
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTriphenylphosphonium-modified catiomers enhance in vivo mRNA delivery through stabilized polyion complexation†
Jumpei
Norimatsu
a,
Hayato L.
Mizuno
b,
Takayoshi
Watanabe
a,
Takumi
Obara
a,
Makoto
Nakakido
ac,
Kouhei
Tsumoto
 acd,
Horacio
Cabral
*ae,
Daisuke
Kuroda
*f and
Yasutaka
Anraku
*abe
acd,
Horacio
Cabral
*ae,
Daisuke
Kuroda
*f and
Yasutaka
Anraku
*abe
aDepartment of Bioengineering, Graduate School of Engineering, The University of Tokyo, 7-3-1 Hongo, Bunkyo-ku, Tokyo 113-8656, Japan. E-mail: horacio@g.ecc.u-tokyo.ac.jp
bDepartment of Materials Science and Engineering, School of Materials and Chemical Technology, Tokyo Institute of Technology, 2-12-1 Ookayama, Meguro-ku, Tokyo 152-8550, Japan. E-mail: anraku.y.aa@m.titech.ac.jp
cDepartment of Chemistry and Biotechnology, School of Engineering, The University of Tokyo, 7-3-1 Hongo, Bunkyo-ku, Tokyo 113-8656, Japan
dThe Institute of Medical Sciences, The University of Tokyo, 4-6-1 Shirokanedai, Minato-ku, Tokyo 108-8639, Japan
eInnovation Center of NanoMedicine, Kawasaki Institute of Industrial Promotion, 3-25-14 Tonomachi, Kawasaki-ku, Kanagawa 210-0821, Japan
fResearch Center of Drug and Vaccine Development, National Institute of Infectious Diseases, 1-23-1 Toyama, Shinjuku-ku, Tokyo 162-8640, Japan. E-mail: dkuroda@niid.go.jp
First published on 10th July 2024
Abstract
Nanocarriers based on cationic materials play a central role in the success of mRNA-based therapies. Traditionally, amine-bearing lipids and polymers have been successfully employed for creating mRNA-loaded nanocarriers, though they still present challenges, such as physical and biological instability, limiting both delivery efficiency and therapeutic potential. Non-amine cations could be a promising avenue in addressing these limitations. However, such alternatives remain notably underexplored. Herein, we introduced triphenylphosphonium (TPP) as an alternative cationic moiety for mRNA delivery, leveraging its advantageous properties for nucleic acid complexation. Through the modification of amine-bearing catiomers, we replaced traditional amine-based counterparts with TPP to create innovative polymeric micelles as mRNA nanocarriers. A comprehensive analysis, encompassing physicochemical, thermodynamic, and computational approaches, revealed that the TPP substitution significantly influenced polymer self-assembly, mRNA binding, and the overall stability of mRNA-loaded polymeric micelles. Upon intravenous injection, TPP-bearing micelles demonstrated a remarkable increase in mRNA bioavailability, facilitating efficient protein production in solid tumors. These findings provide a compelling rationale for substituting amines with TPP, emphasizing their potential for advancing mRNA therapeutics.
New conceptsCurrent non-viral mRNA carriers, composed of polymers or lipids, primarily utilize amine-bearing materials for ionic complexation with mRNA. Although numerous formulations have been explored, the fundamental aspect of ionic complexation, which currently relies on amines, has been overlooked in carrier design, thereby missing significant opportunities to enhance the complex stability. We addressed this gap by substituting traditional amines with triphenylphosphonium (TPP) in catiomers, aiming to develop innovative polymeric micelles with a potent alternative cation. This simple substitution remarkably enhanced the stability of mRNA-loaded micelles, facilitating systemic mRNA delivery. The key to improved performance lies in their distinctive complexation modes. In addition to favorable ionic interaction with mRNA, TPP-bearing catiomers elicited their hydrophobic properties upon mRNA binding, efficiently expelling water molecules from mRNA and forming multivalent networks between polymer–mRNA and polymer–polymer. Consequently, mRNA can be densely packaged within a stabilized core, preserving its function under harsh physiological conditions. Computational analysis further underscored the pivotal role of each structural unit in TPP. Our concept of amine-to-TPP substitution, supported by comprehensive analysis, can be broadly applicable to existing mRNA carriers, such as lipid-based or polymer-based nanoparticles, potentially improving their repertoire and delivery efficacy to advance mRNA therapeutics. |
Introduction
Messenger RNA (mRNA) has garnered significant interest as a potent therapeutic agent for addressing various diseases.1–5 mRNA's inherent instability within the biological milieu, along with its poor cellular uptake, hampers its distribution and function at the intended target site.6,7 As such, carriers for mRNA that provide effective protection and efficient delivery are essential for its therapeutic application. Among the carriers for mRNA delivery, non-viral synthetic nanocarriers comprising polymers or lipids have demonstrated significant advantages, such as biocompatibility, flexible design, and high cargo loading capacity.8–10 However, they still face challenges including their instability in vivo, off-target distribution, and intricacy in their optimal formulation.Most of these nanocarrier formulations predominantly employ amine-based cationic molecules, such as polyamines or amino lipids,11,12 that directly interact with negatively charged phosphates of mRNA to encapsulate it within ionic complexes. However, these complexes alone have shown issues in colloidal,13 physical,14,15 and biological stability.16–18 Thus, for in vivo applications, current carriers primarily combine additional components, such as lipids,13,19 PEG,20,21 or other functional moieties,22–24 along with amine-based materials. While these multi-component systems show promise for in vivo mRNA delivery, the fundamental principle of mRNA complexation, i.e., ionic interactions with mRNA, remains underdeveloped.
Because amine-based cations are popular materials for mRNA, the alternative cations should provide a viable and straightforward approach without the intricate designs associated with multi-component systems. Moreover, the integration of these cationic alternatives into a variety of presently employed amine-based mRNA carriers could enhance their versatility and applicability. In this study, we focused on triphenylphosphonium (TPP) as an alternative cation to conventional amines for effective mRNA complexation. Phosphonium-based cations provide unique ionic properties that favor the interaction with anions, such as their charge distribution and binding force to anions, which are attributed to differences in electronegativity between phosphorus and nitrogen.25–27 Moreover, its three phenyl moieties facilitate hydrophobic interaction, potentially leading to stable mRNA complexation.28,29 Thus, substituting amines with TPP could significantly impact mRNA complexation, thereby influencing mRNA delivery efficiency.
To assess the potential of TPP-modified materials, we used polymeric micelles (PMs) as model mRNA nanocarriers. Compared to other carriers, PMs have several advantages including ease of preparation in aqueous solutions,30 deep tissue penetration due to their small size,31 evasion of the reticuloendothelial system (RES),32 and the potential for targeted delivery when paired with appropriate ligands.33,34 Nucleic acids can be encapsulated into PMs using block copolymers, like PEG-poly(L-lysine) (PEG-PLys), a classic polymer used for gene delivery.12,35,36 Such PM formulates a core–shell structure, in which the core is formed by the electrostatic interaction between lysine-derived amines and nucleic acids, while the PEG forms the outer shell inducing steric hindrance. In this study, we carefully substituted the amines in PEG-PLys with TPP at varying ratios to formulate mRNA-loaded PMs. Through in-depth analysis of TPP-bearing polymers, mRNA-loaded PMs, and the underlying polymer–mRNA interactions, our study rationalized the application of TPP as a cationic alternative in mRNA carriers. Ultimately, we showcased the practical potential of TPP-bearing PMs for in vivo mRNA delivery.
Results and discussion
Preparation of block catiomers
We first synthesized block catiomers bearing triphenylphosphonium (TPP). Poly(ethylene glycol)-poly(L-lysine) (PEG-PLys) was synthesized as previously reported.37 The polymerization degree of PLys was determined to be 68 by 1H- nuclear magnetic resonance (NMR) measurement (Fig. S1 and S2, ESI†). TPP moieties were introduced into the lysine residues of PEG-PLys via an amide coupling reaction (Fig. 1A). By changing the feeding amount of 4-(carboxybutyl)triphenylphosphonium bromide (TPP-COOH), block catiomers having different number of TPP residues [PEG-PLys(TPP)] were successfully prepared. TPP introduction ratio of each polymer was determined to be 26, 45, 75, and 97% of the total amines in the original PEG-PLys by 1H-NMR measurement (Fig. S1, ESI†). In size exclusion chromatography (SEC) analysis, each polymer exhibited a monodisperse peak and its retention time was shifted with TPP introduction, which further confirmed the successful synthesis of PEG-PLys(TPP) (Fig. S3, ESI†). Hereafter, PEG-PLys with X% of total amines substituted by TPP is denoted as PEG-PLys(X% TPP) (X = 26, 45, 75, 97).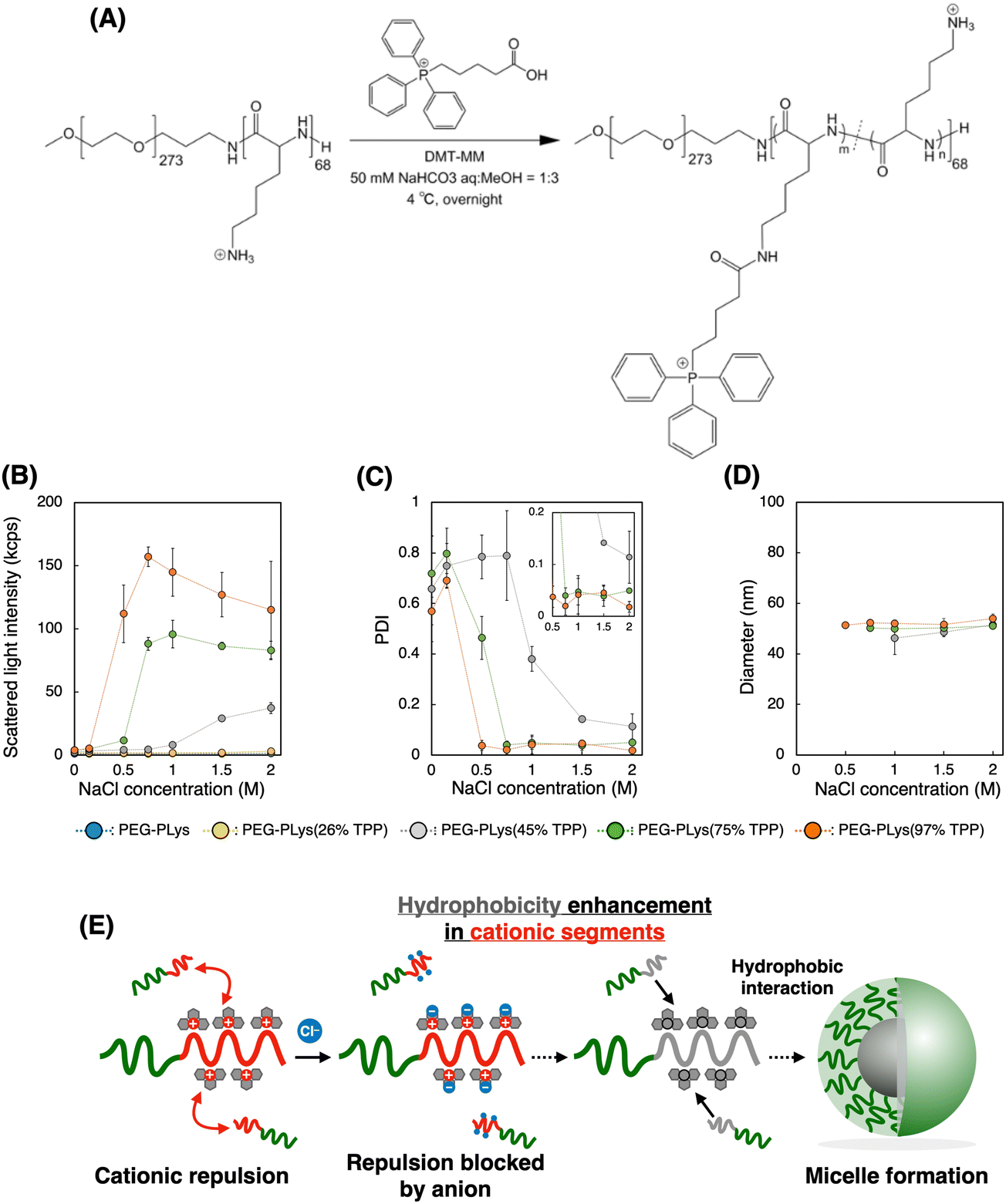 | ||
| Fig. 1 Synthesis and characterization of PEG-PLys(TPP) polymers. (A) Synthesis scheme. (B) Scattered light intensity (SLI), (C) polydispersity index (PDI), and (D) diameters of the polymers dissolved in 10 mM HEPES buffer with various NaCl concentrations were analyzed by DLS. Note that PEG-PLys and PEG-PLys(26% TPP) are not included in (C) and (D) due to their low SLI values. The diameter data presented in (D) are exclusively for polymers exhibiting SLI > 10 and PDI < 0.5, ensuring the inclusion of reliable measurement data. Data are shown as the mean ± S.D. (n = 3). (E) Proposed mechanism of PEG-PLys(TPP) self-assembly induced by NaCl. | ||
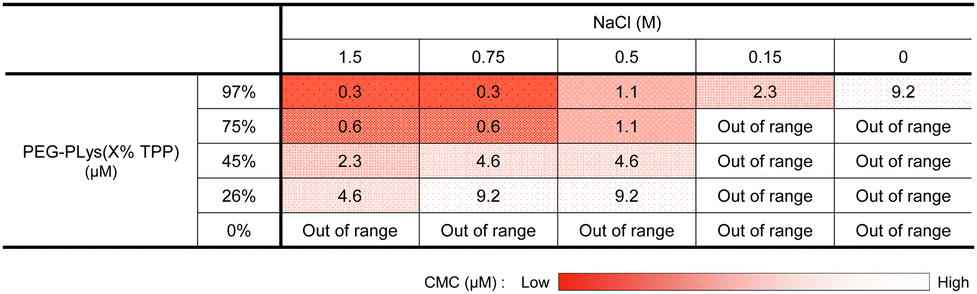
Due to its hydrophobic nature, excess TPP modification may diminish polymer solubility, hindering mRNA/PM formation in an aqueous buffer. Despite this concern, all obtained polymers, including that modified with TPP at the highest degree, PEG-PLys(97% TPP), were well-soluble in 10 mM HEPES buffer without forming aggregates or assemblies, judging from dynamic light scattering (DLS) measurements. Phosphonium-derived cationic repulsion likely prevented polymer association driven by TPP hydrophobicity. Importantly, when ionic strength in 10 mM HEPES buffer was increased with NaCl, PEG-PLys(X% TPP) (X = 45, 75, or 97) showed a gradual increase in scattered light intensity (SLI), while PEG-PLL and PEG-PLys(26% TPP) did not (Fig. 1B). The increase in SLI implies the formation of multimolecular assemblies. Indeed, PEG-PLys(X% TPP) (X = 45, 75, or 97) formed particles around 50 nm with low polydispersity index (PDI) (Fig. 1C and D). In particular, PDI of PEG-PLys(X% TPP) (X = 75 or 97) was below 0.05 (see inset Fig. 1C), suggesting highly monodisperse nanoparticles. Under such a high ionic environment, cationic repulsion is suppressed by chloride anions to allow hydrophobic interaction between polymers, resulting in self-assembly (Fig. 1E). Generally, amphiphilic block copolymers in aqueous solution can self-assemble into micelles or vesicles, depending on the PEG weight fraction (fPEG) of the block copolymer.38 When fPEG > 10%, micelle structure is favored, and our PEG-PLys(TPP) polymers most likely form this structure. Polymer concentration also affected these self-assembling behaviors (Table 1 and Fig. S4, ESI†). Increased polymer concentration enhanced the sensitivity of PEG-PLys(TPP) to NaCl, resulting in their self-assembly at lower NaCl concentrations. Under NaCl-deficient conditions, however, the polymer showed minimal self-assembly, indicating that cationic repulsion predominates over hydrophobicity even at high polymer concentrations. Collectively, NaCl and TPP synergistically lowered the critical micelle concentration (CMC) of PEG-PLys(TPP) polymers.
|
This NaCl concentration-responsive behavior highlights two novel aspects of PEG-PLys(TPP), both of which could be beneficial for mRNA encapsulation. First, the hydrophobicity of block copolymers increases with TPP introduction, while polymer dispersity in an aqueous solution can be maintained because of TPP's cationic nature. Although previous studies have incorporated hydrophobic moieties like cholesterol or alkane into polycations for stabilization,39–41 these approaches often compromise the cationic nature of the polymers, resulting in only moderate incorporation to maintain their dispersion and ability of ionic interaction with nucleic acids. In contrast, TPP contains both hydrophobic and cationic properties within a single residue, potentially overcoming this tradeoff to benefit from both stabilization effects. Second, chloride anions prompt the hydrophobic nature of PEG-PLys(TPP) polymers, driving their assembly (Fig. 1E). In other words, anionic molecules can act as “molecular glue” between PEG-PLys(TPP) polymers. Because mRNA is anionic, it may also promote the interaction between PEG-PLys(TPP) polymers, leading to stable micelle formation.
Preparation of mRNA-loaded polymeric micelles (mRNA/PMs)
We next prepared mRNA/PMs by mixing each block polycation [PEG-PLys or PEG-PLys(TPP)] with mRNA in 10 mM HEPES buffer without NaCl (Fig. 2A). mRNA encoding firefly luciferase (Fluc) was used as a model cargo (∼1.9 kb). Their mixed ratio, described as [cationic moieties in polymers]/[anionic moieties in mRNA] (C/A) ratio, was fixed at 3 to ensure complete mRNA complexation with a slight excess of cationic moieties. Our control polymer, PEG-PLys, has been reported to form compact and monodisperse mRNA complexes at this ratio,42 which was also confirmed in our study (Fig. S5, ESI†). After the formulation of mRNA/PMs by simply mixing the polymer and mRNA solutions at the predetermined ratio, their size distribution and zeta potential were analyzed by DLS and electrophoretic light scattering (ELS) measurement, respectively. All mRNA/PMs exhibited diameters within the range of 50–60 nm with low PDI (below 0.2), having slightly positive charges. (Table 2 and Fig. 2B). Transmission electron microscopy (TEM) observation confirmed their spherical core structures around 20–25 nm in diameter (Fig. 2C), where the PEG shell cannot be observed due to its lower electron density compared to the stained core.43,44 By comparing the diameters measured by DLS and TEM, we can estimate the thickness of the PEG shell, which is around 15–20 nm. Note that the staining degree within the core varied with the TPP introduction. Increased hydrophobicity likely limited the access of staining solutions to the core. Overall, these characteristics indicate the successful formation of monodisperse mRNA/PMs.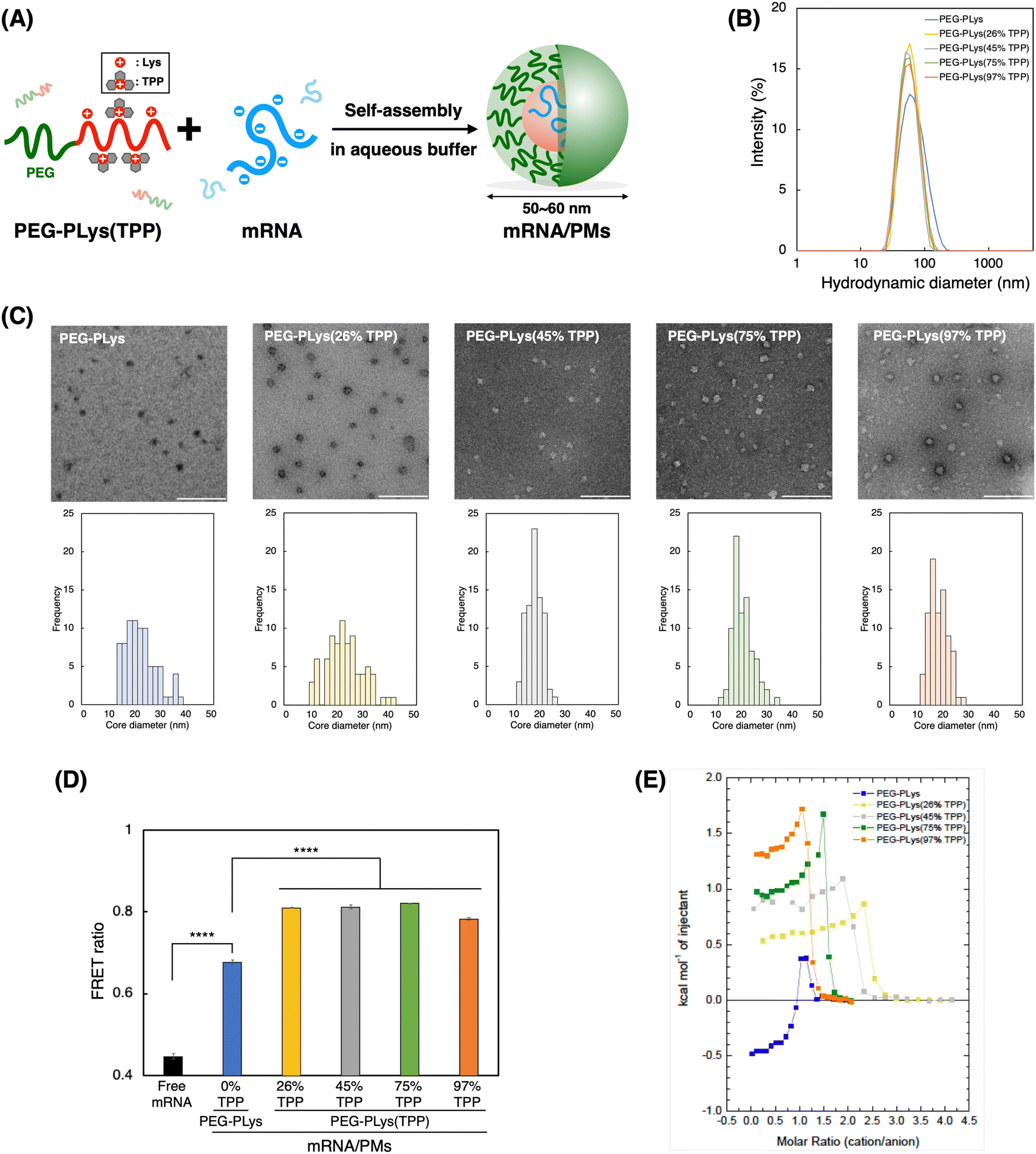 | ||
| Fig. 2 Preparation and characterization of mRNA-loaded polymeric micelles (mRNA/PMs). (A) Schematic representation of mRNA/PMs. (B) Size distribution of mRNA/PMs determined by DLS. (C) Morphology of mRNA-loaded polymeric micelles (mRNA/PMs) observed by transmission electron microscopy (TEM). The scale bar represents 200 nm. Each bottom image displays a size distribution histogram of the mRNA/PM core. (n = 80). (D) Evaluation of mRNA condensation status using fluorescence resonance energy transfer (FRET). Data are shown as the mean ± S.D. (n = 3). Statistical significance was calculated using one-way ANOVA with Bonferroni post hoc test. ****p < 0.0001. (E) Isothermal titration calorimetry (ITC) thermograms of PEG-PLys(TPP) polymers titrated into Fluc mRNA in 10 mM HEPES buffer at 25 °C. The polymer dilution heat was subtracted from each plot. | ||
| Block polycations | mRNA/PMs | |||||||
|---|---|---|---|---|---|---|---|---|
| TPP introduction (%) | DP of Lysa | DP of TPPa | Diameterb (nm) | Polydispersity indexb | Zeta potentialc (mV) | Association number of mRNAd | mRNA encapsulation efficiencye (%) | |
| a Determined by 1H-NMR. b Determined by DLS (mean ± S.D., n = 3). c Determined by ELS (mean ± S.D., n = 4). d Determined by FCS (mean ± S.D., n = 3). e Determined by Qubit RNA HS assay (mean ± S.D., n = 3). | ||||||||
| PEG-PLys | 0 | 68 | 0 | 59 ± 3 | 0.17 ± 0.01 | 3.7 ± 0.9 | 1.7 ± 0.4 | 98.0 ± 0.3 |
| PEG-PLys(26% TPP) | 26 | 50 | 18 | 56 ± 0 | 0.10 ± 0.00 | 2.8 ± 0.2 | 2.0 ± 0.2 | 97.3 ± 0.8 |
| PEG-PLys(45% TPP) | 45 | 37 | 31 | 54 ± 2 | 0.12 ± 0.01 | 2.8 ± 0.9 | 1.6 ± 0.3 | 98.4 ± 1.3 |
| PEG-PLys(75% TPP) | 75 | 17 | 51 | 54 ± 1 | 0.12 ± 0.01 | 3.3 ± 0.7 | 1.8 ± 0.5 | 99.0 ± 1.0 |
| PEG-PLys(97% TPP) | 97 | 2 | 66 | 54 ± 3 | 0.12 ± 0.01 | 3.9 ± 0.2 | 1.8 ± 0.1 | 99.6 ± 0.5 |
To gain more insight into PM structure, we evaluated mRNA condensation status in each mRNA/PM core using fluorescence resonance energy transfer (FRET).45 Cy3/Cy5 double-labeled mRNA was packaged into PMs, and FRET signal was measured as an indicator of mRNA condensation. In this assay, a stronger FRET signal suggests close localization of Cy3 and Cy5, which should be derived from condensed mRNA. As shown in Fig. 2D, all PM formulations exhibited increased FRET signal compared to free mRNA by encapsulation. Of note, PMs from PEG-PLys(TPP) showed a significantly higher FRET ratio than those from PEG-PLys despite containing a similar number of mRNA molecules within the PMs (Table 2), suggesting a more condensed status of mRNA. This FRET analysis indicates that TPP moieties facilitate tight mRNA packaging to form compact and uniform mRNA/PMs. This condensation property inspired us to investigate the encapsulation of longer mRNA into PMs. Generally, encapsulation of longer mRNA is more challenging due to its bulkiness.17 The encapsulation of VEE-GFP mRNA, which is approximately 4 times longer than Fluc mRNA,17,46 with PEG-PLys resulted in broad and large size distribution with some aggregates, whereas PEG-PLys(TPP) achieved compact packaging into uniform structures below 100 nm (Fig. S6A, ESI†). In the case of shorter EGFP mRNA (∼1 kb), both PEG-PLys and PEG-PLys(TPP) could form compact PM with diameters around 50 nm (Fig. S6B, ESI†). Collectively, PEG-PLys(TPP) can package mRNA with a large size window into the compact structure, exhibiting its potential as a versatile platform for mRNA delivery.
Thermodynamic analysis of polymer–RNA complexation
The thermodynamics of polymer binding to mRNA was explored using isothermal titration calorimetry (ITC) to elucidate the fundamental interactions governing mRNA–polymer complexation. Each polymer was titrated into mRNA to monitor their thermal exchange. In case of a simple binding interaction between two molecules, exothermic/endothermic reactions gradually decrease as titrations progress and the enthalpy change upon the titration diminishes at the point where the binding stoichiometry reaches saturation. However, our ITC analyses from all polymers displayed more complicated reactions; i.e. the titrations showed endothermic reactions with gradually increasing positive enthalpy change followed by titrations with dilution heat. (Fig. 2E and Fig. S7, ESI†). These profiles likely originated from mRNA condensation upon complex formation with the polymers, as reported in previous ITC studies involving polymers and nucleic acids.47–49 It is noteworthy that molecular stoichiometry corresponding saturation point for the endothermic reactions differ among the various polymers. Specifically, it emerged around the charge stoichiometric point for PEG-PLys and PEG-PLys(97% TPP), followed by other PEG-PLys(X% TPP) (X = 75, 45, and 26). This sequential pattern suggests that sidechain length influences mRNA interaction. Polymers with uniform sidechain structures, like PEG-PLys and PEG-PLys(97% TPP), may enable all sidechains to participate in mRNA interaction, resulting in mRNA complexation with a minimal number of cationic residues. Conversely, polymers with uneven sidechains, where larger sidechains with TPP predominantly interact with mRNA over residual amines, necessitate more polymers to cover the mRNA surface for complexation. This finding underscores the significance of sidechain uniformity in mediating their interaction with mRNA.When we focus on the initial titrations, PEG-PLys and PEG-PLys(TPP) displayed opposite thermal profiles. PEG-PLys exhibited exothermic reaction, whereas PEG-PLys(TPP) showed endothermic reaction, which became more pronounced at higher TPP introduction ratios. This disparity implies distinct modes of polymer–mRNA interaction. While enthalpy-driven electrostatic interaction may predominate in PEG-PLys, entropy-driven hydrophobic interactions involving solvent exclusion may become dominant with TPP incorporation. These interactions likely occur not only between polymer–RNA but also between polymers, given that anionic mRNA can act as glue between PEG-PLys(TPP) polymers, as evidenced by their salt-responsive self-assembly (Fig. 1E). Our ITC results suggest TPP incorporation significantly affects thermodynamic profiles responsible for polymer–mRNA complexation.
Computational analysis of polymer–RNA complexation
To explore the atomic-level interactions between polymers and RNA, we also conducted molecular dynamics (MD) simulations. Due to the complexity of simulating real experimental systems, our calculations focused on simplified model systems. Specifically, we simulated the interaction between three 10-mer cationic polymers and a 10-mer RNA molecule in the presence of explicit water molecules (Fig. S8, ESI†). Five random RNA sequences were artificially generated to ensure that the proportion of AUCG bases closely matched that found in the coding sequence (CDS) of firefly luciferase (Fluc) (Table S1, ESI†). As 10-mer cationic polymers, we included two additional polycations, denoted as BA and TPA, to bridge the structural gap between Lys and TPP (Fig. 3A). These virtual intermediates highlight the structural components contributing to the superiority of TPP over Lys. These additions include sidechain elongation, phenyl incorporation, and nitrogen replacement with phosphorus. Through sequential comparison of these four structures in molecular simulations, we aim to elucidate the individual effects of each structural difference between Lys and TPP on RNA complexation. | ||
| Fig. 3 Molecular dynamics (MD) simulations of polymer–RNA interactions. (A) Chemical structures of the polymers utilized in this study. (B) Count of atomic contacts between RNA and polymers during the final 100 ns of MD simulations for the RNA–polymer systems. (C) Radial distribution function (RDF) of water molecules with reference to each RNA. (D) Count of atomic contacts between polymers during the final 100 ns of MD simulations for the polymer-only systems. | ||
In the simulations, the cationic polymers BA, TPA, and TPP generally formed more atomic contacts with RNA compared to Lys did, especially with seq02, seq03, and seq04 (Fig. 3B). This higher propensity for interaction is likely attributed to their larger side chains, suggesting that these polymers have enhanced affinity for RNA. We also computed the radial distribution function (RDF) of water molecules relative to each RNA (Fig. 3C). As anticipated, in simulations involving polymers, the RDF of water molecules around each RNA tended to be smaller compared to simulations of RNA alone. Notably, in simulations with TPA and TPP, the RDF was even smaller at distances of around 3 Å or more from the RNA. This suggests that TPA and TPP might hinder water from interacting with RNA molecules, implying a superior RNA encapsulation capability for these polymers. This observation was further corroborated when we calculated the solvent-accessible surface area (SASA) of RNA during the simulations (Fig. S9, ESI†). Compared to Lys and BA, the smaller SASA observed with TPA and TPP indicates more effective shielding of the RNA. This may enhance the RNA's stability against nuclease attack in biological environments.50 Moreover, to evaluate potential interactions between the polymers, we conducted MD simulations of ten 10-mer polymers in the absence of RNA. The number of contacts between the polymers was more pronounced in the TPA and TPP simulations, with TPP showing even greater interactions (Fig. 3D). While the exposed side chain cations of Lys and BA would typically repel each other, the hydrophobic phenyl groups in TPA and TPP likely fostered interactions between the polymers.
Theoretically, the nitrogen in TPA is more electronegative compared to the phosphorus in TPP. This increased ability to attract electrons potentially renders TPA more polarized, leading to a decrease in hydrophobicity compared to TPP. To further elucidate this, we employed QM calculations using the Gaussian software,51 to directly compare the dipole moments of the monomeric forms of TPA and TPP (Fig. 3A). Initial structures were first geometry-optimized using the B3LYP/6-31G(d,p) level, and dipole moments were subsequently calculated from these optimized structures using the same method and basis set (Fig. S10, ESI†). As expected, the dipole moment of TPA (17.07 D) is greater than that of TPP (10.35 D), underscoring the more polarized nature of TPA. Given this data, TPP is likely superior to TPA in encapsulating RNA due to its less polarized and more hydrophobic character.
Taken together, we conducted computational analyses to gain atomic-level insight into RNA complexation by Lys-based and TPP-based polycations. Given the multiple structural differences between Lys and TPP, we included BA and TPA as virtual intermediates to better understand the structure-behavior correlation in the simulations. Our computational results suggest that: (1) sidechain elongation in BA, TPA, and TPP enhances interactions between polycations and RNA, as well as among polycations themselves, thus stabilizing RNA through more effective complexation (Fig. 3B and D); (2) the incorporation of phenyl groups in TPA and TPP reduces the presence of water molecules around RNA compared to Lys and BA, further stabilizing RNA through efficient shielding (Fig. 3C and Fig. S9, ESI†); and (3) the replacement of nitrogen with phosphorus in TPP molecules leads to more frequent interactions among TPP molecules than among TPA molecules, likely due to the more hydrophobic nature of TPP (Fig. 3D and S10, ESI†). Our molecular simulations highlight the potential of TPP-based polycations by pinpointing the pivotal roles of each structural unit within the TPP moiety. These units may synergistically facilitate robust RNA complexation within a solvent-excluded PM core, stabilized via multivalent networks between RNA–polymer and polymer–polymer.
Biological evaluation of mRNA-loaded polymeric micelles (mRNA/PMs)
We evaluated the delivery performance of our mRNA/PMs under both in vitro and in vivo biological settings. It is well-known that polyanion and nuclease, both abundant in the biological milieu, stand as major obstacles for systemic mRNA delivery in vivo by destabilizing poly-ion complex structure or by degrading mRNA, respectively.14,18 Thus, we investigated each mRNA/PM stability against these components.To assess the stability of mRNA/PM against polyanions, we used dextran sulfate (DS) as a model polyanion, and the structural integrity of mRNA/PMs against DS was monitored utilizing FRET signal from Cy3/Cy5 double-labeled mRNA (Fig. 4A and Fig. S11, ESI†). After incubation with DS, the FRET ratio of mRNA/PMs formed by PEG-PLys decreased dramatically to reach the same value as free mRNA, indicating complete mRNA release from PMs due to polyanion exchange. mRNA/PMs formed of PEG-PLys(TPP), on the other hand, maintained higher FRET ratios than free mRNA, although the ratio gradually decreased with DS addition. While the gradual decrease in FRET ratio could infer the loosening of the PM core, the TPP introduction-rate dependent increase in FRET ratio directly indicates TPP's function in maintaining the complex core structure even under the coexistence of polyanions. This protective effect of TPP can be explained as a product of the combinatory electrostatic and hydrophobic interactions, which have made the mRNA/PMs enriched with TPP moieties less susceptible to polyanion exchange, resulting in improved structural integrity. Subsequently, nuclease tolerability was evaluated by incubating mRNA/PMs in 50% serum, followed by remaining mRNA quantification (Fig. 4B). Tolerability increased with TPP introduction and the highest stability was achieved by PEG-PLys(97% TPP). TPP-induced compact PM core, involving efficient solvent exclusion as indicated by ITC and MD simulations (Fig. 2E and 3C), likely prevents nuclease attack toward mRNA.
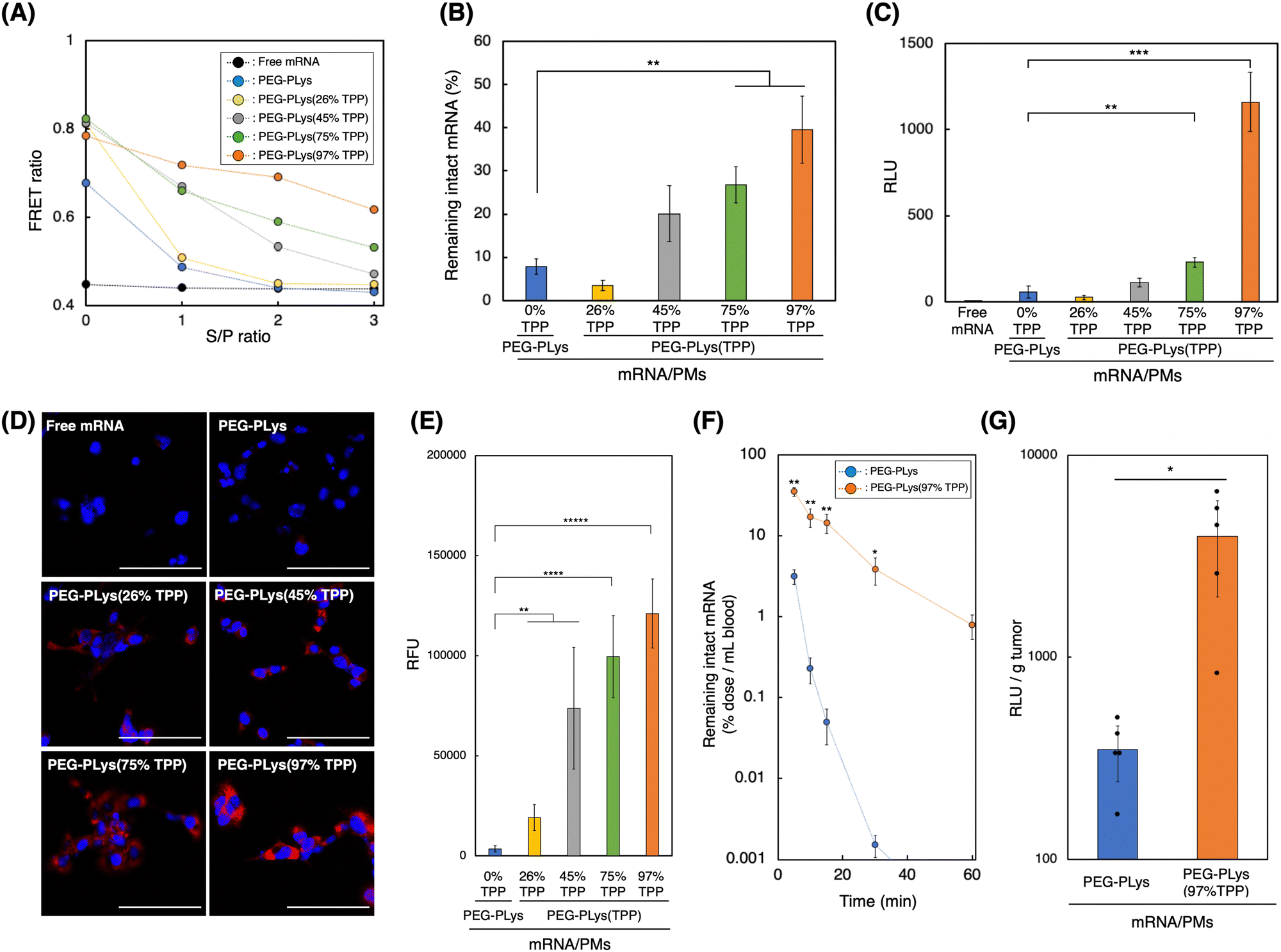 | ||
| Fig. 4 In vitro and in vivo performance of mRNA-loaded polymeric micelles (mRNA/PMs). (A) Tolerability against polyanion exchange reaction. Cy3/Cy5 double-labeled mRNA was packaged into PMs, followed by incubation with dextran sulfate (DS) at various S/P (sulfate groups of DS/Phosphate groups of mRNA) ratio. Structural integrity of mRNA/PMs was analyzed using fluorescence resonance energy transfer (FRET). Data are shown as the mean ± S.D. (n = 3). (B) Tolerability against nuclease. mRNA/PMs were incubated with 50% FBS and the remaining mRNA was quantified using qRT-PCR. Data are shown as the mean ± S.D. (n = 4). (C) Fluc expression in CT26 cells. After 24 h incubation, Fluc protein levels in cell lysate were quantified. Data are shown as the mean ± S.D. (n = 4). (D) Cellular uptake of mRNA by cultured cells. After incubation with CT26 cells for 24 h, internalized Cy5-labeled mRNA was observed by fluorescence microscopy. Blue: nucleus. Red: Cy5-labeled mRNA. The scale bar represents 100 μm. (E) Quanitification of internalized Cy5-labeled mRNA. Data are shown as the mean ± S.D. (n = 5 images) **p < 0.01, ***p < 0.001, ****p < 0.0001 and *****p < 0.00001, determined by one-way ANOVA with Bonferroni post hoc test. (F) Blood circulation profiles of mRNA/PMs after intravenous injection into BALB/c mice. Intact Fluc mRNA remaining in the bloodstream was quantified by qRT-PCR. Data are shown as the mean ± standard error of the mean (s.e.m.) (n = 4). (G) Fluc expression in the tumor. Fluc protein levels were measured 24 h after intravenous injection into tumor-bearing mice. Data are shown as the mean ± standard error of the mean (s.e.m.) (n = 5). *p < 0.05, **p < 0.01, determined by two-tailed Student's t-test. | ||
Before applying our mRNA/PMs to cultured cells, polymer cytotoxicity was evaluated (Fig. S12, ESI†). While all polymers exhibited dose-dependent cytotoxicity, likely due to their cationic nature, TPP incorporation did not enhance, or rather suppressed, the cytotoxicity of the original PEG-PLys, suggesting their biocompatibility. Given the tendency of TPP-bearing polymers to self-assemble in the presence of anionic molecules (Fig. 1E), their cationic moieties might be partially shielded under culture conditions, potentially mitigating the cytotoxicity. We then incubated mRNA/PMs with cultured cells. After 24 hours of incubation, enhanced Fluc expression was observed for mRNA/PMs with PEG-PLys(TPP). In particular, mRNA/PMs with PEG-PLys(97% TPP) exhibited maximum expression, which was approximately 20-fold higher compared to mRNA/PMs with PEG-PLys (Fig. 4C). Enhanced protein expression was consistently observed across multiple cell types, highlighting the versatile potential of TPP-bearing mRNA/PMs (Fig. S13, ESI†). Stabilized mRNA/PMs could deliver more mRNA into cells without being degraded within the supernatant (Fig. 4D and E), which may have led to such high protein expression. As a positive control, we also evaluated the protein expression efficiency of polyethylenimines (PEIs), widely used commercial transfection reagents.52 Given the structural variety of PEIs, we assessed two types: high-molecular-mass linear PEI (jetPEI) and low-molecular-mass branched PEI (bPEI). Both PEIs demonstrated significantly higher protein expression than TPP-bearing mRNA/PMs (Fig. S14, ESI†), with jetPEI showing even higher expression than bPEI, suggesting the impact of PEI configurations.53,54 The highly cationic charge of PEIs may promote cellular uptake via direct ionic interaction with cellular membranes. However, both PEIs also displayed higher cytotoxicity, likely due to cell membrane disruption by the cationic charges, posing a significant concern for in vivo application (Fig. S12, ESI†). Overall, mRNA/PMs and PEIs demonstrated different toxicity-efficiency profiles.
The TPP-bearing PMs achieved functional mRNA delivery without obvious cellular toxicities. However, it should be noted that the incorporation of exogenous mRNA or carrier materials may cause intracellular responses at the molecular level. For example, exogenous mRNA may affect the endogenous protein synthesis machinery through immune stimulation or ribosomal occupation.55,56 Also, given the role of biogenic polycations in cellular RNA function,57 synthetic polycations may also electrostatically interact with endogenous RNAs, potentially affecting their activities. These points are a major concern in nanocarrier design and remain largely unexplored.
Together, mRNA/PMs with TPP moieties improved stability against biological components, as well as protein expression in cells. 97% TPP introduction into PEG-PLys was determined to be the optimal ratio. Thus, for the following experiments, we focused on mRNA/PMs with PEG-PLys(97% TPP) to elucidate the beneficial effect of TPP moieties on systemic mRNA delivery in vivo.
mRNA/PMs were intravenously injected into mice to quantify intact mRNA remaining in the bloodstream. Although PMs with PEG-PLys could improve the stability of naked mRNA in vivo (Fig. S15, ESI†), their mRNA underwent rapid degradation with time, suggesting insufficient stability for systemic application. In contrast, PEG-PLys(97% TPP) efficiently protected mRNA to demonstrate a much longer circulation profile (Fig. 4F). This prolonged circulation in blood can be attributed to the following factors. First, PEG-PLys(97% TPP) formed stable multivalent networks between polymer–mRNA and polymer–polymer, enhancing structural integrity against polyanion exchange with biomacromolecules and reducing disassembly in the bloodstream (Fig. 4A). Second, PEG-PLys(97% TPP) provided tight mRNA condensation and solvent exclusion, possibly limiting RNase access to encapsulated mRNA and improving its stability (Fig. 4B). Overall, enhanced stability against polyanions and RNase likely contributed to the prolonged mRNA circulation in blood.
Finally, protein expression efficiency was evaluated. Because solid tumor has leaky vasculatures,31,58 we hypothesized that our developed mRNA/PMs could deliver intact mRNA into the tumor efficiently due to their size below 100 nm and prolonged circulation profile. To test this hypothesis, mRNA/PMs were intravenously injected into tumor-bearing mice, and protein expression in the tumor was analyzed at 24 h post-injection. As shown in Fig. 4G, mRNA/PMs with PEG-PLys(97% TPP) achieved higher Fluc expression in the tumor by an order of magnitude than mRNA/PMs with PEG-PLys. Biodistribution analysis revealed that PEG-PLys(97% TPP) dramatically increased the intact mRNA distribution across tissues (Fig. S16, ESI†), which likely contributed to the efficient protein production in the tumor. Furthermore, the in vivo safety of mRNA/PMs was assessed by monitoring several biomarkers in plasma after systemic injection. No significant toxicity was observed with mRNA/PMs containing either PEG-PLys or PEG-PLys(97% TPP) (Fig. S17, ESI†). These results highlight the great potential of TPP-bearing PMs for systemic mRNA delivery in terms of both efficiency and safety.
Conclusions
Current non-viral mRNA carriers commonly rely on amine-based materials for mRNA encapsulation, yet the potential of alternative cations remains underexplored. This study investigated the effect of amine substitution with TPP using PM-based nanocarriers. TPP-incorporated polymers exhibited salt-responsive self-assembling properties, a result of a delicate balance between the hydrophobic and cationic properties of TPP, likely beneficial for stable mRNA complexation. These polymers could tightly package mRNA within the PM core. Thermodynamic and computational analysis of mRNA–polymer interactions revealed that TPP residues could preferentially interact with mRNA and polymers themselves, expelling water molecules from mRNA through an entropy-driven process. Such PMs can effectively protect mRNA within physically and biologically stabilized complexes. In addition to the physical and biological stabilities, storage stability is another critical aspect of mRNA carrier development.59 The optimal conditions for the long-term storage of these mRNA/PMs are under investigation in our lab.Our molecular simulations effectively discerned subtle differences in the polycations' side chains, illustrating the potential value of rational design and virtual screening in selecting polycations for forming PMs. This approach could offer new directions for future polymer discovery.
Finally, upon systemic administration, TPP-bearing PMs significantly increased mRNA bioavailability, enhancing protein expression in solid tumors. Our results validate the amine substitution with TPP in mRNA carriers and demonstrate their substantial potential for in vivo mRNA delivery. Given that PMs can be targeted to specific tissues by attaching ligands, TPP-bearing PMs might serve as a robust platform for mRNA delivery across various tissues. Beyond PMs, our simple yet powerful strategy of amine-to-TPP substitution may be broadly applied to diverse amine-based carriers, potentially facilitating stable mRNA complexation in a well-separated phase from the biological milieu without relying on additional materials.
Author contributions
J. N. performed the experiments and wrote the manuscript. H. L. M. assisted with the experiments and edited the manuscript. T. W. and T. O. aided in interpreting and validating the results. M. N. and K. T. supervised the thermodynamic analysis. D. K. conceived and performed the computational studies and edited the manuscript. H. C. and Y. A. conceived the study, supervised the project, and edited the manuscript.Conflicts of interest
Y. A. is a scientific advisor of Braizon Therapeutics, Inc. H. C. is a co-founder of Red Arrow Therapeutics, Inc. Y. A. and H. C. received funding support from Daiichi Sankyo Co., Ltd. for this project. The remaining authors declare no competing financial interests.Acknowledgements
This work was supported by JSPS KAKENHI Grant Numbers JP23H00545 (to Y. A.), JP21H05090 (to Y. A.), JP23K18558 (to Y. A.), JP20H04525 (to Y. A.), JP21K18310 (to D. K. and Y. A.), JP23KJ0601 (to J. N.), JP22KJ3168 (to H. L. M.). This work was also supported by JST FOREST Program (JPMJFR2138 to Y. A.) and JST-Mirai Program (JPMJMI21G6 to Y. A.) from the Japan Science and Technology Agency (JST) and the Research on Development of New Drugs (21483716 to H. C. and Y. A.) from Japan Agency for Medical Research and Development (AMED). This work was also supported by Daiichi Sankyo Co., Ltd. The supercomputing resources in this study were provided in part by the Human Genome Center at the Institute of Medical Science, The University of Tokyo, Japan and by Research Center for Computational Science, Okazaki, Japan (Project: 23-IMS-C078). Table of Contents figure was partially created with https://Biorender.com. The authors are grateful to Ms Y. Takemori at the University of Tokyo for technical assistance.References
- R. R. Goel, M. M. Painter, S. A. Apostolidis, D. Mathew, W. Meng, A. M. Rosenfeld, K. A. Lundgreen, A. Reynaldi, D. S. Khoury, A. Pattekar, S. Gouma, L. Kuri-Cervantes, P. Hicks, S. Dysinger, A. Hicks, H. Sharma, S. Herring, S. Korte, A. E. Baxter, D. A. Oldridge, J. R. Giles, M. E. Weirick, C. M. McAllister, M. Awofolaju, N. Tanenbaum, E. M. Drapeau, J. Dougherty, S. Long, K. D'Andrea, J. T. Hamilton, M. McLaughlin, J. C. Williams, S. Adamski, O. Kuthuru, U. P. C. P. U. dagger, I. Frank, M. R. Betts, L. A. Vella, A. Grifoni, D. Weiskopf, A. Sette, S. E. Hensley, M. P. Davenport, P. Bates, E. T. Luning Prak, A. R. Greenplate and E. J. Wherry, Science, 2021, 374, abm0829 CrossRef.
- L. A. Rojas, Z. Sethna, K. C. Soares, C. Olcese, N. Pang, E. Patterson, J. Lihm, N. Ceglia, P. Guasp, A. Chu, R. Yu, A. K. Chandra, T. Waters, J. Ruan, M. Amisaki, A. Zebboudj, Z. Odgerel, G. Payne, E. Derhovanessian, F. Muller, I. Rhee, M. Yadav, A. Dobrin, M. Sadelain, M. Luksza, N. Cohen, L. Tang, O. Basturk, M. Gonen, S. Katz, R. K. Do, A. S. Epstein, P. Momtaz, W. Park, R. Sugarman, A. M. Varghese, E. Won, A. Desai, A. C. Wei, M. I. D'Angelica, T. P. Kingham, I. Mellman, T. Merghoub, J. D. Wolchok, U. Sahin, O. Tureci, B. D. Greenbaum, W. R. Jarnagin, J. Drebin, E. M. O'Reilly and V. P. Balachandran, Nature, 2023, 618, 144–150 CrossRef CAS.
- S. Qin, X. Tang, Y. Chen, K. Chen, N. Fan, W. Xiao, Q. Zheng, G. Li, Y. Teng, M. Wu and X. Song, Signal Transduction Targeted Ther., 2022, 7, 166 CrossRef CAS.
- C. E. Deal, A. Carfi and O. J. Plante, Vaccines, 2021, 9, 108 CrossRef CAS.
- N. Pardi, M. J. Hogan, F. W. Porter and D. Weissman, Nat. Rev. Drug Discovery, 2018, 17, 261–279 CrossRef CAS.
- H. K. Wayment-Steele, D. S. Kim, C. A. Choe, J. J. Nicol, R. Wellington-Oguri, A. M. Watkins, R. A. Parra Sperberg, P. S. Huang, E. Participants and R. Das, Nucleic Acids Res., 2021, 49, 10604–10617 CrossRef CAS.
- N. B. Tsui, E. K. Ng and Y. M. Lo, Clin. Chem., 2002, 48, 1647–1653 CrossRef CAS.
- K. A. Hajj and K. A. Whitehead, Nat. Rev. Mater., 2017, 2, 17056 CrossRef CAS.
- P. S. Kowalski, A. Rudra, L. Miao and D. G. Anderson, Mol. Ther., 2019, 27, 710–728 CrossRef CAS PubMed.
- S. Uchida, F. Perche, C. Pichon and H. Cabral, Mol. Pharmaceutics, 2020, 17, 3654–3684 CrossRef CAS.
- X. Hou, T. Zaks, R. Langer and Y. Dong, Nat. Rev. Mater., 2021, 6, 1078–1094 CrossRef CAS PubMed.
- W. Yang, L. Mixich, E. Boonstra and H. Cabral, Adv. Healthcare Mater., 2023, 12, 2202688 CrossRef CAS PubMed.
- X. Cheng and R. J. Lee, Adv. Drug Delivery Rev., 2016, 99, 129–137 CrossRef CAS PubMed.
- G. M. Poon and J. Gariepy, Biochem. Soc. Trans., 2007, 35, 788–793 CrossRef CAS PubMed.
- J. E. Zuckerman, C. H. Choi, H. Han and M. E. Davis, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 3137–3142 CrossRef CAS.
- G. Kartha, J. Bello and D. Harker, Nature, 1967, 213, 862–865 CrossRef CAS PubMed.
- K. Koji, N. Yoshinaga, Y. Mochida, T. Hong, T. Miyazaki, K. Kataoka, K. Osada, H. Cabral and S. Uchida, Biomaterials, 2020, 261, 120332 CrossRef CAS.
- A. Yen, Y. Cheng, M. Sylvestre, H. H. Gustafson, S. Puri and S. H. Pun, Mol. Pharmaceutics, 2018, 15, 2268–2276 CrossRef CAS PubMed.
- S. Liu, X. Wang, X. Yu, Q. Cheng, L. T. Johnson, S. Chatterjee, D. Zhang, S. M. Lee, Y. Sun, T.-C. Lin, J. L. Liu and D. J. Siegwart, J. Am. Chem. Soc., 2021, 143, 21321–21330 CrossRef CAS PubMed.
- T. Nomoto, Y. Matsumoto, K. Miyata, M. Oba, S. Fukushima, N. Nishiyama, T. Yamasoba and K. Kataoka, J. Controlled Release, 2011, 151, 104–109 CrossRef CAS PubMed.
- M. Suzuki, Y. Mochida, M. Hori, A. Hayashi, K. Toh, T. A. Tockary, X. Liu, V. Marx, H. Yokoo, K. Miyata, M. Oba and S. Uchida, Small Sci., 2024, 2300258, DOI:10.1002/smsc.202300258.
- W. Yang, P. Chen, E. Boonstra, T. Hong and H. Cabral, Pharmaceutics, 2022, 14, 1205 CrossRef CAS PubMed.
- N. Yoshinaga, S. Uchida, A. Dirisala, M. Naito, K. Koji, K. Osada, H. Cabral and K. Kataoka, Adv. Healthcare Mater., 2022, 11, 2102016 CrossRef CAS.
- H. J. Kim, S. Ogura, T. Otabe, R. Kamegawa, M. Sato, K. Kataoka and K. Miyata, ACS Cent. Sci., 2019, 5, 1866–1875 CrossRef CAS PubMed.
- C. Ornelas-Megiatto, P. R. Wich and J. M. J. Fréchet, J. Am. Chem. Soc., 2012, 134, 1902–1905 CrossRef CAS PubMed.
- S. T. Hemp, M. H. Allen, Jr., M. D. Green and T. E. Long, Biomacromolecules, 2012, 13, 231–238 CrossRef CAS.
- S.-W. Wang, W. Liu and R. H. Colby, Chem. Mater., 2011, 23, 1862–1873 CrossRef CAS.
- N. Yoshinaga, S. Uchida, M. Naito, K. Osada, H. Cabral and K. Kataoka, Biomaterials, 2019, 197, 255–267 CrossRef CAS PubMed.
- Q. Chen, R. Qi, X. Chen, X. Yang, S. Wu, H. Xiao and W. Dong, Mol. Ther., 2017, 25, 92–101 CrossRef CAS.
- S. Kotta, H. M. Aldawsari, S. M. Badr-Eldin, A. B. Nair and K. Yt, Pharmaceutics, 2022, 14, 1636 CrossRef CAS PubMed.
- H. Cabral, Y. Matsumoto, K. Mizuno, Q. Chen, M. Murakami, M. Kimura, Y. Terada, M. R. Kano, K. Miyazono, M. Uesaka, N. Nishiyama and K. Kataoka, Nat. Nanotechnol., 2011, 6, 815–823 CrossRef CAS PubMed.
- H. Cabral, J. Li, K. Miyata and K. Kataoka, Nat. Rev. Bioeng., 2024, 2, 214–232 CrossRef.
- P. Mi, H. Cabral and K. Kataoka, Adv. Mater., 2020, 32, 1902604 CrossRef CAS.
- Y. Anraku, H. Kuwahara, Y. Fukusato, A. Mizoguchi, T. Ishii, K. Nitta, Y. Matsumoto, K. Toh, K. Miyata, S. Uchida, K. Nishina, K. Osada, K. Itaka, N. Nishiyama, H. Mizusawa, T. Yamasoba, T. Yokota and K. Kataoka, Nat. Commun., 2017, 8, 1001 CrossRef CAS.
- H. S. Min, H. J. Kim, M. Naito, S. Ogura, K. Toh, K. Hayashi, B. S. Kim, S. Fukushima, Y. Anraku, K. Miyata and K. Kataoka, Angew. Chem. Int. Ed., 2020, 59, 8173–8180 CrossRef CAS PubMed.
- P. B. D. Michael, W. Konstan, J. S. Wagener, K. A. Hilliard, R. C. Stern, L. J. H. Milgram, T. H. Kowalczyk, S. L. Hyatt, T. L. Fink, C. R. Gedeon, S. M. Oette, J. M. Payne, O. Muhammad, A. G. Ziady, R. C. Moen and Dr. M. J. Cooper, Hum. Gene Ther., 2004, 15, 1255–1269 CrossRef PubMed.
- K. Kataoka, H. Togawa, A. Harada, K. Yasugi, T. Matsumoto and S. Katayose, Macromolecules, 1996, 29, 8556–8557 CrossRef CAS.
- S. Chuanoi, A. Kishimura, W. F. Dong, Y. Anraku, Y. Yamasaki and K. Kataoka, Polym. J., 2014, 46, 130–135 CrossRef CAS.
- S. Uchida, H. Kinoh, T. Ishii, A. Matsui, T. A. Tockary, K. M. Takeda, H. Uchida, K. Osada, K. Itaka and K. Kataoka, Biomaterials, 2016, 82, 221–228 CrossRef CAS.
- C. Sun, T. Tang and H. Uludag, Biomaterials, 2013, 34, 2822–2833 CrossRef CAS.
- W. W. Shen, H. Wang, Y. Ling-Hu, J. Lv, H. Chang and Y. Y. Cheng, J. Mater. Chem. B, 2016, 4, 6468–6474 RSC.
- T. Miyazaki, S. Uchida, S. Nagatoishi, K. Koji, T. Hong, S. Fukushima, K. Tsumoto, K. Ishihara, K. Kataoka and H. Cabral, Adv. Healthcare Mater., 2020, 9, 2000538 CrossRef CAS.
- Y. Mochida, H. Cabral, Y. Miura, F. Albertini, S. Fukushima, K. Osada, N. Nishiyama and K. Kataoka, ACS Nano, 2014, 8, 6724–6738 CrossRef CAS.
- P. Chen, W. Yang, K. Nagaoka, G. L. Huang, T. Miyazaki, T. Hong, S. Li, K. Igarashi, K. Takeda, K. Kakimi, K. Kataoka and H. Cabral, Adv. Sci., 2023, 10, e2205139 CrossRef.
- A. Dirisala, S. Uchida, T. A. Tockary, N. Yoshinaga, J. J. Li, S. Osawa, L. Gorantla, S. Fukushima, K. Osada and K. Kataoka, J. Drug Targeting, 2019, 27, 670–680 CrossRef CAS.
- N. Yoshioka, E. Gros, H. R. Li, S. Kumar, D. C. Deacon, C. Maron, A. R. Muotri, N. C. Chi, X. D. Fu, B. D. Yu and S. F. Dowdy, Cell Stem Cell, 2013, 13, 246–254 CrossRef CAS.
- O. Samsonova, S. Glinca, A. Biela, C. Pfeiffer, E. Dayyoub, D. Sahin, G. Klebe and T. Kissel, Acta Biomater., 2013, 9, 4994–5002 CrossRef CAS.
- L. E. Prevette, M. L. Lynch and T. M. Reineke, Biomacromolecules, 2010, 11, 326–332 CrossRef CAS PubMed.
- W. Kim, Y. Yamasaki, W.-D. Jang and K. Kataoka, Biomacromolecules, 2010, 11, 1180–1186 CrossRef CAS.
- P. Heller, J. Zhou, B. Weber, D. Hobernik, M. Bros, F. Schmid and M. Barz, Small, 2017, 13, 1603694 CrossRef PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision C.02, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- P. Huang, H. Deng, Y. Zhou and X. Chen, Matter, 2022, 5, 1670–1699 CrossRef CAS.
- W. T. Godbey, K. K. Wu and A. G. Mikos, J. Biomed. Mater. Res., 1999, 45, 268–275 CrossRef CAS.
- L. Wightman, R. Kircheis, V. Rössler, S. Carotta, R. Ruzicka, M. Kursa and E. Wagner, J. Gene Med., 2001, 3, 362–372 CrossRef CAS.
- B. R. Anderson, H. Muramatsu, S. R. Nallagatla, P. C. Bevilacqua, L. H. Sansing, D. Weissman and K. Karikó, Nucleic Acids Res., 2010, 38, 5884–5892 CrossRef CAS.
- R. Katz, E. Attias, T. Tuller and M. Margaliot, J. R. Soc., Interface, 2022, 19, 20220535 CrossRef CAS.
- H. L. Lightfoot and J. Hall, Nucleic Acids Res., 2014, 42, 11275–11290 CrossRef CAS PubMed.
- Y. Matsumura and H. Maeda, Cancer Res., 1986, 46, 6387–6392 CAS.
- Y. Cao, Z. He, Q. Chen, X. He, L. Su, W. Yu, M. Zhang, H. Yang, X. Huang and J. Li, Nano Lett., 2022, 22, 6580–6589 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4mh00325j |
| This journal is © The Royal Society of Chemistry 2024 |