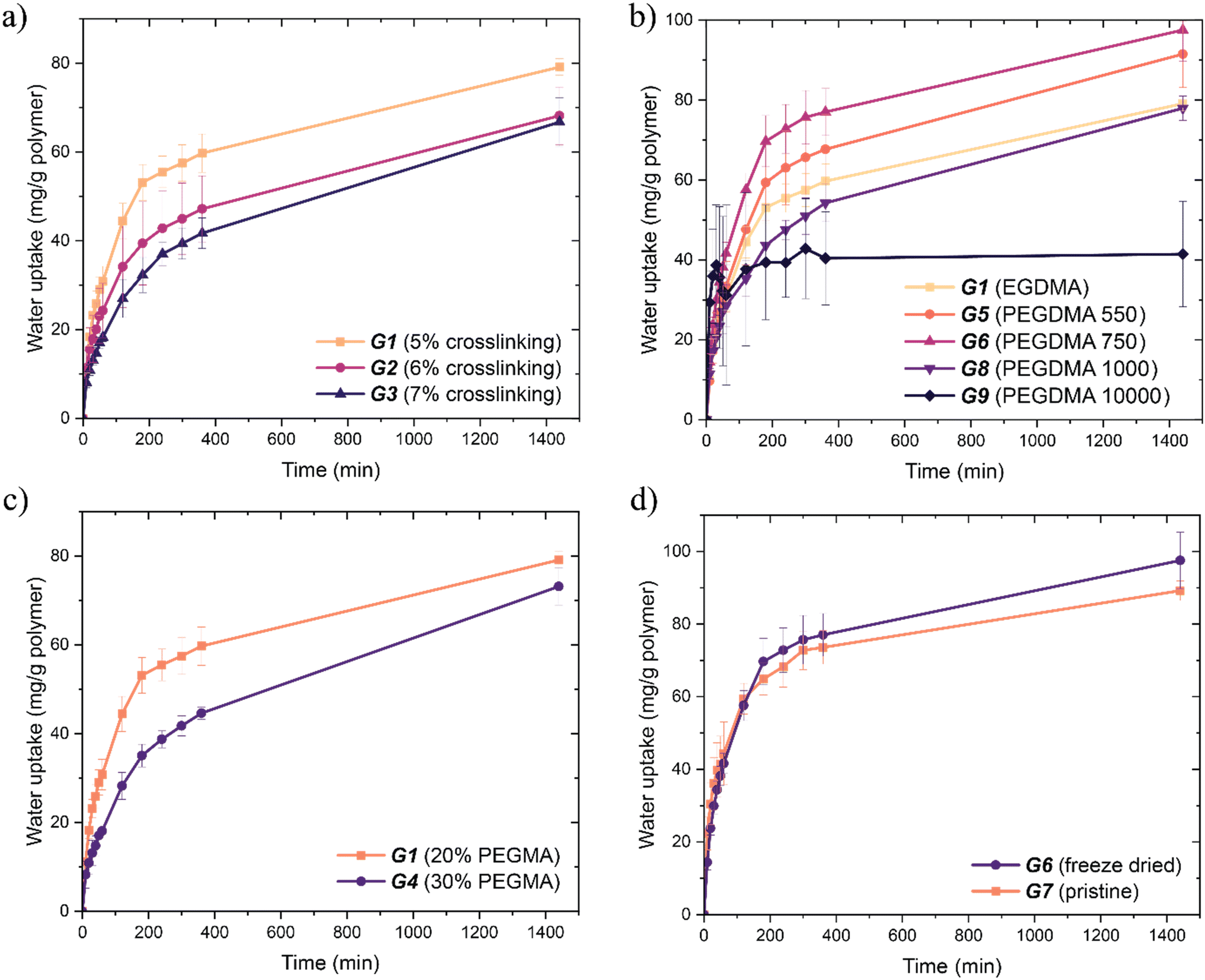 | ||
| Fig. 4 Water uptake over 24 hours of poly(MAA-co-PEGMA)-based hydrogels: a) uptake using G1, G2 and G3, comparing crosslinking density, demonstrating a decrease in crosslinking density increases uptake capacity; b) uptake using G1, G5, G6, G8, and G9, comparing crosslinker length, demonstrating a non-linear relationship between crosslinker length and water uptake; c) uptake using G1 and G4, comparing PEGMA content demonstrating increasing MAA content increases uptake; d) uptake using G6 and G7, comparing effects of freeze-drying demonstrating additional porosity imparted by freeze drying allowed for an slight increase in water uptake after 24 hours. | ||
The effects of crosslinker length on water uptake were also investigated. The intermediate length PEGDMA 550 Da (G5) and 750 Da (G6) crosslinkers showed increased water capacity after 24 hours compared to both shorter and longer crosslinkers. The short chain EGDMA (G1) crosslinked network displayed the next best water uptake kinetics (Fig. 4c). Surprisingly, the gels with the longer PEGDMA crosslinkers (1000 Da and 10000 Da, G8 and G9 respectively) showed the lowest water uptake.
The non-linear change of water uptake with length suggests the combination of several effects. Firstly, there is the positive effect on water uptake from a looser polymer network, as demonstrated above (Fig. 4a). The increase in water uptake can be seen from EGDMA to PEGDMA 750 Da, showing the positive effect of a looser network. However, the positive effect does not increase linearly with length. As water is a small molecule, the increase in the degree of freedom provided to the solvent within the polymer network decreases with crosslinker length. There is therefore a reduced increase of the positive effect on water uptake as crosslinker length increases. At the point of PEGDMA 10000 Da, the positive effect of length on water uptake would be minimal, if not negligible.
There is a simultaneous negative effect on water uptake with crosslinker length. This is due to the mass fraction of MAA units decreasing with the increasing length and molecular weight of the crosslinker. PEGDMA is less hygroscopic than MAA, so as the proportion of PEGDMA by weight increases, decreasing hygroscopicity of the polymer is expected (Fig. S2†). We tested the hypothesis that the low water uptake of G9 was observed because of the smaller mass fraction of MAA units to PEGMA and PEGDMA in the system. A sample of hydrogel replacing the PEGMA in G9 with MAA (leading to a 95:5 feed ratio poly(MAA-co-PEGDMA)) was prepared (GS1). The water uptake of GS1 is increased in comparison to G9 (Fig. S2†). This confirms our assumption that MAA is the hygroscopic element of the polymer system and that the reduced proportion of MAA in G9 was the cause of the reduced water uptake.31,32 The negative effect increases proportionally to the reduction of MAA mass fraction in the polymer, i.e. with increasing crosslinker length. The combination of both positive and negative effects explains the increase then decrease in water uptake with crosslinker length.
We then investigated the effect of composition of PEGMA and MAA on the water absorbing capacity. Gels (G1 and G4) were placed in a humid environment and their mass was recorded periodically. Again, a sharp increase followed by a plateau was observed in the first 24 hours (Fig. 4b). The gel with the highest composition of MAA showed both a faster water absorption and a larger water absorbing capacity. The equivalent copolymers in Fig. 2 showed the opposite results where increasing MAA decreased the water absorbing capacity. A looser network resulting from increased PEGMA content would still be observed in the crosslinked gel, implying a positive correlation between PEGMA content and water uptake. However, MAA is the primary component of the hygroscopic polymer system (Fig. S2†), which implies a positive correlation between MAA content and water uptake. If both correlations occur simultaneously, it can be concluded that the effect of each relationship changes with crosslinking of the poly(MAA-co-PEGMA) system. The loosening of the polymer network is minimal between G1 and G4; the crosslinking density remains the same. Thus, the positive correlation between water uptake and MAA content takes precedence over that with PEGMA, and we observe G1 (with lower PEGMA content, i.e. higher MAA content) having more water capacity than G4.
The effects of porosity on water uptake were also studied. Freeze drying is an effective method to produce porous materials.20 We conducted freeze drying on the majority of the synthesised hydrogels. Freeze drying-induced porosity allows a greater surface area of hydrogel to be exposed to humid air and should increase the rate of water absorption. Increased surface area via porosity is easily observable between G6 and G7: G6 has visible pores in the range of 1–10 μm while G7 is smooth with no discernible pores (Fig. 5). However, there is little difference between the freeze-dried and pristine samples in the first two hours of water uptake (Fig. 4d). Slightly increased water capacity in the freeze-dried sample was observed after two hours, owing to the more rapid absorption and diffusion of water into the system via porosity. Since the hydrogels were hard and brittle, the porosity imparted by freeze drying was appreciable yet could be improved further. Freeze drying is more effective in hydrogels with less crosslinking density (i.e. a smaller proportion of added crosslinker or the use of a longer crosslinker), and this increased porosity results in clearer changes in water harvesting behaviour in the first 6 hours. Increased water uptake is generally attributed to increased surface area of a material.7,9,17 Therefore, these results show more about the material itself rather than the relationship between surface area and water uptake. No substantial difference in water capacity between freeze-dried and pristine samples was expected because the only difference was surface area through porosity. However, there was a 10% increase with 10 mg g−1 difference in water capacity after 24 hours (Fig. 4d). This demonstrates the importance of surface area in water uptake as well as its relationship with diffusivity within the polymer network.
 | ||
| Fig. 5 SEM images of G6 and G7, showing differences in porosity between freeze-dried (top left) and non-freeze-dried samples (top right). Pores in G6 are around 1–10 μm in size (bottom). Scale bars are 20 μm (top left and right) and 2 μm (bottom) respectively. | ||
Next, the water release behaviour of the hydrogels was studied. We initially measured the VPTT by conducting differential scanning calorimetry (DSC) on hydrogel samples up to 100 °C (Table 1), where VPTT can be detected as an endothermic peak.33 This endothermic change in heat flow corresponds to the temperature at which the polymer–water hydrogen bonds become less favourable than the polymer–polymer and water–water hydrogen bonds, resulting in the release of water. Over 100 °C, the vaporisation of water as well as the phase transition of the hydrogel are taken into account. Temperatures beyond this point would not only yield less accurate VPTT measurements, but would also negate the benefits of exploiting the VPTT for water release.
Increasing crosslinker length was associated with a decrease a non-linear change in VPTT, where VPTT lowered with length until PEGDMA 750 Da and then increased up to 71.3 °C for PEGDMA 10000 Da. While increasing length of PEGMA pendant groups is generally associated with increasing lower critical solution temperature (LCST), it should be recognised that different interactions would exist within hydrogels which plausibly gives rise to differing LCST/VPTT behaviour.34–38
There are possibly both negative and positive entropic effects on VPTT with increasing crosslinker length. Dissolution of larger polymers requires the ordering of many solvent molecules along the polymer chain and is generally entropically unfavourable. The effect on entropy increases with the size of the polymer. Lower critical solution temperature (LCST) is entropically controlled, considering TΔS comprises more of the Gibbs free energy of mixing as temperature increases.39 This means that dissolution of larger polymers becomes entropically unfavourable at lower temperatures.40–46 While this explanation directly contradicts existing literature on PEGMA pendant group effects on LCST, it should be noted that these papers do not include pendant chains more than eight or nine ethylene glycol units long. At over two hundred ethylene glycol units in PEGDMA 10000 Da, the ordering of water molecules along the crosslinker chain should have an appreciable negative effect on entropy, thus increasing the VPTT. However, there is a positive effect on entropy that should also be considered as crosslinker length increases. Longer crosslinker chains means an overall looser network, that is, an increased number of configurations of the crosslinker chain.
In a similar vein of logic, both positive and negative enthalpic effects are possible for VPTT with increasing crosslinker length. Increasing hydrophilicity with crosslinker length is analogous to increasing pendant group length in soluble polymers, thus having a positive effect on VPTT. Additionally, as crosslinker length increases, so too does the number of hydrogen bond acceptors within the polymer system, while the proportion of hydrogen bond donating MAA carboxyl units is decreased. At very high crosslinker lengths, the material experiences a large deficit in the number of hydrogen bond donors relative to the hydrogen bond acceptors. This causes the polymer–polymer hydrogen bonds that would occur above the VPTT to be less energetically favourable, thus increasing the VPTT needed to allow the phase transition to occur.47 The closer the number of hydrogen bond donors and acceptors in the polymer system, the less positive (if not outright negative) this enthalpic effect will be. It is currently unclear whether all or some of these entropic and enthalpic effects on VPTT are present. Nonetheless, the observed non-linear trend indicates the interplay of positive and negative effects on VPTT.
We found that increasing crosslinking density also correlated with a decrease in VPTT. Tighter polymer networks have fewer possible configurations in solvent than looser networks, making them less entropically favourable. The temperature at which mixing of polymer and solvent becomes unfavourable has a positive correlation with entropic favourability. Hence, there is a correlation between increasing crosslinking density and decreasing VPTT.
Interestingly, increasing the proportion of PEGMA in polymer composition has negligible effects on VPTT. G1 and G4 have a composition of 75:5:20 and 65:5:30 ratios of MAA:EGDMA:PEGMA, with a VPTT of 65.3 °C and 65.1 °C respectively. A similar change in the soluble copolymer yielded a 14 °C increase in LCST with increased PEGMA content (Table S1†). This result implies the greater influence of crosslinking density on VPTT compared to composition.
Finally, we investigated the cycling of water uptake and release. We demonstrated this property with G6 with multiple cycles of water uptake (70% RH, 24 h) and then water release (60 °C, 1 h). G6 had the highest-measured water uptake after 24 hours, and thus was chosen for this experiment. Cycling experiments conducted showed only a marginal loss in water uptake capacity over multiple uptake/release cycles (Fig. 5). This shows that poly(MAA-co-PEGMA) hydrogels are capable of repeated uptake/release cycles and hence offer a lengthy functional lifespan. Critically, the water release temperature requirement is only above that of the VPTT, which is significantly lower than the energy required to release water from traditional desiccants (Fig. 6).
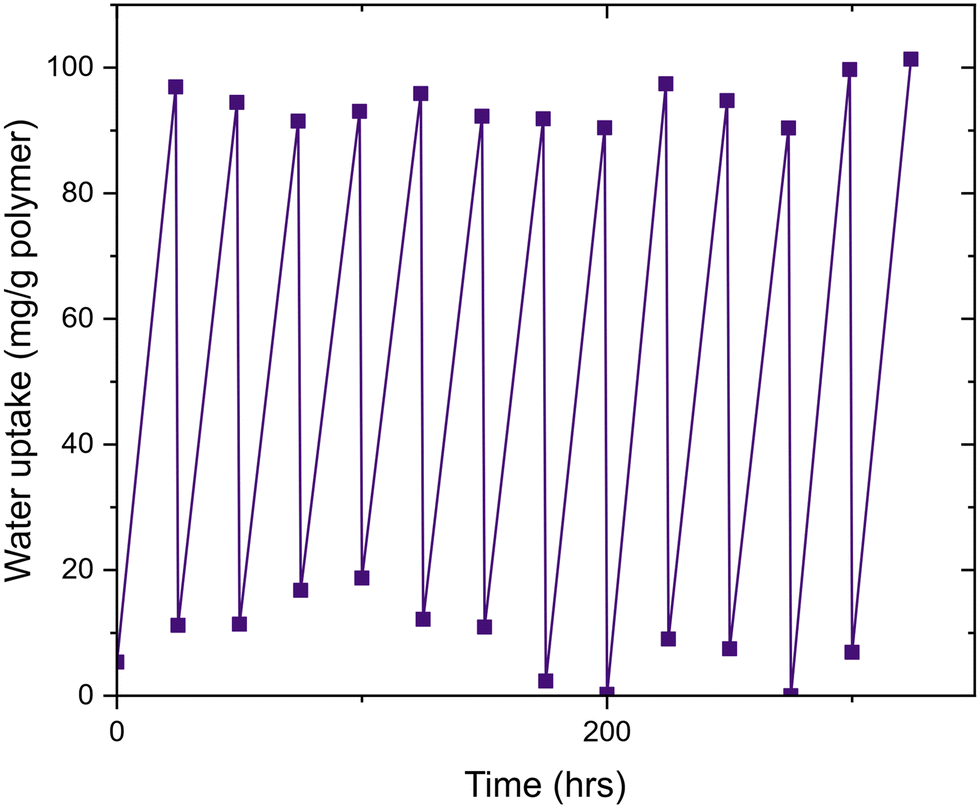 | ||
| Fig. 6 Cycling data of G6, showing no loss in water uptake capacity over 12 uptake/release cycles. Water uptake was consistent at an average of 95 mg g−1 for each cycle. | ||
Conclusion
In summary, we have demonstrated in-depth structure–property relationships for water-harvesting polymer gels. We assessed the water uptake and release capabilities of a library of poly(MAA-co-PEGMA)-based hydrogels. Most notably, longer crosslinkers generally improved the water uptake of hydrogels, up to a total of around 200 mg water (per gram of polymer) after 24 hours exposure to RH 70% air. We also found that this must be balanced with the proportion of hygroscopic material within the hydrogel, which offers insight into the rational design of water-harvesting polymer systems. Porosity of the hydrogels induced by freeze-drying was also observed to promote rapid water uptake in the first 6 hours. The hydrogels were not only thermoresponsive, but retained their water-harvesting capabilities over several water uptake and release cycles. These insights into the structure–property relationships of water-harvesting polymers will allow for a more streamlined approach to their rational design.Experimental section
All chemicals were purchased from Sigma-Aldrich and were used as supplied. Monomers supplied with inhibitors (BHT and MEHQ) were subjected to basic-alumina chromatography to remove inhibitors prior to polymerisation. 1H NMR spectra were obtained at 298 K on a Bruker Ascend 400 at 400 MHz, using DMSO-d6 as solvent. FTIR spectroscopy was conducted on a Perkin Elmer Spectrum 2. DSC experiments were conducted using a Perkin-Elmer DSC8500 apparatus. Humidity was measured inside a custom set-up as shown in Fig. S3.† Humidity and ambient temperature were measured using a RS-1260 humidity temperature meter. The humidity box operated on a constant flow of compressed air (CA) (1). Two adjustable lines of air are controlled by a flow meter (2): the “dry” air coming directly off the CA, and the “wet” air which is percolated through a flask of water (3). The proportions of each air flow were adjusted manually using valves. Field emission electron scanning microscopy (FESEM) images were taken at 3 kV using a Zeiss UltraPlus analytical FESEM. Samples were desiccated and coated with platinum before analysis.Synthesis of poly(MAA-co-PEGMA)
RAFT polymerisation was conducted with MAA and PEGMA 500 monomers, 2,2′-azobis(2-methylpropionitrile) (AIBN) initiator and 4-cyano-4-(phenyl-carbonothioylthio)pentanoic acid (CTP) chain transfer agent in anhydrous DMF. The solution was allowed to react at 65 °C for 24 h before the reaction vessel was exposed to air.A detailed procedure for poly(MAA80-co-PEGMA20) is provided. CTP (40.4 mg, 0.14 mmol), MAA (1.38 g, 16 mmol), PEGMA 500 (2.00 g, 4.0 mmol), AIBN (0.2 M in toluene; 1.64 mg, 0.01 mmol) and 6.0 mL of anhydrous N,N-dimethylformamide (DMF) were charged into a 25 mL round bottom flask. The mixture was maintained at 0 °C and degassed with nitrogen for 20 minutes. The flask was then carefully sealed and stirred at 65 °C in an oil bath for 24 hours. After cooling, the resultant mixture was precipitated into chilled diethyl ether (180 mL), forming a pink, solid mass. After air drying overnight, the product was precipitated in chilled n-hexane (180 mL) using THF as a carrier solvent. The polymer product was dried at 50 °C to afford a pink solid (41%).
1H NMR (poly(MAA60-co-PEGMA40), 400 MHz, DMSO-d6) δ 12.33 (b, 200 × 1H), 4.00 (s, 133 × 2H), 3.51, 3.44, 3.43, 3.24 (m, 133 × 18H), 3.07 (s, 2H), 2.89 (s, 2H), 1.70 (b, 133 × 2H + 200 × 2H), 0.94 (b, 133 × 3H + 200 × 3H).
1H NMR (poly(MAA70-co-PEGMA30), 400 MHz, DMSO-d6) δ 12.33 (b, 156 × 1H), 4.01, 3.60, 3.44, 3.43 (m, 67 × 18H), 3.24 (s, 67 × 3H), 2.90 (s, 2H), 2.74 (s, 2H), 1.71 (b, 67 × 2H + 156 × 2H), 0.94 (b, 67 × 3H + 156 × 3H).
1H NMR (poly(MAA80-co-PEGMA20), 400 MHz, DMSO-d6) δ 12.33 (b, 133 × 1H), 4.00, 3.61, 3.52, 3.44, 3.43 (m, 33 × 18H), 3.24 (s, 33 × 3H), 3.06 (s, 2H), 2.67 (s, 2H), 1.71 (b, 33 × 2H + 133 × 2H), 0.94 (b, 33 × 3H + 133 × 3H).
1H NMR (poly(MAA90-co-PEGMA10), 400 MHz, DMSO-d6) δ 12.34 (b, 150 × 1H), 4.01, 3.62, 3.52, 3.46 (m, 17 × 18H), 3.24 (s, 17 × 3H), 3.07 (s, 2H), 2.74 (s, 2H), 1.70 (b, 17 × 2H + 150 × 2H), 0.94 (b, 17 × 3H + 150 × 3H).
General procedure for the synthesis of crosslinked poly(MAA-co-PEGMA) hydrogels
RAFT polymerisation was conducted using MAA and PEGMA 500 monomers, crosslinker, AIBN initiator and 4-cyano-4-(phenyl-carbonothioylthio)pentanoic acid (CTP) chain transfer agent in anhydrous dioxane. The solution was allowed to react at 65 °C for 24 h, with stirring, before the vessel was opened and exposed to air. The gel product obtained was then freeze-dried for 48 hours.A detailed procedure for poly(MAA-co-EGDMA-co-PEGMA) with a feed ratio of 75:5:20 (G1) is provided. CTP (40.4 mg, 0.14 mmol), MAA (1.29 g, 15 mmol), PEGMA (2.00 g, 4.0 mmol), EGDMA (198 mg, 1.0 mmol), AIBN (0.2 M in toluene; 1.64 mg, 0.01 mmol) and 6 mL of anhydrous dioxane were charged into a 25 mL round bottom flask. The mixture was maintained at 0 °C and degassed with nitrogen for 15 minutes. The flask was then carefully sealed and gently stirred at 65 °C in an oil bath for 24 hours. After mechanically breaking the resultant solid into small chunks, the product was submerged in RO water over two days to leach unreacted monomers, with the water changed daily. After purification, the product was freeze-dried for 2 days to afford an orange or pink glassy solid.
G7 was prepared with a modified version of the above method. After purification, freeze-drying was not conducted. The product was then dried at 50 °C to afford an orange gel.
VPTT measurements
The VPTT of hydrogel products was determined using DSC. The surface of water-swollen gels were blotted dry with filter paper and a small piece (5–15 mg) was loaded into a hermetically sealed aluminium pan. Two cycles were run from ambient temperature to 80 °C, with a heating rate of 3 °C per minute. Endothermic phase transition peaks were defined by the peak maxima of the endotherm. Reported data is from the second cycle.Water uptake
Dried polymer samples were maintained at 50 °C before experiments were conducted. Petri dishes containing samples were placed into the humidity box shown in Fig. S3.† Humidity was adjusted to approximately 70% RH. Samples were removed, weighed and returned to the box as quickly as possible. Water content was determined as mass of water per gram of polymer. For the first hour, measurements were taken every ten minutes, then every hour for the next six hours, then a final measurement 24 hours after commencing the experiment.Cycling experiments
G6 was kept maintained at 50 °C before experiments were conducted. The water uptake method described above was followed, with the exception of measurements only being taken after 24 hours. After this, the sample was placed into an oven maintained at the sample's VPTT (i.e. 60 °C). After 1 hour, the sample was weighed. This uptake-release cycle was repeated for another twelve cycles.Author contributions
M. K. T. and L. C. conceived and designed the experiments. L. C. supervised the project. M. K. T. performed the experiments, analysed experimental results, and wrote the manuscript. P. S., B. L. P. and L. C. edited and revised the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research is supported by an Australian Research Council (DP230100679) and through an Australian Government Research Training Program (RTP) Scholarship. The authors acknowledge the instruments and expertise of Microscopy Australia at the Centre for Advanced Microscopy, Australian National University, a facility enabled by NCRIS and university support.References
- IEA, Water-Energy Nexus, Paris, 2016 Search PubMed.
- T. Oki and S. Kanae, Science, 2006, 313, 1068–1072 CrossRef CAS.
- I. A. Shiklomanov, in Water in Crisis: A Guide to the World's Fresh Water Resources, ed. P. H. Gleick, Oxford University Press, New York, 1993, ch. 2, pp. 13–24 Search PubMed.
- H. E. Gad, A. M. Hamed and I. I. El-Sharkawy, Renewable Energy, 2001, 22, 541–556 CrossRef CAS.
- R. Peeters, H. Vanderschaeghe, J. Rongé and J. A. Martens, Environ. Sci.: Water Res. Technol., 2020, 6, 2016–2034 RSC.
- H. Lu, W. Shi, Y. Guo, W. Guan, C. Lei and G. Yu, Adv. Mater., 2022, 34, 2110079 CrossRef CAS.
- Ş. Ç. Sayılgan, M. Mobedi and S. Ülkü, Microporous Mesoporous Mater., 2016, 224, 9–16 CrossRef.
- J. Y. Wang, R. Z. Wang, L. W. Wang and J. Y. Liu, Energy, 2017, 138, 542–551 CrossRef.
- H. Kim, S. Yang, S. R. Rao, S. Narayanan, E. A. Kapustin, H. Furukawa, A. S. Umans, O. M. Yaghi and E. N. Wang, Science, 2017, 356, 430–434 CrossRef CAS.
- J. Xu, T. Li, J. Chao, S. Wu, T. Yan, W. Li, B. Cao and R. Wang, Angew. Chem., Int. Ed., 2020, 59, 5202–5210 CrossRef CAS PubMed.
- T. Lyu, Z. Wang, R. Liu, K. Chen, H. Liu and Y. Tian, ACS Appl. Mater. Interfaces, 2022, 14, 32433–32443 CrossRef.
- N. Hanikel, M. S. Prévot, F. Fathieh, E. A. Kapustin, H. Lyu, H. Wang, N. J. Diercks, T. G. Glover and O. M. Yaghi, ACS Cent. Sci., 2019, 5, 1699–1706 CrossRef CAS PubMed.
- M. Wei, Y. Gao, X. Li and M. J. Serpe, Polym. Chem., 2017, 8, 127–143 RSC.
- G. C. Patel, V. K. Parmar and P. S. Patel, in Stimuli Responsive Polymeric Nanocarriers for Drug Delivery Applications, ed. A. S. H. Makhlouf and N. Y. Abu-Thabit, Woodhead Publishing, 2019, pp. 463–489, DOI:10.1016/B978-0-08-101995-5.00023-4.
- M. A. Haq, Y. Su and D. Wang, Mater. Sci. Eng., C, 2017, 70, 842–855 CrossRef PubMed.
- S. J. Lee, N. Ha and H. Kim, ACS Sustainable Chem. Eng., 2019, 7, 10561–10569 CrossRef CAS.
- Y. Guo, W. Guan, C. Lei, H. Lu, W. Shi and G. Yu, Nat. Commun., 2022, 13, 2761 CrossRef CAS PubMed.
- K. Matsumoto, N. Sakikawa and T. Miyata, Nat. Commun., 2018, 9, 2315 CrossRef PubMed.
- F. Zhao, X. Zhou, Y. Liu, Y. Shi, Y. Dai and G. Yu, Adv. Mater., 2019, 31, 1806446 CrossRef.
- Z. Jiang, B. Diggle, I. C. G. Shackleford and L. A. Connal, Adv. Mater., 2019, 31, 1904956 CrossRef CAS PubMed.
- Z. Zhang, Y. Wang, Z. Li, H. Fu, J. Huang, Z. Xu, Y. Lai, X. Qian and S. Zhang, ACS Appl. Mater. Interfaces, 2022, 14, 55295–55306 CrossRef CAS.
- D. Zheng, X. Xu, J. Zhu, B. Bai, Q. Wang, W. Shi and J. Li, J. Environ. Chem. Eng., 2023, 11, 109247 CrossRef CAS.
- J. Israelachvili, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 8378–8379 CrossRef CAS PubMed.
- S. Aleid, M. Wu, R. Li, W. Wang, C. Zhang, L. Zhang and P. Wang, ACS Mater. Lett., 2022, 4, 511–520 CrossRef CAS.
- C. Li, J. Wang, C. Deng, R. Wang and H. Zhang, STAR Protoc., 2022, 3, 101780 CrossRef CAS.
- F. Deng, Z. Chen, C. Wang, C. Xiang, P. Poredoš and R. Wang, Adv. Sci., 2022, 9, 2204724 CrossRef CAS PubMed.
- N. Schmeling, K. Hunger, G. Engler, B. Breiten, P. Rölling, A. Mixa, C. Staudt and K. Kleinermanns, Polym. Int., 2009, 58, 720–727 CrossRef CAS.
- B. A. Drain and C. R. Becer, Eur. Polym. J., 2019, 119, 344–351 CrossRef CAS.
- J. A. Jones, N. Novo, K. Flagler, C. D. Pagnucco, S. Carew, C. Cheong, X. Z. Kong, N. A. D. Burke and H. D. H. Stöver, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 6095–6104 CrossRef CAS.
- Y. Zhang, D. K. Nandakumar and S. C. Tan, Joule, 2020, 4, 2532–2536 CrossRef.
- M. Ulasan, E. Yavuz, E. U. Bagriacik, Y. Cengeloglu and M. S. Yavuz, J. Biomed. Mater. Res., Part A, 2015, 103, 243–251 CrossRef PubMed.
- S. P. Victor and C. P. Sharma, J. Biomater. Appl., 2002, 17, 125–134 CrossRef CAS.
- R. Fei, J. T. George, J. Park, A. K. Means and M. A. Grunlan, Soft Matter, 2013, 9, 2912–2919 RSC.
- J.-F. Lutz, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 3459–3470 CrossRef CAS.
- S. Han, M. Hagiwara and T. Ishizone, Macromolecules, 2003, 36, 8312–8319 CrossRef CAS.
- B. Zhao, D. Li, F. Hua and D. R. Green, Macromolecules, 2005, 38, 9509–9517 CrossRef CAS.
- J.-F. Lutz and A. Hoth, Macromolecules, 2006, 39, 893–896 CrossRef CAS.
- A. M. Jonas, K. Glinel, R. Oren, B. Nysten and W. T. S. Huck, Macromolecules, 2007, 40, 4403–4405 CrossRef CAS.
- M. A. Ward and T. K. Georgiou, Polymers, 2011, 3, 1215–1242 CrossRef CAS.
- T. Trongsatitkul and B. M. Budhlall, Colloids Surf., B, 2013, 103, 244–252 CrossRef CAS.
- Z. Li, Y.-H. Kim, H. S. Min, C.-K. Han and K. M. Huh, Macromol. Res., 2010, 18, 618–621 CrossRef CAS.
- S. Furyk, Y. Zhang, D. Ortiz-Acosta, P. S. Cremer and D. E. Bergbreiter, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 1492–1501 CrossRef CAS.
- Y. Zhang, S. Furyk, D. E. Bergbreiter and P. S. Cremer, J. Am. Chem. Soc., 2005, 127, 14505–14510 CrossRef CAS.
- D. Patterson, Macromolecules, 1969, 2, 672–677 CrossRef CAS.
- D. G. Lessard, M. Ousalem and X. X. Zhu, Can. J. Chem., 2001, 79, 1870–1874 CrossRef CAS.
- N. S. Ieong, M. Hasan, D. J. Phillips, Y. Saaka, R. K. O'Reilly and M. I. Gibson, Polym. Chem., 2012, 3, 794–799 RSC.
- A. D. Drozdov and J. d. Christiansen, J. Mech. Behav. Biomed. Mater., 2021, 114, 104215 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3me00132f |
| This journal is © The Royal Society of Chemistry 2024 |