
Optimizing the energy level alignment for achieving record-breaking efficiency in hot exciton deep red OLEDs†
Yujie
Wu‡
abc,
Jiasen
Zhang‡
ab,
Deli
Li‡
d,
Songyu
Du
ab,
Xilin
Mu
ab,
Chunyu
Liu
ab,
Kaibo
Fang
abc,
Tingting
Feng
ab,
Tao
Wang
c,
Wei
Li
 *ab and
Ziyi
Ge
*ab
*ab and
Ziyi
Ge
*ab
aZhejiang Provincial Engineering Research Center of Energy Optoelectronic Materials and Devices, Ningbo Institute of Materials Technology and Engineering, Chinese Academy of Sciences, Ningbo, 315201, P. R. China. E-mail: liwei1987@nimte.ac.cn; geziyi@nimte.ac.cn
bCenter of Materials Science and Optoelectronics Engineering, University of Chinese Academy of Sciences, P. R. China
cSchool of Materials Science and Engineering Zhejiang Sci-Tech University, Hangzhou 310018, P. R. China
dInstitute for Smart Materials & Engineering, University of Jinan, No. 336 Nanxin Zhuang West Road, Jinan 250022, P. R. China
First published on 2nd June 2024
Abstract
To effectively compete with the quenching process in long-wavelength regions like deep red (DR) and near-infrared (NIR), rapid radiative decay is urgently needed to address the challenges posed by the “energy gap law”. Herein, we confirmed that it is crucial for hot exciton emitters to attain a narrow energy gap (ΔES1–T2) between the lowest singlet excited (S1) state and second triplet excited (T2) state, while ensuring that T2 slightly exceeds S1 in the energy level. Two proofs-of-concept of hot exciton DR emitters, namely αT-IPD and βT-IPD, were successfully designed and synthesized by coupling electron-acceptors N,N-diphenylnaphthalen-2-amine (αTPA) and N,N-diphenylnaphthalen-1-amine (βTPA) with an electron-withdrawing unit 5-(4-(tert-butyl) phenyl)-5H-pyrazino[2,3-b]indole-2,3-dicarbonitrile (IPD). Both emitters exhibited a narrow ΔES1–T2, with T2 being slightly higher than S1. Additionally, both emitters showed significantly large ΔET2–T1. Moreover, due to their aggregation-induced emission characteristics, J-aggregated packing modes, moderate strength intermolecular CN⋯H–C and C–H⋯π interactions, and unique, comparatively large center-to-center distances among trimers in the crystalline state, both αT-IPD and βT-IPD emitters exhibited remarkable photoluminescence quantum yields of 68.5% and 73.5%, respectively, in non-doped films. Remarkably, the corresponding non-doped DR-OLED based on βT-IPD achieved a maximum external quantum efficiency of 15.5% at an emission peak wavelength of 667 nm, representing the highest reported value for hot exciton DR-OLEDs.
New conceptsThe efficient development of deep-red (DR) and near-infrared (NIR) emitters encounters considerable obstacles due to their emission efficiency being relatively lower compared to those of visible light emitters, despite their immense potential for various applications. One major quenching mechanism is known as the “energy gap law”, and this nonradiative process experiences significant enhancement in the DR/NIR emitters since they possess smaller emission gaps compared to visible emitters. Fortunately, the “hot exciton” process provides a promising solution to address the challenges. Herein, we confirmed that it is crucial for hot exciton emitters to attain a narrow energy gap (ΔES1–T2) between the lowest singlet excited (S1) state and second triplet excited (T2) state, while ensuring that T2 slightly exceeds S1 in the energy level. Two proofs-of-concept of hot exciton DR emitters exhibited a narrow ΔES1–T2, with T2 being slightly higher than S1. Additionally, both emitters showed significantly large ΔET2–T1. Moreover, due to the aggregation-induced emission characteristics, J-aggregated packing modes, moderate strength intermolecular CN⋯H–C and C–H⋯π interactions, and unique comparatively large center-to-center distances among trimers in the crystalline state, both target emitters exhibited remarkable photoluminescence quantum yields in non-doped films. Remarkably, the corresponding non-doped DR-OLED achieved a maximum external quantum efficiency of 15.5% at an emission peak wavelength of 667 nm, representing the highest reported value for hot exciton DR-OLEDs. |
Introduction
Organic deep-red (DR) and near-infrared (NIR) light-emitting materials are essential for applications of photodynamic therapy, night vision technologies, optical signal processing, bioimaging, etc.1–5 However, the efficient development of DR/NIR emitters encounters considerable obstacles due to their emission efficiency being relatively lower compared to those of visible light emitters, despite their immense potential for various applications. One major quenching mechanism is known as the “energy gap law”,6–8 and this nonradiative process experiences significant enhancement in the DR/NIR emitters since they possess smaller emission gaps compared to visible emitters.Two feasible approaches for suppressing the quenching process and increasing the efficiency of DR/NIR emitters can be formed and described as follows: one approach is to minimize vibrational overlap as much as possible.9 To achieve this, deuterated or fluorinated methods are used to reduce high-frequency vibrations from C–H, O–H, and N–H stretching. Nevertheless, despite these techniques preserving a multitude of additional modes of vibration and their combination, the resulting improvements are only partial or even insignificant. The second approach involves the creation of a shallow or repulsive potential energy surface (PES) in the ground (S0) state, which significantly reduces quenching caused by high-frequency vibrations.10 This type of PES can be formed in excimers/excited oligomers, where interactions between mono-molecules are minimal or absent in the S0 state, and excimers/excited oligomers can form in excited states. Due to the repulsive-like or shallow nature of the PES in the S0 state, the emission of excimers is less affected by vibrational quenching. However, such excimers have only been reported and utilized in Pt-containing complexes with square-planar structures and are challenging to create in typical luminous materials.6–8 Therefore, effectively harnessing triplet excitons in pure DR/NIR emitters while reducing nonradiative decay remains a formidable challenge that requires further investigation.
Fortunately, the “hot exciton” process provides a promising solution by enhancing the rate of radiative decay, effectively competing with the quenching process. Previous studies have demonstrated the presence of high-energy reverse intersystem crossing (hRISC) processes in commonly utilized fluorophores, including isoquinoline, quinoline, naphthalene, anthracene, and their derivatives, as well as tetraphenyl-porphyrin.11–13 In 2012, Ma et al. ingeniously employed this photophysical phenomenon to design organic light-emitting materials and subsequently synthesized a series of novel organic electroluminescent (EL) materials with various colors.14–16 they named this innovative luminescent mechanism the “hot exciton” mechanism. Novel emitters based on this principle are known as hot exciton materials. By implementing efficient hRISC mechanisms, OLEDs based on hot exciton materials achieved a significant enhancement in the utilization efficiency of excitons (EUE).
Firstly, the breakthrough of hot excitons emitters-based OLEDs in overcoming the spin-statistical limit of fluorescent materials relies on the hRISC from high-lying triplet states (Tn, n ≥ 2) to singlet states (Sm, m ≥ 1). Secondly, to attain a high EUE, it is crucial to ensure that the high-energy hRISC rate (khRISC) from Tn to Sm significantly exceeds the rate of internal conversion (kIC) from Tn to T1. According to Fermi's golden rule,17,18
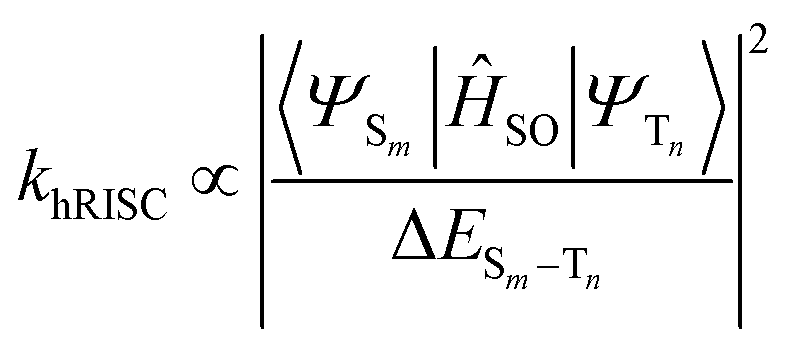
where
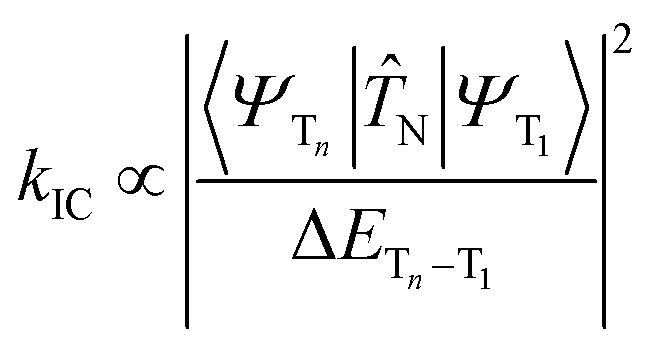
![[T with combining circumflex]](https://www.rsc.org/images/entities/i_char_0054_0302.gif) N represents the electronic coupling factor between the wavefunctions of T1 state (ΨT1) and Tn state (ΨTn), 〈ΨTn∣N∣ΨT1〉 represents the matrix element of internal conversion. Therefore, a small electronic coupling factor (when ΨT1 and ΨTn are perpendicular to each other) coupled with a large ΔETn–T1 can effectively mitigate the kIC enabling effective competition with ISC process. Therefore, the excited states of highly efficient hot exciton materials should possess the following distinctive characteristics: a significantly large ΔETn–T1, a small ΔESm–Tn, and a substantial SOC between Sm and Tn states. However, despite these requirements, the EL performance still falls considerably behind that of OLEDs utilizing thermally activated delayed fluorescence (TADF) and phosphorescent emitters. Thus, it is crucial to propose a more comprehensive hot exciton mechanism for guiding the design of highly efficient materials and bridging this performance gap.
N represents the electronic coupling factor between the wavefunctions of T1 state (ΨT1) and Tn state (ΨTn), 〈ΨTn∣N∣ΨT1〉 represents the matrix element of internal conversion. Therefore, a small electronic coupling factor (when ΨT1 and ΨTn are perpendicular to each other) coupled with a large ΔETn–T1 can effectively mitigate the kIC enabling effective competition with ISC process. Therefore, the excited states of highly efficient hot exciton materials should possess the following distinctive characteristics: a significantly large ΔETn–T1, a small ΔESm–Tn, and a substantial SOC between Sm and Tn states. However, despite these requirements, the EL performance still falls considerably behind that of OLEDs utilizing thermally activated delayed fluorescence (TADF) and phosphorescent emitters. Thus, it is crucial to propose a more comprehensive hot exciton mechanism for guiding the design of highly efficient materials and bridging this performance gap.
To address the challenges, we have developed two efficient DR emitters, namely αT-IPD and βT-IPD, based on the hot exciton mechanism. These emitters are synthesized by combining electron-acceptors αTPA and βTPA with an electron-withdrawing unit IPD (Fig. 1A).20 The intelligent molecular structures of these emitters allow for precise modulation of their energy levels in both singlet and triplet excited states. Both emitters exhibit significant energy gaps, with ΔET2–T1 approximately 0.75 eV and ΔES1–T1 around 0.70 eV, effectively reducing the dissipation of triplet excitons via IC mechanism. Furthermore, when compared to DT-IPD, both αT-IPD and βT-IPD possess larger ΔET2–T1 and ΔES1–T1 values. As a result, αT-IPD and βT-IPD demonstrate reduced kIC from the T2 to the T1 state as well as kISC from the S1 to the T1 state. Consequently, αT-IPD and βT-IPD achieve high photoluminescence quantum yields (ΦPLQYs) of 68.5% and 73.5%, respectively. Remarkably, a groundbreaking maximum external quantum efficiency (EQEmax) of 15.5% was achieved for non-doped DR-OLEDs based on βT-IPD, which is the highest value among non-doped DR-OLEDs employing the hot exciton mechanism.
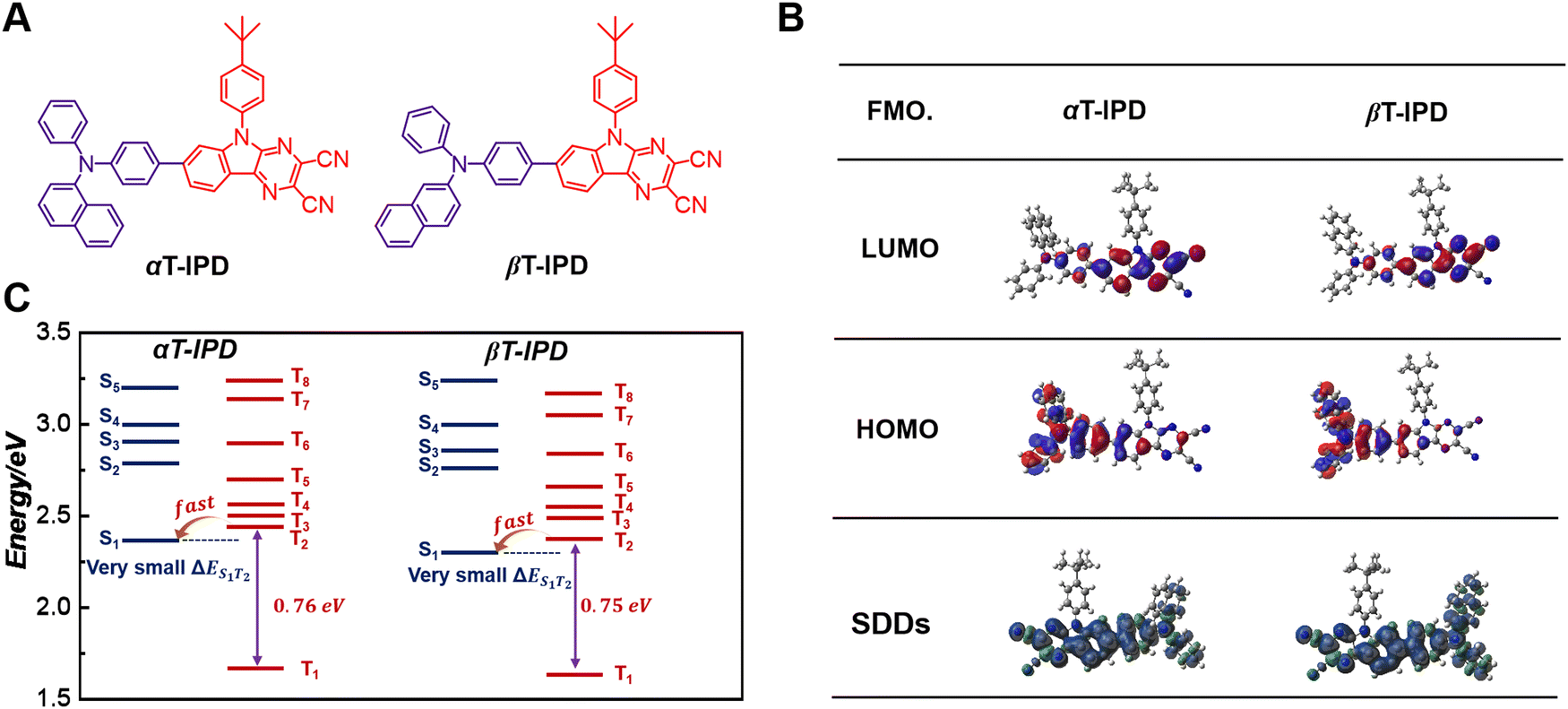 | ||
| Fig. 1 (A) The chemical structures of αT-IPD and βT-IPD. (B) HOMO and LUMO, and T1 states spin-density distributions (TSDDs) of αT-IPD and βT-IPD, respectively. (C) Calculated energy arrangement of singlet states and triplet states of αT-IPD and βT-IPD. | ||
Results and discussion
Identification of chemical structure and thermal stability measurement
The detailed synthetic procedures of αT-IPD and βT-IPD are summarized in the ESI.† The synthesis of the key intermediate 8-bromo-5-(4-(tert-butyl)phenyl)-5H-pyrazino[2,3-b]indole-2,3-dicarbonitrile (IPD-Br) can refer to the related literature.20 Target materials αT-IPD and βT-IPD were identified by 1H NMR and 13C NMR, along with mass spectra (Fig. S1–S6, ESI†). Remarkably, both αT-IPD and βT-IPD demonstrated excellent thermostability and morphological stability, with decomposition temperatures of 452 and 459 °C, along with glass transition temperatures of 144 and 152 °C, respectively (Fig. S7, ESI†). Thus, their outstanding thermal stabilities render them highly suitable for OLED formation via a vacuum evaporation technique.Density functional theory (DFT) and time-dependent density functional theory (TD-DFT) simulations
Initially, the electronic structures of αT-IPD and βT-IPD were comprehensively investigated through DFT and TD-DFT simulations to obtain a comprehensive understanding (Fig. 1B). The highest occupied molecular orbitals (HOMOs) of αT-IPD and βT-IPD were predominantly localized on IPD (except for the tert-butylbenzene subunit), as well as on the benzene ring connected with nitrogen-atoms exhibiting electron-donating characteristics. Conversely, the lowest unoccupied molecular orbitals (LUMOs) primarily reside on αTPA and βTPA moieties, along with the benzene ring linked to the IPD acceptor. Notably, the TSDDs of αT-IPD and βT-IPD significantly encompass the entire IPD acceptor and the αTPA and βTPA donors, suggesting that the T1 state for both emitters are locally excited (LE).To gain a more comprehensive understanding of the electronic configurations in the excited states, the natural transition orbits (NTOs) for both emitters were implemented. In the S1 state, the dihedral angle between the IPD and αTPA/βTPA in both emitters was relatively small (∼17°), resulting in a significant overlap of holes and particles, as well as large oscillator strength (f) values of 1.291 and 1.252 for αT-IPD and βT-IPD, respectively, suggesting that their LE state character dominate in the S1 state with minimal involvement of the charge-transfer (CT) state. Moreover, such large f values exhibited by both emitters are particularly advantageous for long-wavelength luminescent materials, as they enable intense light emission while significantly bolstering their competitive edge against non-radiative decay processes. In the T1 state, both emitters exhibit an even stronger LE character compared to the S1 state due to a smaller dihedral angle.
Investigation of the photophysical properties and exciton dynamics processes of αT-IPD and βT-IPD
In the ultraviolet-visible (UV-vis) absorption spectra, both emitters display two discernible absorption bands (Fig. S10, ESI†). The π–π* transitions of the IPD unit and adjacent benzene ring are accountable for the main absorption peaks around 320 nm. At the same time, the broad absorption peaks with moderate intensities of around 480 nm are attributed to the intramolecular charge transfer (ICT) process from the αTPA and βTPA donors to the IPD acceptor (Fig. S10 and Table S1, ESI†). In the photoluminescence (PL) spectra, αT-IPD and βT-IPD show a pronounced DR emission centered at 639 and 660 nm, respectively. Low-temperature PL and phosphorescent spectra were obtained to determine the energy levels of S1 and T1 in αT-IPD and βT-IPD (Fig. S11, ESI†). These experiments resulted in significant ΔEST values of 0.61 eV, aligning well with the calculated data (Table S1, ESI†). Furthermore, these findings effectively exclude their TADF properties.To further investigate the luminescence mechanism and exciton dynamics process of αT-IPD and βT-IPD, transient PL decay photoluminescence quantum yields (ΦPLQYs) were evaluated in their neat films (Fig. S12, ESI†). As expected, both emitters exclusively exhibit nanosecond lifetimes (Table S1, ESI†), while no delayed lifetimes in the microsecond range could be detected, ruling out their TADF mechanism. Interestingly, αT-IPD and βT-IPD achieved remarkably high ΦPLQY values of 68.5% and 73.5%, respectively, in non-doped films. Such high ΦPLQY values are exceptionally rare in the field of DR light, indicating the effective suppression of various exciton quenching pathways.
To elucidate the underlying factors contributing to the exceptional ΦPLQYs observed in non-doped states, as well as the dynamics of energy transfer for both αT-IPD and βT-IPD emitters, the singlet/triplet energy levels were evaluated using TD-DFT calculations (Fig. 1C). It is worth noting that a remarkable ΔET2–T1 of up to 0.75 eV was achieved for both emitters, leading to a highly effective suppression of the IC process from the T2 to the T1 state. Additionally, both emitters feature a significant ΔES1–T1 value of nearly 0.70 eV, which serves to weaken the rate of ISC (kISC). Furthermore, all the energy levels of the higher-lying Tn states (n ≥ 2) were found to be elevated compared to that of the S1 state, ensuring rapid hRISC processes from Tn to S1. Given the small energy gap between Tn (n = 2, 3, 4) and S1 ΔES1–Tn, multi-channel hRISC is anticipated, facilitating effective utilization of excitons (Fig. 2A). In comparison with DT-IPD, reported by our group the considerably smaller ΔET2–T1 and ΔES1–T1 values of DT-IPD pose a heightened vulnerability of undergoing IC from the T2 to the T1 state, as well as ISC from the S1 to the T1 state. This configuration is unfavorable for the efficient utilization of excitons (Fig. 2B). Ma and coworkers also reported intriguing findings regarding certain blue emitters characterized by the hot exciton mechanism (Fig. 2C).13,21–25 They emphasize that maintaining an n value lower than 4 is crucial at high-lying triplet energy levels to prevent rapid intersystem crossing between triplet states, which can weaken the efficiency of the Tn–S1 conversion process.23 However, according to our current research, in an emitter predominantly governed by the hot exciton mechanism, it is preferable for a minimal energy gap to exist between the high-lying triplet state and S1 state (ΔETn–S1, n ≥ 2) to occur, while ensuring that the energy level of the T2 state slightly surpasses that of the S1 state. This configuration enables the efficient transfer of triplet excitons to the S1 state even during the IC process between high energy states (Tn-T2, n > 2). Therefore, αT-IPD and βT-IPD exhibit a more favorable energy level arrangement for achieving efficient exciton utilization and contributing to exceptional EL characteristics.
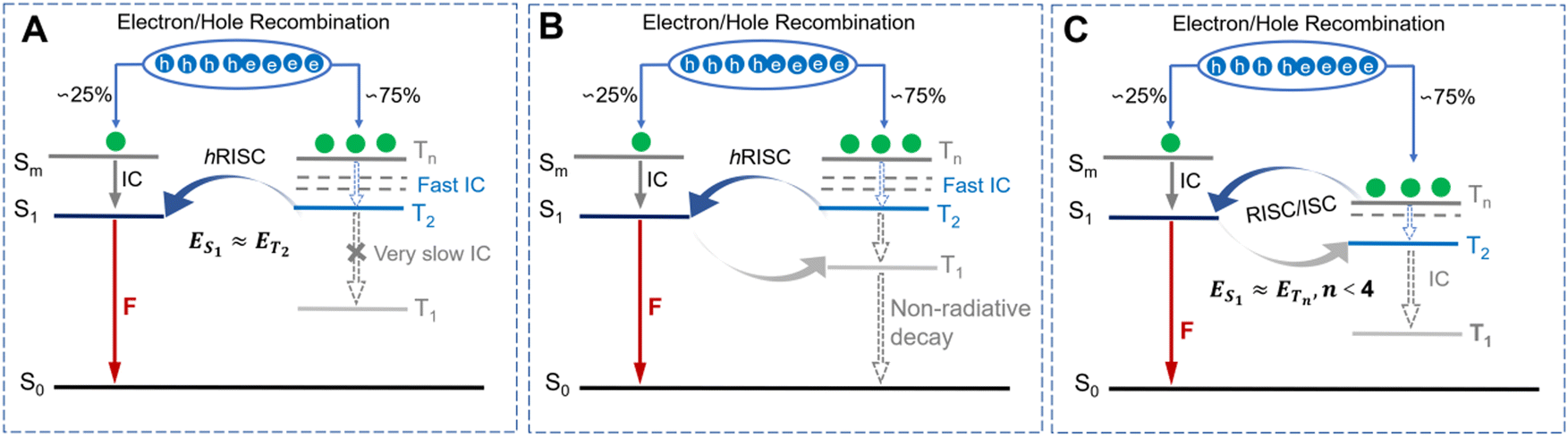 | ||
| Fig. 2 Schematic diagram of exciton dynamics processes for materials based on the hot exciton mechanism. (A) With an optimizing energy level alignment. (B) With relatively small ΔES1–Tn. (C) With small ΔES1–Tn, n ≤ 4. Abbreviations: F is fluorescence; IC denotes internal conversion. DF represents delayed fluorescence. | ||
Luminophores exhibiting aggregation-induced emission (AIE) have attracted considerable attention as promising candidates for non-doped OLED construction due to their exceptional ability to emit bright light and significantly enhance the PLQYs in the aggregate state.26,27 Hence, to delve deeper into the intricate mechanisms governing the ΦPLQY, a comprehensive investigation was conducted on the AIE properties in a mixture of tetrahydrofuran (THF) and water (Fig. 3A–D). Remarkably, both αT-IPD and βT-IPD exhibit minimal emission when the fraction of water (fW) is below 50%. However, the photoluminescence intensities experienced a rapid increase as the fW reached 60%, indicating a pronounced AIE characteristic in the aggregate state.
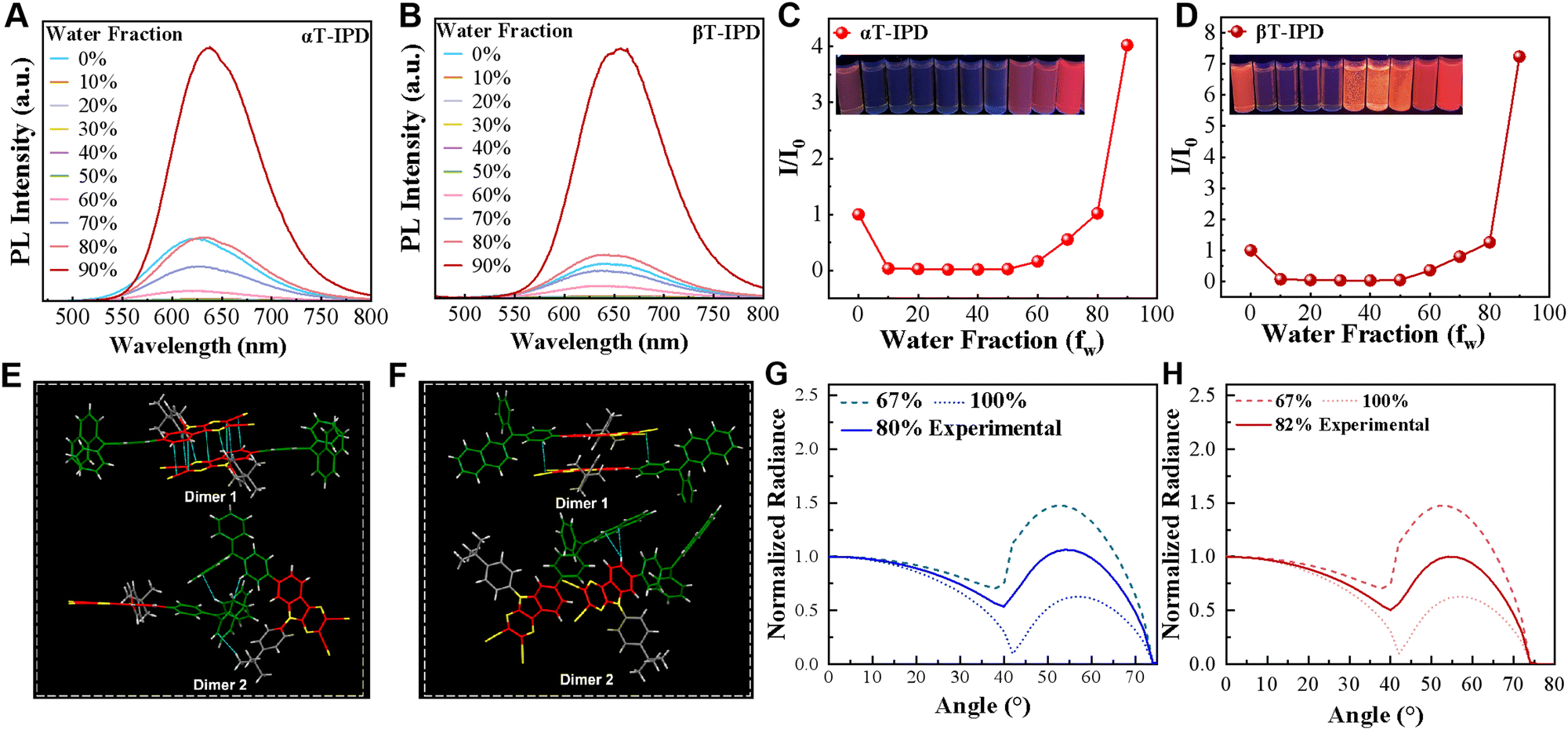 | ||
| Fig. 3 PL intensity of (A) αT-IPD and (B) βT-IPD. The plot of maximum emission intensity of (C) αT-IPD and (D) βT-IPD versus fW. (E) Packing arrangement of αT-IPD with π–π stacking and intermolecular CN⋯H–C interactions. (F) Packing arrangement of βT-IPD with intermolecular CN⋯π interactions. The angle-dependent PL intensity of (G) αT-IPD and (H) βT-IPD in neat films was measured with respect to the emission angle. | ||
The investigation of single crystals plays a pivotal role in acquiring profound insights into the chemical characteristics of materials, as well as understanding the intricate relationship between structure and properties. Therefore, both αT-IPD and βT-IPD crystals were successfully cultivated (Fig. 3E, F, and Table S2, ESI†). Single-crystal analysis revealed that the interlayer slip angles of the two emitter is smaller than 54.7° (Fig. S13, ESI†), thereby adopting J-aggregate stacking frameworks, which can significantly improve the emissive properties. Notably, although the dimers in both crystal structures exhibit a close intermolecular distance, the trimer composed of three monomers is comparatively further apart (Fig. S14, ESI†). This feature holds crucial importance in mitigating exciton annihilation, as a considerable number of long-lived triplet excitons are generated under electrical excitation, and exciton annihilation is primarily governed by short-range multi-molecular processes facilitated by the Dexter energy transfer (DET) mechanism. Therefore, minimizing the distance between multiple molecules effectively reduces the diffusion length of long-lived triplet excitons, thereby mitigating the annihilation process and significantly improving the EL performance of these molecules. Additionally, it is worth mentioning that the PLQY of αT-IPD is marginally lower compared to that of βT-IPD, and this observation appears to be partly attributed to the arrangement of single crystals. The formation of dimers in αT-IPD crystals induces π–π interactions, leading to an aggregation-caused quenching (ACQ) effect to a certain extent, consequently resulting in a reduction in PLQY for αT-IPD (Fig. S14 and Table S1, ESI†).
The optimization of the horizontal alignment (Θ) of the emissive transition dipole moment (TDM) in OLEDs holds significant potential for enhancing light extraction efficiency (ηout), resulting in a possible 50% improvement compared to random TDM orientation. Therefore, actively promoting a horizontal dipole orientation is crucial for bolstering OLEDs’ efficiency. To determine the Θ of both emitters, we measured the intensity of angle-dependent PL spectra in neat thin films (Fig. 3G and H). Remarkably, both αT-IPD and βT-IPD manifested high Θ of 80% and 82%, respectively. These noteworthy values can be attributed to the presence of rigid polycyclic aromatic hydrocarbon acceptors and a planar molecular framework.
OLED characteristics of αT-IPD and βT-IPD
We employ a non-doped OLED architecture to assess the EL performance of αT-IPD and βT-IPD, with the device structure depicted below: ITO/HAT-CN (10 nm)/TAPC (30 nm)/TCTA (5 nm)/EMLs (30 nm)/B3PyPb (70 nm)/LiF (1 nm)/Al (100 nm) were fabricated (Fig. 4A–E, S15, and Table 1). The corresponding material structure of the functional layer is shown in Fig. 4B. The EMLs in these devices only consist of neat films of αT-IPD and βT-IPD. The non-doped OLEDs based on αT-IPD and βT-IPD demonstrate DR emission, with their EL spectral peaks at wavelengths of 660 and 667 nm, respectively (Table 1). Benefitting from optimizing energy level alignment, rapid hRISC processes, favorable packing modes in single-crystal structures, and high ΦPLQYs and Θ values in non-doped films, both αT-IPD and βT-IPD exhibit record-breaking EQEs of 13.4% and 15.5%, respectively, in non-doped OLEDs (Fig. 4F and Table 1).11,20,28–49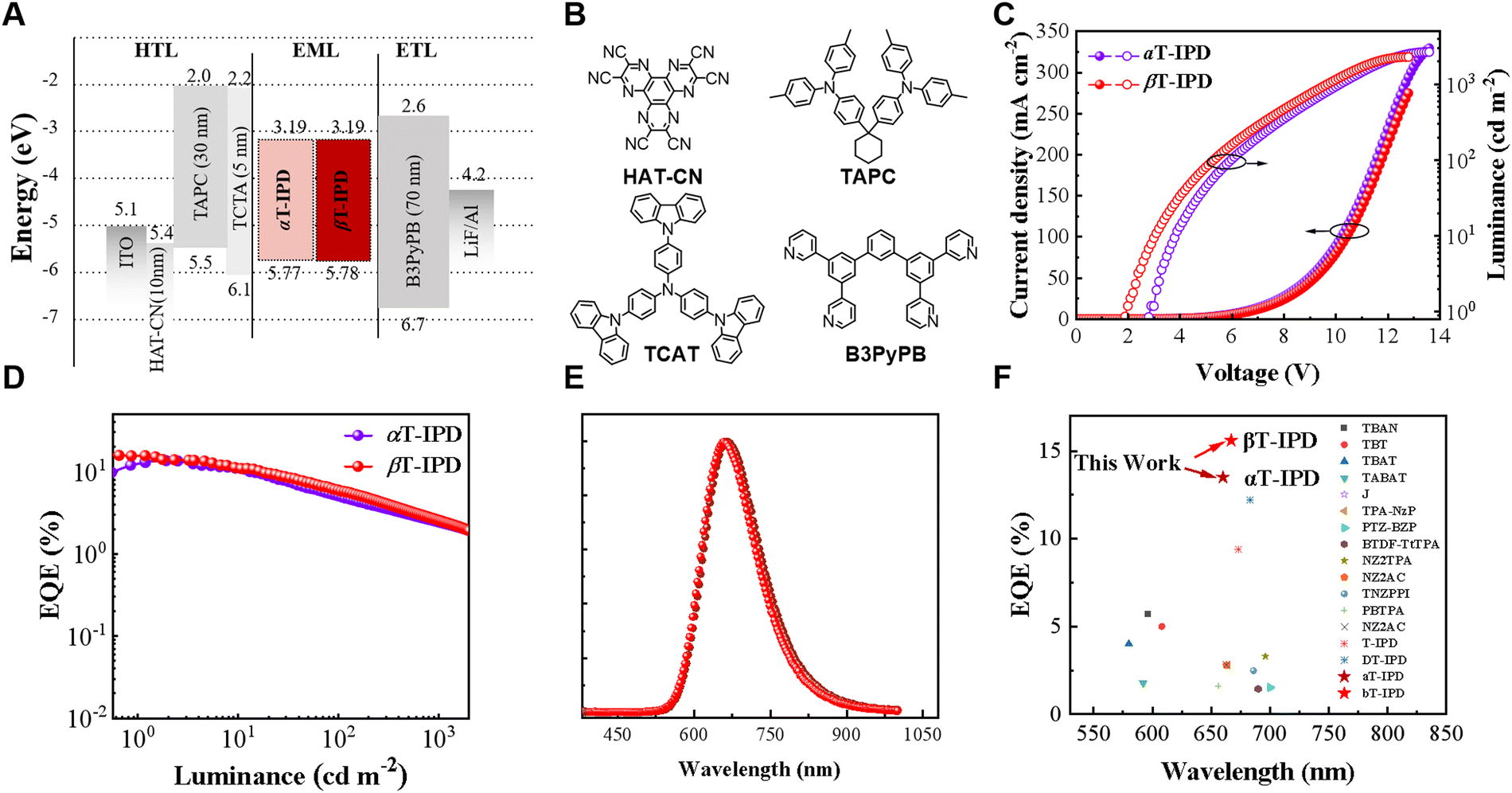 | ||
| Fig. 4 (A) OLED structure, HOMO and LUMO energy diagram. (B) The material structure used in the functional layer, (C) current density (J)–voltage curves (V)–luminance (L), (D) EQE–L curves, and (E) EL spectra of non-doped OLEDs based on αT-IPD and βT-IPD. (F) Reported representative hot exciton DR-OLEDs. | ||
Conclusions
In summary, two new DR luminogens with hot exciton mechanisms have been developed facilely. Intriguingly, αT-IPD and βT-IPD demonstrated much higher ΦPLQYs compared to other emitters with hot exciton mechanisms in non-doped films. These remarkable advantages can be ascribed to the optimized energy level alignment of both singlet and triplet excited states, the AIE effect, the ingenious packing mode of their single crystals with the J-aggregate nature, moderate strength intermolecular CN⋯H–C and C–H⋯π interactions, and unique comparatively large distances between trimers. It is noteworthy that achieving a minimal energy difference between the S1 and T2 states, while ensuring a slightly higher energy level for T2 compared to the S1 state, is crucial for hot exciton emitters. Such considerations are essential for facilitating a more efficient process of hRISC for triplet excitons. Equipped with high ΦPLQYs and horizontal dipolar ratios, The DR-OLED based on βT-IPD achieved a high EQE of 15.5% with an emission peak at 667 nm, making it the highest performing of hot exciton DR-OLED. This research paves the way for developing highly efficient emitters with the hot exciton mechanism.Data availability
Data will be made available on request.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work is financially supported by the Distinguished Young Scholars (21925506), the National Science Fund for National Natural Science Foundation of China (52003088, U21A20331, 22375212, 51773212, 22305086, and 81903743), the Hundred Talents Program of Chinese Academy of Sciences and Ningbo Key Scientific and Technological Project (2022Z124 and 2022Z119).References
- G. Qian and Z. Y. Wang, Chem. – Asian J., 2010, 5, 1006–1029 CrossRef CAS PubMed.
- W. Qin, D. Ding, J. Liu, W. Z. Yuan, Y. Hu, B. Liu and B. Z. Tang, Adv. Funct. Mater., 2011, 22, 771–779 CrossRef.
- B. Stender, S. F. Völker, C. Lambert and J. Pflaum, Adv. Mater., 2013, 25, 2943–2947 CrossRef CAS PubMed.
- Z. Guo, S. Park, J. Yoon and I. Shin, Chem. Soc. Rev., 2014, 43, 16–29 RSC.
- F. Anzengruber, P. Avci, L. F. de Freitas and M. R. Hamblin, Photochem. Photobiol. Sci., 2015, 14, 1492–1509 CrossRef CAS PubMed.
- K. Tuong Ly, R.-W. Chen-Cheng, H.-W. Lin, Y.-J. Shiau, S.-H. Liu, P.-T. Chou, C.-S. Tsao, Y.-C. Huang and Y. Chi, Nat. Photonics, 2016, 11, 63–68 CrossRef.
- Y.-C. Wei, S. F. Wang, Y. Hu, L.-S. Liao, D.-G. Chen, K.-H. Chang, C.-W. Wang, S.-H. Liu, W.-H. Chan, J.-L. Liao, W.-Y. Hung, T.-H. Wang, P.-T. Chen, H.-F. Hsu, Y. Chi and P.-T. Chou, Nat. Photonics, 2020, 14, 570–577 CrossRef CAS.
- S.-F. Wang, B.-K. Su, X.-Q. Wang, Y.-C. Wei, K.-H. Kuo, C.-H. Wang, S.-H. Liu, L.-S. Liao, W.-Y. Hung, L.-W. Fu, W.-T. Chuang, M. Qin, X. Lu, C. You, Y. Chi and P.-T. Chou, Nat. Photonics, 2022, 16, 843–850 CrossRef CAS.
- C. C. Tong and K. C. Hwang, J. Phys. Chem. C, 2007, 111, 3490–3494 CrossRef CAS.
- L. Duan, Z. Liu, Y. Lu, C. Zhang, G. Li, T. Huang, D. Zhang and Y. Zhang, Angew. Chem., Int. Ed., 2021, 60, 20498–20503 CrossRef.
- S. Reindl and A. Penzkofer, Chem. Phys., 1996, 211, 431–439 CrossRef CAS.
- Y. Xu, P. Xu, D. Hu and Y. Ma, Chem. Soc. Rev., 2021, 50, 1030–1069 RSC.
- Y. Xu, X. Liang, X. Zhou, P. Yuan, J. Zhou, C. Wang, B. Li, D. Hu, X. Qiao, X. Jiang, L. Liu, S. J. Su, D. Ma and Y. Ma, Adv. Mater., 2019, 31, 1807388 CrossRef PubMed.
- W. Li, D. Liu, F. Shen, D. Ma, Z. Wang, T. Feng, Y. Xu, B. Yang and Y. Ma, Adv. Funct. Mater., 2012, 22, 2797–2803 CrossRef CAS.
- W. Li, Y. Pan, R. Xiao, Q. Peng, S. Zhang, D. Ma, F. Li, F. Shen, Y. Wang, B. Yang and Y. Ma, Adv. Funct. Mater., 2013, 24, 1609–1614 CrossRef.
- W. Li, Y. Pan, L. Yao, H. Liu, S. Zhang, C. Wang, F. Shen, P. Lu, B. Yang and Y. Ma, Adv. Opt. Mater., 2014, 2, 892–901 CrossRef CAS.
- V. Lawetz, G. Orlandi and W. Siebrand, J. Chem. Phys., 1972, 56, 4058–4072 CrossRef CAS.
- D. Beljonne, Z. Shuai, G. Pourtois and J. L. Bredas, J. Phys. Chem. A, 2001, 105, 3899–3907 CrossRef CAS.
- T. J. Penfold, E. Gindensperger, C. Daniel and C. M. Marian, Chem. Rev., 2018, 118, 6975–7025 CrossRef CAS.
- S. Du, M. Luo, D. Li, L. Lyu, W. Li, M. Zhao, Z. Wang, J. Zhang, D. Liu, Y. Li, S. J. Su and Z. Ge, Adv. Mater., 2023, 35, 2303304 CrossRef CAS PubMed.
- X. Guo, P. Yuan, X. Qiao, D. Yang, Y. Dai, Q. Sun, A. Qin, B. Z. Tang and D. Ma, Adv. Funct. Mater., 2020, 30, 1908704 CrossRef CAS.
- C. Lin, P. Han, S. Xiao, F. Qu, J. Yao, X. Qiao, D. Yang, Y. Dai, Q. Sun, D. Hu, A. Qin, Y. Ma, B. Z. Tang and D. Ma, Adv. Funct. Mater., 2021, 31, 2106912 CrossRef CAS.
- X. Guo, P. Yuan, J. Fan, X. Qiao, D. Yang, Y. Dai, Q. Sun, A. Qin, B. Z. Tang and D. Ma, Adv. Mater., 2021, 33, 2006953 CrossRef CAS PubMed.
- P. Han, C. Lin, E. Xia, J. Cheng, Q. Xia, D. Yang, A. Qin, D. Ma and B. Z. Tang, Angew. Chem., Int. Ed., 2023, 62, e202310388 CrossRef CAS PubMed.
- C. Lin, P. Han, F. Qu, S. Xiao, Y. Li, D. Xie, X. Qiao, D. Yang, Y. Dai, Q. Sun, A. Qin, B. Z. Tang and D. Ma, Mater. Horiz., 2022, 9, 2376–2383 RSC.
- J. Luo, Z. Xie, J. W. Y. Lam, L. Cheng, B. Z. Tang, H. Chen, C. Qiu, H. S. Kwok, X. Zhan, Y. Liu and D. Zhu, Chem. Commun., 2001, 1740–1741 RSC.
- Y. Hong, J. W. Y. Lam and B. Z. Tang, Chem. Soc. Rev., 2011, 40, 5361–5388 RSC.
- L. Yao, S. Zhang, R. Wang, W. Li, F. Shen, B. Yang and Y. Ma, Angew. Chem., Int. Ed., 2014, 53, 2119–2123 CrossRef CAS PubMed.
- T. Liu, L. Zhu, S. Gong, C. Zhong, G. Xie, E. Mao, J. Fang, D. Ma and C. Yang, Adv. Opt. Mater., 2017, 5, 1700145 CrossRef.
- T. Liu, L. Zhu, C. Zhong, G. Xie, S. Gong, J. Fang, D. Ma and C. Yang, Adv. Funct. Mater., 2017, 27, 1606384 CrossRef.
- X. Tang, X.-L. Li, H. Liu, Y. Gao, Y. Shen, S. Zhang, P. Lu, B. Yang, S.-J. Su and Y. Ma, Dyes Pigm., 2018, 149, 430–436 CrossRef CAS.
- Q. Wan, J. Tong, B. Zhang, Y. Li, Z. Wang and B. Z. Tang, Adv. Opt. Mater., 2020, 8, 1901520 CrossRef CAS.
- W. Xie, B. Li, X. Cai, M. Li, Z. Qiao, X. Tang, K. Liu, C. Gu, Y. Ma and S. J. Su, Front. Chem., 2019, 7, 276 CrossRef CAS PubMed.
- Y. Yu, M. Cang, W. Cui, L. Xu, R. Wang, M. Sun, H. Zhou, W. Yang and S. Xue, Dyes Pigm., 2021, 184, 108770 CrossRef CAS.
- D. Zhang, T. Yang, H. Xu, Y. Miao, R. Chen, R. Shinar, J. Shinar, H. Wang, B. Xu and J. Yu, J. Mater. Chem. C, 2021, 9, 4921–4926 RSC.
- S. Kongsabay, P. Funchien, P. Chasing, T. Sudyodsuk and V. Promarak, J. Lumin., 2022, 248, 118921 CrossRef CAS.
- Y. Yu, Z. Yu, Z. Ma, J. Jiang and D. Hu, Dyes Pigm., 2022, 208, 110868 CrossRef CAS.
- Y. Xu, P. Xu, D. Hu and Y. Ma, Chem. Soc. Rev., 2021, 50, 1030–1069 RSC.
- Y. Gao, M. Yao, C. Zhou, H. Liu, S.-T. Zhang and B. Yang, J. Mater. Chem. C, 2022, 10, 4579–4583 RSC.
- Q. Wan, B. Zhang, C. Mao, T. Zhang, Z. Wang and B. Z. Tang, J. Mater. Chem. C, 2022, 10, 1062–1068 RSC.
- L. Yao, S. Zhang, R. Wang, W. Li, F. Shen, B. Yang and Y. Ma, Angew. Chem., 2014, 126, 2151–2155 CrossRef.
- L. Yao, S. Zhang, R. Wang, W. Li, F. Shen, B. Yang and Y. Ma, Angew. Chem., Int. Ed., 2014, 53, 2119–2212 CrossRef CAS PubMed.
- T. Liu, L. Zhu, S. Gong, C. Zhong, G. Xie, E. Mao, J. Fang, D. Ma and C. Yang, Adv. Opt. Mater., 2017, 5, 1700145 CrossRef.
- T. Liu, L. Zhu, C. Zhong, G. Xie, S. Gong, J. Fang, D. Ma and C. Yang, Adv. Funct. Mater., 2017, 5, 1700145 Search PubMed.
- W. Xie, B. Li, X. Cai, M. Li, Z. Qiao, X. Tang, K. Liu, C. Gu, Y. Ma and S.-J. Su, Front. Chem., 2019, 7, 276 CrossRef CAS PubMed.
- X. He, L. Gao, H. Liu, F. Liu, D. Jiang, C. Du, C. Sun and P. Lu, Chem. Eng. J., 2021, 404, 127055 CrossRef CAS.
- Y. Yu, H. Xing, D. Liu, M. Zhao, H. H. Y. Sung, I. D. Williams, J. W. Y. Lam, G. Xie, Z. Zhao and B. Z. Tang, Angew. Chem., Int. Ed., 2022, 61, e202204279 CrossRef CAS PubMed.
- M. Zhao, M. Li, W. Li, S. Du, Z. Chen, M. Luo, Y. Qiu, X. Lu, S. Yang, Z. Wang, J. Zhang, S. J. Su and Z. Ge, Angew. Chem., Int. Ed., 2022, 61, e202210687 CrossRef CAS PubMed.
- Y. Yu, Z. Yu, Z. Ma, J. Jiang and D. Hu, Dyes Pigm., 2023, 208, 110868 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2309641 (αT-IPD) and 2309564 (βT-IPD). For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4mh00441h |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |