
Strong, tough and anisotropic bioinspired hydrogels†
Shu
Wang‡
ab,
Ling
Lei‡
a,
Yuanhao
Tian
c,
Huiming
Ning
*a,
Ning
Hu
 *ad,
Peiyi
Wu
e,
Hanqing
Jiang
f,
Lidan
Zhang
g,
Xiaolin
Luo
h,
Feng
Liu
a,
Rui
Zou
d,
Jie
Wen
d,
Xiaopeng
Wu
a,
Chenxing
Xiang
a and
Jie
Liu
*i
*ad,
Peiyi
Wu
e,
Hanqing
Jiang
f,
Lidan
Zhang
g,
Xiaolin
Luo
h,
Feng
Liu
a,
Rui
Zou
d,
Jie
Wen
d,
Xiaopeng
Wu
a,
Chenxing
Xiang
a and
Jie
Liu
*i
aCollege of Aerospace Engineering, Chongqing University, 174 Shazheng St, Shapingba District, Chongqing, 400044, P. R. China. E-mail: ninghuiming@cqu.edu.cn; ninghu@hebut.edu.cn; Fax: +86-23-65102521; Tel: +86-23-65102527
bState Key Laboratory of Resource Insects, College of Sericulture, Textile and Biomass Sciences, Southwest University, Chongqing 400715, China
cSouthwest Technology and Engineering Research Institute, Chongqing, 400039, P. R. China
dSchool of Mechanical Engineering, Hebei University of Technology, Tianjin, 300401, P. R. China
eState Key Laboratory for Modification of Chemical Fibers and Polymer Materials, College of Chemistry, Chemical Engineering and Biotechnology, Donghua University, Shanghai, 201620, P. R. China
fSchool of Engineering, Westlake University, Hangzhou, 310024, P. R. China
gSchool of Basic Medicine, Chongqing Medical University, 400042, P. R. China
hFirst Teaching Hospital of Tianjin University of Traditional Chinese Medicine, National Clinical Research Center for Chinese Medicine Acupuncture and Moxibustion, Tianjin, 300381, China
iState Key Laboratory of Advanced Design and Manufacturing for Vehicle Body, Changsha, Hunan, 410082, P. R. China. E-mail: liujie@hnu.edu.cn
First published on 20th February 2024
Abstract
Soft materials are widely used in tissue engineering, soft robots, wearable electronics, etc. However, it remains a challenge to fabricate soft materials, such as hydrogels, with both high strength and toughness that are comparable to biological tissues. Inspired by the anisotropic structure of biological tissues, a novel solvent–exchange-assisted wet-stretching strategy is proposed to prepare anisotropic polyvinyl alcohol (PVA) hydrogels by tuning the macromolecular chain movement and optimizing the polymer network. The reinforcing and toughening mechanisms are found to be “macromolecule crystallization and nanofibril formation”. These hydrogels exhibit excellent mechanical properties, such as extremely high fracture stress (12.8 ± 0.7 MPa) and fracture strain (1719 ± 77%), excellent modulus (4.51 ± 0.76 MPa), high work of fracture (134.47 ± 9.29 MJ m−3), and fracture toughness (305.04 kJ m−2) compared with other strong hydrogels and even natural tendons. In addition, excellent conductivity, strain sensing capability, water retention, freezing resistance, swelling resistance, and biocompatibility can also be achieved. This work provides a new and effective method to fabricate multifunctional anisotropic hydrogels with high tunable strength and toughness with potential applications in the fields of regenerative medicine, flexible sensors, and soft robotics.
New conceptsA variety of anisotropic hydrogels with excellent mechanical properties are currently available; however, there are no satisfactory anisotropic hydrogels that meet all the critical requirements of high strength, high toughness, stretchability, transparency, biocompatibility and environmental-stability. Here, we present a new facile method to fabricate highly anisotropic fiber-based hydrogels with all necessary characteristics. The simple method includes oriented wet spinning, soaking and cyclic freezing treatment and the mechanical properties of the obtained hydrogels can be fine-tuned according to the actual requirements. The mechanical properties of the fiber-based hydrogel were pushed up to a high level, whose tensile strength and toughness along the L direction are higher than those of most reported hydrogels to date. As an engineering implementation, we further demonstrate its electrical properties and regulate its electrical conductivity and strain sensing ability by introducing metal ions, which offers superior sensitivity and cycle stability than conventional hydrogels. We believe that our work will inspire further studies in the development of anisotropic hydrogel fields and biological fields such as artificial soft tissue materials and biosensors. |
Introduction
Most natural tissues, such as ligaments, muscles, cartilage and tendons, have highly ordered hierarchical microstructures and exhibit excellent anisotropic mechanical properties, of which the tensile strength is around 10 MPa.1–3 To mimic natural tissues, hydrogels are one of the best candidates due to their excellent flexibility, biocompatibility, and high water content, which can be applied for biomedical and bioengineering applications, such as drug delivery,4,5 soft actuators,6–11 electrochemical devices,12–16 and artificial muscles.17,18 However, most traditional hydrogels tend to have a uniformly disordered network structure and loose crosslinking, resulting in poor mechanical properties and unordered homogeneous structures, which severely hinder their practical applications. Tremendous improvements have been made to improve their mechanical performance by introducing various reinforcing materials19–22 or double-networks.23–31 The mechanical properties of hydrogels with double-networks (DN) can be effectively improved, because they are prepared by combining brittle, rigid and tightly crosslinked networks with the soft, tough and loosely one inspired by living organisms.23–31 However, due to the lack of ordered reinforcing structures, the mechanical properties of these improved isotropic hydrogels are still very weak compared to that of natural tissues.In recent years, a variety of anisotropic hydrogels with enhanced mechanical properties have been prepared by means of directional freezing,32–34 magnetic or electric field,35,36 self-assembly37 and mechanical stretching.38–41 For example, Hua et al. produced a highly anisotropic PVA hydrogel using a freezing-assisted salting-out treatment, which exhibits extremely high strength, toughness and fatigue resistance at high water contents.42 Wei et al. developed anisotropic gel belts via dynamic stretching of hydrogel fibers, which exhibit high elastic moduli and unique anisotropic swelling behaviors.43 In addition, it is found that forming a hierarchically anisotropic hydrogel with similar a fiber and matrix would be promising for making hydrogels with simultaneously high strength, toughness, stretchability and fatigue resistance.42,44,45 Nevertheless, most of these anisotropic hydrogels are still not as strong and tough as natural tissues, thus greatly limiting their application potential in regenerative medicine and tissue engineering. Some anisotropic hydrogels have excellent mechanical properties comparable to those of biological tissues but their transparency or biocompatibility is limited.42,44,45 In addition, the currently used methods for preparing anisotropic hydrogels, such as directional freezing and mechanical cutting,44,45 are usually complex and cannot achieve mass production. Therefore, it remains challenging to create simultaneously strong, tough, stretchable, transparent, biocompatible and environment-adaptable hydrogels with anisotropic structures using a generic and facile approach.
Herein, we explored a new facile method, namely a novel solvent–exchange-assisted wet-stretching strategy, to develop anisotropic fiber-based multifunctional hydrogels with excellent mechanical properties even superior to biological tissues. First, the continuous and uniform PVA fibers prepared by wet spinning were directionally collected to form a tightly arranged fiber aggregate with a certain direction to obtain the anisotropic structure. Secondly, taking advantage of the water solubility and hygroscopicity of PVA, the aggregate of PVA fibers was soaked and swollen in PVA/glycerin aqueous solution to regulate the mechanical properties and simultaneously obtain water retention and anti-freezing properties. Finally, after classical freeze–thaw cycles, an anisotropic fiber-based hydrogel with excellent tunable mechanical properties, certain transparency, good biocompatibility, and long-term environmental stability was obtained. In the traditional dry-stretching, the solid state of the polymer network limited the conformational adjustment of the macromolecular chains, indicating the potential room for further improvement of the mechanical properties of the hydrogel. By contrast, the wet-stretching provided a freer and looser environment for macromolecular movement and allowed construction of a mechanically robust polymer network. The combination of solvent exchange and wet-stretching enabled the hydrogels to have an extremely high tensile strength of 12.8 ± 0.7 MPa and a work of fracture of 134.47 ± 9.29 MJ m−3 with stretchability up to 1719 ± 77%. These superior mechanical properties were attributed to the synergy of the high crystallinity and hydrogen bonds in the hydrogels. The facile strategy is generalizable to other polymers and could expand the applicability of structural hydrogels to conditions involving more demanding mechanical loading.
Results and discussion
Construction of anisotropic structures
To mimic the anisotropic structure of human muscle, the wet spinning method was applied to fabricate uniform fibers using PVA as a model system (Fig. 1a). The obtained single PVA fiber had a microscopic anisotropic structure and excellent mechanical properties (Fig. S1, ESI†).46 This is contributed to the regulation of the rotating force produced by controlling the rotation speed of the coagulation bath and the ejection speed of the syringe, which makes the PVA fibers and molecular chains stretched and aligned. Then a macroscopic anisotropic structure was created by collecting fibers directionally on a rotating spool in circles. In the wet stretching process, the conformation of the macromolecules was adjusted via increased macromolecule interaction, indicating more crystalline domains (Fig. 1). Before swelling in impregnating solutions, the anisotropic fiber bundles were dried and a more compact structure in which the fibers are in closer contact with each other due to loss of water was obtained. Finally, through a solvent–exchange and freeze–thaw process, it was transformed to a strong and tough hydrogel with high crystallinity.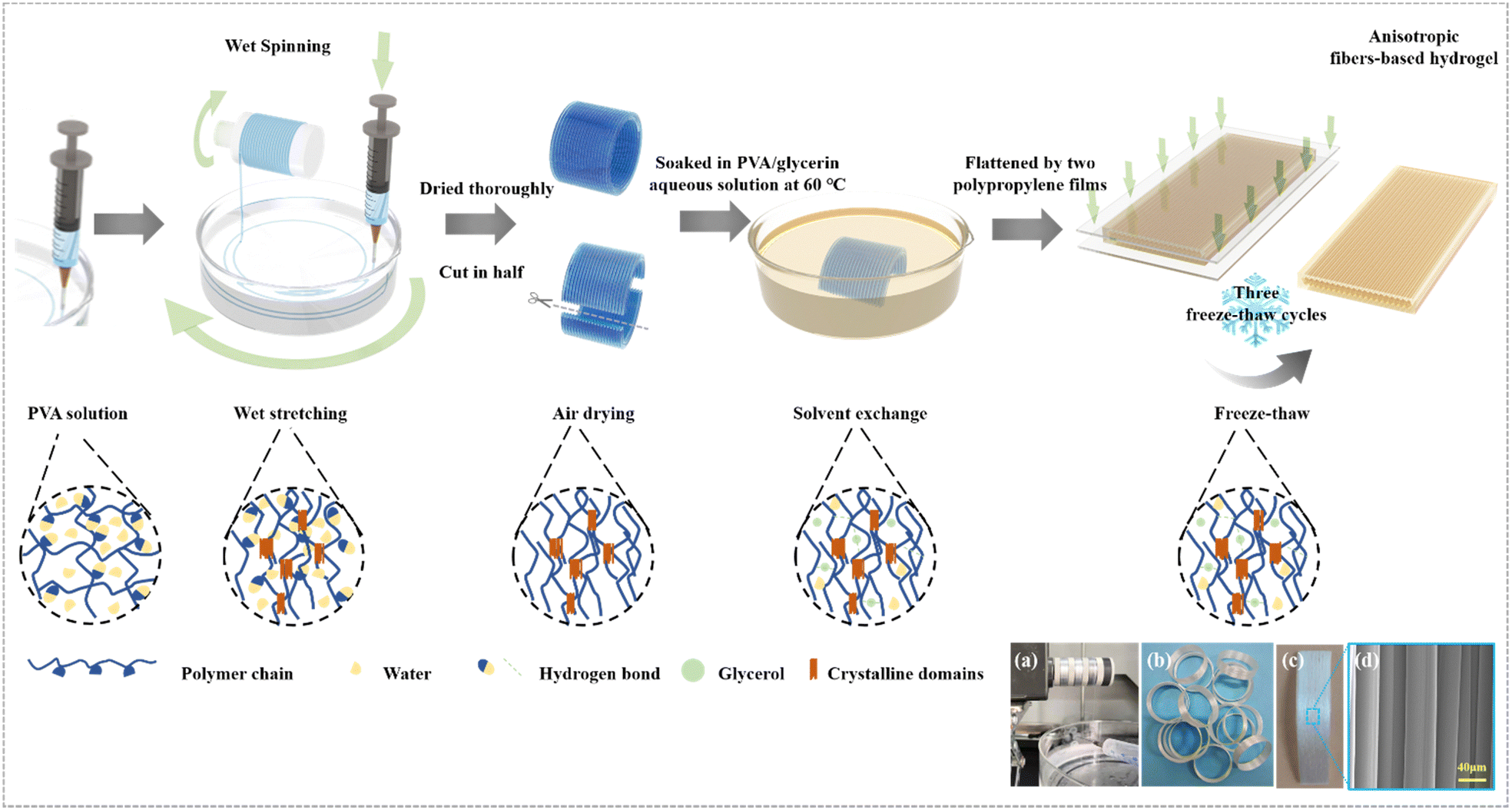 | ||
| Fig. 1 Design strategy. Schematic illustration of the preparation process of anisotropic fiber-based hydrogels. (a) The process of wet spinning (more details are shown in Movie S1, ESI†). (b) PVA fibers after directional collection. (c) PVA fibers were dried and cut after directional collection. (d) SEM image of PVA fibers after directional collection and drying. | ||
We studied the influence of the impregnating solutions on the swelling degree of the hydrogel. Various anisotropic hydrogels after being swollen were tested, and the PVA fiber impregnated by PVA/glycerol solution showed a smaller swelling degree of the PVA fiber impregnated by PVA solution. It was observed that the volume of the PF_10P50G hydrogel (PF denotes the PVA fiber, and xP and yG denote the weight fraction of PVA and glycerol in the impregnated aqueous solution, respectively) was smaller than that of PF_10P after soaking and cyclic freezing (Fig. 2a and b). Hydrogels with a broad tunable microstructure and mechanical properties can be obtained by different swelling effects caused by adjusting impregnating solutions.
 | ||
| Fig. 2 Morphology of hydrogels. Photographs of PF_10P (a) and PF_10P50G (b) hydrogels. Optical images of PF_10P (c) and PF_10P50G (d). Polarizing optical microscopic (POM) images of PF_10P (e) and PF_10P50G (f). SEM images of PF_10P (g) and (i) and PF_10P50G (h) and (j), and the corresponding magnification SEM images (the outlines of fibers are indicated by red dotted lines). Schematic illustration of the chemical structure of PF_10P (k) and PF_10P50G (l). | ||
The structures of anisotropic fiber-based hydrogels are shown in Fig. 2. We performed optical microscopy and polarized optical microscopy (POM) observation on the hydrogel samples to analyze their anisotropic structure and birefrigence. Great orientation of the PVA fiber can be observed in PF_10P and PF_10P50G (Fig. 2c and d). A lattice-like birefringence pattern was observed that can be related to the orientation of PVA molecules that have positive optical properties (Fig. 2e and f). Given the high transparency (Fig. S2, ESI†) of the hydrogel, this lattice-like birefringence is attributed to the overlapping of the several individual birefrigence patterns of the PVA fiber in the hydrogel, as the PVA fibers are aligned in one direction to each other. And PF_10P50G showed better oriented-crystallization than PF_10P. Meanwhile, more hydrogen bonds formed among the PVA molecular chains (Fig. 2k and l). This is due to the glycerol impregnating solution indicating its better mechanical performance.
SEM images in the cross and longitudinal-section exhibit totally different morphologies further conforming its anisotropic structure (Fig. 2g–j). The cross section of fibers was no longer deformed in a regular circular shape due to the interaction between the fibers during directional collection and drying shrinkage and the longitudinal-section images show the oriented layered structure of the hydrogels, which also demonstrate that the PVA fibers align in one direction, and the gaps between the fibers are filled with the hydrogels formed by the impregnating solution. Due to the presence of glycerol in the impregnating solution, the swelling degree of the fibers in the PF_10P50G hydrogel was relatively low, resulting in a dense network structure (Fig. 2h and j). No obvious holes were observed in the internal cross-section of the fibers, just some tiny holes were found between the fibers. For comparison, the PF_10P hydrogel exhibits structural features similar to double network hydrogels, with the dense network within the fibers and the loose network between the fibers (Fig. 2g and i). Due to the structural anisotropy, the prepared hydrogels exhibited significant anisotropic differences in mechanical properties. As shown in Fig. 3d, the mechanical properties of the hydrogel with a collection width of 32 mm were clearly superior in the longitudinal direction than in the transverse direction. However, due to the fiber reinforcement, even in the transverse direction, the mechanical properties of the hydrogels were still superior to most hydrogels reported in the literature,26,28,47,48 with a fracture stress of 1.5 MPa and a fracture strain of 124%. In summary, a unique hierarchical anisotropic structure of hydrogels can be formed through the proposed solvent–exchange-assisted wet-stretching strategy, which is crucial for simultaneously achieving high strength and high toughness (305.04 kJ m−2) of the hydrogel shown in Fig. S10 (ESI†).
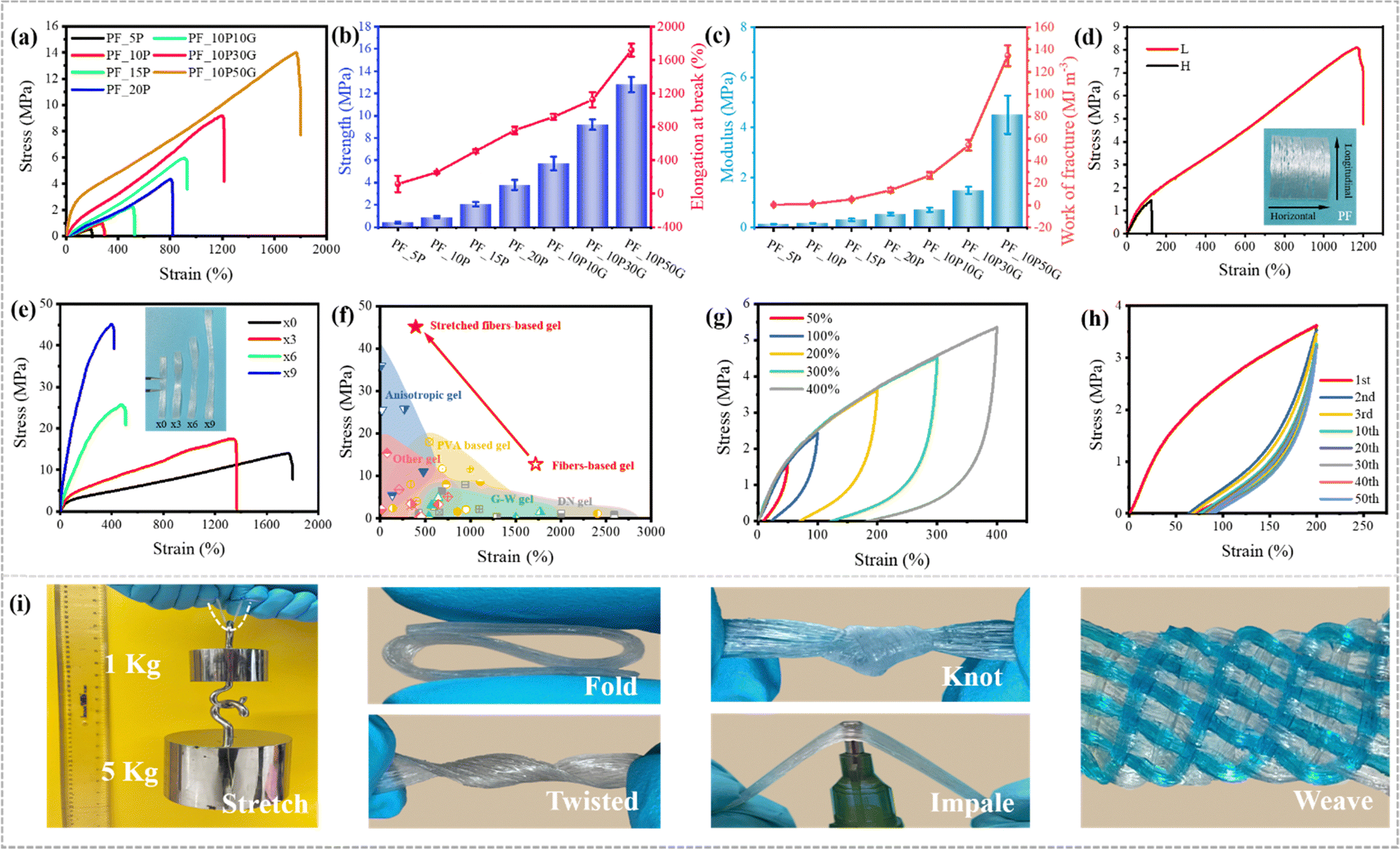 | ||
| Fig. 3 Mechanical properties. (a) Tensile stress–strain curves of the hydrogels prepared by impregnating PVA/glycerin aqueous solutions at different concentrations. (b) The tensile strength and elongation at break of various fiber-based hydrogels. (c) The elastic modulus and work of fracture of various fiber-based hydrogels. (d) The longitudinal and horizontal (along and perpendicular to the fiber orientation direction) tensile stress–strain curves of the PF_10P50G hydrogel. (e) Tensile stress–strain curves of PF_10P50G hydrogels after mechanical pre-stretching treatments. (f) Comparison between our work and other reported tough hydrogels, including anisotropic hydrogels,3,43,49–51 PVA based hydrogels,52–62 glycerol–water (G–W) hydrogels,47,48,63–65 double network (DN) hydrogels23–31 and other hydrogels.66–74 (g) Tensile loading–unloading stress–strain curves of the PF_10P50G hydrogel at various strains. (h) Tensile stress–strain curves of the PF_10P50G hydrogel for 50 successive tensile cycles. (i) Excellent strength and toughness of the PF_10P50G hydrogel were confirmed by stretching, folding, twisting, knotting, impaling and weaving (some samples were stained by methylene blue). | ||
Mechanical properties
We can tune the mechanical properties of the PVA hydrogels by changing their PVA ratio, impregnating solution, pre-stretching ratio and width (Fig. 3a–e). As control samples, the PVA hydrogels prepared by PVA impregnating solution alone or other different ratios of PVA/glycerol impregnating solutions showed lower strength, toughness and stretchability than the PF_10P50G hydrogel (Table S1 and Fig. 3b, c, ESI†). As shown in Fig. 3a, the properties of PF_10P were found to be considerably weak in comparison to PF_10P10G. Fig. S7 (ESI†) demonstrates the impact of PVA and glycerol on the mechanical properties. It is evident that the strength increased with an increase in the addition of PVA and glycerol individually. However, a substantial enhancement in fracture stress was observed when PVA and glycerol were introduced simultaneously. These outcomes underscore the importance of both PVA and glycerol. Moreover, the combination of PVA and glycerol (PVA–glycerol) exhibited greater significance in comparison to their individual counterparts. The tensile strength, elongation at break, modulus, and work of fracture of the PF_10P50G hydrogel are up to 12.8 ± 0.7 MPa, 1719 ± 77%, 4.51 ± 0.76 MPa, and 134.47 ± 9.29 MJ m−3, respectively, which are 14 times, 7 times, 24 times, and 93 times higher than those of the PF_10P hydrogel in turn. However, excess glycerol decreases the solubility of PVA, resulting in a heterogeneous impregnating solution, which ultimately adversely affects the performance of the hydrogel.To further optimize the mechanical properties of the hydrogels, the effects of width on the mechanical properties, as a critical issue, were investigated. As the width decreased, the collected fibers became denser, resulting in better orientation of the prepared hydrogels, which exhibited higher tensile stress and strain (Fig. S3, ESI†). Mechanical pre-stretching is an effective way to improve the degree of orientation. We pre-stretched the PF_10P50G hydrogel, and found that the tensile strength of the hydrogel gradually increased with the increase of the stretching ratio. When the stretching ratio was 9, the maximum tensile strength of the hydrogel reached an impressive 45 MPa at a strain of 394% (Fig. 3e), which is attributed to the fact that mechanical pre-stretching not only improved the orientation of the anisotropic fiber-based hydrogel but also further enhanced the internal interaction between the fibers. In a word, the mechanical properties of hydrogels were affected by the composition and content of impregnating solutions and the orientation of its internal structure. Among them, the PF_10P50G hydrogel and pre-stretched PF_10P50G hydrogel showed outstanding stress, strain, modulus and toughness, far superior to many reported tough hydrogels (Fig. 3f and Table S2, ESI†).
We further studied the reversibility of the fiber-based hydrogels by conducting multiple loading–unloading tests. Mechanical hysteresis was observed for the PF_10P50G hydrogel at different strains (Fig. 3g), which indicated the presence of sacrificial bonds that broke during deformation. The dissipative energy increased with the strain. PF_10P50G showed dissipative energy as high as 0.21 MJ m−3 and 11.97 MJ m−3, respectively, when the strain increased from 50% to 400%. In addition, the energy dissipation rate was 50.3% and 86.9%, respectively. The tensile cycles of PF_10P50G was tested at a fixed strain (200%) with 50 cycles (Fig. 3h). A clear hysteresis was observed in the first cycle, indicating that the network structure of the hydrogel changed during this process. While in the second cycle, the loop area decreased significantly. After more than 20 cycles, the hysteresis curves overlapped and no obvious loop area decay was observed, indicating that the PF_10P50G hydrogel has good cycle stability. This also indicated that the most sacrificial bonds (ionic bonds and hydrogen bonds) were reversible. All the ionic bonds, hydrogen bonds, and the network entanglement of PVA chains and fibers are contributed to toughen the hydrogels.
Remarkably, the high strength and flexibility of the PF_10P50G hydrogel were demonstrated under stretching, folding, twisting, knotting, puncturing and weaving (Fig. 3i). We can easily lift a weight of 6 kg (more than 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 times of the hydrogel) by the hydrogel with a width of 8 mm and a thickness of 0.8 mm. The as-prepared hydrogels can withstand various deformations such as folding, twisting, knotting and puncturing, indicating their superior flexibility, stiffness and toughness. In addition, the prepared anisotropic fiber-based hydrogels can be woven into complex patterns and structures without rupturing, reducing their limitations in different applications.
000 times of the hydrogel) by the hydrogel with a width of 8 mm and a thickness of 0.8 mm. The as-prepared hydrogels can withstand various deformations such as folding, twisting, knotting and puncturing, indicating their superior flexibility, stiffness and toughness. In addition, the prepared anisotropic fiber-based hydrogels can be woven into complex patterns and structures without rupturing, reducing their limitations in different applications.
To further reveal the mechanism of their excellent mechanical property, Fourier transform infrared (FTIR) spectroscopy was conducted. In the FTIR spectra (Fig. S5, ESI†), the absorption peak at 1086 cm−1 representing C–O of PVA was related to the formation of a crystalline domain. The PF_10P50G_0.5Na and PF_10P50G hydrogels exhibited larger peak intensity than PF_10P and PF, suggesting that the solvent–exchange strategy formed more crystalline domains. The glycerol and water introduced more noncovalent interactions in the PVA network of fibers as well as in the PVA network between the fibers, which is also confirmed by the FTIR tests (Fig. S5, ESI†), and these noncovalent interactions act as sacrificial bonds to effectively dissipate the external energy during the deformation process, thus greatly improving the strength and toughness of the hydrogel (Fig. 4a).
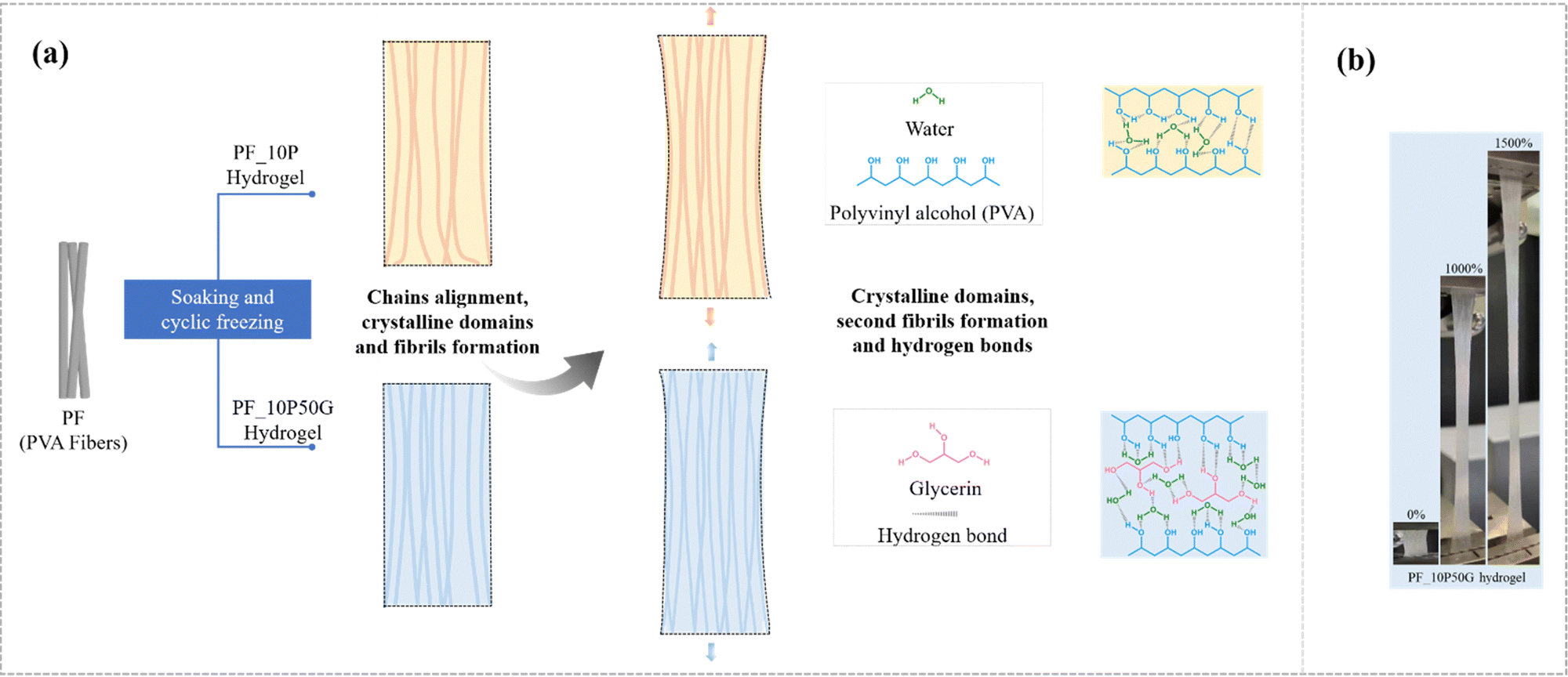 | ||
| Fig. 4 (a) Toughening mechanisms of the anisotropic fiber-based hydrogel. (b) The state of the PF_10P50G hydrogel during stretching (more details are shown in Movie 2, ESI†). | ||
The excellent mechanical properties of the PF_10P50G hydrogel including high strength and toughness may originate from crystallinity and stretching-induced chain alignment, which are schematically illustrated in Fig. 4(a). The PVA fibers have good mechanical properties and reinforcing effects (Fig. S1, ESI†) due to their oriented molecular chains, which promotes stress transfer between individual fibers and greatly improves the mechanical properties of the hydrogel along the fiber direction. As mentioned, wet-stretching can induce possible conformation of macromolecules and enhanced crystallinity in the crystalline region. In the crystalline regions, the dense crystalline domains recognized as tight crosslinkers physically strengthen the PVA fiber hydrogel. The higher the crystallinity, the larger the stress required to break the arranged macromolecular chains42 (Fig. 4a). And the higher the fiber content, the better the mechanical properties of hydrogels (Fig. S6, ESI†). Compared with the PVA solution, the PVA–glycerin solution as the impregnating solution reduces the swelling degree of the PVA fiber bundles (as shown in Fig. S8, ESI†), making the internal network of the fibers denser and the contact between the fibers tighter, resulting in stronger interface adhesion and molecular chain entanglement (Fig. 4a). In addition, macromolecular chains in both amorphous and crystalline regions tend to align during wet-stretching, decreasing the distance between the individual chains and thus facilitating the formation of dense nanofibrils, which can effectively enhance the strength and toughness of hydrogels. PVA impregnating solution can act as an interfacial adhesive providing better interfacial properties, indicating improving mechanical properties. By contrast, when PVA was not present in the impregnating solution, the fiber-based hydrogels became loose due to the lack of interaction between fibers, resulting in poor mechanical properties (Fig. S9, ESI†). For example, the PF_50G hydrogel exhibited a progressive failure mode during stretching, characterized by progressive fracture fibers. At the micro-scale, the addition of glycerol results in the formation of hydrogen interaction with the PVA chain. These hydrogen bonds effectively crosslink the PVA chains and promote their binding, resulting in higher strength, modulus, and toughness of PF. Meanwhile, hydrogen bonds (introduced by glycerol) can facilitate dynamic fracture and re-crosslinking, allowing the fractured bonds to reconnect. This also contributes to the improved mechanical properties especially the fracture resistance and toughness of PF. Moreover, glycerol is able to improve the effective crosslinking density of the hydrogels,75 and the hydrogen bond zones can be utilized to achieve crack propagation insensitivity.76 At the macro-scale, the PVA matrix, as a binder, can connect the PVA fiber together, facilitating its fracture stress. And the PVA fiber reinforced the PVA matrix structure, and can also promote its mechanical property to some extent.
Environmental stability and biocompatibility
In addition to the relatively weak mechanical properties, the poor long-term stability is another critical issue for hydrogel materials, which makes it difficult to cope with changes in the daily environment. Many attempts have been made to improve the working temperature range of hydrogels,77–79 in which the introduction of organic solvents or inorganic salts into the hydrogel system can effectively improve their water retention and anti-freezing performance.80–83 Glycerol is a well-known non-toxic antifreeze with hygroscopic and anticorrosive properties, which can form strong hydrogen bonds with water molecules to disrupt ice lattice formation at low temperatures and prevent water evaporation at high temperatures.47,48,63–65 And glycerol has been used to endow hydrogels with excellent anti-freezing and anti-drying properties.84,85 Although significant progress has been made in the field of anisotropic hydrogels, it is still very challenging to construct mechanically excellent anisotropic hydrogels that are environmentally adaptable.Thus, glycerol was used to construct environmentally adaptable anisotropic hydrogels. Strong hydrogen bonds between water, glycerin and PVA ensure that free water molecules do not evaporate easily (water retention capacity) and inhibit free water molecules from freezing at low temperatures (anti-freezing performance). Both water retention performance and anti-freezing performance of hydrogels are important in response to environmental changes during daily use. To evaluate the water retention capacity, the hydrogels were placed in an environment with a constant temperature and humidity for 12 days (T = 25 °C, RH = 50%). In Fig. 5a, both PF_10P50G and PF_10P50G_0.5Na hydrogels show excellent water retention capacity due to the presence of glycerol. On the fourth day, their dehydration did not exceed 12% and 10%, respectively, far below 70% (dehydration of the PF_10P hydrogel). Even on day 12, PF_10P50G showed no obvious change in appearance, while PF_10P showed significant shrinkage and volume reduction. The TGA results (Fig. S11, ESI†) showed that the weight loss rates of PF_10P50G and PF_10P50G_0.5Na were significantly lower than that of PF_10P, which further confirms that glycerin improved the water retention performance of the hydrogels.
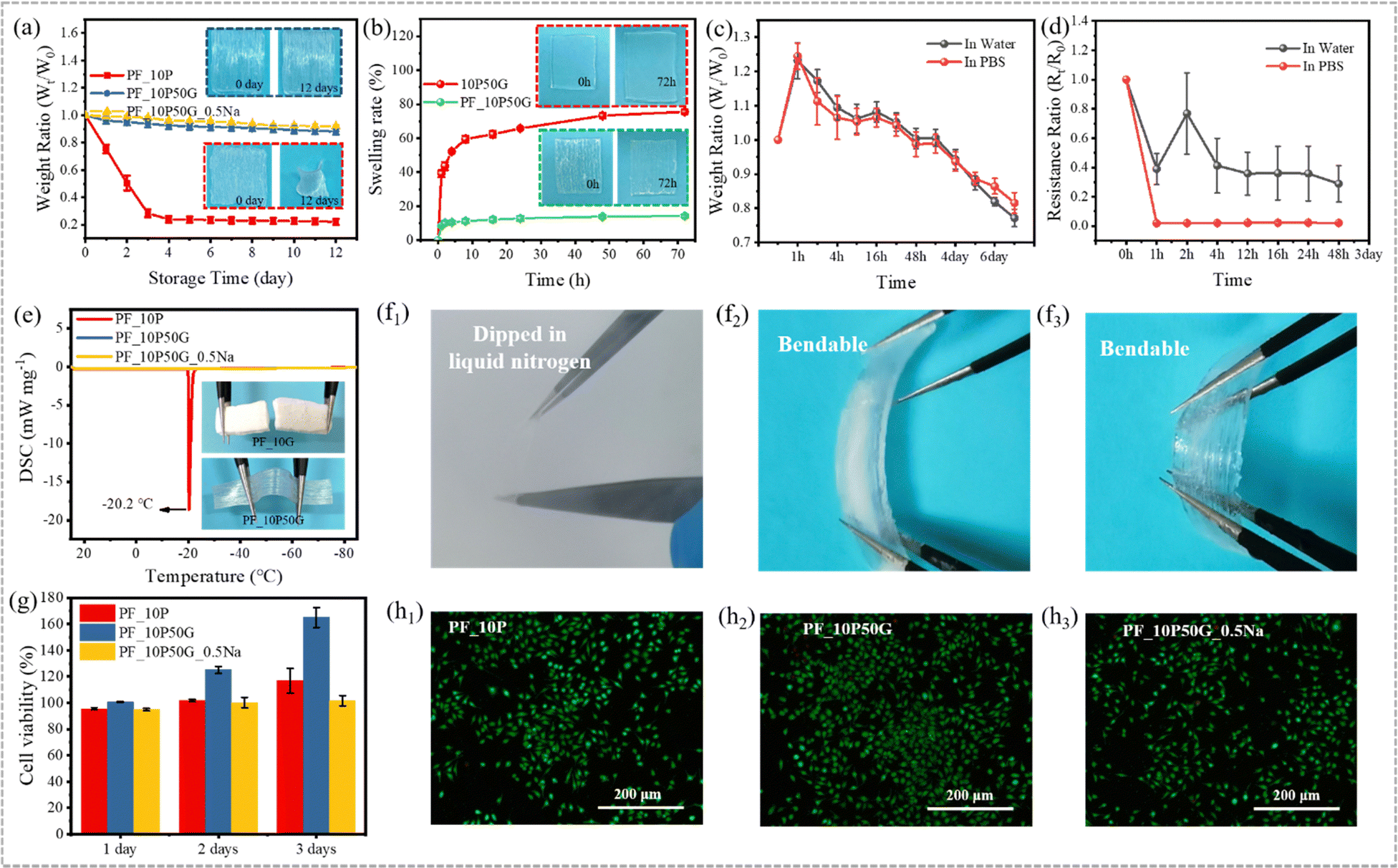 | ||
| Fig. 5 Environmental stability and biocompatibility. (a) The water retention capacity characterized by weight changes of hydrogels in an environmental chamber with 50% relative humidity (RH) at 25 °C for 12 days. (b) The anti-swelling performance characterized by weight changes of 10P50G and PF_10P50G hydrogels soaked in DI water for 72 h. (c) The weight ratio of PF_10P50G hydrogels soaked in DI water and PBS for 7 days. (d) The resistance ratio of PF_10P50G hydrogels soaked in DI water and PBS for 48 h. (e) The anti-freezing performance characterized by differential scanning calorimetry (DSC) of PF_10G, PF_10P50G and PF_10P50G_0.5Na hydrogels from −85 °C to 25 °C. (f1)–(f3) The state of PF_10P50G hydrogels after being dipped in liquid nitrogen. (g) The cell viability of L929 cells cultured in hydrogel leaching media after 1, 2 and 3 days. Confocal fluorescence microscopy images of cells cultured in PF_10G (h1), PF_10P50G (h2) and PF_10P50G_0.5Na (h3) hydrogel leaching media for 3 days. | ||
The excellent anti-swelling properties endow the hydrogels with a range of in vivo applications, such as drug carriers and bone repair materials. In Fig. 5b, the 10P50G hydrogel swelled rapidly in water due to the high water-absorption of PVA. After soaking in water for 1 h, the swelling rate reached 40%, and then slowed down until the maximum swelling rate reached 76% after 72 h. However, the swelling rate of PF_10P50G in water was always less than 15%, which contributes to its large number of tightly arranged fibers and the interaction between fibers. As shown in Fig. 5b, the volume of PF_10P50G remained basically unchanged, while the volume of 10P50G increased significantly, which demonstrate its excellent anti-swelling properties intuitively. The swelling behavior of hydrogels is related to their hydrophilic network and internal structure. To further evaluate the anti-swelling properties of PF_10P50G hydrogels, we tested their weight ratio and resistance ratio after placing the PF_10P50G hydrogels into water and PBS solution. The weight ratio increased in the first hour and then deceased until the 7 day (Fig. 5c). Nevertheless, the weight ratio is still higher than 0.75 on the 7th day, indicating its great anti-swelling properties. In contrast, the resistance deceased suddenly and then tended to be steady until the 2 day (Fig. 5d), suggesting its stability in water and PBS solution.
To evaluate the anti-freezing performance of hydrogels, differential scanning calorimetry (DSC) was conducted to evaluate their anti-freezing performance qualitatively and the results are shown in Fig. 5e. The PF_10P50G and PF_10P50G_0.5Na hydrogels exhibited no exothermic peaks in the temperature range from 25 °C to −85 °C, indicating that both had good freezing resistance, which is attributed to the glycerol. While for comparison, PF_10P showed an exothermic peak at −20.2 °C, indicating poor anti-freezing performance, which was caused by the freezing of free water molecules. To demonstrate the excellent freezing resistance more intuitively, the hydrogels were placed at −20 °C for 24 h. And it was found that PF_10P50G hardly changed and still showed good flexibility even after 24 h, while PF_10P was frozen, becoming brittle and difficult to stretch and deform (Fig. 5e). In addition, PF_10P50G hydrogels were dipped in liquid nitrogen and it was found that the hydrogels are still bendable (Fig. 5f1–f3), indicating excellent anti-freezing performance.
The biocompatibility of anisotropic fiber-based hydrogels was tested by the MTS assay to explore their application potentials as artificial soft tissues. As shown in Fig. 5g, the PF_10P, PF_10P50G and PF_10P50G_0.5Na hydrogels can promote the production and proliferation of L929 cells on day 1, day 2 and day 3, and the cell viability was higher than 94%. Compared with PF_10P and PF_10P50G_0.5Na, PF_10P50G had a significant promoting effect on cell proliferation. The calcein-AM/ETHD-I double staining method was used to demonstrate the effects of hydrogels on cell proliferation. The fluorescence microscopy images show that a large number of live cells with green fluorescence were observed and just a small number of dead cells with red fluorescence existed, indicating that L929 cells can successfully adhere and proliferate on the surface of the fiber-based hydrogels (Fig. 5g and h1–h3). The results indicate that the anisotropic fiber-based hydrogels had good biocompatibility and might be used in biological applications.
Electrical properties
As a muscle-inspired material, the fiber-based hydrogel not only exhibited excellent environmental stability and mechanical properties, but also demonstrates excellent strain-sensing performance, suggesting potential bio-applications. Both the PF_10P50G_Na hydrogel and 10P50G_Na hydrogel were prepared by adding a small amount of NaCl to the impregnating solution, demonstrating good ion transport capacity. The ionic conductivity of the hydrogels increased gradually with the increase of the NaCl content (Fig. S12, ESI†). However, the ionic conductivity of PF_10P50G_Na was lower than that of 10P50G directly prepared from its impregnating solution. This may be due to the fact that the incompletely oriented fibers inside the anisotropic fiber-based hydrogels hindered the ion transport channel and reduced its ionic conductivity. Nevertheless, as shown in Fig. 6a, PF_10P50G_0.5Na can be used as the wire to light up a LED light, and the brightness of the LED light gradually decreased with the increase of the stretching length of the hydrogel, indicating that its resistance gradually increased during the stretching process.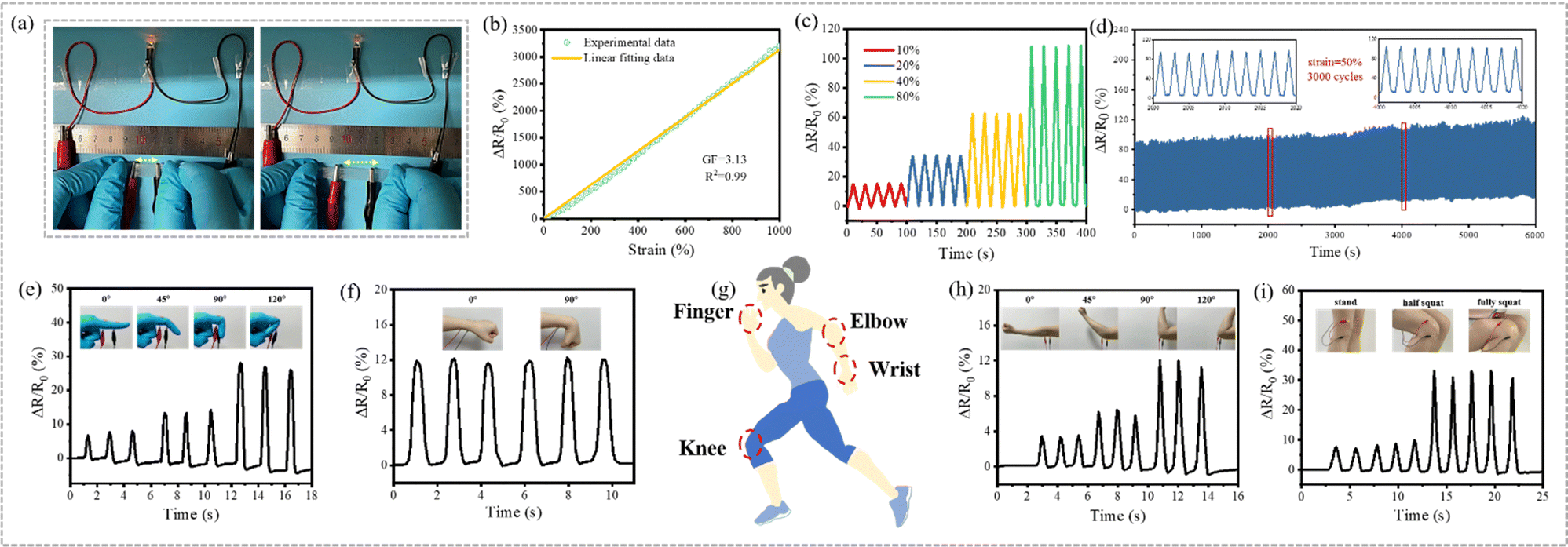 | ||
| Fig. 6 Electrical properties. (a) The PF_10P50G_0.5Na hydrogel was used as a wire to light up LED lights (more details are shown in Movie 3). (b) The relative change of resistance of the PF_10P50G_0.5Na hydrogel. (c) The relative change of resistance of the PF_10P50G_0.5Na hydrogel under various tensile strains (10%, 20%, 40%, and 80%). (d) The relative change of resistance of the PF_10P50G_0.5Na hydrogel during 3000 cycles of continuous loading and unloading. (g) Functional demonstration of the PF_10P50G_0.5Na hydrogel as a biosensor. The relative change of resistance of the PF_10P50G_0.5Na hydrogel used as a biosensor to monitor real-time signals of human body movement including the bending of fingers (e), wrists (f), elbows (h) and knees (i). | ||
As strain sensors, the PF_10P50G_Na and 10P50G_Na hydrogels with the same NaCl content show nearly uniform RCR (the relative change of resistance) in the range of 200% tensile strain. However, PF_10P50G_Na hydrogels demonstrate a larger response range (up to 1000%) than the 10P50G_Na hydrogel which exhibits poor mechanical properties due to the absence of PVA fibers (Fig. S13, ESI†). Among the PF_10P50G hydrogels with different NaCl contents in the impregnating solution, the PF_10P50G_0.5Na hydrogel with 0.5 wt% NaCl exhibits the best sensing performance (Fig. 6b), with a sensitivity (GF) of 3.13 and a corresponding linearity (R2) of 0.999 over the strain range of 0–1000%, higher than that of the most reported hydrogel-strain sensor.60,63,77,78,86
To further demonstrate its strain-sensing performance, the response times were investigated. As shown in Fig. S14 (ESI†), the PF_10P50G_0.5Na hydrogel sensor was stretched and released at 30% strain in a short time. Obviously, the sensor had negligible hysteresis, and the response times during the loading and unloading were 22 ms and 16 ms, respectively. The response stability at different strains was also evaluated, and it was found that the PF_10P50G_0.5Na hydrogel sensor can accurately identify the strain of 10%-80% and shows excellent reproducibility and reliability under the cyclic strain (Fig. 6c). In addition, PF_10P50G_0.5Na hydrogels exhibited good dynamic stability in 3000 continuous loading–unloading cyclic stretching under 50% strain (Fig. 6d). In summary, the PF_10P50G_0.5Na hydrogels, as a strain sensor, had many advantages such as high sensitivity, good stability and a wide detection range of strain. Thus, we used the PF_10P50G_0.5Na hydrogel as a biosensor to monitor real-time signals of human body movements, including bending of fingers, wrists, elbows and knees (Fig. 6e–i). As the body parts bent at different angles, the hydrogel sensors deformed, and the motion signals were converted into corresponding electrical signals. The signals were repeatable and clear, indicating that the hydrogels have great potential as biological sensors. In addition, the PF_10P50G hydrogel showed higher transmittance than the PF_10P hydrogel (Fig. S2, ESI†), which facilitates their applications in electro-optical fields, such as control panels and flexible displays.
Conclusions
We have reported a synergistic strategy of solvent–exchange-assisted wet-stretching for developing a strong, tough, highly anisotropic fiber-based hydrogel inspired by muscles. This strategy breaks through the limitations of the existing dry-stretching method on the adjustment of polymer conformation and crystallization simultaneously improving the strength and toughness. Considering that the design concept of solvent–exchange-assisted wet-stretching can be applied for various polymers, we are convinced that the present strategy is versatile. In the future, the explanation of fiber-based hydrogel's excellent mechanical properties would be further investigated, and the mimicking-hierarchical structure of human muscles can be extended to different scales. As a proof-of-concept, we demonstrate that the fiber-based hydrogel can be used as a biosensor benefiting from its excellent flexibility, strain sensing capability, long-term stability, and biocompatibility. It shows excellent linearity and the gauge factor is 3.13 in a wide range of 1000%. These merits make the anisotropic fiber-based hydrogels highly attractive for a wide range of applications including biological fields such as artificial soft tissue materials and biosensors for human movement monitoring.Experimental section
Experimental details are provided in the ESI.†Author contributions
S. W., L. L., M. H. N., N. H. and J. L., conceived the idea, designed the study, and wrote the paper. L. L. and S. W. performed the experiments. All the authors analyzed and discussed the results.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant No. 12302192, 12227801, 32300666, and 52305602), the National Key Research and Development Program of China (2019YFC0840709), the Fund for Innovative Research Groups of Natural Science Foundation of Hebei Province (A2020202002), the Key Program of Research and Development of Hebei Province (202030507040009), the Natural Science Foundation of Chongqing (cstc2021jcyj-msxmX0199, cstc2021jcyj-msxmX0241, and cstb2023nscq-msx0303), the Fundamental Research Funds for the Central Universities (Grant No. SWU-KQ22025), the Science and Technology Research Program of Chongqing Municipal Education Commission (Grant No. KJQN202300222), the Xinjiang Production and Construction Corps Regional Innovation Guidance Program (2022BB004), and the Hebei Province Military-civilian Integration Science and Technology Innovation Project (SJMYF2022X15). With this acknowledgement, we would like to thank Junchao Huang (Lanzhou University) for his helpful conversations on the creation and modification of the work.References
- H. Fan and J. P. Gong, Fabrication of bioinspired hydrogels: challenges and opportunities, Macromolecules, 2020, 53(8), 2769–2782 CrossRef CAS.
- S. Ling, D. L. Kaplan and M. J. Buehler, Nanofibrils in nature and materials engineering, Nat. Rev. Mater., 2018, 3(4), 18016 CrossRef CAS PubMed.
- L. Geng, S. Hu, M. Cui, J. Wu, A. Huang, S. Shi and X. Peng, Muscle-inspired double-network hydrogels with robust mechanical property, biocompatibility and ionic conductivity, Carbohydr. Polym., 2021, 262, 117936 CrossRef CAS PubMed.
- P. Wang, J. Sun, Z. Lou, F. Fan, K. Hu, Y. Sun and N. Gu, Assembly-Induced Thermogenesis of Gold Nanoparticles in the Presence of Alternating Magnetic Field for Controllable Drug Release of Hydrogel, Adv. Mater., 2016, 28(48), 10801–10808 CrossRef CAS PubMed.
- C. Ma, Y. Shi, D. A. Pena, L. Peng and G. Yu, Thermally responsive hydrogel blends: a general drug carrier model for controlled drug release, Angew. Chem., 2015, 127(25), 7484–7488 CrossRef.
- B. P. Lee and S. Konst, Novel hydrogel actuator inspired by reversible mussel adhesive protein chemistry, Adv. Mater., 2014, 26(21), 3415–3419 CrossRef CAS PubMed.
- M. K. Shin, G. M. Spinks, S. R. Shin, S. I. Kim and S. J. Kim, Nanocomposite hydrogel with high toughness for bioactuators, Adv. Mater., 2009, 21(17), 1712–1715 CrossRef CAS.
- H. Yuk, S. Lin, C. Ma, M. Takaffoli, N. X. Fang and X. Zhao, Hydraulic hydrogel actuators and robots optically and sonically camouflaged in water, Nat. Commun., 2017, 8(1), 1–12 CrossRef PubMed.
- Z. Chen, J. W. To, C. Wang, Z. Lu, N. Liu, A. Chortos, L. Pan, F. Wei, Y. Cui and Z. Bao, A three-dimensionally interconnected carbon nanotube-conducting polymer hydrogel network for high-performance flexible battery electrodes, Adv. Energy Mater., 2014, 4(12), 1400207 CrossRef.
- S. J. Benight, C. Wang, J. B. Tok and Z. Bao, Stretchable and self-healing polymers and devices for electronic skin, Prog. Polym. Sci., 2013, 38(12), 1961–1977 CrossRef CAS.
- B. C. Tee, C. Wang, R. Allen and Z. Bao, An electrically and mechanically self-healing composite with pressure and flexion-sensitive properties for electronic skin applications, Nat. Nanotechnol., 2012, 7(12), 825–832 CrossRef CAS PubMed.
- Y. Zhao, B. Liu, L. Pan and G. Yu, 3D nanostructured conductive polymer hydrogels for high-performance electrochemical devices, Energy Environ. Sci., 2013, 6(10), 2856–2870 RSC.
- Y. Shi, L. Pan, B. Liu, Y. Wang, Y. Cui, Z. Bao and G. Yu, Nanostructured conductive polypyrrole hydrogels as high-performance, flexible supercapacitor electrodes, J. Mater. Chem. A, 2014, 2(17), 6086–6091 RSC.
- Y. Shi, C. Ma, L. Peng and G. Yu, Conductive “smart” hybrid hydrogels with PNIPAM and nanostructured conductive polymers, Adv. Funct. Mater., 2015, 25(8), 1219–1225 CrossRef CAS.
- Y. Shi, L. Peng and G. Yu, Nanostructured conducting polymer hydrogels for energy storage applications, Nanoscale, 2015, 7(30), 12796–12806 RSC.
- Y. Shi, M. Wang, C. Ma, Y. Wang, X. Li and G. Yu, A conductive self-healing hybrid gel enabled by metal–ligand supramolecule and nanostructured conductive polymer, Nano Lett., 2015, 15(9), 6276–6281 CrossRef CAS PubMed.
- J. Liu, C. S. Y. Tan, Z. Yu, Y. Lan, C. Abell and O. A. Scherman, Biomimetic supramolecular polymer networks exhibiting both toughness and self-recovery, Adv. Mater., 2017, 29(10), 1604951 CrossRef PubMed.
- Z. Liu and P. Calvert, Multilayer hydrogels as muscle-like actuators, Adv. Mater., 2000, 12(4), 288–291 CrossRef CAS.
- K. Phogat, S. Kanwar, D. Nayak, N. Mathur, S. B. Ghosh and S. Bandyopadhyay-Ghosh, Nano-enabled poly(vinyl alcohol) based injectable bio-nanocomposite hydrogel scaffolds, J. Appl. Polym. Sci., 2020, 137(23), 48789 CrossRef CAS.
- W. Yang, E. Fortunati, F. Bertoglio, J. S. Owczarek, G. Bruni, M. Kozanecki, J. M. Kenny, L. Torre, L. Visai and D. Puglia, Polyvinyl alcohol/chitosan hydrogels with enhanced antioxidant and antibacterial properties induced by lignin nanoparticles, Carbohydr. Polym., 2018, 181, 275–284 CrossRef CAS PubMed.
- P. Pirahmadi, M. Kokabi and G. Alamdarnejad, Polyvinyl alcohol/chitosan/carbon nanotubes electroactive shape memory nanocomposite hydrogels, J. Appl. Polym. Sci., 2020, 138(11), 1–17 Search PubMed.
- W. Chen, N. Li, Y. Ma, M. L. Minus, K. Benson, X. Lu, X. Wang, X. Ling and H. Zhu, Superstrong and Tough Hydrogel through Physical Cross-Linking and Molecular Alignment, Biomacromolecules, 2019, 20(12), 4476–4484 CrossRef CAS PubMed.
- L. Zhou, X. Pei, K. Fang, R. Zhang and J. Fu, Super tough, ultra-stretchable, and fast recoverable double network hydrogels physically crosslinked by triple non-covalent interactions, Polymer, 2020, 192 Search PubMed.
- Y. Zhang, M. Li, X. Han, Z. Fan, H. Zhang and Q. Li, High-strength and highly electrically conductive hydrogels for wearable strain sensor, Chem. Phys. Lett., 2021, 769 Search PubMed.
- Y. Wang, Y. Xue, J. Wang, Y. Zhu, Y. Zhu, X. Zhang, J. Liao, X. Li, X. Wu, Y. X. Qin and W. Chen, A Composite Hydrogel with High Mechanical Strength, Fluorescence, and Degradable Behavior for Bone Tissue Engineering, Polymers, 2019, 11(7), 1–12 Search PubMed.
- G. Su, S. Yin, Y. Guo, F. Zhao, Q. Guo, X. Zhang, T. Zhou and G. Yu, Balancing the mechanical, electronic, and self-healing properties in conductive self-healing hydrogel for wearable sensor applications, Mater. Horiz., 2021, 8(6), 1795–1804 RSC.
- X. Lu, L. Cao, X. Yin, Y. Si, J. Yu and B. Ding, Stretchable, tough and elastic nanofibrous hydrogels with dermis-mimicking network structure, J. Colloid Interface Sci., 2021, 582, 387–395 CrossRef CAS PubMed.
- Z. Jing, A. Xu, Y. Q. Liang, Z. Zhang, C. Yu, P. Hong and Y. Li, Biodegradable Poly(acrylic acid-co-acrylamide)/Poly(vinyl alcohol) Double Network Hydrogels with Tunable Mechanics and High Self-healing Performance, Polymers, 2019, 11(6), 952 CrossRef CAS PubMed.
- H. Yin, D. R. King, T. L. Sun, Y. Saruwatari, T. Nakajima, T. Kurokawa and J. P. Gong, Polyzwitterions as a Versatile Building Block of Tough Hydrogels: From Polyelectrolyte Complex Gels to Double-Network Gels, ACS Appl. Mater. Interfaces, 2020, 12(44), 50068–50076 CrossRef CAS PubMed.
- Y. Wang, D. Liang, Z. Suo and K. Jia, Synergy of noncovalent interlink and covalent toughener for tough hydrogel adhesion, Extreme Mech. Lett., 2020, 39, 100797 CrossRef.
- Y. N. Ye, M. Frauenlob, L. Wang, M. Tsuda, T. L. Sun, K. Cui, R. Takahashi, H. J. Zhang, T. Nakajima, T. Nonoyama, T. Kurokawa, S. Tanaka and J. P. Gong, Tough and Self-Recoverable Thin Hydrogel Membranes for Biological Applications, Adv. Funct. Mater., 2018, 28, 31 Search PubMed.
- Q. Luo, Y. Shan, X. Zuo and J. Liu, Anisotropic tough poly(vinyl alcohol)/graphene oxide nanocomposite hydrogels for potential biomedical applications, RSC Adv., 2018, 8(24), 13284–13291 RSC.
- X. Peng, C. He, J. Liu and H. Wang, Biomimetic jellyfish-like PVA/graphene oxide nanocomposite hydrogels with anisotropic and pH-responsive mechanical properties, J. Mater. Sci., 2016, 51(12), 5901–5911 CrossRef CAS.
- C. Su, Y. Su, Z. Li, M. A. Haq, Y. Zhou and D. Wang, In situ synthesis of bilayered gradient poly(vinyl alcohol)/hydroxyapatite composite hydrogel by directional freezing-thawing and electrophoresis method, Mater. Sci. Eng., C, 2017, 77, 76–83 CrossRef CAS PubMed.
- M. Liu, Y. Ishida, Y. Ebina, T. Sasaki, T. Hikima, M. Takata and T. Aida, An anisotropic hydrogel with electrostatic repulsion between cofacially aligned nanosheets, Nature, 2015, 517(7532), 68–72 CrossRef CAS PubMed.
- Q. L. Zhu, C. F. Dai, D. Wagner, M. Daab, W. Hong, J. Breu, Q. Zheng and Z. L. Wu, Distributed Electric Field Induces Orientations of Nanosheets to Prepare Hydrogels with Elaborate Ordered Structures and Programmed Deformations, Adv. Mater., 2020, 32(47), e2005567 CrossRef PubMed.
- H. Qin, T. Zhang, N. Li, H. P. Cong and S. H. Yu, Anisotropic and self-healing hydrogels with multi-responsive actuating capability, Nat. Commun., 2019, 10(1), 2202 CrossRef PubMed.
- R. Bai, J. Yang, X. P. Morelle and Z. Suo, Flaw-Insensitive Hydrogels under Static and Cyclic Loads, Macromol. Rapid Commun., 2019, 40(8), e1800883 CrossRef PubMed.
- K. Chen, Q. Chen, T. Zong, S. Liu, X. Yang, Y. Luo and D. Zhang, Effect of Directional Stretching on Properties of PVA-HA-PAA Composite Hydrogel, J. Bionic Eng., 2021, 18, 1202–1214 CrossRef.
- K. Chen, T. Zong, Q. Chen, S. Liu, L. Xu and D. Zhang, Preparation and characterization of polyvinyl alcohol/sodium alginate/carboxymethyl cellulose composite hydrogels with oriented structure, Soft Mater., 2021, 1–10 Search PubMed.
- M. T. I. Mredha, H. H. Le, V. T. Tran, P. Trtik, J. Cui and I. Jeon, Anisotropic tough multilayer hydrogels with programmable orientation, Mater. Horiz., 2019, 6(7), 1504–1511 RSC.
- M. Hua, S. Wu, Y. Ma, Y. Zhao, Z. Chen, I. Frenkel, J. Strzalka, H. Zhou, X. Zhu and X. He, Strong tough hydrogels via the synergy of freeze-casting and salting out, Nature, 2021, 590(7847), 594–599 CrossRef CAS PubMed.
- P. Wei, T. Chen, G. Chen, K. Hou and M. Zhu, Ligament-Inspired Tough and Anisotropic Fibrous Gel Belt with Programed Shape Deformations via Dynamic Stretching, ACS Appl. Mater. Interfaces, 2021, 13(16), 19291–19300 CrossRef CAS PubMed.
- C. Xiang, Z. Wang, C. Yang, X. Yao, Y. Wang and Z. Suo, Stretchable and fatigue-resistant materials, Mater. Today, 2020, 34, 7–16 CrossRef CAS.
- Z. Wang, C. Xiang, X. Yao, P. Le Floch, J. Mendez and Z. Suo, Stretchable materials of high toughness and low hysteresis, Proc. Natl. Acad. Sci. U. S. A., 2019, 116(13), 5967–5972 CrossRef CAS PubMed.
- C. Jing, W. Liu, H. Hao, H. Wang, F. Meng and D. Lau, Regenerated and rotation-induced cellulose-wrapped oriented CNT fibers for wearable multifunctional sensors, Nanoscale, 2020, 12(30), 16305–16314 RSC.
- Y. Wei, L. Xiang, H. Ou, F. Li, Y. Zhang, Y. Qian, L. Hao, J. Diao, M. Zhang, P. Zhu, Y. Liu, Y. Kuang and G. Chen, MXene-Based Conductive Organohydrogels with Long-Term Environmental Stability and Multifunctionality, Adv. Funct. Mater., 2020, 30(48), 2005135 CrossRef CAS.
- Z. You, Y. Dong, X. Li, P. Yang, M. Luo, Z. Zhu, L. Wu, X. Zhou and M. Chen, One-pot synthesis of multi-functional cellulose-based ionic conductive organohydrogel with low-temperature strain sensitivity, Carbohydr. Polym., 2021, 251, 117019 CrossRef CAS PubMed.
- Y. Wang, S. Liu and W. Yu, Bioinspired anisotropic chitosan hybrid hydrogel, ACS Appl. Bio Mater., 2020, 3(10), 6959–6966 CrossRef CAS PubMed.
- W. Kong, C. Wang, C. Jia, Y. Kuang, G. Pastel, C. Chen, G. Chen, S. He, H. Huang, J. Zhang, S. Wang and L. Hu, Muscle-Inspired Highly Anisotropic, Strong, Ion-Conductive Hydrogels, Adv. Mater., 2018, 30(39), e1801934 CrossRef PubMed.
- M. T. I. Mredha, Y. Z. Guo, T. Nonoyama, T. Nakajima, T. Kurokawa and J. P. Gong, A Facile Method to Fabricate Anisotropic Hydrogels with Perfectly Aligned Hierarchical Fibrous Structures, Adv. Mater., 2018, 30(9), 1704937 CrossRef PubMed.
- J. Yang, X. Yu, X. Sun, Q. Kang, L. Zhu, G. Qin, A. Zhou, G. Sun and Q. Chen, Polyaniline-Decorated Supramolecular Hydrogel with Tough, Fatigue-Resistant, and Self-Healable Performances for All-In-One Flexible Supercapacitors, ACS Appl. Mater. Interfaces, 2020, 12(8), 9736–9745 CrossRef CAS PubMed.
- X. Xiang, G. Chen, K. Chen, X. Jiang and L. Hou, Facile preparation and characterization of super tough chitosan/poly(vinyl alcohol) hydrogel with low temperature resistance and anti-swelling property, Int. J. Biol. Macromol., 2020, 142, 574–582 CrossRef CAS PubMed.
- J. Wang, Y. Lin, A. Mohamed, Q. Ji and H. Jia, High strength and flexible aramid nanofiber conductive hydrogels for wearable strain sensors, J. Mater. Chem. C, 2021, 9(2), 575–583 RSC.
- X. Sun, C. Luo and F. Luo, Preparation and properties of self-healable and conductive PVA-agar hydrogel with ultra-high mechanical strength, Eur. Polym. J., 2020, 124 Search PubMed.
- F. Lin, Z. Wang, J. Chen, B. Lu, L. Tang, X. Chen, C. Lin, B. Huang, H. Zeng and Y. Chen, A bioinspired hydrogen bond crosslink strategy toward toughening ultrastrong and multifunctional nanocomposite hydrogels, J. Mater. Chem. B, 2020, 8(18), 4002–4015 RSC.
- A. Li, Y. Si, X. Wang, X. Jia, X. Guo and Y. Xu, Poly(vinyl alcohol) Nanocrystal-Assisted Hydrogels with High Toughness and Elastic Modulus for Three-Dimensional Printing, ACS Appl. Nano Mater., 2019, 2(2), 707–715 CrossRef CAS.
- H. Jing, J. Shi, P. Guoab, S. Guan, H. Fu and W. Cui, Hydrogels based on physically cross-linked network with high mechanical property and recasting ability, Colloids Surf., A, 2021, 611 Search PubMed.
- Y. Guo, X. An and Z. Fan, Aramid nanofibers reinforced polyvinyl alcohol/tannic acid hydrogel with improved mechanical and antibacterial properties for potential application as wound dressing, J. Mech. Behav. Biomed. Mater., 2021, 118, 104452 CrossRef CAS PubMed.
- X. Di, Q. Ma, Y. Xu, M. Yang, G. Wu and P. Sun, High-performance ionic conductive poly(vinyl alcohol) hydrogels for flexible strain sensors based on a universal soaking strategy, Mater. Chem. Front., 2021, 5(1), 315–323 RSC.
- K. Chen, S. Liu, X. Wu, F. Wang, G. Chen, X. Yang, L. Xu, J. Qi, Y. Luo and D. Zhang, Mussel-inspired construction of Ti6Al4V-hydrogel artificial cartilage material with high strength and low friction, Mater. Lett., 2020, 265 Search PubMed.
- X. Liu, C. Steiger, S. Lin, G. A. Parada, J. Liu, H. F. Chan, H. Yuk, N. V. Phan, J. Collins, S. Tamang, G. Traverso and X. Zhao, Ingestible hydrogel device, Nat. Commun., 2019, 10(1), 493 CrossRef CAS PubMed.
- J. Gu, J. Huang, G. Chen, L. Hou, J. Zhang, X. Zhang, X. Yang, L. Guan, X. Jiang and H. Liu, Multifunctional Poly(vinyl alcohol) Nanocomposite Organohydrogel for Flexible Strain and Temperature Sensor, ACS Appl. Mater. Interfaces, 2020, 12(36), 40815–40827 CrossRef CAS PubMed.
- S. Pan, M. Xia, H. Li, X. Jiang, P. He, Z. Sun and Y. Zhang, Transparent, high-strength, stretchable, sensitive and anti-freezing poly(vinyl alcohol) ionic hydrogel strain sensors for human motion monitoring, J. Mater. Chem. C, 2020, 8(8), 2827–2837 RSC.
- Q. Yu, Z. Qin, F. Ji, S. Chen, S. Luo, M. Yao, X. Wu, W. Liu, X. Sun, H. Zhang, Y. Zhao, F. Yao and J. Li, Low-temperature tolerant strain sensors based on triple crosslinked organohydrogels with ultrastretchability, Chem. Eng. J., 2021, 404 Search PubMed.
- H. Yang, M. Ji, M. Yang, M. Shi, Y. Pan, Y. Zhou, H. J. Qi, Z. Suo and J. Tang, Fabricating hydrogels to mimic biological tissues of complex shapes and high fatigue resistance, Matter, 2021, 4(6), 1935–1946 CrossRef CAS.
- J. Kim, G. G. Zhang, M. X. Shi and Z. G. Suo, Fracture, fatigue, and friction of polymers in which entanglements greatly outnumber cross-links, Science, 2021, 374(6564), 212 CrossRef CAS PubMed.
- T. Nonoyama, Y. W. Lee, K. Ota, K. Fujioka, W. Hong and J. P. Gong, Instant Thermal Switching from Soft Hydrogel to Rigid Plastics Inspired by Thermophile Proteins, Adv. Mater., 2020, 32(4), e1905878 CrossRef PubMed.
- F. Zhu, S. Y. Zheng, J. Lin, Z. L. Wu, J. Yin, J. Qian, S. Qu and Q. Zheng, Integrated multifunctional flexible electronics based on tough supramolecular hydrogels with patterned silver nanowires, J. Mater. Chem. C, 2020, 8(23), 7688–7697 RSC.
- F. Luo, T. L. Sun, T. Nakajima, T. Kurokawa, Y. Zhao, K. Sato, A. B. Ihsan, X. Li, H. Guo and J. P. Gong, Oppositely charged polyelectrolytes form tough, self-healing, and rebuildable hydrogels, Adv. Mater., 2015, 27(17), 2722 CrossRef CAS PubMed.
- B. Lu, H. Yuk, S. Lin, N. Jian, K. Qu, J. Xu and X. Zhao, Pure PEDOT:PSS hydrogels, Nat. Commun., 2019, 10(1), 1043 CrossRef PubMed.
- J. Ni, S. Lin, Z. Qin, D. Veysset, X. Liu, Y. Sun, A. J. Hsieh, R. Radovitzky, K. A. Nelson and X. Zhao, Strong fatigue-resistant nanofibrous hydrogels inspired by lobster underbelly, Matter, 2021, 4(6), 1919–1934 CrossRef CAS.
- X. Liu, J. Wu, K. Qiao, G. Liu, Z. Wang, T. Lu, Z. Suo and J. Hu, Topoarchitected polymer networks expand the space of material properties, Nat. Commun., 1622, 2022(13), 1 Search PubMed.
- F. Luo, T. L. Sun, T. Nakajima, T. Kurokawa, X. Li, H. Guo, Y. Huang, H. Zhang and J. P. Gong, Tough polyion-complex hydrogels from soft to stiff controlled by monomer structure, Polymer, 2017, 116, 487–497 CrossRef CAS.
- X. Zhu, Y. Zhang, J. Deng and X. Luo, Effect of Glycerol on the Properties of the Cross-Linked Polyvinyl Alcohol Hydrogel Beads, ChemistrySelect, 2018, 3(2), 467–470 CrossRef CAS.
- W. Li, X. Wang, Z. Liu, X. Zou, Z. Shen, D. Liu, L. Li, Y. Guo and F. Yan, Nanoconfined polymerization limits crack propagation in hysteresis-free gels, Nat. Mater., 2023, 1–8 Search PubMed.
- L. Wu, L. Li, M. Qu, H. Wang and Y. Bin, Mussel-Inspired Self-Adhesive, Antidrying, and Antifreezing Poly(acrylic acid)/Bentonite/Polydopamine Hybrid Glycerol-Hydrogel and the Sensing Application, ACS Appl. Polym. Mater., 2020, 2(8), 3094–3106 CrossRef CAS.
- J. Wen, J. Tang, H. Ning, N. Hu, Y. Zhu, Y. Gong, C. Xu, Q. Zhao, X. Jiang, X. Hu, L. Lei, D. Wu and T. Huang, Multifunctional Ionic Skin with Sensing, UV-Filtering, Water-Retaining, and Anti-Freezing Capabilities, Adv. Funct. Mater., 2021, 31(21), 2011176 CrossRef CAS.
- M. Guo, J. Yan, X. Yang, J. Lai, P. An, Y. Wu, Z. Li, W. Lei, A. T. Smith and L. Sun, A transparent glycerol-hydrogel with stimuli-responsive actuation induced unexpectedly at subzero temperatures, J. Mater. Chem. A, 2021, 9(12), 7935–7945 RSC.
- X. F. Zhang, X. Ma, T. Hou, K. Guo, J. Yin, Z. Wang, L. Shu, M. He and J. Yao, Inorganic Salts Induce Thermally Reversible and Anti-Freezing Cellulose Hydrogels, Angew. Chem., Int. Ed., 2019, 58(22), 7366–7370 CrossRef CAS PubMed.
- D. Ma, X. Wu, Y. Wang, H. Liao, P. Wan and L. Zhang, Wearable, Antifreezing, and Healable Epidermal Sensor Assembled from Long-Lasting Moist Conductive Nanocomposite Organohydrogel, ACS Appl. Mater. Interfaces, 2019, 11(44), 41701–41709 CrossRef CAS PubMed.
- X. Han, M. Li, Z. Fan, Y. Zhang, H. Zhang and Q. Li, PVA/Agar Interpenetrating Network Hydrogel with Fast Healing, High Strength, Antifreeze, and Water Retention, Macromol. Chem. Phys., 2020, 221(22), 2070049 CrossRef.
- J. Chen, D. Shi, Z. Yang, K. Dong, D. Kaneko and M. Chen, Hand-extended noodle inspired physical conjoined-network organohydrogels with anti-freezing, high stiffness and toughness properties, J. Mater. Sci., 2021, 56(14), 8887–8899 CrossRef CAS.
- L. Han, K. Liu, M. Wang, K. Wang, L. Fang, H. Chen, J. Zhou and X. Lu, Mussel-Inspired Adhesive and Conductive Hydrogel with Long-Lasting Moisture and Extreme Temperature Tolerance, Adv. Funct. Mater., 2018, 28(3), 1704195 CrossRef.
- H. Sun, Y. Zhao, S. Jiao, C. Wang, Y. Jia, K. Dai, G. Zheng, C. Liu, P. Wan and C. Shen, Environment Tolerant Conductive Nanocomposite Organohydrogels as Flexible Strain Sensors and Power Sources for Sustainable Electronics, Adv. Funct. Mater., 2021, 31(24), 2101696 CrossRef CAS.
- J. Zhang, L. Wan, Y. Gao, X. Fang, T. Lu, L. Pan and F. Xuan, Highly Stretchable and Self-Healable MXene/Polyvinyl Alcohol Hydrogel Electrode for Wearable Capacitive Electronic Skin, Adv. Electron. Mater., 2019, 5(7), 1900285 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3mh02032k |
| ‡ Shu Wang and Ling Lei contributed equally. |
| This journal is © The Royal Society of Chemistry 2024 |