
Two decades of ceria nanoparticle research: structure, properties and emerging applications
Ali
Othman†
ab,
Akshay
Gowda†
b,
Daniel
Andreescu
 a,
Mohamed H.
Hassan
a,
S. V.
Babu
b,
Jihoon
Seo
*b and
Silvana
Andreescu
*a
a,
Mohamed H.
Hassan
a,
S. V.
Babu
b,
Jihoon
Seo
*b and
Silvana
Andreescu
*a
aDepartment of Chemistry and Biomolecular Science, Clarkson University, Potsdam, New York 13699-5810, USA. E-mail: eandrees@clarkson.edu
bDepartment of Chemical and Biomolecular Engineering, Clarkson University, Potsdam, New York 13699, USA. E-mail: jseo@clarkson.edu
First published on 26th April 2024
Abstract
Cerium oxide nanoparticles (CeNPs) are versatile materials with unique and unusual properties that vary depending on their surface chemistry, size, shape, coating, oxidation states, crystallinity, dopant, and structural and surface defects. This review encompasses advances made over the past twenty years in the development of CeNPs and ceria-based nanostructures, the structural determinants affecting their activity, and translation of these distinct features into applications. The two oxidation states of nanosized CeNPs (Ce3+/Ce4+) coexisting at the nanoscale level facilitate the formation of oxygen vacancies and defect states, which confer extremely high reactivity and oxygen buffering capacity and the ability to act as catalysts for oxidation and reduction reactions. However, the method of synthesis, surface functionalization, surface coating and defects are important factors in determining their properties. This review highlights key properties of CeNPs, their synthesis, interactions, and reaction pathways and provides examples of emerging applications. Due to their unique properties, CeNPs have become quintessential candidates for catalysis, chemical mechanical planarization (CMP), sensing, biomedical applications, and environmental remediation, with tremendous potential to create novel products and translational innovations in a wide range of industries. This review highlights the timely relevance and the transformative potential of these materials in addressing societal challenges and driving technological advancements across these fields.
 Ali Othman | Ali Othman received his PhD in Chemistry from Clarkson University (Potsdam, NY, USA) in 2019. He was a Postdoctoral Associate and Adjunct Assistant Professor at Clarkson University (2019–2022). His research interests focus on the development of novel functional nanomaterials and their applications for (bio)sensing, bioanalytical, chemical mechanical planarization (CMP), and environmental remediation applications. He has co-authored over 25 peer-reviewed papers, 4 patents and 2 book chapters. Currently, Dr Othman is a Research Scientist leading and working on the development of materials for gas/liquid purification at Pall Corporation, USA. |
 Akshay Gowda | Dr Akshay Gowda is a Process Integration Engineer at Intel Corporation, Rio Rancho, NM, USA. He received his PhD in Chemical and Biomolecular Engineering from Clarkson University, Potsdam, NY, USA in 2020. His PhD thesis focused on the physico-chemical properties of ceria particles and developing cleaning chemistries for their removal from silicon dioxide film surfaces for wafer cleaning applications post-chemical mechanical planarization (CMP) process in semiconductor industries. After working as a CMP Process Development Engineer for two years, he currently works on developing and integrating processes for photonics-based chips. |
 Daniel Andreescu | Daniel Andreescu is Associate Professor in the Department of Chemistry and Biomolecular Science at Clarkson University. He received his MS and PhD degrees in Chemistry from the University of Bucharest, Romania in 2002. Between 1996 and 2001, he was researcher at the National Research and Development Institute for Industrial Ecology. In 2003, he joined Clarkson University, where he currently works on the synthesis and characterization of nanosize metallic and metal-composite particles and their applications in sensing and environmental remediation. |
 Mohamed H. Hassan | Dr Mohamed H. Hassan is a postdoctoral research fellow in the Department of Materials Science and Engineering at the University of Pennsylvania. He earned his PhD in Chemistry from Clarkson University in 2023. His primary research endeavors revolve around investigating charge transport in metal–organic materials, with a specific emphasis on their practical applications in water remediation, energy storage, and conversion technologies. |
 S. V. Babu | Dr Babu is Professor Emeritus at Clarkson University, where he was the Director (1999–2016) of the NY State Center for Advanced Materials Processing (CAMP) and Vice Provost, Research (2001–04). He is now CTO, ChEmpower Corp. His PhD (Physics) is from SUNY, Stony Brook (1971). He held research appointments at Niels Bohr Institute, Copenhagen, International Center for Theoretical Physics, Trieste and NYU. He started at IIT, Kanpur, in 1972, and moved to Clarkson in 1981. He published over 290 papers, is the co-author of 33 patents, and co-edited four books. Babu supervised 50 PhD and 40 MS students. |
 Jihoon Seo | Dr Jihoon Seo is an Assistant Professor of Chemical and Biomolecular Engineering at Clarkson University in NY, USA. He holds a PhD in Energy Engineering and a Bachelor of Engineering in Materials Science and Engineering from Hanyang University in South Korea. His research focuses on novel planarization and cleaning technologies in manufacturing processes. Dr Seo collaborates with semiconductor manufacturers and equipment suppliers, advancing CMP through the development of cutting-edge processes and materials. Currently, Prof. Seo is leading the CMP team at Clarkson University. |
 Silvana Andreescu | Dr Silvana Andreescu is the Egon Matijević Endowed Chair in Chemistry in the Department of Chemistry and Biomolecular Science at Clarkson University. She received her PhD in 2002 from the University of Bucharest, Romania and University of Perpignan, France. Dr Andreescu's research program integrates electroanalytical, biochemical and materials science advances to develop innovative sensing technology for human and environmental health. Her recent work features the development of easy-to-use chemical and biological sensors based on nanoceria chemistry for the field detection of clinical, food and environmental targets. |
Wider impactCeria nanoparticles or nanoceria is a versatile material with unique and unusual optical, mechanical and catalytic properties. The co-existence of two-oxidation states Ce3+/Ce4+ at the nanoscale level facilitates the formation of oxygen vacancies and defect states, which confer an extremely high reactivity and the ability to act as a catalyst for both oxidation and reduction reactions. Due to its unique features, we now see a broad interest in the implementation of this material across a wide array of industries. However, its unique features are a function of the chemical, structural, surface defects, size, shape and doping effects, which are currently not fully understood. This review provides a comprehensive, critical, and accessible resource of general interest to the materials community, highlighting the structure and fundamental properties of this material and translation of its distinct features into applications. Scientists can use this resource as a starting point to explore this material's properties and pursue avenues for creating new products and breakthrough innovations. |
1. Introduction
Cerium, a member of the lanthanide series, is the most abundant of all rare earth metals. Unlike other elements in the series that typically exist in the +3 state, cerium adopts between +3 and +4 oxidation states, exhibiting unique redox properties and reactivity. Its oxide form, e.g. ceria, cerium oxide or cerium dioxide (CeO2), has gained significant interest in both industry and academia. Ceria-based materials, composed of CeO2, embody an array of distinctive features that make them highly desirable across many scientific areas and a wide range of industries. Nanosized ceria has gained significant attention over the past twenty years owing to its exceptional properties at the nanoscale. The unique crystal structure, characterized by the presence of oxygen vacancies and cerium cations with variable oxidation states, confers a plethora of properties including high oxygen storage capacity, redox activity, high surface functionality and catalytic power. Notably, nanoscale ceria can accommodate more oxygen vacancies in its crystal structure than its bulk counterpart.1 This attribute enables ceria nanoparticles (CeNPs) to exhibit very high oxygen mobility and exceptional oxygen storage capacity (OSC), making them adept at releasing and storing oxygen depending on the redox environment. These distinctive features have spurred intense investigation into CeNPs’ research and exploration of their properties in applications ranging from catalysis, energy conversion, semiconducting industries to environmental remediation, biosensing and biomedicine.Ceria-based materials are used in catalytic converters, solid oxide fuel cells (SOFCs), catalysts for volatile organic compound (VOC) oxidation, sensors, and chemical mechanical planarization (CMP). More recently, the scope of CeNPs has expanded to water and CO2 splitting for hydrogen production and biomedical applications as enzyme mimetics, theranostic probes and therapeutic materials. Over 30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 reports have been published on the synthesis, properties, and applications of ceria-based materials,2–7 many focusing on their role in catalysis, energy and environment. An emerging trend is the exploration of CeNPs in fields such as pharmacology, medicine, biosensing and bioimaging.8,9 However, challenges like biodistribution, cross-reactivity with clinically relevant species, and potential toxicity constrain their commercial potential.10,11
000 reports have been published on the synthesis, properties, and applications of ceria-based materials,2–7 many focusing on their role in catalysis, energy and environment. An emerging trend is the exploration of CeNPs in fields such as pharmacology, medicine, biosensing and bioimaging.8,9 However, challenges like biodistribution, cross-reactivity with clinically relevant species, and potential toxicity constrain their commercial potential.10,11
This review aims to offer readers a comprehensive understanding of critical physicochemical properties of the CeNPs, focusing on surface modification, doping and structure–activity relationships, followed by an exploration of their significant applications. Throughout this review, we will highlight the importance of CeNPs’ structural design parameters, interactions, and reactivity in the context of next-generation devices. We will commence by examining the structural and shape-dependent properties of CeNPs, followed by a discussion of size-dependent characteristics, with a particular emphasis on the Ce3+/Ce4+ ratio and oxygen vacancies, along with the mechanisms underlying oxygen vacancy formation. Ceria doping is a pivotal focus as it can substantially enhance ceria's properties, opening doors to innovative applications. We delve into the intricacies of the doping process and the role of doped ceria in catalytic reactions. Subsequently, we address the antioxidant properties of CeNPs and their interactions with reactive oxygen species (ROS), touching upon their potential applications in catalysis, sensing, and biomedicine. A dedicated section will cover surface functionalization and its crucial role in modulating interactions between NPs and their surrounding environment. Additionally, we will explore the incorporation of CeNPs into composite and nanohybrid structures interfaced with other metals or rare-earth metal oxides,12 enhancing their performance and augmenting their area of applicability. Finally, we will delineate applications and future directions, encompassing catalysis, CMP, biosensing, and environmental remediation.
2. Physicochemical properties of ceria nanoparticles
Physicochemical properties, including size, shape, crystal plane, and surface chemistry, have significant influence over the reactivity of ceria particles. Over the past two decades, substantial efforts have been dedicated to modulating oxygen vacancy levels and Ce3+/Ce4+ ratio by manipulating the synthesis conditions. A significant body of research has been dedicated to the creation of ceria particles with enhanced reactivity and higher Ce3+ concentration at the surface. This can be achieved through variations in the surface size and by doping with rare earth (RE), transition, or noble metals. This interest is driven by CeNPs’ remarkable capacity to neutralize free radicals, which has garnered considerable attention in the field of biomedicine. It has led to the development of innovative ceria-based antioxidant therapeutics. The recognized antioxidant activity of CeNPs is strongly linked to their redox activity, the Ce3+ levels present at the surface, and the presence of oxygen vacancies. In this section, our focus will be on elucidating how the structural and physicochemical properties of ceria influence over oxygen vacancies and surface reactivity (Fig. 1).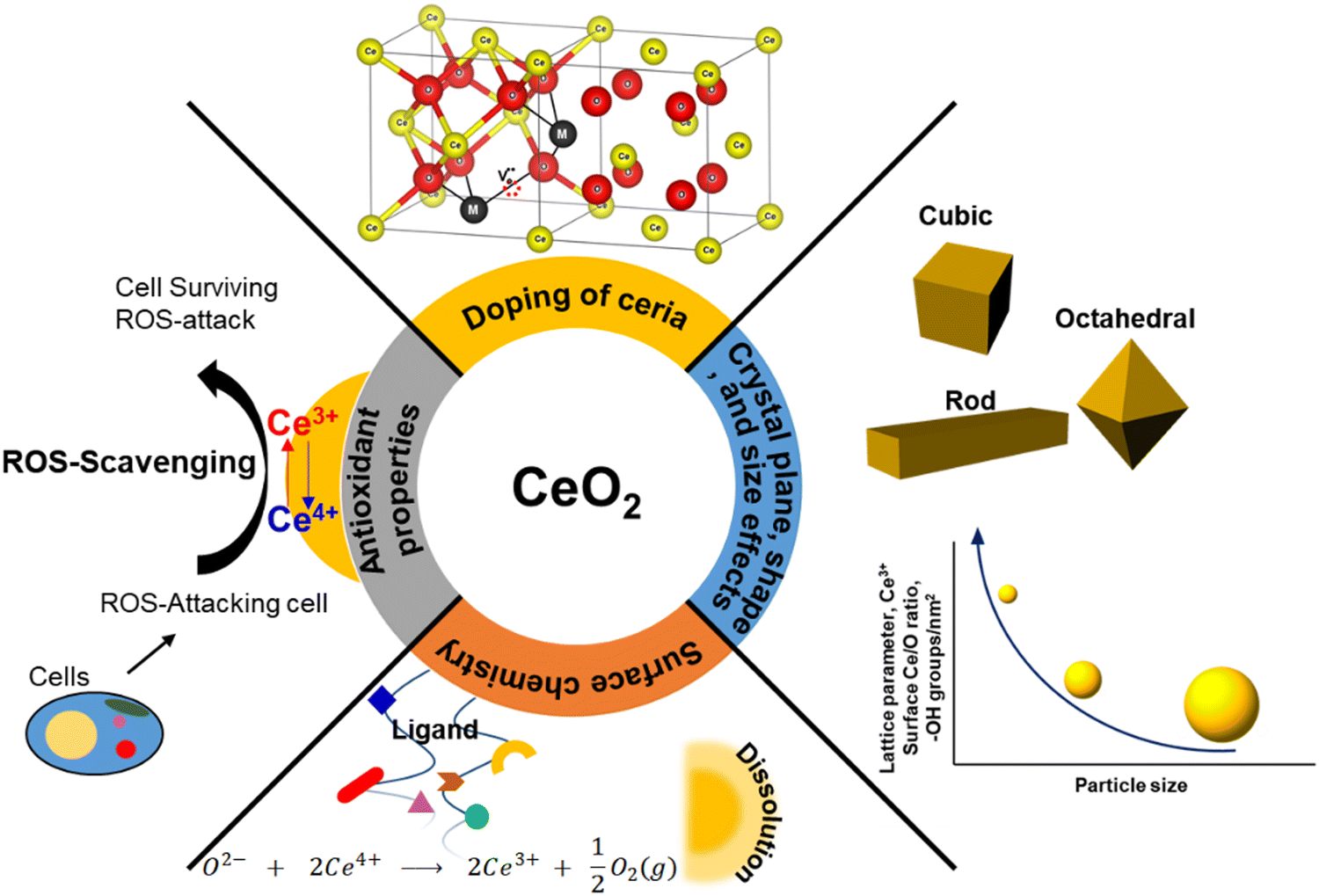 | ||
| Fig. 1 Summary of physicochemical properties that influence the reactivity of CeNPs. | ||
2.1. Structural and defect properties
Bulk cerium oxide or ceria has a fluorite crystal structure with face-centered cubic unit cell (space group Fm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m) and a lattice parameter of 5.411 Å at room temperature. In this arrangement, each Ce4+ ion (yellow spheres) is surrounded by eight nearest-neighbor O2− ions (red spheres), forming a cube structure. Each O2− ion is tetrahedrally coordinated to four closest Ce4+ ions, as depicted in Fig. 2a.
m) and a lattice parameter of 5.411 Å at room temperature. In this arrangement, each Ce4+ ion (yellow spheres) is surrounded by eight nearest-neighbor O2− ions (red spheres), forming a cube structure. Each O2− ion is tetrahedrally coordinated to four closest Ce4+ ions, as depicted in Fig. 2a.
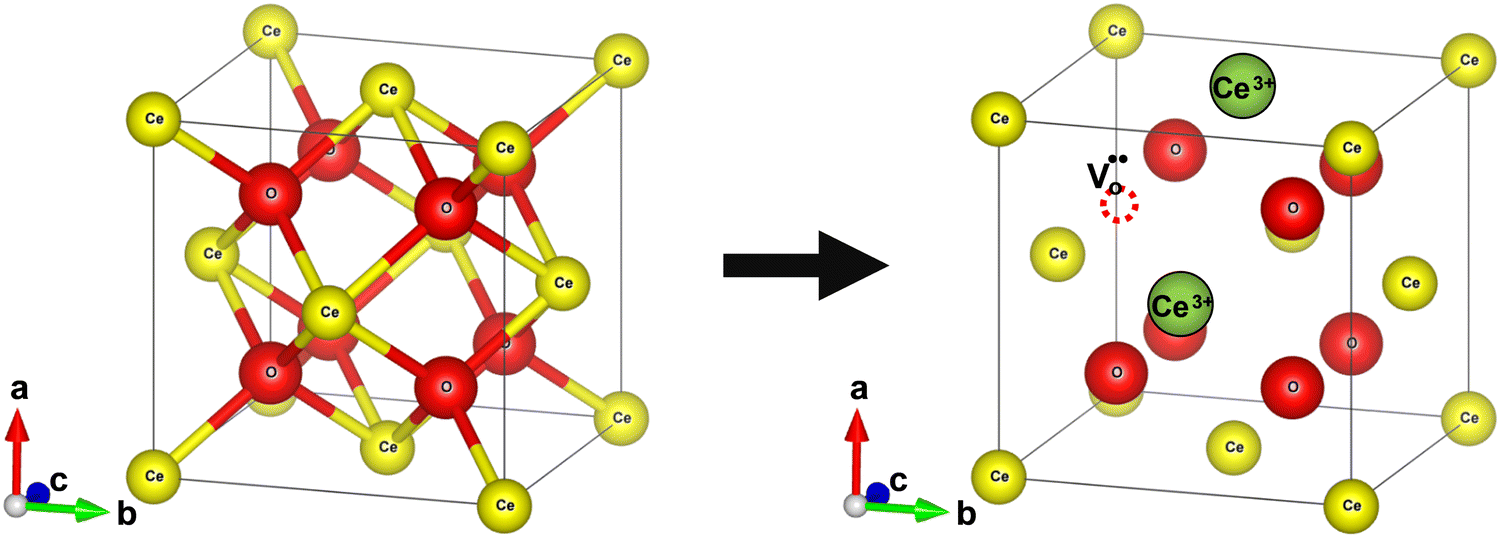 | ||
Fig. 2 Schematic diagram of reversible lattice distortion in a cubic ceria unit cell. (a) Fluorite crystal structure of bulk ceria. (b) Distorted crystal structure of nanoceria due to the formation of oxygen vacancy and concomitant reduction of Ce4+ ions to Ce3+ ions. Red and yellow-colored spheres represent O−- and Ce4+ ions, respectively, while green spheres represent 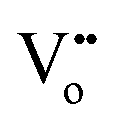 . . | ||
Defects within ceria can be introduced by changing temperature, oxygen partial pressure, electrical field or surface stress, or through the incorporation of other ions, and these defects can be broadly categorized into intrinsic and extrinsic types.1 Intrinsic defects primarily arise due to thermal fluctuations or interaction with the surrounding environment such as redox processes. In contrast, extrinsic defects emerge from impurities or the introduction of dopants1 (will be covered in Section 2.4). The two most noteworthy intrinsic defects observed in ceria are oxygen vacancies13,14 and anion Frenkel defect. In an anion Frenkel defect, an oxygen ion is displaced from its lattice position to an interstitial position, resulting in a defect at the interstitial location and a vacancy at the original position.15 These defects are typically present in low concentrations and do not impact the lattice change.
In oxygen vacancy type defect, an oxygen vacancy is created due to the liberation of an oxygen ion from a lattice site. The two excess electrons generated by the vacancy created are localized on two neighboring cerium ions, thereby reducing Ce4+ to Ce3+.16,17 This process can be represented as follows.
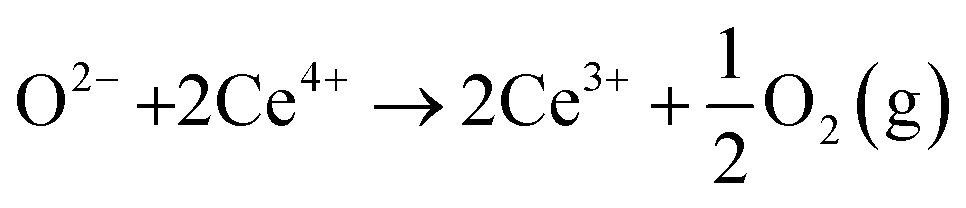 | (2.1) |
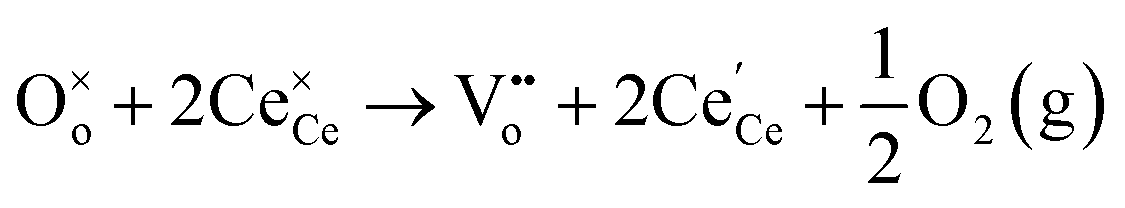 | (2.2) |
Eqn (2.1), which represents the vacancy formation reaction, is written using Kröger–Vink defect notation in eqn (2.2), where 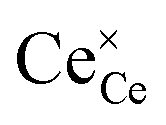 represents a Ce4+ ion on a Ce lattice site,
represents a Ce4+ ion on a Ce lattice site, 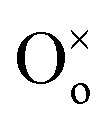 represents an O2− ion on an O lattice site,
represents an O2− ion on an O lattice site, 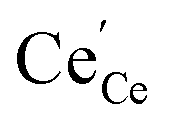 represents a Ce3+ ion on a Ce lattice site and
represents a Ce3+ ion on a Ce lattice site and 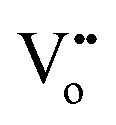 represents a neutral oxygen vacancy site.
represents a neutral oxygen vacancy site.
The formation of an oxygen vacancy results in a decrease in the coordination number of Ce4+ to O2− ions and introduces Ce3+ ions into the crystal lattice, as shown in Fig. 2b. Ce3+ ions have a larger ionic radius (1.034 Å) as compared to Ce4+ ions (0.92 Å).18 Therefore, the introduction of Ce3+ ions and oxygen vacancies results in a distortion (dislocation of atoms from their equilibrium lattice points) of the local symmetry and generates strain in the lattice. Ceria particles release strain by undergoing lattice expansion,19,20 as shown in Fig. 2b. Different distortions can be induced due to different Ce3+ localization motifs with correspondingly different vacancy formation energies. The localization of electrons upon vacancy formation occurs either on the cerium ions neighboring the vacancy (i.e., Ce–O) or cerium ions next to the nearest neighbor (i.e., Ce–Ce).21,22
Yang et al. reported vacancy formation energies of 3.39 and 3.21 eV for surface and subsurface oxygen vacancies, respectively, on a (111) surface, implying that the energies for the formation of surface and subsurface vacancies are relatively close to each other.27 Using state-of-the-art STM as well as DFT calculations, Esch and co-workers showed in an elegant study that the surface oxygen vacancies on the (111) ceria surface are immobile at room temperature and that direct diffusion (movement of lattice oxygen) needs higher temperatures (>400 °C).24 Note that with increasing annealing time, the number of surface defects should decrease due to the small diffusion barrier, making it highly mobile at the elevated temperatures, in which the clustering of surface vacancies is energetically unfavorable.28
Fig. 3a and b show the two types of vacancies, namely, surface and subsurface oxygen vacancies, identified on a partially reduced ceria surface in filled-state and empty-state, respectively. Both types of vacancies were present with similar coverages. This observation was indeed in line with DFT calculations, which predicted the same vacancy formation energy for both the defects, as shown in Fig. 3c. Furthermore, on a fully reduced ceria surface, three configurations of surface vacancies were identified: single surface or subsurface vacancies, vacancy dimers and vacancy trimers. The structural models of dimers and trimers of oxygen vacancies are summarized in Fig. 3d. The superposition of single surface and subsurface vacancies results in the formation of a vacancy dimer. Double LSVCs form after the removal of another surface oxygen atom. Two possible variations of vacancy trimers, of which only the one that exposes Ce3+ ions (SVT 1 in Fig. 3d), was observed.
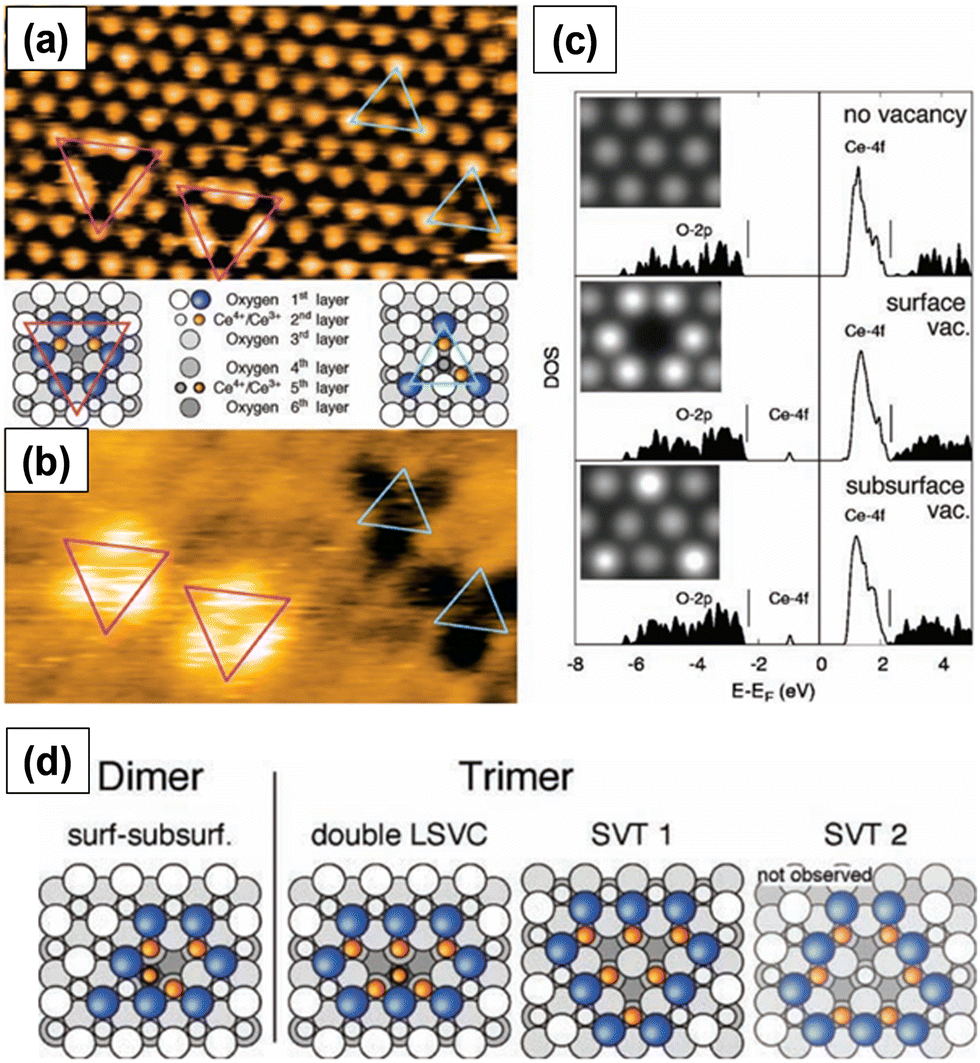 | ||
| Fig. 3 STM images of surface and subsurface oxygen vacancies in (a) filled-state and (b) empty-state. Pink and blue triangles represent surface and subsurface vacancies, respectively. (c) Density of states (DOS) calculations along with STM images in filled-state (inset). Structures of the observed vacancy dimers and trimers. Reprinted with permission from ref. 24. | ||
Both surface and subsurface oxygen vacancies on a partially reduced (111) ceria surface were observed by several other researchers.29–31 Oxygen vacancies are located at the third surface atomic layer. When the concentration of subsurface oxygen vacancies is very high, the defects form arrays with a propensity to form linear motifs.29 The oxygen vacancies initially form triangular clusters and upon annealing form line defects. The clustering of oxygen vacancy defects on ceria surface is energetically more favorable than the formation of isolated surface oxygen vacancy defects.30
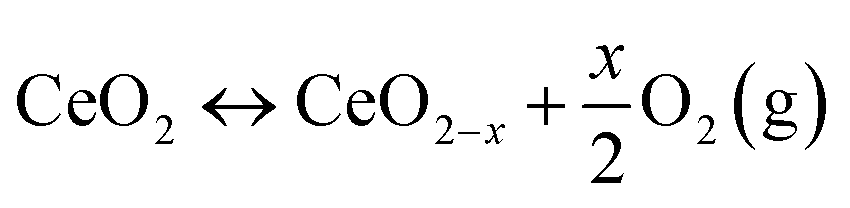 | (2.3) |
Different types of CeO2−x oxide phases are identified in the literature. However, some conflicting results exist on the structures and compositions of intermediate phases. The traditional powder X-ray diffraction (XRD) method has limitations in determining the structural characteristics of nonstoichiometric oxides.3 Due to the low X-ray scattering power of O atoms, it is difficult to obtain the detailed sublattice structure of O from the XRD technique.13 Thus, neutron and electron diffraction techniques have been employed to overcome the limitations of the XRD technique.33,34
The so-called α-phase non-stoichiometric oxides have compositions in the range of 0 < x < 0.286 for CeO2−x (CeO1.714–CeO2). The α-phase shows a disordered nonstoichiometric fluorite-related structure that is stable at high temperatures (above 685 °C). The XRD patterns obtained at high temperatures do not show superstructures and the lattice parameter of the cubic phase increases with x.34 At lower temperatures, however, the α-phase transforms into a series of fluorite related phases described by the general formula CenO2n−2m. The lattice parameters at low temperatures are close to those reported for α-phase, although the α-phase is disordered and low temperature phases show structural changes.35–37 This implies that variation in oxygen partial pressure and temperature does not alter the positions of cerium ions and that the change in symmetry arising due to the reduction of ceria is primarily due to the formation of oxygen vacancies. Some examples of these phases are Ce10O18 (the ε phase), Ce6O11 (the β phase, monoclinic) and Ce11O20 (the δ phase, triclinic).37,38
The CeO2−x oxides with the composition x > 0.286 at high temperature are dominated by the σ-phase, a nonstoichiometric phase with body-centered cubic crystal structure. The C-type sesquioxide (Ce2O3), end member of the σ-phase, shows bixbyite crystal structure (space group Ia3).35 The structures of CeO2 and Ce2O3 are closely related in that their cerium ion arrays are almost identical and oxygen ions occupy tetrahedral positions in both cases. The oxygen ions occupy all available locations in the fluorite structure while only three-quarters are occupied in the bixbyite structure.19,36 As a result, the lattice parameter of Ce2O3 is almost twice that of CeO2. The nonstoichiometric σ-phase is very difficult to distinguish from the C-type sesquioxide.
2.2. Crystal plane and shape-dependent properties
The reactivity of ceria-based materials is significantly affected by the morphology, oxidation states and the exposed crystal planes.39–41 In this section, we will discuss the three thermodynamically stable low-index crystallographic planes of ceria, namely (100), (110) and (111),42–44 as shown in Fig. 4a–c. Some of the important characteristics of these three crystal planes are summarized in Table 1. Other ceria surfaces like (211), (210) and (310) are less stable and suffer severe reconstruction.40 The (110) surface is a type 1 surface that exposes both cerium and oxygen ions, and each layer has zero charge as the anions and cations are present in stoichiometric proportions in each plane (six coordinate cerium ions and three coordinate oxygen ions).45,46 The (111) ceria surface is a type 2 surface, which is terminated with oxygen ions. There is no net dipole moment as the three O–Ce–O layers (stacked one on top of the other) maintain charge neutrality. The coordination number is three and seven for oxygen and cerium, respectively.44 The (100) surface of ceria is a type 3 surface, which is intricate because an ideal surface could have either oxygen or cerium ions in the top surface, thereby leading to a polar and unstable surface.46 Oxygen and cerium have coordination numbers of two and six, respectively.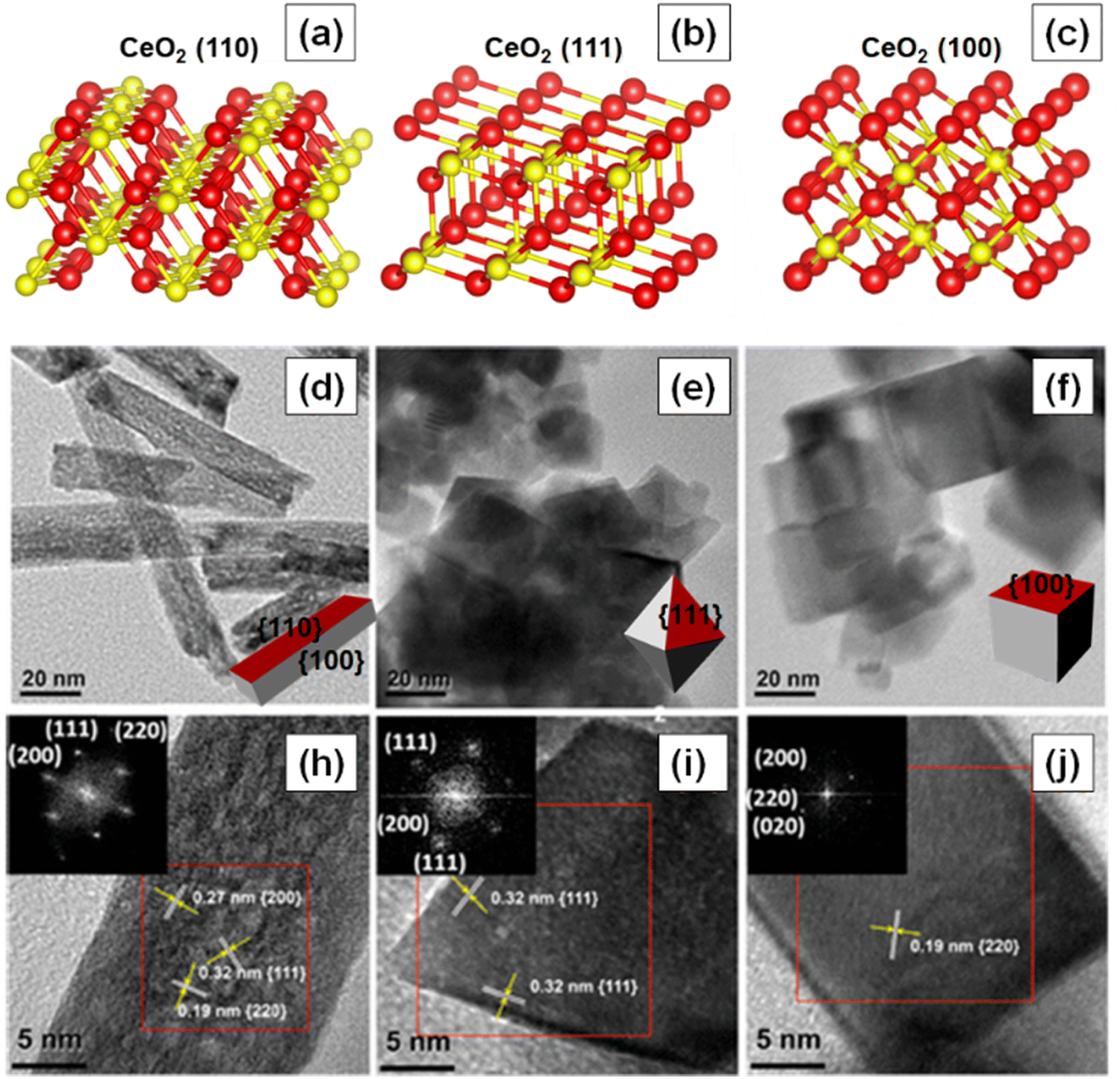 | ||
| Fig. 4 Atomic structures of the three low index crystal planes of ceria. (a) 110, (b) 111, and (c) 100. Red and yellow-colored spheres represent O−- and Ce4+ ions, respectively. TEM and HRTEM images of the three different ceria nanoshapes (d) and (h) nanorods, (e) and (i) nanooctahedra and (f) and (j) nanocubes. Fourier transform patterns of the selected areas of the samples are shown in the insets of HRTEM images. Fig. 4d–j reprinted with permission from ref. 39. Copyright 2018 American Chemical Society. | ||
| Property/crystallographic plane | (110) | (111) | (100) |
|---|---|---|---|
| Coordination number | Ce(6) | Ce(7) | Ce(6) |
| O(3) | O(3) | O(2) | |
| Atom/s exposed by first layer | Ce and O | O | O |
| Relaxed surface energy (eV) | 1.01 | 0.68 | 1.41 |
| Unrelaxed surface energy (eV) (estimated from DFT calculations) | 1.26 | 0.69 | 2.05 |
| Oxygen vacancy formation energy (eV) (calculated from DFT corrected for on-site Coulomb interactions) | 1.99 | 2.6 | 2.27 |
The structures and stabilities of the three low-index lattice planes of ceria have been extensively investigated by many researchers. Although the results vary, the stability generally follows the order (111) > (110) > (100).27,47 Vacancies are the active sites for oxygen activation in oxidation reactions in many catalytic applications. (110) and (111) ceria surfaces have the lowest and highest vacancy formation energies, respectively. The (111) surface is less prone to accommodate a vacancy and hence the energy needed for vacancy formation on its surface is the highest among the three.47 The subsurface vacancy site is the most stable vacancy on the (111) surface.48 In (110) surface, the vacancy with one Ce3+ ion in both the surface and first subsurface layers is determined to be the most stable. In (100) surface, two Ce3+ ions neighboring the vacancy site are the most stable; the next most stable site is 0.30 eV higher in energy.22 In case of (110) and (100) ceria surfaces, for the same oxygen vacancy, more than one distribution of Ce3+ sites are observed.
The morphology and crystal plane of ceria can be affected by the synthesis method and the cerium precursor salt.49–53 Ceria with well-defined lattice planes and morphologies can be obtained by modulating some of the critical synthesis parameters. Synthesis of ceria nanomaterials with different shapes like cube, octahedron, tetrahedron, sphere, rod and plate have been reported in the literature. The most widely employed synthesis procedure to produce ceria in industrial applications is by chemical precipitation, which results in the formation of NPs. Factors like pH, base concentration, reaction temperature and time affect the physicochemical properties of NPs including the shape and size.54–58 Facet engineering can be used as a powerful tool to enhance the modification of CeNPs, for example, silanization through the favorable adsorption of tetraethyl orthosilicate (TEOS) on the (100) facets and spontaneous breakage of the Si–O bonds of TEOS as the rate determining step for silanization.59
Among various other methods, the hydrothermal process has been used to prepare different ceria nanoshapes. Mai and collaborators showed that ceria nanocubes, nanorods and nanopolyhedra can be selectively synthesized by varying the base concentration and temperature during hydrothermal synthesis. First, anisotropic Ce(OH)3 nuclei form from the reaction of Ce3+ precursor ions with the base (NaOH). At low base concentration (0.01 mol L−1) and temperature of 100 °C, the rate of dissolution/recrystallization is very low. Consequently, the driving force for the anisotropic growth of Ce(OH)3 nuclei is also low, and hence, nanopolyhedra exposing (111) and (100) crystal planes are formed. At a much higher base concentration (6 mol L−1) and 100 °C temperature, the dissolution/recrystallization rate is enhanced. Ce(OH)3 nuclei grow anisotropically to form nanorods exposing (110) and (100) surfaces. The increase in synthesis temperature to 180 °C led to the oxidation of Ce(OH)3 to CeO2 and nanocubes exposing (100) planes were formed. Besides NaOH, PO43−, urea and H2O2 can also be used for the hydrothermal synthesis of ceria nanomaterials.60–65
Surfactant- or organic-assisted synthesis, with organic molecules used as coating agents, are often used to control the particle growth as the organic material adsorbs on specific planes of nanocrystals and directs the oriented growth. As mentioned above, the concentration of surfactant and cerium precursor salt, reaction temperature and time are some of the crucial factors that affect the morphology of ceria nanomaterials.66–70 Pan et al. used a surfactant-assisted method to synthesize nanorods, nanoplates and nanotubes52 using cetyltrimethylammonium bromide (CTAB). CTA+ adsorbs on ceria nanocrystals and interacts with the (111) and (100) facets. The exposed surfaces couple to reduce the surface energy to form nanoplates. Due to the low coating ability at lower CTAB concentrations, nanoplates are transformed into nanorods to lower the surface energy by an anisotropic growth mechanism. At high CTAB concentrations, the nanoplates transform into nanotubes due to a rolling mechanism. Sometimes, the combination of different organic molecules can also be used to selectively fabricate ceria nanomaterials with specific shapes. For example, the addition of oleic acid (OA) as a co-surfactant to a solution of Ce(NO3)3 and diphenyl ether in oleylamine at 320 °C in the sol–gel synthesis method led to the generation of nanowires and tadpole-shaped ceria nanocrystals due to the anisotropic growth of the nuclei.71
Surfactants or surface coating agents play an important role in the synthesis and later dictate the properties of the CeNPs. For instance, nanospheres were obtained using diethylene glycol as the reaction solvent and polyvinylpyrrolidone (PVP) as the surfactant, but when the synthesis was carried out in the absence of PVP, agglomerated and irregular shaped nanoparticles were obtained.72 The charge of the surfactant can affect the morphology and the stability of ceria nanomaterials and should be considered. Anionic surfactants can adsorb on the positively charged ceria and effectively stabilize them while cationic surfactants repel away from cerium cations and result in the agglomeration of nanocrystals. The ceria nanomaterials prepared by surfactant-assisted synthesis usually have good dispersibility and uniform size distribution. This technique can be employed to fabricate different shapes ceria, which cannot be obtained using the conventional methods. Nonetheless, the use of organic solvents and surfactants can results in impurities and depending of the solvent and surfactant used can increase the manufacturing costs. The effect of other synthesis methods on different ceria nanoshapes and the associated formation mechanisms are presented in several comprehensive reviews, and the readers can refer to them for additional information.73–75 The anions of the cerium precursor salt can selectively interact with specific crystal planes and hence form nanocrystals with different morphologies. The Br−, I−, Cl− and SO42− counter anions of cerium precursor salt form nanorods, whereas NO3− and BrO3− anions lead to the formation of nanocubes and NPs, respectively.50 The anisotropic growth of Ce(OH)3 nuclei formed by Br−, I−, Cl− and SO42− results in nanorod formation. In the presence of NO3− and BrO3−, CeO2 nuclei are formed due to the oxidizing ability of these anions, and the growth of isotropic CeO2 nuclei results in the formation of nanocubes/NPs. CeCl3 precursor salt exposes (100) and (110) planes due to the formation of nanorods while Ce(NO3)3 exposes (100) planes due to the formation of nanocubes.51
The shape of nanoceria can also affect the concentration of oxygen vacancy defects and hence their reactivity. Numerous studies have indeed investigated the effect of shape on vacancy concentration and catalytic activity. For instance, Cao and collaborators studied the effect of shape or crystal plane on the catalytic activity of ceria for CO oxidation.39 Different morphologies, e.g., nanorods, nanooctahedra and nanocubes, with all having similar crystallite sizes were synthesized. The high-resolution transmission electron microscopy (HRTEM) images of the synthesized ceria nanoshapes are shown in Fig. 4d–j. The ceria nanocubes and nanorods exposed (100) crystal plane while the nanooctahedra exposed (111) crystal plane. The concentration of Ce3+ ions, however, was similar in all three nanoshapes. Electron paramagnetic resonance (EPR) spectra suggested the presence of isolated vacancies on the (100) facets of ceria nanorods and nanocubes and vacancy clusters on the (111) facets of ceria octahedra. Furthermore, the type of oxygen vacancies and surface properties of ceria nanoshapes were correlated to their catalytic activity.
Ceria octahedra expose eight (111) planes while ceria cubes expose six (100) planes. Ceria nanorods expose (100) and (110) surfaces.76 The reactivity of different facets, as determined by theoretical calculations, follows the order (110) > (100) > (111).1,77,78 This order, in fact, is in agreement with the experimental results on the activity of different crystal planes of ceria. Nevertheless, different vacancy formation energies in a given surface can be obtained, which is due to some of the limitations of techniques used for theoretical calculations, discussed in detail elsewhere.1,44
2.3. Size impact on the reactivity and catalytic activity properties
Particle size has a significant impact on the reactivity and catalytic activity of ceria nanomaterials. The effect of particle size was first experimentally probed by Tsunekawa's group. In a series of publications, they showed that the lattice parameter (calculated from electron diffraction patterns) increases with the decrease in the particle size from 6.7 to 2.1 nm.18,19 Three possible models were proposed to explain the increase in the lattice parameter value based on the fact that oxygen forms peroxide, O22−, and superoxide, O2−, on the surface of reduced CeO2. One model suggests that CeNPs consist of a layer of Ce2O3 with an estimated thickness of 0.561 nm on the outside while the core is CeO2. Several studies also observed size-induced lattice expansion in CeNPs, particularly for sizes below 20 nm, due to the increase in the oxygen vacancies and Ce3+.Wu et al. determined the concentration of Ce3+ as a function of particle size using electron energy loss spectroscopy (EELS).79 The fraction of Ce3+ ions rapidly increased with decreasing particle size below 15 nm. Interestingly, their EELS spectra revealed completely reduced ceria, Ce2O3, at a diameter of 3 nm. This reduced ceria was found to have a fluorite structure like that of bulk CeO2. Also, the EELS spectra acquired from the edge and the center of the particles showed that for larger particles, the reduction of Ce4+ to Ce3+ happens mainly at the surface, resulting in a Ce2O3 surface layer but leaving the core as CeO2. Hailstone and collaborators studied the size-dependent lattice expansion of colloidal ceria NPs using TEM.80 The lattice constant increased with a decrease in particle size, with 1.1 and 11.8 nm particles having values of 0.578 and 0.547 nm, respectively. The corresponding lattice expansion was found to be about 6.8% and 1.1% for 1.1 and 11.8 nm CeNPs, respectively. The diffraction patterns for all the three different-sized particles were found to be consistent with that of the fluorite crystal structure of CeO2 and not with the predicted cubic or the hexagonal structure of Ce2O3. Most of the surface ions were Ce3+ and, correspondingly, there were substantial oxygen vacancies. The larger radius of Ce3+ along with the associated oxygen vacancies corroborate the higher lattice constant and hence the greater lattice expansion observed for smaller particles.
As the size of ceria NP decreases, more and more oxygen vacancies are formed and hence the concentration of Ce3+ increases, as shown in Fig. 5a. Since Ce3+ ions have a larger ionic radius than Ce4+ ions, the particle size reduction leads to lattice distortion and introduces strain in the lattice. This strain is liberated after lattice expansion; therefore, the lattice parameter increases (Fig. 5a).4,6,8,16,17,19 Xu et al. studied the size-dependent structural, chemical and electronic properties of ceria.81 The lattice parameter increased with the decrease in particle size, as shown in Fig. 5b, but their results did not provide any proof of increase in Ce3+ concentration and oxygen vacancies. Nonetheless, a dramatic increase in the concentration of reducible oxygen was observed at sizes below 5 nm due to peroxide formation on ceria surface. It is now established that the CeNP size influences the degree of hydroxylation and hydroxyl stability, with particles <5 nm having a higher density of more thermally stable hydroxyl groups, as compared to larger particles.82 These functional groups dictate the surface reactions and the behavior of the CeNPs in aqueous environments, which is relevant for many processes, e.g., pro- or anti-oxidant activity and surface sorption. This also suggests that, beyond the varying Ce3+/Ce4+ ratios, the surface hydroxyl groups also play a critical role in the CeNP reactivity.
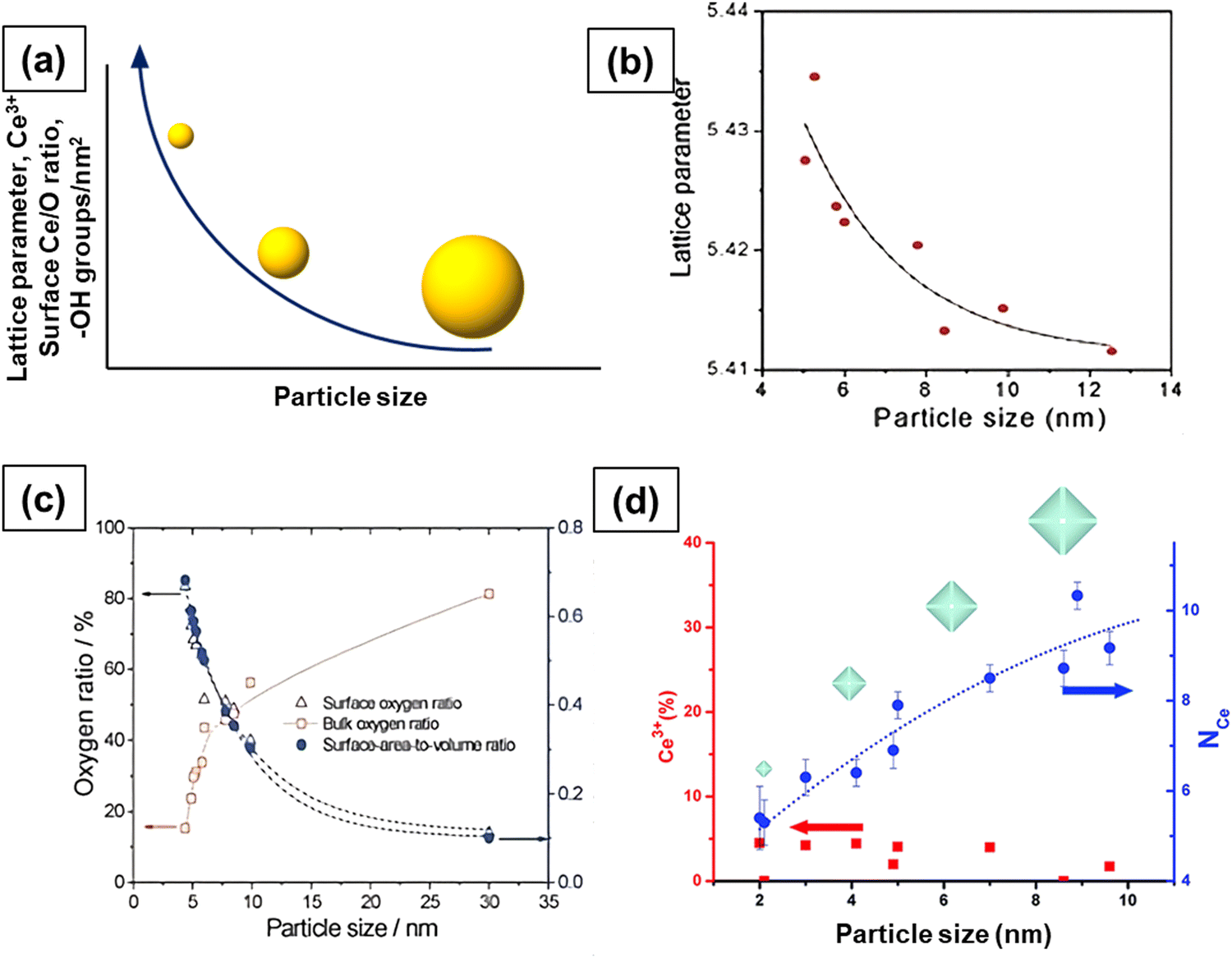 | ||
| Fig. 5 (a) Effect of particle size on lattice parameter, concentration of Ce3+, oxygen vacancies and hydroxyl species. More and more oxygen vacancies form with decrease in size and this leads to an increase in the concentration of Ce3+ ions and lattice parameter. (b) Increase in lattice parameter with the decrease in size. (c) The concentration of surface oxygen and bulk oxygen as a function of particle size. (d) Concentration of Ce3+ and Ce–Ce coordination as a function of particle size. Fig. 5b and c reproduced from ref. 81 with permission from the Royal Society of Chemistry. Fig. 5d reprinted with permission from ref. 83. Copyright 2012 American Chemical Society. | ||
The surface oxygen concentration varies inversely with the bulk oxygen concentration as a function of particle size, as shown in Fig. 5c. The concentration of surface oxygen increases at the expense of the concentration of bulk oxygen. It is likely that the significant increase in the concentration of reducible/surface oxygen is due to the increased Ce3+ concentration as Ce3+ ions interact with O2 and form cerium superoxide (Ce4+O2−). A decrease in particle size leads to increased Ce3+ species and hence a higher concentration of Ce4+O2− species. The formation of cerium superoxide (Ce4+O2−) and cerium peroxide (Ce3+O22−) species will be discussed in Section 2.6. The size-dependent lattice parameter and relative Ce3+ concentration values of ceria particles reported by several published studies are summarized in Table 2.
| Particle size range (nm) | [Ce3+] range (%) | Preparation method | Lattice parameter range (nm) | Ref. |
|---|---|---|---|---|
| 2–7 | 17.3–42.5 | Hydrothermal | 0.5453–0.5560 | 18 and 19 |
| 4–60 | — | Precipitation (semi-batch reactor) | 0.5401–0.5419 | 84 |
| 3–20 | — | Thermal evaporation | 0.5402–0.5615 | 79 |
| 6–15 | — | Precipitation | 0.5413–0.5433 | 85 |
| 1–12 | — | Precipitation | 0.547–0.578 | 80 |
| 4–10 | 29.4–29.5 | Microemulsion | 0.5415–0.5435 | 81 |
| 3–30 | 17–44 | Precipitation | 0.548–0.560 | 17 |
| 10–90 | 15–26.3 | Precipitation | — | 86 |
| 10–235 | 19.3–27.6 | Supercritical solvothermal | 0.5406–0.5425 | 87 |
| ∼10 | 34 | Green synthesis | — | 88 |
| Bulk | — | — | 0.5403 | 89 |
Vayssilov et al. showed that 2 nm ceria particles prepared by thermal evaporation contained only 2% Ce3+.90 Using X-ray absorption near edge structure spectroscopy (XANES), Nachimuthu et al. estimated that the concentration of Ce3+ ions was very low (<5%) even for 2 nm ceria particles.91 Paun et al. studied the size-dependent properties of ceria particles using TEM, XRD, X-ray spectroscopy (XPS), extended X-ray absorption fine structure (EXAFS) and XANES.83 These analyses revealed that the Ce3+ concentration does not vary with size under ambient conditions although the lattice parameter increases with the decrease in particle size. As can be seen in Fig. 5d, the Ce3+ concentration remained at the same ∼4% value with the decrease in particle size from 8 nm to 2 nm. This value of 4% is way lower than that reported in other studies, which are in the range of 20–30% for about the same particle size. The presence of Ce3+ ions in traces was attributed to the use of Ce3+ precursors used for synthesis.
While most studies reported lattice expansion in the range of 0.1–0.5% for particles smaller than 5 nm, Tsunekawa et al.19 and Wu et al.79 observed lattice expansion in the range of 2–3.5% for similar particle sizes. Hailstone and collaborators reported a very high lattice expansion of 7% for 1 nm CeNPs.80 These dramatic differences in lattice expansion is potentially attributed to the effect of surface stabilizers used in the synthesis to control the particle growth. Another aspect that is worth highlighting is the relative Ce3+ concentration. Majority of the studies reported Ce3+ concentrations greater than 15% for particles smaller than 5 nm. Nevertheless, a few studies reported very low (<5%) relative Ce3+ concentration values for particles smaller than 5 nm. These apparent differences could be due to variations in the preparation method, synthesis conditions, surface coating agents, or the characterization method employed to estimate Ce3+ concentration. A thorough knowledge of the particle synthesis chemistry is necessary to understand the effect of synthesis method on Ce3+ concentration. In this regard, it is critical to create standards for the synthesis method and the determination of relative Ce3+ concentration.
The size-dependent oxygen vacancy formation energies in (CeO2)n with n values in the range of 20–140 was investigated using DFT calculations.92–94 The removal of a low-coordinate O atom from ceria needs the least energy, consistent with the results reported for other metal oxides. The removal of such O atoms from nanosized particles is more favored than that from the extended surfaces. The increase in particle size is associated with a marked decrease in the oxygen vacancy formation energy, indicating that this energy reaches a minimum at certain sizes. The size dependence of the oxygen vacancy formation energy is controlled by electrostatics. The bandwidth of the unoccupied density of states projected on to the cerium 4f orbital levels is a crucial factor that affects the energy of vacancy formation. The presence of corner cerium atoms is identified as the structural pattern necessary for a significant reduction of vacancy formation energy.
By summarizing these results, the following conclusions can be drawn. (1) With a decrease in particle size, more and more oxygen vacancies are formed and, consequently, the Ce3+ concentration is enhanced. (2) An increase in Ce3+ concentration increases the lattice parameter due to the lattice expansion as Ce3+ ions have higher ionic radius than Ce4+ ions. (3) Most of these changes (formation of oxygen vacancies and generation of Ce3+ ions) occur on the surface. As a result, the surface is very similar to that of Ce2O3 while the bulk remains as CeO2. When the particle size decreases to about a nanometer or two, the particle transforms from CeO2 to Ce2O3, as shown by several studies, and the particle mostly consists of Ce3+ ions. The conversion from CeO2 to Ce2O3 C-type sesquioxide does not require a change in the crystal structure. Hence, in some studies, even ∼1–2 nm particles showed fluorite structure as the C-type is a combination of three types of fluorite like unit cells. (4) The oxygen vacancy formation is thermodynamically more favorable on CeNP’ surfaces than in the bulk. (5) The corresponding formation energies of the Ce3+ ions are also lower on the nanoceria surfaces than in the bulk. Clearly, these findings suggest that the concentration of surface Ce3+ ions, the surface functional groups and the associated oxygen vacancies and, hence, the reactivity of NPs increases with a decrease in particle size, while the oxygen vacancy formation energy diminishes markedly.
2.4. Modification by doping
Due to the increased surface area to volume ratio and relative ease of vacancy defect formation, CeNPs show enhanced reactivity and outstanding catalytic activities enabling their use in many commercial applications. However, pure CeNPs are associated with some drawbacks like the deactivation of OSC and catalytic activity and a loss of surface reactivity due to thermal sintering at high temperatures, which limit their use in some commercial applications.95,96 It is, therefore, of interest to modify the properties of CeNPs and alter the surface energies to facilitate their use in such applications. Any chemical modification or doping of CeNPs involving an increase in the concentration of oxygen vacancies should in principle increase their reactivity. This is true only if (a) the chemical modification process does not lead to a significant reduction in the concentration of active redox species and (b) the defects formed at high degrees of reduction do not cluster, making ion transport difficult.One of the important strategies to overcome these limitations is to incorporate other metal ions into the crystal structure of CeNPs by doping. The addition of dopants results in higher resistance to sintering at elevated temperatures and an enhancement of reducibility and OSC of CeNPs and therefore improves the overall catalytic performance.97,98 The concept of modifying CeNPs by doping with foreign materials indeed led to enhanced thermal stability and improved catalytic activity. In a typical doping process, cerium atoms in the crystal lattice are replaced with noble metals, transition metals or rare earth metals.
Doping is done during synthesis and the dopants substitute Ce4+. The result is that oxygen vacancies are introduced in the crystal structure of ceria for charge compensation,99 as shown in Fig. 6a. Doped CeNPs show very high oxygen mobility through a vacancy diffusion mechanism and hence high ionic conductivity.100,101 Given the importance of oxygen vacancy defects of CeNPs in many applications, this topic has garnered a lot of attention. Most reported works explored the use of rare earth (Y3+, Gd3+, Sc3+, Sm3+, Er3+, Eu3+, La3+, etc.), noble (Pt, Pd, Au, and Rh) and transition metals (Cu, Co, Ni, Mn, Zr, Zn, Fe, etc.) as dopant materials to modify the physicochemical properties of CeNPs (Fig. 6b). The dopants are further categorized into two types, isovalent and aliovalent, based on their oxidation state.102,103 Isovalent dopants are the ones that have a 4+ oxidation state (same as that of the host Ce4+), while aliovalent dopants have an oxidation state that is different from that of the host. The substitution of Ce4+ with the isovalent dopants in the crystal lattice of ceria introduces intrinsic oxygen vacancies and reduces the oxygen vacancy formation energy because of structural distortion.72 On the other hand, substitution with aliovalent dopants incorporates both extrinsic and intrinsic defects and reduces vacancy formation energy due to structural distortion and electronic modifications.
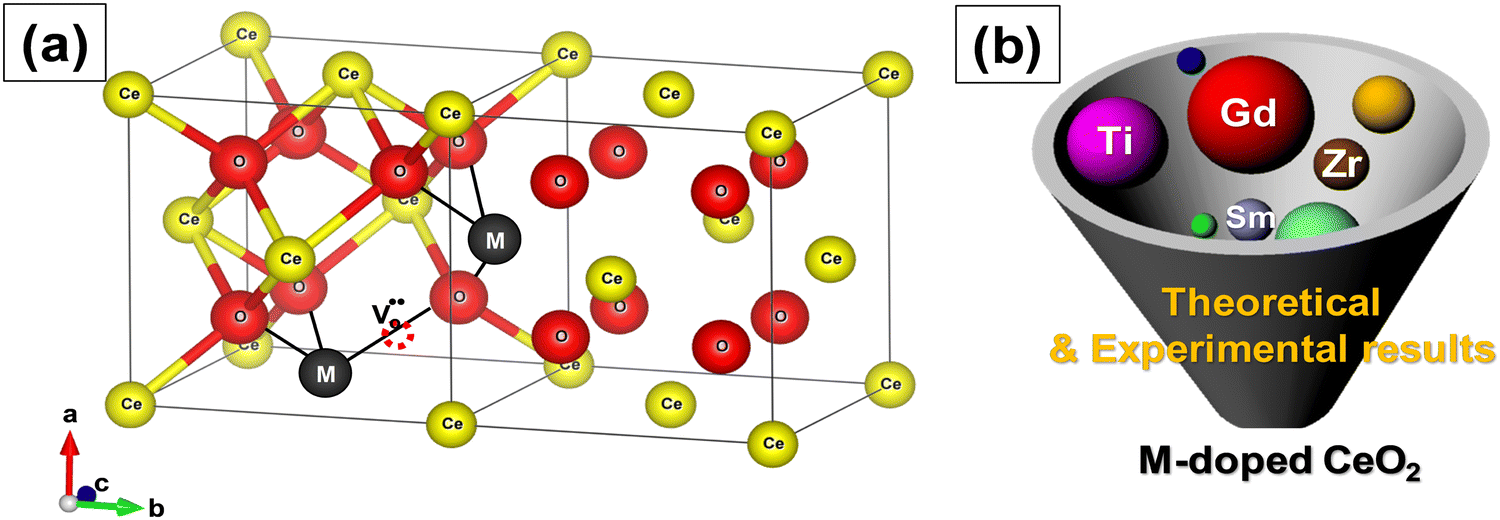 | ||
| Fig. 6 (a) Schematic of the crystal structure of ceria with metal ion dopant M. Addition of dopants (rare earth, transition or noble metal ions) during synthesis results in the formation of oxygen vacancies. (b) Development strategy for ceria with metal ion dopant M. | ||
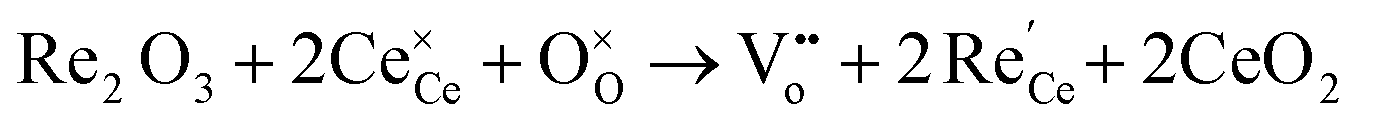 | (2.4) |
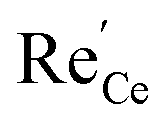 represents a Re3+ ion on a Ce lattice site.
represents a Re3+ ion on a Ce lattice site.
The increase in the concentration of Gd from 0 to 15% increased the OSC of CeNPs but with a further increase, the OSC decreased.104 The enhanced OSC was attributed to the increased specific surface area and decreased particle size. In contrast, Hennings et al. reported that the OSC decreased after with Gd.105 Nevertheless, the concentration of oxygen vacancies increased with the increase in Gd concentration. They argued that the drop in OSC was due to the replacement of reducible Ce4+ by non-reducible Gd3+ and the suppressed reduction of Ce4+. Trivalent La, Sm, Y and Gd increase the concentration of Ce3+ ions on the surface of particles.106 An increase in Ce3+ concentration enhances oxygen vacancies and facilitates the transport of oxygen from the bulk to the surface. La and Pr doping boosted the concentration of surface reducible Ce4+ ions and shifted the reduction peak of CeNPs to lower temperatures.107 It is important to point out that the optimal concentration of dopant required to obtain the maximum increase in the concentration of Ce3+ and oxygen vacancies depends upon the type of the dopant. For instance, in one study, it was shown that the optimal concentration of La and Pr was about 5% and 50%, respectively.108
Of all RE elements, Pr yields the maximum concentration of oxygen vacancies. In case of Pr doping, the presence of both Ce3+ and Pr3+ ions increased the concentration of oxygen vacancies due to the reduction of Pr4+ to Pr3+ and Ce4+ to Ce3+.107 Of Nd- and La-doped CeNPs prepared using the microemulsion method, the oxygen vacancy defect concentration was higher for La-doped ceria than for Nd-doped ceria, and this difference was attributed to the larger ionic radius of La as compared to Nd.109 Y, Sm and Gd doping increased the Ce3+ and oxygen vacancy defect concentration while Yb doping reduced it due to the smaller ionic radius of Yb3+ as compared to that of the other three ions.110 Er and Ho also decreased the density of oxygen vacancies due to a similar reason.111 Using DFT calculations, Kim et al. established a trend between the radius of the RE metal ion and oxygen vacancy formation energy, which is related to the catalytic activity.112 The relationship between the oxygen vacancy formation energy and the ionic radius of RE metal, and the highest reaction energy and ionic radius of RE metal ion (Fig. 7a and b) showed an increase in the vacancy formation energy with the ionic radius, whereas the maximum reaction energy (for the case of CO oxidation considered as an example) decreases with the ionic radius. The contribution of the dopant to oxygen vacancy formation energy was also investigated. The ionic radius of the dopant (Rion) was also found to influence the RE–O interaction, which in turn altered the oxygen vacancy formation energy (Evf), by measuring how easily oxygen vacancies are formed. While most research suggests that doping CeNPs with RE elements typically results in expansion, the radius and coordination number of the dopant ion also play an important role and can lead to variations in RE–O bonds length and different Evf. The charge density distribution between RE–O was studied by calculating the RE–O bond strength. The charge of cerium ion in pure CeO2 is 4+ and the ionic radius of Ce4+ is 114.3 pm. In contrast, the charge of the other RE elements in CeO2 is 3+ and the ionic radius of the RE ion is relatively smaller than that of Ce4+ with La being an exception. As a result, structural stress is induced along RE–O. The stress created by the dopant ion having a smaller ionic radius weakens the strength of the RE–O, as shown in Fig. 7c. Therefore, dopants having a lower ionic radius lead to a lower Evf.
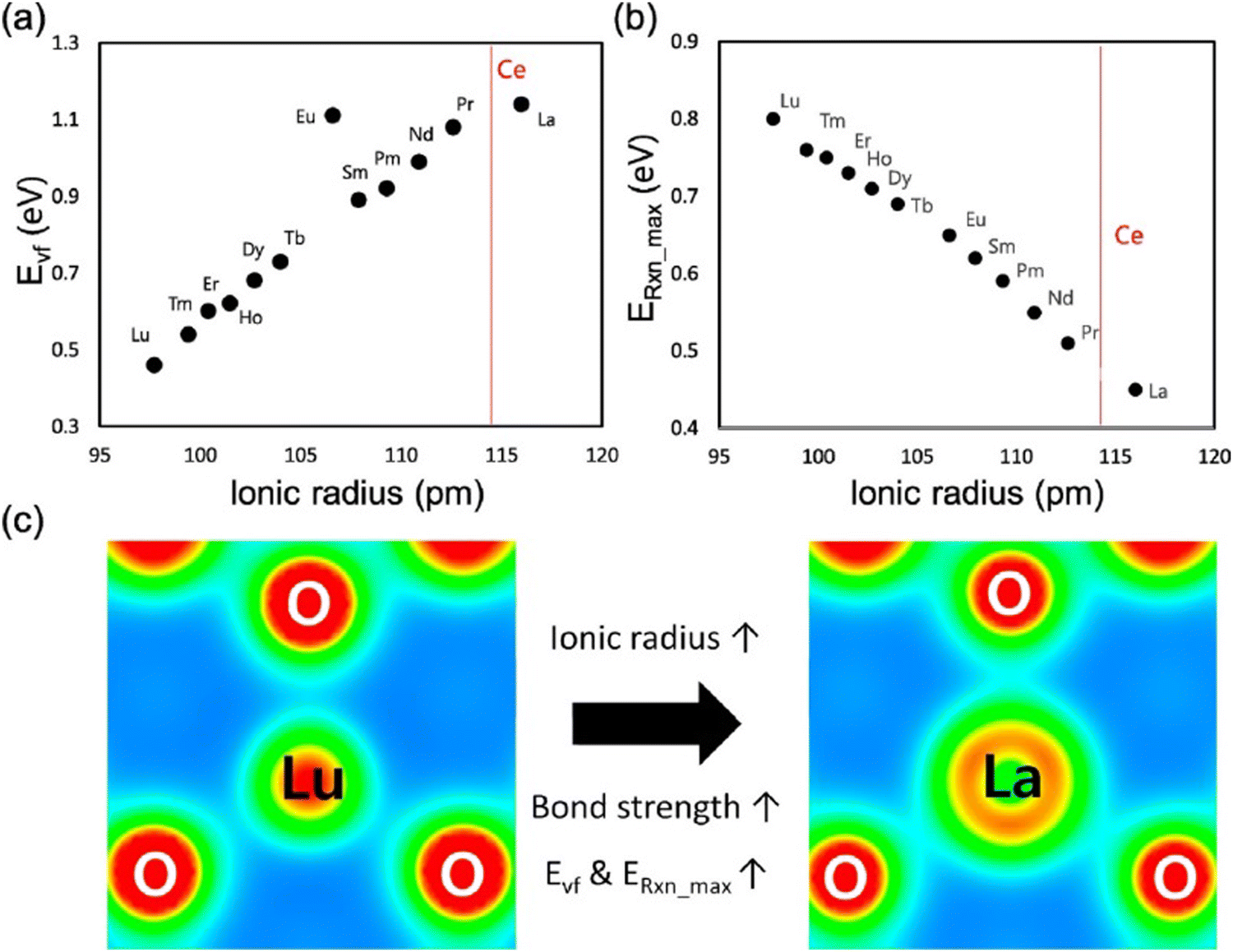 | ||
| Fig. 7 Relationship between (a) oxygen vacancy formation energy and radius of RE ion and (b) maximum reaction energy and radius of RE ion. (c) Charge density distribution in RE ion having largest ionic radius (La) and with smallest ionic radius (Lu) bonded with O atoms in RE doped ceria. Reprinted from permission from ref. 112. Copyright 2017 American Chemical Society. | ||
Because lanthanide complexes have excellent luminescence, these structures can be widely used in biosensing and bioimaging applications.113,114 CeNPs show weak emission characteristics that restrict their use in fluorescence-based imaging and sensing. Europium (Eu), one of the rare earth metals, has a strong red light emission upon doping and is considered a suitable dopant to enhance emission in CeNPs as the ionic radius of Eu3+ ion (0.1066 Å) is higher than that of Ce4+ (0.097 Å) but lower than that of Ce3+ (0.1143 Å) and shows excitation from ultraviolet to visible region.115 Therefore, Eu doping intensifies the photoluminescence properties of CeNPs, opening up many possible opportunities for applications in molecular imaging. The appearance of a broad band in the excitation spectrum of Eu-doped CeNPs is due to the charge-transfer from O2− to Ce4+.116 Both Ce3+ and oxygen vacancy concentration increase with an increase in Eu concentration. The conflicting roles of Ce3+ ions and oxygen vacancy defects in influencing the photoluminescence response of doped CeNPs have been reported. Kumar et al. found that the increase in Ce3+ concentration enhanced the photoluminescence properties, whereas an increase in vacancy defect concentration adversely affected the photoluminescence by interfering with the radiative route of emission.117 Erbium (Er) is another rare-earth element that has been used by several investigators as a dopant to impart fluorescence in ceria particles.118,119
Not all trivalent rare-earth metals increase the concentration of Ce3+ and oxygen vacancies. An important question that then needs to be answered is what cations can be used for doping ceria particles? Several studies suggested that dopants having ionic radii lesser than that of Ce4+ ion can be doped into the ceria lattice. However, this is not always the case as evidenced from many contradictory results reported in the literature.2,120 When the dopant substitutes a Ce4+ ion in the crystal lattice of ceria, the periodic electrostatic field distribution changes due to different electronegativity, oxidation state and ionic radius of the dopant with respect to the Ce ion. The thermodynamics (enthalpy) of dopant incorporation and other thermodynamically relevant factors can be potentially helpful in identifying the right dopant. Computational techniques can also provide some guidance in understanding the thermodynamics of dopant addition to the ceria lattice.
Among composite structures, zirconia-doped ceria (ceria–zirconia) has been studied intensively due to its significance in various industrial catalytic reactions. Ceria forms a substitutional type solid solution with zirconia. When CeNPs are doped with zirconia, the smaller Zr4+ ion causes lattice distortion, leading to an increase in oxygen mobility. After introducing Zr4+ ions into the crystal lattice of ceria, both Ce–O–Zr and Ce–O–Ce bond lengths in CexZr1−xO2 lattice decrease and oxygen shows a centro-symmetric eight-fold coordination.121 This change in the local environment of oxygen around Ce4+ and Zr4+ cations leads to the formation of active oxygen species that plays an important role in the OSC property and therefore its catalytic activity. The reduction of ceria in ceria–zirconia solid solutions can be described by the following chemical reaction.
| CexZr1−xO2 + H2 ↔ CexZr1−xO2−δ + H2O + Vo | (2.5) |
Ceria–zirconia catalysts have gradually replaced pure ceria whose properties do not meet the requirements of high conversion efficiency and thermal resistance needed to sustain stringent emission regulations. Additionally, a critical need for more thermally stable materials to improve the performance of TWCs during cold start led to the development of several strategies. Among them, the doping of ceria–zirconia with rare earth elements like La, Pr, Y, Sm and Nd resulted in an increased thermal stability and OSC.125 The cerium precursor salt was found to have a significant influence on the catalytic properties. The ceria–zirconia catalysts prepared using cerium salt having +4 oxidation state, (NH4)2Ce(NO3)6, was found to have higher Ce3+ concentration than those prepared using cerium salt having +3 oxidation state Ce(NO3)3·6H2O.126 Also, the catalysts prepared with Ce4+ precursor salt had higher Zr concentration and showed improved oxygen mobility than those prepared by the Ce3+ precursor. The catalytic activity of ceria–zirconia catalysts is dependent on both the BET surface area and Ce/Zr surface atomic ratio. The Ce/Zr value in turn depends on the synthesis technique and therefore the catalytic activity strongly depends on the preparation method. Thus, the BET surface area of the catalyst prepared using Ce(NO3)3·6H2O was higher than that prepared using (NH4)2Ce(NO3)6; the latter showed higher catalytic activity, which was attributed to the higher Zr concentration on its surface.
In other reports, the addition of Cu to the ceria lattice increased its OSC and redox properties by reducing the activation energy for Ce4+ reduction, as showed by Hu et al.127,128 Their results showed that the Cu/Ce ratio in the catalyst strongly influenced the catalytic and redox properties. The pore structure and morphology of Cu-doped ceria NPs can be affected by the precursor used for synthesis. CuCeOx nanofiber catalysts prepared by the electrospinning technique showed higher catalytic activities as compared to those prepared by other conventional methods.129 The enhanced catalytic activity of nanofibers was ascribed to the large specific surface area and increased concentration of Ce3+ ions and oxygen vacancy defects. The doping of CeNPs with Cu also increased the overall surface area and improved the redox properties.
Mn doping into ceria lattice has also shown to increase the concentration of oxygen vacancy defects. XPS analysis of the surface indicated that Ce is present in +4 or +3 oxidation state while Mn exists in +3 or +2 oxidation state.130 EPR analysis indicated the presence of Mn2+ and Mn3+ species in the defect sites of the ceria lattice, interstitial spots and on the surface of ceria. Doping with Mn decreased the particle size of calcined CeNP powders and dramatically increased the BET surface area.131 Impedance spectroscopy measurements have revealed that the Mn ions enter into the solid solution and increased the conductivity by decreasing the apparent activation energy. DRIFTS results showed that a fraction of Mn ions remain segregated on the particle surface. The remarkable enhancement in the catalytic activity of Mn-doped ceria at low temperature is ascribed to the good dispersion of Mn2+/Mn3+ ions in the ceria matrix, high OSC, increased redox properties and synergistic interaction between the Mn and Ce. Among co-precipitation (CP), sol–gel (SG) and hydrothermal methods used to prepare Mn-doped ceria, the catalyst synthesized by the HT method showed very high catalytic activity at low temperature.131 The larger surface area, increased reducibility, higher concentration of surface adsorbed active oxygen species and an increased number of oxygen vacancy sites are the possible reasons for the enhanced catalytic activity of the mixed catalyst prepared by the hydrothermal method.
In other structures, the presence of Ni3+ ions and a very high concentration of oxygen vacancy defect sites in Ni-doped ceria NPs (Ni0.1Ce0.9O2−x) has showed an increase in the surface reducibility of these doped structures as compared to pure ceria.132 Ni-doped ceria showed superior electrochemical properties due to the increased concentration of Ce3+, Ni3+ and oxygen vacancy defects used to boost faradaic surface redox reactions.133 Doping with Ni leads to additional structural stress and lattice expansion and extrinsic defects on the particle surface and near grain boundaries. It also results in enhanced surface oxygen reactivity and thus more active reduction sites at their surface. Other reports explored the use of Co to increase the catalytic properties of CeO2 and found the enhanced performance of the Co–CeO2 catalyst as compared to that of bulk Co3O4 or ceria due to a synergetic effect between Co and CeO2, leading to the increased concentration of oxygen vacancy defects and enhanced reducibility.134 The preferential exposure of the (112) crystal plane of Co–CeO2, which contained a large concentration of Co3+ active sites, was also suggested to be responsible for the higher catalytic activity. The increase in cobalt dopant concentration also increased the band gap energy and reducibility of the catalyst.135
Fe doping of ceria enhanced the catalytic activity due to the increased concentration of surface Ce3+ and chemisorbed reactive oxygen species.136 The addition of iron into the crystal lattice of ceria decreased the crystallite size and increased the specific surface area.137 Fe-doped ceria favors sintering at lower temperatures, enhances the reducibility and increases the number and strength of basic sites.138 The doping of ceria with iron can also improve the conductivity, decrease the activation energy and enhance the dynamic oxygen storage capacity (DOSC). Therefore, the Fe-doped ceria catalyst showed low temperature activity and increased total oxygen storage capacity (TOSC). It was suggested that the incorporation of Fe into the crystal lattice of ceria strongly modifies the kinetics of oxygen diffusion and enhances the OSC.
Ag, Au, Pt and Pd doping are other suitable approaches to enhance the catalytic activity of CeNPs. The addition of Ag favors the formation of reducible oxygen species that is suggested to be responsible for the improved catalytic activity.139 The synergistic interaction between Ag and ceria is the key for the low-temperature reduction of ceria.140 Three different methods used to prepare Pt-doped ceria catalyst resulted in different values of ionic Pt/Pt0 ratios.141 Furthermore, these three different catalysts were found to achieve varying catalytic activities, increasing with the increasing concentration of ionic Pt species. DFT calculations suggested that the presence of ionic Pt species activated the oxygen atoms on the cerium next to it, resulting in decreased activation energy for dissociative methane adsorption. As a result, the novel square-planar configuration of PdO4 in Pd-doped ceria is shown to be more reactive.142 The creation of an oxygen vacancy in this structure is energetically more favorable. The structural and chemical attributes of Pd–O–Ce moieties were found to contribute to the higher catalytic activity of Pd-doped ceria.143 All Ce ions in the crystal are in 4+ oxidation and they are not reduced to 3+ state upon the formation of oxygen vacancies, as evident from the density of electron states, where the filled Ce 4f gap states associated with the reduced ceria are not present. The physicochemical properties of rare earth, transition and noble metal-doped ceria particles are summarized in Fig. 8.
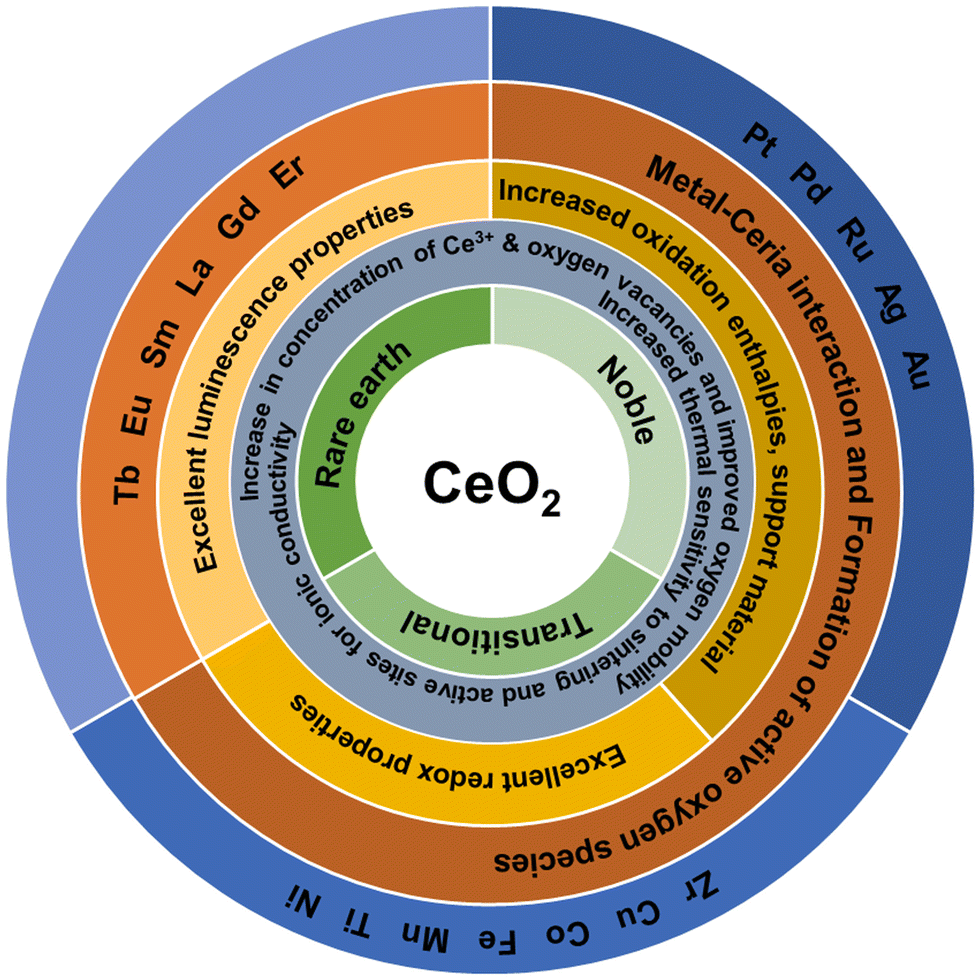 | ||
| Fig. 8 Schematic of the physicochemical properties of rare earth, noble and transition metal-doped ceria particles used to obtain mixed and doped structures with CeO2. | ||
The improved catalytic activity of doped CeNPs has been generally attributed to an increased concentration of oxygen vacancies and Ce3+ and OSC. Other factors like large surface area, improved redox activity, enhanced thermal resistance and the synergistic interaction between the metal and ceria also contribute to the enhanced activity. Almost all transition and noble metals investigated were reported to enhance the catalytic activity. Nevertheless, other factors like the preparation method, concentration of the dopant, type of dopant, oxidation state, and crystal structure also influence the catalytic properties of doped ceria.144–146
Experimental studies provide critical information on the characteristics of the catalyst, oxidation reactions, surface processes and its mechanism. The selection of appropriate characterization methods is essential in gaining mechanistic details on important aspects of the ceria's activity such as metal–ceria interactions, role of the dopant, oxygen vacancy defect and surface oxygen species.125 In addition to experimental tools, theoretical studies based on computer simulations can provide an atomistic understanding of the structure of ceria-based catalysts, the role of dopant, oxygen vacancy defect and Ce3+ ions on the catalytic properties of catalysts.
Detailed structural analysis of Ce1−xZrxO2 catalyst suggests that the formation of long and short Zr–O bonds in fluorite structure is mainly responsible for enhanced OSC observed for ceria–zirconia.147–149 The substitution of Ce4+ ion with Zr4+ increases the reducibility of Ce4+ in ceria, although ZrO2 is not a reducible oxide. The addition of reducible ions like Sn4+ and Ti4+ into the ceria lattice can enhance the OSC of Ce1−xSnxO2 and Ce1−xTixO2, respectively. Along with the Ce4+/Ce3+ couple, the Sn4+/Sn2+ and Ti4+/Ti3+ redox couples can also contribute to and therefore increase the reducibility of CeNPs. The coordination around Ti4+ or Sn4+ ion is different from the fluorite structure of ceria due to the lower ionic radius (0.74 Å for Ti4+ and 0.69 Å for Sn4+) of these ions as compared to that of Ce4+ ion (0.97 Å) and this results in distortion of the ceria lattice.150–152
Computational methods were used to study the properties of doped ceria materials.72,153,154 The distortion of the oxygen sublattice leads to long and short Ce–O and Ti–O, Zr–O, Fe–O, and Zn–O bonds in CeO2–TiO2, CeO2–ZrO2, CeO2–Fe2O3 and CeO2–ZnO2 solid solutions. The increased reducibility of these materials was explained by weak longer bonds.155,156 The Ce4+ coordination gets distorted to 4+4 type coordination from its ideal 8-fold coordination in these different materials. Transition and noble metal ion doping significantly enhanced the reducibility of Ce1−xMxO2−δ (M = Co, Mn, Cu, Ni, Fe, Ru, Pt, and Pd), whereas doping with rare earth metal ions Ce1−xAxO2−δ (A = La, Y) was seen to have little effect in increasing the OSC and reducibility of ceria. The optimized structure obtained by computer simulations exhibited a deviation in the bond length of cation–oxygen from the ideal bond length value of 2.34 Å (for Ce–O in CeO2).155 For instance, simulation results for Ce28Mn4O62 structure showed that the mean Mn–O bond length was 2.0 Å in 4+2 coordination. Doping with other transition and noble metal ions also changed the coordination of Ce4+ and formed longer Ce–O bonds. The addition of Pd in Ce1−xMxO2−δ (M = Mn Ni, Fe, Cu and Co) resulted in a further increment in OSC; correspondingly, the model calculations revealed a further increment in the bond length. These enhancements have been attributed to the improved reducibility of both the host and dopant ions because of lattice distortion in the presence of the dopant.72 In the case of rare earth ion doping, the calculations showed a very little increase in bonds lengths from the fluorite structure; therefore, the absence of longer Ce–O and RE–O (rare earth or RE = La and Y) bonds make the resulting structure less susceptible to reduction.153
Dopants like Zr, Hf, Ti, Nb and Ta in the (110) ceria surface reduce the oxygen vacancy formation energy.157 Pentavalent dopants like Nb and Ta in the (110) ceria surface enhance reducibility by charge transfer from the dopant ion to a cerium ion in the surface. The doping of ceria with Pd and Pt also lowers the oxygen vacancy formation energy, attributable to the gap states formed above the valence band and below the empty Ce 4f states.158 Alfredsson and Catlow compared the adsorption energies of Pt and Pd on (111) zirconia and ceria surfaces using periodic DFT analysis.159 They found higher adsorption energies for Pt layer (400 kJ mol−1) on both ceria and zirconia than Pd layer (200 kJ mol−1).
Krcha et al. used density functional theory (DFT+U) to investigate the structural and electronic effects of transition metal dopants belonging to groups IV–XII in the (111) surface of ceria.160 The dopant can have an oxidation state anywhere between 3+ and 8 +, and, in such a case, the Ce4+ ions are reduced to Ce3+ only when the dopant has an oxidation state higher than 4+. Fig. 9a summarizes the oxidation state of dopant in both the completely oxidized and oxygen vacant surfaces. The transition metal ions in groups IV and V change the surface reducibility and the ones in groups X–XII become the reduction center. Metal ions in groups IV and V are associated with their stable oxidation states of 4+ and 5+, respectively, in the oxidized as well as reduced surfaces. Metal ions of groups X–XII are associated with 4+ oxidation state in the oxidized surface and a 2+ oxidation state in the reduced surface. Au, however, is an exception as it has a 3+ oxidation state in the reduced surface. There is no clear trend in metal ions of groups VI through IX. Group metals have a 6+ oxidation state and the metals in the 4th row of groups VII, VIII and IX (Co, Fe and Mn) have a 4+ oxidation state in the oxidized surface and a 3+ oxidation state in the reduced surface. They also showed that the oxygen vacancy formation energy decreases with the group number, as shown in Fig. 9b. The oxygen vacancy formation energies are usually higher when the dopant reduces as compared to only when Ce reduces. Oxygen vacancy formation increases more or less linearly with M–O bond energy, as shown in Fig. 9c.
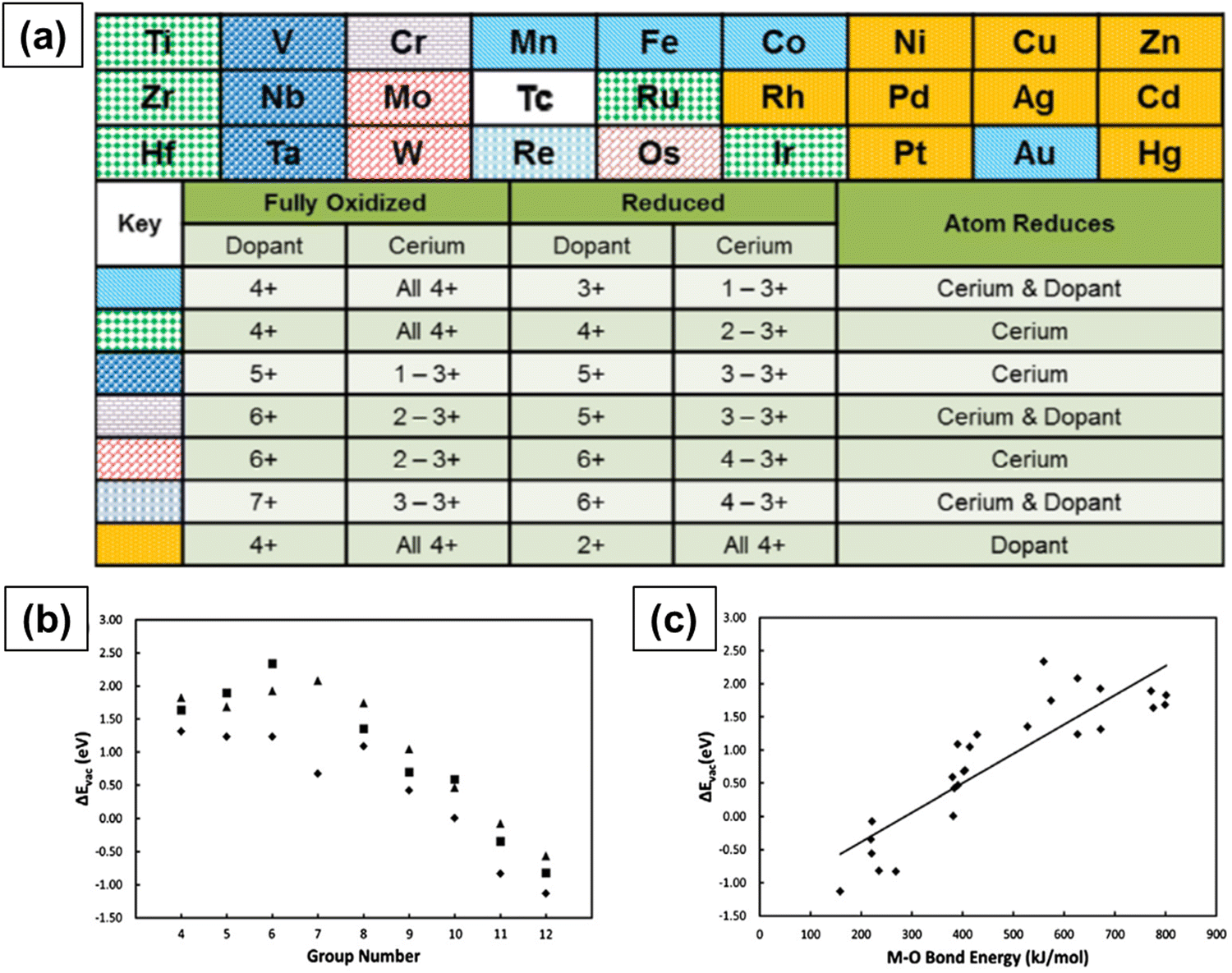 | ||
| Fig. 9 (a) Trends in the electronic behavior of M-doped CeO2 upon the formation of a surface oxygen vacancy in the nearest neighbor. Relationship between (b) oxygen vacancy formation energy and metal ion dopant column in the periodic table of elements and (c) oxygen vacancy formation energy and M–O bond dissociation energy. Reprinted from ref. 160. | ||
Structural changes or effects modifying the Ce–O chemical bonding are local effects while the global electronic structure is considered as a global effect.101 To identify whether a particular dopant exhibited a local or global effect, the dopant ion was placed next to and far away from the oxygen vacancy and the vacancy formation energy was calculated. If the energy of forming an oxygen vacancy far away from the dopant is equal to the energy of forming a vacancy on nondoped oxide, then the dopant has a local effect. On the other hand, if the energy needed to form a vacancy next to the dopant is equal to that needed to form a vacancy far from it, then the dopant has a global effect. Dopants like La and Y reduce the energy needed for the formation of an oxygen vacancy next to them or far from them.72 Such dopant ions affect vacancy formation energy due to the creation of a hole in the valence band maximum. When an oxygen vacancy is formed, one of the two electrons fills the hole and the other electron occupies a Ce 4f state. As the Ce 4f gap states are energetically higher than the hole states, La or Y doping lowers the vacancy formation energy. This is considered as a global effect as this mechanism is found to be independent of the distance between the vacancy formation site and the dopant. In case of non-reducible dopant like Zr, since Zr4+ is smaller than Ce4+, structure relaxation effects drive the lowering of the vacancy formation energy and hence the energies of the doped and defective surfaces are about the same. On the contrary, reducible dopants like Pt, Ru, Nb and Ta exhibit both local and global effects and hence the energy required to form a vacancy near the dopant is lower than that required to create a vacancy defect away from it. The local effect with these dopants is related to the chemical interaction, i.e., dopant-oxygen binding.
The catalytic properties at the interface of ceria–metal have been extensively studied as this interface is the favored spot for the exchange of oxygen and is recognized as the active site responsible for the enhanced catalytic activity.161,162 An interfacial reaction mechanism was proposed in which CO is suggested to be adsorbed on the metal and then oxidized by oxygen transfer from ceria, which in turn is oxidized by H2O.163,164 More detailed information has been obtained on the ceria–metal interaction, in which ceria is supported on the metal.165 Additional proof of the interfacial activity involving the dissociation of CO-like molecules on oxygen vacancies on the ceria surface and a systematic investigation of ceria–metal properties has been demonstrated using advanced characterization techniques.166–168 Some of the important fundamental observations of these studies explain the enhanced catalytic activities due to (a) the interplay between metal and oxygen vacancy, with the metal enabling the creation of oxygen vacancies on ceria; (b) the primary role of interface spots in the creation of reaction intermediates, therefore describing the different behaviors of metal–ceria systems and the ceria or metal alone; and (c) the presence of Ce3+ species under transient reaction conditions in CO oxidation mechanism.
Theoretical studies using DFT calculations have unraveled a great deal of information on the structure of ceria-based catalysts, the mechanism and the role of oxygen vacancy defects and dopants in enhancing the catalytic activity. These results are in line with those of experimental studies and hence are helpful in gaining a fundamental understanding of the catalytic attributes of ceria-based materials in industrially relevant applications. While extensive research has been dedicated to this topic over the past twenty years, there remain several questions that still have to be explored. (i) Will different sized doped-ceria materials behave differently – does the size of the doped-ceria particle have an influence on catalytic activity; (ii) can any dopant enhance the catalytic activity – can dopants be grouped into different categories like dopants that have a very little effect and the ones that have significant effect on the catalytic activity; (iii) will doping with multiple metals enhance the catalytic activity greatly? If yes, what combination of metals can do this; (iv) will surface coating on doped ceria particles alter their catalytic properties (iiv); is surface modification by the addition of metal oxide clusters possible? If yes, what impact will it have on the catalytic activity?
2.5. Free radical scavenging
The distinctive free radical scavenging and regenerative antioxidant properties of the CeNPs have attracted considerable interest in a broad range of applications in biomedicine.169–171 In 2007, Seal's group first showed that CeNPs can efficiently scavenge superoxide radical anions and attributed this scavenging activity to the ability of Ce to switch between 3+ and 4+ oxidation states.172 Subsequently, Pirmohamed and coworkers reported that the CeNPs show catalase mimetic activity.173 Over the past decade, CeNPs have been investigated intensively as a potential therapeutic tool to inactivate free radicals and ameliorate causes of oxidative stress in a variety of in vitro and in vivo models, including cells, bacteria and whole animals. CeNPs can show antioxidant enzyme-mimetic activity, scavenge reactive oxygen species (ROS) and reactive nitrogen species (RNS) and protect against radiation.174–178 They were also found to be effective in treating many diseases including cardiovascular diseases and neurodisorders.170,179,180 Here, we review the reaction mechanisms and discuss the physicochemical properties of CeNPs that are responsible for their ROS/RNS scavenging activities and enable them to be used in pharmaceutical and biomedical applications.| O2˙− + 2H+ + (Cu+)-SOD → H2O2 + (Cu2+)-SOD | (2.6) |
| O2˙− + (Cu2+)-SOD → O2 + (Cu+)-SOD | (2.7) |
| 2O2˙− + 2H+ → H2O2 + O2 (overall) | (2.8) |
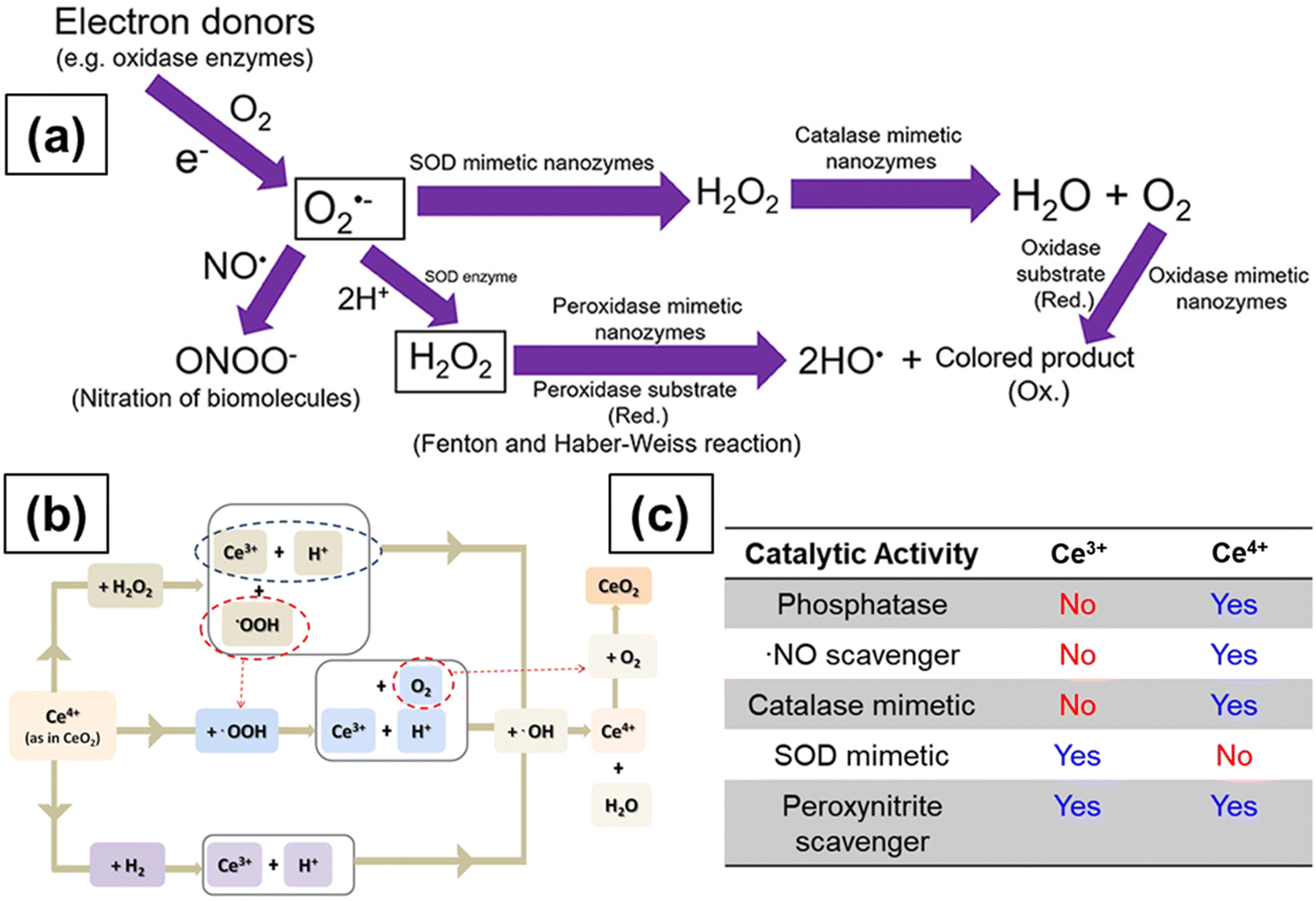 | ||
| Fig. 10 (a) Schematic representation showing the reactions involved in antioxidant enzyme-mimetic and ROS and RNS scavenging properties of ceria particles. Superoxide (O2˙−) anions are first produced by single electron donors and the O2˙− anions react with ˙NO and H+ to form OONO− and H2O2, respectively. The O2˙− anions are converted to H2O2 by SOD and SOD mimetic enzymes. Catalase mimetic enzymes can further convert H2O2 into water and oxygen. Hydroxyl radicals can be generated from H2O2 in the presence of peroxidase mimetic enzymes, and oxidase mimetic enzymes can oxidize the substrate and form a colored product without the presence of H2O2. Reprinted from ref. 199. (b) Transformation of Ce3+ to Ce4+ and regeneration of Ce3+ from Ce4+. Reprinted from ref. 193. (c) Effect of SOD and catalase mimetic properties and ˙NO and OONO− scavenging properties of ceria particles on the relative surface Ce3+ concentration of ceria. Reprinted with permission from ref. 200. Copyright 2013 American Chemical Society. | ||
The SOD enzyme works by accepting electrons from or losing electrons to O2˙−. In reaction 2.6, the reduced (Cu+)-SOD catalyzes the oxidation of O2˙− to H2O2 while in reaction 2.7, the oxidized (Cu2+)-SOD catalyzes the reduction of O2˙− to O2. Reduced (Cu+)-SOD is regenerated after reaction 2.7 and the cycle begins again. Overall, for every molecule of H2O2 formed, 2O2˙− molecules are dismutated. A similar mechanism for superoxide radical anion scavenging by ceria NPs has been proposed involving the oxidoreduction of Ce3+/Ce4+ states at the NP surface, as shown below.170,180
| O2˙− + Ce3+ + 2H+ → H2O2 + Ce4+ (oxidation of Ce3+ to Ce4+) | (2.9) |
| O2˙− + Ce4+ → O2 + Ce3+ (reduction of Ce4+ to Ce3+) | (2.10) |
The above two reactions indicate that the Ce3+/Ce4+ redox couple of ceria NPs can be regenerated. In the first step, superoxide anions bind to oxygen vacancies around two Ce3+ species and an electron is transferred from Ce3+ ion to an oxygen atom. In the second step, two protons in the solution bind to two oxygen atoms and form a molecule of H2O2. The second superoxide anion then binds to the remaining oxygen vacancy and a second H2O2 molecule is formed and Ce3+ is oxidized to Ce4+. Furthermore, the two electrons produced will reduce the Ce4+ ions to Ce3+ ions and, in this case, H2O2 acts as a reducing agent. However, while these reactions have been postulated, the mechanisms driving the SOD-mimetic activity of ceria NPs are still subject to debate and their behavior in biological environments are subject of numerous scientific investigations. In some studies, CeNPs having higher Ce3+ concentration were more efficient scavengers of O2˙− than those with lower Ce3+ concentration.172 In another study, CeNPs treated with H2O2 and hence having lower Ce3+ concentration showed a decrease in the SOD mimetic activity as compared to bare ceria, implying that the surface Ce3+ species play a key role in the SOD mimetic activity,187 as indicated in Fig. 10c. These processes take place at the surface of the metal oxide and are affected by conditions such as pH and temperature.169
High H2O2 levels are considered detrimental to cellular homeostasis as compared to high levels of O2˙− as H2O2 forms highly toxic ˙OH radicals via the Fenton reaction with metals, as shown in Fig. 10a. Enzymes like glutathione peroxidase, catalase and peroxiredoxins all lower the H2O2 levels in cells, but catalase is the most efficient enzyme in disproportionating H2O2 into innocuous H2O and O2,188 as shown in Fig. 10a. The catalase mimetic activity of CeNPs was first demonstrated based on the shift in the peak in the luminescence spectra of ceria NPs after the addition of H2O2 to astrocyte cells.174 The luminescence spectra of the CeNPs reversed to its original form (the peak shifted back to the original wavelength) after the decomposition of H2O2. The CeNPs protect cells from oxidative stress due to their catalase mimetic and SOD mimetic activities. The catalase mimetic activity depends strongly on the concentration of Ce3+ with ceria having low Ce3+ concentration, showing improved catalase activity as compared to those with high concentration of Ce3+,173 as indicated in Fig. 10c.
Besides, Ce3+ concentration, factors like particle size, buffer species like PO43−, and solution pH can all affect the catalase and SOD-mimetic activities of ceria.189,190 The presence of PO43− was shown to diminish the SOD-mimetic activity and enhance the catalase activity due to the ability of Ce3+ ions to form strong coordination complexes on reaction with PO43−, which bind preferentially to ceria NPs with high Ce3+ concentration. The mechanism by which phosphate changed the properties of ceria NPs was attributed to the formation of CePO4-like complexes that blocks the redox activity of ceria NPs.189 Since the ability of ceria particles to cycle between 3+ and 4+ oxidation states is the key to their catalytic activity, the catalase mimetic activity diminishes in the presence of any compound that can block Ce3+ and stabilize the ceria NPs in the 3+ state. Therefore, the ability of CeNPs to swiftly change between 3+ and 4+ oxidation states is crucial in maintaining their SOD-mimetic activity. These studies indicate the need to comprehensively study and understand the influence of phosphate ions and other ions on SOD mimetic activity of ceria, particularly because cells contain high concentrations of phosphate ions, and other ions and their presence can potentially affect the biological activities of ceria NPs.
Contradictory results were reported in the literature on the influence of pH on the antioxidant activity of CeNPs with some studies showing no effect of pH and other studies, revealing marked effects on both the SOD mimetic activity and Ce3+ concentration on the particle surface. Singh et al.189 and Xue et al.176 showed that the dispersion stability and SOD mimetic activity of ceria NPs are not affected by pH. In contrast, using XPS spectroscopy, Karakoti et al. showed that the Ce3+ concentration changed with pH.191 Perez et al. investigated the effect of pH on the antioxidant properties of CeNPs.190 Dextran coated-ceria NPs exhibited catalase mimetic activity in alkaline conditions but not in acidic conditions (pH 4). They argued that the antioxidant ability of ceria NPs is dependent on their ability to cycle between 3+ and 4+ oxidation states and that the Ce3+ ions are converted to Ce4+ ions during the H2O2 scavenging process. The regeneration of Ce3+ occurs through chemical reactions between Ce4+ ions on the surface of NPs and H+ ions in solution, as shown in Fig. 10b. The low pH of the solution inhibited the ability of ceria NPs to scavenge more free radicals and therefore diminished the antioxidant activity due to the high concentration of H+ ions. These conflicting results could be due to different synthesis methods and hence varying surface Ce3+ concentrations. Moreover, the effect of surface coating or surface reactions, the potential binding of peroxide and superoxide species to the ceria surface, and the stabilizing effects of coexisting ions have not been taken into account in these studies and can have significant effects in biological environments. Systematic studies of the effect of surface ligands, surface faceting and a potential reorganization of ceria NPs are the essential next steps needed to understand these processes, the interfacial reactions and their contribution to the ceria's unique reactivity.
CeNPs can also efficiently scavenge ˙OH (hydroxyl radical), a strong oxidant. Hydroxyl radicals in cells are usually scavenged by two methods. The first method involves preventing the initiation of ˙OH by enzymes like SOD, catalase and glutathione peroxidase. The second method involves the breaking of the chain reaction of ˙OH by nonenzymatic antioxidants.192 The ˙OH scavenging ability of CeNPs was first shown by Das et al., who first demonstrated that CeNPs were able to scavenge ˙OH formed from H2O2.175 When CeNPs were treated with H2O2, the color changed from yellow to orange, indicating that Ce3+ (yellow) reacts with the ˙OH generated from H2O2 and gets oxidized to Ce4+ (orange), as shown in Fig. 10a. In a later report, Xue et al. demonstrated that CeNPs scavenge ˙OH by providing direct experimental evidence via a methyl violet assay.176 With the decrease in the size of CeNPs and a corresponding increase in the concentration of surface Ce3+, the efficiency of CeNPs in scavenging ˙OH and preventing a reduction in the optical absorption of methyl violet increased. The ˙OH scavenging activity of CeNPs was attributed to a two-step mechanism involving the ability of ˙OH to reversibly cycle between Ce3+ and Ce4+. In the first step, ˙OH oxidizes Ce3+ to Ce4+, and in the second step, Ce4+ is reduced back to Ce3+. The mechanism can be described by the chemical reactions 2.9 and 2.10 and is also schematically represented193 in Fig. 10b. The morphology of CeNPs was also found to have a significant impact on their scavenging activity to the different crystal planes exposed by different morphologies. The antioxidant activity of different morphologies follows the order: nanowires > nanobars > NPs.194 A better understanding of the catalytic mechanisms and the fundamental parameters affecting the ‘enzyme-like’ activity will enable the future development of high performance nanoenzymes195,196 as alternatives to natural oxidase and peroxidase enzymes, providing advantages such as a stability, robustness and low cost, making them suitable for many applications.
| Ce4+ + ˙NO → (Ce4+NO ↔ Ce3+NO+) | (2.11) |
Fig. 10c summarizes the SOD and catalase mimetic properties and ˙NO and OONO− scavenging properties of CeNPs and the effect of the relative surface Ce3+ concentration on varying radicals.
| Ce3+ + H2O2 + H+ → Ce4+ + ˙OH + H2O | (2.12) |
| ˙OH + H2O2 → HO2− + H2O | (2.13) |
| Ce4+ + HO2− → O2 + Ce3+ + H+ | (2.14) |
CeNPs can exhibit peroxidase-like catalytic activity over a wide range of temperatures and pH values, making them more stable substitutes to natural enzymes.201
A phosphatase is an enzyme that hydrolyzes phosphoric acid monoesters into phosphate ions.202 Phosphatase enzymes play a significant role in many biological processes, including signal transduction and cellular regulation. Some metal ions and their complexes were shown to enhance the rate of phosphate ester hydrolysis through different processes like nucleophile activation, Lewis acid activation and leaving group activation.203 Lanthanide ions and their complexes are efficient catalysts for hydrolyzing phosphate esters.204 CeNPs have been shown to effectively mediate the dephosphorylation of phosphopeptides205 in less than ten minutes. The dephosphorylation activity of CeNPs was attributed to the Lewis acidity of Ce3+/Ce4+ surface sites capable of coordinating oxygen atoms of phosphate groups and lowering the P–O bond scission energy by promoting nucleophilic attack from hydroxyl groups on the surface. Ceria NPs can also be used for the dephosphorylation of biologically active amino acids.206 Kuchma and collaborators investigated the reactivity of CeNPs towards phosphate ester bonds of biologically relevant molecules like ATP, p-nitrophenylphosphate (pNPP), DNA and o-phospho-L-tyrosine.207 The dephosphorylation mechanism was investigated using DFT calculations, and the reaction was found to proceed through phosphate group inversion similar to an SN2 mechanism. Because of the ability of CeNPs to interact with the phosphate ester bonds of biologically relevant molecules, they can be used as potential therapeutics.
The ability of CeNPs to eliminate surplus levels of O2˙− and H2O2 from cells makes them ideal SOD and catalase-mimetic. Nevertheless, the exact mechanisms by which CeNPs act as antioxidant enzyme-mimetic in the cells have been a topic of debate. The variability in the types of CeNPs used in the in vitro and in vivo studies can strongly influence the surface Ce3+ concentration and hence their surface reactivity. Limitations of existing characterization tools and lack of sophisticated techniques to analyze such interactions in situ hinder the fundamental understanding of the enzyme-mimetic activity of the CeNPs. The apparent differences in results can also arise from the different synthetic procedures of the particles. Moreover, the limited data on the detailed characterization of CeNPs exposed to biological environments that would take into account the effect of the medium composition, surface binding and non-specific adsorption on the particle reactivity makes it difficult to draw fundamental conclusions based on a direct comparison with the reported mimetic activity in simple aqueous solutions. Ideally, standards for the synthesis techniques of CeNPs with a full characterization of their physicochemical properties should be established to prevent the misinterpretation of results. Advances in different microscopy spectroscopic methods have been very helpful in the analytical characterization of these materials under biological conditions. Establishing a set of characterization tools that enable comprehensive correlation between the physicochemical properties of CeNPs and their antioxidant/prooxidant activity is necessary and, in principle, can be helpful in tuning the properties of the particles. A schematic of the SOD and catalase mimetic activities of pure ceria, triphenylphosphonium (TPP) coated ceria208 and Gd-doped ceria209 is shown in Fig. 11, indicating the effect of surface ligands on CeNPs activity.
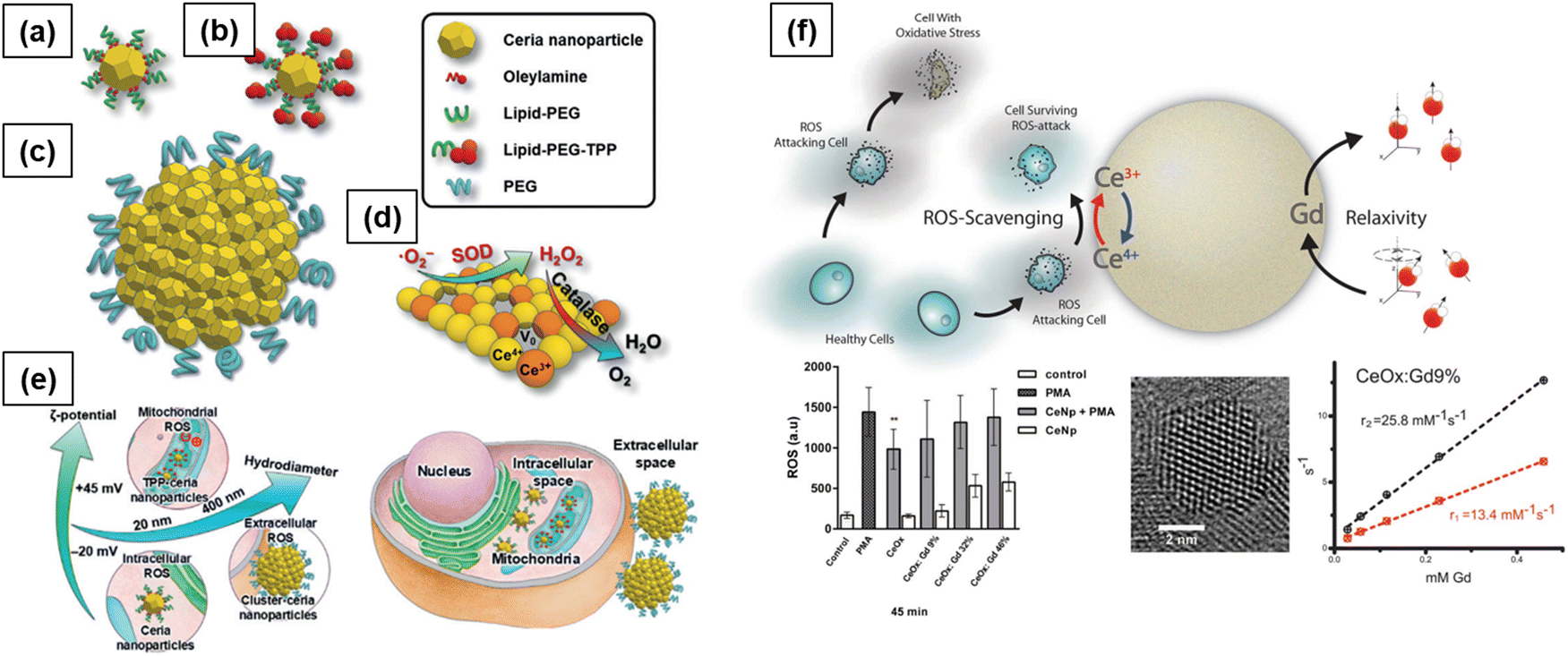 | ||
| Fig. 11 Schematic showing (a) ceria particle, (b) triphenylphosphonium (TPP) coated ceria particle, (c) ceria particle cluster, (d) SOD- and catalase-mimetic activities of ceria particles and associated ROS scavenging reactions, (e) ROS scavenging activities of ceria, TPP-coated ceria and ceria particle cluster in cells and (f) top: Gd-doped ceria particles with antioxidant and MRI contrast enhancing properties; bottom: antioxidant assay, HRTEM image and relaxivity measurement results (from left to right). Reprinted from ref. 208 and 209. | ||
2.6. Interactions with molecular oxygen and H2O2
The characterization of different types of oxygen species formed by the adsorption of O2 on CeNPs is important as these species play a key role in many oxidation reactions. One of the major applications of CeNPs is as an oxygen storage material in TWC converter in automotive exhausts. Ceria acts as a buffer to supply O2 during O2-deficient conditions of engine operation and extract O2 when there is surplus O2 in the exhaust stream, thus helping to maintain the concentration of oxygen. The mechanism of oxidation reactions on ceria surfaces involves the reaction of the adsorbate with the O2 present in the substrate, which is then replenished from the gas phase O2.125 Studies to understand the adsorption and desorption of O2 on ceria surfaces are thus critical for TWC applications. Using both experimental methods and theoretical models, it has been established that the reactivity of stoichiometric ceria (unreduced ceria) towards molecular oxygen is very low as compared to that of reduced ceria. O2 physisorbs on the surface of unreduced ceria NPs by a weak interaction dominated by dispersion forces.210–212 DFT calculations of oxygen adsorption revealed very low adsorption energies and showed that O2 adsorbs only at very low temperatures on unreduced ceria. On the other hand, molecular oxygen binds very strongly on reduced ceria surfaces.213–215The formation mechanism involves the creation of an oxygen vacancy, which results in the formation of two Ce3+ ions; hence, the vacancy site is a two-electron donor center. O2 interacts with the oxygen vacancy and adsorbs such that one oxygen atom fills the vacancy and the other points out from the surface.210 O2 adsorption at an oxygen vacancy site therefore removes the defect and results in the formation of a surface peroxo species (O22−).216 Diamagnetic O22− can be detected by Raman and FTIR spectroscopy. The peaks related to O22− typically occur in the 820–880 cm−1 region.217 O2− species, on the other hand, are formed when O2 interacts with low-coordinated Ce3+ ions (steps, edges, dislocations, etc.), which are one-electron donors.218 In this case, oxygen vacancies are not involved and O2− species are formed in the vicinity of exposed Ce3+ ions.213 The fingerprint of the O2− species is, besides the characteristic EPR signal, a Raman or IR peak in the region 1110–1150 cm−1.216
Early experimental studies used FTIR spectroscopy to investigate the adsorption of O2 on ceria. The adsorption of O2 on partially reduced ceria resulted in an IR peak at 1128 cm−1 attributed to O2− species and at 883 cm−1 related to peroxide species. O2− and O22− are the intermediates formed during oxygen dissociation/re-oxidation that occurs according to the following scheme.219
| O2(aq/g) ↔ O2(ads) ↔ O2(ads)− ↔ O2(ads)2− ↔ 2O(ads)− ↔ 2O(lattice)2− | (2.15) |
As can be seen from Eq. 2.15, first, the O2− is formed after exposure to O2. The O2− is then converted into peroxide and finally to lattice oxygen by gaining more electrons from the surface. As a result of this re-oxidation, the surface will become electron-deficient and further electron transfer from the surface to O2 does not occur. Similar bands were also observed in studies that used Raman spectroscopy to probe the adsorption of O2 on ceria. Pushkarev et al. studied the adsorption of O2 on CeNPs using in situ Raman spectroscopy and demonstrated that O2 adsorbs on one- and two-electron defects and forms O2− and O22− species, respectively.215 Two bands each in ranges of 1120–1140 cm−1 and 830–880 cm−1, related to superoxide and peroxide species, respectively, were observed. The two different bands can be ascribed to disparate configurations of the binding of superoxide species to ceria and to the peroxide species bound to defects with different geometries. Temperature-programmed experiments revealed the re-oxidation of the surface by adsorbed oxygen species. The O2− that form first due to the adsorption of O2 are unstable and transform into peroxides, which then translate into lattice oxygen, similar to what was reported previously.
Wu et al. investigated the adsorption of O2 on ceria nanocubes, nanorods and nanooctahedra exposing (100), (110)/(100) and (111)/(100) crystal planes, respectively.220 They illustrated the mechanism of oxygen activation and transformation on these surfaces, which is pivotal in understanding the mechanism of oxidation reactions catalyzed by nanoscale ceria. The adsorption of O2 on ceria gave rise to a band at 1139 cm−1, which they attributed to the O–O stretching of O2−. The other observed peaks at 830 and 860 cm−1 were ascribed to peroxide species on isolated and clustered two-electron defects. The amount of adsorbed oxygen species on reduced ceria nanoshapes changes with the degree of reduction. The weak bands of adsorbed oxygen species in ceria nanorods suggest the presence of small amounts of defect sites. Temperature-dependent oxygen adsorption experiments revealed that the adsorbed O2− species were stable up to 350 K while the peroxide species were stable until 473 K. Based on their results, they suggested that the re-oxidation of ceria occurs, as shown in Eq. 2.15. 20% of adsorbed oxygen desorbs into gas phase through disproportionation reactions of O22− and O2− species, as shown below.
| O2 ads− + O2 ads− → O2 ads2− + O2 | (2.16) |
| O2 ads2− + O2 ads2− → 2Olattice2− + O2 | (2.17) |
The adsorption of oxygen on the (111) surface of ceria was studied using DFT calculations. O2 does not adsorb on oxidized surfaces and binds weakly to one-electron defect sites.210 O2− forms on the hollow sites of the (111) surface in a side-on configuration above Ce4+ ions. The O2− preferentially adsorbs on Ce4+ surface sites due to electrostatic interaction; hence, Ce4+O2− is predicted to be the reactive intermediate as Ce3+O2 is more stable. Partially-reduced ceria exposed to O2 resulted in the formation of O2− and O22−, and the associated Raman peaks were observed at 1131 and 825 cm−1, respectively.217 The energies of O2 adsorption on unreduced ceria surfaces were found to be endothermic while those on reduced surfaces were exothermic. The position of oxygen vacancies can affect the formation and dissociation of oxygen species.
Most of the experimental and theoretical results indicate that oxygen vacancy sites and Ce3+ play crucial roles in the formation of peroxide and superoxide species, respectively, and that these adsorbed oxygen species are active intermediates in the subsequent oxidation reactions.221,222 The possible geometries of adsorbed oxygen species at different active sites on the reduced and unreduced surfaces of ceria are depicted in Fig. 12.
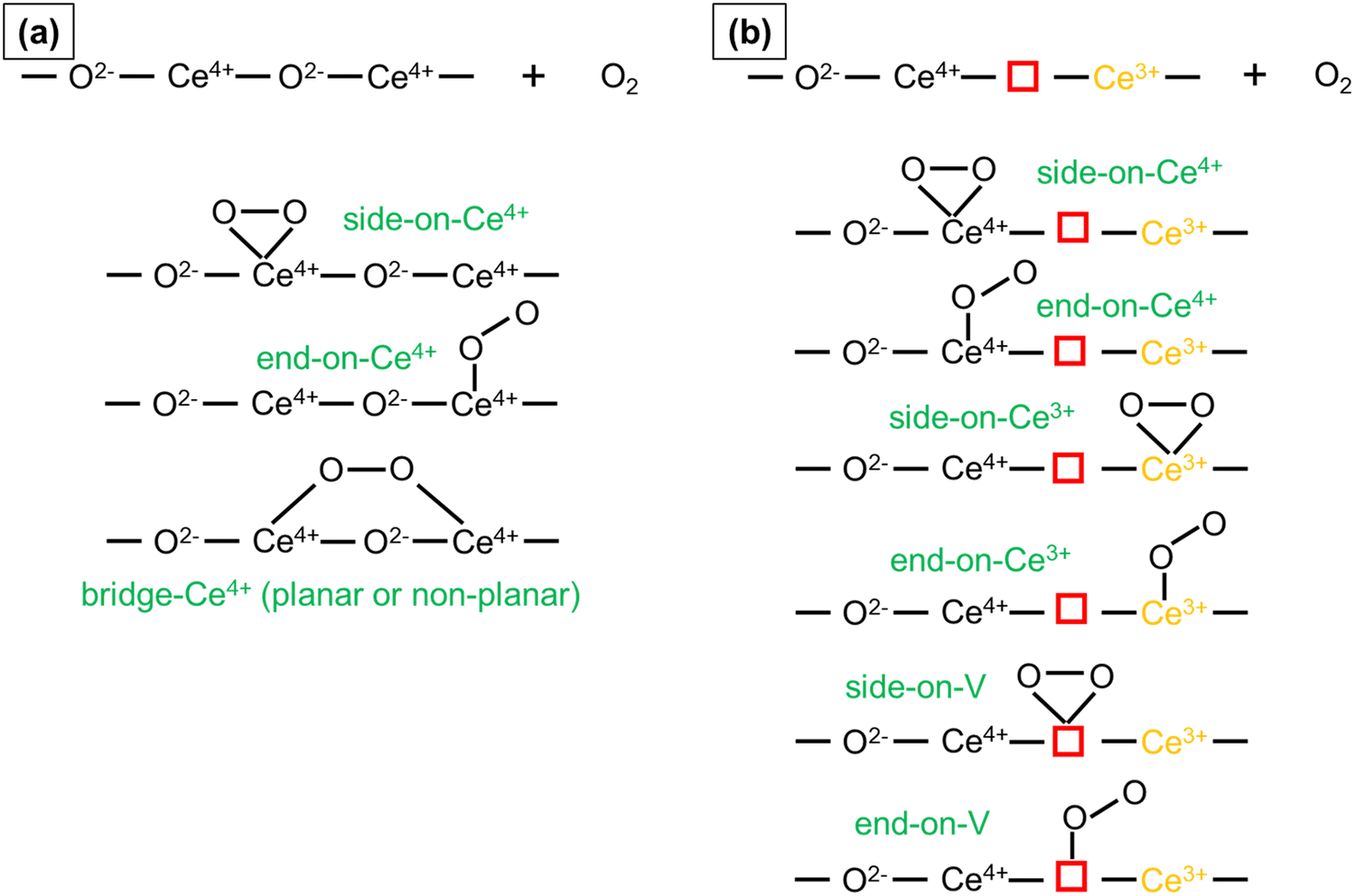 | ||
| Fig. 12 Possible complexes of adsorbed oxygen species formed due to the interaction of molecular O2 with ceria NPs. | ||
The adsorption energies, vibrational frequencies, partial charges and relevant geometrical parameters for different configurations have been calculated by several theoretical studies.210,213 The end-on-V configuration of peroxide adsorption on ceria is energetically the most favorable one of all the possible structures. The side-on-V structure of superoxide adsorption is the second stable structure. O2 physisorbs (adsorbs very weakly) on Ce4+ of an unreduced surface in an end-on configuration. The values of energies suggest that O2 binds very strongly above one-(Ce3+) and two-electron defects (oxygen vacancies), implying that the dissociation process is energetically more favorable on the reduced ceria surface than on an oxidized surface.223
In summary, O2 adsorbs very strongly on the surface oxygen vacancy and forms diamagnetic peroxide species since two electrons from 4f orbitals of Ce3+ ions are transferred to the half-filled π2p* orbitals of O2.223 O22− species are formed from the adsorption of O2 at low-coordinated Ce3+ ion, which is quite far away from the oxygen vacancy site.213 O2 preferentially binds in a side-on configuration, with the end-on mode being less favorable. This interaction results in the charge transfer of one of the two electrons in the 4f orbital of Ce3+ to the πg* orbital of O2. Simultaneously, one Ce3+ ion is oxidized to Ce4+ ion and a complex (Ce4+O2−) is formed through ionic interaction.217
The interaction of H2O2 with CeNPs was also studied in great detail in many reports on antioxidant activity relevant to biomedical applications. Nonetheless, the mechanism of reaction of H2O2 with CeNPs is still not completely understood. Postulating that H2O2 oxidizes Ce3+ to Ce4+, it was shown that CeNPs exposed to H2O2 and having a lower concentration of Ce3+ suppressed the SOD mimetic activity.187 Later, several studies demonstrated that the oxidation of Ce3+ to Ce4+ is reversible and that the reduction happens after a long period (15 days) in aqueous solution following the degradation of H2O2.180,190 The mechanism of the reduction reaction was investigated, and it was proposed that H2O2 reacts with Ce4+ ions and reduces them to Ce3+ ions while it simultaneously gets oxidized to molecular O2. Some controversies exist regarding the mechanism of oxidation/surface complexation of CeNPs, and the impact of surface coating on the oxidation/complexation in the presence of H2O2. Most of the studies ascribed the change in the color of CeNPs in the presence of H2O2 to the conversion of Ce3+ to Ce4+ while some suggested that the color change was due to the formation of Ce3+O22− and/or Ce4+O2− or Ce4+.224 With respect to surface coating, some studies showed that coating does not have any influence on the reaction of H2O2 with the particles while others showed apparent differences in the reactivity depending on the surface coating.225,226 In a comparative study, the 8.2 nm ceria NPs coated with a thick layer of polyethylene imine or polymaleicanhydride-alt-1-octadecene were found to be less reactive than the smaller 3.8 nm with a thin layer of poly(lactic acid).225
Yu et al. developed an E-pH diagram for the CeO2–H2O–H2O2 system to understand the role of H2O2 in the preparation of cerium conversion coating processes.227 Their results indicated that H2O2 could perform dual functions, either oxidize Ce3+ to Ce4+ or reduce Ce4+ to Ce3+. It was shown that H2O2 acts as a reducing agent at lower pH values (pH < 2) and as an oxidizing agent at higher pH values. Ce4+ can exist in several hydrolyzed forms depending on pH and the most stable ones are Ce(OH)3+, Ce(OH)4, Ce(OH)22+ and polymerized form. The overall reaction for reducing the action of H2O2 on Ce4+, Ce(OH)3+ and Ce(OH)22+ in the pH regions <−0.7, −0.7 to 0.7 and 0.7–2.1, respectively, can be written by the following equations.
| 2Ce4+ + H2O2→ 2Ce3+ + O2 + 2H+ | (2.18) |
| 2Ce(OH)3+ + H2O2→ 2Ce3+ + O2 + 2H2O | (2.19) |
| 2Ce(OH)22+ + H2O2 + 2H+→ 2Ce3+ + O2 + 4H2O | (2.20) |
| 2Ce3+ + H2O2 + xH2O → 2Ce(OH)1+x/2(3−x/2)+ + xH+ | (2.21) |
Ce3+ is the more stable form in the lower pH range and Ce4+ can be reduced to Ce3+ by H2O2. Conversely, Ce4+ is the more stable form at higher pH values. Ce3+ is soluble over a wide pH range (pH < 11) as compared to Ce4+, which is soluble at pH < 4. During the oxidation of Ce3+ to Ce4+ with H2O2, the concentration of the intermediate Ce3+/Ce4+ complexes is very high.
Scholes and co-workers performed the titration of cerium-based conversion coating solutions to investigate the role of H2O2.228 The titration results indicated the formation of a Ce(III) peroxo complex like Ce(H2O2)3+, and successive deprotonation led to the formation of Ce3+O22−, which on oxidation gave Ce(IV) peroxo species like Ce(IV)(O2)(OH)2. Finally, the Ce(IV)peroxo complex converted to CeO2 after several months and the authors attributed the conversion to the decomposition of the complex. The use of ceria NPs as colorimetric probes for the detection of glucose has been explored by several studies. The addition of H2O2 to a dilute dispersion of 20 nm ceria NPs (colorless) changed the color of the particles to reddish-orange,229 as shown in Fig. 13. The color change indicates the modification in surface properties and chemical composition of the ceria NPs. FTIR analysis confirmed the presence of peroxo species in the presence of H2O2, and XPS analysis indicated a slight change in the concentration of Ce4+. Ultraviolet-visible (UV-Vis) absorption analysis shows a red-shift in the spectra of CeNPs in the presence of H2O2 with the extent of red shift increasing with the concentration of H2O2.230,231 The coordination number of Ce ions after H2O2 addition was found to increase based on analysis of the X-ray absorption fine structure (XAFS) measurements. However, the coordination number decreased back nearly to the original value at the end of the reaction (after 9 hours); the increase in the coordination number was attributed to the formation of surface-adsorbed peroxide complexes.
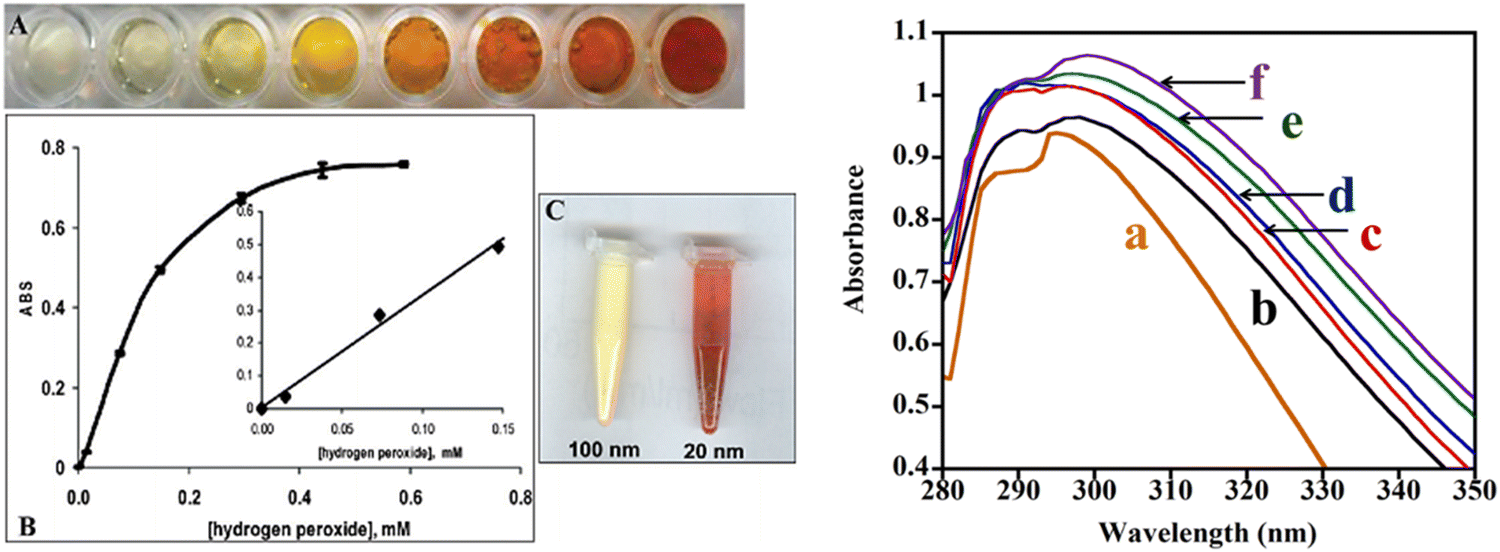 | ||
| Fig. 13 Left. Colorimetric analysis of colloidal ceria nanoparticle dispersion treated with H2O2 as a function of H2O2 concentration at neutral pH, indicating color change proportional with the concentration of H2O2: (A) photos of ceria dispersions showing color with increasing intensity as a function of H2O2 concentration. (B) Calibration curve obtained by plotting optical absorption as a function of H2O2 concentration. Inset shows the linear region. (C) Effect of H2O2 addition to ceria dispersion on particle size (20 and 100 nm ceria). Reprinted with permission from ref. 229. Copyright 2011 American Chemical Society. Right. UV spectra illustrating the CeO2 NPs peaks of Ce3+ (289 nm) and Ce4+ (297 nm) for unreduced CeO2 NPs (no H2O2) (a) and CeO2 NPs reduced for 30 min with different H2O2 concentrations of 5 mM (b), 10 mM (c), 20 mM (d), 50 mM (e) and 100 mM (f). Reprinted with permission from ref. 230. Copyright 2013 American Chemical Society. | ||
The interaction of H2O2 with oleate-capped CeNPs in dichloromethane (DCM) was investigated.232Fig. 14 shows the UV-Vis spectra of CeNPs treated with H2O2 as a function of time in the wavelength range from 200 to 700 nm. H2O2 addition was found to increase the absorbance at 2 wavelengths (λ1 = 285 nm) and (λ2 = 380 nm). The deconvolution of the UV-Vis spectra of the H2O2 adsorption by CeNPs revealed the appearance of two peaks corresponding to two processes: the peak at 285 nm was related to the oxidation of Ce3+ to Ce4+ while the peak at 380 nm was due to the formation of surface-adsorbed peroxide species. Further, the FTIR peak at 840 cm−1 indicated the presence of peroxide/hydroperoxide species.
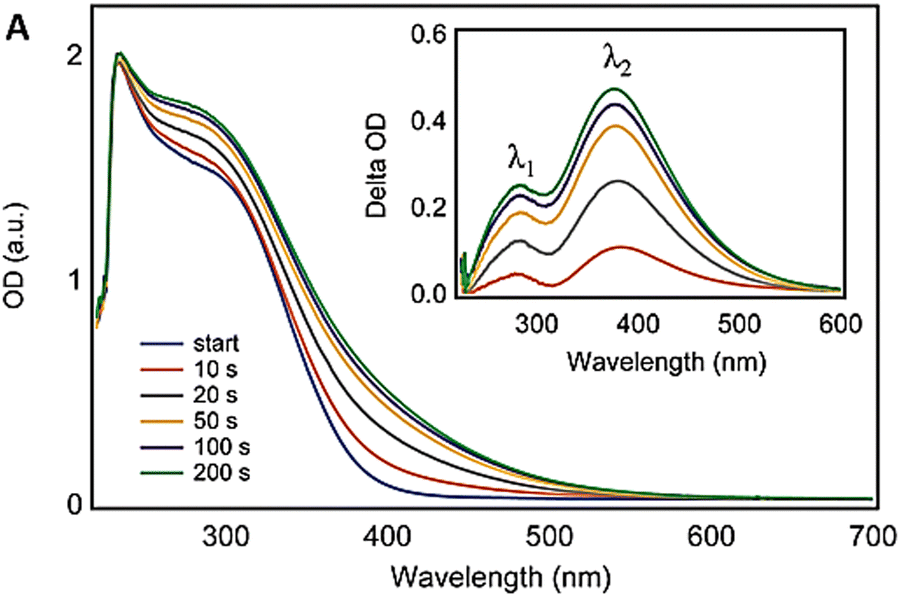 | ||
| Fig. 14 UV-Vis spectra of the CeNPs dispersion treated with H2O2 obtained as a function of time. Reproduced from ref. 232 with permission from the Royal Society of Chemistry. | ||
It should be noted that in an aqueous medium, ceria can exist in different hydrolyzed forms like Ce(OH)22+, Ce(OH)3, and Ce(OH)4, and after the addition of H2O2, it can form cerium(III) peroxide (Ce3+O22−), cerium(IV) peroxide (Ce4+O22−) and cerium hydroperoxide (Ce(OH)2(O2)). Certainly, the ratio of these different species is dependent on the size, surface coating, preparation method, pH and ionic strength. These complexes are reversible and the particles can recover their activity after several days/weeks at room temperature or within few minutes at high temperatures of 70–100 °C due to the decomposition of the surface-adsorbed peroxide and hydroperoxides.5
2.7. Dissolution of ceria-based systems as a promising strategy for regeneration
Every year, large quantities of ceria-based materials are used in TWC converters in automotive exhausts and CMP of dielectric films during integrated circuit (IC) fabrication. Recent reports suggest record high increases in the price of cerium-based materials due to more stringent export rules of the world's largest producer of rare earth materials and scarcity of cerium materials.233,234 A threefold approach, which includes recycling and reuse of these materials and development of alternative materials having less hazardous ecological impacts, has been proposed. The selective dissolution of ceria-based systems appears as a promising strategy to recover cerium; hence, thorough knowledge about the fundamental aspects of ceria dissolution is essential.235 Herein, we review the methods proposed for ceria dissolution and the impact of several parameters on dissolution.The dissolution of ceria is not thermodynamically favorable (ΔrG° = 40 kJ mol−1) even in concentrated mineral acid solutions.236,237 However, the reductive dissolution of ceria is feasible due to the highly positive redox potential of the Ce4+/Ce3+ couple (1.4 V per SHE).238 A solution containing a very strong mineral acid and a reducing agent can effectively dissolve ceria.239 While any strong mineral acid can be used, sulfuric acid and nitric acid are especially known to be effective towards ceria dissolution. As far as reducing agent is concerned, a green reducing agent like ascorbic acid, citric acid, or oxalic acid would be very advantageous. Choosing the appropriate mineral acid and the reducing agent is paramount since mixing a mineral acid with reducing agents like H2O2 (which acts as a reducing agent under highly acidic pH conditions) is an exothermic reaction and hence deleterious.
Early work on ceria dissolution was reported in patents on cleaning ceria particles after polishing glass or silicon dioxide films with slurries containing ceria particles and other chemical additives. Mitani and Saito proposed compositions containing an acid and a reducing agent to clean ceria particles adhered to glass substrates after CMP.240 Several reducing agents were tested in solutions containing sulfuric acid/nitric acid to enhance the dissolution of ceria particles. A solution containing 0.1 M HNO3 and 1 M H2O2 yielded the highest dissolution rate. However, the pH of the solutions containing H2SO4/HNO3 and H2O2 was very low (<0); hence, such compositions caused problems with subsequently deposited materials during IC fabrication and corroded the polymeric brushes used for post-CMP cleaning. Later, significant efforts were made to developing cleaning compositions that were compatible with the consumables (polymeric pads and brushes) used for post-CMP cleaning process. Avanzino and Shonauer proposed cleaning compositions containing a reducing agent (like phosphorus acid, hypophosphoric acid, oxalic acid and L-ascorbic acid) and a complexing agent (like ascorbic acid, citric acid, tartaric acid, malic acid and glutamic acid) for removing ceria particles on silicon dioxide and silicon nitride films.241 In an embodiment of their invention, they showed that the dissolution of ceria particles increased with the increase in phosphorus acid and ascorbic acid concentrations chosen as prototype additives.
Of all the mineral acids, nitric acid and sulfuric acid are known to yield high dissolution rates.242 The dissolution rate in the presence of acids increases with acid concentration. Temperature has a strong influence on dissolution, and the dissolution rate increases gradually with the rise in temperature.243,244 Several studies have reported an enhancement in dissolution rate by ∼3–4 orders of magnitude between 20 °C and 90 °C. The surface of ceria reacts with H+ ions in solution and the particle surface area decreases due to the reduction in particle radius with time.242 The dissolution of ceria in dilute HNO3 or H2SO4 can be catalyzed by Pt NPs. Ultrasound energy can also boost the dissolution of ceria in various solutions.238 pH has a significant impact on ceria dissolution, and the dissolution is significant only below pH 5.245 The dissolution rate of ceria, however, is independent of the mixing rate. The dissolution of ceria in nitric acid is controlled by chemical reaction and not by diffusion as the dissolution rate did not change with the mixing rate.244
The dissolution of CeNPs depends strongly on the chemical composition. For instance, the incorporation of 10% trivalent lanthanide ions increased the dissolution rate by an order of magnitude,243 as shown in Fig. 15a. The crystal lattice weakening because of oxygen vacancies formed to balance the charge deficit after the incorporation of lanthanide ions was found to be the key reason for the increase in the dissolution rate. The modification of the crystal structure of ceria did not lead to any change in the dissolution rate. While an increase in temperature and concentration of acid increased the dissolution rate by one and half orders of magnitude, respectively, changing the reducing agent did not have any impact on the dissolution rate, as shown in Fig. 15a. Beaudoux et al. showed that ceria particles can be dissolved at room temperature in a mixture of ascorbic acid and a dilute mineral acid.237 The dissolution rate of ceria particles having a specific surface area of 15 m2 g−1 increased with the concentration of sulfuric acid or nitric acid in a solution containing 0.5 M ascorbic acid, as shown in Fig. 15b. Ceria particles having different specific surface areas, i.e., different particle sizes, showed significantly different dissolution rates.237 Regardless of the different conditions employed, CeNPs were completely dissolved in 0.5 M ascorbic acid and dilute nitric acid or sulfuric acid. A three-step mechanism is proposed for the dissolution process in ascorbic acid and nitric acid mixture. The first step involves the adsorption of reducing agent on the reactive Ce3+ surface sites on ceria. The second step is the reduction of Ce ions from Ce4+ to Ce3+ by the reducing agent, and the third step involves the release of Ce3+ species via Ce–O bound acid hydrolyses.
 | ||
| Fig. 15 (a) Effect of different physicochemical parameters on the dissolution rate of Ce1−xLnxO2−x/2. Reprinted with permission from ref. 243. Copyright 2012 American Chemical Society. (b) Effect of acid concentration on the dissolution rate of ceria particles in a solution containing 0.5 M ascorbic acid. Reproduced from ref. 237 with permission from the Royal Society of Chemistry. | ||
Recently, the dynamics of dissolution of CeNPs using in situ liquid cell TEM coupled with high intensity electron-beam irradiation of nanoparticle dispersions246 were studied. Very high dissolution rates, exceeding previously reported values at room temperature by several orders of magnitude, were reported by the authors. It was suggested that the radicals, ions and electrons were generated by electron–water interaction and that these species assist in the reductive dissolution of CeNPs. Several procedures have been demonstrated for recovering cerium from post-polish CMP slurry. The recovery process involves selectively dissolving ceria in a mixture containing nitric acid and H2O2.247–250 Cerium from the solution is precipitated post-dissolution as cerium(III) carbonate, which is subsequently converted into ceria by annealing in a rotary kiln. However, the dissolution rates reported in most of the studies are too low to consider any practical application.
Evidently, severe conditions are required to dissolve CeNPs. Published research suggests that sulfuric acid and nitric acid yield high dissolution rates than other mineral acids. There are many unanswered questions regarding this topic; hence, there is still a need to understand the dissolution of CeNPs from a fundamental perspective. In particular, it is pivotal to study the effect of the preparation method, particle size and surface coating, among other factors, on the dissolution of ceria particles as the results of these experiments would provide some valuable insights relevant to the dissolution of ceria particles used in commercial applications.
3. Controlled synthesis of ceria-based hybrid nanomaterials
The synthesis conditions and the ability to control the surface properties are essential aspects influencing the morphology, crystallinity and surface chemistry of CeNPs, ultimately dictating their catalytic, redox and other functional properties. Variations in ceria surface have been obtained by incorporating of a variety of ligands or by binding or mixing them to form composite hybrid structures with other molecules. The size of ceria nanocrystals and variations in their morphologies play a critical role in determining the surface oxidation states, the CeO2−x stoichiometry, stability and catalytic performance.73,251 Various synthesis methods have been employed to tailor these properties, ranging from classical precipitation routes252 to more advanced methods such as hydrothermal,253 sol–gel254 and microwave-assisted synthesis.255 The selection of solvents and capping agents is also essential, particularly for achieving ultrasmall CeNPs. For example, ultrasmall (2.5 ± 0.2 nm) mixed valence state (Ce3+/Ce4+) CeNPs have been obtained via a direct room temperature synthesis using ethylenediamine as a capping agent.256 The ultimate choice of the method governs the size, shape and surface functions. Surface functionalization techniques including doping with other metals or metal oxides or organic ligands, surface oxidation/reduction treatments and deposition of metal NPs257 allow for tailoring the surface chemistry and electronic structure to enhance the catalytic activity, stability and dispersibility in various media. More recent advances include the use of green solvents and biosynthesis methods to produce less toxic NPs compatible with living tissues.258 Furthermore, post synthesis treatments such as annealing, calcination or reduction can modify surface defects, oxygen vacancy concentration, and surface area, thus affecting the redox behavior and reactivity.259Such modifications generate novel interesting properties, allowing them to be used in a variety of applications. Many emerging applications of CeNPs in biomedicine, sensing and environment require surface functionalization to make them biocompatible for target binding and molecular recognition. However, the interfacial behavior of functional particles is sensitive to changes in the environment such as pH, ionic strength, temperature, natural organic materials, and concentration, often leading to aggregation,260 and many functionalized NPs exhibit differential behavior and reactivity profiles for practical applications. In this section, we highlight the most common strategies to create functional CeNPs and CeNPs-based hybrid materials (Fig. 16). In particular, CeNPs used in conjunction with carbon-based materials, polymers, metal organic frameworks (MOFs), metals and metal oxides, and supramolecular assemblies are particularly attractive due to their chemical structures, tailorability and synergistic properties, enabling the effective utilization of these materials in different applications.
 | ||
| Fig. 16 Representation of the modular synthesis of CeNPs, with size and morphology control, and the surface functionalization and creation of hybrid structures with various types of materials. | ||
3.1. Ceria–carbon nanostructures
CeNPs stabilized on the surface of carbon-based materials such as graphene (GR), graphene oxide (GO) and carbon nanotubes (CNT) have enabled the design of a new class of hybrid two-dimensional materials that combine the catalytic properties of ceria with the large surface area and conductivity of carbon-based nanostructures. Graphene has been extensively studied due to its excellent electrical mobility, extremely large specific surface area, high thermal conductivity, and mechanical properties.261 Two different procedures can be used to control the synthesis and graft CeNPs on graphene-based nanostructures. The first involves the adsorption of the precursor salt on the surface of the graphene matrix, followed by the in situ growth of the NPs with control of the size and shapes. The second consists of the anchoring or deposition of the as-synthesized CeNPs onto the surface of the graphene matrix (self-assembly approach).262 CeO2–graphene nanostructures synthesized using a single step (in situ growth) hydrothermal method demonstrated the ability to control growth of CeNPs on graphene sheets (Fig. 17). The combination of defects in CeNPs with an optimal amount of two-dimensional graphene sheets had a significant effect on the properties of the resulting hybrid, providing improved optical, photocatalytic, and photocapacitive performance.263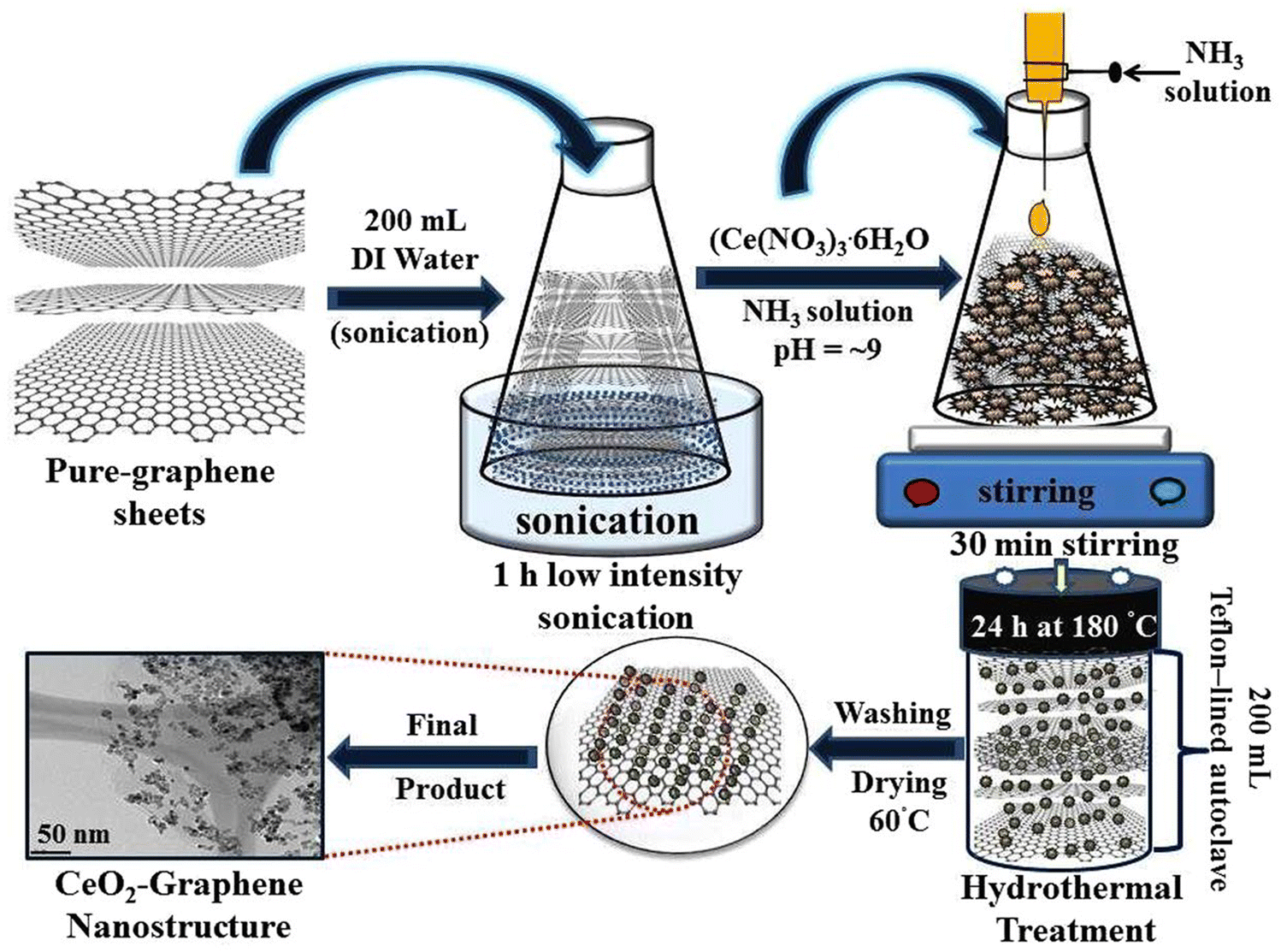 | ||
| Fig. 17 Schematic representation for the synthesis steps of the CeO2–graphene nanostructures. Reprinted from ref. 263. | ||
The intercalation of CeNPs within the layers of graphene nanosheets allowed the nanosheets to maintain their high surface area and prevent reduced graphene oxide (RGO) from restacking.264 Anchoring crystalline ceria onto RGO sheets revealed strong electrostatic interaction between the ceria NPs and RGO. Therefore, the interaction of mobile holes and the increased density of the oxygen vacancies from Ce4+ to Ce3+ in the RGO resulted in a transformation in the n-type CeO2–RGO composites, which are suitable for applications in energy devices, batteries and sensors.265 Li et al.266 utilized an in situ growth method to synthesize a CeO2–graphene nanocomposite, which showed a specific capacitance of 208 F g−1 and a maximum power density of 18 kW kg−1 in supercapacitors. Dezfuli et al.264 developed CeO2–RGO nanocomposite electrodes, which exhibited excellent supercapacitive behavior with high specific capacitance (211 F g−1 at 2 mV s−1 and 185 F g−1 at 2.0 A g−1) and reversibility. Combining the redox characteristics of CeNPs with graphene is an effective approach to design materials for redox supercapacitors.266 However, their wide band gap semiconductor (∼3.4 eV) limits their use as an electronic material and photocatalyst.267 Tuning the band structure by chemical functionalization or attachment to conducting supports is an excellent method to enable their implementation in electronic and optical applications. In particular, defects, such as oxygen vacancies, generate localized electrons on the CeO2 surface, which may interact with the functional group in the RGO sheets, thus improving the properties of RGO.268 Such composites also show high adsorption capacities as adsorbent materials for preventing environmental pollution with contaminants such as heavy metal ions and phosphates, thus enabling applications in environmental remediation.269 To improve the selectivity (towards cationic or anionic species), sorption capacity, and overcome limitations such as the leaching of the NPs or stacking of the C-sheets, the surface of these materials can be modified to enable specific applications. For example hydrous ceria-modified graphene has been used as a novel adsorbent for arsenic removal from aqueous solutions.270 In other works, GO decorated with ceria showed selectivity in determination of trace metal ions and speciation of selenium.271 Ceria-GO composites fabricated using one-pot synthesis exhibited an almost complete (>99.9%) and quick removal of arsenic species within 0.1 mg L−1 of the initial concentration.272 Additional uses have been demonstrated for preparing ceria-carbon based catalysts for solid oxide fuel cells (SOFCs) and field effect transistors. The chemical vapor deposition (CVD) growth of single-walled carbon nanotubes (SWNTs) is normally performed at 800–1000 °C.273 When CeO2, which stores oxygen, was used, the ceria-supported catalyst enabled the selective growth of SWCNTs on substrates due to the oxidative environment provided by the ceria.274
3.2. Polymeric ceria-based hybrid materials
The combination of the metal oxide NPs with polymers is increasingly used to overcome limitations such as NP aggregation, dispersibility, lack of specificity for reactions in complex systems, and weak mechanical strength.275 The properties of such hybrid materials depend on the nature of the guest NPs and the structure and the functional groups of the host polymer. This approach allows tuning the composition, surface functionality, and colloidal stability of the dispersed NPs. Colloidal carrier systems or so-called smart carrier systems, such as thermosensitive microgels, have been developed, which encapsulate and stabilize the particles.276 Polymeric materials are commonly used to stabilize CeNPs for applications in biomedicine. However, the surface charge and their surface functionalization have been found to play a critical role in their uptake mechanism and subcellular localization. Results with CeNP functionalized with different polymeric coatings (poly(acrylic acid), aminated poly(acrylic acid) or dextran, Fig. 18), endowing positive, negative or neutral changes, showed that CeNPs with a positive or neutral charge enters most cells, while those negatively charged internalize mostly in cancer cell lines.277 In other works, polymers like polyethylene glycol and polyvynilpyrrolidone create functionalized CeNPs with biocompatible coatings that show enhanced autophagic clearance.278 Recent advances in the surface functionalization of ceria involve green synthesis methods using room-temperature aqueous and solvent-free solutions. CeNPs prepared by metal-free organocatalyzed photoinduced electron transfer radical polymerization (O-PET-ATRP) generated zwitterion-functionalized NPs that retained their shape and prevented protein adsorption. The resulting passivation of the particle surface generated higher uptake and enhanced redox properties under physiological conditions. The functionalization using this environment-friendly approach directly enhanced the intracellular delivery for cancer therapeutics.279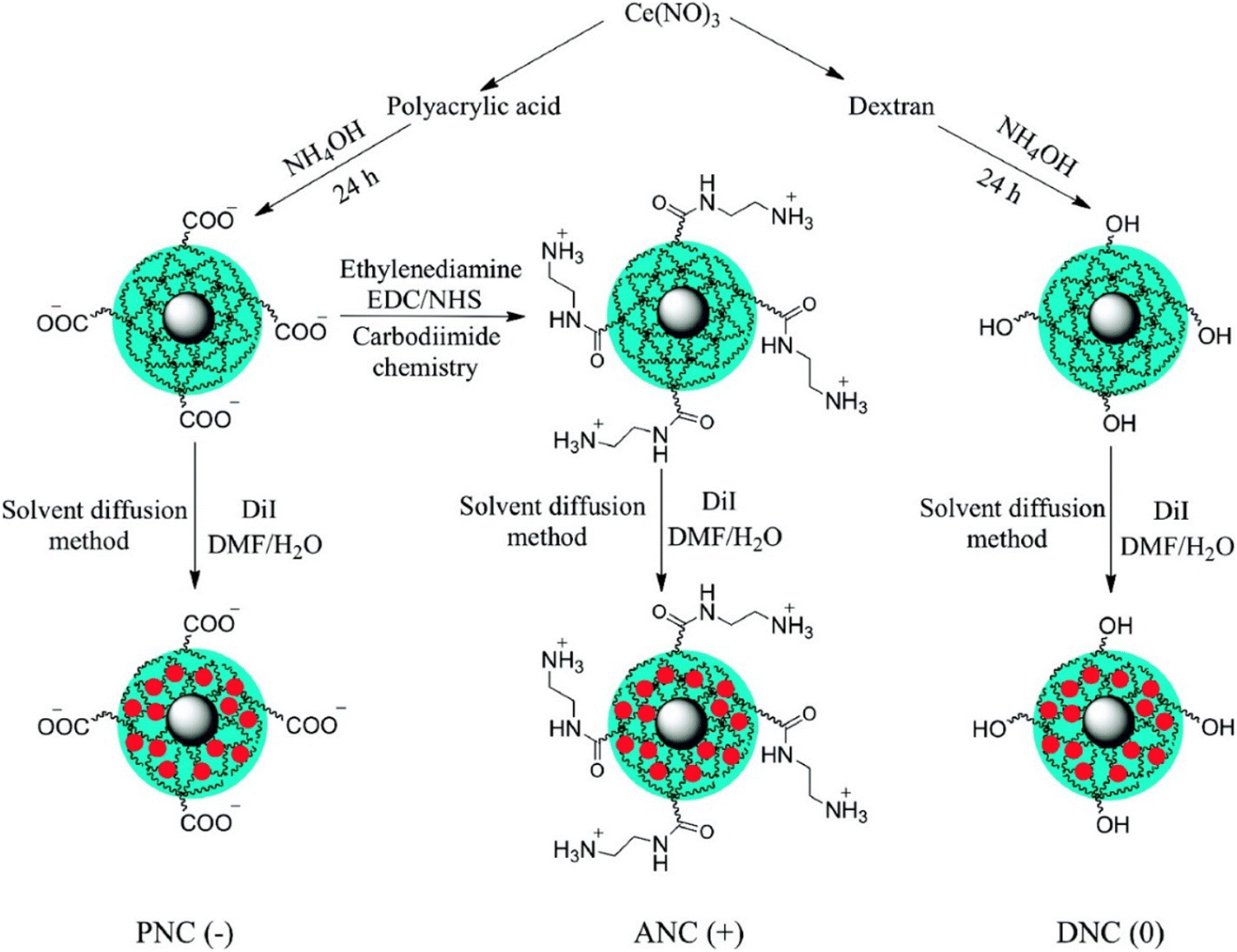 | ||
| Fig. 18 Surface functionalization of ceria NPs with different polymer coatings and surface modifications were synthesized to yield nanoparticles with negative [PNC(−)], positive [ANC(+)], and neutral [DNC(0)] charge, where PNC is poly(acrylic acid), ANC is aminated poly(acrylic acid) and DNC is dextran. A fluorescent dye (DiI, red circle) was encapsulated using a modified solvent diffusion method (reprinted with permission from ref. 277. Copyright 2010 American Chemical Society). | ||
Polymers can also be used to fabricate hybrid CeNPs–polymer composites in which the polymer acts as a supporting material or dispersing agent. CeNPs can also be crystallized in a controlled manner on the surface of polymer NPs.280 Hybrid polymeric core–shell nanoparticles, i.e., CeNPs/polystyrene nanocapsules containing a model fluorophore with encapsulated fluorescent dyes, were developed by free radical microemulsion polymerization.281 The use of CeNPs generated a significant enhancement of the fluorescence properties by suppressing the quenching of the encapsulated dye by molecular oxygen. Cellulose, a sustainable and renewable biopolymer, was used to incorporate CeNPs to obtain transparent films with UV shielding properties.282
Other studies have shown the use of coordination polymers employing lanthanide ions instead of d-block metal ions to generate supramolecular architectures with distinctive structures and accessible metal coordination sites.283,284 Using a sonochemical method, NPs of a Ce(III) supramolecular compound with fluorescent proepties have been synthesized as a precursor for nanoceria.285 Polymeric materials can be used to facilitate the self-assembly of NPs and create structured supramolecular assemblies.260,286 The surface of CeNPs has been shown to form stable surface complexes with molecules incorporating phosphate, phosphonate, or carboxylate moiety.287 Several reports demonstated the synthesis of CeNPs using coordination polymers with tunable shell tickness and surface functions.284,288–290
3.3. Ceria-based metal organic frameworks
Metal–organic frameworks (MOFs) have emerged as a promising class of materials for gas storage, gas purification and separations, and catalysis.291 A large number of organic linkers and metal ions provide versatility for creating tailorable structures with open metal sites that can act as catalytic or binding sites for a variety of reactions. Stabilizing CeNPs within MOF frameworks can be used as an effective strategy to enhance their reactivity by preventing aggregation and providing high accessibility to the catalytic sites through the open channels. A cerium-MOF based on MIL-125 with open-metal sites292 was shown to retain the porosity and enhance the overall adsorption of SO2 and CO2. The synthesis of Ce(IV)-based UiO-66 (referred to as Ce-BDC) with different pore sizes and functionalities was reported to stabilize the [Ce6O4(OH)4]12+ cluster during MOF formation, preventing metal-hydroxo precipitation.293,294Several studies have been reported on introducing Ce to the MOF framework by doping to increase the number of active sites for gas adsorption. Ce-doped MOF ((Ce-UiO-66(Zr))) exhibits over 25% increase in the amount of NO2 adsorbed under dry conditions in comparison with the unmodified MOF.295 Support materials or core shell structures have been shown to improve the catalytic activity of CeNPs.296 Kim et al.297 developed a simple method to synthesize nanoporous CeO2 by the thermal conversion of aliphatic carboxylate ligand-based Ce-MOF into hierarchical metal oxides with nanocrystalline frameworks. It was found that the pore size of the prepared framework could be tuned, providing an increase in the adsorption capacity. A 3D network CeO2-M was prepared by a Ce-MOF-template method and K+, doping which resulted in an increased oxygen species and superior adsorption and oxygen storage capacity of ceria.298 The solid catalyst was evaluated for o-xylene oxidation at low temperature. Inspired by these studies, a new generation of porous CeNPs/Au@SiO2 core–shell nanocatalysts has been prepared by the pyrolysis of Ce-based MOFs (Fig. 19).299 The CeO2/Au@SiO2 catalysts possess a porous nanotube structure, resulting in high catalytic activity for the reduction of 4-nitrophenol.
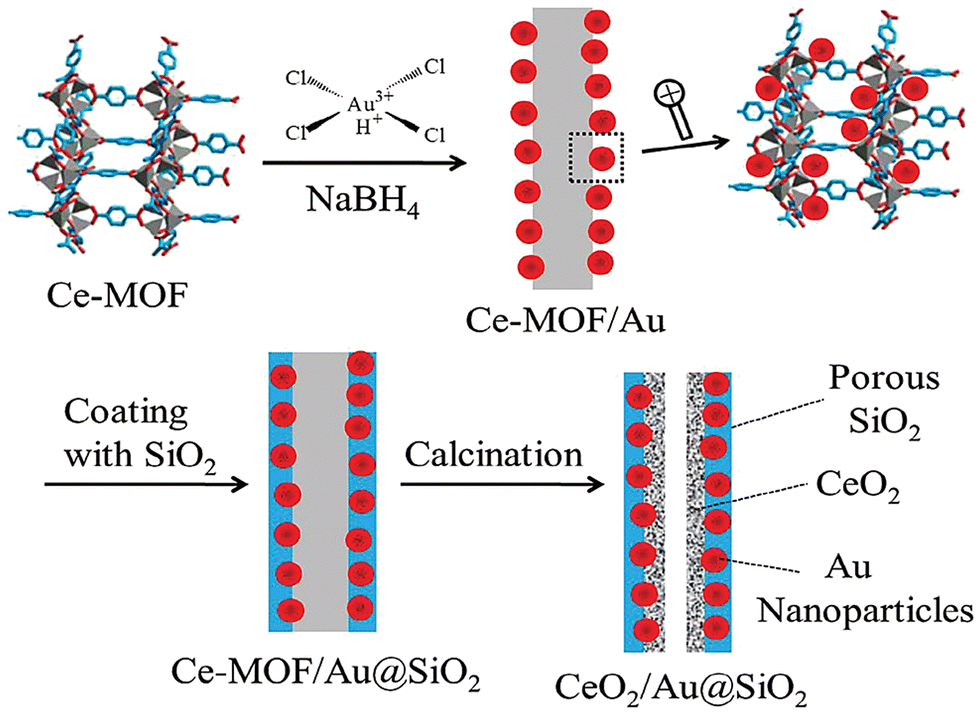 | ||
| Fig. 19 The synthesis procedure for the CeO2/Au@SiO2 nanocomposites. Reproduced from ref. 299 with permission from the Royal Society of Chemistry. | ||
The enhanced reactivity of CeNPs can be obtained by creating ultrasmall (1–2 nm) CeNPs within a Ce-MOF via the in situ etching of the parent NPs (Fig. 20). This strategy enhanced the activity of the CeNPs by preventing NP aggregation, stabilizing them in a dispersed form within the porous framework.300 At the same time, the guest molecule has high accessibility to the active sites through the porous network. The resulting CeNPs-MOF showed fast degradation kinetics of the nerve agent, p-nitrophenylphosphate (DMNP), providing superior activity as compared to the parent CeNPs. The ability of the CeO2-MOF to degrade DMNP was explained by the increased abundance of Ce3+/Ce4+ neighboring sites and oxygen vacancies, facilitating the nucleophilic attack of the hydroxyl groups of the DMNP phosphate, leading to P–O bond cleavage and further degradation. This method provides a strategy to enhance the reactivity of CeNPs, making them more effective catalysts.
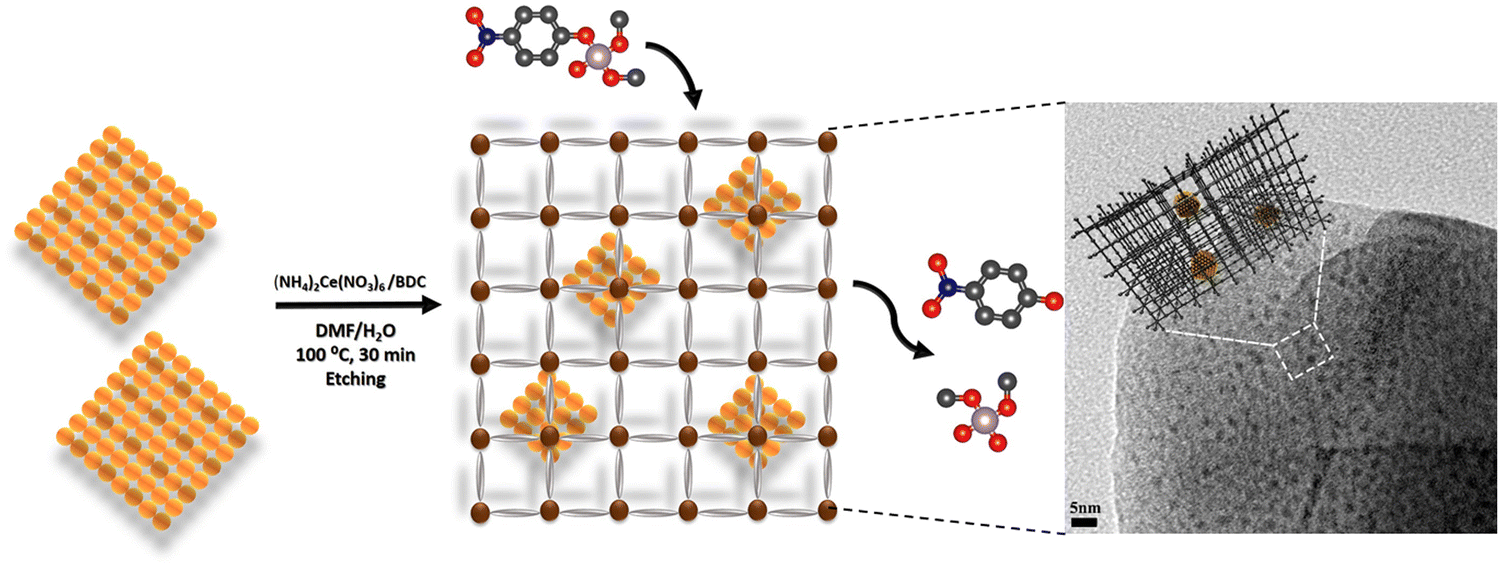 | ||
| Fig. 20 Schematic of CeNPs immobilization inside Ce-MOF for DMNP degradation and the subsequent CeNP–MOF structure analysed by TEM (reprinted with permission from ref. 300. Copyright 2020 American Chemical Society). | ||
3.4. Ceria-based metals and metal oxides heterostructures
Noble metals (M) and metal oxides (MO) can be used in conjunction with CeNPs to create hybrid materials with lower cost and superior characteristics for applications. For example, platinum-group metals, Pd, Rh and Pt, serve as active species in TWA exhaust catalysts and the price of these metals represents around 40% of the manufacturing cost of the catalysts.301 Reducing the use of expensive Pt Pt-MO heterostructures has been explored. Enhanced activity can be obtained by encapsulating noble metals inside ceria, e.g., Pt@CeO2 core–shell hybrid structures, or by loading the MNPs on the surface of hollow CeO2 nanostructures. This strategy has been used to create a variety of Au, Pt, Ag@CeO2 hybrid nanostructures,302,303 providing enhanced catalytic activity by stabilizing small size noble metal NPs and creating hybrid junctions at the two-phase interface.304 Doping CeNPs with metals has emerged as a promising approach to produce supported catalysts with atomically dispersed metals.305 The size, morphology, stability and catalytic activity of these materials can be controlled by varying the dopant, ligand used and the reducing and oxidizing conditions. Studies to assess the effects of metal dopants on the reducibility of bulk and extended surfaces of CeNPs have been extensively reported using both experimental and theoretical approaches.142,158,306,307The enhanced properties provided by these hybrid materials have found unique applications in catalysis, environmental remediation, sensing and biomedicine. For example, Ag@CeO2 has demonstrated the inactivation of Escherichia coli through the formation of reactive oxygen species (ROS).308 Similarly, CeO2–CdO binary metal oxide nanocomposites showed antibacterial activity and growth inhibition towards P. aeruginosa.309 The properties of CeNPs were also integrated with the photoinduced electron transfer of quantum dots (QDs), providing materials with significantly improved optoelectronic properties. A synthesized ceria–carbon quantum dots/RGO nanohybrid showed improved photocatalytic properties, enabling the rapid decomposition of organic pollutants.310
4. Applications and future directions
The unique properties of nanoscale ceria quickly inspired their translation in a variety of applications. The broad applicability and versatility are a consequence of the redox properties of CeNPs originating from the presence of oxygen vacancies and the Ce3+/Ce4+ mixed valence states. As a result, CeNPs are increasingly used in applications such as catalysis, chemical mechanical polishing, fuel cells, oxygen sensors, ultraviolet absorbents, sensing and biosensing, drug delivery and bio-medical diagnostics.311–316 In the following sections, we provide examples of the representative use cases of CeNPs in catalysis, chemical mechanical polishing, chemical sensing and biosensing, and environmental remediation.4.1. Catalysis
The thermal stability, high OSC and proton transfer abilities make CeNPs particularly useful in catalysis.165,317 The 3+/4+ reversibility and oxygen mobility, which is higher compared with other oxides such as SiO2, Al2O3, and ZrO2, enable the use of CeNPs as a catalyst in redox, CO oxidation, and acid–base reactions3,318,319 among others. The main concerns related to CeNPs are related to the deterioration of the active surface area due to sintering and segregation processes, resulting in the loss of catalytic activity.320 Therefore, much attention has been paid to develop CeNP-based catalysts with enhanced structural characteristics to resist degradation, such as combining binary metal oxide catalysts. For example, CeO2–MnO2 showed enhanced performance for processes such as CO oxidation,321 selective catalytic reduction of NOx with NH3,322 and the oxidation of volatile organic compounds.323 Further optimization has been accomplished using a Ce-based MOF to maximize the exposed active sites and enhance the porosity of the system.324 Ceria is an excellent support for high-performance metallic catalysts, enhancing the stability and catalytic performance, as shown for example with PtFe nanocrystals hybridized by CeOx, e.g., PtFe–PtxFeyCezOj.12 A particular case is the use of mesoporous ceria, characterized by well-defined pores, typically ranging from 2 to 50 nm, and a high surface area, which provides unique capabilities for catalysis,325 gas sensing and energy storage. The high surface area provides enhanced accessibility to active sites and facilitates the transport of reactants and products. Additionally, the oxygen vacancies facilitate switching between CeO2 and CeO2−x. Therefore, they showed improved performance in catalytic reactions including oxidation, hydrogenation and pollutant removal. A review summarizing the synthesis and catalytic applications of mesoporous ceria can be found in the ref. 325.Doping CeNPs with transition metals or rare earth metal such as Cu, Cr, Ti, Y, and La have been adopted as a promising approach to tune the catalytic activity of these materials.326–330 The substitution process causes structural defects and creates more oxygen vacancies on the catalyst surface, which affects the generation and mobility of charged species such as electrons and oxygen anions. Furthermore, doped ceria can also tolerate high temperature annealing treatments due to the cooperative nature of the cations of the binary oxides.331 The electronegativity and ionic radius of Ce4+ are 1.12 and 0.97 Å, respectively. A synergetic dopant would ideally have electronegativity and ionic radius close to these numbers. For instance, samarium (Sm) has an electronegativity and ionic radius of 1.17 and 1.07 Å, respectively. It is also soluble in the ceria sublattice and can stabilize the fluorite structure of ceria at low dopant concentration. A nanocrystalline CeO2–Sm2O3 (CS) solid solution has been easily synthesized, yielding a promising catalyst for the oxidation of CO at lower temperatures owing to the large number of lattice defects, high OSC and improved redox properties.331 Sm-doped CeO2 nanopostructures have been widely used as a catalyst in methane selective oxidation,332 allylic oxidation333 and fuel cells applications,334 while Gd-doped CeO2 was used for gas sensing applications.335 The Gd-doped CeO2 gas sensor had better sensitivity, good stability and lower operating temperature over the nondoped one with a detection concentration of 800 ppm CO2 gas. In addition, Ni-doped CeNPs have been hydrothermally prepared and showed an increased magnetization and electrical conductivity with increasing dopant concentration.336
As diesel emission produces soot, which have grown concerns for human health and the environment, several emission control strategies have been implemented to reduce environmental impact.337 For example, over 90% of soot can be trapped and then be continuously or periodically combusted to CO2 using the diesel particulate filter (DPF) technology. However, the periodic regeneration of DPF is not passive, which makes it complex and expensive. Ceria-based materials have been developed to decrease the soot combustion temperature (∼600 °C compared to diesel exhaust temperature (∼175–400 °C)) and have been extensively studied as catalysts and catalysts promoter in this research area.338 Nascimento et al.339 studied the performance of binary oxides CeOx/FeOy catalysts modified with AgNPs for the control of diesel soot combustion. Catalytic tests showed a significant reduction of about 300 °C in the soot oxidation temperature with CeOx/FeOy compared to CeO2. Shen et al.340 also investigated the catalytic performances of Fe-doped ceria catalysts with diffeent Fe contents for the soot oxidation reaction with O2 or O2/NO gases. They demonstrated that the addition of iron increased the number of oxygen vacancies on the surface and therefore the catalytic activities for NO oxidation and NO2-assisted soot combustion.
Uniform anchoring/decoration of CeNPs on the surface of graphene sheets improves the photocatalytic and photocapacitive performance of the nanostructures for effective charge transfer and catalytic degradation processes.263 For example, the photocatalytic activity was examined by monitoring their ability to degrade Congo red ∼94.5% and methylene blue dye ∼98% under visible light irradiation. These developments indicate the broad applicability of CeNPs-based nanostructures for future energy and environment-related applications.
4.2. Chemical mechanical planarization (CMP)
CMP is a surface smoothing process that is used in the semiconductor fabrication process and in a number of other manufacturing processes. It has become a critical and enabling process for the multilevel metallization necessary for interconnecting the immense number of active devices created on silicon substrates and in the device fabrication process sequence at the gate level.312,341 Silicon dioxide CMP, or simply oxide CMP, in which a colloidal slurry consisting of ceria abrasives is commonly used, is one of the many critical processes in the fabrication of integrated circuits (ICs).341–344 CeNPs are widely used as abrasives for oxide CMP since they interact strongly with the oxide, leading to high removal rates (RRs). With appropriate additives to ceria slurry, it is possible to achieve a highly selective polishing of the oxide with respect to the underlying silicon nitride films, as required in the planarization of shallow trench isolation structures.344–346 Ce3+ species present on the surface of CeNPs particles react with the hydrated oxide surface (Si–O−), forming strong Ce–O–Si bonds and ultimately rupturing the underlying and weaker bonds in the oxide film, leading to its continual removal.344,347,348 There are extensive discussions of the reaction mechanisms involved in the polishing of oxide and nitride films.344,349,350Two methods, namely, solid-state displacement and solution-growth, are commonly used for the preparation of CeNPs particles for CMP applications.341,351 In the solid-state displacement method, particles are prepared by calcining cerium carbonate at more than 600 °C under controlled air flow conditions, which is subsequently wet milled to obtain the desired particle size.352 CeNPs with less than 100 nm size can be obtained by controlling the pre-treatment of cerium salt, calcination temperature, air flow conditions, and bead size of the milling process.352–354 However, the particles produced by this method are faceted and have sharp edges, corners, and apexes as well as a very wide size distribution (Fig. 21a). These features tend to scratch the films during polishing and generate many unacceptable defects. Consequently, the usage of these types of particles is being avoided, while the solution-grown CeO2 particles, despite of their higher cost and potential for contamination, have gained wider acceptance.87,355,356
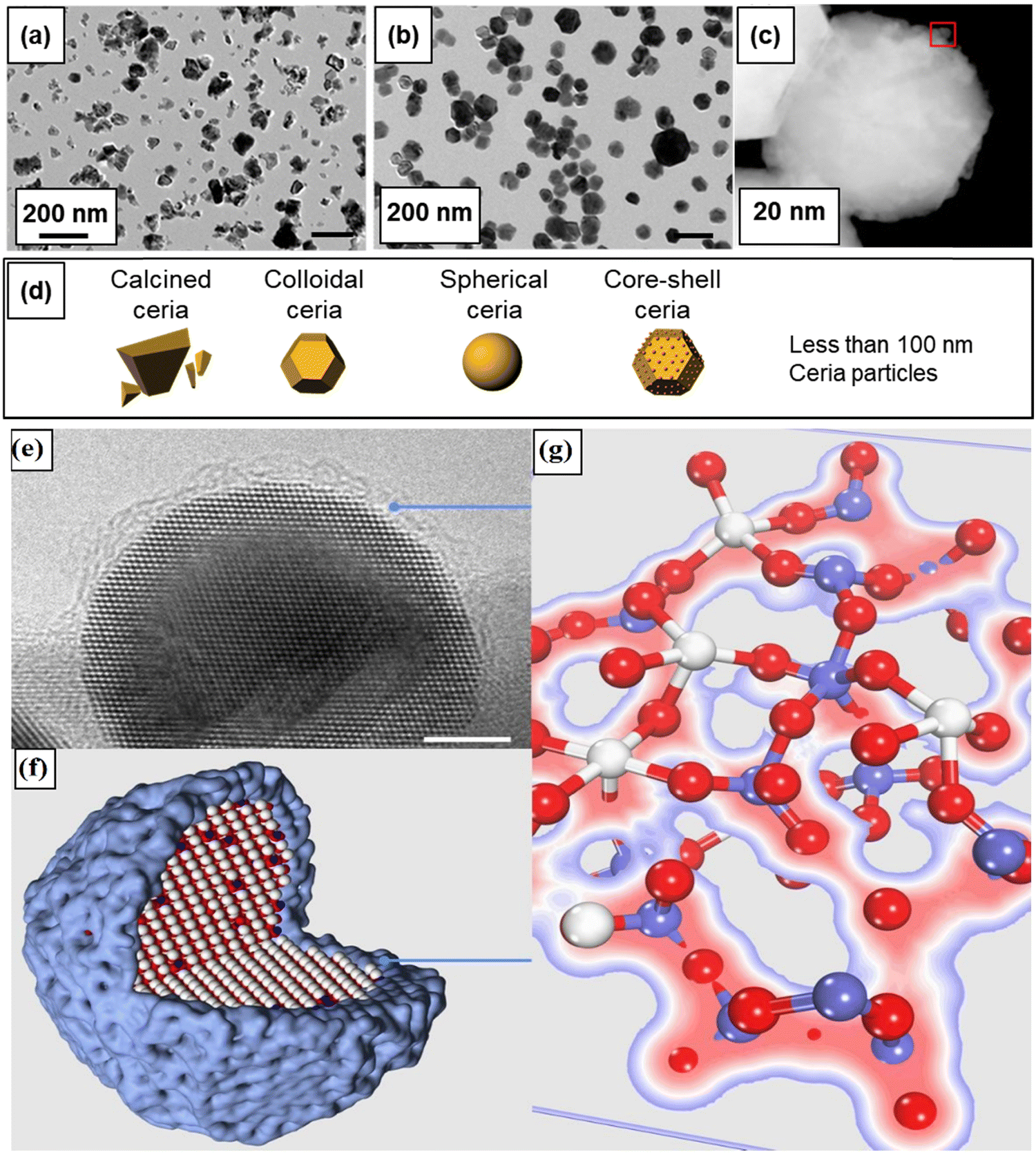 | ||
| Fig. 21 Different types of CeNPs used in recent CMP slurries; (a) calcined ceria, (b) colloidal ceria, and (c) core/shell ceria particle, and (d) their schematic illustration. HRTEM image of Ti-doped ceria single-crystalline nanoparticle (e), model of a Ti-doped CeO2 sphere (f), and a molecular model of the TiO2 amorphous shell on the nanoparticle (g). (a) and (b) Reprinted with permission from ref. 351. Copyright 2014 American Chemical Society. (c) reprinted with permission from ref. 364. (e) and (f) Reprinted with permission from ref. 371. | ||
In the solution growth method, cerium hydroxide (Ce(OH)3) is precipitated from a solution of cerium nitrate mixed with ammonium hydroxide and transformed to CeO2 in the presence of oxygen (Ce(OH)3 + 1/2O2 → CeO2(s) + 3H2O). These particles are separated and washed repeatedly for removing residual precursors until the desired conductivity of the suspension is obtained.357,358 Particles with a very narrow size distribution can be produced by this method, as shown in Fig. 21b. These relatively uniform-sized CeNP abrasives have polyhedral and nearly spherical shape, which is expected to minimize the generation of scratches because of fewer sharp edges and corners.
Coating ceria abrasives with a layer of different materials and chemical compositions makes it is possible to change the physicochemical properties of the core abrasive, including the shape, surface chemistry, surface charge, porosity, and hardness, and can be tailored to increase their reactivity with films. To benefit from this strategy, core/shell type CeO2 abrasives have been investigated and have showed increased removal rates as well as improved surface roughness of polished oxide films. Core/shell structured composite particles, comprising of mesoporous silica,359,360 dendritic silica,361 polymethylmethacrylate (PMMA),362 and polystyrene (PS)363 as core materials with metal-doped CeO2 particles making up the shell, generated relatively high oxide removal rates with low surface scratch and roughness values. This was ascribed to the resilient spring-like effect of the softer core silica during polishing. Also, since smaller particles tend to produce a higher quality surface finish, the coating of core CeO2 particles (∼100 nm) with a layer of smaller (5 nm or so) CeO2 shell has been proposed and was shown to achieve high oxide polish rates (Fig. 21c).364 In principle, these types of composite abrasives can result in significant cost savings for high volume manufacturing due to the higher throughput, lower abrasive concentrations, and lower waste treatment cost. However, all these composite ceria abrasives are prone to form broken particles as well as peeling-off and/or brittle collapse of the shells. Therefore, further advancements in the field are necessary to avoid these issues. Various types and shapes of pure and composite CeO2 particles used in most CMP slurry formulations are summarized in Fig. 21d.
As NAND technology has evolved from 2D planar to 3D vertical, new CMP steps with higher planarization efficiencies and much higher oxide polish rates have become necessary. This is due to the larger initial step heights and much thicker dielectric layers than those in conventional ILD or STI CMP.365–367 To meet these challenges, CeNPs-based slurries having the necessary very high oxide polish rates need to be developed. One of the current development approaches is to prepare Ce3+-rich CeO2 particles through either metal-doping or coating. Doping with lanthanide (La, Sm, Gd, Nd, and Yb) elements increases Ce3+ concentration by increasing oxygen vacancies at the surface, thus enhancing the removal rates of the oxide film.368–370 Praveen et al. reported that La-doped CeO2 particles showed ∼20% higher removal rates of SiO2 films when compared to the non-doped CeO2 particles.368 Similarly, Cheng et al. showed that Nd doping increases Ce3+ concentration on the particles surface and thus produced ∼30% higher removal rates of the oxide film at pH 9.5.369 The removal rates of oxide films were well correlated with Ce3+ concentrations in metal-doped particles. For example, Feng et al. prepared single-crystal Ti-doped CeNPs spheres with a size distribution of 50–150 nm through the liquid-phase flame spray pyrolysis of solutions of cerium and titanium precursors.371 The CeO2 core of the particles were encapsulated by a 1–2 nm shell of TiO2 to minimize the surface energy, resulting in a nearly spherical morphology (Fig. 21e–g). The polishing results showed that these particles reduced the CMP-caused defects by 80% and increased the removal rates of the oxide film by 50%.371
4.3. Chemical and biological sensing
The field of chemical sensors and biosensors has witnessed a radical transformation due to the rapid advancement and innovations in functional materials and biomaterials. Metal oxide nanomaterials can serve for both recognition and transduction purposes372 alone or in conjunction with biomolecules such as DNA or enzymes. Enzyme-based sensors are considered to be one of the most reliable sensing techniques due to their high sensitivity, selectivity and fast response.373–375 However, an enzyme's stability can be severely compromised with temperature and pH changes. For these reasons, researchers have focused on using nanotechnology tools to develop more robust sensors by engineering materials that mimic the function and complexities of natural enzymes.376–379 Metals and metal oxides NPs and nanocomposites,374,380,381 metal organic frameworks,382,383 and carbon-based nanomaterials384–386 have demonstrated promising potential as artificial nanoenzymes195 with peroxidase or oxidase-like mimetic activity, which could be used as receptors or transduction materials for sensing.CeNPs have emerged as a promising enzyme-mimetic material that possesses intrinsic oxidase or peroxidase-like properties due to the mixed oxidation states (Ce3+/Ce4+), which provides the ability to catalyze many chemical and biologically relevant reactions.375 Upon the reduction of Ce4+ to Ce3+, the CeNPs mimics catalase activity by inducing H2O2 oxidation, generating molecular O2. On the contrary, oxidation of Ce3+ to Ce4+ mimics superoxide dismutase (SOD) activity by reducing O2− to H2O2. The efficiency of CeNPs as a peroxidase mimic is strongly dependent on the low ratio of (Ce3+/Ce4+).387 The high catalytic activity, oxygen rich surface and low toxicity175 make them excellent candidates for enzyme-mimetic and enzyme immobilization purposes.229,388 Furthermore, CeNPs have been used as a material with therapeutic potential to reduce harmful ROS/RNS due to the unique regenerative ability to scavenge ROS/RNS species.200,389 The combination of sensing and therapeutic functions makes CeNPs a powerful theranostic platform for biomedical applications.26,390 An ROS-scavenging tissue adhesive nanocomposite was synthesized by immobilizing ultrasmall ceria nanocrystals on the surface of uniform mesoporous silica (MSN).391 The ceria nanocrystals-decorated MSN does not only have a strong tissue adhesion strength but they also help minimize the ROS-mediated deleterious effects, thus efficiently accelerating the wound healing process, as evidenced by marked skin appendage morphogenesis and limited scar formation.
Various studies have shown the ability of CeNPs to reduce symptoms of oxidative stress-related diseases, such as chronic inflammation,392 endometriosis,393 cancer,394,395 and diabetes.396,397 However, with the diversity of methods and synthetic procedures used to synthesize these particles, each producing different types, shapes, sizes, coatings and Ce4+/Ce3+ ratios, the antioxidant effects can vary significantly. Controlling the surface chemistry to efficiently regulate the reducing and oxidizing efficiency (e.g., antioxidant vs. pro-oxidant) in biological systems remains a challenge, as is the tendency of CeNPs to agglomerate in biological fluids.170 In some cases, CeNPs have been found toxic, but the mechanism responsible for CeNPs toxicity, if present, is still inclear and subject to debate.11 Therefore, it is important to develop new kinds of CeNPs with high dispersibility and enhanced catalytic activity that are able to maintain their redox properties in biological fluids. Several strategies have been reported in the literature to improve these properties. For example, CeNPs have been coupled with materials such as Cu2O,398 TiO2,399 and Eu400,401 to enhance their catalytic and enzyme mimetic properties. The use of CeNPs in conjunction with TiO2 nanotubes increased the peroxidase mimetic activity.402 In another study, adding Eu to the nanocrystal structure imparted fluorescence and increased the oxidase-like activity of ultra-small (2–5 nm diameter) CeNPs.400 The decoration of graphene sheets with metal oxide NPs such as CeNPs improved the mechanical, electrical and thermal properties of both materials. In this hybrid structure, CeNPs have a wide band gap (3.4 eV) with a high isoelectric point (IEP, 9.0) and excellent enzyme binding ability,403 while graphene has high electrical conductivity. Therefore, CeNP–graphene composites that synergistically combine the electrical conductivity of graphene with the catalytic properties of CeNPs are excellent materials for electrochemical biosensors. The CeNP-reduced graphene oxide (CeO2–rGO) composite prepared by a facile one-pot hydrothermal method used in combination with horseradish peroxidase (HRP) enabled the detection of H2O2 at very low levels. The CeNPs–rGO composite provided a well-defined micro-environment for HRP immobilization and enhanced the direct electron transfer (DET) between the enzyme and the CeO2–rGO at the electrode surface, significantly improving performance for H2O2 detection. Other works reported a 100-fold increase in the oxidase mimicking activity of naked CeNPs particles by fluoride capping. This mechanism has been used for the ultrasensitive sensing of fluoride (0.64 μM).404
Hybrid nanomaterials offer several advantages for chemical sensing and biosensing due to their ability to provide synergistic catalytic functions due to the formation of hybrid junctions associated with rich redox reactions, thus enhancing their catalytic performance.373 Artificial intelligence and machine learning, through the use of computer-aided designs of the CeNPs, could in the future be used to screen and rationally design particles with the desired properties for applications.406,407 Using these tools, it is now possible to predict the optimal CeNP shape that delivers the best catalytic performance in some specific reactions.406 For instance, the facet engineering of CeNPs with the help of computational calculations made it possible to understand the crystal facet effect that plays a crucial role in determining the surface energy and reactivity.408 CeO2 nanorods with a dominant (110) facet showed the highest peroxidase-mimetic activity due to the rich defect chemistry on their surface (Fig. 22). These studies provided an increased understanding of the fundamental CeNPs properties that drive their unique reactivity. This knowledge is essential to rationally design and tailor their activity.
 | ||
| Fig. 22 CeO2 with different shapes and exposed facets (a)–(i), and H2O2 detection scheme using HRP/CeO2–rGO-modified glassy carbon electrode (j). Reprinted with permission from ref. 405. Copyright 2022 American Chemical Society. | ||
Several types of chemical and biological sensing systems where CeNPs have been used as receptors for gases, enzyme mimetic materials, co-catalyst or as a label for quantifying affinity recognition reactions have been developed (Fig. 23). Applications include gas, biomimetic or enzyme-based sensors, immuno, aptamer and DNA-based sensors. Examples for each of these classes are discussed in the section below.
 | ||
| Fig. 23 Summary of chemical sensing and biosensing involving CeNPs-based materials and their applications. | ||
Highly porous yolk–shell CeNP nanospheres prepared by microwave-assisted solvothermal synthesis showed a two times increase in the sensitivity for CO2 detection as compared to commercial CeNPs (Fig. 24) due to increased porosity and permeability for gas adsorption though the hollow structure.411 Similar structures were used to build sensors for acetone, with detection in the ppm concentration range, and a response and recovery time of 6 and 11 s, respectively.412 The doping of CeNPs with lanthanum was shown to strongly influence the redox (Ce3+/Ce4+) ratio and gas adsorption properties for CO. Under optimized conditions, the sensor provided fast response at a temperature of 380 °C under CO exposure associated with Ce4+ reduction and vacancy generation.416
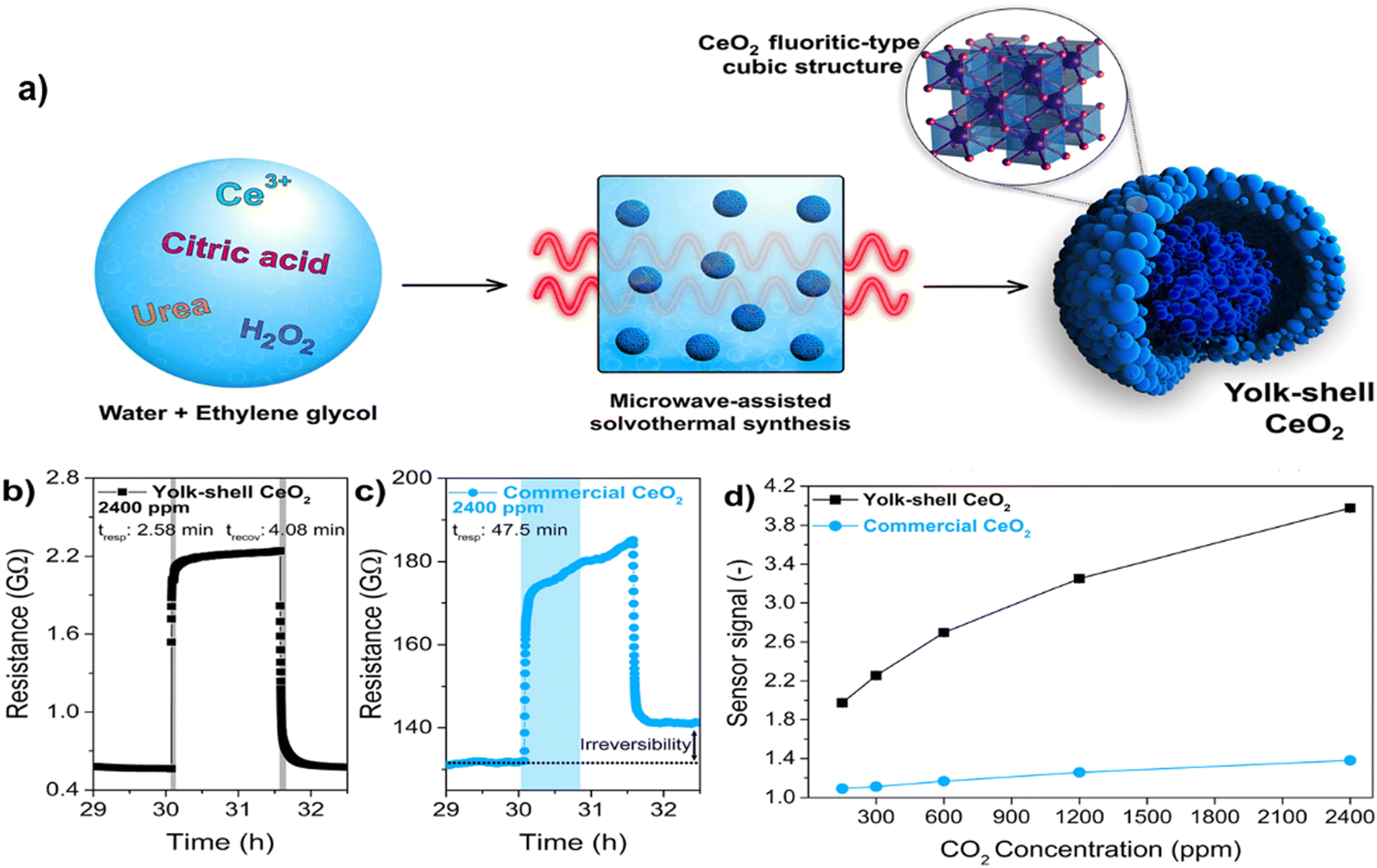 | ||
| Fig. 24 Microwave-assisted solvothermal synthesis of yolk–shell CeNPs (a), response and recovery times to 2400 ppm of the sensor prepaid with yolk–shell CeNPs (b) as compared to the commercial CeNPs (c), and comparative sensor signal in response to CO2 concentrations (d) (reprinted with permission from ref. 411. Copyright 2020 American Chemical Society). | ||
Sensors with optical detection measure changes in the optical properties associated with redox processes at the CeNPs surface. It is known that CeNPs form charge transfer complexes with H2O2224 and catecholate compounds,420 generating a distinctive concentration-dependent optical response. Based on these principles, a variety of colorimetric sensors have been reported using CeNPs as color-generating probes (e.g., as a replacement to commonly used soluble dyes) or to enhance the colorimetric signals of redox dyes such as 3,3′,5,5,-tetramethylbenzidine (TMB) or azino-bis(3-ethylbenzothiazoline-6-sulfonic acid) (ABTS). The high stability and rich surface functionalities of the CeNPs makes them amenable to patterning on solid surfaces, providing opportunities for the development of portable and inexpensive sensors.
One of the first demonstration of CeNPs as colorimetric probes was reported in 2011 for the detection of H2O2 and oxidase enzyme substrates such as glucose.224 The advantage of this system over conventional colorimetric assays is evident when comparing the two types of assays in Fig. 25A and B. When CeNPs is used as a color probe and peroxidase mimetic, in the presence of H2O2, the while/yellowish color of the CeNPs changes into bright yellow/orange due to the oxidation of Ce3+ to Ce4+ and formation of peroxide-Ce complexes at the NP surface, producing a unique concentration-dependent response.224 When CeNPs are co-immobilized with glucose oxidase on filter paper, H2O2 is generated on exposure to glucose, and the enzymatically generated H2O2 is then measured as a color change. Conventional enzyme-based colorimetric assays for oxidase enzymes involve the use of two enzymes, one for the conversion of substrate to H2O2 (e.g., glucoseoxidase), and one for the conversion of the enzymatically generated H2O2, typically a peroxidase, and a soluble redox dye. Because these reagents are soluble, they are generally used in solution or stored on specially designed reservoirs with reactions being accomplished though microfluidic channels. Conventional dyes are expensive and maintaining their stability is a challenge. Since CeNPs is a stable inorganic material that can replace both the peroxidase enzyme and the organic dye, they can be easily affixed to a solid platform such as paper, plastic or textiles for creating stand-alone portable sensors. Such reagent-less sensors have significantly enhanced the stability and portability and are low-cost as compared to solution-based assays (Fig. 25C). For example, a CeNP-based biosensor for glucose developed on filter paper displayed a limit of detection of 0.5 mM with a linear range up to 100 mM glucose. This concept can be extended as a general platform for any oxidase enzyme system that involves the generation of H2O2 as a reaction product, such as lactate, glutamate, cholesterol, uric acid, ethanol and hypoxhantine (Fig. 25D).
 | ||
| Fig. 25 Detection of glucose using glucose oxidase (GOx) enzyme by conventional colorimetric assay with GOx, peroxidase and a redox dye (A) and the CeNP-based sensor (B). Immobilization of CeNPs on paper using 3-aminopropyltriethoxysilane (APTS) and chitosan biopolymer and ImageJ quantification of biosensor response (C) with examples of biosensor targets developed based on CeNPs used as colorimetric probes (A, B reproduced with permission from ref. 224. Copyright 2011 American Chemical Society). | ||
Similar types of colorimetric sensors based on CeNPs have been designed to detect enzyme activities, such as alkaline phosphatase (ALP). For example, a CeNPs-based sensor was developed based on the color changes of CeNPs reacting with hydrolytic products of ALP-catalyzed reactions, from substrates such as catechol monophosphate, ascorbic 2-phosphate and hydroquinone diphosphate. The method, adapted to a paper-based platform, had a detection limit of 0.04 U L−1 ALP with a linear range up to 2 U L −1when ascorbic 2-phosphate was used as the substrate.421 More recently, the activity of beta-galactosidase (beta-Gal) was determined using a procedure involving CeNP-induced oxidation of 4-aminophenol (4-AP) produced in the hydrolysis of the 4-aminophenyl-beta-D-galactopyranoside substrate (4-APG) by beta-Gal. The approach enhanced the detection sensitivity and enabled beta-Gal measurements in the visible range, reaching a detection limit of 0.06 U L−1 and a linear range up to 2.0 U L−1. A 30-fold amplification as compared to a commercially available beta-Gal assay was reported using the CeNP assay.422 In addition to colorimetric sensors, opportunities exist to design CeNP-based fluorescence and imaging assays by doping CeNPs with Eu. For example, Eu-doped CeNPs (5 nm diameter doped with 2% Eu) exhibit bright and stable fluorescence in aqueous media (Fig. 26). Their combined catalysis and fluorescent properties can serve as a universal concept to design fluorescent assays with the ability to monitor enzymatic reactions and quantify enzymatic activity and substrates such as AlP activity, dopamine, glucose and lactate with increased selectivity and sensitivity.400
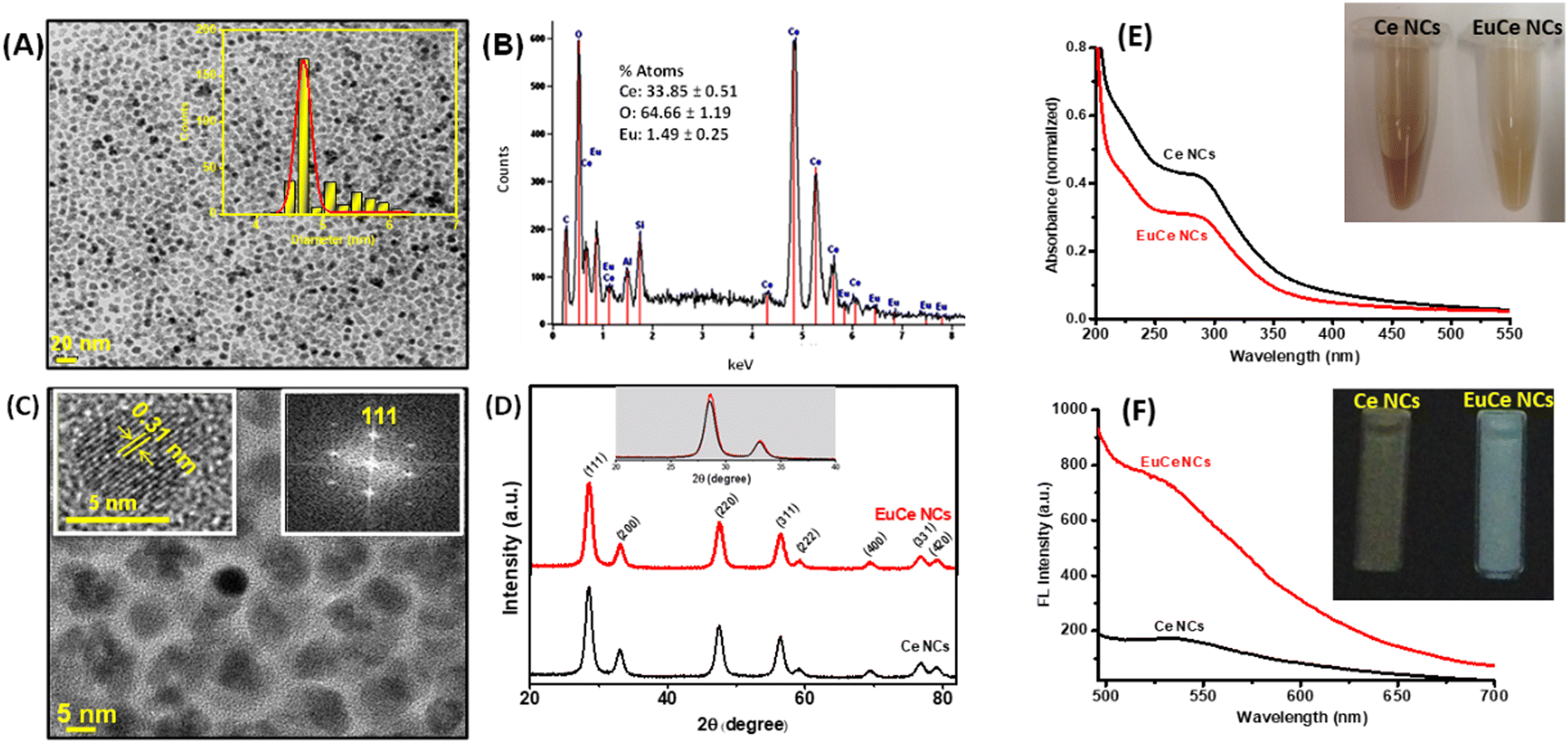 | ||
| Fig. 26 Morphological, structural and catalytic activity characterization of Eu-doped CeNPs, (A) The HR-TEM (inset: particle size distribution). (B) EDX, (C) HR-TEM at high magnification (inset: dimension of single spherical particle (left) and electron diffraction (right)), (D) XRD patterns of CeNPs and Eu-CeNPs. (E) UV-vis spectra of CeNPs and Eu-CeNPs showing emission at λex, 466 nm, and λem 522 nm, inset: photographs of Eu-CeNPs under UV (365 nm) (reprinted with permission from ref. 400. Copyright 2018 American Chemical Society). | ||
The distinctive changes in the optical properties of the CeNPs reacting with compounds with phenolic or catecholoamine moieties were used to develop paper-based sensors for the detection of phenolic antioxidants, with the reported detection limits in the range of 20–400 μM, depending on the structure of the compound. These sensors have the capability to provide a unique optical signature generated when CeNPs interact with a single or a mixture of antioxidants. The method enabled field assessment and classification of the antioxidant capacity in tea,423 wine,424 medical mushrooms425 and cosmetic products.426 For these measurements, the CeNPs stabilized onto a paper platform form the actual sensing platform (e.g., sensing spot or label), enabling portability and reagent-less operation. Similar to a pH paper, analysis is performed by adding the sample to the sensor; the antioxidants in the sample produce an immediate color change that is visible to the naked eye. Quantification is achieved by comparison with a pre-calibrated detection scheme. For more precise quantification, analysis can be performed with a color reader such as a cell phone with a color reading app; no additional reagents are needed (other than the sample). In comparison with other assays for antioxidant analysis, the CeNPs sensors have very low cost and are stable at room temperature and field deployable. Finally, biosensing systems based on CeNPs have practical utility, as demonstrated by the development of several standalone devices for the detection of ethanol in breath,427 phenolic antioxidants for evaluating antioxidant activity in teas and wines,425 and measuring the hypoxanthine content to assess the freshness of fish and meat,428 are summarized in Fig. 27.
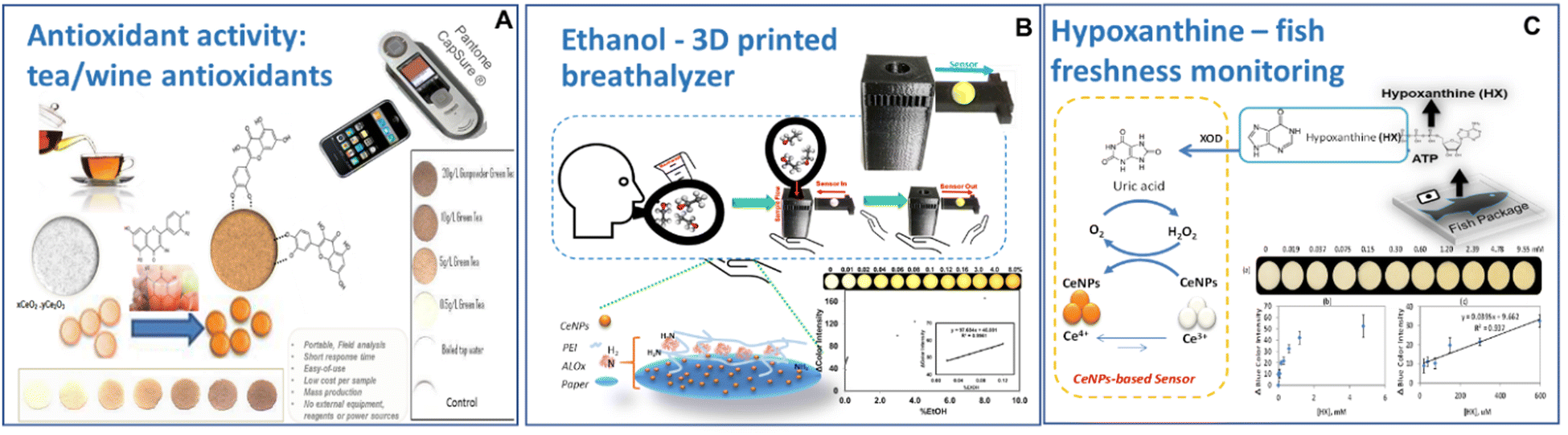 | ||
| Fig. 27 Examples of practical CeNP-based paper sensors for the detection of antioxidant activity in tea (A), ethanol in breath using a 3D printed device (B) and hypoxanthine to assess the freshness of fish (C). Reprinted with permission from Refs for (A),425 (B)427 and (C).428 Copyright 2021 American Chemical Society. | ||
Several hybrid configurations of CeNPs-based enzyme biosensors have been developed in which the CeNPs are used in conjunction with oxidase enzymes to catalytically amplify the enzymatic reaction or the product of an enzymatic reaction, significantly enhancing the sensitivity of the biosensor. Moreover, the ability of the CeNPs to store and release oxygen has been used as an oxygen reservoir to fuel oxygen to oxidase enzymes, enabling them to operate in oxygen-restrictive environments such as tissue hypoxia. This principle has been demonstrated effectively for improving the sensitivity of implantable electrochemical microbiosensors fabricated by co-immobilizing the enzyme with the CeNPs at the surface of a microelectrode, typically a Pt wire or a carbon fiber brush, with diameters ranging from 100 to 150 μm. Successful developments have been demonstrated with CeNPs-based biosensors for the detection of dopamine,429 glutamate,430 lactate431 and glucose.432 Enhanced selectivity has been obtained with Pt-doped CeNPs, which imparts conductivity and amplifies the electrocatalytic signal for H2O2 oxidation, as shown in Fig. 28A for glucose detection. The lactate biosensor, for example, had a detection limit of 37 μM in implantable conditions and was able to quantify the real time release of lactate in vivo during cerebral ischemia and reperfusion (Fig. 28B).431 This lactate biosensor operated in a continuous mode due to the ability of the enzymatically generated H2O2 to react with the CeNPs, producing oxygen in the process. This released oxygen is then utilized by the enzyme, facilitating its functionality in the hypoxic medium. Notable, the biosensors without CeNPs do not provide any quantifiable response due to the lack of oxygen, a necessary cofactor for ensuring enzyme function. Other possibilities to enhance the conductivity for electrochemical biosensors are to use the CeNP in conjunction with graphene oxide and PtNPs,433 conductive polymers such as polyaniline,434 or mesoporous carbon,435 leading to enhanced sensitivities. CeNPs can also be designed in the form of a dual-tethered nanosystem in conjunction with fullerene, which combines the properties of the two materials, leading to hybrid structures with high antioxidant and UV-shielding properties.436
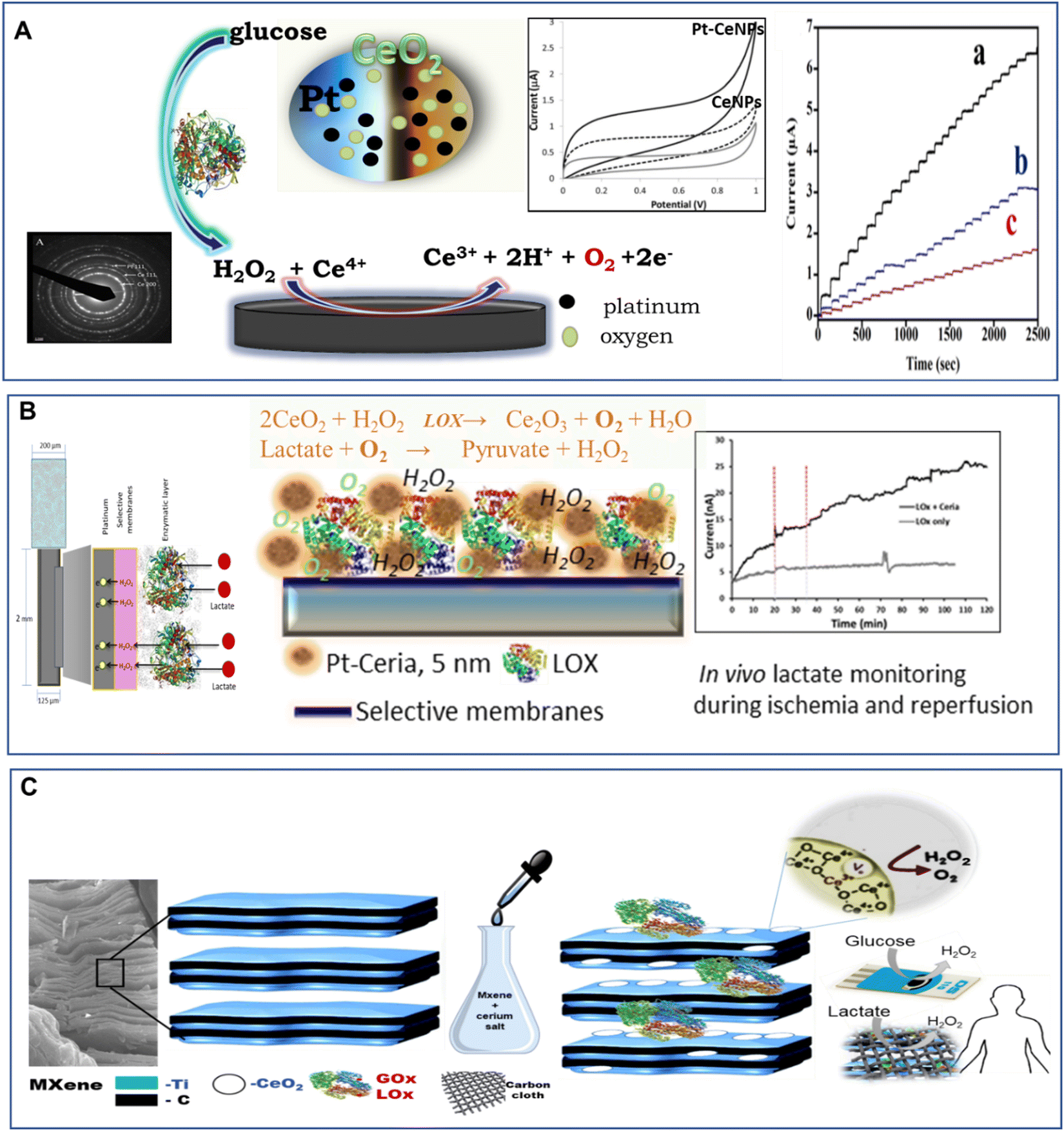 | ||
| Fig. 28 (A) Electrochemical CeNP-based implantable microbiosensors using Pt-doped CeNP (5 nm), which electrocatalyzes H2O2 oxidation, further enhancing glucose detection; significantly enhanced responses were obtained when Pt–CeNPs were used (a) as compared to bare CeNPs, (b) or no CeNPs (c) (with permission from ref. 432); (B) implantable microbiosensor configuration for lactate detection during ischemia and reperfusion showing quantifiable responses when Pt-doped CeNPs were immobilized with lactate oxidase (reprinted with permission from ref. 431. Copyright 2015 American Chemical Society.); (C) catalytic MXene-CeNPs biosensing platform for wearable biosensors (with permission from ref. 437). | ||
Lastly, significant improvements can be achieved by interfacing CeNPs with high surface area materials such as 2D layered MXenes, producing catalytically active materials that can be used in a variety of applications. In a recent work (Fig. 28C), MXene decorated with CeNPs enabled the development of an electrochemical biosensor platform broadly applied to a variety of enzymes and enzyme substrates, achieving detection limits of 0.8 μM H2O2, 0.49 μM glucose, 3.6 μM lactate and 1.7 μM hypoxanthine.437 These materials can be integrated within flexible platforms such as carbon cloth and be used to construct wearable devices, as demonstrated for lactate monitoring in sweat.437
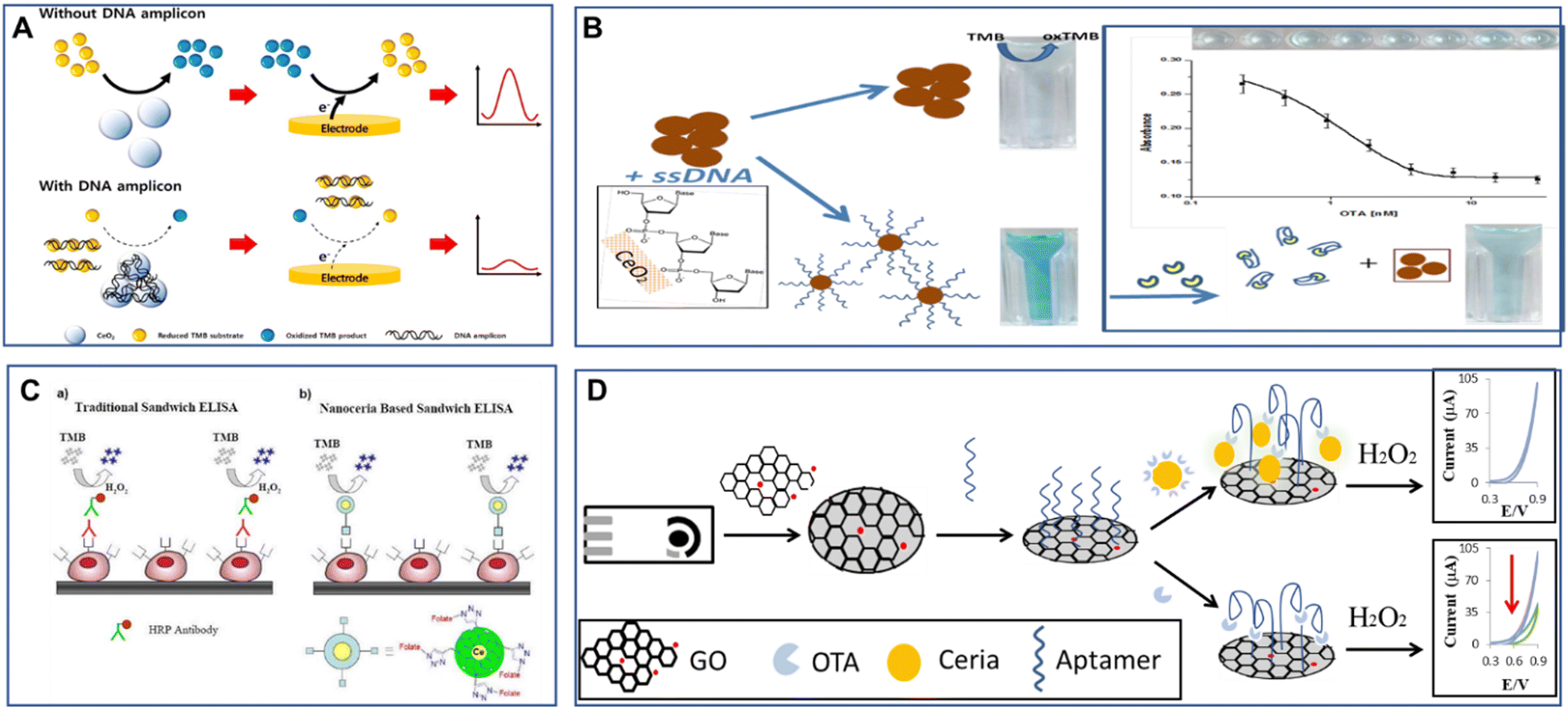 | ||
| Fig. 29 Examples of bioaffinity interactions with CeNPs as an enzyme-mimetic nanocatalyst with electrochemical (A) and (D) and colorimetric (B) and (C) detection: (A) electrochemical detection of DNA amplicons by quantifying the catalytic activity of CeNPs using TMB (reprinted with permission from ref. 438), (B) colorimetric detection of target binding to DNA-aptamer-modified CeNPs with the detection of target-induced conformational changes and aggregation (with permission from ref. 440), (C) comparison between traditional immunoassay (ELISA, a) and CeNPs-based sandwich assay with CeNP labels mimicking the activity of oxidase enzyme. (Reprinted with permission from ref. 441), and (D) electrochemical sensor using CeNP tag and graphene oxide for the detection of aptamer targets through the measurement of the CeNP's-catalyzed H2O2 oxidation with and without target (reprinted from ref. 442 with permission from the Royal Society of Chemistry). | ||
Another interesting application is to use CeNPs as a replacement of the enzyme label in conventional affinity assays (e.g., ELISA) in lieu of oxidase or peroxidase enzymes. One of the first reports suggesting that polymer (polyacrylic acid)-coated CeNPs can be used as an oxidase enzyme label in immunoassayas was published by Asati et al. in 2009, who demonstated that conjugation of CeNPs with targetting ligands can be used as a nanocatalyst with dual function as target binding and catalytic detector via the oxidation of the colorimetric dye TMB (Fig. 29C).441 Enhanced detection for electrochemical aptasensors has been achieved by immobilizing the aptamer on a graphene-modified screen-printed electrode.442 The target was captured by the immobilized aptamer and determined via a competitive mechanism between the free and the CeNPs labelled target by monitoring the H2O2 oxidation, amplified by the CeNP tag. Detection limits in the low nM range (0.1 nM), with a linearity range up to 180 nM, were obtained for the detection of a mycotoxin, ochratoxin A. Such strategies can be successfully implemented in point-of-care diagnostic devices.443
DNA-functionalized CeNPs can be used for a variety of biosensing applications such as to probe the oxidation of phosphorous compounds444 and H2O2.445 Modifications of the CNP surface can be made to enhance the oxidase-like activity and enable the detection of ions such as fluoride, quantified indirectly using ABTS dye.404 Moreover, the CeNP's surface chemistry and surface ligand can be modulated to immobilize or displace DNA from the CeNPs’ surface for the detection of substrates such as H2O2 and glucose in serum.445 When CeNPs were coupled with glucose oxidase, a detection limit of 8.9 mM glucose was obtained. More recently, an integrated system of CeNPs/glucose oxidase was used to produce bio-nanoenzyme catalytic cascades with oxidase-like activity.446 Inhibition of the oxidase-like activity of CeNPs by ions such as arsenate and arsenite447 or phosphate due to blocking of Ce3+ sites448 was also reported, opening up new avenues for the development of biomimetic CeNPs inhibition-based sensors. Such engineering designs of the CeNPs surface could increase the performance of CeNPs, making them powerful nanoenzymes for many practical applications.
More recently, hybrid materials and supramolecular structures such as Ce-based MOFs, traditionally known for their electrolytic performances,449,450 have begun to be explored in biosensing. A bimetallic Ce/Cu-MOF (CeO2/CuOx@mC) aptamer nanocomposite was prepared for the detection of trace tobramycin (TOB) in human serum and milk.451 Similarly, a nanohybrid of covalent organic framework (COF) and Ce-based (Ce-MOF@COF) nanostructure showed high binding affinity toward the oxytetracycline (OTC)-targeted aptamer.452 The aptasensor displayed good reproducibility, selectivity, stability, and acceptable applicability for detecting OTC in various samples, including milk, wastewater, and urine. The future development of Ce-based mesoporous materials such as Ce-MOFs, which possess high porosity, could lead to the development of enhanced catalysts for next-generation sensing.
4.4. Environmental remediation
The presence of contaminants, such as metals, organic dyes, pesticides, nutrients and toxins, is a worldwide concern due to their long-term environmental effects.396 Because of their thermal stability, optical properties, and oxygen transport capacity, CeNP-based materials can be used as sorbents in environmental remediation for water and wastewater treatment.453–455 CeNPs can be used alone, in binary mixtures or deposited on various supports such as activated carbon, graphene, polymeric nanofibers, or mesoporous structures (Fig. 30). Their removal properties are attributed to the high surface charge density,456 hydroxilated surface and rich surface defects that facilitate the rapid complexation of oxyanions such as phosphates and arsenates.5,457 To improve handling, CeNPs has been added to conventional adsorbents such as activated carbon or carbon cryogel,458 patterned on surfaces, or used in conjunction with porous materials such as mesoporous silica,459 hierarchical porous CeO2–ZrO2 nanospheres,460 or MOFs.300,457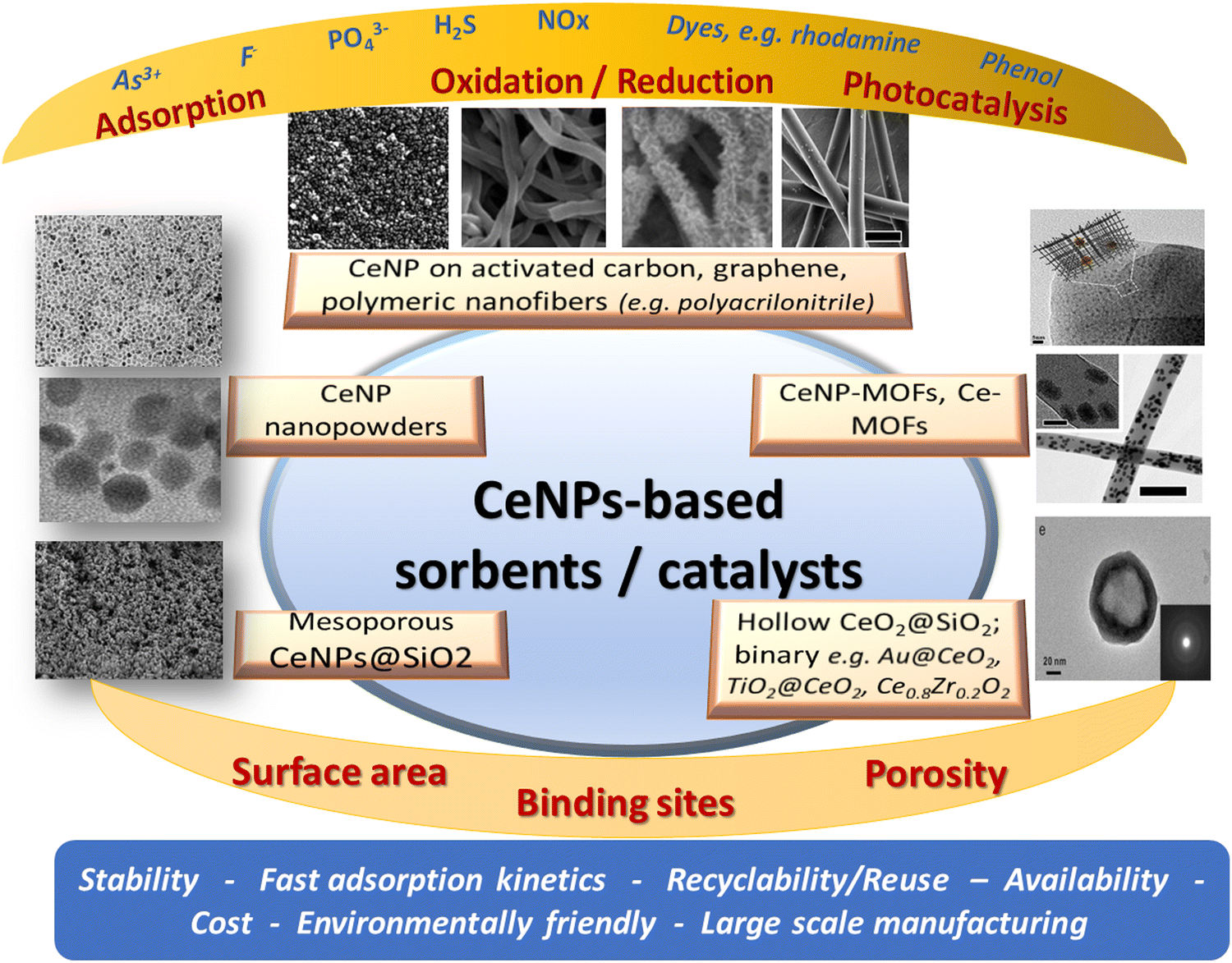 | ||
| Fig. 30 Summary of CeNPs-based materials for environmental remediation. | ||
Ceria-coated activated carbon was synthesized in a single step and used in water purification, where it demonstrated high efficiency for the removal of As(III) and As(V).461 Additionally, a nanocomposite prepared from graphene nanosheets and CeNPs (G/CeO2) was used for the extraction of Se(IV), As(V), As(III), Cu(II) and Pb(II) from water.462 The maximum adsorption capacity of G/CeO2 was extrapolated from the Langmuir model and found to be 8.4 for As(V), 14.1 for Se(IV), 50.0 for Cu(II) and 75.6 for Pb(II) (expressed in mg g−1). These also showed good selectivity towards Se(IV) in the presence of Se(VI). CeNP-based materials were also used for ultratrace metal ions detection and speciation via electrochemistry.271 Enhaced arsenic removal was achieved using porous CeO2–ZrO2 hollow nanospheres due to the high sorption capacity and selectivity for As; adsorption capacities of 110.7 and 145.3 mg g−1 were reported for As3+ and As5+, respectively.460 As a dual Ce3+/Ce4+ oxidation state material, CeNPs can participate in electron exchange reactions with the As3+ ions present in the solution. The mechanism of As adsorption and redox processes occurring between the surface Ce3+/Ce4+ and As3+ at different pH conditions was demonstarted at single particle levels using nanoscale electrochemistry by monitoring the oxidation of As3+ to As5+ at 0.8 V vs. Ag/AgCl or the reduction of As3+ to As0 at −0.3 V (vs. Ag/AgCl).463 The highest sorption capacity was observed at pH 8. In acidic environments (pH < 4), a small fraction of As3+ was oxidized to As5+ by surface Ce4+ and further adsorbed onto the CeO2 surface as an As5+ bidentate complex (Fig. 31). Adsorption was common in basic conditions, confirming the strong adsorption of As3+ onto CeNPs. These findings pave the way for rationally selecting the optimal material structure and conditions to increase the sorption capacity for environmental sensing and remediation applications.
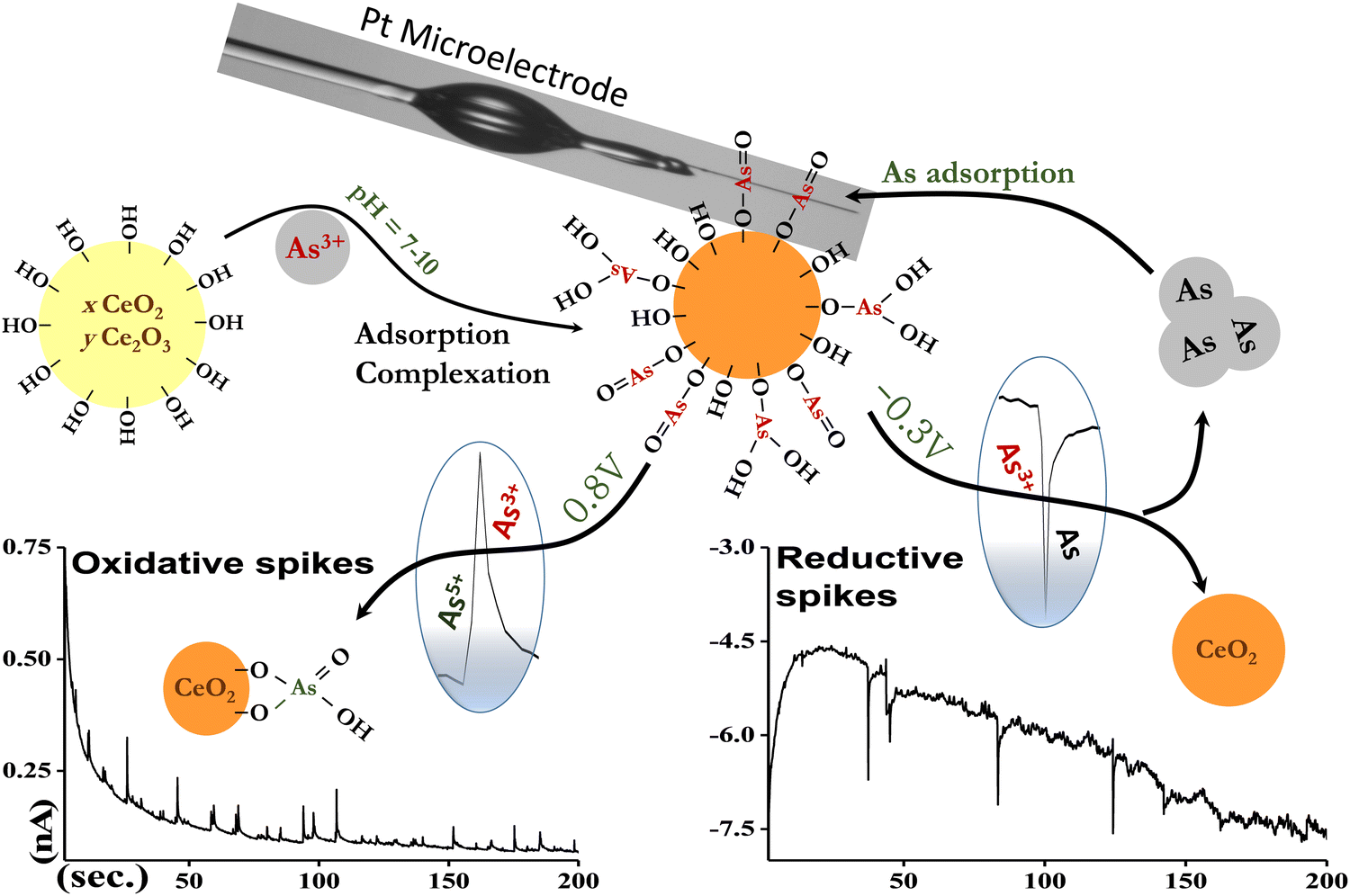 | ||
| Fig. 31 Mechanism of As3+ adsorption on CeNPs quantified by nanoscale electrochemistry by measuring current transients measured in reductive (−0.3 V) and oxidative (0.8 V vs. Ag/AgCl) conditions in PB at pH 8. Reproduced with permission from ref. 463. Copyright © 2019, American Chemical Society. | ||
CeNPs exhibit high selectivity for fluoride (F−) adsorption and can be used as an adsorbent for F− removal from water. F is one of the essential elements in the human body for the formation of dental enamel and normal mineralization of bones.464 However, the excessive intake of F− is harmful to the body and can lead to various health issues ranging from mild dental fluorosis to crippling skeletal fluorosis. While CeNPs are effective adsorbents and could be used in large scale column purification technologies for water defluoridation, their small size is impractical as it could cause column blockage, high pressure drop, and low flow rate. To overcome these limitations, binary oxides and hydroxides such as CeO2–ZrO2 nanocages465 and Ce–La binary hydroxide466 were explored as alternative sorbents with good F− uptake but slow adsorption rate. Further improvements were achieved using CeNPs incorporated on active supports like silica and organic polymers, which provide a large specific surface, fast diffusion kinetics, low cost, high mechanical strength and low pressure drop in a packed adsorption column.467–470 F− adsorption onto a cubical ceria nanoadsorbent has shown pH-responsive behavior during the adsorption process.471 At lower solution pH, a release of OH− was observed during the adsorption of fluoride by inner-sphere complex formation. At higher solution pH, the pH was found to decrease due to the exchange of Na+ added in the form of NaOH by outer-sphere complex formation with H+ ions adsorbed electrostatically on the adsorbent surface.471
Other environmental applications of CeNPs are as catalysts to remove exhaust emission,472 and volatile organic chemicals (VOCs) and degrade organic pollutants from the environment. For example, CeNPs have shown high performance towards the reduction of NOx and the catalytic oxidation of NH3 from engine exhaust.473 Owing to their oxygen storage/release and the Ce4+/Ce3+ redox cycle, CeO2-based catalysts are excellent candidates for the selective catalytic reduction of nitrogen oxides (NOx) and carbon monoxide (CO), which contribute to acid rain and photochemical smog. In a typical catalytic convertor, ceria acts as a buffer adsorbing and releasing oxygen species depending on the operating conditions and temperature. It was reported that the use of CeNPs (30 nm) as an additive to fuel resulted in reduced fuel consumption by 2.5%, as well as reduction in NOx emission by 15.7% and smoke opacity by 34.7% compared to the additive-free fuel.474 Enhanced catalytic performance for selective NOx reduction by NH3 was obtained with the CeNP-based catalyst on oxide supports such as Al2O2, TiO2 and WO3 with the highest activation ability for Ce–O–W through rich Brønsted acid sites, redox ability and strong electronic CeO2–support interactions.475 NPs of CeO2, CuO/CeO2 and Pt-MnO2/CeO2 have shown high activity for the oxidation of phenol intermediates.476–478 Nanocomposites of TiO2/CeO2 on carbon nanotubes have demonstrated photo-oxidation capabilities for the degradation of organic content in agricultural wastewaters.479 Pd/CeO2 with carefully designed morphology and optimized shapes were effective catalysts for methanol decomposition to syngas with excellent activity at low temperatures.480 Mn-doped CeNPs prepared by hydrothermal synthesis have shown a drastic increase in their capacity to adsorb oxygen due to the intercalation of Mn3+ and the decrease in the Ce3+/(Ce3+ + Ce4+) content in the doped samples by replacement with Mn3+. The doped samples had a significantly higher activity for toluene oxidation.481
CeNPs have also been studied for the photocatalytic degradation of reactive dyes, at which more than 95% of the dyes were destroyed under solar irradiation.482 Recent work showed the ability of CeNPs to effectively degrade organic dyes such as fluorescein, rhodamine B, brilliant blue and Coomassie blue, with enhanced degradability in the presence of fluoride at ambient temperature.483 The enhanced degradability in the presence of fluoride was attributed to the increased oxygen vacancy and the strong acidic environment. Hybrid CeNPs-lanthanide oxides demonstrated H2S adsorption and dissociation with subsequent oxygen vacancy regeneration, enabling their use for the desulfurization of biomass effluents.484 It was demonstrated that adding Ce to metal oxide catalysts enhanced the catalytic activity and SO2 resistance of Mn/Ti catalysts for the NO reduction with NH3 by inhibiting the accumulation of ammoniom sulfates and preventing sulfidation.485 The addition of a small amount of Ce (Ce/Ti = 0.05) was shown to improve the oxygen storage/release of MnOx–CeO2/TiO2, thus enhancing the activity. Binary nanofibrous structures based on anisotropic Au/CeNPs embedded in poly(vinyl alcohol) (PVA) have shown potential as photocatalytic materials, promoting both oxidation and reduction of model waste products for environmental remediation processes such as the catalytic reduction of 4-nitrophenol to 4-aminophenol and of phenol red to bromophenol as well as the degradation rhodamine B in a flow-through reactor (Fig. 32).486
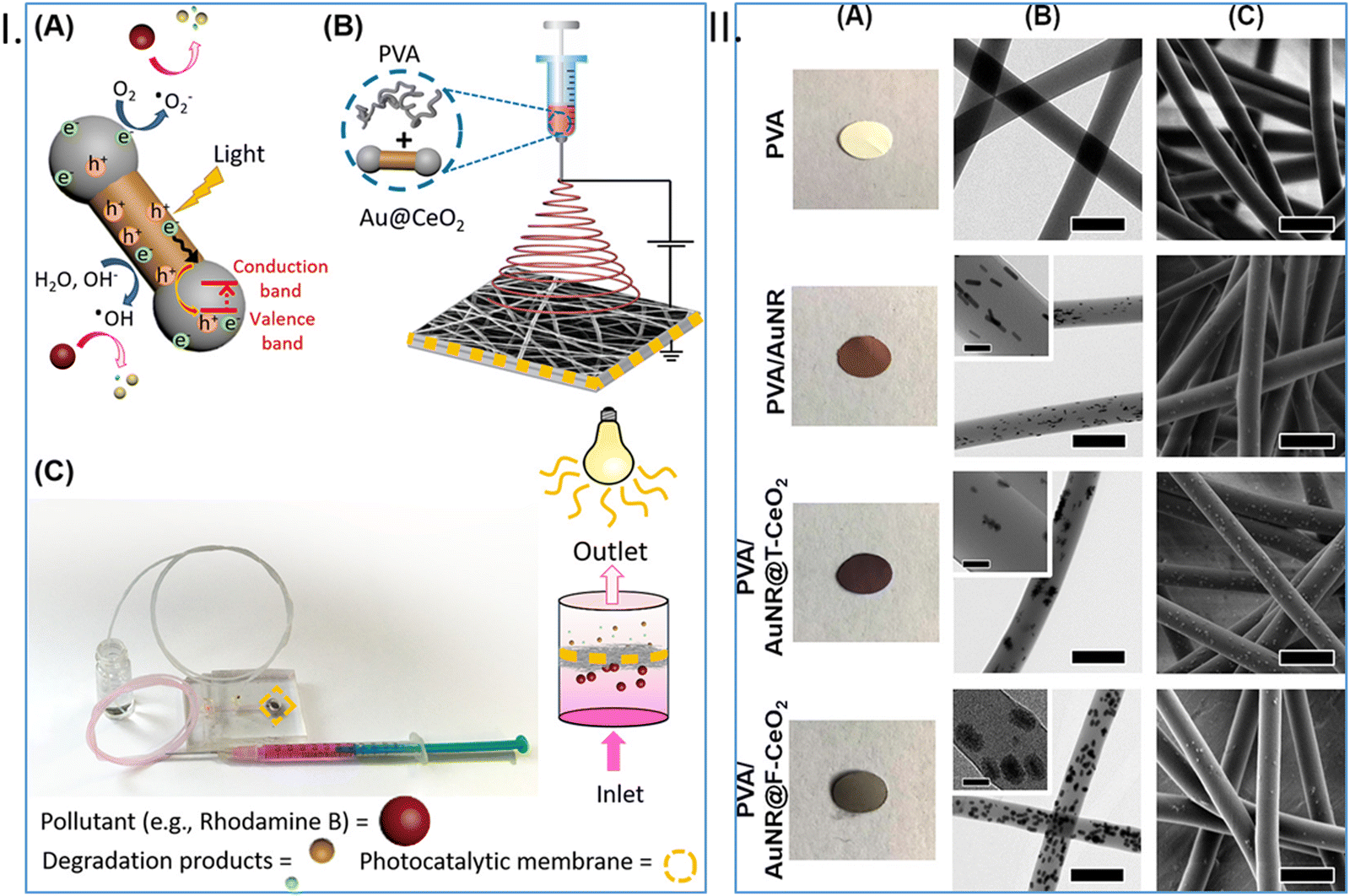 | ||
| Fig. 32 (I) Photocatalytic membranes prepared by the colloidal electrospinning of a suspension of poly(vinyl alcohol) (PVA) and Au–CeNPs for use in a flow-through fluidic reactor showing (A) photocatalytic process, (B) Preparation of Au@CeO2/PVA nanofibers by electrospinning, and (C) photocatalysis with the flow-through reactor containing the nanofibrous membrane; and (II) images (A), TEM (B) and SEM (C) of electrospun hybrid membranes made with PVA, PVA/AuNR, PVA/T-AuNR@CeO2, and PVA/F-AuNR@CeO2. Reprinted with permission from ref. 486. Copyright 2019 American Chemical Society. | ||
In other environmental applications, CeNPs have shown tremendous potential as sorbents for the removal of phosphorus to prevent water eutrophication. Eutrophication has become a global environmental problem due to the unnatural enrichment of water bodies with phosphorus (P) and nitrogen (N), which leads to the excessive growth of harmful algae blooms and depletion of dissolved oxygen levels, creating dead zones.269,457 Thus, removing the excess P and N is essential for preventing the negative consequences of eutrophication.269,487 CeO2-coated nanofibers have shown high adsorption capacity for the removal of P from aqueous solutions488 with an adsorption capacity of 17 and 12 mmol g−1 at low pH (2–6) and neutral pH. The removal process involved the formation of CePO4 on the nanofiber surface. In other works, a recyclable adsorbent based on CeNPs grafted on mesoporous silica beads (CeO2–SiO2) has shown reversible adsorption behavior for P, demonstrating potential for both the removal and recovery of this important nutrient from eutrophic waters (Fig. 33).459 The adsorption mechanism involved the binding of PO43− through ion exchange and Lewis-acid based interactions, which provided a sorption/desorption capacity of 110 mg g−1 with 99% removal within 60 min, significantly higher as compared to other sorbents. In the follow-up work with this system, ultrafast removal was achieved using a Ce(IV)-based MOF, Ce(IV) UiO-ss, and Xe 1,4-benzenedicarboxylate (Ce-BDC), which provided high surface area due to the open pores and high binding affinity of its open metal sites for phosphate. A removal capacity of 179 mg g−1 within 4 min and in a broad range of pH (2–12) was reported.457 Other enhancements have been obtained using binary oxides with properly designed ratios,489,490 optimized to enhance surface defects and facilitate complexation, thus increasing the adsorption capacity as compared to the single bare oxide. Su et al. developed a solvothermal process to synthesize a series of Ce/Zr binary oxide nanoadsorbents. A sorption capacity of 112.2 mg g−1 was reported with an optimized oxides ratio of Ce0.8Zr0.2O2.491 In order to facilitate the recovery of the adorbents after the treatment, core–shell CeNPs (Fe3O4@SiO2–CeO2)492 and (Fe3O4@SiO2@mCeO2)493 were designed to magnetically separate and remove these sorbents from the water environment. These structures can be utilized in a variety of applications such as membranes for phosphate separation and purification, sorbents for solid phase extraction or as P-selective materials for environmental sensing. Recently, it was shown that the use of Ce-metal nodes in the UiO-66-NH2 MOF structure enables the fluorimetric sensing of phosphate, with a detection limit of 4.5 μM in a single-step measurement procedure.494
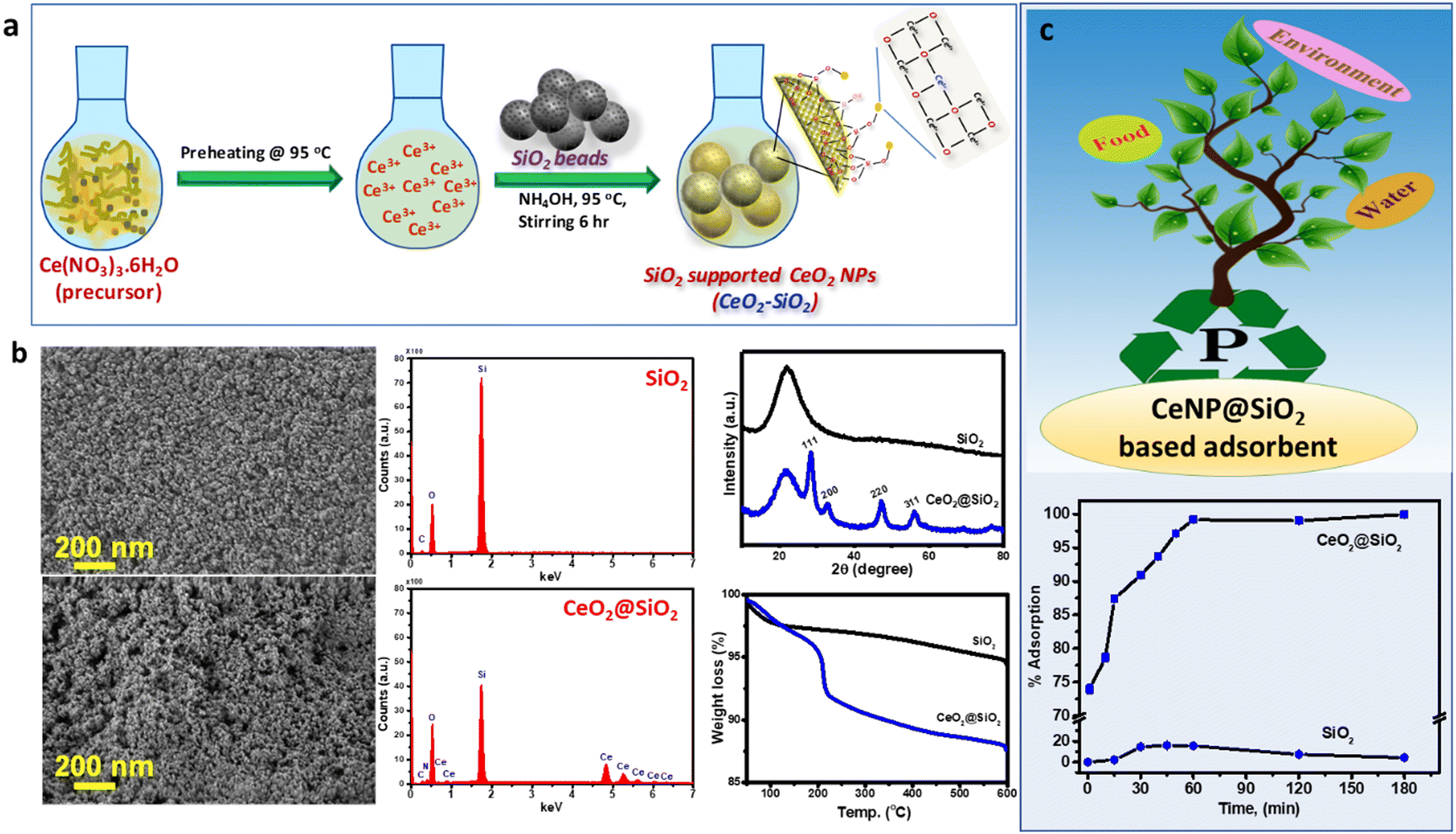 | ||
| Fig. 33 Synthesis of CeO2@SiO2 adsorbent (a) with SEM, XRD and TGA characterization (b) and adsorption isotherms for the removal of phosphate (c) showing the significantly enhanced adsorption capacity of CeO2@SiO2 as compared to SiO2 (reprinted with permission from ref. 459. Copyright 2019 American Chemical Society). | ||
While the properties of CeNPs are vastly explored, their potential environmental and toxicological effects are still under investigation. The release of CeNPs into the environment is expected to increase as they start to be implemented in real-world applications.493,495 Their use as polishers in the semiconducting industry496 and as diesel fuel additives for catalytic convertors to control auto exhaust emission497 are just two examples that can the result in the release and accumulation of CeNPs in the environment. The toxicology studied suggest that CeNPs by themselves are not particularly toxic; however, they can become toxic by the adsorption of toxic ions (e.g., As3+) or organics on their surface.463 Therefore, understanding the fate, transport, and toxicity of these NPs exposed to contaminants is becoming increasingly important and will be critical in further studies to examine their environmental and health impacts.5,498 The green synthesis of CeNPs can provide alternative routes to prepare CeNP-based materials without the need of using environmentally harmful reagents. For example, CeNPs were synthesized using plant-mediated synthesis wherein different plants such as Gloriosa superba, Acalypha indica and Aloe vera were used.499–501 Additionally, honey, egg white and fungus-mediated synthesis were also used.502–504 These plant extracts and natural components act as capping and reducing agents to produce nanocrystalline CeNPs in different sizes and morphology. These methods also offer advantages such as the low cost, large-scale production, and more importantly, their environment-friendly nature, making them interesting candidates for use in biomedicine and pharmaceutical applications. A detailed review on the green synthesis of CeNPs and their biological applications has been published.505
4.5. Biomedical applications
The ability of CeNPs to catalyze the detoxification of free radicals and act as artificial antioxidants provides a broad range of opportunities for using this system in biomedicine, particularly for treating disease conditions in which excess free radical formation is thought to play a role.506 These range from relatively rare diseases, such as Amyotrophic Lateral Sclerosis (ALS), to more common conditions, such as stroke, cancer,507 traumatic brain injury, Parkinson's and Alzheimer's disease to the ubiquitous normal processes of aging. CeNPs possess a number of advantages over other antioxidants. CeNPs have been shown to provide antioxidant, immunomodulatory, antimicrobial and antiviral, and anticancer properties, and their ability to be used as theranostic probes.8 When administered systemically, CeNPs appear to cross the blood brain barrier.508Their properties can be enhanced by modification with other drugs, as demonstrated with edaravone attached using poly(ethylene glycol); the combined effect provided enhanced neuroprotective action for stroke treatment through the inactivation of ROS and enhanced accessibility to cross the blood–brain barrier.509 The regenerative behavior of the CeNPs, enabling them to regain the antioxidant activity upon interaction with ROS, provide additional opportunities for these particles to remain active for long periods of time and be used in tissue engineering to support neural survival and growth.510 Thus, they could be administered preventatively or early in a chronic disease process. Other applications include utilization in spinal cord repair as neuroprotective agents,511,512 support cell survival,513,514 and scavenging ROS in the eyes to prevent degenerative retinal disorders.513 When cells were exposed to H2O2, CeNPs reduced oxidative damage by over 60%.9,511,512
CeNPs holds potential as a drug delivery platform for targeted therapy. For example, they can be conjugated with other therapeutically-effective molecules to improve drug delivery515 or modified with specific receptors and biomolecules for targeted delivery516 and wound healing.517 The tunable surface and large surface-to-volume ratio facilitate enhanced loading and potential for release, making them a versatile platform for personalized medicine.518 They can also be incorporated in hydrogels or other biomaterial matrices and used to support cell adhesion and proliferation, and promote wound healing.519 The tunable catalytic activity of the CeNPs has been shown to be effective in preventing chemotherapy-induced acute kidney injury and restore redox homeosthasis.520 Biomedical imaging is another area in which CeNPs can find exciting emerging applications such as contrast agents and imaging modalities for magnetic resonance imaging (MRI) and computed tomography. This can be achieved by the surface modification of the CeNPs with dyes or other imaging agents or by doping with Eu or Gd, enabling the visualization of biological processes and monitoring of therapeutic response, thus facilitating the early detection and monitoring of diseases.521,522 While the biomedical applications of CeNPs are rapidly emerging, the rational design and better control of the structure, properties and dose response is still needed to establish conditions that can effectively reduce the level of free radicals without impairing the normal physiological functions. Several reviews expanding on the biomedical and theranostic applications of CeNPs can be found in the literature.8,169,171,523
5. Summary and outlook
The unique structure, tunable reactivity and rich surface chemistry make CeNP an extremely versatile material for a wide variety of applications. Herein, we have comprehensively reviewed the progress made in the development of CeNPs-based materials, their structure, properties and emerging applications over the past two decades. The synthesis and control of the surface properties represent key strategies for customizing the physicochemical characteristics of these materials to meet the requirements of diverse applications and advancing new products and innovations. Understanding the structural properties at the atomic levels provides the fundamental knowledge necessary to engineer structures with controlled reactivity and surface functions. Such studies are still needed to identify key parameters in the synthetic design that can be used to predict the reactivity and generate CeNP-based materials and devices with the desired properties for a given target. Further improvements in the synthesis, surface functionalization and the use of in situ characterization techniques will help achieve better control of the CeNPs properties (e.g., antioxidant vs. pro-oxidant) and develop predictable structures with tailored reactivity and surface binding characteristics. Moving forward, we expect development in the following areas.• First, fundamental work is still needed to identify the origin of CeNPs reactivity (surface defects, complexation, Ce3+/Ce4+ ratio, size, shape and surface ligand effects) and how these relate to the physicochemical properties and reactivity of the materials. Considerable progress is expected in modulating the surface chemistry and developing structure/function relations, some of which may require computational design for understanding the complex CeNPs chemistry and predicting activity and selectivity for a given target.
• Secondly, recent work has shown that combining two or more materials and creating hybrid and composite structures can significantly enhance the properties. Understanding the effect of interfacial structures and interactions between two or more types of materials (chemical bonds, surface energy, interface mechanics, etc.) is needed to more rationally design structures that take advantage of the synergistic effects and complementarity of their mixed components. Examples include noble metals (Pt, Au)-CeNPs, polymeric materials-CeNPs, metal oxides-CeNPs, in particular those with catalytic and magnetic properties (TiO2, ZnO, Fe2O3), as well as bio-interfaces by conjugating CeNPs with enzymes, aptamers, DNA, antibodies, etc., for obtaining improved catalytic and sensing functions.
• Third, as the applications of these materials continue to grow, the manufacturing and integration of CeNPs in devices, membranes and coatings will constitute an area of intensive research. The use of advanced manufacturing practices such as 2D and 3D printing are of great interest to produce low-cost high throughput structures. Moreover, integrating CeNPs into functional and practical devices will be needed to effectivelly translate these innovations into practice and consumer applications.
• Fourth, as more CeNPs are implemented into consumer products, understanding their environmental transformation, fate, transport, toxicity and their transformed products will be needed, particularly in areas where there is a risk of exposure such as semiconducting manufaring plants, where large amounts of CeNPs-containing waste is generated, or in high trafic areas, where small CeNPs additives could be released from diesel engine emissions. Tailoring the surface to minimize potential harmful effects is also needed to prevent toxicity on the natural environment and humans. Given their reactivity, the use of CeNPs should be considered with a full understanding of their unique properties in order to reduce potential risks and ensure their safe utilization and implementation into the practice.
• Finally, we expect exciting developments in several areas, highlighting the versatility of these materials in addressing many societal challenges ranging from semiconducting industries to healthcare, environmental remediation and consumer products. Among these, new generations of CeNPs with controlled morphology and surface chemistry will be needed to be used as polishers in CMP to address the needs for smaller and greener semiconducting manufacturing processes. CeNP-based photocatalysts and sorbents will continue to attract interest for environmental remediation and energy conversion. Particularly, the tunable redox chemistry and electrochemical stability make them interesting candidates for electrode materials, electrolytes and catalysis, while the catalytic activity towards pollutants such as VOCs, NOx, metals and organics make them well suited for promoting detoxification and removing contaminants through redox and surface adsorption reactions. An array of products and applications are expected to emerge from the exploration of the antioxidant activity and ability to protect cells from oxidative damage, with impact in drug delivery, biomedical imaging, neuroprotection and the cosmetic industry as anti-aging and UV-protection agents in skin-care. Continued research and development will be needed to explore and optimize the performance of CeNPs in these diverse applications.
In summary, it is increasingly clear that CeNPs have huge potential, as demonstrated by an already increasing number of applications in many areas of science and technology and industries such as catalysis, sensing, semiconductor and environment. The review has tried to provide the most significant advancements to date in a systematic and comprehensive manner. Scientists and engineers can use this overview as a starting point to explore this material's unique and unusual properties and pursue new avenues into the future.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This material is based upon work supported by the National Science Foundation under Grants 20425544, 1336493, 0954919, and the United States Department of Agriculture (USDA) under Grant 2023-67021-39750. Any opinions, findings, and conclusions or recommendations expressed in this material are those of the author(s) and do not necessarily reflect the views of the National Science Foundation, or the USDA. SA acknowledges all past and present members of Andreescu's group for their contribution to the study of various aspects of ceria chemistry.References
- J. Paier, C. Penschke and J. Sauer, Chem. Rev., 2013, 113, 3949–3985 CrossRef CAS PubMed.
- R. Schmitt, A. Nenning, O. Kraynis, R. Korobko, A. I. Frenkel, I. Lubomirsky, S. M. Haile and J. L. M. Rupp, Chem. Soc. Rev., 2020, 49, 554–592 RSC.
- T. Montini, M. Melchionna, M. Monai and P. Fornasiero, Chem. Rev., 2016, 116, 5987–6041 CrossRef CAS PubMed.
- E. Ozkan, P. Cop, F. Benfer, A. Hofmann, M. Votsmeier, J. M. Guerra, M. Giar, C. Heiliger, H. Over and B. M. Smarsly, J. Phys. Chem. C, 2020, 124, 8736–8748 CrossRef.
- D. Andreescu, G. Bulbul, R. E. Ozel, A. Hayat, N. Sardesai and S. Andreescu, Environ. Sci.: Nano, 2014, 1, 445–458 RSC.
- J. Zhou, R. T. Guo, X. F. Zhang, Y. Z. Liu, C. P. Duan, G. L. Wu and W. G. Pan, Energy Fuel, 2021, 35, 2981–2998 CrossRef CAS.
- E. Grulke, K. Reed, M. Beck, X. Huang, A. Cormack and S. Seal, Environ. Sci.: Nano, 2014, 1, 429–444 RSC.
- S. Banavar, A. Deshpande, S. Sur and S. Andreescu, JPhys Mater., 2021, 4, 042003 CrossRef CAS.
- M. A. Saifi, S. Seal and C. Godugu, J. Controlled Release, 2021, 338, 164–189 CrossRef CAS PubMed.
- A. Hayat, D. Andreescu, G. Bulbul and S. Andreescu, J. Colloid Interface Sci., 2014, 418, 240–245 CrossRef CAS PubMed.
- R. A. Yokel, S. Hussain, S. Garantziotis, P. Demokritou, V. Castranova and F. R. Cassee, Environ. Sci.: Nano, 2014, 1, 406–428 RSC.
- J. Ma, X. Tong, J. Wang, G. Zhang, Y. Lv, Y. Zhu, S. Sun, Y.-C. Yang and Y. Song, Electrochim. Acta, 2019, 229, 80–88 CrossRef.
- A. Trovarelli, Catalysis by Ceria and Related Materials, World Scientific Publishing Co Pte Ltd, 2002 Search PubMed.
- E. Mamontov and T. Egami, J. Phys. Chem. Solids, 2000, 61, 1345–1356 CrossRef CAS.
- T. S. Sakthivel, D. L. Reid, U. M. Bhatta, G. Möbus, D. C. Sayle and S. Seal, Nanoscale, 2015, 7, 5169–5177 RSC.
- B. Choudhury and A. Choudhury, Mater. Chem. Phys., 2012, 131, 666–671 CrossRef CAS.
- S. Deshpande, S. Patil, S. V. Kuchibhatla and S. Seal, Appl. Phys. Lett., 2005, 87, 133113 CrossRef.
- S. Tsunekawa, R. Sivamohan, S. Ito, A. Kasuya and T. Fukuda, Nanostruct. Mater., 1999, 11, 141–147 CrossRef CAS.
- S. Tsunekawa, R. Sahara, Y. Kawazoe and K. Ishikawa, Appl. Surf. Sci., 1999, 152, 53–56 CrossRef CAS.
- V. K. Paidi, L. Savereide, D. J. Childers, J. M. Notestein, C. A. Roberts and J. Van Lierop, ACS Appl. Mater. Interfaces, 2017, 9, 30670–30678 CrossRef CAS PubMed.
- H.-Y. Li, H.-F. Wang, X.-Q. Gong, Y.-L. Guo, Y. Guo, G. Lu and P. Hu, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 79, 193401 CrossRef.
- M. Nolan, Chem. Phys. Lett., 2010, 499, 126–130 CrossRef CAS.
- M. Nolan, S. Grigoleit, D. C. Sayle, S. C. Parker and G. W. Watson, Surf. Sci., 2005, 576, 217–229 CrossRef CAS.
- F. Esch, S. Fabris, L. Zhou, T. Montini, C. Africh, P. Fornasiero, G. Comelli and R. Rosei, Science, 2005, 309, 752 CrossRef CAS PubMed.
- C. Zhang, A. Michaelides, D. A. King and S. J. Jenkins, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 79, 075433 CrossRef.
- S. C. Shirbhate, A. K. Yadav and S. A. Acharya, Appl. Phys. Lett., 2016, 108, 143501 CrossRef.
- Z. Yang, T. K. Woo, M. Baudin and K. Hermansson, J. Chem. Phys., 2004, 120, 7741–7749 CrossRef CAS PubMed.
- J. Kullgren, M. J. Wolf, C. Castleton, P. Mitev, W. J. Briels and K. Hermansson, Phys. Rev. Lett., 2014, 112, 156102 CrossRef CAS PubMed.
- S. Torbrügge, M. Reichling, A. Ishiyama, S. Morita and Ó. Custance, Phys. Rev. Lett., 2007, 99, 056101 CrossRef PubMed.
- H. Nörenberg and G. A. D. Briggs, Surf. Sci., 1999, 424, L352–L355 CrossRef.
- Y. Namai, K.-i Fukui and Y. Iwasawa, J. Phys. Chem. B, 2003, 107, 11666–11673 CrossRef CAS.
- G.-y Adachi and N. Imanaka, Chem. Rev., 1998, 98, 1479–1514 CrossRef CAS PubMed.
- P. Knappe and L. Eyring, J. Solid State Chem., 1985, 58, 312–324 CrossRef CAS.
- M. Yashima, D. Ishimura, Y. Yamaguchi, K. Ohoyama and K. Kawachi, Chem. Phys. Lett., 2003, 372, 784–787 CrossRef CAS.
- M. Zinkevich, D. Djurovic and F. Aldinger, Solid State Ionics, 2006, 177, 989–1001 CrossRef CAS.
- O. T. Sørensen, J. Solid State Chem., 1976, 18, 217–233 CrossRef.
- E. Kümmerle and G. Heger, J. Solid State Chem., 1999, 147, 485–500 CrossRef.
- S. Ray and D. Cox, J. Solid State Chem., 1975, 15, 333–343 CrossRef CAS.
- Y. Cao, L. Zhao, T. Gutmann, Y. Xu, L. Dong, G. Buntkowsky and F. Gao, J. Phys. Chem. C, 2018, 122, 20402–20409 CrossRef CAS.
- A. Trovarelli and J. Llorca, ACS Catal., 2017, 7, 4716–4735 CrossRef CAS.
- W. Wei-Wei, Y. Wen-Zhu, D. Pei-Pei, X. Hui, J. Zhao, S. Rui, M. Chao, S. Shuo, J. Chun-Jiang and Y. Chun-Hua, Crystal Plane Effect of Ceria on Supported Copper Oxide Cluster Catalyst for CO Oxidation: Importance of Metal–Support Interaction, 2017 Search PubMed.
- J. Conesa, Surf. Sci., 1995, 339, 337–352 CrossRef CAS.
- M. M. Branda, R. M. Ferullo, M. Causa and F. Illas, J. Phys. Chem. C, 2011, 115, 3716–3721 CrossRef CAS.
- M. V. Ganduglia-Pirovano, A. Hofmann and J. Sauer, Surf. Sci. Rep., 2007, 62, 219–270 CrossRef CAS.
- D. R. Mullins, Surf. Sci. Rep., 2015, 70, 42–85 CrossRef CAS.
- D. R. Mullins, P. M. Albrecht and F. Calaza, Top. Catal., 2013, 56, 1345–1362 CrossRef CAS.
- M. Nolan, S. C. Parker and G. W. Watson, Surf. Sci., 2005, 595, 223–232 CrossRef CAS.
- M. V. Ganduglia-Pirovano, J. L. F. Da Silva and J. Sauer, Phys. Rev. Lett., 2009, 102, 026101 CrossRef PubMed.
- D. Zhang, X. Du, L. Shi and R. Gao, Dalton Trans., 2012, 41, 14455–14475 RSC.
- Q. Wu, F. Zhang, P. Xiao, H. Tao, X. Wang, Z. Hu and Y. Lü, J. Phys. Chem. C, 2008, 112, 17076–17080 CrossRef CAS.
- X. Liu, K. Zhou, L. Wang, B. Wang and Y. Li, J. Am. Chem. Soc., 2009, 131, 3140–3141 CrossRef CAS PubMed.
- C. Pan, D. Zhang and L. Shi, J. Solid State Chem., 2008, 181, 1298–1306 CrossRef CAS.
- S. Yang and L. Gao, J. Am. Chem. Soc., 2006, 128, 9330–9331 CrossRef CAS PubMed.
- H.-I. Chen and H.-Y. Chang, Solid State Commun., 2005, 133, 593–598 CrossRef CAS.
- N. Du, H. Zhang, B. Chen, X. Ma and D. Yang, J. Phys. Chem. C, 2007, 111, 12677–12680 CrossRef CAS.
- J.-G. Li, T. Ikegami, Y. Wang and T. Mori, J. Am. Ceram. Soc., 2002, 85, 2376–2378 CrossRef CAS.
- H.-Y. Chang and H.-I. Chen, J. Cryst. Growth, 2005, 283, 457–468 CrossRef CAS.
- M. Cabus-Llaurado, Y. Cesteros, F. Medina, P. Salagre and J. Sueiras, Microporous Mesoporous Mater., 2007, 100, 167–172 CrossRef CAS.
- H. Y. Ma, H. J. Ren, P. Koshy, C. C. Sorrell and J. N. Hart, J. Phys. Chem. C, 2020, 124, 2644–2655 CrossRef CAS.
- B. Tang, L. Zhuo, J. Ge, G. Wang, Z. Shi and J. Niu, Chem. Commun., 2005, 3565–3567 RSC.
- Z. Yang, J. Wei, H. Yang, L. Liu, H. Liang and Y. Yang, Eur. J. Inorg. Chem., 2010, 3354–3359 CrossRef CAS.
- W.-Q. Han, L. Wu and Y. Zhu, J. Am. Chem. Soc., 2005, 127, 12814–12815 CrossRef CAS PubMed.
- H.-X. Mai, L.-D. Sun, Y.-W. Zhang, R. Si, W. Feng, H.-P. Zhang, H.-C. Liu and C.-H. Yan, J. Phys. Chem. B, 2005, 109, 24380–24385 CrossRef CAS PubMed.
- G. Li, K. Chao, H. Peng, K. Chen and Z. Zhang, J. Phys. Chem. C, 2008, 112, 16452–16456 CrossRef CAS.
- Z.-R. Tang, Y. Zhang and Y.-J. Xu, RSC Adv., 2011, 1, 1772–1777 RSC.
- Z. Guo, F. Du, G. Li and Z. Cui, Inorg. Chem., 2006, 45, 4167–4169 CrossRef CAS PubMed.
- Y. Zhang, F. Hou and Y. Tan, Chem. Commun., 2012, 48, 2391–2393 RSC.
- K. W. Kolasinski, Curr. Opin. Solid State Mater. Sci., 2006, 10, 182–191 CrossRef CAS.
- D. Wang, Y. Kang, V. Doan-Nguyen, J. Chen, R. Küngas, N. L. Wieder, K. Bakhmutsky, R. J. Gorte and C. B. Murray, Angew. Chem., Int. Ed., 2011, 50, 4378–4381 CrossRef CAS PubMed.
- X. Wang, J. Zhuang, Q. Peng and Y. Li, Adv. Mater., 2006, 18, 2031–2034 CrossRef CAS.
- T. Yu, J. Joo, Y. I. Park and T. Hyeon, Angew. Chem., Int. Ed., 2005, 44, 7411–7414 CrossRef CAS PubMed.
- D. Zhang, F. Niu, H. Li, L. Shi and J. Fang, Powder Technol., 2011, 207, 35–41 CrossRef CAS.
- C. Sun, H. Li and L. Chen, Energy Environ. Sci., 2012, 5, 8475–8505 RSC.
- Q. Yuan, H.-H. Duan, L.-L. Li, L.-D. Sun, Y.-W. Zhang and C.-H. Yan, J. Colloid Interface Sci., 2009, 335, 151–167 CrossRef CAS PubMed.
- K. Wu, L. D. Sun and C. H. Yan, Adv. Energy Mater., 2016, 6, 1600501 CrossRef.
- I. Florea, C. D. Feral-Martin, J. R. Majimel, D. Ihiawakrim, C. Hirlimann and O. Ersen, Cryst. Growth Des., 2013, 13, 1110–1121 CrossRef CAS.
- M. Nolan, J. Mater. Chem., 2011, 21, 9160–9168 RSC.
- M. Nolan, V. S. Verdugo and H. Metiu, Surf. Sci., 2008, 602, 2734–2742 CrossRef CAS.
- L. Wu, H. J. Wiesmann, A. R. Moodenbaugh, R. F. Klie, Y. Zhu, D. O. Welch and M. Suenaga, Phys. Rev. B: Condens. Matter Mater. Phys., 2004, 69, 125415 CrossRef.
- R. K. Hailstone, A. G. DiFrancesco, J. G. Leong, T. D. Allston and K. J. Reed, J. Phys. Chem. C, 2009, 113, 15155–15159 CrossRef CAS.
- J. Xu, J. Harmer, G. Li, T. Chapman, P. Collier, S. Longworth and S. C. Tsang, Chem. Commun., 2010, 46, 1887–1889 RSC.
- M. K. Ghosalya, X. S. Li, A. Beck, J. A. Van Bokhoven and L. Artiglia, J. Phys. Chem. C, 2021, 125, 9303–9309 CrossRef CAS.
- C. Paun, O. V. Safonova, J. Szlachetko, P. M. Abdala, M. Nachtegaal, J. Sa, E. Kleymenov, A. Cervellino, F. Krumeich and J. A. van Bokhoven, J. Phys. Chem. C, 2012, 116, 7312–7317 CrossRef CAS.
- X.-D. Zhou and W. Huebner, Appl. Phys. Lett., 2001, 79, 3512–3514 CrossRef CAS.
- F. Zhang, S.-W. Chan, J. E. Spanier, E. Apak, Q. Jin, R. D. Robinson and I. P. Herman, Appl. Phys. Lett., 2002, 80, 127–129 CrossRef CAS.
- J. Seo, A. Gowda and S. Babu, ECS J. Solid State Sci. Technol., 2018, 7, P243–P252 CrossRef CAS.
- J. Seo, J. Moon, J. H. Kim, K. Lee, J. Hwang, H. Yoon, D. K. Yi and U. Paik, Appl. Surf. Sci., 2016, 389, 311–315 CrossRef CAS.
- K. M. Kumar, M. Mahendhiran, M. C. Diaz, N. Hernandez-Como, A. Hernandez-Eligio, G. Torres-Torres, S. Godavarthi and L. M. Gomez, Mater. Lett., 2018, 214, 15–19 CrossRef CAS.
- B. Choudhury, P. Chetri and A. Choudhury, J. Exp. Nanosci., 2015, 10, 103–114 CrossRef CAS.
- G. N. Vayssilov, Y. Lykhach, A. Migani, T. Staudt, G. P. Petrova, N. Tsud, T. Skála, A. Bruix, F. Illas, K. C. Prince, V. R. Matolın, K. M. Neyman and J. Libuda, Nat. Mater., 2011, 10, 310 CrossRef CAS PubMed.
- P. Nachimuthu, W.-C. Shih, R.-S. Liu, L.-Y. Jang and J.-M. Chen, J. Solid State Chem., 2000, 149, 408–413 CrossRef CAS.
- A. Migani, G. N. Vayssilov, S. T. Bromley, F. Illas and K. M. Neyman, J. Mater. Chem., 2010, 20, 10535–10546 RSC.
- A. Bruix and K. M. Neyman, Catal. Lett., 2016, 146, 2053–2080 CrossRef CAS.
- M. A. Sk, S. M. Kozlov, K. H. Lim, A. Migani and K. M. Neyman, J. Mater. Chem. A, 2014, 2, 18329–18338 RSC.
- A. Ruiz Puigdollers, P. Schlexer, S. Tosoni and G. Pacchioni, ACS Catal., 2017, 7, 6493–6513 CrossRef CAS.
- M. Boaro, S. Colussi and A. Trovarelli, Front. Chem., 2019, 7, 28 CrossRef CAS PubMed.
- H. J. Kim, M. G. Jang, D. Shin and J. W. Han, ChemCatChem, 2020, 12, 11–26 CrossRef CAS.
- L. Mei, L. Zhaogang, H. Yanhong, W. Mitang and L. Hangquan, J. Rare Earths, 2008, 26, 357–361 CrossRef.
- C. Gunawan, M. S. Lord, E. Lovell, R. J. Wong, M. S. Jung, D. Oscar, R. Mann and R. Amal, ACS Omega, 2019, 4, 9473–9479 CrossRef CAS PubMed.
- D. A. Andersson, S. I. Simak, N. V. Skorodumova, I. A. Abrikosov and B. Johansson, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 3518–3521 CrossRef CAS PubMed.
- M. Coduri, S. Checchia, M. Longhi, D. Ceresoli and M. Scavini, Front. Chem., 2018, 6, 526 CrossRef CAS PubMed.
- T. Vinodkumar, B. G. Rao and B. M. Reddy, Catal. Today, 2015, 253, 57–64 CrossRef CAS.
- D. Mukherjee and B. M. Reddy, Catal. Today, 2018, 309, 227–235 CrossRef CAS.
- C. Veranitisagul, N. Koonsaeng, N. Laosiripojana and A. Laobuthee, J. Ind. Eng. Chem., 2012, 18, 898–903 CrossRef CAS.
- U. Hennings and R. Reimert, Appl. Catal., A, 2007, 325, 41–49 CrossRef CAS.
- D. N. Durgasri, T. Vinodkumar, P. Sudarsanam and B. M. Reddy, Catal. Lett., 2014, 144, 971–979 CrossRef CAS.
- K. Krishna, A. Bueno-López, M. Makkee and J. Moulijn, Appl. Catal., B, 2007, 75, 189–200 CrossRef CAS.
- A. Bueno-López, Appl. Catal., B, 2014, 146, 1–11 CrossRef.
- S. Patil, S. Seal, Y. Guo, A. Schulte and J. Norwood, Appl. Phys. Lett., 2006, 88, 243110 CrossRef.
- S. Babu, R. Thanneeru, T. Inerbaev, R. Day, A. E. Masunov, A. Schulte and S. Seal, Nanotechnology, 2009, 20, 085713 CrossRef PubMed.
- N. Shehata, K. Meehan, M. Hudait, N. Jain and S. Gaballah, J. Nanomater., 2014, 2014, 156 Search PubMed.
- K. Kim, J. D. Yoo, S. Lee, M. Bae, J. Bae, W. Jung and J. W. Han, ACS Appl. Mater. Interfaces, 2017, 9, 15449–15458 CrossRef CAS PubMed.
- I. Hemmilä, V.-M. Mukkala and H. Takalo, J. Alloys Compd., 1997, 249, 158–162 CrossRef.
- Q. Ma, J. Wang, Z. Li, X. Lv, L. Liang and Q. Yuan, Small, 2019, 15, 1804969 CrossRef PubMed.
- J. Wu, G. Wang, D. Jin, J. Yuan, Y. Guan and J. Piper, Chem. Commun., 2008, 365–367 RSC.
- L. Li, H. K. Yang, B. K. Moon, Z. Fu, C. Guo, J. H. Jeong, S. S. Yi, K. Jang and H. S. Lee, J. Phys. Chem. C, 2009, 113, 610–617 CrossRef CAS.
- A. Kumar, S. Babu, A. S. Karakoti, A. Schulte and S. Seal, Langmuir, 2009, 25, 10998–11007 CrossRef CAS PubMed.
- K. Woan, Y.-Y. Tsai and W. Sigmund, Nanomedicine, 2010, 5, 233–242 CrossRef CAS PubMed.
- I. Celardo, M. De Nicola, C. Mandoli, J. Z. Pedersen, E. Traversa and L. Ghibelli, ACS Nano, 2011, 5, 4537–4549 CrossRef CAS PubMed.
- N. Yang, P. Orgian, E. D. Bartolomeo, V. Foglietti, O. Piero Torelli, E. V. Levlev, G. Rossi, S. Licoccia, G. Balestrino, S. V. Kalinin and C. Aruta, J. Phys. Chem. C, 2017, 121, 8841–8849 CrossRef CAS.
- N. Novik, V. Konakov and I. Y. Archakov, Rev. Adv. Mater. Sci., 2015, 40 Search PubMed.
- S. Damyanova, B. Pawelec, K. Arishtirova, M. V. M. Huerta and J. L. G. Fierro, Appl. Catal., A, 2008, 337, 86–96 CrossRef CAS.
- T. Masui, T. Ozaki, K.-I. Machida and G.-Y. Adachi, J. Alloys Compd., 2000, 303–304, 49–55 CrossRef CAS.
- D. Devaiah, L. H. Reddy, S.-E. Park and B. M. Reddy, Catal. Rev., 2018, 60, 177–277 CrossRef CAS.
- E. Aneggi, M. Boaro, S. Colussi, C. de Leitenburg and A. Trovarelli, in Handbook on the Physics and Chemistry of Rare Earths, ed. J.-C. G. Bünzli and V. K. Pecharsky, Elsevier, 2016, vol. 50, pp. 209–242 Search PubMed.
- N. Guillén-Hurtado, I. Atribak, A. Bueno-López and A. García-García, J. Mol. Catal. A: Chem., 2010, 323, 52–58 CrossRef.
- C. Hu, Q. Zhu, Z. Jiang, L. Chen and R. Wu, Chem. Eng. J., 2009, 152, 583–590 CrossRef CAS.
- C. Hu, Chem. Eng. J., 2011, 168, 1185–1192 CrossRef CAS.
- R. Qin, J. Chen, X. Gao, X. Zhu, X. Yu and K. Cen, RSC Adv., 2014, 4, 43874–43881 RSC.
- P. Venkataswamy, D. Mukherjee, D. Devaiah, M. Vithal and B. M. Reddy, Curr. Nanomater., 2018, 3, 103–113 CrossRef CAS.
- S. Ramana, B. G. Rao, P. Venkataswamy, A. Rangaswamy and B. M. Reddy, J. Mol. Catal. A: Chem., 2016, 415, 113–121 CrossRef CAS.
- W. Derafa, F. Paloukis, B. Mewafy, W. Baaziz, O. Ersen, C. Petit, G. Corbel and S. Zafeiratos, RSC Adv., 2018, 8, 40712–40719 RSC.
- A. J. M. Araújo, V. D. Silva, A. R. O. Sousa, J. P. F. Grilo, T. A. Simões, D. A. Macedo, R. M. Nascimento and C. A. Paskocimas, Ceram. Int., 2019, 45, 7157–7165 CrossRef.
- A. Jha, D.-W. Jeong, Y.-L. Lee, I. W. Nah and H.-S. Roh, RSC Adv., 2015, 5, 103023–103029 RSC.
- A. A. Ansari, J. Labis, M. Alam, S. M. Ramay, N. Ahmad and A. Mahmood, Phase Trans., 2016, 89, 261–272 CrossRef CAS.
- Y. Wang, F. Wang, Y. Chen, D. Zhang, B. Li, S. Kang, X. Li and L. Cui, Appl. Catal., B, 2014, 147, 602–609 CrossRef CAS.
- M. O. Mazan, J. Marrero-Jerez, A. Soldati, P. Núñez and S. A. Larrondo, Int. J. Hydrogen Energy, 2015, 40, 3981–3989 CrossRef CAS.
- J. Wang, B. Zhang, M. Shen, J. Wang, W. Wang, J. Ma, S. Liu and L. Jia, J. Sol-Gel Sci. Technol., 2011, 58, 259–268 CrossRef CAS.
- L. Kundakovic and M. Flytzani-Stephanopoulos, J. Catal., 1998, 179, 203–221 CrossRef CAS.
- A. A. Ansari, J. P. Labis, M. Alam, S. M. Ramay, N. Ahmed and A. Mahmood, Anal. Lett., 2017, 50, 1360–1371 CrossRef CAS.
- W. Tang, Z. Hu, M. Wang, G. D. Stucky, H. Metiu and E. W. McFarland, J. Catal., 2010, 273, 125–137 CrossRef CAS.
- Y.-Q. Su, I. A. W. Filot, J.-X. Liu and E. J. M. Hensen, ACS Catal., 2018, 8, 75–80 CrossRef CAS PubMed.
- S. Colussi, A. Gayen, M. Farnesi Camellone, M. Boaro, J. Llorca, S. Fabris and A. Trovarelli, Angew. Chem., Int. Ed., 2009, 48, 8481–8484 CrossRef CAS PubMed.
- E. Aneggi, C. de Leitenburg and A. Trovarelli, Catal. Today, 2012, 181, 108–115 CrossRef CAS.
- I. Atribak, A. Bueno-Lopez and A. Garcia-Garcia, Top. Catal., 2009, 52, 2088 CrossRef CAS.
- M. Konsolakis, S. A. C. Carabineiro, G. E. Marnellos, M. F. Asad, O. S. G. P. Soares, M. F. R. Pereira, J. J. M. Órfão and J. L. Figueiredo, Inorg. Chim. Acta, 2017, 455, 473–482 CrossRef CAS.
- D. Marrocchelli, S. R. Bishop, H. L. Tuller and B. Yildiz, Adv. Funct. Mater., 2012, 22, 1958–1965 CrossRef CAS.
- R. Grau-Crespo, H. de Leeuw Nora, S. Hamad and V. Waghmare Umesh, Proc. R. Soc. A, 2011, 467, 1925–1938 CrossRef CAS.
- M. Yashima, J. Phys. Chem. C, 2009, 113, 12658–12662 CrossRef CAS.
- F. Capel, C. Moure and P. Durán, Ceram. Int., 2002, 28, 627–636 CrossRef CAS.
- M. Fu, L. Wei, Y. Li, X. Zhou, S. Hao and Y. Li, Solid State Sci., 2009, 11, 2133–2137 CrossRef CAS.
- X. Feng, D. C. Sayle, Z. L. Wang, M. S. Paras, B. Santora, A. C. Sutorik, T. X. T. Sayle, Y. Yang, Y. Ding, X. Wang and Y.-S. Her, Science, 2006, 312, 1504 CrossRef CAS PubMed.
- A. Gupta, U. V. Waghmare and M. S. Hegde, Chem. Mater., 2010, 22, 5184–5198 CrossRef CAS.
- U. Castanet, C. Feral-Martin, A. Demourgues, R. L. Neale, D. C. Sayle, F. Caddeo, J. M. Flitcroft, R. Caygill, B. J. Pointon, M. Molinari and J. Majimel, ACS Appl. Mater. Interfaces, 2019, 11, 11384–11390 CrossRef CAS PubMed.
- A. Gupta, A. Kumar, U. V. Waghmare and M. S. Hegde, Chem. Mater., 2009, 21, 4880–4891 CrossRef CAS.
- G. Dutta, U. V. Waghmare, T. Baidya, M. S. Hegde, K. R. Priolkar and P. R. Sarode, Chem. Mater., 2006, 18, 3249–3256 CrossRef CAS.
- M. Nolan, J. Chem. Phys., 2009, 130, 144702 CrossRef PubMed.
- Z. Yang, Z. Lu, G. Luo and K. Hermansson, Phys. Lett. A, 2007, 369, 132–139 CrossRef CAS.
- M. Alfredsson and C. R. A. Catlow, Phys. Chem. Chem. Phys., 2002, 4, 6100–6108 RSC.
- M. D. Krcha, A. D. Mayernick and M. J. Janik, J. Catal., 2012, 293, 103–115 CrossRef CAS.
- A. Romero-Núñez and G. Díaz, RSC Adv., 2015, 5, 54571–54579 RSC.
- S. Gatla, D. Aubert, G. Agostini, O. Mathon, S. Pascarelli, T. Lunkenbein, M. G. Willinger and H. Kaper, ACS Catal., 2016, 6, 6151–6155 CrossRef CAS.
- H. Zhu, Y. Chen, Z. Wang, W. Liu and L. Wang, RSC Adv., 2018, 8, 14888–14897 RSC.
- M. Mittal, A. Gupta and O. P. Pandey, Sol. Energy, 2018, 165, 206–216 CrossRef CAS.
- S. D. Senanayake, D. Stacchiola and J. A. Rodriguez, Acc. Chem. Res., 2013, 46, 1702–1711 CrossRef CAS PubMed.
- K. Mudiyanselage, S. D. Senanayake, L. Feria, S. Kundu, A. E. Baber, J. Graciani, A. B. Vidal, S. Agnoli, J. Evans, R. Chang, S. Axnanda, Z. Liu, J. F. Sanz, P. Liu, J. A. Rodriguez and D. J. Stacchiola, Angew. Chem., Int. Ed., 2013, 52, 5101–5105 CrossRef CAS PubMed.
- X. Wang, J. A. Rodriguez, J. C. Hanson, D. Gamarra, A. Martínez-Arias and M. Fernández-García, J. Phys. Chem. B, 2006, 110, 428–434 CrossRef CAS PubMed.
- R. Kopelent, J. A. van Bokhoven, J. Szlachetko, J. Edebeli, C. Paun, M. Nachtegaal and O. V. Safonova, Angew. Chem., Int. Ed., 2015, 54, 8728–8731 CrossRef CAS PubMed.
- A. S. Karakoti, N. A. Monteiro-Riviere, R. Aggarwal, J. P. Davis, R. J. Narayan, W. T. Self, J. McGinnis and S. Seal, JOM, 2008, 60, 33–37 CrossRef CAS PubMed.
- S. Das, J. M. Dowding, K. E. Klump, J. F. McGinnis, W. Self and S. Seal, Nanomedicine, 2013, 8, 1483–1508 CrossRef CAS PubMed.
- C. Walkey, S. Das, S. Seal, J. Erlichman, K. Heckman, L. Ghibelli, E. Traversa, J. F. McGinnis and W. T. Self, Environ. Sci.: Nano, 2015, 2, 33–53 RSC.
- C. Korsvik, S. Patil, S. Seal and W. T. Self, Chem. Commun., 2007, 1056–1058 RSC.
- T. Pirmohamed, J. M. Dowding, S. Singh, B. Wasserman, E. Heckert, A. S. Karakoti, J. E. S. King, S. Seal and W. T. Self, Chem. Commun., 2010, 46, 2736–2738 RSC.
- B. A. Rzigalinski, K. Meehan, R. M. Davis, Y. Xu, W. C. Miles and C. A. Cohen, Nanomedicine, 2006, 1, 399–412 CrossRef CAS PubMed.
- M. Das, S. Patil, N. Bhargava, J.-F. Kang, L. M. Riedel, S. Seal and J. J. Hickman, Biomaterials, 2007, 28, 1918–1925 CrossRef CAS PubMed.
- Y. Xue, Q. Luan, D. Yang, X. Yao and K. Zhou, J. Phys. Chem. C, 2011, 115, 4433–4438 CrossRef CAS.
- C. Li, X. Shi, Q. Shen, C. Guo, Z. Hou and J. Zhang, J. Nanomater., 2018, 2018, 12 Search PubMed.
- B. C. Nelson, M. E. Johnson, M. L. Walker, K. R. Riley and C. M. Sims, Antioxidants, 2016, 5, 15 CrossRef PubMed.
- F. Pagliari, C. Mandoli, G. Forte, E. Magnani, S. Pagliari, G. Nardone, S. Licoccia, M. Minieri, P. Di Nardo and E. Traversa, ACS Nano, 2012, 6, 3767–3775 CrossRef CAS PubMed.
- C. Xu and X. Qu, NPG Asia Mater., 2014, 6, e90 CrossRef CAS.
- C. López-Alarcón and A. Denicola, Anal. Chim. Acta, 2013, 763, 1–10 CrossRef PubMed.
- B. Drew and C. Leeuwenburgh, Ann. N. Y. Acad. Sci., 2002, 959, 66–81 CrossRef CAS PubMed.
- B. Uttara, A. V. Singh, P. Zamboni and R. T. Mahajan, Curr. Neuropharmacol., 2009, 7, 65–74 CrossRef CAS PubMed.
- J. M. McCord and I. Fridovich, J. Biol. Chem., 1969, 244, 6049–6055 CrossRef CAS PubMed.
- S. Singh, Front. Chem., 2019, 7, 46 CrossRef CAS PubMed.
- T. Fukai and M. Ushio-Fukai, Antioxid. Redox Signaling, 2011, 15, 1583–1606 CrossRef CAS PubMed.
- E. G. Heckert, A. S. Karakoti, S. Seal and W. T. Self, Biomaterials, 2008, 29, 2705–2709 CrossRef CAS PubMed.
- A. Karakoti, S. Singh, J. M. Dowding, S. Seal and W. T. Self, Chem. Soc. Rev., 2010, 39, 4422–4432 RSC.
- S. Singh, T. Dosani, A. S. Karakoti, A. Kumar, S. Seal and W. T. Self, Biomaterials, 2011, 32, 6745–6753 CrossRef CAS PubMed.
- J. M. Perez, A. Asati, S. Nath and C. Kaittanis, Small, 2008, 4, 552–556 CrossRef CAS PubMed.
- A. S. Karakoti, P. Munusamy, K. Hostetler, V. Kodali, S. Kuchibhatla, G. Orr, J. G. Pounds, J. G. Teeguarden, B. D. Thrall and D. R. Baer, Surf. Interface Anal., 2012, 44, 882–889 CrossRef CAS PubMed.
- B. Lipinski, Oxid. Med. Cell. Longevity, 2011, 2011 Search PubMed.
- V. Prabhakaran, C. G. Arges and V. Ramani, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 1029–1034 CrossRef CAS PubMed.
- Y. Zhang, K. Zhou, Y. Zhai, F. Qin, L. Pan and X. Yao, RSC Adv., 2014, 4, 50325–50330 RSC.
- Y. Y. Huang, J. S. Ren and X. G. Qu, Chem. Rev., 2019, 119, 4357–4412 CrossRef CAS PubMed.
- D. W. Jiang, D. L. Ni, Z. T. Rosenkrans, P. Huang, X. Y. Yan and W. B. Cai, Chem. Soc. Rev., 2019, 48, 3683–3704 RSC.
- L. A. Ridnour, D. D. Thomas, D. Mancardi, M. G. Espey, K. M. Miranda, N. Paolocci, M. Feelisch, J. Fukuto and D. A. Wink, Biol. Chem., 2004, 385, 1 CAS.
- J. M. Dowding, T. Dosani, A. Kumar, S. Seal and W. T. Self, Chem. Commun., 2012, 48, 4896–4898 RSC.
- S. Singh, Front. Chem., 2019, 7 Search PubMed.
- J. M. Dowding, S. Das, A. Kumar, T. Dosani, R. McCormack, A. Gupta, T. X. T. Sayle, D. C. Sayle, L. von Kalm, S. Seal and W. T. Self, ACS Nano, 2013, 7, 4855–4868 CrossRef CAS PubMed.
- X. Jiao, H. Song, H. Zhao, W. Bai, L. Zhang and Y. Lv, Anal. Methods, 2012, 4, 3261–3267 RSC.
- P. Cohen, Nat. Cell Biol., 2002, 4, E127 CrossRef CAS PubMed.
- J. Chin, Curr. Opin. Chem. Biol., 1997, 1, 514–521 CrossRef CAS PubMed.
- S. J. Franklin, Curr. Opin. Chem. Biol., 2001, 5, 201–208 CrossRef CAS PubMed.
- F. Tan, Y. Zhang, J. Wang, J. Wei, Y. Cai and X. Qian, J. Mass Spectrom., 2008, 43, 628–632 CrossRef CAS PubMed.
- A. J. Patil, R. K. Kumar, N. J. Barron and S. Mann, Chem. Commun., 2012, 48, 7934–7936 RSC.
- M. H. Kuchma, C. B. Komanski, J. Colon, A. Teblum, A. E. Masunov, B. Alvarado, S. Babu, S. Seal, J. Summy and C. H. Baker, Nanomedicine, 2010, 6, 738–744 CrossRef CAS PubMed.
- H. J. Kwon, D. Kim, K. Seo, Y. G. Kim, S. I. Han, T. Kang, M. Soh and T. Hyeon, Angew. Chem., Int. Ed., 2018, 57, 9408–9412 CrossRef CAS PubMed.
- P. Eriksson, A. A. Tal, A. Skallberg, C. Brommesson, Z. Hu, R. D. Boyd, W. Olovsson, N. Fairley, I. A. Abrikosov, X. Zhang and K. Uvdal, Sci. Rep., 2018, 8, 6999 CrossRef PubMed.
- M. Huang and S. Fabris, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 75, 081404 CrossRef.
- X. Huang and M. J. Beck, Chem. Mater., 2015, 27, 5840–5844 CrossRef CAS.
- Z. Wu, M. Li, J. Howe, H. M. Meyer III and S. H. Overbury, Langmuir, 2010, 26, 16595–16606 CrossRef CAS PubMed.
- G. Preda, A. Migani, K. M. Neyman, S. T. Bromley, F. Illas and G. Pacchioni, J. Phys. Chem. C, 2011, 115, 5817–5822 CrossRef CAS.
- S. Fabris, G. Vicario, G. Balducci, S. de Gironcoli and S. Baroni, J. Phys. Chem. B, 2005, 109, 22860–22867 CrossRef CAS PubMed.
- V. V. Pushkarev, V. I. Kovalchuk and J. L. d'Itri, J. Phys. Chem. B, 2004, 108, 5341–5348 CrossRef CAS.
- C. Li, K. Domen, K.-I. Maruya and T. Onishi, J. Catal., 1990, 123, 436–442 CrossRef CAS.
- Y. M. Choi, H. Abernathy, H.-T. Chen, M. C. Lin and M. Liu, ChemPhysChem, 2006, 7, 1957–1963 CrossRef CAS PubMed.
- C. Loschen, A. Migani, S. T. Bromley, F. Illas and K. M. Neyman, Phys. Chem. Chem. Phys., 2008, 10, 5730–5738 RSC.
- C. Li, K. Domen, K. Maruya and T. Onishi, J. Am. Chem. Soc., 1989, 111, 7683–7687 CrossRef CAS.
- Z. Wu, M. Li, J. Howe, H. M. Meyer and S. H. Overbury, Langmuir, 2010, 26, 16595–16606 CrossRef CAS PubMed.
- F. Vindigni, M. Manzoli, A. Damin, T. Tabakova and A. Zecchina, Chem. – Eur. J., 2011, 17, 4356–4361 CrossRef CAS PubMed.
- C. Yang, X. Yu, S. Heissler, P. G. Weidler, A. Nefedov, Y. Wang, C. Woell, T. Kropp, J. Paier and J. Sauer, Angew. Chem., Int. Ed., 2017, 56, 16399–16404 CrossRef CAS PubMed.
- H.-T. Chen, J.-G. Chang, H.-L. Chen and S.-P. Ju, J. Comput. Chem., 2009, 30, 2433–2442 CrossRef CAS PubMed.
- M. Ornatska, E. Sharpe, D. Andreescu and S. Andreescu, Anal. Chem., 2011, 83, 4273–4280 CrossRef CAS PubMed.
- S. S. Lee, W. Song, M. Cho, H. L. Puppala, P. Nguyen, H. Zhu, L. Segatori and V. L. Colvin, ACS Nano, 2013, 7, 9693–9703 CrossRef CAS PubMed.
- S. Saraf, C. J. Neal, S. Das, S. Barkam, R. McCormack and S. Seal, ACS Appl. Mater. Interfaces, 2014, 6, 5472–5482 CrossRef CAS PubMed.
- P. Yu, S. A. Hayes, T. J. O’Keefe, M. J. O’Keefe and J. O. Stoffer, J. Electrochem. Soc., 2006, 153, C74–C79 CrossRef CAS.
- F. H. Scholes, C. Soste, A. E. Hughes, S. G. Hardin and P. R. Curtis, Appl. Surf. Sci., 2006, 253, 1770–1780 CrossRef CAS.
- M. Ornatska, E. Sharpe, D. Andreescu and S. Andreescu, Anal. Chem., 2011, 83, 4273–4280 CrossRef CAS PubMed.
- N. P. Sardesai, D. Andreescu and S. Andreescu, J. Am. Chem. Soc., 2013, 135, 16770–16773 CrossRef CAS PubMed.
- Y.-J. Wang, H. Dong, G.-M. Lyu, H.-Y. Zhang, J. Ke, L.-Q. Kang, J.-L. Teng, L.-D. Sun, R. Si, J. Zhang, Y.-J. Liu, Y.-W. Zhang, Y.-H. Huang and C.-H. Yan, Nanoscale, 2015, 7, 13981–13990 RSC.
- D. Damatov and J. M. Mayer, Chem. Commun., 2016, 52, 10281–10284 RSC.
- K. Binnemans, P. T. Jones, K. Van Acker, B. Blanpain, B. Mishra and D. Apelian, JOM, 2013, 65, 846–848 CrossRef.
- G. A. Campbell, Miner. Econ., 2014, 27, 21–31 CrossRef.
- K. Binnemans, P. T. Jones, B. Blanpain, T. Van Gerven, Y. Yang, A. Walton and M. Buchert, J. Cleaner Prod., 2013, 51, 1–22 CrossRef CAS.
- S. Jakab, S. Picart, B. Tribollet, P. Rousseau, H. Perrot and C. Gabrielli, Anal. Chem., 2009, 81, 5139–5145 CrossRef CAS PubMed.
- X. Beaudoux, M. Virot, T. Chave, G. Durand, G. Leturcq and S. I. Nikitenko, Green Chem., 2016, 18, 3656–3668 RSC.
- M. Virot, T. Chave, D. Horlait, N. Clavier, N. Dacheux, J. Ravaux and S. I. Nikitenko, J. Mater. Chem., 2012, 22, 14734–14740 RSC.
- S. Tamilmani, V. Lowalekar, S. Raghavan and R. Small, 7th International Symposium on Ultra Clean Processing of Silicon Surfaces, UCPSS 2004, 2005.
- US Pat., US6568995B1, 2003 Search PubMed.
- US Pat., US6326305 B1, 2001 Search PubMed.
- N. Um, M. Miyake and T. Hirato, Green Energy and Technology, 2011, 66, DOI:10.1007/978-4-431-53910-0_22.
- D. Horlait, N. Clavier, S. Szenknect, N. Dacheux and V. Dubois, Inorg. Chem., 2012, 51, 3868–3878 CrossRef CAS PubMed.
- K. Dhamodharan, T. Aneesh and A. Pius, J. Radioanal. Nucl. Chem., 2019, 319, 1127–1133 CrossRef CAS.
- J. T. Dahle, K. Livi and Y. Arai, Chemosphere, 2015, 119, 1365–1371 CrossRef CAS PubMed.
- M. S. A. Asghar, B. J. Inkson and G. Möbus, ChemPhysChem, 2017, 18, 1247–1251 CrossRef CAS PubMed.
- P. Janos, P. Kuran, M. Kormunda, V. Stengl, T. M. Grygar, M. Dosek, M. Stastny, J. Ederer, V. Pilarova and L. Vrtoch, J. Rare Earths, 2014, 32, 360–370 CrossRef CAS.
- P. Janoš, J. Ederer and M. Došek, Nova Biotechnol. Chim., 2014, 13, 148 Search PubMed.
- P. Janoš, P. Kuráň, J. Ederer, M. Šťastný, L. Vrtoch, M. Pšenička, J. Henych, K. Mazanec and M. Skoumal, Adv. Mater. Sci. Eng., 2015, 2015, 241421 Search PubMed.
- W. Kim, B. Kim, D. Choi, T. Oki and S. Kim, J. Hazard. Mater., 2010, 183, 29–34 CrossRef CAS PubMed.
- Y. Xu, S. S. Mofarah, R. Mehmood, C. Cazorla, P. Koshy and C. C. Sorrell, Mater. Horiz., 2021, 8, 102–123 RSC.
- Y. Shlapa, S. Solopan, V. Sarnatskaya, K. Siposova, I. Garcarova, K. Veltruska, I. Timashkov, O. Lykhova, D. Kolesnik, A. Musatov, V. Nikolaev and A. Belous, Colloids Surf., B, 2022, 220, 112960 CrossRef CAS PubMed.
- M. Nyoka, Y. Choonara, P. Kumar, P. P. D. Kondiah and V. Pillay, Nanomaterials, 2020, 10(2), 242 CrossRef CAS PubMed.
- T. Yu, J. Joo, Y. I. Park and T. Hyeon, Angew. Chem., 2005, 44, 7411–7414 CrossRef CAS PubMed.
- V. D. Araújo, W. Avansi, H. B. de Carvalho, M. L. Moreira, E. Longo, C. Ribeirod and M. I. B. Bernardia, CrystEngComm, 2012, 14, 1150–1154 RSC.
- S. Kar, C. Patel and S. Santra, J. Phys. Chem. C, 2009, 113, 4862–4867 CrossRef CAS.
- A. F. Zedan, S. Moussa and M. S. El-Shall, Mater. Res. Express, 2023, 10 Search PubMed.
- K. R. Singh, V. Nayak, T. Sarkar and R. P. Singh, RSC Adv., 2020, 10, 27194–27214 RSC.
- R. Olbrich, G. E. Murgida, V. Ferrari, C. Barth, A. M. Llois, M. Reichling and M. V. Ganduglia-Pirovano, J. Phys. Chem. C, 2017, 121, 6844–6851 CrossRef CAS.
- Z. Nie, A. Petukhova and E. Kumacheva, Nat. Nanotechnol., 2009, 5, 15 CrossRef PubMed.
- W.-J. Ong, L.-L. Tan, S.-P. Chai and S.-T. Yong, Chem. Commun., 2015, 51, 858–861 RSC.
- Z. Ji, X. Shen, M. Li, H. Zhou, G. Zhu and K. Chen, Nanotechnology, 2013, 24, 115603 CrossRef PubMed.
- M. E. Khan, M. M. Khan and M. H. Cho, Sci. Rep., 2017, 7, 5928 CrossRef PubMed.
- A. S. Dezfuli, M. R. Ganjali, H. R. Naderi and P. Norouzi, RSC Adv., 2015, 5, 46050–46058 RSC.
- D. Joung, V. Singh, S. Park, A. Schulte, S. Seal and S. I. Khondaker, J. Phys. Chem. C, 2011, 115, 24494–24500 CrossRef CAS.
- Y. Wang, C. X. Guo, J. Liu, T. Chen, H. Yang and C. M. Li, Dalton Trans., 2011, 40, 6388–6391 RSC.
- M. M. Khan, S. A. Ansari, D. Pradhan, D. H. Han, J. Lee and M. H. Cho, Ind. Eng. Chem. Res., 2014, 53, 9754–9763 CrossRef CAS.
- D. Joung, V. Singh, S. Park, A. Schulte, S. Seal and S. I. Khondaker, J. Phys. Chem. C, 2011, 115, 24494–24500 CrossRef CAS.
- A. Othman, E. Dumitrescu, D. Andreescu and S. Andreescu, ACS Sustainable Chem. Eng., 2018, 6, 12542–12561 CrossRef CAS.
- L. Yu, Y. Ma, C. N. Ong, J. Xie and Y. Liu, RSC Adv., 2015, 5, 64983–64990 RSC.
- A. Baranik, R. Sitko, A. Gagor, I. Queralt, E. Marguí and B. Zawisza, Anal. Chem., 2018, 90, 4150–4159 CrossRef CAS PubMed.
- T. S. Sakthivel, S. Das, C. J. Pratt and S. Seal, Nanoscale, 2017, 9, 3367–3374 RSC.
- A. Szabó, C. Perri, A. Csató, G. Giordano, D. Vuono and J. B. Nagy, Materials, 2010, 3, 3092–3140 CrossRef.
- X. Qin, F. Peng, F. Yang, X. He, H. Huang, D. Luo, J. Yang, S. Wang, H. Liu, L. Peng and Y. Li, Nano Lett., 2014, 14, 512–517 CrossRef CAS PubMed.
- S. Sarkar, E. Guibal, F. Quignard and A. K. SenGupta, J. Nanopart. Res., 2012, 14, 715 CrossRef.
- S. Wu, J. Dzubiella, J. Kaiser, M. Drechsler, X. Guo, M. Ballauff and Y. Lu, Angew. Chem., Int. Ed., 2012, 51, 2229–2233 CrossRef CAS PubMed.
- A. Asati, S. Santra, C. Kaittanis and J. M. Perez, ACS Nano, 2010, 4, 5321–5331 CrossRef CAS PubMed.
- W. Song, S. Soo Lee, M. Savini, L. Popp, V. L. Colvin and L. Segatori, ACS Nano, 2014, 8, 10328–10342 CrossRef CAS PubMed.
- R. Mehmood, S. S. Mofarah, A. Rawal, F. Tomasetig, X. C. Wang, J. L. Yang, P. Koshy and C. C. Sorrell, ACS Sustainable Chem. Eng., 2019, 7, 9189–9201 CrossRef CAS.
- M. Mari, B. Müller, K. Landfester and R. Muñoz-Espí, ACS Appl. Mater. Interfaces, 2015, 7, 10727–10733 CrossRef CAS PubMed.
- K. Katta, D. Busko, Y. Avlasevich, K. Landfester, S. Baluschev and R. Muñoz-Espí, Beilstein J. Nanotechnol., 2019, 10, 522–530 CrossRef CAS PubMed.
- M. E. Culica, A. L. Chibac-Scutaru, V. Melinte and S. Coseri, Materials, 2020, 13 Search PubMed.
- R. Cao, D. Sun, Y. Liang, M. Hong, K. Tatsumi and Q. Shi, Inorg. Chem., 2002, 41, 2087–2094 CrossRef CAS PubMed.
- R. Mohammadinasab, M. Tabatabaee, B.-M. Kukovec and H. Aghaie, Inorg. Chim. Acta, 2013, 405, 368–373 CrossRef CAS.
- P. Gohari Derakhshandeh and J. Soleimannejad, Ultrason. Sonochem., 2016, 31, 122–128 CrossRef CAS PubMed.
- A. K. Boal, F. Ilhan, J. E. DeRouchey, T. Thurn-Albrecht, T. P. Russell and V. M. Rotello, Nature, 2000, 404, 746–748 CrossRef CAS PubMed.
- L. Qi, J. Fresnais, P. Muller, O. Theodoly, J. F. Berret and J. P. Chapel, Langmuir, 2012, 28, 11448–11456 CrossRef CAS PubMed.
- Z. Shen, J. Liu, F. Hu, S. Liu, N. Cao, Y. Sui, Q. Zeng and Y. Shen, CrystEngComm, 2014, 16, 3387–3394 RSC.
- P. G. Derakhshandeh, J. Soleimannejad and J. Janczak, Ultrason. Sonochem., 2015, 26, 273–280 CrossRef CAS PubMed.
- Z. Shen, S. He, P. Yao, X. Lao, B. Yang, Y. Dai, X. Sun and T. Chen, RSC Adv., 2014, 4, 12844–12848 RSC.
- A. U. Czaja, N. Trukhan and U. Müller, Chem. Soc. Rev., 2009, 38, 1284–1293 RSC.
- W. P. Mounfield, U. Tumuluri, Y. Jiao, M. Li, S. Dai, Z. Wu and K. S. Walton, Microporous Mesoporous Mater., 2016, 227, 65–75 CrossRef CAS.
- M. Lammert, M. T. Wharmby, S. Smolders, B. Bueken, A. Lieb, K. A. Lomachenko, D. D. Vos and N. Stock, Chem. Commun., 2015, 51, 12578–12581 RSC.
- T. Islamoglu, A. Atilgan, S.-Y. Moon, G. W. Peterson, J. B. DeCoste, M. Hall, J. T. Hupp and O. K. Farha, Chem. Mater., 2017, 29, 2672–2675 CrossRef CAS.
- A. M. Ebrahim and T. J. Bandosz, ACS Appl. Mater. Interfaces, 2013, 5, 10565–10573 CrossRef CAS PubMed.
- J. Qi, J. Chen, G. Li, S. Li, Y. Gao and Z. Tang, Energy Environ. Sci., 2012, 5, 8937–8941 RSC.
- T. K. Kim, K. J. Lee, J. Y. Cheon, J. H. Lee, S. H. Joo and H. R. Moon, J. Am. Chem. Soc., 2013, 135, 8940–8946 CrossRef CAS PubMed.
- H. Wang, M. Liu, S. Guo, Y. Wang, X. Han and Y. Bai, Mol. Catal., 2017, 436, 120–127 CrossRef CAS.
- Z. Zhang, H. Shi, Q. Wu, X. Bu, Y. Yang, J. Zhang and Y. Huang, New J. Chem., 2019, 43, 4581–4589 RSC.
- M. H. Hassan, D. Andreescu and S. Andreescu, ACS Appl. Nano Mater., 2020, 3, 3288–3294 CrossRef CAS.
- J. M. Thomas, J. Chem. Phys., 2008, 128, 182502 CrossRef PubMed.
- J. Zhang, L. P. Li, X. S. Huang and G. S. Li, J. Mater. Chem., 2012, 22, 10480–10487 RSC.
- J. Qi, J. Chen, G. D. Li, S. X. Li, Y. Gao and Z. Y. Tang, Energy Environ. Sci., 2012, 5, 8937–8941 RSC.
- S. Song, X. Wang and H. Zhang, NPG Asia Mater., 2015, 7, e179 CrossRef CAS.
- A. Figueroba, A. Bruix, G. Kovács and K. M. Neyman, Phys. Chem. Chem. Phys., 2017, 19, 21729–21738 RSC.
- N. Acerbi, S. Golunski, S. C. Tsang, H. Daly, C. Hardacre, R. Smith and P. Collier, J. Phys. Chem. C, 2012, 116, 13569–13583 CrossRef CAS.
- E. A. Derevyannikova, T. Y. Kardash, L. S. Kibis, E. M. Slavinskaya, V. A. Svetlichnyi, O. A. Stonkus, A. S. Ivanova and A. I. Boronin, Phys. Chem. Chem. Phys., 2017, 19, 31883–31897 RSC.
- L. Wang, H. He, Y. Yu, L. Sun, S. Liu, C. Zhang and L. He, J. Inorg. Biochem., 2014, 135, 45–53 CrossRef CAS PubMed.
- C. M. Magdalane, K. Kaviyarasu, J. J. Vijaya, B. Siddhardha and B. Jeyaraj, J. Photochem. Photobiol., B, 2016, 163, 77–86 CrossRef CAS PubMed.
- R. Kannan, A. R. Kim, S. K. Eo, S. H. Kang and D. J. Yoo, Ceram. Int., 2017, 43, 3072–3079 CrossRef CAS.
- Z. Kebin and L. Yadong, Angew. Chem., Int. Ed., 2012, 51, 602–613 CrossRef PubMed.
- M. Krishnan, J. W. Nalaskowski and L. M. Cook, Chem. Rev., 2010, 110, 178–204 CrossRef CAS PubMed.
- S. Park, J. M. Vohs and R. J. Gorte, Nature, 2000, 404, 265 CrossRef CAS PubMed.
- P. Jasinski, T. Suzuki and H. U. Anderson, Sens. Actuators, B, 2003, 95, 73–77 CrossRef CAS.
- L. Truffault, M.-T. Ta, T. Devers, K. Konstantinov, V. Harel, C. Simmonard, C. Andreazza, I. P. Nevirkovets, A. Pineau, O. Veron and J.-P. Blondeau, Mater. Res. Bull., 2010, 45, 527–535 CrossRef CAS.
- F. Caputo, M. Mameli, A. Sienkiewicz, S. Licoccia, F. Stellacci, L. Ghibelli and E. Traversa, Sci. Rep., 2017, 7, 4636 CrossRef PubMed.
- U. Tumuluri, G. Rother and Z. Wu, Ind. Eng. Chem. Res., 2016, 55, 3909–3919 CrossRef CAS.
- J. A. Rodriguez, D. C. Grinter, Z. Liu, R. M. Palomino and S. D. Senanayake, Chem. Soc. Rev., 2017, 46, 1824–1841 RSC.
- A. Trovarelli, Catal. Rev., 1996, 38, 439–520 CrossRef CAS.
- K. Polychronopoulou, A. F. Zedan, M. AlKetbi, S. Stephen, M. Ather, M. S. Katsiotis, J. Arvanitidis, D. Christofilos, A. F. Isakovic and S. AlHassan, J. Environ. Chem. Eng., 2018, 6, 266–280 CrossRef CAS.
- J. Zhang, Y. Cao, C.-A. Wang and R. Ran, ACS Appl. Mater. Interfaces, 2016, 8, 8670–8677 CrossRef CAS PubMed.
- S. H. Kim, B. C. Park, Y. S. Jeon and Y. K. Kim, ACS Appl. Mater. Interfaces, 2018, 10, 32112–32119 CrossRef CAS PubMed.
- J. Quiroz, J.-M. Giraudon, A. Gervasini, C. Dujardin, C. Lancelot, M. Trentesaux and J.-F. Lamonier, ACS Catal., 2015, 5, 2260–2269 CrossRef CAS.
- L. Song, T. Xu, D. Gao, X. Hu, C. Li, S. Li and G. Chen, Chem. – Eur. J., 2019, 25, 6621–6627 CrossRef CAS PubMed.
- M. Dubey, S. Wadhwa, A. Mathur and R. Kumar, Appl. Surf. Sci. Adv., 2022, 12, 100340 CrossRef.
- Y. Mordekovitz, L. Shelly, B. A. Rosen and S. Hayun, J. Am. Ceram. Soc., 2021, 104, 2337–2347 CrossRef CAS.
- S. Zhang, C. Zhao, Y. Liu, W. Li, J. Wang, G. Wang, Y. Zhang, H. Zhang and H. Zhao, Chem. Commun., 2019, 55, 2952–2955 RSC.
- I. Y. Habib, J. Burhan, F. Jaladi, C. M. Lim, A. Usman, N. T. R. N. Kumara, S. C. E. Tsang and A. H. Mahadi, Catal. Today, 2021, 375, 506–513 CrossRef CAS.
- K. Polychronopoulou, A. A. AlKhoori, A. M. Efstathiou, M. A. Jaoude, C. M. Damaskinos, M. A. Baker, A. Almutawa, D. H. Anjum, M. A. Vasiliades, A. Belabbes, L. F. Vega, A. F. Zedan and S. J. Hinder, ACS Appl. Mater. Interfaces, 2021, 13, 22391–22415 CrossRef CAS PubMed.
- H. A. Aleksandrov, I. Z. Koleva, K. M. Neyman, T. T. Tabakova and G. N. Vayssilov, RSC Adv., 2018, 8, 33728–33741 RSC.
- K. Kuntaiah, P. Sudarsanam, B. M. Reddy and A. Vinu, RSC Adv., 2013, 3, 7953–7962 RSC.
- A. Chien, N. Ye, C. W. Huang and I. H. Tseng, Catalysts, 2021, 11, 731 CrossRef CAS.
- N. Sutradhar, A. Sinhamahapatra, S. Pahari, M. Jayachandran, B. Subramanian, H. C. Bajaj and A. B. Panda, J. Phys. Chem. C, 2011, 115, 7628–7637 CrossRef CAS.
- J. Liu and C. Zhu, Nanomaterials, 2021, 11(9), 2231 CrossRef CAS PubMed.
- A. A. Aboud, H. Al-Kelesh, W. M. A. E. Rouby, A. A. Farghali, A. Hamdedein and M. H. Khedr, J. Mater. Res. Technol., 2018, 7, 14–20 CrossRef CAS.
- G. Jayakumar, A. Albert Irudayaraj, A. Dhayal Raj, S. John Sundaram and K. Kaviyarasu, J. Phys. Chem. Solids, 2022, 160, 110369 CrossRef CAS.
- M. Makkee and J. A. Moulijn, Catal. Rev., 2001, 43, 489–564 CrossRef.
- P. Sudarsanam, K. Kuntaiah and B. M. Reddy, New J. Chem., 2014, 38, 5991–6001 RSC.
- L. F. Nascimento, J. F. Lima, P. C. de Sousa Filho and O. A. Serra, Chem. Eng. J., 2016, 290, 454–464 CrossRef CAS.
- Q. Shen, G. Lu, C. Du, Y. Guo, Y. Wang, Y. Guo and X. Gong, Chem. Eng. J., 2013, 218, 164–172 CrossRef CAS.
- B. Suryadevara, Advances in chemical mechanical planarization (CMP), Woodhead Publishing, 2016 Search PubMed.
- P. V. Dandu, B. Peethala and S. Babu, J. Electrochem. Soc., 2010, 157, H869–H874 CrossRef CAS.
- N. K. Penta, B. Peethala, H. Amanapu, A. Melman and S. Babu, Colloids Surf., A, 2013, 429, 67–73 CrossRef CAS.
- R. Srinivasan, P. V. Dandu and S. Babu, ECS J. Solid State Sci. Technol., 2015, 4, P5029–P5039 CrossRef CAS.
- P. D. Veera, S. Peddeti and S. Babu, J. Electrochem. Soc., 2009, 156, H936–H943 CrossRef.
- J. Seo, J. Moon, J.-Y. Bae, K. S. Yoon, W. Sigmund and U. Paik, J. Nanosci. Nanotechnol., 2014, 14, 4351–4356 CrossRef CAS PubMed.
- R. Sabia and H. J. Stevens, Mach. Sci. Technol., 2000, 4, 235–251 CrossRef CAS.
- H. Doi, M. Suzuki and K. Kinuta, Proceedings of International Conference on Planarization/CMP Technology 2014, 2014.
- L. M. Cook, J. Non-Cryst. Solids, 1990, 120, 152–171 CrossRef CAS.
- S. R. Alety, K. V. Sagi and S. Babu, ECS J. Solid State Sci. Technol., 2017, 6, P898 CrossRef CAS.
- J. Seo, J. W. Lee, J. Moon, W. Sigmund and U. Paik, ACS Appl. Mater. Interfaces, 2014, 6, 7388–7394 CrossRef CAS PubMed.
- J.-S. Kim, H.-G. Kang, M. Kanemoto, U. Paik and J.-G. Park, Jpn. J. Appl. Phys., 2007, 46, 7671 CrossRef CAS.
- P. Janoš, J. Ederer, V. Pilařová, J. Henych, J. Tolasz, D. Milde and T. Opletal, Wear, 2016, 362, 114–120 CrossRef.
- S.-K. Kim, Y.-H. Kim, U. Paik and J.-G. Park, J. Electrochem. Soc., 2007, 154, H642–H646 CrossRef CAS.
- D. Ryuzaki, Y. Hoshi, Y. Machii, N. Koyama, H. Sakurai and T. Ashizawa, 2009 Symposium on VLSI Technology, 2009.
- H.-G. Kang, J.-D. Koh, S.-W. Han, J. W. Lee, B.-G. Lee, S.-H. Pyi, B.-S. Lee, J.-W. Kim, B. Reiss and J.-D. Jeong, ICPT 2012-International Conference on Planarization/CMP Technology, 2012.
- USA Pat., 2014 Search PubMed.
- USA Pat., 2012 Search PubMed.
- A. Chen, J. Long, Z. Li and Y. Chen, J. Mater. Sci.: Mater. Electron., 2018, 29, 11466–11477 CrossRef CAS.
- Y. Chen, C. Zuo, Z. Li and A. Chen, J. Alloys Compd., 2018, 736, 276–288 CrossRef CAS.
- Y. Chen, W. Cai, W. Wang and A. Chen, J. Nanopart. Res., 2019, 21, 226 CrossRef CAS.
- Y. Chen, Z. Li and N. Miao, Tribol. Int., 2015, 82, 211–217 CrossRef CAS.
- Y. Chen and R. Long, Appl. Surf. Sci., 2011, 257, 8679–8685 CrossRef CAS.
- K. Kim, J. Seo, M. Lee, J. Moon, K. Lee, D. K. Yi and U. Paik, J. Mater. Res., 2017, 32, 2829–2836 CrossRef CAS.
- N. D. Urban, D. Dickmann, B. Her and B. Santora, ECS Trans., 2016, 72, 37–42 CrossRef CAS.
- J. Choe, Comparison of 20 nm & 10 nm-class 2D Planar NAND and 3D V-NAND Architecture, Techinsights, 2015 Search PubMed.
- T. C. Li, B. Reiss, S. P. Kuttiatoor, V. Lam, J.-D. Lee and R. Jia, 2015 International Conference on Planarization/CMP Technology (ICPT), 2015.
- B. Praveen, B.-J. Cho, J.-G. Park and S. Ramanathan, Mater. Sci. Semicond. Process., 2015, 33, 161–168 CrossRef CAS.
- J. Cheng, S. Huang, Y. Li, T. Wang, L. Xie and X. Lu, Appl. Surf. Sci., 2020, 506, 144668 CrossRef CAS.
- N. Shehata, K. Meehan, M. Hudait and N. Jain, J. Nanopart. Res., 2021, 14, 1173 CrossRef.
- X. Feng, D. C. Sayle, Z. L. Wang, M. S. Paras, B. Santora, A. C. Sutorik, T. X. Sayle, Y. Yang, Y. Ding and X. Wang, Science, 2006, 312, 1504–1508 CrossRef CAS PubMed.
- B. W. Liu and J. W. Liu, TrAC, Trends Anal. Chem., 2019, 121 Search PubMed.
- D. Jampaiah, T. Srinivasa Reddy, V. E. Coyle, A. Nafady and S. K. Bhargava, J. Mater. Chem. B, 2017, 5, 720–730 RSC.
- Y. Tao, E. Ju, J. Ren and X. Qu, Adv. Mater., 2015, 27, 1097–1104 CrossRef CAS PubMed.
- A. S. Finny, A. Othman and S. Andreescu, in Cerium Oxide (CeO2): Synthesis, Properties and Applications, ed. S. Scirè and L. Palmisano, Elsevier, 2020, pp. 259–277 Search PubMed.
- A. Othman, A. Karimi and S. Andreescu, J. Mater. Chem. B, 2016, 4, 7178–7203 RSC.
- A. Karimi, A. Othman, A. Uzunoglu, L. Stanciu and S. Andreescu, Nanoscale, 2015, 7, 6909–6923 RSC.
- J. Wu, X. Wang, Q. Wang, Z. Lou, S. Li, Y. Zhu, L. Qin and H. Wei, Chem. Soc. Rev., 2019, 48, 1004–1076 RSC.
- H. Wei and E. Wang, Chem. Soc. Rev., 2013, 42, 6060–6093 RSC.
- L. Song, C. Huang, W. Zhang, M. Ma, Z. Chen, N. Gu and Y. Zhang, Colloids Surf., A, 2016, 506, 747–755 CrossRef CAS.
- Y.-Z. Li, T.-T. Li, W. Chen and Y.-Y. Song, ACS Appl. Mater. Interfaces, 2017, 9, 29881–29888 CrossRef CAS PubMed.
- C. Hou, Y. Wang, Q. Ding, L. Jiang, M. Li, W. Zhu, D. Pan, H. Zhu and M. Liu, Nanoscale, 2015, 7, 18770–18779 RSC.
- L. Ai, L. Li, C. Zhang, J. Fu and J. Jiang, Chem. – Eur. J., 2013, 19, 15105–15108 CrossRef CAS PubMed.
- S. Kumar, P. Bhushan and S. Bhattacharya, RSC Adv., 2017, 7, 37568–37577 RSC.
- Y.-W. Bao, X.-W. Hua, H.-H. Ran, J. Zeng and F.-G. Wu, J. Mater. Chem. B, 2019, 7, 296–304 RSC.
- H. Sun, Y. Zhou, J. Ren and X. Qu, Angew. Chem., Int. Ed., 2018, 57, 9224–9237 CrossRef CAS PubMed.
- I. Celardo, J. Z. Pedersen, E. Traversa and L. Ghibelli, Nanoscale, 2011, 3, 1411–1420 RSC.
- A. A. Ansari, P. R. Solanki and B. D. Malhotra, J. Biotechnol., 2009, 142, 179–184 CrossRef CAS PubMed.
- J. M. Yong, L. Fu, F. Tang, P. Yu, R. P. Kuchel, J. M. Whitelock and M. S. Lord, ACS Biomater. Sci. Eng., 2022, 8, 512–525 CrossRef CAS PubMed.
- C. Ispas, J. Njagi, M. Cates and S. Andreescu, J. Electrochem. Soc., 2008, 155(8), F169 CrossRef CAS.
- H. Wu, F. Li, S. Wang, J. Lu, J. Li, Y. Du, X. Sun, X. Chen, J. Gao and D. Ling, Biomaterials, 2018, 151, 66–77 CrossRef CAS PubMed.
- S. M. Hirst, A. S. Karakoti, R. D. Tyler, N. Sriranganathan, S. Seal and C. M. Reilly, Small, 2009, 5, 2848–2856 CrossRef CAS PubMed.
- K. Chaudhury, N. Babu K, A. K. Singh, S. Das, A. Kumar and S. Seal, Nanomedicine, 2013, 9, 439–448 CrossRef CAS PubMed.
- D. Zahra, A. Javaid, M. Iqbal, I. Akbar and U. A. Ashfaq, Crit. Rev. Ther. Drug Carrier Syst., 2021, 38, 1–26 CrossRef PubMed.
- Y. Gao, K. Chen, J.-L. Ma and F. Gao, OncoTargets Ther., 2014, 7, 835–840 CrossRef CAS PubMed.
- N. Pourkhalili, A. Hosseini, A. Nili-Ahmadabadi, S. Hassani, M. Pakzad, M. Baeeri, A. Mohammadirad and M. Abdollahi, World J. Diabetes, 2011, 2, 204–210 CrossRef PubMed.
- R. Najafi, A. Hosseini, H. Ghaznavi, S. Mehrzadi and A. M. Sharifi, Brain Res. Bull., 2017, 131, 117–122 CrossRef CAS PubMed.
- H. Bao, Z. Zhang, Q. Hua and W. Huang, Langmuir, 2014, 30, 6427–6436 CrossRef CAS PubMed.
- L. Artiglia, S. Agnoli, M. C. Paganini, M. Cattelan and G. Granozzi, ACS Appl. Mater. Interfaces, 2014, 6, 20130–20136 CrossRef CAS PubMed.
- A. Othman, A. Hayat and S. Andreescu, ACS Appl. Nano Mater., 2018, 1, 5722–5735 CrossRef CAS.
- E. O. Gubernatorova, X. Liu, A. Othman, W. T. Muraoka, E. P. Koroleva, S. Andreescu and A. V. Tumanov, Adv. Healthcare Mater., 2017, 6, 1700176 CrossRef PubMed.
- H. Zhao, Y. Dong, P. Jiang, G. Wang and J. Zhang, ACS Appl. Mater. Interfaces, 2015, 7, 6451–6461 CrossRef CAS PubMed.
- A. A. Ansari, P. R. Solanki and B. D. Malhotra, Appl. Phys. Lett., 2008, 92, 263901 CrossRef.
- B. W. Liu, Z. C. Huang and J. W. Liu, Nanoscale, 2016, 8, 13562–13567 RSC.
- S. Radhakrishnan and S. J. Kim, RSC Adv., 2015, 5, 12937–12943 RSC.
- M. Molinari, A. R. Symington, D. C. Sayle, T. S. Sakthivel, S. Seal and S. C. Parker, ACS Appl. Bio Mater., 2019, 2, 1098–1106 CrossRef CAS PubMed.
- P. Pentyala, V. Singhania, V. K. Duggineni and P. A. Deshpande, Mol. Catal., 2022, 522, 112190 CrossRef CAS.
- M. Chen, X. Zhou, C. Xiong, T. Yuan, W. Wang, Y. Zhao, Z. Xue, W. Guo, Q. Wang, H. Wang, Y. Li, H. Zhou and Y. Wu, ACS Appl. Mater. Interfaces, 2022, 14, 21989–21995 CrossRef CAS PubMed.
- A. Kolmakov and M. Moskovits, Annu. Rev. Mater. Res., 2004, 34, 151–180 CrossRef CAS.
- C. Schilling and C. Hess, J. Phys. Chem. C, 2018, 122, 2909–2917 CrossRef CAS.
- C. A. Zito, T. M. Perfecto, A. C. Dippel, D. P. Volanti and D. Koziej, ACS Appl. Mater. Interfaces, 2020, 12, 17745–17751 CrossRef CAS PubMed.
- L. Lyu, Q. Xie, Y. Y. Yang, R. R. Wang, W. F. Cen, S. Y. Luo, W. S. Yang, Y. Gao, Q. Q. Xiao, P. Zou and Y. Yang, Appl. Surf. Sci., 2022, 571 Search PubMed.
- P. Jasinski, T. Suzuki and H. U. Anderson, Sens. Actuators, B, 2003, 95, 73–77 CrossRef CAS.
- H. J. Beie and A. Gnorich, Sens. Actuators, B, 1991, 4, 393–399 CrossRef CAS.
- T. Divya, M. P. Nikhila, M. Anju, T. V. A. Kusumam, A. K. Akhila, Y. T. Ravikiran and N. K. Renuka, Sens. Actuators, B, 2017, 261, 85–93 CrossRef CAS.
- L. S. R. Rocha, M. Cilense, M. A. Ponce, C. M. Aldao, L. L. Oliveira, E. Longo and A. Z. Simoes, Phys. B, 2018, 536, 280–288 CrossRef CAS.
- F. Charbgoo, M. Ramezani and M. Darroudi, Biosens. Bioelectron., 2017, 96, 33–43 CrossRef CAS PubMed.
- A. Hayat, J. Cunningham, G. Bulbul and S. Andreescu, Anal. Chim. Acta, 2015, 885, 140–147 CrossRef CAS PubMed.
- A. Hayat and S. Andreescu, Anal. Chem., 2013, 85, 10028–10032 CrossRef CAS PubMed.
- G. Bulbul, A. Hayat, X. B. Liu and S. Andreescu, RSC Adv., 2016, 6, 60007–60014 RSC.
- A. Hayat, G. Bulbul and S. Andreescu, Biosens. Bioelectron., 2014, 56, 334–339 CrossRef CAS PubMed.
- F. Mustafa, S. Liebich and S. Andreescu, Anal. Chim. Acta, 2021, 1186 Search PubMed.
- E. Sharpe, R. Bradley, T. Frasco, D. Jayathilaka, A. Marsh and S. Andreescu, Sens. Actuators, B, 2014, 193, 552–562 CrossRef CAS PubMed.
- V. Andrei, E. Sharpe, A. Vasilescu and S. Andreescu, Talanta, 2016, 156, 112–118 CrossRef PubMed.
- E. Sharpe, T. Frasco, D. Andreescu and S. Andreescu, Analyst, 2013, 138, 249–262 RSC.
- A. Othman, L. Norton, A. S. Finny and S. Andreescu, Talanta, 2020, 208 Search PubMed.
- F. Mustafa, M. Carhart and S. Andreescu, ACS Appl. Nano Mater., 2021, 4, 9361–9369 CrossRef CAS.
- F. Mustafa, A. Othman and S. Andreescu, Sens. Actuators, B, 2021, 332 Search PubMed.
- J. Njagi, M. M. Chernov, J. C. Leiter and S. Andreescu, Anal. Chem., 2010, 82, 989–996 CrossRef CAS PubMed.
- R. E. Ozel, C. Ispas, M. Ganesana, J. C. Leiter and S. Andreescu, Biosens. Bioelectron., 2014, 52, 397–402 CrossRef CAS PubMed.
- N. P. Sardesai, M. Ganesana, A. Karimi, J. C. Leiter and S. Andreescu, Anal. Chem., 2015, 87, 2996–3003 CrossRef CAS PubMed.
- N. P. Sardesai, A. Karimi and S. Andreescu, ChemElectroChem, 2014, 1, 2082–2088 CrossRef CAS.
- Z. H. Yang, Y. Zhuo, R. Yuan and Y. Q. Chai, Biosens. Bioelectron., 2015, 69, 321–327 CrossRef CAS PubMed.
- M. B. Gumpu, N. Nesakumar, S. Sethuraman, U. M. Krishnan and J. B. B. Rayappan, Sens. Actuators, B, 2014, 199, 330–338 CrossRef CAS.
- E. Kang, J. Lee, B. Y. Won, S. Kim, S. Shin, M. I. Kim and H. G. Park, RSC Adv., 2015, 5, 78747–78753 RSC.
- A. Pinna, E. Cali, G. Kerherve, G. Galleri, M. Maggini, P. Innocenzi and L. Malfatti, Nanoscale Adv., 2020, 2, 2387–2396 RSC.
- R. Khan and S. Andreescu, Biosens. Bioelectron., 2024, 248, 115975 CrossRef CAS PubMed.
- H. Y. Kim, J. K. Ahn, M. I. Kim, K. S. Park and H. G. Park, Electrochem. Commun., 2019, 99, 5–10 CrossRef CAS.
- H. Y. Kim, K. S. Park and H. G. Park, Theranostics, 2020, 10, 4507–4514 CrossRef CAS PubMed.
- G. Bubul, A. Hayat and S. Andreescu, Adv. Healthcare Mater., 2016, 5, 822–828 CrossRef PubMed.
- A. Asati, S. Santra, C. Kaittanis, S. Nath and J. M. Perez, Angew. Chem., Int. Ed., 2009, 48, 2308–2312 CrossRef CAS PubMed.
- G. Bulbul, A. Hayat and S. Andreescu, Nanoscale, 2015, 7, 13230–13238 RSC.
- R. Wang, Y. Du, Y. Fu, Y. Guo, X. Gao, X. Guo, J. Wei and Y. Yang, ACS Sens., 2023, 8, 4442–4467 CrossRef CAS PubMed.
- X. Z. Wang, B. W. Liu and J. W. Liu, Langmuir, 2018, 34, 15871–15877 CrossRef CAS PubMed.
- B. W. Liu, Z. Y. Sun, P. J. J. Huang and J. W. Liu, J. Am. Chem. Soc., 2015, 137, 1290–1295 CrossRef CAS PubMed.
- J. Y. Zhang, J. Wang, J. Liao, Y. Lin, C. B. Zheng and J. W. Liu, ACS Appl. Mater. Interfaces, 2021, 13, 50236–50245 CrossRef CAS PubMed.
- Y. Y. Chang, Q. S. Chen, B. W. Liu, Z. J. Zhang, M. Liu and J. W. Liu, Inorg. Chem. Commun., 2021, 134 Search PubMed.
- R. N. McCormack, P. Mendez, S. Barkam, C. J. Neal, S. Das and S. Seal, J. Phys. Chem. C, 2014, 118, 18992–19006 CrossRef CAS.
- M. Al Sharabati, R. Sabouni and G. A. Husseini, Nanomaterials, 2022, 12, 277 CrossRef CAS PubMed.
- Y. Ma, X. Qu, C. Liu, Q. Xu and K. Tu, Front. Mol. Biosci., 2021, 8, 805228 CrossRef CAS PubMed.
- S. Wang, Z. Li, F. Duan, B. Hu, L. He, M. Wang, N. Zhou, Q. Jia and Z. Zhang, Anal. Chim. Acta, 2019, 1047, 150–162 CrossRef CAS PubMed.
- N. Zhou, Y. Ma, B. Hu, L. He, S. Wang, Z. Zhang and S. Lu, Biosens. Bioelectron., 2019, 127, 92–100 CrossRef CAS PubMed.
- X. Ouyang, W. Li, S. Xie, T. Zhai, M. Yu, J. Gan and X. Lu, New J. Chem., 2013, 37, 585–588 RSC.
- S. Olivera, K. Chaitra, K. Venkatesh, H. B. Muralidhara, A. Inamuddin, A. M. Asiri and M. I. Ahamed, Environ. Chem. Lett., 2018, 16(4), 1233–1246 CrossRef CAS.
- G. K. Sarma, S. Sen Gupta and K. G. Bhattacharyya, Environ. Sci. Pollut. Res., 2019, 26, 6245–6278 CrossRef CAS PubMed.
- M. Lürling, G. Waajen and F. van Oosterhout, Water Res., 2014, 54, 78–88 CrossRef PubMed.
- M. H. Hassan, R. Stanton, J. Secora, D. J. Trivedi and S. Andreescu, ACS Appl. Mater. Interfaces, 2020, 12, 52788–52796 CrossRef CAS PubMed.
- T. M. Arsic, A. Kalijadis, B. Matovic, M. Stoiljkovic, J. Pantic, J. Jovanovic, R. Petrovic, B. Jokic and B. Babic, Process. Appl. Ceram., 2016, 10, 17–23 CrossRef CAS.
- A. Othman, P. Vargo and S. Andreescu, ACS Appl. Nano Mater., 2019, 2, 7008–7018 CrossRef CAS.
- W. Xu, J. Wang, L. Wang, G. Sheng, J. Liu, H. Yu and X. J. Huang, J. Hazard. Mater., 2013, 260, 498–507 CrossRef CAS PubMed.
- R. Sawana, Y. Somasundar, V. S. Iyer and B. Baruwati, Appl. Water Sci., 2017, 7, 1223–1230 CrossRef CAS.
- A. Baranik, A. Gagor, I. Queralt, E. Marguí, R. Sitko and B. Zawisza, Microchim. Acta, 2018, 185, 264 CrossRef CAS PubMed.
- A. Karimi, S. Andreescu and D. Andreescu, ACS Appl. Mater. Interfaces, 2019, 11, 24725–24734 CrossRef CAS PubMed.
- G. Zhang, Z. He and W. Xu, Chem. Eng. J., 2012, 183, 315–324 CrossRef CAS.
- J. Wang, W. Xu, L. Chen, Y. Jia, L. Wang, X.-J. Huang and J. Liu, Chem. Eng. J., 2013, 231, 198–205 CrossRef CAS.
- P. Zhang, T. Ouyang, S. Chen, R. Li and L. Lai, 2010 4th International Conference on Bioinformatics and Biomedical Engineering, 2010.
- J. Lin, Y. Wu, A. Khayambashi, X. Wang and Y. Wei, Adsorpt. Sci. Technol., 2018, 36, 743–761 CrossRef CAS.
- K. Mukhopadhyay, A. Ghosh, S. K. Das, B. Show, P. Sasikumar and U. Chand Ghosh, RSC Adv., 2017, 7, 26037–26051 RSC.
- M. Chigondo, H. Kamdem Paumo, M. Bhaumik, K. Pillay and A. Maity, J. Colloid Interface Sci., 2018, 532, 500–516 CrossRef CAS PubMed.
- E. E. Merodio-Morales, H. E. Reynel-Ávila, D. I. Mendoza-Castillo, C. J. Duran-Valle and A. Bonilla-Petriciolet, Int. J. Environ. Sci. Technol., 2019, 115–128 Search PubMed.
- A. Dhillon, T. Kumar Sharma, S. K. Soni and D. Kumar, RSC Adv., 2016, 6, 89198–89209 RSC.
- J. G. Dale, S. S. Cox, M. E. Vance, L. C. Man and M. F. Hochella, Environ. Sci. Technol., 2017, 51, 1973–1980 CrossRef CAS PubMed.
- L. Zhang, J. Pierce, V. L. Leung, D. Wang and W. S. Epling, J. Phys. Chem. C, 2013, 117, 8282–8289 CrossRef CAS.
- P. Dinesha, S. Kumar and M. A. Rosen, Biofuel Res. J., 2021, 8, 1374–1383 CrossRef CAS.
- J. X. Chen, Y. X. Chen, M. J. Zhou, Z. W. Huang, J. Y. Gao, Z. Ma, J. M. Chen and X. F. Tang, Environ. Sci. Technol., 2017, 51, 473–478 CrossRef CAS PubMed.
- A. F. Santiago, J. F. Sousa, R. C. Guedes, C. E. Jeronimo and M. Benachour, J. Hazard. Mater., 2006, 138, 325–330 CrossRef CAS PubMed.
- L. Chang, I. P. Chen and S. S. Lin, Chemosphere, 2005, 58, 485–492 CrossRef CAS PubMed.
- S. S. Lin, C. L. Chen, D. J. Chang and C. C. Chen, Water Res., 2002, 36, 3009–3014 CrossRef CAS PubMed.
- D. Wilson, W. Wang and R. J. G. Lopes, Appl. Catal., B, 2012, 123, 273–281 CrossRef.
- F. Carraro, A. Fapohunda, M. C. Paganini and S. Agnoli, ACS Appl. Nano Mater., 2018, 1, 1492–1501 CrossRef CAS.
- S. Kurajica, I. K. Ivkovic, G. Drazic, V. Shvalya, M. Duplancic, G. Matijasic, U. Cvelbar and K. Muzina, Ceram. Int., 2022, 33, 29451–29458 Search PubMed.
- P. K. Sane, S. Tambat, S. Sontakke and P. Nemade, J. Environ. Chem. Eng., 2018, 6, 4476–4489 CrossRef CAS.
- Y. W. Wang, T. T. Liu and J. W. Liu, ACS Appl. Nano Mater., 2020, 3, 842–849 CrossRef CAS.
- A. D. Mayernick, R. Li, K. M. Dooley and M. J. Janik, J. Phys. Chem. C, 2011, 115, 24178–24188 CrossRef CAS.
- Q. L. Wang, J. J. Zhou, J. C. Zhang, H. Zhu, Y. H. Feng and J. Jin, Aerosol Air Qual. Res., 2020, 20, 477–488 CAS.
- Y. Z. Guo and H. Therien-Aubin, ACS Appl. Mater. Interfaces, 2021, 13, 37578–37588 CrossRef CAS PubMed.
- D. J. Conley, H. W. Paerl, R. W. Howarth, D. F. Boesch, S. P. Seitzinger, K. E. Havens, C. Lancelot and G. E. Likens, Science, 2009, 323, 1014–1015 CrossRef CAS PubMed.
- Y. G. Ko, T. Do, Y. Chun, C. H. Kim, U. S. Choi and J.-Y. Kim, J. Hazard. Mater., 2016, 307, 91–98 CrossRef CAS PubMed.
- M. Teng, L. Luo and X. Yang, Microporous Mesoporous Mater., 2009, 119, 158–164 CrossRef CAS.
- Y. Zhang, M. Yang, X.-M. Dou, H. He and D.-S. Wang, Environ. Sci. Technol., 2005, 39, 7246–7253 CrossRef CAS PubMed.
- Y. Su, W. Yang, W. Sun, Q. Li and J. K. Shang, Chem. Eng. J., 2015, 268, 270–279 CrossRef CAS.
- J. Liu, J. Cao, Y. Hu, Y. Han and J. Zhou, Water Sci. Technol., 2017, 76, 2867–2875 CrossRef CAS PubMed.
- H. Ding, Y. Zhao, Q. Duan, J. Wang, K. Zhang, G. Ding, X. Xie and C. Ding, J. Rare Earth., 2017, 35, 984–994 CrossRef CAS.
- M. H. Hassan and S. Andreescu, Inorg. Chem., 2023, 62(51), 20970–20979 CrossRef CAS PubMed.
- A. Othman, D. Andreescu, D. P. Karunaratne, S. V. Babu and S. Andreescu, ACS Appl. Mater. Interfaces, 2017, 9, 12893–12905 CrossRef CAS PubMed.
- E. Dumitrescu, D. P. Karunaratne, S. V. Babu, K. N. Wallace and S. Andreescu, Chemosphere, 2018, 192, 178–185 CrossRef CAS PubMed.
- J. J. Zhang, K. B. Lee, L. He, J. Seiffert, P. Subramaniam, L. Yang, S. Chen, P. Maguire, G. Mainelis, S. Schwander, T. Tetley, A. Porter, M. Ryan, M. Shaffer, S. Hu, J. Gong and K. F. Chung, Environ. Sci.: Processes Impacts, 2016, 18, 1333–1342 RSC.
- B. Collin, M. Auffan, A. C. Johnson, I. Kaur, A. A. Keller, A. Lazareva, J. R. Lead, X. M. Ma, R. C. Merrifield, C. Svendsen, J. C. White and J. M. Unrine, Environ. Sci.: Nano, 2014, 1, 533–548 RSC.
- A. Arumugam, C. Karthikeyan, A. S. Haja Hameed, K. Gopinath, S. Gowri and V. Karthika, Mater. Sci. Eng., C, 2015, 49, 408–415 CrossRef CAS PubMed.
- S. Kannan and M. Sundrarajan, Int. J. Nanosci., 2014, 13, 1450018 CrossRef CAS.
- G. S. Priya, A. Kanneganti, K. A. Kumar, K. V. Rao and S. Bykkam, Int. J. Sci. Res. Publ., 2014, 4, 199–224 Search PubMed.
- S. Maensiri, C. Masingboon, P. Laokul, W. Jareonboon, V. Promarak, P. L. Anderson and S. Seraphin, Cryst. Growth Des., 2007, 7, 950–955 CrossRef CAS.
- M. Darroudi, S. J. Hoseini, R. Kazemi Oskuee, H. A. Hosseini, L. Gholami and S. Gerayli, Ceram. Int., 2014, 40, 7425–7430 CrossRef CAS.
- S. A. Khan and A. Ahmad, Mater. Res. Bull., 2013, 48, 4134–4138 CrossRef CAS.
- F. Charbgoo, M. B. Ahmad and M. Darroudi, Int. J. Nanomed., 2017, 12, 1401–1413 CrossRef CAS PubMed.
- F. Zeng, Y. Wu, X. Li, X. Ge, Q. Guo, X. Lou, Z. Cao, B. Hu, N. J. Long, Y. Mao and C. Li, Angew. Chem., Int. Ed., 2018, 130, 25485–25492 Search PubMed.
- J. L. Y. Tang, S. S. Moonshi and H. T. Ta, Cell. Mol. Life Sci., 2023, 80, 46 CrossRef CAS PubMed.
- W. Yang, M. Zhang, J. He, M. Gong, J. Sun and X. Yang, Regener. Biomater., 2022, 9, rbac037 CrossRef CAS PubMed.
- Q. Bao, P. Hu, Y. Xu, T. Cheng, C. Wei, L. Pan and J. Shi, ACS Nano, 2018, 12(7), 6794–6805 CrossRef CAS PubMed.
- Y. Malyukin, P. Maksimchuk, V. Seminko, E. Okrushko and N. Spivak, J. Phys. Chem. C, 2018, 122, 16406–16411 CrossRef CAS.
- R. W. Tarnuzzer, J. Colon, S. Patil and S. Seal, Nano Lett., 2005, 5, 2573–2577 CrossRef CAS PubMed.
- M. Das, N. Bhargava, S. Patil, L. Riedel, P. Molnar, S. Seal and J. J. Hickman, In Vitro Cell. Dev. Biol.: Anim., 2005, 41, 9A–9A Search PubMed.
- G. A. Silva, Nat. Nanotechnol., 2006, 1, 92–94 CrossRef CAS PubMed.
- D. Schubert, R. Dargusch, J. Raitano and S. W. Chan, Biochem. Biophys. Res. Commun., 2006, 342, 86–91 CrossRef CAS PubMed.
- J. M. Dowding, W. Song, K. Bossy, A. Karakoti, A. Kumar, A. Kim, B. Bossy, S. Seal, M. H. Ellisman, G. Perkins, W. T. Self and E. Bossy-Wetzel, Cell Death Differ., 2014, 21, 1622–1632 CrossRef CAS PubMed.
- D.-W. Kang, C. K. Kim, H.-J. Jeong, M. Soh, T. Kim, I. Choi, C.-K. Ki, D. Kim, W. Yang, T. Hyeon and S.-H. Lee, NanoResearch, 2017, 10, 2743–2760 CrossRef CAS.
- H. Nosrati, M. Heydari and M. Khodaei, Mater. Today Bio, 2023, 23, 100823 CrossRef CAS PubMed.
- A. P. Dayanandan, W. J. Cho, H. Kang, A. B. Bello, B. J. Kim, Y. Arai and S. H. Lee, Biomater. Res., 2023, 27, 68 CrossRef CAS PubMed.
- Y. Xue, F. Yang, L. Wu, D. Xia and Y. Liu, Adv. Healthcare Mater., 2024, 13, e2302858 CrossRef PubMed.
- Q. Weng, H. Sun, C. Fang, F. Xia, H. Liao, J. Lee, J. Wang, A. Xie, J. Ren, X. Guo, F. Li, B. Yang and D. Ling, Nat. Commun., 2021, 12 CrossRef PubMed.
- A. García, J. A. Cámara, A. M. Boullosa, M. Gustà, L. Mondragón, S. Schwartz, E. Casals, I. Abasolo, N. J. Bastús and V. Puntes, Nanomaterials, 2023, 2208 CrossRef PubMed.
- P. C. Naha, J. C. Hsu, J. Kim, S. Shah, M. Bouche, S. Si-Mohamed, D. N. Rosario-Berrios, P. Douek, M. Hajfathalian, P. Yasini, S. Singh, M. A. Rosen, M. A. Morgan and D. P. Cormode, ACS Nano, 2020, 14, 10187–10197 CrossRef CAS PubMed.
- S. Kargozar, F. Baino, S. J. Hoseini, S. Hamzehlou, M. Darroudi, J. Verdi, L. Hasanzadeh, H. W. Kim and M. Mozafari, Nanomedicine, 2018, 13, 3051–3069 CrossRef CAS PubMed.
Footnote |
| † Contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |