
Mechanically robust, self-reporting and healable polyurethane elastomers by incorporating symmetric/asymmetric chain extenders†
Haitao
Wu
a,
Hao
Wang
a,
Mi
Luo
b,
Zhaoyang
Yuan
a,
Yiwen
Chen
b,
Biqiang
Jin
a,
Wenqiang
Wu
a,
Bangjiao
Ye
b,
Hongjun
Zhang
 b and
Jinrong
Wu
*a
b and
Jinrong
Wu
*a
aState Key Laboratory of Polymer Materials Engineering, College of Polymer Science and Engineering, Sichuan University, Chengdu 610065, China. E-mail: wujinrong@scu.edu.cn; Fax: +86-28-85468433; Tel: +86-28-85468433
bState Key Laboratory of Particle Detection and Electronics, University of Science and Technology of China, Hefei 230026, China
First published on 24th January 2024
Abstract
Self-healing elastomers usually show poor mechanical properties and environmental stability, and they cannot self-report mechanical/chemical damage. Herein, an innovative design strategy is reported that combines symmetric/asymmetric chain extenders to create large yet disordered hard domains within polyurethane (PU) elastomers, enabling the integration of mechanical robustness and self-reporting and self-healing capabilities to overcome both mechanical and chemical damage. Specifically, large yet disordered hard domains were created by governing the molar contents of asymmetric fluorescent 2-(4-aminophenyl)-5-aminobenzimidazole (PABZ) and symmetric 4-aminophenyl disulfide (APDS). Such a structural feature led to a small free-volume fraction, prominent strain-induced crystallization (SIC), and high energy of dissipation, enabling the PU elastomer to display outstanding mechanical strength (60.7 MPa) and toughness (177.9 MJ m−3). Meanwhile, the loose stacking of disordered hard domains imposed small restriction on network chains and imparted the network with high relaxation dynamics, leading to high healing efficiency (97.8%). More importantly, the fluorescence intensity was stimulus-responsive and thus the PU elastomer could self-report mechanical/chemical damage and healing processes. The PU elastomer also showed potential application prospects in information encoding and encryption. Furthermore, selecting polydimethylsiloxane as one of the soft segments could effectively endow the PU elastomer with intrinsic hydrophobicity. Therefore, this work provides valuable guidance for designing multi-functional materials with anti-counterfeiting, self-reporting, and healing properties as well as high mechanical properties and hydrophobicity.
New conceptsThe fast relaxation dynamics of self-healing elastomer networks are contradicted by high cohesive energy. As a result, self-healing elastomers typically exhibit poor mechanical properties and environmental stability, and they are unable to self-report mechanical/chemical damage. Herein, we propose an innovative design strategy that combines symmetric/asymmetric fluorescent chain extenders to create large yet disordered hard domains within polyurethane (PU) elastomers. On the one hand, such phase separation leads to outstanding reinforcement and toughening effects, since hard domains are the largest, and strain-induced crystallization (SIC) and energy dissipation are the most prominent. On the other hand, this phase structure can facilitate network reorganization, as the loosest hard domains exhibit the highest relaxation dynamics and the lowest cohesive energy. Meanwhile, the fluorescence intensity is stimulus-responsive and thus the PU elastomer can self-report the mechanical/chemical damage and healing processes as well as information encryption and decryption. Therefore, this innovative design strategy enables the integration of mechanical robustness, self-reporting and self-healing capabilities for both mechanical and chemical damage and anti-counterfeiting functions. |
Introduction
Elastomers are widely used in military, aerospace, and other applications owing to their outstanding mechanical strength,1 elastic resilience,2 dimensional stability,3 and environmental stability,4 rendering them indispensable across a wide variety of applications. Nevertheless, despite these excellent mechanical and physical properties, elastomers inevitably undergo mechanical/chemical damage during practical applications. Unfortunately, conventional elastomers lack intrinsic healing dynamics and responsive capabilities, resulting in difficulties in self-detecting and self-healing mechanical and chemical damage. Consequently, these elastomers are discarded after failure, leading to severe environmental pollution. Thus, it is imperative to develop elastomers with self-reporting and self-healing abilities. Although there are many examples of self-healing elastomers based on the reorganization of supramolecular interactions or dynamic covalent bonds, such as hydrogen bonds,5–9 ionic bonds,10–12 metal–coordination bonds,13,14 disulfide bonds,15 and Diels–Alder bonds,16 as well as single-function elastomers with self-reporting capabilities through the incorporation of self-diagnosis micro- and nano-capsules. However, to the best of our knowledge, the integration of self-reporting and self-healing capabilities into an artificial elastomer has rarely been studied.Recently, Wang et al.17 designed a class of hierarchical ionic skins (HI-skins) to self-report and self-repair mechanical damage (e.g., fractures or punctures) underwater. When HI-skins were mechanically damaged underwater, pre-embedded calcium peroxide was exposed to and reacted with the water to generate hydrogen peroxide, which further activated a peroxyoxalate chemiluminescence reaction to report the damage. Xia et al.18 reported a tough, self-reporting and self-healing polyurethane (PU) elastomer with near-infrared (NIR) light-induced shape memory. A radically exchangeable covalent bond was used as a dynamic covalent crosslink in the elastomer, endowing it with mechanochromism, thermochromism, and shape-memory behaviours, which collectively enabled the self-reporting of mechanical failure and the thermal status. Liu et al.19 constructed a self-healing and damage-reporting PU elastomer through a biomimetic design strategy. Mechanical damage detection within the elastomer was facilitated by the interaction between phenanthroline groups and Fe2+ ions, resulting in a noticeable red coloration, thereby achieving the self-reporting function of the bionic elastomer. Kim et al.20 fabricated novel dual-stimulus-responsive self-reporting thiol-epoxy thermoset (DSRTET) coatings that could detect both crack occurrence and pH variation. However, despite these meaningful strategies, the reported materials typically exhibited inferior mechanical properties (less than 30 MPa), which is undesirable during practical applications. Thus, it is still a big challenge to fabricate mechanical robust polymeric materials that combine self-reporting and self-healing properties.
If a straightforward design approach could enable polymers to attain mechanical robustness, self-reporting and healable capabilities, it could potentially become a universally significant tool. Herein, we report an innovative design strategy that combines symmetric/asymmetric chain extenders to create large yet disordered hard domains within PU elastomers, enabling the integration of mechanical robustness, self-reporting and self-healing capabilities for both mechanical and chemical damage. Here, we utilized 4-aminophenyl disulfide (APDS), as APDS derivatives are well-known for exhibiting self-healing properties and forming symmetric dynamic bonds, and 2-(4-aminophenyl)-5-aminobenzimidazole (PABZ), as PABZ monomer derivatives are recognized for their fluorescence properties and the formation of asymmetric dynamic bonds, as chain extenders. With this in mind, we designed a series of PU elastomers by adjusting the molar ratio of the symmetric/asymmetric chain extenders (Fig. 1(a)). On the one hand, such phase separation led to outstanding reinforcement and toughening effects, since the hard domains were the largest, and the strain-induced crystallization (SIC) and energy dissipation were most prominent. On the other hand, this phase structure could facilitate network reorganization, as the loosest hard domains showed the highest relaxation dynamics and the lowest cohesive energy. As such, the optimized PU elastomer showed excellent mechanical strength (60.7 MPa) and toughness (177.9 MJ m−3), high healing efficiency (97.8%) and admirable self-recovery ability. More importantly, the fluorescence feature of PABZ allowed the PU elastomer not only to self-report mechanical/chemical damage and the healing processes, but also to be used in information encoding and encryption. Moreover, selecting polydimethylsiloxane as one of soft segments could effectively solve the intrinsic hydrophilicity of the PU elastomer. Therefore, this work not only provides a valuable strategy for the development of robust healable materials, but it can also be extended to the fabrication of flexible and smart materials.
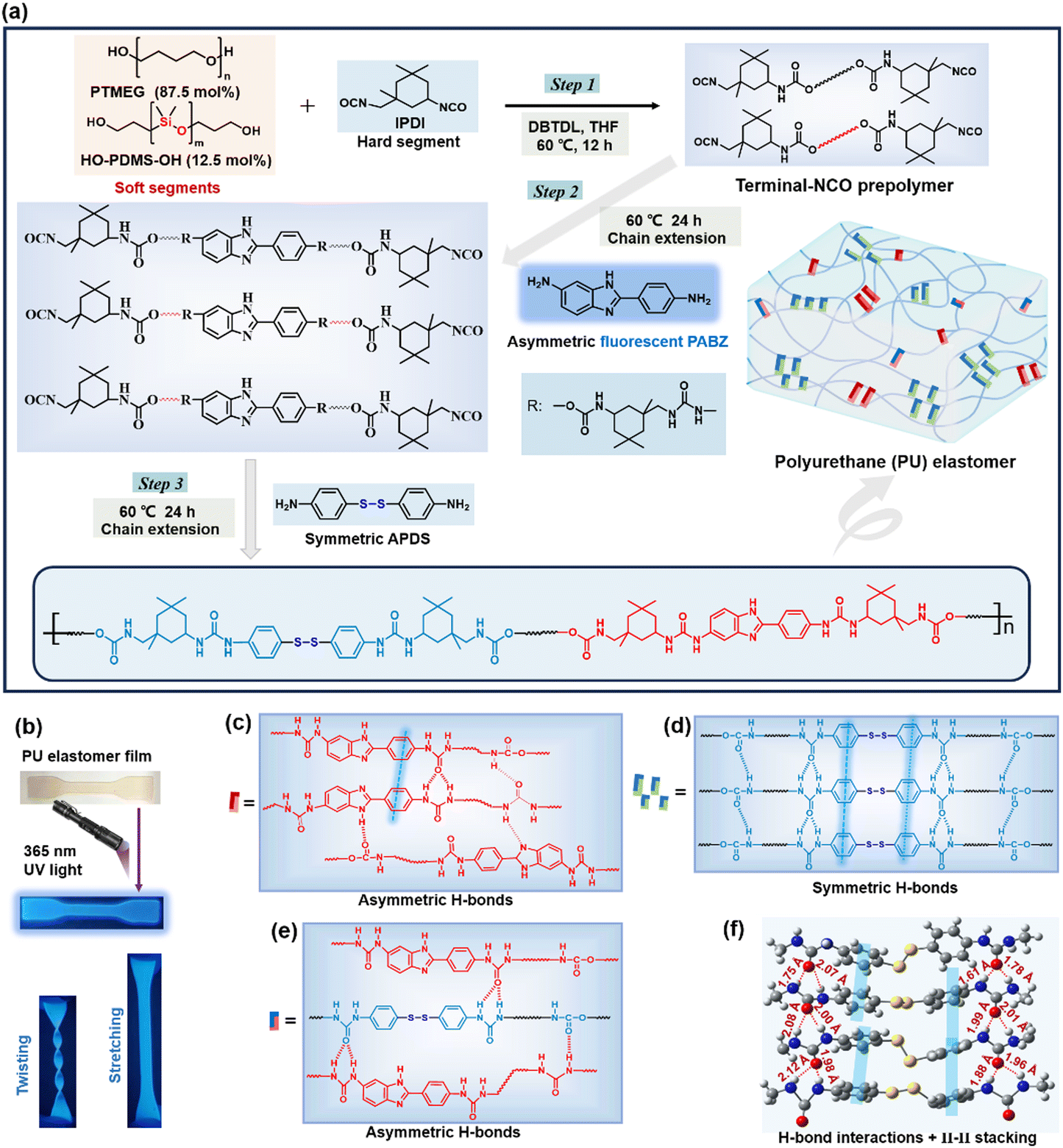 | ||
| Fig. 1 Molecular structure. (a) Schematic diagram of the preparation procedure of polyurethane (PU) elastomer. (b) PU elastomer shows attractive blue fluorescent emission under 365 nm UV light, and even in different deformation states. (c)–(e) Schematic diagram of the H-bond and π–π interactions in the PU network (dotted red lines represent H-bond interactions, and dotted blue lines represent π–π interactions). (f) Optimal conformation and the interactions between the APDS tetramers proved via DFT calculations using small molecule models (grey, white, red, blue, and yellow denote C, H, O, N, and S atoms, respectively). | ||
Results and discussion
The synthetic route for the healable PU-X/Y elastomer (where X and Y denote the molar quantities of the chain extenders APDS and PABZ, respectively) is shown in Fig. 1(a) and Fig. S1 (ESI†). The hydroxy groups of the soft segments polytetrahydrofuran (PTMEG) and polydimethylsiloxane (PDMS) react with the NCO groups of isophorone diisocyanate (IPDI) to form urethane groups, while the amino groups of the chain extenders APDS and PABZ react with the NCO groups of IPDI to form urea groups. According to the GPC results (Table S1, ESI†), the number-average molecular weight (Mn) of the PU elastomers with different APDS/PABZ ratios was about 105 g mol−1. Fourier transform infrared spectroscopy with the attenuated total reflectance mode (ATR-FTIR) was utilized to study the molecular structure of the PU elastomers (Fig. S2, ESI†). The results revealed the existence of urea and urethane groups in the PU elastomers. Energy dispersive X-ray analysis (EDX) was used to prove the presence of sulfur (disulfide bonds) in the PU elastomers (Fig. S3, ESI†). Simultaneously, dynamic mechanical analysis (DMA) was performed to determine the glass transition temperature (Tg) of the PU elastomers (Fig. S4(a), ESI†).Moreover, the temperature-variable FTIR spectra in the absorption region between 1750 and 1600 cm−1, attributed to different C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching bands, were collected, as shown in Fig. 2(a). Evidently, there was a decrease in the peak intensity at 1704 cm−1 (H-bonded CO urethane, ordered) and an increase in the peak intensity at 1722 cm−1 (free CO urethane) upon heating.21 Meanwhile, the peak intensities at 1635 cm−1 (H-bonded CO urea, ordered), 1652 cm−1 (H-bonded CO urea, disordered), and 1668 cm−1 (H-bonded CO urea, disordered) decreased, with the peak at the higher wavenumber at 1682 cm−1, assigned to the free CO urea, becoming stronger.21 In addition, the peak intensities at 1557 cm−1 (H-bonded –NH– groups) and 1563 cm−1 (H-bonded –NH– between imidazole ring and carbamate/urea groups) gradually decreased,22 while the peak intensity at 1506 cm−1 (free –NH– groups) was remarkably increased (Fig. S4(b), ESI†). These results revealed the existence of multiple H-bonds (carbamate H-bonds, urea H-bonds, carbamate/urea H-bonds, and imidazole ring and carbamate/urea H-bonds) in the PU elastomer (Fig. 1(c)–(e)) and these interactions gradually dissociated at elevated temperatures.
O stretching bands, were collected, as shown in Fig. 2(a). Evidently, there was a decrease in the peak intensity at 1704 cm−1 (H-bonded CO urethane, ordered) and an increase in the peak intensity at 1722 cm−1 (free CO urethane) upon heating.21 Meanwhile, the peak intensities at 1635 cm−1 (H-bonded CO urea, ordered), 1652 cm−1 (H-bonded CO urea, disordered), and 1668 cm−1 (H-bonded CO urea, disordered) decreased, with the peak at the higher wavenumber at 1682 cm−1, assigned to the free CO urea, becoming stronger.21 In addition, the peak intensities at 1557 cm−1 (H-bonded –NH– groups) and 1563 cm−1 (H-bonded –NH– between imidazole ring and carbamate/urea groups) gradually decreased,22 while the peak intensity at 1506 cm−1 (free –NH– groups) was remarkably increased (Fig. S4(b), ESI†). These results revealed the existence of multiple H-bonds (carbamate H-bonds, urea H-bonds, carbamate/urea H-bonds, and imidazole ring and carbamate/urea H-bonds) in the PU elastomer (Fig. 1(c)–(e)) and these interactions gradually dissociated at elevated temperatures.
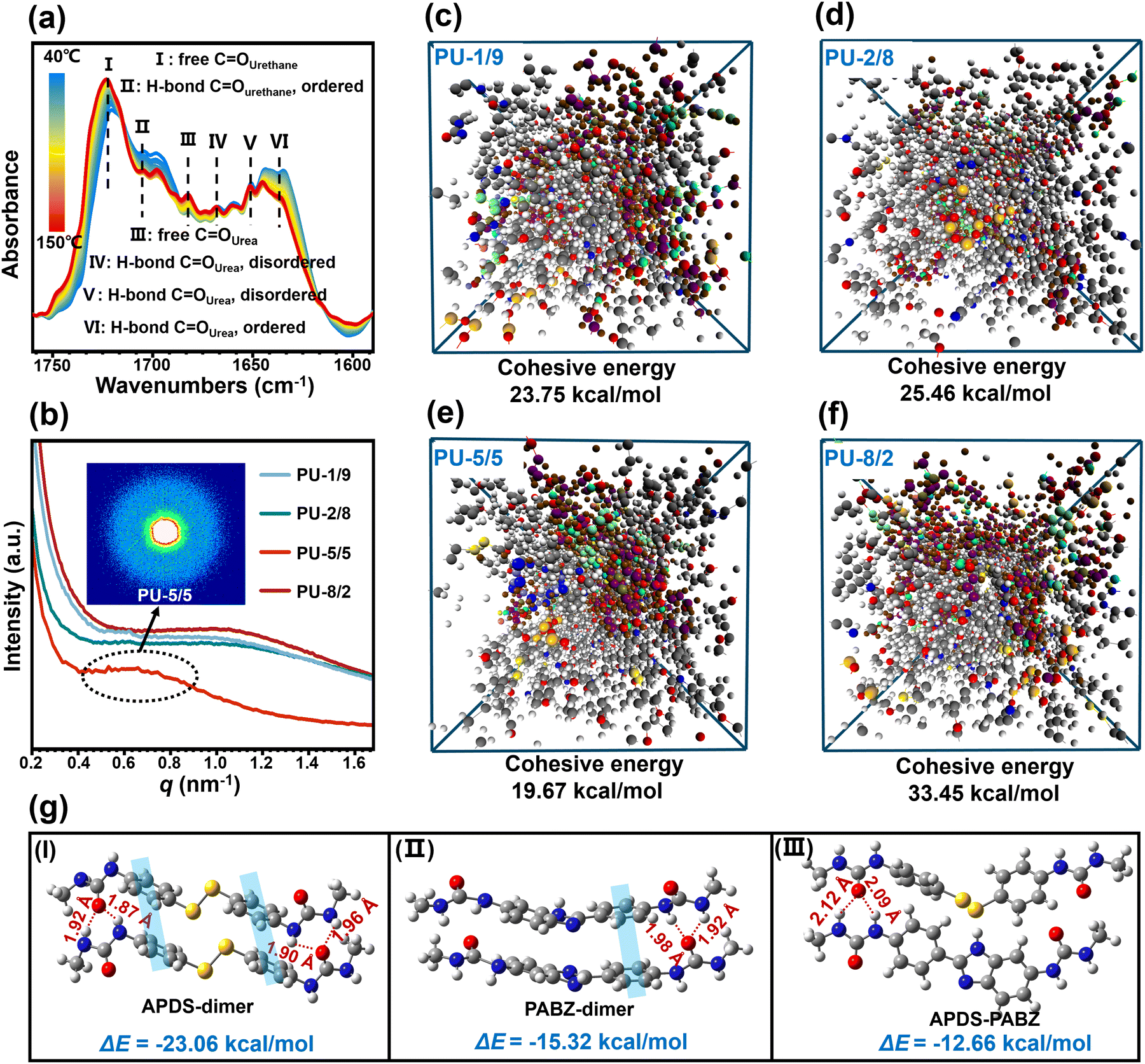 | ||
| Fig. 2 (a) Temperature-dependent FTIR spectra in the CO region of 1590–1760 cm−1 upon heating from 40 °C to 150 °C. (b) One-dimensional (1D) SAXS curves of the PU elastomers and two-dimensional (2D) SAXS pattern of PU-5/5. Molecular dynamics (MD) simulations. (c)–(f) Snapshots of the PU elastomer simulation models in this system in equilibrium conformation and their cohesive energy of the network. Density functional theory (DFT) calculations. (g) Interaction energies (ΔE) between different chain extender dimers (APDS-dimers, PABZ-dimers, and APDS-PABZ) (grey, white, red, blue, and yellow denote C, H, O, N, and S atoms, respectively). | ||
The microphase-separated morphology of the PU elastomers was investigated by SAXS (Fig. 2(b)). Clearly, all the elastomers had wide scattering peaks on the SAXS curves, revealing the existence of phase separation between the soft and hard phases. The intensities of the scattering peaks for the samples with APDS/PABZ ratios of 1/9, 2/8, and 8/2 were nearly the same. Interestingly, when the APDS/PABZ ratio was 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5, the scattering peak showed an abnormal decrease in intensity. The intensity of the scattering peak represents the stacking degree of the hard domains, whereby a smaller intensity corresponds to looser-stacked hard domains.23 Thus, the SAXS result indicated that PU-5/5 had the most loosely packed hard domains compared to the other PU elastomers. Meanwhile, it should be noted that PU-5/5 also showed the smallest scattering vector (0.67 nm−1). The periodicity (d) reflects the average distance between the hard domains, and was calculated to be 9.37 nm for PU-5/5, according to Bragg's equation;21 while that of the other PU elastomers was about 6.34 nm. The SAXS result demonstrated that PU-5/5 possesses the largest but loosest hard domains, i.e., the disordered hard domains. We can envision that such a morphology will bring about two benefits. On the one hand, the largest hard domains should be beneficial for strengthening the elastomer; and on the other hand, the loosest stacking of hard domains should be conducive to network relaxation and reorganization, thereby accelerating the healing dynamics.
:5, the scattering peak showed an abnormal decrease in intensity. The intensity of the scattering peak represents the stacking degree of the hard domains, whereby a smaller intensity corresponds to looser-stacked hard domains.23 Thus, the SAXS result indicated that PU-5/5 had the most loosely packed hard domains compared to the other PU elastomers. Meanwhile, it should be noted that PU-5/5 also showed the smallest scattering vector (0.67 nm−1). The periodicity (d) reflects the average distance between the hard domains, and was calculated to be 9.37 nm for PU-5/5, according to Bragg's equation;21 while that of the other PU elastomers was about 6.34 nm. The SAXS result demonstrated that PU-5/5 possesses the largest but loosest hard domains, i.e., the disordered hard domains. We can envision that such a morphology will bring about two benefits. On the one hand, the largest hard domains should be beneficial for strengthening the elastomer; and on the other hand, the loosest stacking of hard domains should be conducive to network relaxation and reorganization, thereby accelerating the healing dynamics.
To demonstrate the formation of disordered hard domains in the PU network at the atomic level, density functional theory (DFT) calculations were performed on small molecule models. Here, all the structures were fully optimized at the M06-2X/6-311+g(d,p)//B3LYP/6-311+g(d,p) level (see details in the supporting information). Fig. 2(g) shows the optimal conformations and the interaction energies between the different chain extenders dimers (APDS-dimers, PABZ-dimers, and APDS-PABZ). A regular topological conformation could be formed either between APDS-dimers or between PABZ-dimers. This was due to the synergy of the π–π stacking and H-bond receptors and donors. By contrast, the optimal conformation between APDS-PABZ was extremely irregular compared to the APD and PABZ dimers. Meanwhile, the interaction energies of the different dimers followed the sequence of: APDS-dimers (23.06 kcal mol−1) > PABZ-dimers (15.32 kcal mol−1) > APDS-PABZ (12.66 kcal mol−1). Therefore, the DFT calculation results revealed that the equivalent molar contents of APDS and PABZ is beneficial for the formation of disordered hard domains. To demonstrate the validity of the concept proposing disordered hard domains, the molecular models of tetramers were also used in DFT calculations (Fig. 1(f) and Fig. S5, ESI†). Identically, the calculation results of the tetramers unveiled the same rule as those of the dimers.
The phase separation can affect the free volume of the PU elastomers. It has been reported that the free volume of a PU elastomer at room temperature is mainly governed by the soft domains in the rubbery state, while the hard domains in the glassy state have negligible contribution to the free volume.24 Therefore, the free volume of the PU elastomers was examined by positron annihilation lifetime spectroscopy (PALS) (see details in the ESI†). The lifetime and intensity of o-Ps are directly related to the size and number of the free-volume hole within the polymer bulk.25Fig. 3(a) shows the o-Ps lifetimes (τ3) and free-volume hole sizes (vf) of all the PU elastomers. Evidently, with the increase in APDS/PABZ molar ratio, both τ3 and vf decreased first and then increased gradually. PU-5/5 showed the lowest vf among all the PU elastomers. The underlying reason is that PU-5/5 had the largest hard domains compared with the other samples, due to the disordered hard domains involving a proportion of soft segments. Meanwhile, the smaller free volume corresponds to the higher probability of o-Ps annihilation. Thus, the PU-5/5 had the lowest τ3. As a result, the PALS result indirectly indicated that PU-5/5 had the largest degree of phase separation.
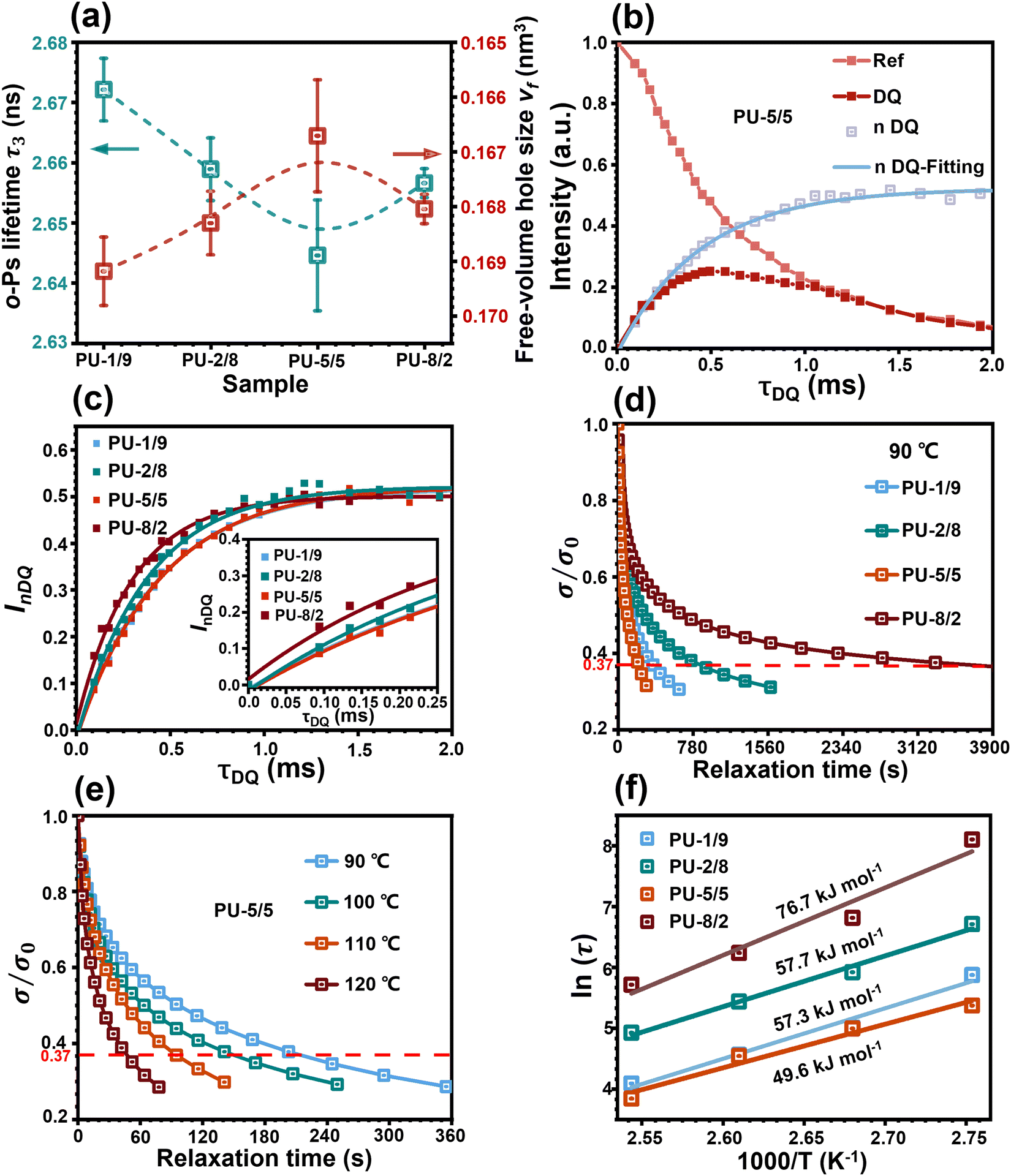 | ||
| Fig. 3 (a) o-Ps lifetimes (τ3) and free-volume hole sizes (vf) of all the PU elastomers. Solid-state nuclear magnetic resonance (NMR). (b) Normalized double-quantum (nDQ) build-up curve and reference signal decay of PU-5/5. (c) Normalized double-quantum (nDQ) signal intensities as a function of DQ excitation time for all the PU elastomers. Network relaxation. (d) Normalized stress-relaxation curves of all the PU elastomers at 90 °C. (e) Normalized stress-relaxation curves of PU-5/5 from 90 °C to 120 °C. (f) Fitting the relaxation activation energies of all the PU elastomers using Arrhenius's law. | ||
Proton multiple-quantum (MQ) nuclear magnetic resonance (NMR) has been proven to be a robust method for probing the segmental dynamics of polymeric materials.26 Thus, the proton MQ NMR spectra were collected to investigate the effect of hybridization of the chain extenders on the restriction of network relaxation. As shown in Fig. 3(b) and Fig. S6, ESI,† the obtained double-quantum (DQ) intensity and reference signal decay as a function of the DQ excitation time could be normalized following a normalization protocol.27 The normalized double-quantum (nDQ) signal intensities as a function of the DQ excitation time for all the PU elastomers are shown in Fig. 3(c). In general, the nDQ signal build-up rate is proportional to the crosslinking density,26 which further reflects the restriction of network relaxation. PU-5/5 possessed the slowest nDQ signal build-up rate, indicating its fastest network relaxation dynamics among all the PU elastomers. This was due to the lowest network crosslinking density formed by the most loosely stacked hard domains. Moreover, the stress-relaxation spectra of the PU elastomers were also collected at 90 °C to investigate the network relaxation dynamics. The normalized stress as a function of time of the PU elastomers is shown in Fig. 3(d). The characteristic relaxation time (τ) of network is defined as the time required for the polymer to reach 1/e (37%) of the initial stress/modulus.28 Strikingly, the PU-5/5 elastomer had a minimum relaxation time of 202 s, indicating the fastest relaxation rate of network, which could be attributed to the most loosely stacked hard domains in the elastomer. Then, the dependence of the network relaxation rate on temperature was further investigated (Fig. 3(e) and Fig. S7(a)–(c), ESI†). With the increase in temperature, the network relaxation times of all the PU elastomers greatly decreased. In particular, the relaxation time of PU-5/5 was reduced to 46 s as the temperature increased to 120 °C. This result indicates that the network relaxation rate largely depends on the temperature. The dependence of the relaxation time on temperature could be further fitted by Arrhenius's law.28 As shown in Fig. 3(f), PU-5/5 had the lowest activation energy (Ea) (49.6 kJ mol−1). Such a low energy barrier can facilitate the rearrangement of the network topology and thus potentially promote the healing dynamics of the damaged PU elastomers. Combining these results, it can be concluded that PU-5/5 had the most active hard domains.
To theoretically elucidate the effect of hybridization of the chain extenders on the network cohesive energy in this PU system, all-atom molecular dynamics (MD) simulation was performed at the atomic level. The simulated systems comprised five polymer chains, with each polymer chain consisting of four hard and five soft segments (see details in the ESI†). The cohesive energy is defined as the average energy per chain required to separate all the polymer chains in a condensed state into infinite distance from each other. The cohesive energies of the PU elastomers were calculated by the following equation:21
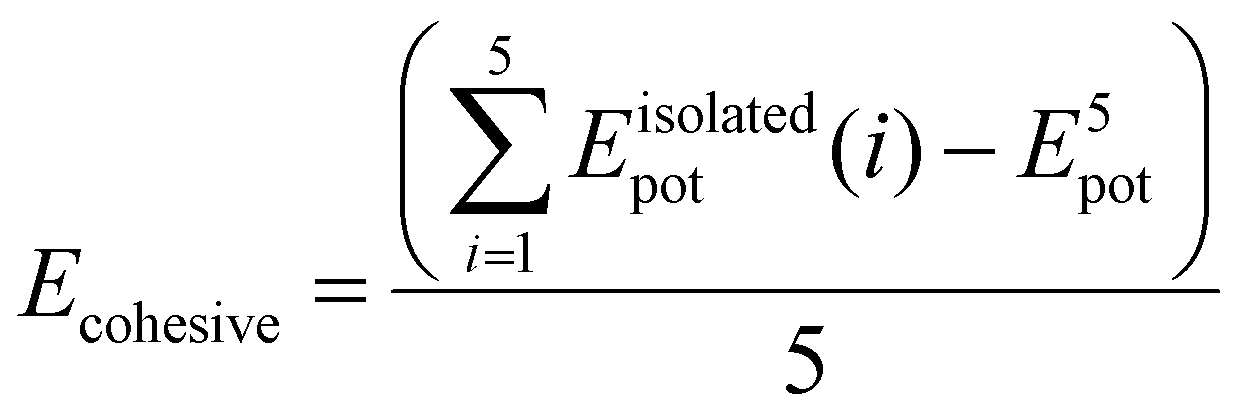
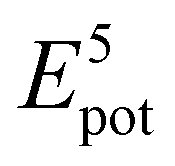 is the average potential energy of the condensed system consisting of five polymer chains. Fig. 2(c)–(f) show the dynamic equilibrium configurations and their cohesive energies in the PU elastomer networks. With the increase in the APDS/PABZ molar ratio, the cohesive energy of the PU elastomer networks decreased at first and then increased gradually. Clearly, the cohesive energy was the lowest when the degree of hybridization of the chain extenders (APDS/PABZ = 5:5) was the maximum. Thus, the MD simulations revealed that PU-5/5 had the lowest cohesive energy of network. The theoretical calculation results agreed well with the experimental results. Evidently, these results reveal that tuning the APDS/PABZ molar ratio can not only tailor the aggregated morphologies of hard domains but also control their cohesive energy. Generally, the dependence of the mechanical properties and network relaxation dynamics on hard domains is mutually exclusive, thus it would be extremely promising to construct large but disordered hard domains to reconcile this competing relationship.
is the average potential energy of the condensed system consisting of five polymer chains. Fig. 2(c)–(f) show the dynamic equilibrium configurations and their cohesive energies in the PU elastomer networks. With the increase in the APDS/PABZ molar ratio, the cohesive energy of the PU elastomer networks decreased at first and then increased gradually. Clearly, the cohesive energy was the lowest when the degree of hybridization of the chain extenders (APDS/PABZ = 5:5) was the maximum. Thus, the MD simulations revealed that PU-5/5 had the lowest cohesive energy of network. The theoretical calculation results agreed well with the experimental results. Evidently, these results reveal that tuning the APDS/PABZ molar ratio can not only tailor the aggregated morphologies of hard domains but also control their cohesive energy. Generally, the dependence of the mechanical properties and network relaxation dynamics on hard domains is mutually exclusive, thus it would be extremely promising to construct large but disordered hard domains to reconcile this competing relationship.
The typical stress–strain curves of the PU elastomers are shown in Fig. 4(a), and the corresponding parameters of the mechanical properties are summarized in Fig. 4(b) and Table S2 (ESI†). The PU-1/9 sample had a mechanical strength of 45.2 MPa and a fracture strain of 891.3%, resulting in a toughness of 124.1 MJ m−3. With the increase in the APDS/PABZ molar ratio, the mechanical strength of the PU elastomers increased at first and then decreased. Obviously, PU-5/5 had the highest mechanical strength (60.7 MPa) and fracture strain (1172%). On the one hand, the largest hard domains gave the most efficient reinforcement effect; and on the other hand, a strain hardening phenomenon, i.e., an obvious abrupt increase in strength as the strain reached a certain value, was observed in the stress–strain curves. To investigate the underlying mechanism of strain hardening, in situ wide-angle X-ray diffraction (WAXD) tensile tests were performed for all the PU elastomers. Fig. S8 (ESI†) shows the two-dimensional (2D) WAXD patterns at different strains of all the PU elastomers. The evolution of the diffraction ring from round to shuttle shaped with the increase in strain indicated that the PTMEG chains were oriented along the stretching direction. With further increasing the strain, two small diffraction spots appeared in the 2D WAXD patterns, signifying the strain-induced crystallization (SIC) due to the alignment of the PTMEG chains along the stretching direction.21 The onset strains of SIC were about 670%, 680%, 720%, and 520% for PU-1/9, PU-2/8, PU-5/5, and PU-8/2, respectively. Notably, PU-5/5 had the largest onset strain. Meanwhile, PU-5/5 showed the brightest diffraction spots when fractured (Fig. 4(d)), indicating it had the highest crystallinity. Meanwhile, the disordered hard domains of PU-5/5 displayed an easier relaxation and reorganization among all the PU elastomers. As a result, the excellent mechanical strength of PU-5/5 could be ascribed to the synergistic effect of the disordered hard domains and the newly-formed crystals contributed by SIC.
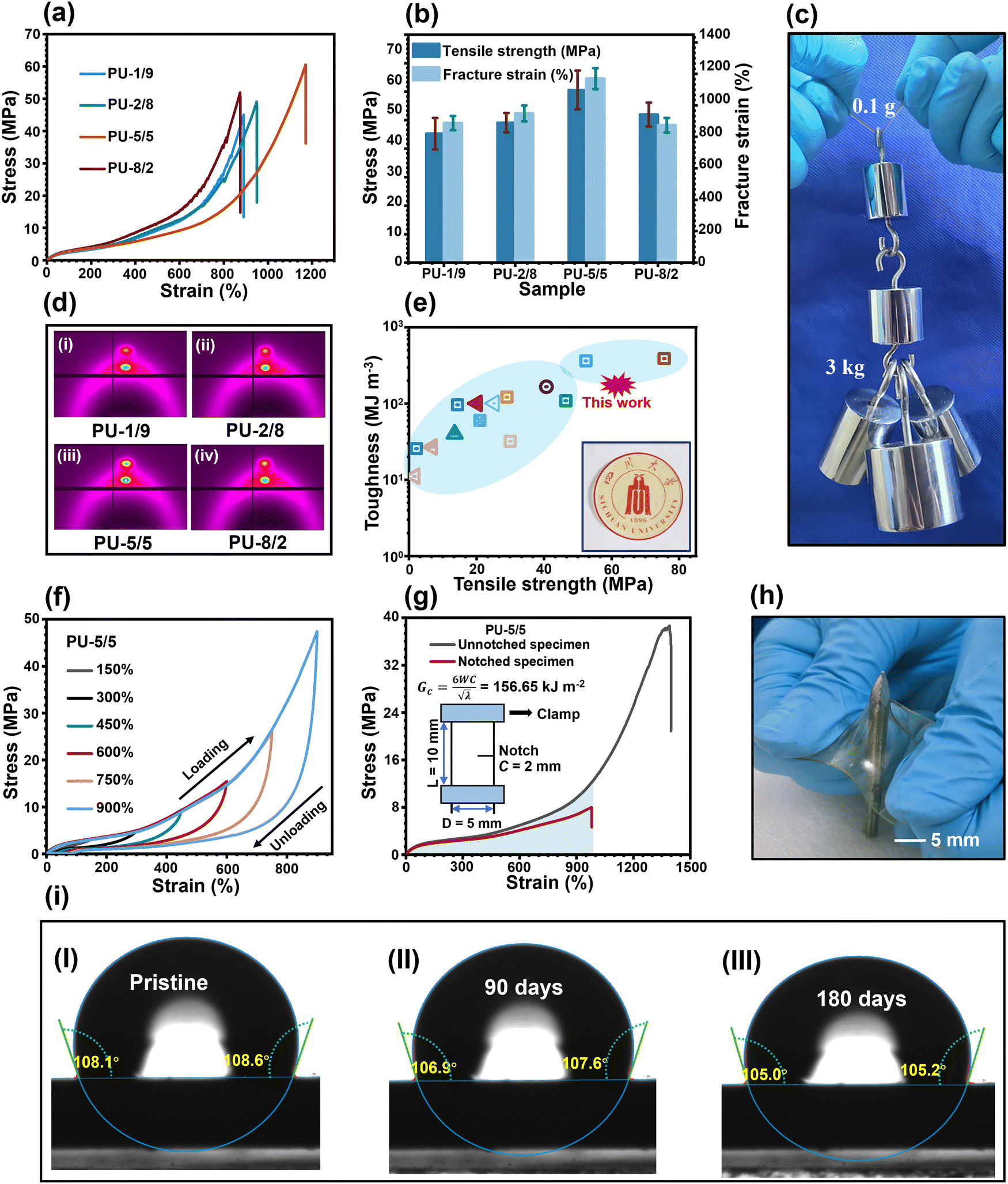 | ||
| Fig. 4 Mechanical properties. (a) Typical stress–strain curves of all the PU elastomers. (b) Summary of the mechanical properties of all the PU elastomers. (c) Five objects (3.0 kg) easily lifted by a film of PU-5/5 (0.01 g). (d) Two-dimensional (2D) WAXD patterns of all the PU elastomers at fracture. (e) Plot showing the superior mechanical robustness of the as-prepared elastomers compared with that of previous reported works. (f) Cyclic tensile curves of PU-5/5 in successive loading–unloading cycles with increasing strains but no relaxing time. (g) Stress–strain curves of notched and non-notched PU-5/5. (h) Image showing the puncture resistance of PU-5/5 by pressing the film against a nail. (i) Water contact angles of PU-5/5 at 0, 90, and 180 days at a temperature of 26 °C and relative humidity (RH) of 54%, respectively. | ||
Remarkably, the toughness of PU-5/5 (177.9 MJ m−3) was much higher than that of many reported synthetic materials, including carbon fibres (25 MJ m−3),29 Kevlar 49 (50 MJ m−3),30 nylon 6,6 (80 MJ m−3),29,30 high-density polyethylene (151.2 MJ m−3),31 and even typical natural spider silk (160 MJ m−3).21 Such superior mechanical robustness of PU-5/5 were validated by the fact that a thin strip of the sample weighing 0.01 g could easily hold a weight of 3.0 kg, that is 300000 times its own weight (Fig. 4(c)).5,11,22,32–39 Meanwhile, its superior mechanical robustness was comparable with that of many previously reported works (Fig. 4(e)). The high toughness was closely related to the energy dissipation ability, which could be examined by cyclic loading–unloading tests to different maximum strains for the PU-5/5 elastomer (Fig. 4(f) and Fig. S9(a), ESI†). The efficiency of energy dissipation, which is defined as the ratio of the hysteresis loop to the toughness under the same strain, of PU-5/5 was up to 65.4% at the maximum strain, which was higher than that of the other samples (Fig. S9(b), ESI†). This could be attributed to the fact that the largest but loosest hard domains have the fastest network relaxation and reorganization rates. Thanks to their efficient energy dissipation ability, the pre-damaged PU elastomers showed high crack tolerance (Fig. 4(g) and Fig. S10, ESI†). In particular, PU-5/5 possessed a fracture energy of 156.7 kJ m−2, thus it could outperform most reported healable and unhealable materials (Fig. S11, ESI†). Concurrently, PU-5/5 also showed a good puncture-resistance property (Fig. 4(h)).
More importantly, selecting the hydrophobic HO-PDMS-OH as one of the soft segments could efficiently solve the intrinsic hydrophilicity of the dynamic networks formed by urethane and urea bonds. As shown in (Fig. 4(i) and Fig. S12, ESI†), after 180 days in a humid environment with a temperature of 26 °C and a relative humidity of 54%, their water contact angle and weight only experienced negligible changes. Meanwhile, despite the mechanical strength of PU-5/5 decreasing from 60.7 to 47.2 MPa (Fig. S13, ESI†), the mechanical strength was still comparable to or even better than most hydrophobic materials. The results reveal that this strategy can greatly enhance the hydrophobicity of PU elastomers and thus provides a strong guarantee for their long-term applications in humid environments.
The large but disordered hard domains with high mobility could be expected to impart the PU elastomers with an effective healing ability. Since PU-5/5 possessed the best mechanical properties and the highest mobility of hard domains, we next focused on its healing ability. To prove the healing ability, cut-off PU-5/5 strips were reconnected and healed for 2 h at 90 °C. The healed strips could hold a weight of 400 g without breaking (Fig. 5(a)). We then used an optical microscope to track the healing process (Fig. S14, ESI†). An artificial scratch became invisible after 2 h at 90 °C on the surface of PU-5/5, indicating that the damaged regions were well healed by H-bonds and disulfide bonds within the disordered hard domains. To quantitatively investigate the healing properties, dumbbell-shaped PU-5/5 strips were initially cut into two damaged parts using a fresh razor blade, followed by recombining and healing for different time periods at different temperatures. The healing efficiency (%) is defined as the ratio of the tensile strength of the healed part to that of the pristine one. The stress–strain curves of the healed PU-5/5 are shown in Fig. 5(b), and the corresponding healing efficiencies are summarized in Fig. 5(c). The healed PU-5/5 showed an acceptable recovered mechanical strength of 23.5 MPa and fracture strain of 670.2% at 50 °C. Elevating the temperature could facilitate the healing dynamics and thus enhance the healing efficiency. Healing at 90 °C led to a healing efficiency of 75%, revealing that the breakage and reformation of the H-bonds and disulfide bonds within the disordered hard domains could be activated at elevated temperatures.
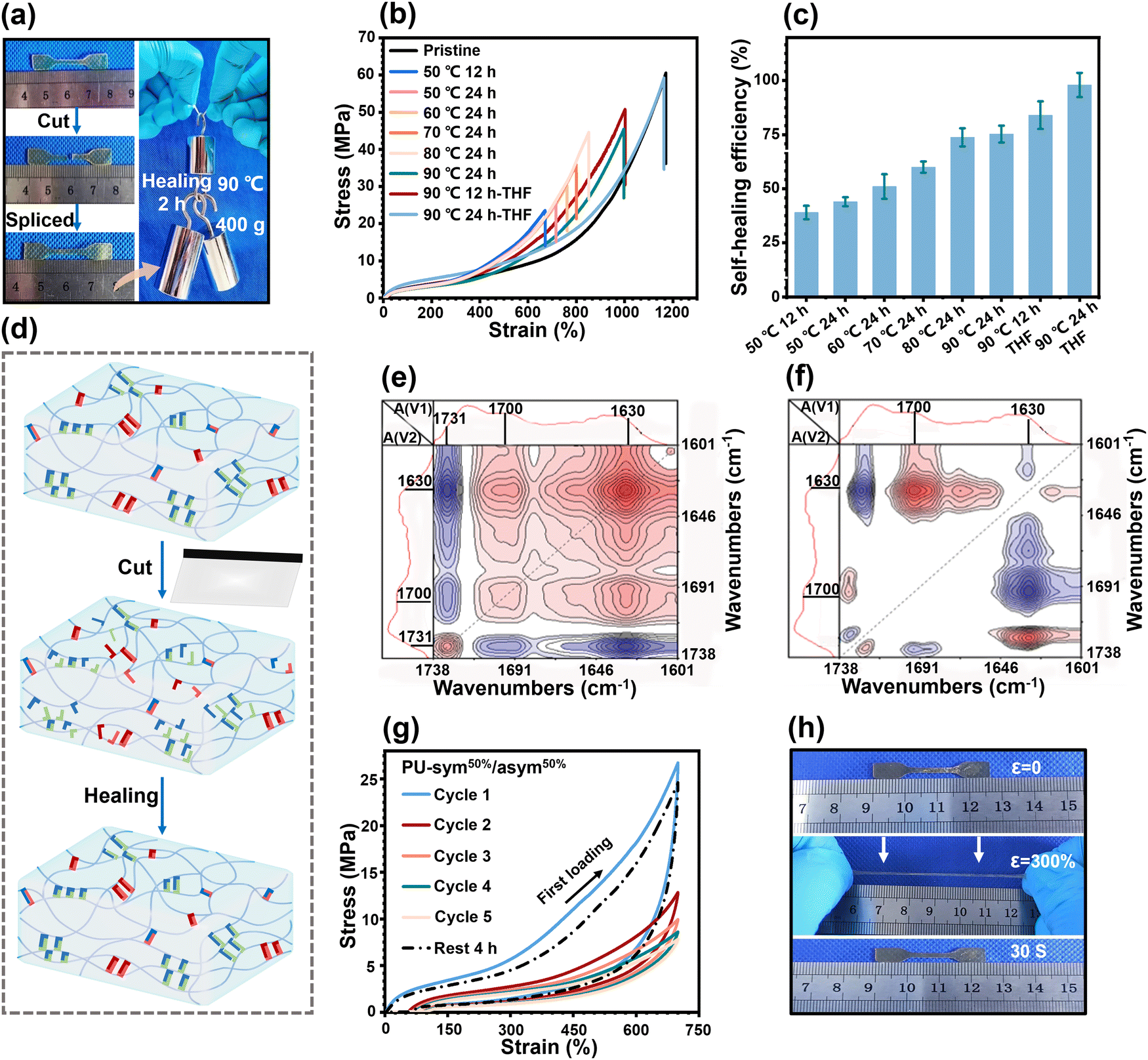 | ||
| Fig. 5 Self-healing properties. (a) Image showing the healing feature of cut-off PU-5/5 strips for healing for 2 h at 90 °C and then lifting a 400 g load. (b) Stress–strain curves for the healed PU-5/5 after different healing times at different temperatures. (c) Summary of the healing efficiencies of the healed PU-5/5. (d) Schematic diagram of the network structural healing. (e) Synchronous and (f) asynchronous maps of PU-5/5 in the region 1738–1601 cm−1 during the heating process from 80 °C to 150 °C. (g) Cyclic stress–strain curves of PU-5/5 with five successive loading–unloading cycles and one cycle after relaxing for 4 h. (h) Images showing the self-recovery ability of the PU-5/5 elastomer after manually stretching a sample. | ||
Moreover, it was found that the healing efficiency could be drastically enhanced by exposing the damaged region to THF. As presented in Fig. 5(b), with the assistance of THF, healing at 90 °C for 24 h led to a recovered fracture strain of 1164.5%, a mechanical strength of 59.4 MPa, and a satisfactory mechanical strength healing efficiency of 97.9%. These improved results could be attributed to the fact that THF acts as a temporary plasticizer to trigger the exchange recombination kinetics of H-bonds and disulfide bonds in damaged regions (Fig. 5(d)). To examine the kinetics of the rupture and recombination of different H-bonds during the healing process, two-dimensional (2D) correlation analysis spectra were generated from variable-temperature FTIR spectroscopy (from 80 °C to 150 °C). The details of the moving sequential order of different H-bonded CO groups can be judged by Noda's rule8 in both synchronous and asynchronous spectra (Fig. 5(e) and (f)). Here, two auto-peaks at [1630, 1630 cm−1] and [1700, 1700 cm−1] could be clearly observed in the synchronous spectra, corresponding to the vibration peaks of the H-bonded CO moieties of urea and urethane groups, respectively. The positive peaks at [1700, 1630 cm−1] showed the motion directions of the urethane and urea groups were identical. Meanwhile, positive peaks at [1700, 1630 cm−1] were also observed in the off-diagonal positions in the asynchronous spectra. According to the rule of 2D-FTIR analysis, the peak at 1700 cm−1 moved first, indicating that the H-bonded CO moieties at urethane groups were disassociated prior to the urea groups.
Elastomers usually experience repeated loading, thus it is necessary for them to have a rapid self-recovery ability, which can effectively prevent the deterioration of the mechanical properties. Thus, cyclic loading–unloading tensile tests of PU-5/5 at a fixed strain of 700% were performed to investigate its self-recovery ability (Fig. 5(g)). The first loading–unloading curve showed a pronounced hysteresis loop (41.5 MJ m−3), revealing a significant energy dissipation due to the force-induced rupture of the H-bonds and disulfide bonds within the disordered hard domains. An obvious reduction of the hysteresis loop during the second loading–unloading was observed owing to the lack of sufficient time for the recombination of the ruptured H-bonds and disulfide bonds. The hysteresis loop was lowered slightly after the second cycle, indicating the delayed and sluggish response of the rupture and recombination of H-bonds and disulfide bonds. Although the hysteresis loop could not be restored immediately, it could still recover well given sufficient time. The 6th loading–unloading curve could nearly recover to the original state after a waiting time of 240 min. The residual strain was also eliminated and reached a negligible value of 5.3%. Fig. 5(h) further indicated the remarkable self-recovery ability of PU-5/5. The dumbbell-shaped strip, after being manually stretched to 3.0 times its original length, was observed to recover to its original state within 30 s.
Interestingly, the high-performance PU elastomers showed an attractive blue fluorescence under 365 nm UV light (Fig. 1(b)). According to a previously reported work,40 the chain extender PABZ is a typical quenching fluorescent molecule. Thus, the blue fluorescence of the PU elastomer was ascribed to the aggregation-induced quenching (ACQ) mechanism of PABZ. Two-dimensional (2D) fluorescence spectroscopy was used to examine the optimum emission peak position (Fig. 6(a)). In the fluorescence spectroscopy analysis, 425 nm wavelength was selected as the emission peak for the investigation of the fluorescence performance of the PU elastomers. Fig. 6(b) shows the fluorescence intensity emission curves of the PU elastomers with different APD/PABZ ratios. Notably, PU-5/5 displayed the highest fluorescence intensity owing to its loosest hard domains. Meanwhile, variable-temperature fluorescence spectra were collected to investigate the relationship between the fluorescence intensity and hard domains (Fig. 6(c)). It could be seen that the normalized fluorescence intensity decreased with the increase in temperature. This could be attributed to the fact that the hard domains tended to be disrupted at elevated temperatures, making non-radiative transitions easier, leading to a reduced fluorescence efficiency and thus a decrease in the fluorescence intensity. Thus, the variation of the fluorescence intensity could indirectly reflect changes in the PU elastomer networks. Consequently, the fluorescent PU elastomer showed great potential for self-reporting mechanical/chemical damage. To demonstrate this ability, the fluorescent PU elastomer was utilized to real-time report microscopic network changes. For example, during network relaxation, the H-bonds within hard domains are gradually dissociated, thus the fluorescence intensity becomes stronger. The normalized procedure is provided in detail in the ESI.† The variation of the normalized fluorescence intensity was consistent with the previous stress-relaxation results (Fig. 6(d) and Fig. S15, ESI†). Here, PU-5/5 had the fastest stress-relaxation rate, corresponding to the fastest change in fluorescence intensity. As a result, the network relaxation could be self-detected sensitively by the fluorescence intensity.
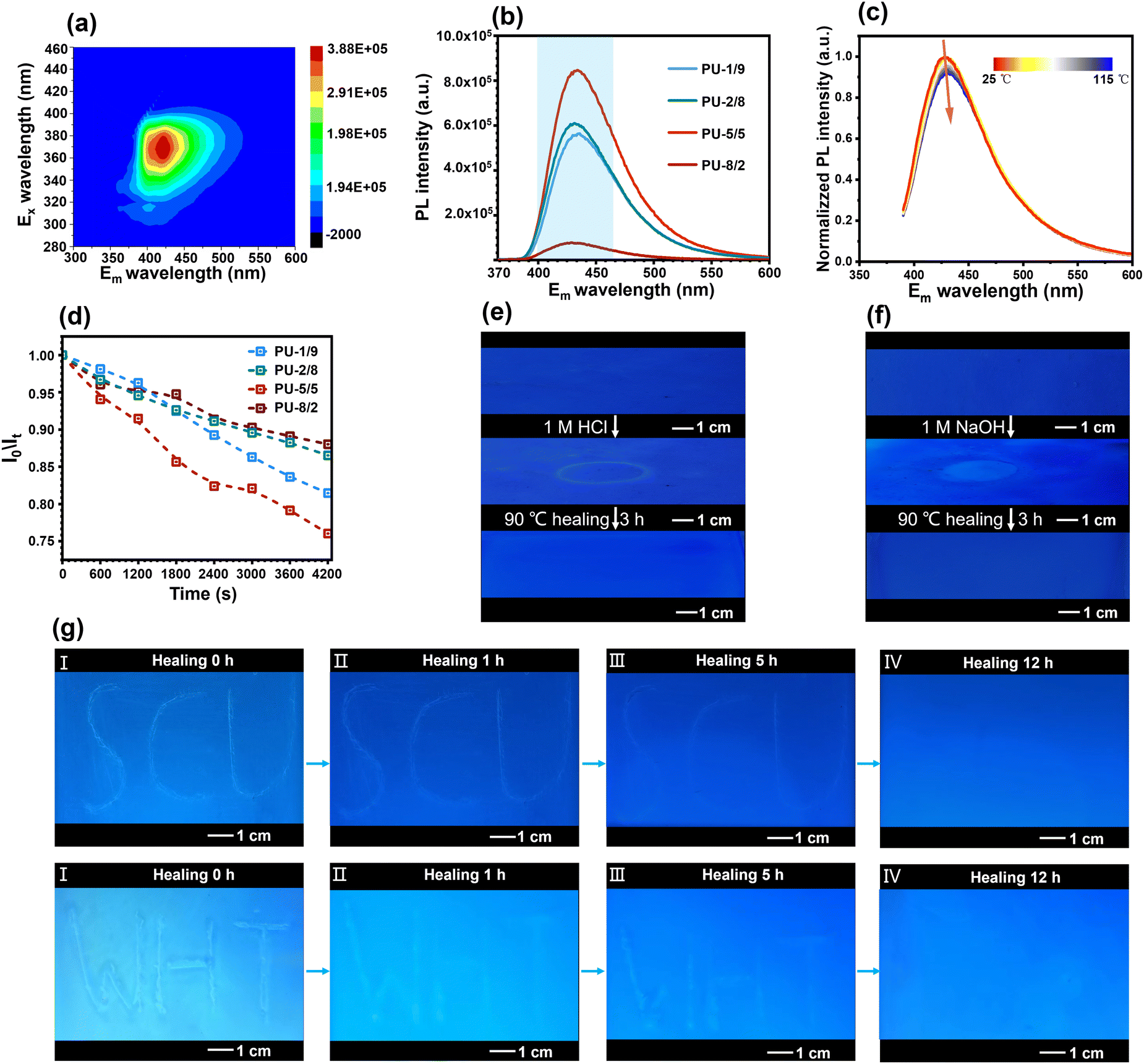 | ||
| Fig. 6 Stimulus-responsive fluorescent feature. (a) Two-dimensional (2D) fluorescence spectroscopy result for examining the optimum emission peak position. (b) Fluorescence intensity emission curves of the PU elastomers with different APDS/PABZ ratios. (c) Normalized variable-temperature fluorescence spectra of PU-5/5 for self-reporting of mechanical/chemical damage. (d) Normalized fluorescence intensity relaxation curves of the PU elastomers with different APDS/PABZ ratios. Healing process of “ring”-shaped micro-crack patterns generated by (e) HCl and (f) NaOH solutions on the surface of the PU films at 90 °C. (g) Micro-crack patterns in the shapes of “SCU” and “WHT” on the surface of the PU film healed for (I) 0, (II) 1, (III) 5, and (IV) 12 h at 90 °C. | ||
This fluorescence tracer in the PU elastomer can also serve as a self-reporter for micro-cracks and to track the entire healing process. To demonstrate this, micro-crack patterns in the shapes of “SCU” and “WHT” were created on the surface of the PU film. The film was then illuminated with 365 nm UV light, resulting in a higher intensity of blue fluorescence on the micro-crack patterns (Fig. 6(g)). This phenomenon indicated that the damage could induce a decreased packing efficiency of molecular chains, which could increase the fluorescent intensity. Meanwhile, the change in fluorescence intensity could also be used to easily track the healing process of micro-cracks. The intensity of blue fluorescence of micro-crack patterns gradually became weaker with the healing process at 90 °C. After 12 h, the micro-crack patterns completely disappeared and the intensity of the blue fluorescence returned to its original state. Similarly, chemical damage could also be easily self-reported by the fluorescence intensity, and the fluorescence intensity could be used to track the healing process. Micro-crack patterns in the shape of a “ring” were created on the surface of the PU film through 1 M hydrochloric acid (HCl) or sodium hydroxide (NaOH) solution. As shown in Fig. 6(e)–(f), The micro-crack patterns showed a higher intensity of blue fluorescence due to a reduction in the stacking density of molecular chains. The micro-crack patterns were entirely healed at 90 °C after 3 h, leading to the intensity of blue fluorescence recovering to its original state. The diagnosis of mechanical/chemical damage and their healing processes provides an early warning system for potential malfunctions or failures, which can improve the overall safety and longevity of materials.
Additionally, the PU elastomer also showed great potential in smart fluorescence-based anti-counterfeiting materials, such as information encoding and encryption. Here, an experiment was performed using an aerosol bottle filled with PU elastomer solution as a fluorescence ink. The PU ink was sprayed on a nonfluorescent paper and quickly formed a layer of an ultrathin film. As shown in Fig. 7(a) and (b), customized molds were used to create different shapes of patterns, including an apple, love, five-pointed star, school badge, panda, and even complicated QR codes. These sprayed patterns were nearly invisible and indistinguishable on the nonfluorescent paper under daylight, but they appeared as blue fluorescence under 365 nm UV light. After turning off the UV light, the fluorescent patterns disappeared completely, as shown in Fig. 7(c) and (d). This ability to simulate the process of information encryption and decryption makes the PU elastomer ideal for anti-counterfeiting applications. Under daylight, the PU ultrathin film could be used as a brand anti-counterfeiting mark on laptops, as shown in Fig. 7(e). By scanning the QR code with a smartphone under 365 nm UV light, the authenticity of the product could be quickly verified. Meanwhile, the excellent hydrophobicity of the PU thin film can effectively prevent the deterioration of its fluorescent performance, making it ideal for use in humid environments for an extended period. Thus, the fluorescent PU elastomers show significant potential for applications in a wide variety of flexible and smart devices due to their anti-counterfeiting function.
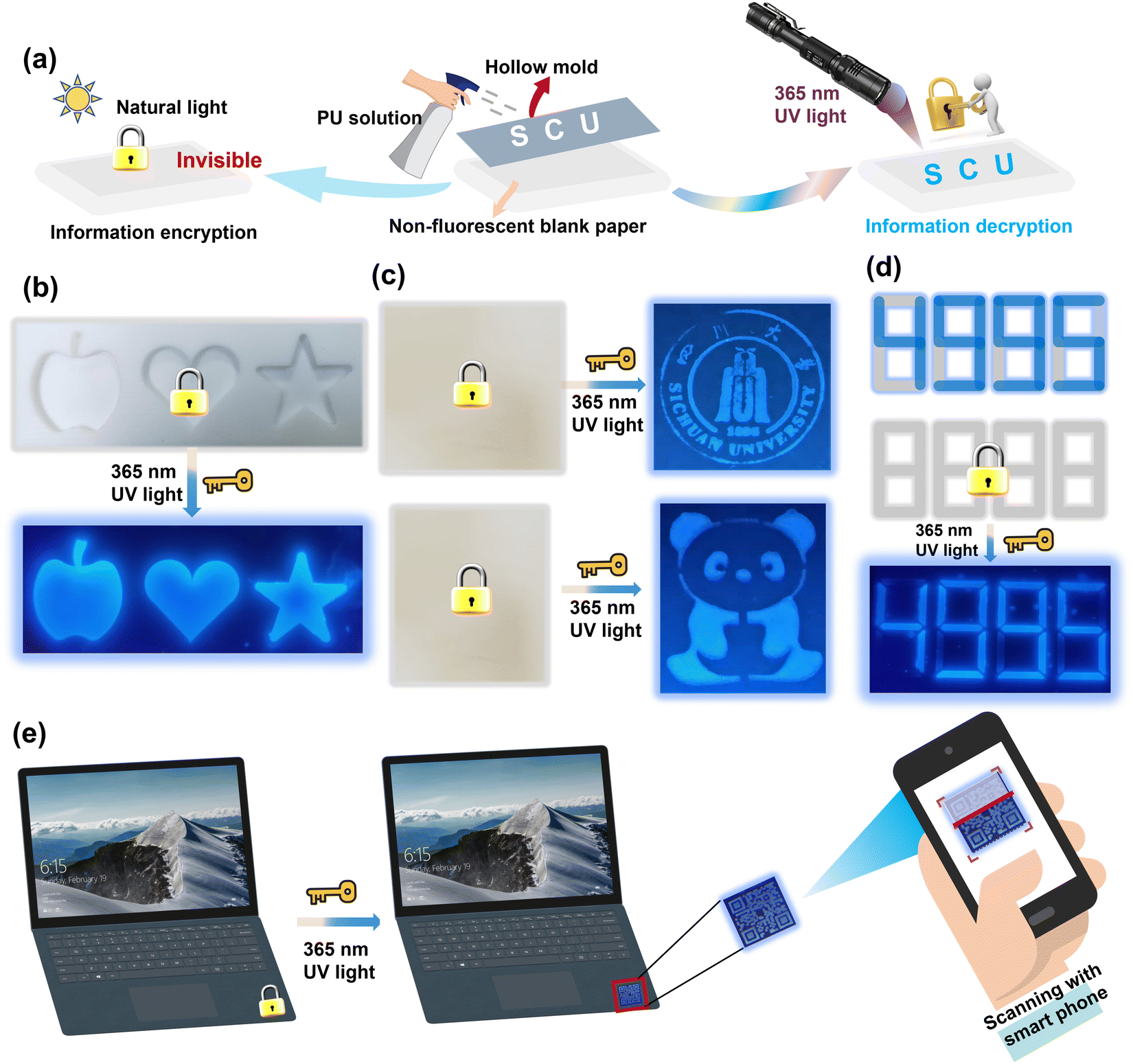 | ||
| Fig. 7 Information encoding and encryption. (a)–(c) Test process showing a dilute solution of the PU elastomer was used to form an ultrathin film with a certain shape on the filter paper through a mold of apple, love, five-printed star, school badge, and panda shapes. (d) Encryption–decryption process of digital and analogue information under 365 nm UV light. (e) Design of QR code graphics with information encryption to serve as an anti-counterfeiting mark for a laptop. | ||
Conclusions
In summary, we propose an innovative design strategy based on creating large yet disordered hard domains to fabricate a mechanically robust, self-reporting, and healable PU elastomer with excellent hydrophobicity. The disordered hard domains were created by symmetric APDS and asymmetric fluorescent PABZ, which showed a large but loose feature. Such phase separation led to a small free-volume fraction, prominent SIC, high energy dissipation, and high network relaxation dynamics. As a result, the optimized PU-5/5 elastomer exhibited exceptional mechanical strength (60.7 MPa), toughness (177.9 MJ m−3), and a high healing efficiency (97.8%) and admirable self-recovery ability. More importantly, this stimulus-responsive fluorescence trait could commendably achieve the self-reporting of mechanical/chemical damage and the healing processes. Meanwhile, the attractive blue fluorescence imparts the PU elastomers with potential application in information encoding and encryption. Furthermore, selecting HO-PDMS-OH as one of the soft segments can effectively solve the intrinsic hydrophilicity of the dynamic networks formed by urethane and urea groups. This work provides valuable guidance in the development of robust multi-functional materials that could be utilized in a wide variety of flexible and smart electronic device systems.Experimental sections
All the procedures for the experiments are given in the ESI.†Author contributions
J. R. Wu conceived and supervised the project and revised the paper. B. J. Ye and H. J. Zhang provided the PALS platform and analysed the PALS data. H. T. Wu designed the experiments, carried out the experiments and theory calculations, analysed the data and wrote the paper and edited the manuscript under the supervision of J. R. Wu. M. Luo and Y. W. Chen measured the PALS data. H. Wang helped the 2D-FTIR characterization and analysed the data. Z. Y. Yuan, B. Q. Jin and W. Q. Wu assisted with the characterization. All authors discussed the results and commented on the manuscript.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was supported by National Natural Science Foundation of China (Grant No. 51873110), Sichuan Science and Technology Program (Grant No. 2021YFS0402 and 2021JDJQ0018), Sichuan Science and Technology Program-Natural Science Foundation of Sichuan Province (2022NSFSC1982) and State Key Laboratory of Polymer Materials Engineering, Sichuan University.References
- G. Zhang, H. Feng, K. Liang, Z. Wang, X. Li, X. Zhou, B. Guo and L. Zhang, Sci. Bull., 2020, 65, 889–898 CrossRef CAS PubMed.
- Q. Chen, S. Liang and G. A. Thouas, Prog. Polym. Sci., 2013, 38, 584–671 CrossRef CAS.
- J. H. Wu, C. H. Li, Y. T. Wu, M. T. Leu and Y. Tsai, Compos. Sci. Technol., 2010, 70, 1258–1264 CrossRef CAS.
- M. Das, A. R. Parathodika, P. Maji and K. Naskar, Eur. Polym. J., 2023, 186, 111844 CrossRef CAS.
- Q. Zhong, X. Chen, Y. Yang, C. Cui, L. Ma, Z. Li, Q. Zhang, X. Chen, Y. Cheng and Y. Zhang, Mater. Chem. Front., 2021, 5, 5371–5381 RSC.
- Y. Peng, S. Gu, Q. Wu, Z. Xie and J. Wu, Acc. Mater. Res., 2023, 4, 323–333 CrossRef CAS.
- H. Wu, B. Jin, H. Wang, W. Wu, Z. Cao and J. Wu, Front. Chem., 2020, 8, 1–11 CrossRef PubMed.
- H. Wang, H. Liu, Z. Cao, W. Li, X. Huang, Y. Zhu, F. Ling, H. Xu, Q. Wu, Y. Peng, B. Yang, R. Zhang, O. Kessler, G. Huang and J. Wu, Proc. Natl. Acad. Sc. U. S. A., 2020, 117, 11299–11305 CrossRef CAS PubMed.
- W. Li, H. Liu, H. Wang, Y. Chen, Y. Peng, H. Wu, Y. Hou, Y. Huang, Z. Yuan, B. Ye, H. Zhang and J. Wu, Chem. Mater., 2023, 35, 682–691 CrossRef CAS.
- Y. Peng, Y. Hou, Q. Wu, Q. Ran, G. Huang and J. Wu, Mater. Horiz., 2021, 8, 2553–2561 RSC.
- Y. Peng, L. Zhao, C. Yang, Y. Yang, C. Song, Q. Wu, G. Huang and J. Wu, J. Mater. Chem. A, 2018, 6, 19066–19074 RSC.
- L. Zhang, H. Xiong, Q. Wu, Y. Peng, Y. Zhu, H. Wang, Y. Yang, X. Liu, G. Huang and J. Wu, Sci. China Mater., 2021, 64, 1780–1790 CrossRef CAS.
- J. Xu, X. Wang, X. Zhang, Y. Zhang, Z. Yang, S. Li, L. Tao, Q. Wang and T. Wang, Chem. Eng. J., 2023, 451, 138673 CrossRef CAS.
- H. Liu, Q. Shen, L. Zhang, S. Gu, Y. Peng, Q. Wu, H. Xiong, H. Zhang, L. Zhao, G. Huang and J. Wu, Sci. China Mater., 2022, 65, 1943–1951 CrossRef CAS.
- Y. Lai, X. Kuang, P. Zhu, M. Huang, X. Dong and D. Wang, Adv. Mater., 2018, 30, 1802556 CrossRef PubMed.
- Y. Chen, Z. Tang, Y. Liu, S. Wu and B. Guo, Macromolecules, 2019, 52, 3805–3812 CrossRef CAS.
- L. Jia, J. Xiao, J. Cui, J. Hao and X. Wang, Mater. Horiz., 2022, 9, 2128–2137 RSC.
- L. Yang, P. Ouyang, Y. Chen, S. Xiang, Y. Ruan, W. Weng, X. He and H. Xia, Giant, 2021, 8, 100069 CrossRef CAS.
- X. Vtfe, B. T. B. Qspufdujwf, N. Gps, U. I. F. Nfubmt, B. Obdsf, I. B. T. Bo, F. Bsnps, U. Jt and B. Mbzfsfe, Nano Res., 2023, 16, 10587–10596 CrossRef.
- T. H. Lee, Y. K. Song, S. H. Park, Y. Il Park, S. M. Noh and J. C. Kim, Appl. Surf. Sci., 2018, 434, 1327–1335 CrossRef CAS.
- Z. Li, Y. L. Zhu, W. Niu, X. Yang, Z. Jiang, Z. Y. Lu, X. Liu and J. Sun, Adv. Mater., 2021, 33, 2101498 CrossRef CAS PubMed.
- Y. Li, W. Li, A. Sun, M. Jing, X. Liu, L. Wei, K. Wu and Q. Fu, Mater. Horiz., 2021, 8, 267–275 RSC.
- D. Wang, J. H. Xu, J. Y. Chen, P. Hu, Y. Wang, W. Jiang and J. J. Fu, Adv. Funct. Mater., 2019, 30, 1907109 CrossRef.
- Y. Ma, H. Rong, Y. Zhang and X. Lu, Chem. Eng. J., 2023, 456, 141137 CrossRef CAS.
- H. J. Zhang, S. Sellaiyan, T. Kakizaki, A. Uedono, Y. Taniguchi and K. Hayashi, Macromolecules, 2017, 50, 3933–3942 CrossRef CAS.
- K. Saalwächter, B. Herrero and M. A. López-Manchado, Macromolecules, 2005, 38, 9650–9660 CrossRef.
- R. Zhang, S. Yu, S. Chen, Q. Wu, T. Chen, P. Sun, B. Li and D. Ding, J. Phys. Chem. B, 2014, 118, 1126–1137 CrossRef CAS PubMed.
- H. T. Wu, B. Q. Jin, H. Wang, W. Q. Wu, Z. X. Cao, Z. Y. Yuan, Y. Huang, W. H. Li, G. S. Huang, L. S. Liao and J. R. Wu, Chin. J. Polym. Sci., 2021, 39, 1299–1309 CrossRef CAS.
- M. Heim, D. Keerl and T. Scheibel, Angew. Chem., Int. Ed., 2009, 48, 3584–3596 CrossRef CAS PubMed.
- J. L. Yarger, B. R. Cherry and A. Van Der Vaart, Nat. Rev. Mater., 2018, 3, 1–11 CrossRef.
- Y. Wang, X. Huang and X. Zhang, Nat. Commun., 2021, 12, 1291 CrossRef CAS PubMed.
- Y. Eom, S. M. Kim, M. Lee, H. Jeon, J. Park, E. S. Lee, S. Y. Hwang, J. Park and D. X. Oh, Nat. Commun., 2021, 12, 621 CrossRef CAS PubMed.
- P. Zhao, C. Yin, Y. Zhang, X. Chen, B. Yang, J. Xia and L. Bian, J. Mater. Chem. A, 2020, 8, 12463–12471 RSC.
- Z. Liu, L. Zhang, Q. Guan, Y. Guo, J. Lou, D. Lei, S. Wang, S. Chen, L. Sun, H. Xuan, E. M. Jeffries, C. He, F. L. Qing and Z. You, Adv. Funct. Mater., 2019, 29, 1901058 CrossRef.
- F. Wang, Z. Yang, J. Li, C. Zhang and P. Sun, ACS Macro Lett., 2021, 10, 510–517 CrossRef CAS PubMed.
- X. Liu, X. Liu, W. Li, Y. Ru, Y. Li, A. Sun and L. Wei, Chem. Eng. J., 2021, 410, 128300 CrossRef CAS.
- D. Fu, W. Pu, Z. Wang, X. Lu, S. Sun, C. Yu and H. Xia, J. Mater. Chem. A, 2018, 6, 18154–18164 RSC.
- J. Liu, J. Liu, S. Wang, J. Huang, S. Wu, Z. Tang, B. Guo and L. Zhang, J. Mater. Chem. A, 2017, 5, 25660–25671 RSC.
- L. Zhang, Z. Liu, X. Wu, Q. Guan, S. Chen, L. Sun, Y. Guo, S. Wang, J. Song, E. M. Jeffries, C. He, F. L. Qing, X. Bao and Z. You, Adv. Mater., 2019, 31, 1901402 CrossRef PubMed.
- L. Singh and N. Ranjan, J. Am. Chem. Soc., 2023, 145, 2745–2749 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3mh01987j |
| This journal is © The Royal Society of Chemistry 2024 |