
Microcage flame retardants with complete recyclability and durability via reversible interfacial locking engineering†
Furong
Zeng
,
Lei
He
,
Jianwen
Ma
,
Danxuan
Fang
,
Zhiwei
Zeng
,
Tongyu
Bai
,
Rong
Ding
,
Bowen
Liu
,
Haibo
Zhao
 * and
Yuzhong
Wang
*
* and
Yuzhong
Wang
*
School of Chemical Engineering, The Collaborative Innovation Center for Eco-Friendly and Fire-Safety Polymeric Materials (MoE), National Engineering Laboratory of Eco-Friendly Polymeric Materials (Sichuan), State Key Laboratory of Polymer Materials Engineering, Sichuan University, No. 24, South Section 1, Yihuan Road, Chengdu, Sichuan 610064, China. E-mail: haibor7@163.com; polymers@vip.126.com
First published on 29th February 2024
Abstract
Flame retardants are effective in protecting materials from fire but pose environmental challenges due to limited recyclability. Urgently needed for circular material economy are new flame retardants that are chemically recyclable and durable. Here, we report a new facile and scalable strategy for engineering reversible microcages with infinite chemical recyclability to starting monomers, exceptional durability, and versatile flame retardancy. This is achieved through a highly synergistic hierarchical assembly of easily obtainable phosphoric acid and Cu2+ monomers. By leveraging dynamic reversible assembly networks, microcages can be circularly and infinitely dissociated into starting monomers via eco-friendly pH adjustment. Remarkable recovery rates of 92% for phosphoric acid and 96.2% for Cu2+ monomers are achieved, while the separated virgin matrix undergoes conventional chemical recycling, facilitating reformulation and seamless reintroduction into new supply chains as needed. Notably, when integrated with matrix-like surfaces, microcage clasp matrices tightly engage through in situ formed interfacial locking structures, showcasing outstanding flame-retardant efficiency, prolonged durability in hydrothermal aging, and extensive applicability across diverse polymeric materials such as polyurethane, epoxy resin, and polycarbonate. This study emphasizes a novel, straightforward, and scalable chemical platform, utilizing reversible interfacial locking engineering, for the development of flame retardants that are not only infinitely recyclable but also durable and broadly applicable.
New conceptsFlame retardants are crucial in protecting materials from fire, but their environmental impact and extensive applicability, stemming from restricted recyclability and versatility, present a significant challenge. There is an urgent necessity for novel flame retardants characterized by chemical recyclability and long-term durability for advancing circular material economy, underscoring a formidable task that demands immediate attention. In this study, a new scalable interfacial locking engineering strategy is demonstrated to enabling microcages with infinite chemical recyclability to starting monomers, exceptional durability and versatile flame retardancy. Highly synergetic dynamic interactions of microcages facilitate the remarkable infinite recycling, reformulation, and seamless reintroduction of starting monomers into new supply chains as needed, while the separated virgin matrix undergoes conventional chemical recycling. Meanwhile, the mesostructured matrix-like surfaces contribute additional pores and areas, enabling tight interfacial locking and achieving prolonged durability under harsh environments. The reversible microcages exhibit outstanding flame-retardant efficiency and extensive applicability across diverse polymers, including polyurethane, epoxy resin, and polycarbonate. The straightforward and scalable design strategy enables the creation of infinitely recyclable, durable, and broadly applicable flame retardants, promising for tackling the circular sustainability of waste flame-retardant plastics. |
Introduction
Flame retardants, widely recognized as pivotal additives that enhance fire safety across diverse sectors, such as electronics, decorations, transportation, and aerospace, experienced a substantial global consumption of 3.0 million tons in 2021.1–3 Projections indicate a remarkable market surge to 22.7 billion RMB by 2029, reflecting a compound annual growth rate of 6.1%.4 However, the environmental and economic challenges associated with the low durability and diverse composition of flame retardants present critical issues in their lifecycle management. The environmental threat posed by discarded flame retardants has evolved into a global concern.5–7 Unfortunately, the circular use of a substantial portion of waste flame-retardant materials is challenging due to economic limitations in sorting and separating, coupled with the degradation of material performance.8–11 The remaining challenges include the following: (1) the widespread migration of flame retardants in materials often leads to reduced flame retardancy and long-term environmental concerns; (2) impractical separation of starting precursors from the matrix results in nonrepeatable reformulation and utilization, causing ecological harm and depleting nonrenewable resources; and (3) the presence of flame retardants significantly hampers the plastic degradation process and impedes chemical recycling. Shunning or replacing flame retardants, such as unconventional novel homologs, is not a viable option at present, as there are no mature and straightforward alternatives that can match their exceptional performance.12–14 Consequently, there is a growing emphasis on developing appropriate designs for recyclable flame retardants to address the inherent trade-offs between durability, circular sustainability, and functionality, simultaneously preventing plastics from becoming waste.Stable yet stimuli-reversible interpenetrating networks within dynamic covalent polymers represent a promising alternative to nonrecyclable thermosets, offering both outstanding environmental durability and recyclability (Fig. 1(a)).15–17 These materials have demonstrated the ability to depolymerize into their original flame-retardant molecules through stimulus-triggered reversibility, employing dynamic Diels–Alder adducts and disulfide, imine, and acetal bonds.18–20 However, the widespread adoption of these materials faces challenges due to the expense and complexity of their designs, coupled with limitations imposed by finite functional groups. This particularly impacts the universal applicability of flame-retardant material fabrication methods, especially for conventional polymers. To address these technical hurdles, recent initiatives have shifted toward exploring supramolecular reversible assembly approaches, such as P-containing phenolic nanospheres, inorganic nanocoatings, and P-doped organosiloxane coatings, through interfacial engineering (Fig. 1(b)). By incorporating environmentally friendly solvents and accommodating pH levels, these advancements enable the separate recycling of flame retardants and original plastics.21–23 This achievement established a self-sustained closed-loop system that completed the feedstock-product-waste cycle. Despite these achievements, many existing recycling systems rely on presynthesized precursors and intricate assembly control. In particular, there has been insufficient emphasis on returning to starting monomers, limiting the potential for refabrication and diverting from the true essence of a “circular materials economy”.24–26
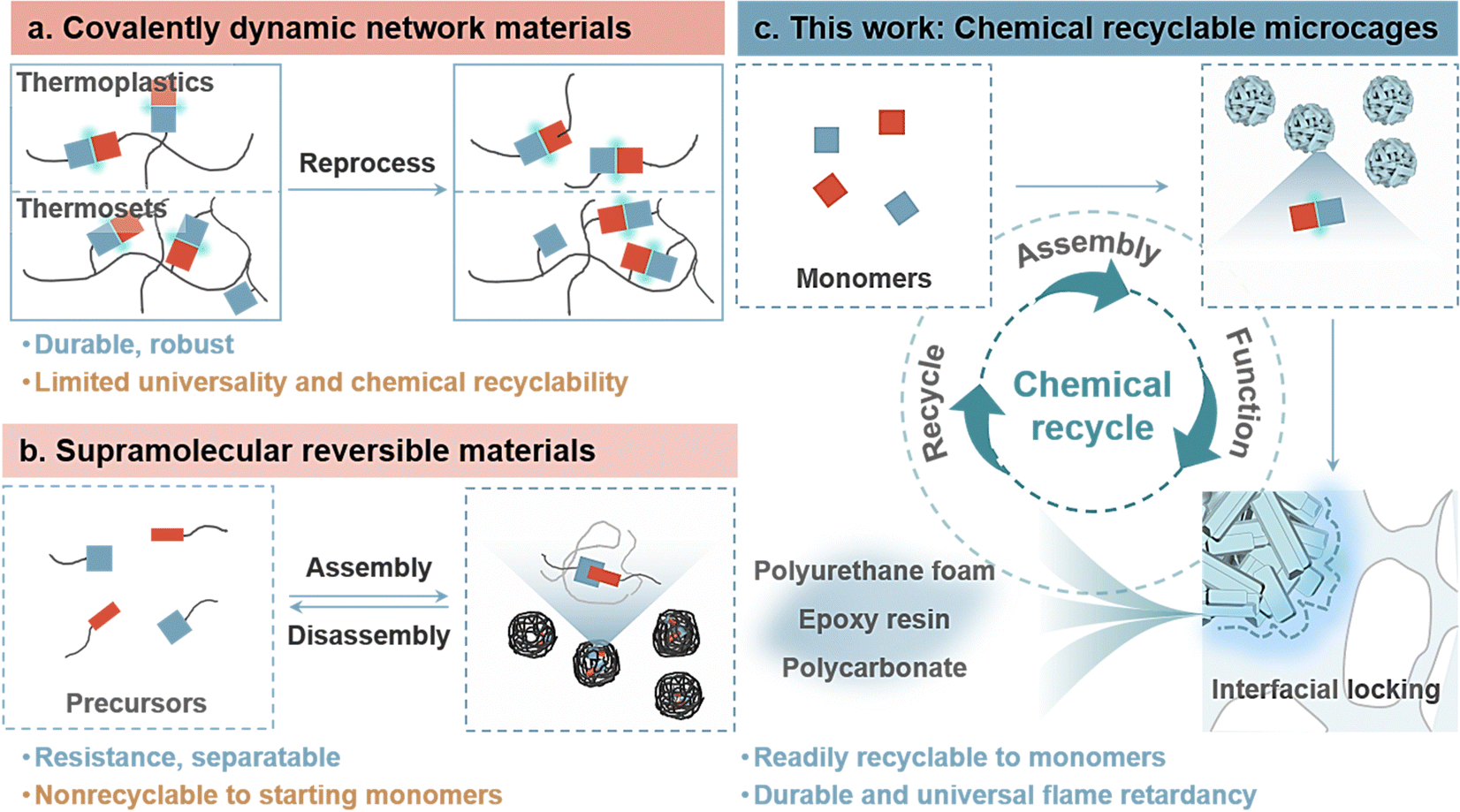 | ||
| Fig. 1 Schematic of traditional dynamic reversible assembly and proposed chemically recyclable flame-retardant systems in this study. (a) Traditional covalent dynamic network materials and (b) supramolecular reversible assemblies. (c) Proposed chemically recyclable microcage that emphasizes the return to starting monomers via dynamic reversible interfacial locking engineering. | ||
Significant strides have been made in advancing circular plastics, demonstrating the capability for infinite recycling back to starting monomers through processes such as acidolysis, catalysis, and solvolysis.27–33 The regenerated plastics, derived from recovered monomers, retain the properties of the original material, and they can serve as versatile building blocks for the creation of new materials with unique characteristics. However, parallel efforts in the realm of chemically recyclable flame retardants are scarce, due to challenges related to the chemo-selectivity required for complex material compositions and the feasibility of cleaving covalent bonds in starting monomers. Compared with recyclable flame retardants relying on presynthesized precursors, the recovered starting monomers derived from wastes retain the unblemished features of raw materials, allowing them to serve as blocks for reentering new supply chains without restricting their application areas. In addition, chemical treatments of starting monomers are necessary to match the requirements of flame-retardant mechanisms or reactivities for various polymers, while presynthesized flame retardants are often applicable to one. Hence, recycling of starting monomers from waste flame-retardant materials offers the benefits of extensive reformulation capability and applicability. The quest for chemically recyclable flame retardants that facilitate the infinite recovery of starting monomers while ensuring high durability and flame retardancy is an attractive yet ongoing strategy.
Here, we present a chemically recyclable and durable microcage flame retardant via reversible interfacial locking engineering, which is applicable to various thermosets/plastics (polyurethane, epoxy resin, polycarbonate, etc.) (Fig. 1(c)). The strategic assembly of phosphoric acid and Cu2+ through highly synergetic interactions allows for the controlled macroscopic assembly of nanosheets into hierarchical mesostructured microcage particles. The dynamically reversible destabilization–refabrication of microcage assembly networks facilitates efficient and infinite recycling of starting monomers via environmentally friendly pH adjustment. The recycled phosphoric acid and Cu2+ monomers from the polymer matrices exhibited impressive recovery rates of 92% and 96.2%, respectively, offering opportunities for reformulation in a supply chain with differentiated properties. Plastics with removed flame retardants can also undergo chemical recycling. The micron-/nanostructures derived from nanosheets contribute additional surfaces and pores to the matrix-like microcage, enabling interfacial locking and tight clasps resembling gears. The significantly improved durability and fire safety of various plastics underscore the excellent flame-retardant efficiency and utility of microcage. The simplicity of preparation, coupled with chemical recyclability, reversible interfacial locking, and high flame-retardant efficiency, position this microcage as a promising avenue for developing future functional circular flame retardants.
Results and discussion
Design and structural characterization
The circular life, durability, and availability of flame retardants should be considered when overcoming the typical trade-offs between long-term service and ring-closing sustainability. Employing simple small feedstocks that are capable of facilitating covalent hierarchical assemblies is desirable for the abovementioned multifunctional integration. Structurally ordered and functional assemblies are generally fabricated by multiple highly synergetic molecular interactions.34,35 We observe that aryl phosphoric acids exhibit the unique ability to engage in multiple covalent and noncovalent interactions concurrently. These interactions include electrostatic bonding with metal ions, π–π stacking, and hydrophobic interactions involving aromatic groups.36,37 Notably, interactions with varying affinities serve as valuable structural cues for these small building blocks, contributing to their recognition. This, in turn, plays a pivotal role in guiding and regulating the assembly process across different scales. These capabilities provide novel prospects for engineering intricate micron-/nanostructured assemblies.Structurally ordered microcages (PM) can be easily obtained by deprotonating phosphoric acid with NaOH and subsequently performing hierarchical assembly with Cu2+ monomers in EtOH/H2O on several scales. As shown in Fig. 2(a), macroscopically, the PM is constructed of centrally interconnected and loosely aggregated brick-like chunks with a thickness of 1.2–2.4 μm, maintaining a uniform size and distribution with a particle diameter of approximately 20–24 μm. Fracture surface and elemental mapping were further conducted to inspect the detailed microstructure of PM (Fig. 2(b) and Fig. S1, ESI†). Interestingly, PM possesses a special nucleocapsid structure that consists of a petaloid shell and a spherical core. Microthick brick-like chunks randomly radiate, synchronously grow outwardly, and loosely pack, pointing to the same core to form a petaloid shell of unset length (∼5.8 μm in Fig. 2(b)). Due to the high spatial hindrance and nucleation rate, the spherical core, which has a consistent composition with the petaloid shell, is relatively independent and shares varying radii (∼6.4 μm in Fig. S1(a), ESI†). Eventually, spherical caged particles with matrix-like porous surfaces are formed. The emergence of new peaks corresponding to P–O–Cu (1052 cm−1) and Cu–O (571 cm−1), the absence of O–P–OH (1180 cm−1), and the shift of O–P–O from 961 cm−1 to 1130 cm−1 confirmed the robust interactions between phosphoric acid (PA) and Cu2+ monomers (Fig. 2(c)). Correspondingly, under the influence of potent electrostatic interactions, the electron binding energies of both phosphorus and oxygen underwent noticeable shifts, and new diffraction peaks were detected (Fig. 2(d) and Fig. S2, S3, ESI†).38
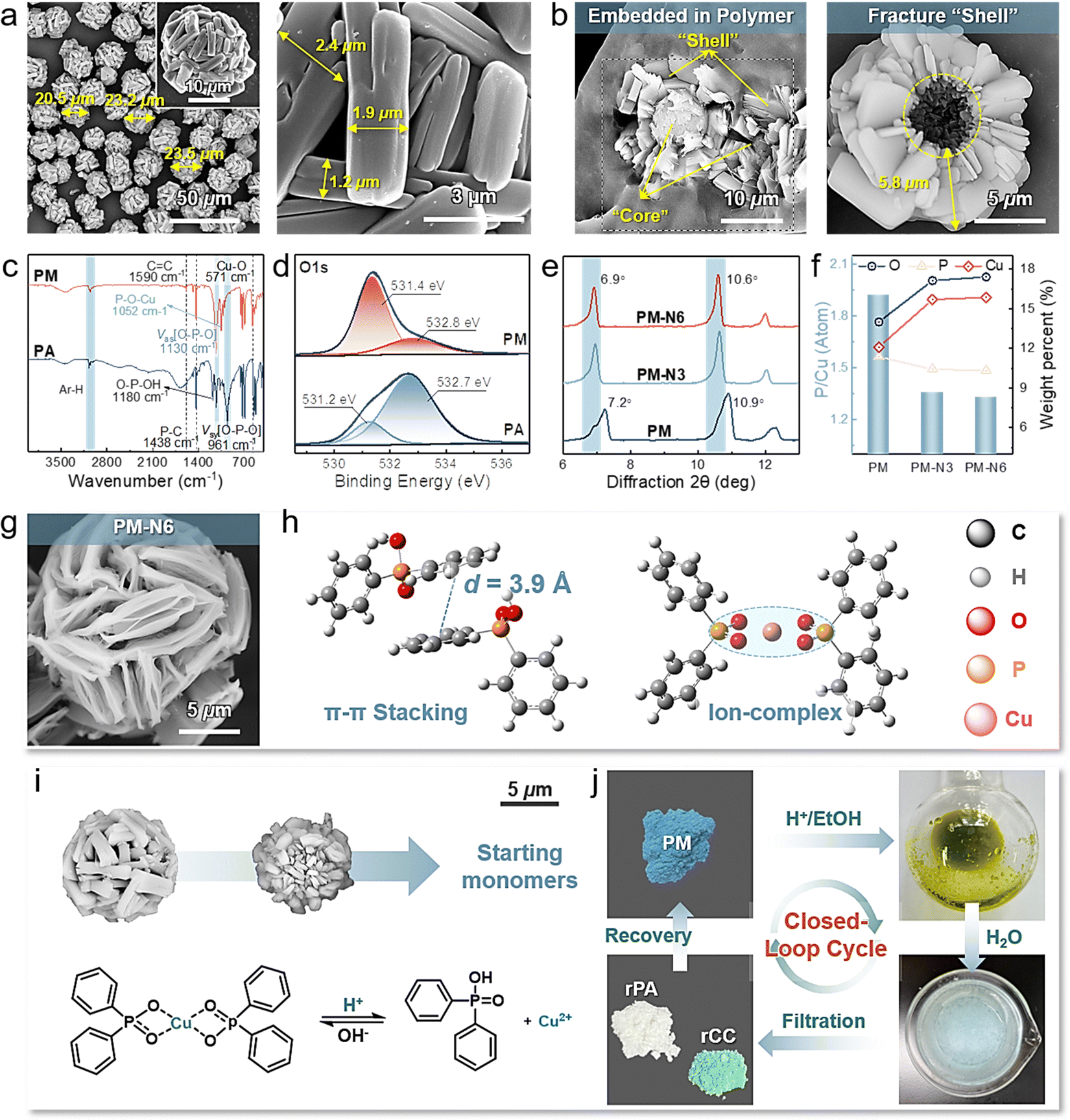 | ||
| Fig. 2 Structural analysis, hierarchal assembly behaviors, and chemical recycling process of PM. (a) SEM images of PM. (b) Localized SEM image of the cross section of PM. (c) FT-IR spectra of the original feedstock PA and resulting PM. (d) High-resolution O1s spectra of PA and PM. (e) XRD patterns and (f) selected elemental contents of PM, PM-N3, and PM-N6 treated with 50 mM NaOH in an ethanol solution for 30 min and 60 min, respectively. (g) SEM images of multiscale-structured PM-N6. (h) Optimized structures showing molecular details of aromatic stacking in EtOH and ion interactions between Cu2+ and deprotonated PA molecules. (i) pH-dependent morphology changes and schematic illustration of the structural transition of PM. (j) Graphical schemes showing the closed-loop chemical recycling of PM under environmentally friendly conditions. | ||
To gain insight into the assembly process and mechanism of these mesostructured microcages, we monitored their formation behavior and time-dependent environmental stability and responsiveness. The solvent nature usually significantly influences the strength and synergism of electrostatic and π–π stacking interactions, which results in modulation of the assembly behavior.39–41 Herein, EtOH and H2O were used as the sole solvents for comparison. As shown in Fig. S4 (ESI†), the mixture of deprotonated PA and Cu2+ monomers formed small particles in several seconds in EtOH, which was quicker than that in H2O. PM-H2O consists of loosely packed amorphous chunks, exhibiting uniform sphericity and large sizes ranging from 37 μm to 40 μm (Fig. S5(a), ESI†). However, the resulting PM-EtOH consists of massive interconnected particles with a diameter of 7–9 μm, showing poor monodispersity (Fig. S5(b), ESI†). In addition, the high resistance of PM to several environments (e.g., EtOH, DMF, CH3CN, and 100 mM urea) but relatively weak structural stability in H2O or 100 mM NaCl aqueous solution reveal that electrostatic interactions are susceptible to H2O (Fig. S6 and S7, ESI†).34 Therefore, we speculate that the stronger electrostatic interactions in EtOH than in H2O admittedly induce rapid nucleation of tiny crystallites and random aggregation of chunks, thus leading to a smaller size and irregular morphology.
Experiments and molecular dynamics simulations further highlight that the primary assembly driving forces are highly synergistic ionic interactions and π–π stacking interactions. PM was incubated in NaOH ethanol solution for 30 min and 60 min to form PM-N3 and PM-N6, respectively (Fig. S8(a), ESI†). The stoichiometry and chemical constitution of PM changed since the P-containing anions were readily replaced by OH−, exposing the subunit structures of neatly aligned flakes. As demonstrated in Fig. 2(e) and (f), the diffraction peaks shift from 7.2° for PM to 6.9° for PM-N6, demonstrating an increase in the lattice space. Coupled with the decrease in P-containing anions, the contents of C and P significantly decreased from 57.06% to 50.24% and 11.37% to 10.32%, respectively, while O and Cu increased to 17.40% and 15.86%, respectively (Table S1 and Fig. S8(b), (c), ESI†). Supported by the microscopic findings shown in Fig. 2(g) and Fig. S9 (ESI†), the layered chunks in PM tend to form strips and basically exist in the form of neatly aligned nanosheets, revealing detailed hierarchical flaky agglomeration.39 Notably, matrix-like three-dimensional Cu(OH)2 and CuO particles are generated when the OH− content and incubation time increase, indicating the complete ion intercalation between P-containing anions and OH− (Fig. S10, S11 and Table S2, ESI†).
Combined with the simulation results, as shown in Fig. 2(h), Fig. S12 and Table S3 (ESI†), the horizontally arranged phenyl rings of PA exhibit a layer spacing of 3.9 Å and a lower binding energy in H2O (−50.43 kJ mol−1) than in EtOH (−37.82 kJ mol−1), indicating stronger π–π interactions when H2O is used as an assembly mediator.21 Cu2+ is coordinated by four oxygen atoms stemming from two deprotonated PA molecules and forms an asymmetric planar structure (Fig. 2(h)). Therefore, the dominant interactions leading to rough chunk surfaces and a larger size of PM-H2O are water-susceptible electrostatic interactions and π–π stacking interactions, respectively.42 The interconnection in PM-EtOH is mainly due to strong ionic interactions, as H2O or NaCl aqueous solution is the only mediator that can disconnect them into independent particles and destroy their surface structure, likely leading to the screening of electrostatic interactions (Fig. S5(c) and Fig. S7, ESI†).34 H2O is used as a green and indispensable modulating agent in this work for balancing multiple synergistic interactions and precisely controlling assembly behavior.
The molecular scale impacts on stoichiometry and macroscopic appearance collectively provide insight into the assembly process and driving forces of PM. It is speculated that the matrix-like PM follows a spatiotemporal hierarchical assembly process. Specifically, driven by relatively strong ion and π–π stacking interactions, ion complexes (elementary structures) develop into initial nanoaggregates, followed by seizing neighbors to form ion complex monolayers (secondary structures). Subsequently, the secondary monolayers sprout into nanosheets (tertiary structures). Resulting from aryl and steric hindrances, the tertiary flakes are neatly aligned and packed into microthick layered chunks (quaternary structure). Finally, the chunks spontaneously grow outward into a matrix-like microcage (quintenary structure) that is mainly coupled and stabilized by π–π stacking interactions. Highly synergetic molecular interactions between PA and Cu2+ facilitate the tailoring of hierarchical macroscopic assemblies of nanosheets into spherical and mesostructured microcages on different scales.
Notably, owing to the simplicity and feasibility, this approach may provide a new system for fabricating programmable hierarchically ordered assemblies for multiple functional integrations. The in situ formed special matrix-like porous structures of PM minimize the restacking of microchunks and offer facile access for liquids to penetrate inside, such as the raw materials of polymers, facilitating robust interfacial locking during the processing of polymeric materials. Consequently, PM that is stabilized by dynamic reversible physical interpenetration can serve long term as a functional filler due to the interdigitated interface, thus achieving the desired trade-offs between durability and recyclability.
Infinitely chemical recyclability and reconfigurability
As expected, the stimuli-triggered reversibility of dynamic covalent networks and differences in the physicochemical properties of the assembled precursors (PA and Cu2+ monomers) collectively endow PM with infinite chemical reusability to form starting feedstocks rather than mediators, thus realizing self-sustained closed-loop recyclability and reconfigurability for new materials. As depicted in Fig. 2(i), (j) and Fig. S13 (ESI†), the stoichiometry of the P-containing anions and Cu2+ changed when triggered by the H+ ethanol solution, and PM was gradually dissolved to form a mixed starting monomer solution. Subsequently, feedstocks PA and Cu2+ monomers can be separately recovered via sequential precipitation and filtration processes, achieving high yields of 98.8% and 99.4%, respectively. The FT-IR and XRD results demonstrated that the chemical structures of recycled PA and Cu2+ monomers (denoted as rPA and rCC, respectively) were consistent with those of the starting monomers (Fig. S14, ESI†). Notably, recovering virgin monomers is considerably feasible for use in reassembling new PMs or reconfiguring other new materials with differentiated properties (Fig. 2(j)).Flexible polyurethane foam (PUF, the foam with the largest market share) was chosen as a typical representative to validate the feasibility of chemical recycling of PM from waste flame-retardant materials to starting monomers.43,44 As shown in Fig. 3(a) and Table S4 (ESI†), PM was physically introduced into PUF during foaming. The resulting foam (PM-F) exhibited a continuous, unblemished, and respectable open-cell morphology comparable to that of neat foam (N-F) (Fig. S15, ESI†), in which the PM was well embedded in the matrices. At the end-of-life stage of PM-F, PM can be efficiently recycled back to its starting monomers and recovered via facile and ecofriendly processes. Specifically, the foam with 2.8 wt% PM was soaked in 0.1 M HCl ethanol solution for 10 min, and the embedded PM was rapidly disassembled into starting monomers via destabilization of dynamic assembly networks and completely diffused away from the foam matrix, which enabled the separate collection of PM solution and virgin foam matrices (vPM-F) (Fig. 3(a)–(c) and (f)). Phosphoric acid and Cu2+ monomers can be regenerated from mixtures via facile sequential filtration and precipitation, realizing imperative recycling rates of 92.0% and 96.2%, respectively (denoted as rPA-F and rCC-F) (Fig. 3(h), (i) and Fig. S16, S17, ESI†). In addition, the remaining foam matrices exhibited an intact cellular structure and skeleton that contained microsized holes resulting from the departure of PM, which can be handled by conventional chemical degradation processes (Fig. 3(j)). Compared with traditional flame retardants, the controllable real-time separation and chemical recyclability of starting monomers of PM show great advantages for effectively alleviating environmental toxicity and wasting resources while offering opportunities for reformulation in reentering supply chains on differentiated demands.
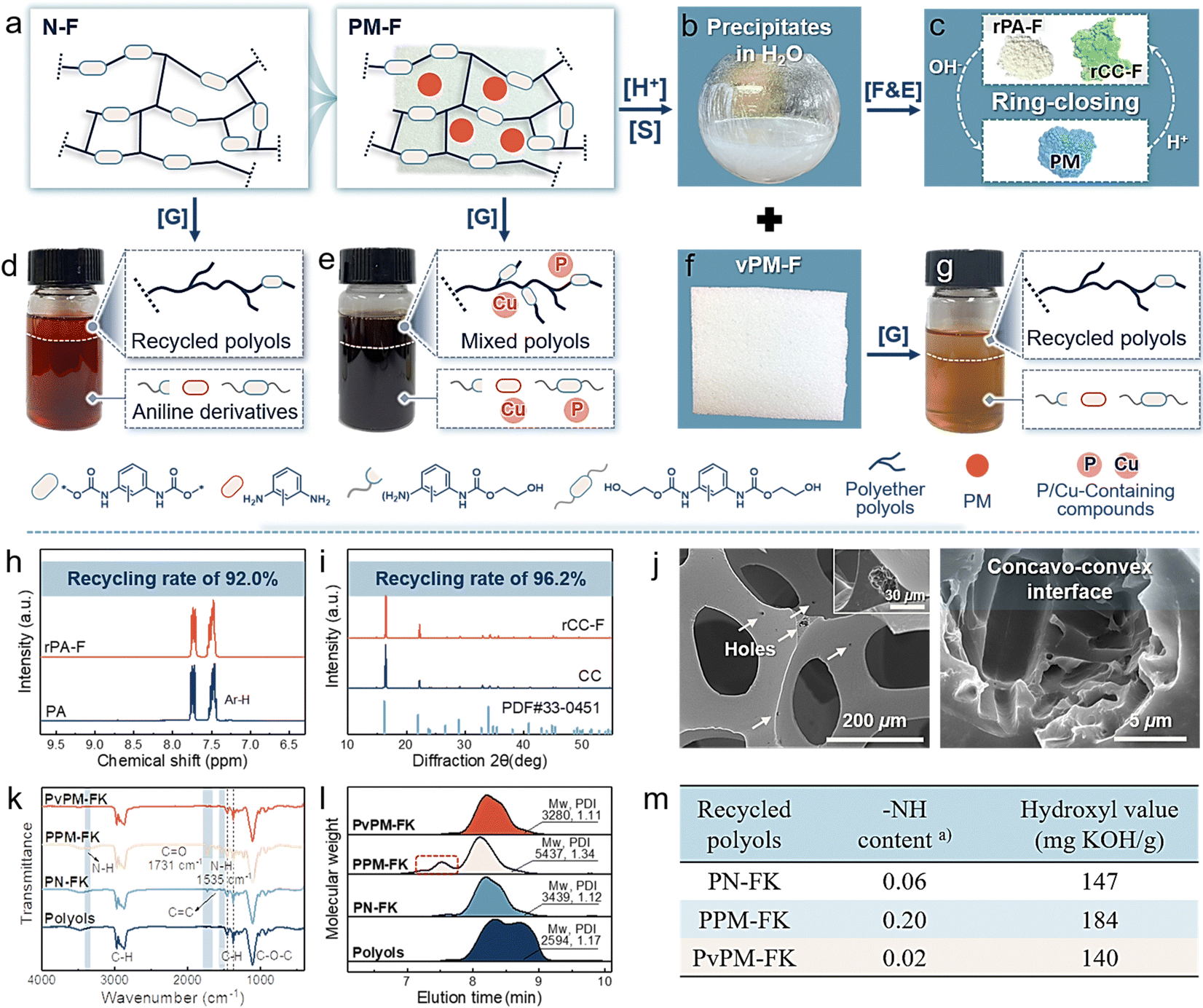 | ||
| Fig. 3 Chemical recyclable behavior of PM-containing PUF. (a) Schematic representation of N-F and PM-F. (b) The separated PM derivatives dispersion from PM-F. (c) Full separation and recovery of starting monomers from solution (b). (d)–(g) Photographs and schematic illustration of the chemical recycling of polyols from N-F, PM-F, and vPM-F in the presence of catalyst. (h) 1H NMR spectrum of rPA-F. (i) XRD patterns of rCC-F. (j) SEM image of vPM-F. (k) FT-IR spectra, (l) representative GPC traces, and (m) functional group analysis of recycled polyols PN-FK, PPM-FK, and PvPM-FK obtained by glycolysis. [G] Glycolysis of foams with 1 wt% potassium propionate as the catalyst. [S] Separation of PM and the foam matrices upon exposure to H+ triggers. [F&E] Sequential filtration and evaporation. (a) Calculated from Fig. S20 (ESI†). | ||
Glycolysis of flame-retardant foams
Chemical recycling is the most attractive and developed management method for waste PUF.45 Here, glycolysis was used as a representative method to elucidate the chemical recyclability of foams with or without flame retardant PM. As shown in Fig. 3(d), (e) and (g), foams were completely dissolved in ethylene glycol (EG) upon heating to 200 °C for 3 h, after which the upper polyol phase was isolated for further analysis. The degradation rate of the foams followed the sequence N-F ≈ vPM-F > PM-F, revealing the inhibitory effect of PM on the conversion of urethane bonds even in the presence of potassium propionate (PK) as a catalyst. As presented in Fig. 3(k) and Fig. S18 and S19 (ESI†), intense peaks centered at 1237 cm−1 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) and 1535 cm−1 (N–H), accompanied by visible proton signals located at 8.4–9.6 ppm (N–H), 4.0–4.1 ppm (O–CH2*–CH2OH), and 6.2–7.6 ppm (Ph–H), were detected in recycled polyols PPM-FK obtained from PM-F catalyzed by potassium propionate.46,47 The intensities of the signals of the amidated and EG-functionalized polyols were significantly greater than those of PN-FK and PvPM-FK obtained from N-F and vPM-F, respectively (Fig. S19(c), ESI†). Accordingly, as shown in Fig. 3(l), (m) and Fig. S20, and Table S5 (ESI†), PPM-FK exhibited an extremely high mass average molar mass (Mw, 5437 kDa), distribution (Đ, 1.34), NH content (0.20), and hydroxyl value (184), while relatively low values were observed for PN-FK (3439 kDa, 1.12, 0.06, and 147) and PvPM-FK (3280 kDa, 1.11, 0.02, and 140). These results jointly demonstrate the suppressive effects of PM on the chemical degradation of foams. Additionally, P/Cu-based impurities inevitably pollute recycled polyols, for which 10.3% of P and 2.2% of Cu residuals were detected (Fig. S21, ESI†). Interestingly, PM had little effect on the degradation process without potassium propionate (Fig. S22–S25 and Table S5, ESI†). Thus, we speculate that the inhibitory effect of PM is mainly achieved by interfering with the catalytic process.
O) and 1535 cm−1 (N–H), accompanied by visible proton signals located at 8.4–9.6 ppm (N–H), 4.0–4.1 ppm (O–CH2*–CH2OH), and 6.2–7.6 ppm (Ph–H), were detected in recycled polyols PPM-FK obtained from PM-F catalyzed by potassium propionate.46,47 The intensities of the signals of the amidated and EG-functionalized polyols were significantly greater than those of PN-FK and PvPM-FK obtained from N-F and vPM-F, respectively (Fig. S19(c), ESI†). Accordingly, as shown in Fig. 3(l), (m) and Fig. S20, and Table S5 (ESI†), PPM-FK exhibited an extremely high mass average molar mass (Mw, 5437 kDa), distribution (Đ, 1.34), NH content (0.20), and hydroxyl value (184), while relatively low values were observed for PN-FK (3439 kDa, 1.12, 0.06, and 147) and PvPM-FK (3280 kDa, 1.11, 0.02, and 140). These results jointly demonstrate the suppressive effects of PM on the chemical degradation of foams. Additionally, P/Cu-based impurities inevitably pollute recycled polyols, for which 10.3% of P and 2.2% of Cu residuals were detected (Fig. S21, ESI†). Interestingly, PM had little effect on the degradation process without potassium propionate (Fig. S22–S25 and Table S5, ESI†). Thus, we speculate that the inhibitory effect of PM is mainly achieved by interfering with the catalytic process.
To validate the inherent inhibition mechanism, the ionization ability of PM and potassium propionate and the catalytic degradation activity of potassium diphenylphosphate (DK) are explored. As depicted in Fig. S26 and S27 (ESI†), deprotonated phosphoric acid and propanoic acid exhibit similar Mulliken charges, and both PM and potassium propionate can rapidly ionize to form anions and cations in ethylene glycol at high temperature. The dissociative K+ can react with ethylene glycol to form initiator alkoxide, which further promotes the cleavage of urethane bonds via nucleophilic substitution by coordination/insertion and exchange, subsequently releasing polyols (Fig. S28(a) and (b), ESI†).48 As expected, potassium diphenylphosphate exhibit similar catalytic activity to that of potassium propionate. Compared with those in recycled polyols PN-F without a catalyst, the characteristic signals of N–H and CO in the recycled polyols PN-F1PK degraded by PK and PN-F1DK degraded by DK sharply decrease, revealing the minor effect of anions on chemical degradation (Fig. S29, ESI†). The coordination of metallic species with carbonyl groups in urethane lowers the electron density, thus enhancing the nucleophilic insertion of alkoxides. However, the coordination capability of Cu2+ is greater than that of K+ because of its smaller atomic radius and lower polarizability.48 As illustrated in Fig. S28(b) and (c) (ESI†), Cu2+ competed with K+ to coordinate with carbonyl groups, inhibiting the nucleophilic insertion of oxygen anions and the release of polyols.
Overall, PM suppresses the nucleophilic attack of alkoxides by competing with K+ to coordinate carbonyl groups, which lowers the glycolysis efficiency and deteriorates the quality of recycled polyols. Separating and removing PM from polymeric matrices in advance by manipulating reversible interfacial locking through a specific stimulus provides a simple and efficient way to address the above-mentioned problems. These findings not only enrich urethane-based glycolysis chemistry but also underpin the sustainable design of flame-retardant polymeric materials.
Widespread applicable flame retardancy and durability
Incorporating flame retardants is the most effective way to reduce the fire risk of highly flammable polymeric materials, but most existing flame retardants feature the main disadvantages of low efficiency, limited universality, and easy aging during long-term service.49–51 Fortunately, the developed PM is suitable for various polymeric materials for significantly and simultaneously improving fire safety and durability due to its high radical scavenging ability and robust interfacial locking structure. As shown in Fig. 4(a), Fig. S30, and Table S6 (ESI†), N-F was readily ignited and violently burnt with severe melt dripping. The resulting foams with 1.1 wt% PM achieved the ability to self-extinguish instantly upon flame removal, attaining a highly improved fire safety that was superior to that of its previous counterparts (Table S7 and Movies S1, ESI†). Notably, an accelerated aging test was performed to imitate the aging resistance of PM-F during long-term use. As depicted in Fig. 4(b) and Fig. S31 (ESI†), aged PM-F exhibited a consistent rapid self-extinguishing ability and constant appearance comparable to those of untreated PM-Fs.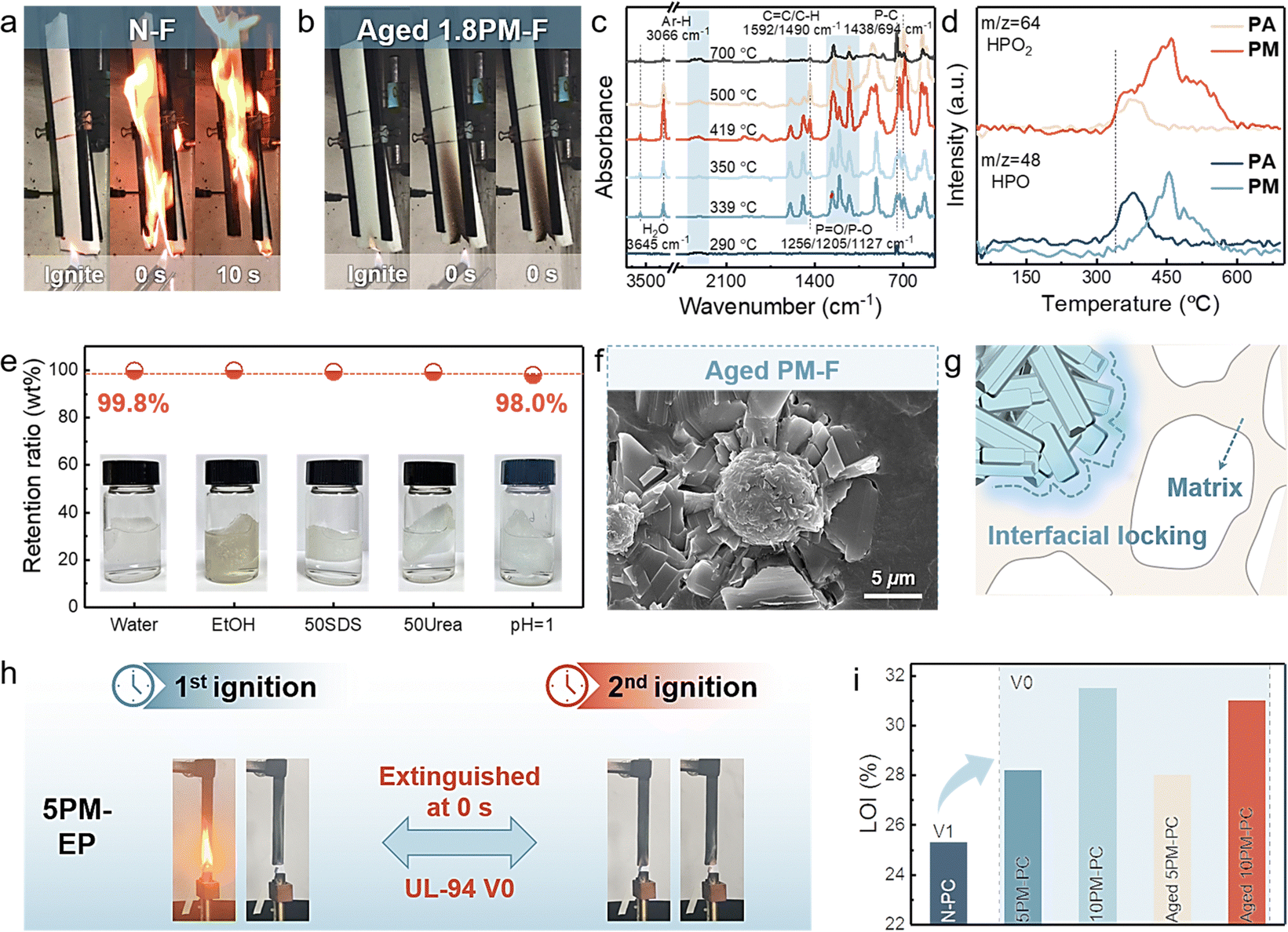 | ||
| Fig. 4 Durable flame retardancy of PM for various polymeric materials. Digital photographs at different burning times of (a) N-F and (b) aged PM-F. (c) FT-IR spectra of gaseous products at selected temperatures of PM. (d) P-containing gaseous decomposition products of PM tracked by TG-MS. (e) Retention rate of weight for PM-F after incubation in different media: H2O, EtOH, 50 mM SDS, 50 mM urea, and HCl aqueous solution (pH = 1) for 24 h. (f) SEM image of the strut joint for aged PM-F. (g) Schematic illustration of the structural gomphosis between PM and the matrix allowing long-term durability. (h) UL-94 results for epoxy resin with 5 wt% PM. (i) LOI values of polycarbonates with PM. | ||
Radical scavenging capability and secure interfacial locking are responsible for the excellent flame retardancy and durability. As shown in Fig. 4(c), (d) and Fig. S33 (ESI†), characteristic peaks for aromatics (Ph–H/CC, 3066/1592 cm−1) and P-containing derivatives (PO/P–O, 3066/1592 cm−1) were detected during heating, in which partial P-containing species were confirmed to be HPO2 (m/z = 64) and HPO (m/z = 48). Compared with N-F, PM-F had a lower ID/IG value for graphite residuals, initial decomposition temperature, and gaseous isocyanate derivatives (–NCO, 2274 cm−1) but a greater ID/IG value for hydrocarbons/ethers (2800–3000 cm−1/1112 cm−1) (Fig. S34 and S35, ESI†).52 Hence, the high radical scavenging capability achieved by P-containing species and the diminished fuel supply in the early combustion stage jointly endow PUF with high self-extinguishing efficiency, while the violent pyrolysis of the matrix subsequently leads to complete burn.53,54
Notably, the loosely packed matrix-like porous surfaces of PM contribute additional pores and areas and offer facile access for liquid raw materials of polymers to penetrate inside, which facilitate robust interfacial locking and tight clasps resembling gears into matrices during the processing of polymeric materials, thus achieving high durability. As shown in Fig. S15 (ESI†), PM is intimately embedded in the foam skeleton without visible phase interfaces, allowing the resilience of PM-F to be comparable to that of neat foam (Fig. S36 and Table S10, ESI†). In addition, PM-F exhibits excellent resistance to harsh environments and maintained a mass rate higher than 99.3% after incubation in H2O, EtOH, SDS, urea solutions, etc. (Fig. 4(e)). Evidently, the well-preserved PM in aged PM-F further revealed its high stability during long-term service (Fig. 4(f) and (g)). Owing to the chemical and morphological characteristics of PM, the dynamically reversible interfacial locking that occurs during foaming provides reinforced durability and infinite chemical recyclability, while the P-containing species generated upon heating or burning work as efficient radical scavengers in the gas phase to significantly improve the fire safety of foams.
Interestingly, PM shows widespread applicable flame retardancy and harsh environmental resistance. Herein, epoxy resin (EP) and polycarbonate (PC) are chosen as representative materials for assessing its universality and resistance. As depicted in Fig. 4(h) and Fig. S37–S40 (ESI†), neat EP (N-EP), with a low limiting oxygen index (LOI) of 25%, was flammable and generated molten droplets during combustion. The resulting 5PM-EP with only 5 wt% PM passes the UL-94 V0 rating and achieves a high LOI value of 29%, which can be further increased to 35.0% (Movies S2, ESI†). Also, PM is suitable for effectively improving the fire safety of thermoplastic PC. As shown in Fig. 4(i), Fig. S30, and Movies S3 (ESI†), a relatively satisfactory LOI value of 28.2% and UL-94 V0 rating were obtained for only 5 wt% PM loading. The corresponding LOI values further increased to 31.5% upon 10 wt% PM loading, indicating highly improved fire safety. Notably, owing to the robust environmental resistance and interfacial locking structures between the PM and polymer matrices, both PM-EP and PM-PC show excellent resistance to hot water (60 °C, 12 h), manifesting commensurate LOI values and self-extinguishing performances, thus achieving long-term durability (Fig. 4(i) and Fig. S39, ESI†). Overall, its facile preparation, chemical recyclability, reversible interfacial locking, and durable/universal flame retardancy make PM a promising candidate for future circular flame retardants.
Conclusions
In summary, we demonstrate a chemically recyclable microcage flame retardant with infinite chemical recyclability, durability, and widespread applicable flame retardancy via reversible interfacial locking engineering, which provides a strategy for combating plastic waste and advancing the ring-closing sustainability of flame-retardant materials. The highly synergetic interactions between phosphoric acid and Cu2+ monomers facilitate the hierarchical macroscopic assembly of nanosheets into microcages, which minimize the restacking of chunks and result in structurally ordered matrix-like porous surfaces. Remarkably, PM, which features dynamically reversible covalent networks, easily destabilizes upon ecofriendly H+ triggers and further allows the chemical recycling of starting monomers from foam with high efficiency and yields (92% for phosphoric acid and 96.2% for Cu2+ monomers). The recovered starting monomers provide fantastic opportunities for infinitely reassembling or reconfiguring new materials on demand, achieving a true “circular material economy”. As expected, the separated virgin matrix can be chemically recycled as a neat matrix via conventional recycling methods. Notably, the matrix-like porous PM enables superior long-term durability by forming unique and robust interfacial locking structures within the matrix. PM endows various thermosets and thermoplastics with highly improved fire safety. Rapid self-extinguishing, V0 ratings, and highly improved LOI values are achieved at relatively low PM loadings, which allow PM to outperform the existing flame retardants. This new facile strategy for designing chemically recyclable, reconfigurable, durable, and widely applicable flame retardants will stand out in the future for circular flame-retardant materials.Experimental
Synthesis of hierarchal microcages PM
In a typical experiment, diphenylphosphinic acid (PA, 0.06 mol) and NaOH (0.06 mol) were added to 500 mL of EtOH under ambient conditions. After stirring for 12 h, 400 mL copper chloride dihydrate (CC, 0.06 mol) solution (VH2O/VEtOH = 3/37) was added rapidly. The resulting mixture was stirred for another 5 s, followed by incubation overnight. The blue precipitates, denoted as PM, were filtered, washed with water and EtOH, and then dried at 80 °C.Time and environmental dependent stability
To analyze the multiscale structures, driving processes/forces, and resistance to various environments of PM, an HO− mediated ion exchange reaction was performed first, and 0.1 g of PM was incubated in 20 mL of 50 mM NaOH ethanol solution for 30 min and 60 min, respectively. PM-N3 and PM-N6 were thoroughly washed with ethanol and dried at 80 °C. The structural and morphological transitions were subsequently characterized via EA, ICP-OES, XPS, XRD, SEM, and elemental mapping. Subsequently, the morphological transitions of PM in the presence of H2O, EtOH, DMF, CH3CN, 100 mM NaCl aqueous solution, and 100 mM urea aqueous solution were also monitored via SEM.Chemical recycling and refabrication of PM
The separation and recovery of starting monomers (phosphoric acid PA and Cu2+ monomers) and the refabrication of PM were conducted to evaluate the circular chemical recyclability of PM via a pH-triggered responsive disassembly-reassembly process. Specifically, a 1.0 M HCl solution was added to the ethanol dispersion of PM (50 mg mL−1) to form a green transparent solution. After EtOH evaporation and precipitation in H2O, the recovered starting monomers rPA and rCC were obtained from the precipitation and evaporation of the filtrate, respectively. High yields of 98.8% for rPA and 99.4% for rCC were achieved, and these products were successfully reused for refabricating new PM with constant quality.Preparation of flame-retardant foams
Flame-retardant polyurethane foams (PM-F) and neat foam (N-F) were prepared through a one-pot and free foaming method according to the ingredients listed in Table S4 (ESI†). For example, PM, a blowing agent (deionized water), catalysts (A33 and T9), and a surfactant (L580) were mixed well with polyether polyols (GEP 560 s) in a beaker. TDI 80/20 was added to the above mixture under vigorous stirring, and the mixture was immediately poured into a cast mold after 5–8 s. The samples were further cured for 72 h under ambient conditions to obtain the final flame-retardant foams (denoted as PM-F).Preparation of flame-retardant epoxy resins
Flame-retardant epoxy resins (PM-EPs) and neat epoxy resins (N-EP) were synthesized via bulk curing of bisphenol A diglycidyl ether (DGEBA) with 4,4′-diaminodiphenylmethane (DDM). Briefly, DGEBA was first mixed with PM and stirred at 80 °C until a homogeneous mixture was obtained. DDM was added to the mixture, which was stirred for 5 min and subsequently cured sequentially at 100 °C and 150 °C for 2 h in a Teflon mold.Preparation of flame-retardant polycarbonates
Flame-retardant polycarbonates with different loadings of PM (PM-PCs) were obtained by the solvent method. PC granules were first dissolved in DCM, while PM was subsequently added and well dispersed. The mixed solution was then moved to the enamel plate and left at room temperature for 6 h to allow slow evaporation of the solvent.Chemical recycling of PM to starting monomers from polymeric materials
Typically, 2.8PM-F was washed with ethanol 3 times and then immersed in freshly prepared 100 mL of 0.1 M HCl at room temperature for 5 min. The yellowish-green solution and virgin foam matrix were collected separately. The obtained solution was further concentrated and precipitated in H2O, followed by filtration to form the white powder rPA-F (yield of 92.0%). Subsequently, the recovery of rCC-F (yield of 96.2%) was achieved by sequential alkalization, filtration, acidization, vacuum distillation, and dying steps. The chemical structures of recycled rPA-F and rCC-F were characterized via NMR, FT-IR, and XRD, and the separated virgin foam was characterized via SEM.Chemical recycling of flame-retardant foams via glycolysis
Typically, 15 g of ethylene glycol and a predetermined amount of catalyst potassium propionate (1 wt% of the mass of foam) were charged in a glass flask equipped with a magnetic stirrer at a temperature of 200 °C under nitrogen. 5 g of PM-F with a diameter of 0.5–1.0 cm was continuously put into the reactor for 30 min and stirred for another 3 h. Then, the upper polyol phase was isolated and collected for further analysis. In addition, glycolysis was also evaluated in foams without a catalyst for comparison. The hydroxyl number of the recycled polyols was determined by standard titration methods according to GB/T 12008.3-2009.Author contributions
Conceptualization and methodology: F. Z. and H. Z. Formal analysis: F. Z. and L. H. Software, visualization: F. Z., D. F., and J. M. Investigation: F. Z., T. B., and R. D. Writing – original draft: F. Z. Writing – review and editing: H. Z. Z. Z., and B. L. Supervision, project administration, resources, funding acquisition: Y. W.Conflicts of interest
The authors declare no competing interests.Acknowledgements
This work was supported by the National Natural Science Foundation of China (51991351, 52303061, 2023M742445, U2037206), Sichuan Science and Technology Program (2023NSFSC1943), and Institutional Research Fund from Sichuan University (2021SCUNL201).References
- B. W. Liu, H. B. Zhao and Y. Z. Wang, Adv. Mater., 2022, 2107905 CrossRef CAS PubMed.
- China Business Intelligence Network, https://www.yhresearch.cn/news/3786/flame-retardant#:~:text=%E6%8D%AE%E8%B0%83%E7%A0%94%E6%9C%BA%E6%9E%84%E6%81%92%E5%B7%9E%E8%AF%9A,GR%E4%B8%BA6.1%25%E3%80%82.
- Z. W. Ma, X. C. Liu, X. D. Xu, L. Liu, B. Yu, C. Maluk, G. B. Huang, H. Wang and P. A. Song, ACS Nano, 2021, 15, 11667–11680 CrossRef CAS PubMed.
- Hengzhou Chengsi, https://www.yhresearch.cn/news/3786/flame-retardant.
- C. Yao, H. P. Yang and Y. Li, Sci. Total Environ., 2021, 795, 148837 CrossRef CAS PubMed.
- M. C. Rillig, S. W. Kim, T.-Y. Kim and W. R. Waldman, Environ. Sci. Technol., 2021, 55, 2717–2719 CrossRef CAS PubMed.
- J. N. Hahladakis, C. A. Velis, R. Weber, E. Iacovidou and P. Purnell, J. Hazard. Mater., 2018, 344, 179–199 CrossRef CAS PubMed.
- H. A. Leslie, P. E. G. Leonards, S. H. Brandsma, J. de Boer and N. Jonkers, Environ. Int., 2016, 94, 230–234 CrossRef CAS PubMed.
- J. R. Peeters, P. Vanegas, L. Tange, J. Van Houwelingen and J. R. Duflou, Resour., Conserv. Recycl., 2014, 84, 35–43 CrossRef.
- C. C. Zhang and F. S. Zhang, J. Hazard. Mater., 2020, 382, 121140 CrossRef CAS PubMed.
- Y. B. Zhao, X. D. Lv and H. G. Ni, Chemosphere, 2018, 209, 707–720 CrossRef CAS PubMed.
- H. B. Zhao and Y. Z. Wang, Macromol. Rapid Commun., 2017, 38, 1700451 CrossRef PubMed.
- B. W. Liu, L. Chen, D. M. Guo, X. F. Liu, Y. F. Lei, X. M. Ding and Y. Z. Wang, Angew. Chem., Int. Ed., 2019, 58, 9188–9193 CrossRef CAS PubMed.
- M. Šilhavík, P. Kumar, Z. A. Zafar, R. Král, P. Zemenová, A. Falvey, P. Jiříček, J. Houdková and J. Červenka, ACS Nano, 2022, 16, 19403–19411 CrossRef PubMed.
- W. Wu Klingler, A. Bifulco, C. Polisi, Z. Huang and S. Gaan, Composites, Part B, 2023, 258, 110667 CrossRef CAS.
- W. Wu Klingler, V. Rougier, Z. Huang, D. Parida, S. Lehner, A. Casutt, D. Rentsch, K. B. Hedlund, G. A. Barandun, V. Michaud and S. Gaan, Chem. Eng. J., 2023, 466, 143051 CrossRef CAS.
- F. Lossada, D. Jiao, D. Hoenders and A. Walther, ACS Nano, 2021, 15, 5043–5055 CrossRef CAS PubMed.
- T. T. Guan, X. H. Wang, X. Zhao, X. Y. Lu, X. L. Wang, Y. Z. Wang and J. Q. Sun, CCS Chem., 2023, 1–12 Search PubMed.
- T. Liu, J. Y. Peng, J. Liu, X. L. Hao, C. G. Guo, R. X. Ou, Z. Z. Liu and Q. W. Wang, Composites, Part B, 2021, 224, 109188 CrossRef CAS.
- F. Wei, J. H. Zhang, C. Wu, M. Luo, B. J. Ye, H. J. Zhang, J. S. Wang, M. H. Miao, T. C. Li and D. H. Zhang, Macromolecules, 2023, 56, 5290–5305 CrossRef CAS.
- Z. H. Wang, B. W. Liu, F. R. Zeng, X. C. Lin, J. Y. Zhang, X. L. Wang, Y. Z. Wang and H. B. Zhao, Sci. Adv., 2022, 8, eadd8527 CrossRef CAS PubMed.
- F. R. Zeng, B. W. Liu, Z. H. Wang, J. Y. Zhang, X. L. Chen, H. B. Zhao and Y. Z. Wang, ACS Mater. Lett., 2023, 5, 1692–1702 CrossRef CAS.
- J. Y. Zhang, F. R. Zeng, B. W. Liu, Z. H. Wang, X. C. Lin, H. B. Zhao and Y. Z. Wang, Mater. Horiz., 2023, 10, 4551–4561 RSC.
- G. Q. Xu and Q. G. Wang, Green Chem., 2022, 24, 2321–2346 RSC.
- C. Jehanno, J. W. Alty, M. Roosen, S. De Meester, A. P. Dove, E. Y. X. Chen, F. A. Leibfarth and H. Sardon, Nature, 2022, 603, 803–814 CrossRef CAS PubMed.
- L. Anderson, E. W. Sanders and M. G. Unthank, Mater. Horiz., 2023, 10, 889–898 RSC.
- Q. Zhang, Y. X. Deng, C. Y. Shi, B. L. Feringa, H. Tian and D. H. Qu, Matter, 2021, 4, 1352–1364 CrossRef CAS.
- A. R. Epstein, J. Demarteau, B. A. Helms and K. A. Persson, J. Am. Chem. Soc., 2023, 145, 8082–8089 CrossRef CAS PubMed.
- M. Häußler, M. Eck, D. Rothauer and S. Mecking, Nature, 2021, 590, 423–427 CrossRef PubMed.
- W. Li, H. Q. Wang, W. T. Gao, Z. X. Wang, P. Xu, H. B. Ma and C. H. Li, CCS Chem., 2022, 4, 3781–3797 CrossRef CAS.
- X. Y. Lu, P. Xie, X. Li, T. Q. Li and J. Q. Sun, Angew. Chem., Int. Ed., 2024, e202316453 CAS.
- T. Habets, G. Seychal, M. Caliari, J.-M. Raquez, H. Sardon, B. Grignard and C. Detrembleur, J. Am. Chem. Soc., 2023, 145, 25450–25462 CrossRef CAS PubMed.
- B. Qin, S. y Liu and J. F. Xu, Angew. Chem., Int. Ed., 2023, 62, e202311856 CrossRef CAS PubMed.
- X. L. Qiu, X. L. Wang, Y. X. He, J. Y. Liang, K. Liang, B. L. Tardy, J. J. Richardson, M. Hu, H. Wu, Y. Zhang, O. J. Rojas, I. Manners and J. L. Guo, Sci. Adv., 2021, 7, eabh3482 CrossRef CAS PubMed.
- Q. Zhang, Y. X. Deng, H. X. Luo, C. Y. Shi, G. M. Geise, B. L. Feringa, H. Tian and D. H. Qu, J. Am. Chem. Soc., 2019, 141, 12804–12814 CrossRef CAS PubMed.
- Y. X. Guo, Q. Sun, F. G. Wu, Y. L. Dai and X. Y. Chen, Adv. Mater., 2021, 33, 2007356 CrossRef CAS PubMed.
- J. J. Zhou, Z. X. Lin, M. Penna, S. J. Pan, Y. Ju, S. Y. Li, Y. Y. Han, J. Q. Chen, G. Lin, J. J. Richardson, I. Yarovsky and F. Caruso, Nat. Commun., 2020, 11, 4804 CrossRef CAS PubMed.
- X. C. Lin, S. L. Li, W. X. Li, Z. H. Wang, J. Y. Zhang, B. W. Liu, T. Fu, H. B. Zhao and Y. Z. Wang, Adv. Funct. Mater., 2023, 33, 2214913 CrossRef CAS.
- J. K. Hwang, R. Walczak, M. Oschatz, N. V. Tarakina and B. V. K. J. Schmidt, Small, 2019, 15, 1901986 CrossRef PubMed.
- J. J. Zhou, M. Penna, Z. X. Lin, Y. Y. Han, R. P. M. Lafleur, Y. Qu, J. J. Richardson, I. Yarovsky, J. V. Jokerst and F. Caruso, Angew. Chem., Int. Ed., 2021, 60, 20225–20230 CrossRef CAS PubMed.
- Y. Q. Wang, M. Cao, B. W. Liu, F. R. Zeng, Q. Fu, H. B. Zhao and Y. Z. Wang, Mater. Horiz., 2024, 11, 978–987 RSC.
- B. Guo, E. Middha and B. Liu, ACS Nano, 2019, 13, 2675–2680 CrossRef CAS PubMed.
- MarketandMarkets. Polyurethane Foam Market by Type (Rigid Foam, Flexible Foam, Spray Foam), End-Use Industry Building & Construction, Bedding & Furniture, Automotive, Electronics, Footwear, Packag.
- J. Y. Zhang, B. W. Liu, Y. Z. Wang and H. B. Zhao, Acta Polym. Sin., 2022, 53(7), 842–855 CAS.
- B. Liu, Z. Westman, K. Richardson, D. Lim, A. L. Stottlemyer, T. Farmer, P. Gillis, V. Vlcek, P. Christopher and M. M. Abu-Omar, ACS Sustainable Chem. Eng., 2023, 11, 6114–6128 CrossRef CAS.
- T. Vanbergen, I. Verlent, J. De Geeter, B. Haelterman, L. Claes and D. De Vos, ChemSusChem, 2020, 13, 3835–3843 CrossRef CAS PubMed.
- H. W. He, H. Su, H. J. Yu, K. M. Du, F. Yang, Y. F. Zhu, M. Ma, Y. Q. Shi, X. J. Zhang, S. Chen and X. Wang, ACS Sustainable Chem. Eng., 2023, 11, 5515–5523 CrossRef CAS.
- C. Molero, A. de Lucas and J. F. Rodríguez, Polym. Degrad. Stab., 2009, 94, 533–539 CrossRef CAS.
- F. R. Zeng, X. F. Men, M. J. Chen, B. W. Liu, Q. W. Han, S. C. Huang, H. B. Zhao and Y. Z. Wang, Chem. Eng. J., 2023, 454, 140023 CrossRef CAS.
- Z. C. Fu, L. P. Feng, Y. Qin, X. H. Mu, X. Q. Zhong, Z. Y. Wang, T. Wang, J. N. Deng, J. F. Li and M. J. Chen, Org. Chem. Front., 2024, 11, 270–276 RSC.
- Z. C. Fu, F. Y. Bu, Z. P. Li, T. Wang, J. N. Deng, H. B. Zhao, S. C. Huang, Y. Z. Wang and M. J. Chen, Chem. Eng. J., 2024, 454, 147935 CrossRef.
- P. Li, B. Wang, Y. J. Xu, Z. M. Jiang, C. H. Dong, Y. Liu and P. Zhu, ACS Sustainable Chem. Eng., 2019, 7, 19246–19256 CrossRef CAS.
- X. L. Chen, F. R. Zeng, W. X. Li, L. Zhang, C. Deng, Y. Tan, M. J. Chen, S. C. Huang, B. W. Liu, Y. Z. Wang and H. B. Zhao, J. Mater. Sci. Technol., 2023, 162, 179–188 CrossRef CAS.
- L. Zhang, G. Q. Zheng, X. L. Chen, S. Q. Guo, F. R. Zeng, B. W. Liu, X. L. Zeng, X. S. Lan, Y. Z. Wang and H. B. Zhao, ACS Mater. Lett., 2023, 5, 2398–2407 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4mh00116h |
| This journal is © The Royal Society of Chemistry 2024 |