
Designing a photocatalytic and self-renewed g-C3N4 nanosheet/poly-Schiff base composite coating towards long-term biofouling resistance†
Saijun
Wu
ab,
Minglong
Yan
 *a,
Yinghao
Wu
a,
Yangmin
Wu
a,
Xijian
Lan
a,
Jianjun
Cheng
a and
Wenjie
Zhao
*a
*a,
Yinghao
Wu
a,
Yangmin
Wu
a,
Xijian
Lan
a,
Jianjun
Cheng
a and
Wenjie
Zhao
*a
aKey Laboratory of Advanced Marine Materials, Ningbo Institute of Materials Technology and Engineering, Chinese Academy of Sciences, Ningbo, China. E-mail: yanminglongivip@163.com; zhaowj@nimte.ac.cn
bUniversity of Chinese Academy of Sciences, Beijing, 100049, China
First published on 24th June 2024
Abstract
Inhibiting the adhesion and growth of marine microorganisms through photocatalysis is a potentially efficient and environmentally friendly antifouling strategy. However, the undesired “shading effect” caused by resin coatings and microbial deposition reduces the utilization of the catalysts and leads to a failure in the antifouling active substance on the coating surface. Here, we successfully developed a composite coating (DPC-x) combining g-C3N4 nanosheet (g-C-NS) photocatalysts with degradable green poly-Schiff base resins, which integrates the dual functions of enhanced dynamic self-renewal and photocatalytic antibacterial activities towards long-term anti-biofouling. The controllable and complete degradability of the poly-Schiff base polymer chains and the self-renewal mechanism of the DPC-x coating exposed the internal g-C-NS, which provided a constant stream of photocatalytic reactive interfaces for 100% utilization and release of the photocatalysts. g-C-NS were homogeneously dispersed in the degradable resin coating, significantly enhancing and adjusting the self-renewal rate of the poly-Schiff base resin coating in visible light. The degradation reaction rate of DPC-0.2 (20 wt% g-C-NS) was 40 times that of DPC, thus improving the capabilities of surface self-renewal and fouling-release. Due to the synergistic antifouling mechanism of the efficient antibacterial properties and the enhanced degradation/self-renewal, the antimicrobial rates of DPC and DPC-0.2 were 94.58% and 99.31% in the dark, and 98.2% and 99.87% in visible light. DPC-x has excellent all-weather antimicrobial efficacy and could offer a new perspective on eco-friendly marine antifouling strategies.
New conceptsWe demonstrate a new long-term anti-biofouling composite coating combining g-C3N4 nanosheet photocatalysts with degradable green poly-Schiff base resins, which integrates the dual functions of enhanced dynamic self-renewal and photocatalytic antibacterial activities. Research on improving the antifouling properties of coatings has been underway for a while, and the combination with antimicrobial agents is generally recognized as an effective approach, but the use of potent antimicrobial agents is prone to environmental damage. g-C3N4 nanosheets, as representative promising green nonmetallic photocatalysts, have rarely been explored as potential alternatives to toxic antimicrobial agents. The reason is that they are unable to maintain long-term stable and efficient photocatalytic activity in real marine and aquatic environments, especially under dark and weak light conditions. This study highlights the enhanced dynamic self-renewal properties of poly-Schiff base resin surfaces to synergize sustained and highly efficient photocatalytic antimicrobial interfaces under visible light, significantly enhancing the light absorption and utilization of photocatalysts. This new composite coating exhibits excellent antimicrobial efficiency under all-weather conditions. We further suggest that the degradation rate of the coatings can also be controlled depending on the light setting and the amount of photocatalyst, thus promising long-term prevention of biofouling on underwater equipment surfaces. |
1. Introduction
Biofouling has become a worldwide challenge due to the widespread harmfulness and complexity of its prevention.1,2 In particular, equipment and ship hull surfaces that are in direct contact with the aquatic or marine environment are susceptible to microbial settlement, which leads to corrosion or increased resistance when moving through the water. Marine biofouling is estimated to cause global economic losses amounting to tens of billions of dollars annually, highlighting its significance as a global issue.3 To effectively combat biofouling, it is necessary to adopt a diversified range of antibacterial strategies that take into account economic, environmental, and safety factors comprehensively. For common physical or chemical methods, antifouling coatings are extensively applied due to their general applicability and cost-effectiveness.1,2,4 However, traditional anti-fouling coatings cause severe damage to the marine ecosystem due to the release of potent and toxic agents. Ever since the global prohibition of coatings containing organic tin in 2008, there has been a surge in the development of environmentally friendly antifouling coatings, such as silicones,5,6 hydrogels,7,8 slippery liquid-infused surfaces,9,10 tin-free self-polishing coatings,1 and biodegradable polymer coatings.11,12Specifically, degradable polymer coatings have received much attention due to their attractive bio-friendliness. The antimicrobial principle is based on the hydrolysis of the polymers to form dynamic self-renewal surfaces, significantly diminishing biofouling adhesion and biofilm formation under static conditions. Yan et al. reported degradable poly-Schiff base antifouling coatings, which will spontaneously degrade into small molecules with aldehydes and amino terminals in the water due to the reversibility of the imine covalent bond (–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N–) in main chains, which also prevents the emergence of microplastics.13 Nevertheless, the intrinsic antifouling capabilities of these coatings are limited and often require augmentation through the incorporation/grafting of supplementary antifouling agents. In previous studies, we have demonstrated that the addition of metal ions to the poly-Schiff base metal complex materials effectively improves anti-biofouling properties.13 Furthermore, a Schiff base metal composite coating (Fe/TOB-PR)n synthesized from tobramycin (TOB) and protocatechuic aldehyde (PR) has shown excellent anti-bacterial and anti-algae adhesion properties.14 However, such metallic or organic antifouling agents or antifouling materials are potentially harmful to microorganisms/organisms and are prone to develop resistance toward bacteria. Therefore, the development of efficient antimicrobial agents that do not cause secondary pollution to the marine environment is imminent.
N–) in main chains, which also prevents the emergence of microplastics.13 Nevertheless, the intrinsic antifouling capabilities of these coatings are limited and often require augmentation through the incorporation/grafting of supplementary antifouling agents. In previous studies, we have demonstrated that the addition of metal ions to the poly-Schiff base metal complex materials effectively improves anti-biofouling properties.13 Furthermore, a Schiff base metal composite coating (Fe/TOB-PR)n synthesized from tobramycin (TOB) and protocatechuic aldehyde (PR) has shown excellent anti-bacterial and anti-algae adhesion properties.14 However, such metallic or organic antifouling agents or antifouling materials are potentially harmful to microorganisms/organisms and are prone to develop resistance toward bacteria. Therefore, the development of efficient antimicrobial agents that do not cause secondary pollution to the marine environment is imminent.
Photocatalysis is one of the most promising environmental antifouling technologies that does not cause secondary pollution to the marine environment, providing a potential alternative to traditional biocidal coatings.15–17 In this regard, graphitic carbon nitride (g-C3N4), as a representative non-metallic photocatalyst, has been gradually utilized in photocatalytic antifouling studies and gained widespread attention due to its visible light absorption properties, high chemical stability, and low cost.18,19 However, the bulk g-C3N4 materials synthesized via thermal polymerization usually ascribe their poor photocatalytic efficiency to the interlayer stacking structure and large grain size.20–22 Therefore, it is necessary to separate these stacking layers to achieve enhanced photocatalytic antibacterial efficiency. Recently, exfoliated bulk g-C3N4 with ultrathin thickness has been considered a promising candidate for solving these problems. Ultrathin g-C3N4 two-dimensional (2D) nanosheets (g-C-NS) have been shown to enhance photocatalytic activity, attributed to their prolonged photogenerated charge lifetime, atomic thickness, and extremely high percentage of exposed active sites.23–25 Unfortunately, heterogeneous photocatalysts in the powder state have limitations for practical applications in marine and aquatic environments.26 To address this issue, researchers have explored several kinds of solid inert carriers or substrates to strongly immobilize g-C3N4, such as epoxy resin,27 PDMS,28 TC4 titanium alloy,29 PS,26 and hydrogels.30,31 However, subsequent problems, including the possibility that the carrier leads to a reduction in the active center of the catalyst and a significant loss in the photocatalytic activity due to the shading effect created by fouling deposits, resin coatings, and self-deactivation, hinder the long-term antifouling efficacy of the coatings.32
The poly-Schiff base coating, as a fully degradable resin, offers the advantages of a dynamic self-renewal surface and environmentally friendly antifouling performance, but further enhancement of its antifouling efficiency is warranted. The g-C-NS possess excellent photocatalytic antibacterial ability, combined with non-metallic environmental characteristics. However, improving their efficient utilization in the coating system and preventing deactivation of the photocatalytic activity are imperative. Herein, by combining their respective advantages, we innovatively integrate ultrathin g-C-NS into the degradable poly-Schiff base resin to create a composite coating (DPC-x). The DPC-x, on the one hand, leverages the completely degradable feature of poly-Schiff base resin to gradually expose the internal g-C-NS to the antifouling interface, thereby circumventing the shading effect and ensuring the 100% utilization and on-demand release of g-C-NS. On the other hand, it utilizes the dual-functional antifouling mechanism of enhanced self-renewal of the poly-Schiff base polymer and the highly efficient photocatalytic antibacterial activity of g-C-NS to achieve excellent biofouling resistance under all-weather conditions. Therefore, DPC-x shows great prospects for long-term anti-biofouling applications in real aquatic and marine environments.
2. Experimental
2.1. Materials
Melamine, toluene and anhydrous ethanol were all obtained from Sinopharm Chemical Reagent Co. Ltd. Terephthalaldehyde (TPA), N-(3-aminopropyl)-N-dodecylpropane-1,3-diamine (Y12D), Na2SO4, KCl, Nafion solution (5 wt%), isopropyl alcohol (IPA) and 5,5-dimethyl-1-pyrroline N-oxide (DMPO) were obtained from Macklin Co. Ltd. Isophoron diamine (IPDA) was purchased from Shanghai Aladdin Bio-Chem Technology Co. Ltd. Phosphate buffered saline (PBS) was purchased from Beijing Solarbio Science & Technology Co. Ltd. Tween-80 was obtained from Bkmam Biotechnology Co. Ltd. Epoxy resin (E51) was purchased from Shandong Yousuo Chemical Engineering Technology Co. Ltd. Curing agent (1618) was obtained from Nanjing Haozhuo Materials Technology Co. Ltd. ITO (indium tin oxide) glass was obtained by Luoyang Tengjing Glass Co. Ltd. E. coli (ATCC8739) was acquired from Shanghai Luwei Technology Co. Ltd. All the chemical reagents were analytically pure and used without further purification.2.2. Preparation of graphitic carbon nitrides
The bulk g-C3N4 (g-C) yellow powders were synthesized by thermal treatment of melamine with a weight of 15 g at 550 °C for 3 h with a heating rate of 5 °C min−1 in a muffle furnace with a yield of 51.8% followed by washing and drying at 90 °C for 8 h.33 The pale-yellow fluffy g-C-T powders were obtained by calcining g-C with a weight of 5 g at 550 °C for 3 h with a heating rate of 5 °C min−1 followed by washing and drying at 90 °C for 8 h, with a yield of 89.3%.34 Then, 1.2 g of g-C-T powders were ground in an agate mortar. After that, the powders were added into 300 mL isopropanol (IPA) for an ultrasonic bath for 12 h.24,25 Then, the mixed solution was centrifuged at 3000 rpm min−1 for 5 min to remove the undispersed particles. Ultrathin graphitic carbon nitride nanosheets (g-C-NS) were collected by centrifugation at 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm min−1 for 5 min and then dried at 90 °C for 8 h, with a yield of 87.6%. All reactions were operated under an air atmosphere (Scheme 1A).
000 rpm min−1 for 5 min and then dried at 90 °C for 8 h, with a yield of 87.6%. All reactions were operated under an air atmosphere (Scheme 1A).
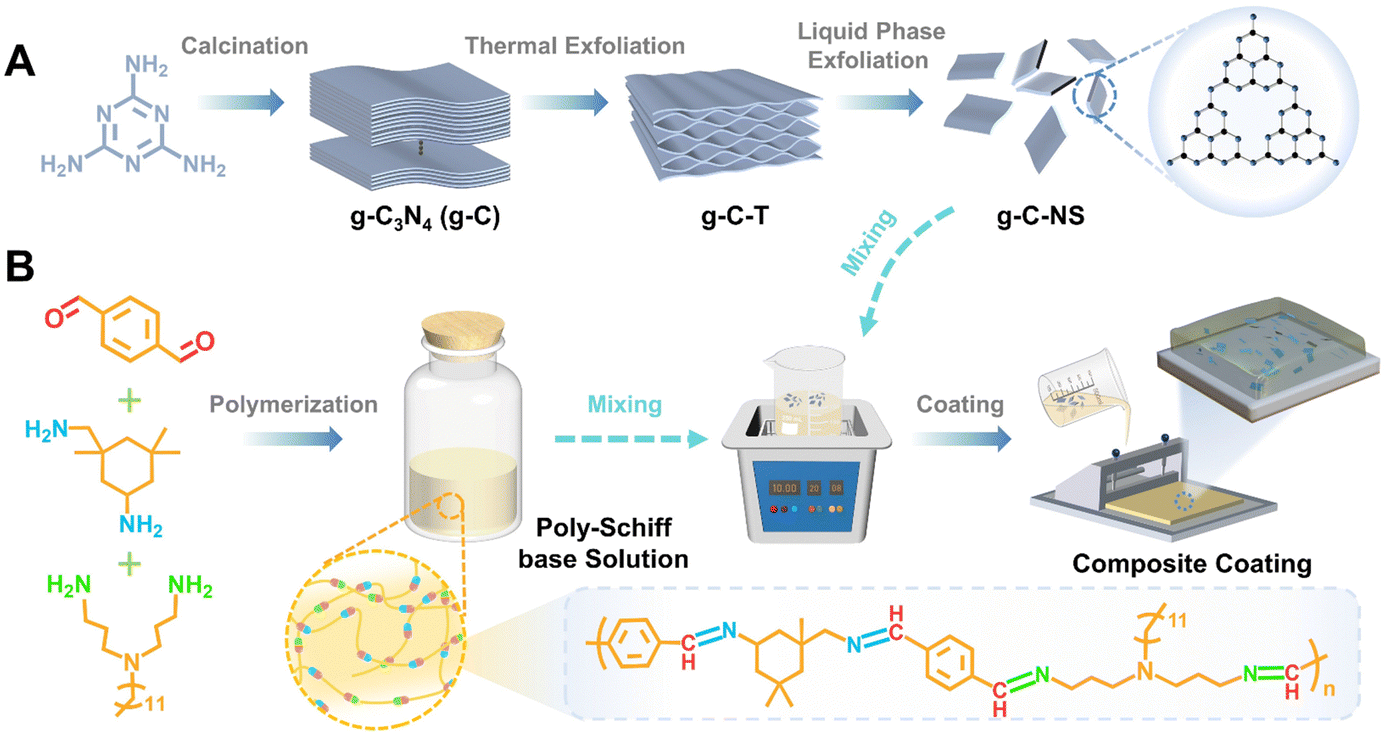 | ||
| Scheme 1 Schematic illustrating the preparation process of the g-C-NS/poly-Schiff base composite coating. (A) Preparation process of g-C, g-C-T, and g-C-NS. (B) Preparation of poly-Schiff base solution and g-C-NS/poly-Schiff base composite coating. | ||
2.3. Synthesis of the poly-Schiff base resin
The aldehyde monomer (TPA 26.826 g) and toluene were first added to the reaction flask, stirred to form a mixed solution, and then the mixture of ammonia monomers (Y12D, 29.954 g, IPDA, 7.03 g) and toluene was added drop by drop. The mixture solution was stirred at 90 °C for 24 h under a N2 atmosphere, and the by-product water was removed by vacuum distillation and separation treatment. After condensation reactions were complete, the resin solution was cooled to room temperature and stored (Scheme 1B).132.4. Preparation of g-C-NS/poly-Schiff base composite coatings (DPC-x)
The amount of g-C-NS was adjusted to control the ratio of the g-C-NS/poly-Schiff base resin. g-C-NS (0 g, 0.15 g, 0.3 g, and 0.45 g) were added to the poly-Schiff base resin (1.5 g) in toluene (6 mL). After 10 min of ultrasonic treatment, about 400 μL of diluted solution was evenly coated on the glass surface (the coating area was 2.5 cm × 2.5 cm) and dried at room temperature until most of the solvent evaporated. Then it was dried under vacuum at 130 °C for 12 h to cure completely. The obtained coating samples were named DPC, DPC-0.1, DPC-0.2, and DPC-0.3, respectively (Scheme 1). As a control, DPC-g-C0.2 was prepared by adding g-C (0.3 g) to the poly-Schiff base resin (1.5 g) using the same procedure.As a comparison, epoxy resin-based coating (EP) samples were also synthesized. The epoxy resin and curing agent were evenly mixed at a ratio of 5:3 and coated on the polished glass surface (the coating area was 2.5 cm × 2.5 cm), and the residual air bubbles inside the resin were pumped under vacuum for 15 min. A follow-up test was carried out after the resin was completely cured at room temperature.
2.5. Material characterization
The chemical structure of photocatalysts and polymer coatings was determined using a Fourier transform infrared spectrophotometer FTIR (Nicolet 6700) and Micro-FTIR (Cary660 + 620), respectively. 1H NMR spectra of organics were obtained using a nuclear magnetic resonance spectrometer (400 MHz AVANCE III) with deuterium-chloroform as the solvent. The chemical compositions of the photocatalysts and coatings were analyzed via X-ray photoelectron spectroscopy XPS (Axis Ultra DLD). The microstructure and morphology of the samples were characterized using a SEM (Regulus-8230), SPM system (dimension 3100), and TEM (Tecnai F20) system. Transmission electron microscopy (TEM), high-resolution transmission electron microscopy (HRTEM), selected area electron diffraction (SAED) and energy dispersive X-ray spectra (EDS) analysis were performed using a Tecnai F20 200 kV microscope from Philips. The X-ray diffraction (XRD) pattern of the photocatalysts was determined using an X-ray diffractometer (Bruker Germany, D8 Advance Davinci) with a scanning range (2θ) of 10° to 90° and a scanning speed of 5° min−1 under Cu-Kα radiation. The UV-vis diffuse reflectance spectroscopy (DRS) of the photocatalysts was carried out using a UV-vis-NIR Spectrometer (PerkinElmer, Lambda950). The photoluminescence (PL) spectra were obtained using a FL3-111 spectrometer (Horiba), with a Xe lamp (excitation at 365 nm) as the light source. A nano photometer spectrophotometer (IMPLEN, NP80) was used to detect the absorbance of the coating degradation products. Thermogravimetry (TGA209F1) was used to detect the thermal stability of the samples (all samples were 5 mg in mass), which was carried out in an argon gas stream heated to 800 °C at a heating rate of 10 °C min−1. The Brunauer–Emmett–Teller (BET) specific surface areas of the photocatalysts were measured by nitrogen adsorption on a Physisorption Analyzer (ASAP 2020 HD88). A contact angle tester (DCAT 21) was used to measure the wettability of the coating surface before and after immersion, and the contact angle of the photocatalysts was measured by pressing them into sheets. The bacterial status and quantity were documented using SEM and a Bacterial Colony Counter (SCAN300). Electron spin resonance spectroscopy (Bruker Germany, E500) was used to detect the electron paramagnetic resonance (EPR) spectral signal of the catalyst. The electrochemical impedance spectroscopy (EIS) and Mott–Schottky curves were obtained using a CHI 760E electrochemical workstation with a standard three-electrode cell at room temperature.2.6. Degradation test
Degradation test: the DPC, DPC-x, DPC-g-C0.2, and EP coating were individually placed in a quartz tube containing 30 mL of deionized water, ensuring complete submersion of the coating. Degradation products in the aqueous solution were monitored using a 10 mm quartz cuvette and a UV spectrophotometer. To evaluate their degradation behavior, quartz tubes wrapped in opaque tinfoil simulated a dark environment, while quartz tubes without any treatment simulated a light environment. The samples were simultaneously incubated at 20 °C in a constant temperature intelligent light incubator (with a illuminance of 1.375 lm cm−2, λ > 400 nm). The absorption intensity of the solution in the quartz tube is measured every 24 h in the 200–900 nm wavelength range. The structure of the degradation products was analyzed by the radiation absorption of substance molecules in the UV region. The concentration of solute (TPA) degraded into solution by the coating was determined by fitting the absorbance to a calibration curve (Fig. S8, ESI†). Consequently, the degradation reaction rate of the coating can be compared and calculated. Epoxy coating was used as control samples. All samples were treated under identical conditions, except for the visible light factor.2.7. Photocatalytic antibacterial test
E. coli was used as the model organism for antibacterial experiments. All vessels and culture media were sterilized at 121 °C under high pressure for 20 min prior to testing. To activate the bacteria, a single colony was selected and cultured in 100 mL of LB broth at 37 °C in a constant temperature incubator with a shaking speed of 120 rpm for 24 h. The culture was then centrifuged at 4000 rpm for 5 min, and then the obtained cell pellet was washed three times with phosphate buffer solution (PBS, 0.01 M, pH = 7.2–7.4) to eliminate the metabolites and culture medium components. The bacterial suspension was diluted with PBS to yield a cell count of approximately 109 colony-forming units per milliliter (CFU mL−1).Antibacterial rate of the coating/photocatalysts:
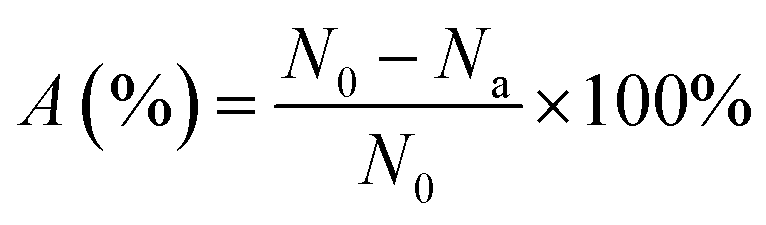
2.8. The morphological observation of bacteria
SEM imaging was employed to examine the morphological changes of the coating before and after service in the light/dark. These coatings were immersed in deionized water for 10 min, and then the bacteria were fixed with 2.5% glutaraldehyde for 30 min. Then, the coatings were immersed in deionized water for 10 min and dried naturally at room temperature. SEM was also utilized to observe the changes of bacterial morphology in the photocatalyst suspensions. A droplet of 2 mL of suspensions from the quartz conical bottle was placed on a silicon wafer. After washing the silicon wafer with deionized water, the bacteria were fixed with 2.5% glutaraldehyde for 30 min. The cells were subsequently dehydrated and sequentially immersed in 25%, 50%, 75%, and 100% ethanol for 15 min. Finally, the samples were air-dried at room temperature and sputter-coated with a gold layer for SEM imaging.2.9. In situ EPR free radical capture test
In the EPR capture test, DMPO was used as a trapping reagent for superoxide (O2˙−) and hydroxyl radicals (˙OH). 10 mg of g-C, g-C-T and g-C-NS photocatalysts were dispersed into 1 mL methanol and 1 mL deionized water to form a homogeneous suspension, respectively. 20 μL DMPO methanol solution was added into 30 μL of the suspension. Then, the prepared solution was immediately collected using a capillary tube. The EPR signals were detected using a Bruker E500 electron spin resonance with irradiation/no irradiation (λ > 420 nm) in situ for 1 min, 5 min, 10 min, and 20 min.2.10. Electrochemical test
A three-electrode system was used for electrochemical testing on a CHI 760E electrochemical workstation with a Pt electrode and an Ag/AgCl electrode as reference electrodes, and the electrolyte was 0.5 M Na2SO4 solution. The working electrode is made of indium tin oxide (ITO) glass. The g-C, g-C-T and g-C-NS with a weight of 5 mg were dispersed in 1 mL ethanol and ultrasonically treated for 20 min to form a suspended ink, respectively. Then, we dropped the ink onto ITO glass with an effective surface area of 1 cm2. Open circuit potential (OCP), electrochemical impedance spectroscopy (EIS) and impedance potential (IMPE) measurements were performed after the working electrode was dried in air at 50 °C. The start and end potentials of the Mott–Schottky curve were set within the range of ±(0.5–1) V with the OCP as the center, and the amplitude was 0.005 V. Three frequencies of 1000 Hz, 1500 Hz and 2000 Hz were selected for measurement, respectively.3. Results and discussion
3.1. Structure and morphology characterization
The poly-Schiff base polymer was synthesized by dehydration condensation reaction with TPA, Y12D, and IPDA. The molecular structure of the polymer was characterized by 1H NMR (Fig. 1A). The new peaks at chemical shifts 8.19 to 8.39 ppm (labeled “r”) were attributed to the imine (–CN–) group attached to the aryl group. The characteristic peak at 10.13 ppm (labeled “a”) corresponded to the aldehyde (–CHO) group of TPA. The signals between 1.34 to 1.48 ppm (labeled “i”) were associated with the amino (–NH2) group on Y12D, while the signals between 1.4 to 1.68 ppm (labeled “s” and “j”) were attributed to the –NH2 group of IPDA. The disappearance of the –CHO and –NH2 signals and the appearance of the –CN– signal in the DPC indicated that the functional groups –CHO and –NH2 condensed to form the polymer structure; evidence that the designed poly-Schiff base polymer was successfully synthesized.
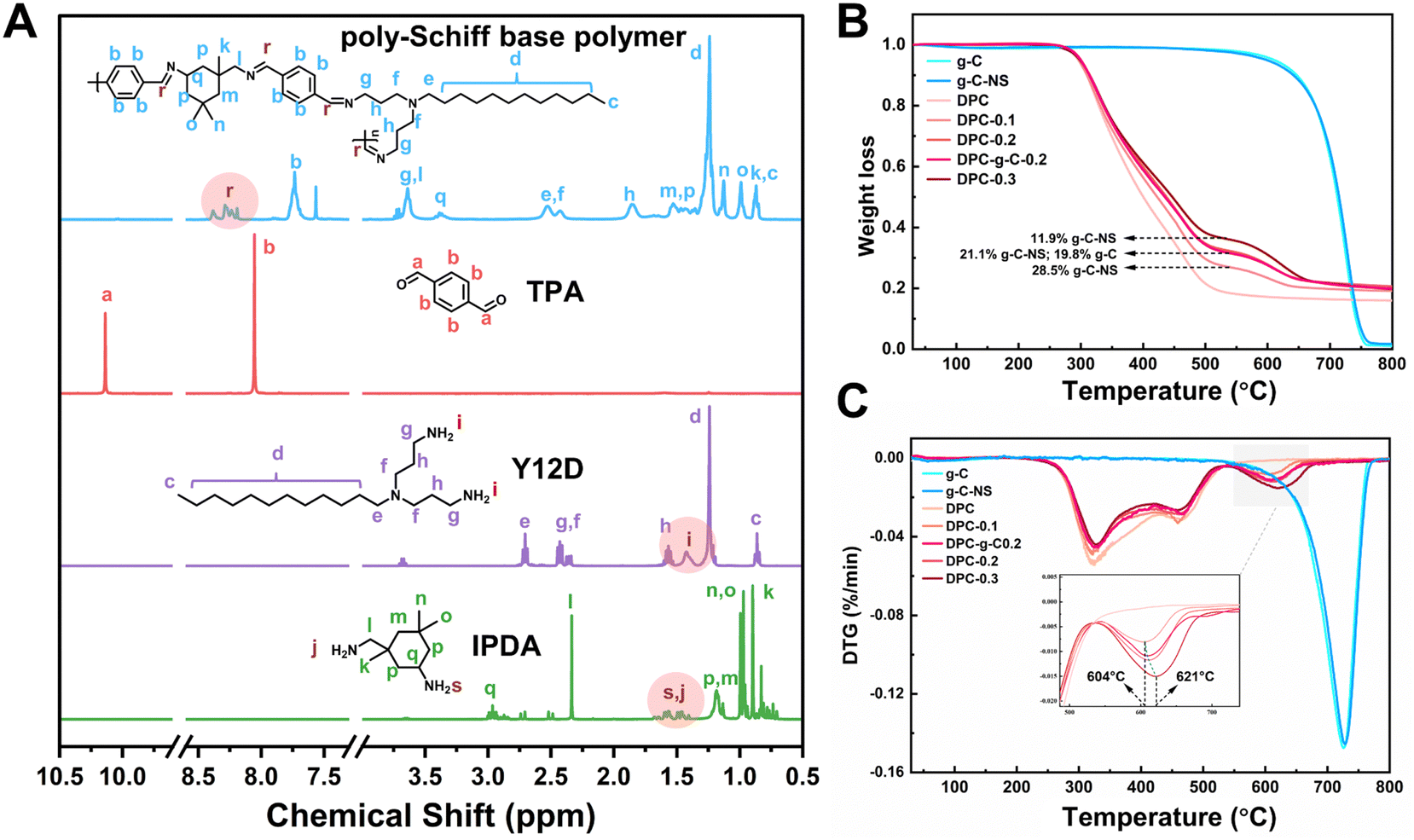 | ||
| Fig. 1 (A) NMR absorption spectra of the synthesized poly-Schiff based polymers. (B) TG analyses and (C) DTG curves for the g-C, g-C-NS and composite coatings. | ||
Furthermore, the thermal stability and chemical composition of the coatings (DPC, DPC-g-C0.2, DPC-x, where x = 0.1, 0.2, 0.3) were evaluated by TG and DTG. The thermal decomposition profiles of g-C and g-C-NS were essentially identical, with a char yield of 1.07% and 1.7% at 800 °C, indicating that the ultrathin carbon nitride nanosheets retained the original thermal stability (Fig. 1B). The DTG curve delineated the rate of mass change in the coatings as a function of temperature (Fig. 1C). The mass loss of DPC between 200 °C and 540 °C was mainly attributed to the decomposition of the poly-Schiff base polymer, with a char yield of 18.4%. As shown in the TG and DTG curves of composite coatings, the thermal degradation occurred in two stages. The second stage of the reaction (540 °C to 680 °C) was due to the thermal degradation of g-C/g-C-NS incorporated in the poly-Schiff base resin. The char residues of DPC-0.1 and g-C-NS at 540 °C were 26.9% and 97.6%, respectively, so the content of g-C-NS was calculated to be 11.9%. Similarly, the real ratios of g-C-NS/g-C to poly-Schiff base resin in DPC-0.2 and DPC-0.3/DPC-g-C0.2 were 21.1% and 28.5%/19.8%, respectively. Therefore, it was indicated that the amount of photocatalyst in the composite coating was consistent with its addition. Furthermore, as shown in the inset of Fig. 1C, the peak decomposition temperature of g-C-NS in the composite coatings increased from 604 °C to 621 °C with increasing catalyst content, which suggested that the thermal decomposition of the poly-Schiff base polymer might affect the decomposition process of g-C3N4 in the coating.
The morphology and microstructure of g-C, g-C-T, and g-C-NS were characterized using HRTEM and SEM, and their thickness was measured using SPM (Fig. 2). The melamine-derived g-C consisted of densely packed two-dimensional (2D) layers (Fig. 2A, D and G). After thermal exfoliation, the original bulk structure was etched into a loose 2D layered structure, as depicted in Fig. 2B, E and H. After liquid-phase exfoliation, the exfoliated nanosheets (g-C-NS) became thinner, as shown in Fig. 2C. All the amorphous powders showed a layered and flaky structure at the edges in the insets of Fig. 2A–C. The SEM images clearly demonstrate the denser stacking structure of the bulk g-C (Fig. 2D–F). The diameter of the ultrathin g-C-NS was about 300 nm. The average thicknesses of g-C and g-C-T were about 33.18 nm and 6.24 nm, whereas that of g-C-NS was about 1.15 nm (Fig. 2G–I). This meant that the thickness of g-C-NS was less than four atomic layers, which effectively validated the successful exfoliation for ultrathin nanosheets. Moreover, the g-C-NS has been uniformly dispersed and maintained stability in isopropanol solution for over five months (Fig. S1, ESI†).
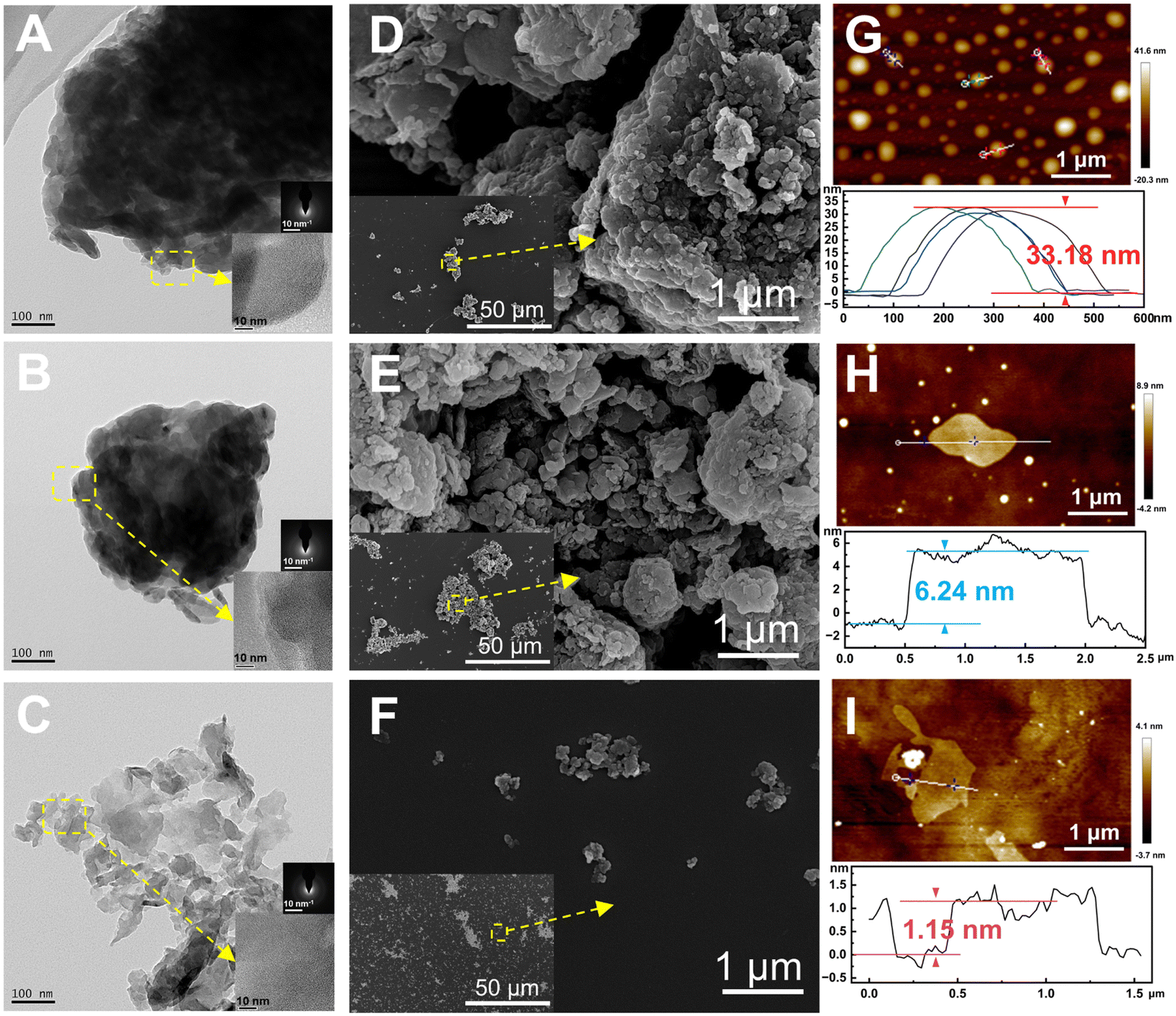 | ||
| Fig. 2 TEM images and the corresponding SAED and HRTEM (scale bars, 10 nm) of (A) g-C, (B) g-C-T, and (C) g-C-NS. SEM images of (D) g-C, (E) g-C-T, and (F) g-C-NS and their corresponding low magnification images. SPM images of (G) g-C, (H) g-C-T, and (I) g-C-NS and their corresponding height profiles. | ||
Subsequently, we explored the surface elemental composition and states of g-C, g-C-T, g-C-NS, and g-C-NS-L using XPS analysis (g-C-NS-L was obtained by immersing g-C-NS in deionized water and irradiating with visible light for 24 h). The XPS spectra showed that all samples contained mainly two elements, C and N (Fig. 3A), and their contents were generally consistent among them (Table S1, ESI†). The C 1s and N 1 s spectra were deconvoluted into three peaks, respectively. The peaks corresponding to binding energies near 284.6 eV, 286 eV, and 287.8 eV represented the C–C, C–N, and sp2 C ((N–)2CN), respectively (Fig. S2, ESI†). In the analysis for N 1s, peaks located at around 398.3 eV, 400 eV, and 401 eV were assigned to sp2 N (CN–C), sp3 N (N–(C)3), and C–NHx (NH2 or NH), respectively (Fig. 3B). The positions and shapes of such peaks were in agreement with previous reports, suggesting the successful synthesis of g-C3N4-based photocatalysts.35 There was no obvious shift in the C 1s and N 1s spectra of g-C-T, g-C-NS, and g-C-NS-L compared to bulk g-C, indicating that the chemical framework of g-C-NS was not disrupted by the exfoliation or light processes (Fig. 3B and Fig. S2, ESI†).
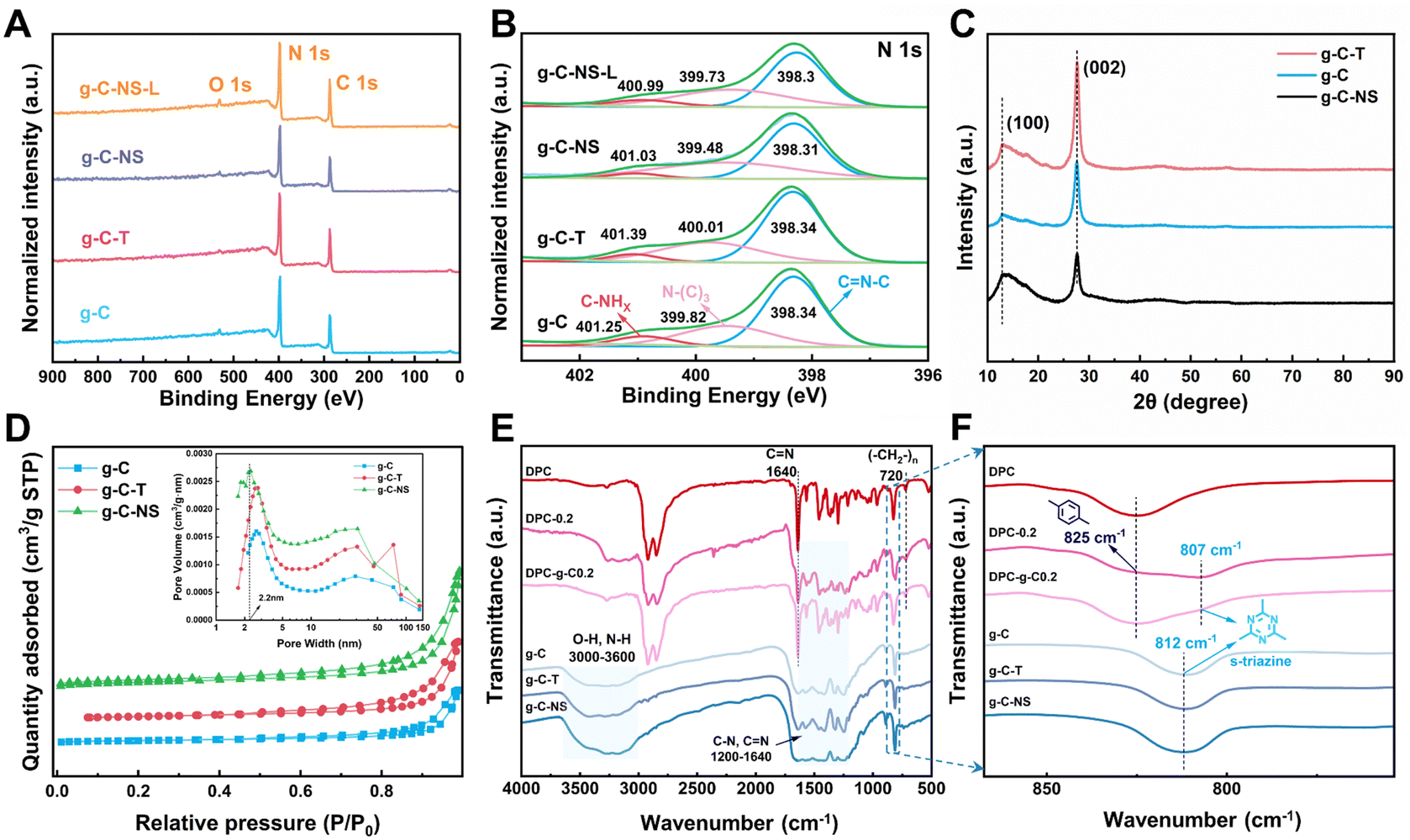 | ||
| Fig. 3 (A) XPS spectra and (B) N 1s XPS spectra. (C) XRD pattern. (D) N2 sorption isotherms of g-C, g-C-T and g-C-NS (inset: pore size distribution). (E) FT-IR spectra of the prepared photocatalysts and coatings and (F) further detail with an enlarged scale from 760 cm−1 to 870 cm−1. | ||
The crystal structures of bulk g-C, g-C-T, and g-C-NS were obtained using XRD. The characteristic peaks located at 12.8° and 27.6° were attributed to the (100) and (002) crystal planes, showing a typical graphic layered structure (Fig. 3C).36–38 These peaks represented the in-plane repeating tri-s-triazine units and the interlayer stacking of conjugated aromatic carbon–nitrogen repeating units, respectively.39 No obvious shift in the (002) peak for g-C-T and g-C-NS suggested that they had equivalent interlaminar distances.40 Conversely, a remarkable decrease in the intensity of the (002) peak implied that the stacking structure of g-C-NS was disrupted due to the two-step exfoliation, consistent with the SPM images.41 In addition, we further measured the specific surface area (SBET) and pore size distribution of the g-C3N4-based photocatalysts, as shown in Fig. 3D. The SBET for g-C, g-C-T, and g-C-NS were 12.58, 19.60, and 26.92 m2 g−1, respectively. All samples demonstrated adsorption–desorption isotherms characteristic of type IV mesoporous structures according to the IUPAC classification. The mesoporous structures were secondary pores formed by cracks between the flakes according to TEM images. g-C-NS exhibited the highest pore volume and the most extensive pore size distribution. The average pore diameter of g-C-NS was approximately 2.2 nm, which might provide more reaction sites for light absorption and photocatalysis.
The chemical structures of g-C, g-C-T and g-C-NS were further characterized using the FT-IR spectra. The absorption bands in the range of 1200–1640 cm−1 correspond to the stretching vibrations of carbon-nitrogen heterocycles (C(sp3)–N, C(sp2)N), as shown in Fig. 3E. The peaks at 812 cm−1 were attributed to the typical breathing mode of s-triazine units. The intensity of stretching vibrations between the adsorbed O–H and N–H gradually increased at 3000–3640 cm−1, confirming that some nitrogen atoms in the g-C-T and g-C-NS were partially hydrogenated.25 The results showed an increase in the content of N–H groups (–NHx) in g-C-NS due to the preparation process. This illustrated that some of the C–N or CN bonds were broken, while some nitrogen defects were formed in the g-C-NS.42 An in-plane wobble of the long-chain methylene at 720 cm−1, C–H out-of-plane bending vibration of the disubstituted toluene at 825 cm−1, and typical CN characteristic absorption peaks at 1640 cm−1 were found in the coatings, which corresponded to the internal structural features of the poly-Schiff base polymers.13,43 Meanwhile, the CO stretching vibration at 1701 cm−1 and the C–H stretching at 2735 cm−1 were attributed to –CHO in the poly-Schiff base resin coatings (Fig. S3C, ESI†). The characteristic peaks of the g-C/g-C-NS and poly-Schiff base polymer were detected in the FT-IR spectra of all composite coatings, which further proved the success of the composite. For DPC-x, the intensity of CO and N–H stretching vibrational gradually increased with the increasing g-C-NS content, while the s-triazine rings were redshifted from 812 cm−1 to 807 cm−1 compared to g-C (Fig. 3F and Fig. S3, ESI†). This peak enhancement may be attributed to hydrogen bond interactions between the –CHO and –NH2 of the poly-Schiff base and the –OH and –NH2 of g-C and g-C-NS (Fig. S3C, ESI†). However, the redshift may be attributed to interactions between conjugated imine networks (–CN–) and the s-triazine ring.44 The poly-Schiff base polymer affected the s-triazine ring π–π conjugation system of g-C and g-C-NS through ultrasound-assisted intercalation. The stretching vibration of N–H in 3000–3270 cm−1 further increased after immersion, suggesting that the hydrolysis of the polymer increased the content of –NH2 and –CHO groups on the composite coatings (Fig. S3D, ESI†). The positions and shapes of the absorption peaks were essentially the same before and after degradation, which meant that the molecular structure of the coating had not been destroyed and the chemical state remained stable.
3.2. Light absorption and photoelectric properties of the photocatalysts
The light absorption properties of g-C, g-C-T, and g-C-NS were characterized using UV-vis DRS spectra and UV spectroscopy. The absorption edge of all the g-C3N4-based photocatalysts was around 460 nm (Fig. 4A). The bandgaps of g-C, g-C-T, and g-C-NS were estimated to be 2.59 eV, 2.55 eV, and 2.56 eV, respectively, based on Tauc plots (the inset of Fig. 4A).25 As depicted in Fig. 4B, the absorbance of all samples was below 0.3 at a dispersion concentration of 0.1 mg mL−1. Conversely, at 0.2 mg mL−1, the absorbance of g-C-NS was 4 times that of g-C or g-C-T. An optical path length was significantly extended by the 2D layered and pore structure of g-C-NS, consequently enhancing the light absorption and trapping. The color of g-C-NS suspensions showed an opaque milky white color and remained dispersed for almost 24 h, whereas g-C and g-C-T were completely precipitated within 3 h (Fig. 4C). These observations showed that the ultrathin g-C-NS with a 2D layered structure enhanced their light absorption and dispersion in aqueous solution. This exceptional light absorption capability was deemed crucial for enhancing photocatalytic performance.45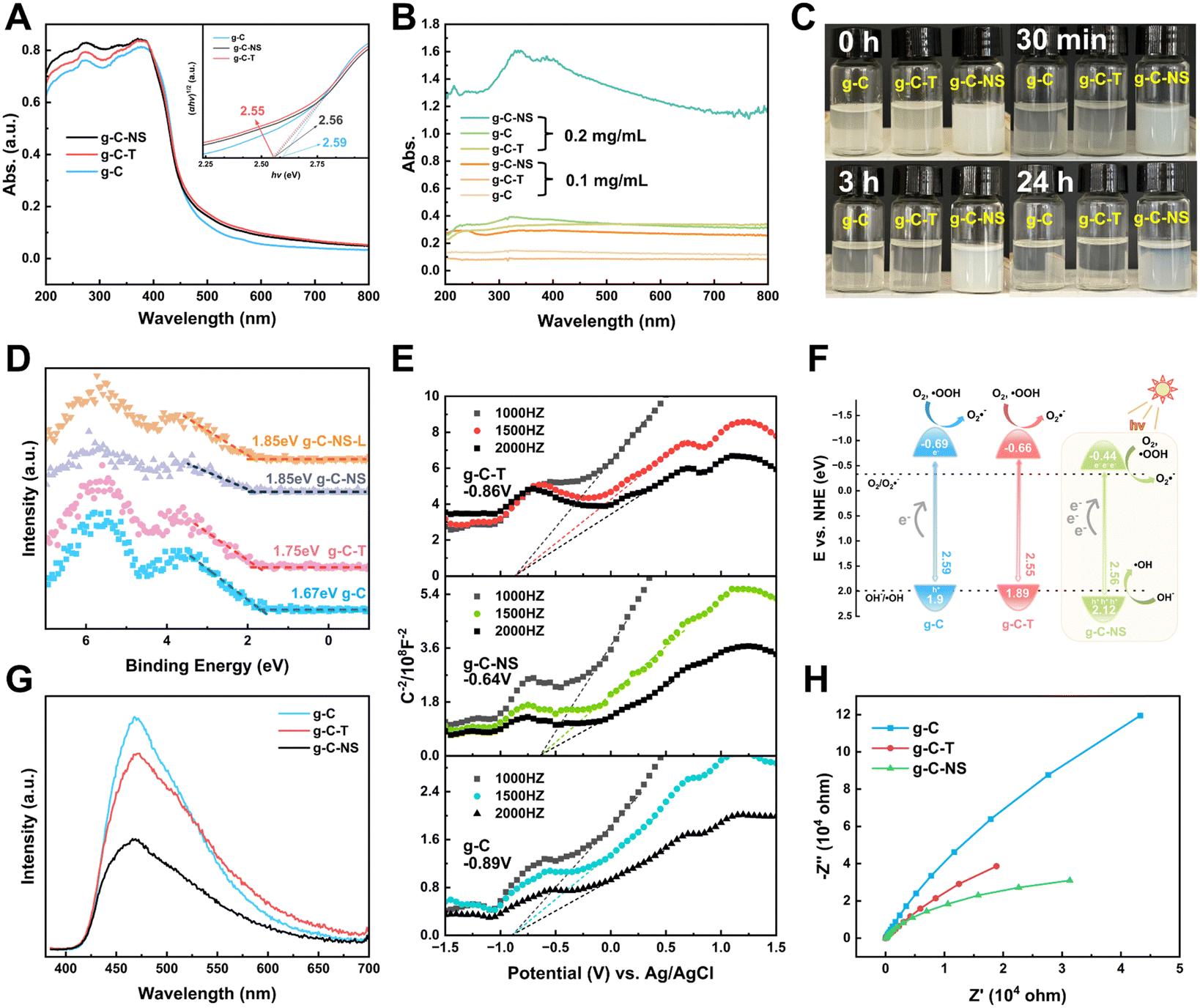 | ||
| Fig. 4 (A) UV-vis DRS spectra (inset: plots of (αhν)1/2versus hν). (B) Absorption spectra of photocatalyst suspensions. (C) Photocatalyst suspensions. (D) XPS-VB spectra. (E) Mott–Schottky plots. (F) Band energy levels. (G) PL spectra. (H) Electrochemical impedance spectra (EIS). | ||
We employed XPS-VB and Mott–Schottky plots to ascertain the relative positions of the bandgap, valence band (VB), and conduction band (CB) of the photocatalysts. The XPS-VB spectra revealed the highest occupied molecular orbital (HOMO) corresponding to the position of the maximum VB (VBM) (Fig. 4D). Surprisingly, the VBM values of g-C, g-C-T, g-C-NS, and g-C-NS-L were 1.67 eV, 1.75 eV, 1.85 eV, and 1.85 eV, respectively. The VBM values of g-C-NS and g-C-NS-L were not altered by short-term light. It has been reported that the VB edge of the density of states (DOS) for single-layer g-C3N4 nanosheets was significantly higher than that of bulk g-C, which derived from an enhancement in the p orbital hybridization of nitrogen.46 Some defects (nitrogen defects and oxygen doping) arising from high-temperature treatment or intercalation may alter the electron distribution in g-C-NS, introducing more reactive sites with dangling bonds (–NHx, –OH) and mid-gap states between the VB and CB, which in turn affected the shapes and positions of the VB spectra. The mid-gap states absorbed photons (hν) with energies below the bandgap, thereby enhancing the light absorption and augmenting the driving force for redox reactions.47,48
The lowest unoccupied molecular orbital (LUMO) of the samples, also known as the position of the minimum CB (CBM), was determined from the Mott–Schottky plots. The Schottky plots of the g-C, g-C-T, and g-C-NS indicated flat potential values of approximately −0.89 V, −0.86 V, and −0.64 V vs. Ag/AgCl (the CB potentials were −0.69 eV, −0.66 eV, and −0.44 eV vs. NHE), respectively (Fig. 4E).34,40 The results showed that the calculated VB potentials were in close agreement with the XPS-VB (Fig. 4F). The redox potentials of photocatalysts were usually expressed in the positions of the HOMO and LUMO energy levels. Importantly, the reduction potential of all three samples satisfied the thermodynamic condition of reducing O2 to O2˙− (−0.33 eV vs. NHE),49 a process capable of oxidizing and decomposing organic compounds into non-toxic and benign small molecules.50,51 In addition, g-C-NS met the thermodynamic conditions for oxidation of OH− to ˙OH (+1.99 eV vs. NHE), which produced more antimicrobial reactive species.52
Additionally, we employed the EPR system to characterize the reactive oxygen species (ROS) produced by g-C, g-C-T, and g-C-NS in the dark/in in situ illumination (λ > 420 nm, for 1, 5, 10, and 20 min). No formed adduction was detected in the methanol solution in a dark environment (Fig. S4, ESI†). However, after light for 1 min, distinct signals of the DMPO–˙OOH and the triplet-peak of DMPOO adducts appeared.53 It had been reported that deprotonation of hydroperoxyl radicals (˙OOH) led to the formation of superoxide radicals (O2˙−), which was identical with the reduction potential.54 The formation of DMPOO suggested the presence of a potent oxidizing agent in the system capable of oxidizing DMPO to DMPOO. The highest levels of ROS generation were obtained by g-C-NS in visible light among all systems. After lighting for 5 min, the signal of DMPO–˙CH2OH adducts took shape. After lighting for 10 min, the intensity of the DMPO–˙OOH signal reduced significantly, accompanied by an enhancement in the DMPOO and DMPO–˙CH2OH signals, indicating that these radicals were transformed. The generation of ˙OOH preceded that of hydroxylated methyl radical (˙CH2OH), suggesting that the latter might be accompanied by side reactions:55
 | (1) |
| O2 + e− + H+ → ˙OOH | (2) |
| ˙OOH ⇌ O2˙− + H+ | (3) |
| h+ + OH− → ˙OH | (4) |
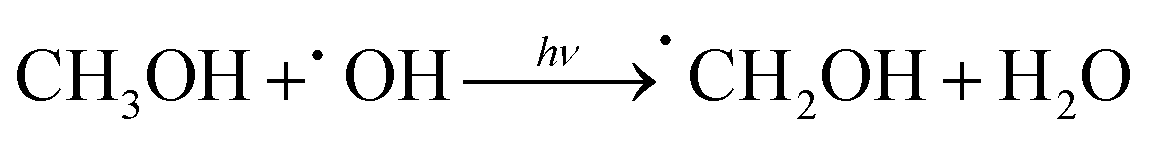 | (5) |
In the water, DMPO–˙OH adducts (1:2:2:1 intensity) and the DMPOO signals were generated after 5 min of light in the g-C-NS system, whereas the g-C and g-C-T systems exhibited negligible signals of radicals, which was in accordance with the oxidation potential.
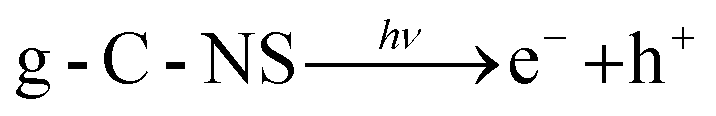 | (6) |
| OH− + h+ → ˙OH | (7) |
The experimental findings also showed that the g-C-NS could effectively and rapidly generate ROS with a strong redox potential, specifically ˙OOH, and ˙OH. These radicals can attack organic compounds through radical addition, electron transfer, or hydrogen abstraction mechanisms.56 Based on CBM, bandgap values, and ROS, the band structures of g-C, g-C-T, and g-C-NS are shown in Fig. 4F.57
The photocatalyst performance is not only influenced by light absorption but also by carrier separation and transfer efficiency. We assessed the photogenerated electron–hole (e−–h+) pair recombination and the charge separation and transfer of the samples by photoluminescence (PL) and electrochemical impedance spectroscopy (EIS). At an excitation wavelength of 365 nm, as depicted in Fig. 4G, g-C-NS exhibited the lowest fluorescence response intensity at 467 nm, indicating the lowest recombination efficiency of photogenerated carriers, followed by g-C-T and g-C.49 It was a signal for the efficient separation of e− and h+. g-C-NS has more defects and dangling bonds with a large number of charge-capture sites than g-C and g-C-T, trapping the photo-induced e− and h+.58 Therefore, the spatial-temporal separation of radiative charges was accelerated to a large extent, which might improve the photocatalytic performance. The radius of the Nyquist plot corresponded to the charge transfer resistance (RCT) of the photocatalysts, and the equivalent circuit diagram are shown in Fig. S5 and Table S2 (ESI†). In the dark, g-C-NS exhibited the smallest Nyquist plot radius and the value of RCT among the tested samples, which indicated a higher electron conductivity (Fig. 4H).59 The 2D layered structure and large specific surface area shortened the time for charges to reach the reaction center on the g-C-NS surface, which enhanced the charge migration efficiency. Combined with the enhanced light absorption, electron separation and transfer, it is evident that the g-C-NS have excellent photocatalytic properties.
3.3. Degradation properties of coatings
N bonds by water molecules, and it can be fully degraded into small molecules, avoiding microplastic contamination, which presents the prominent potential for antifouling applications.13 XPS spectra were utilized to analyze the elemental composition and state of the DPC and DPC-0.2 surface before and after degradation. Notably, the oxygen-containing groups on the degraded coating surface increased, as evidenced by the significant enhancement of its O 1s peak (Fig. 5A). The C 1s spectra were separated into three peaks at 284.7 eV, 285.6 eV, and 287.9 eV, assigned to C–C, CC, and –CO, respectively (Fig. 5B and D). The N 1s peaks at 398.7 eV, 399.9 eV, and 401.8 eV, correspond to CN, N–C, and –NH2, respectively (Fig. 5C and E). After degradation, the content of –CO corresponding to –CHO groups on the DPC surface increased from 3.8% to 10.3%, and for the DPC-0.2 surface, it rose from 2.8% to 10.6% (Table S3, ESI†). The content of CN on the DPC surface decreased from 62.4% to 34.9%, whereas that of the –NH2 elevated from 10.3% to 12.2%. Importantly, the content of CN on the DPC-0.2 surface dramatically reduced from 81.7% to 47%, while the content of –NH2 increased from 3.1% to 9.4%. These findings validated the breaking of imine bonds and the generation of –CHO and –NH2 on the poly-Schiff base coating surface. DPC-0.2 had a higher degree of hydrolysis compared to DPC, which was consistent with the results from FT-IR.
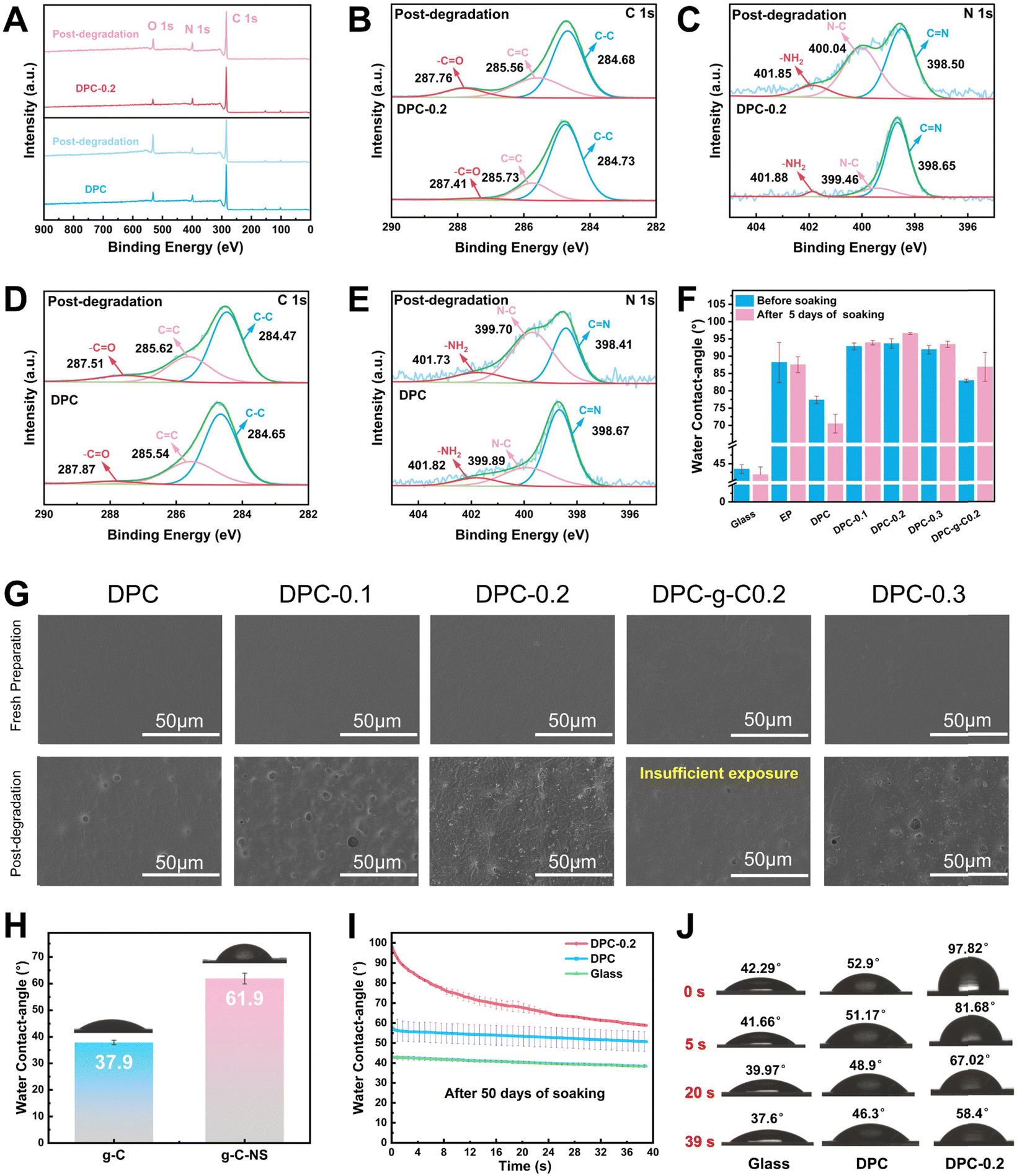 | ||
| Fig. 5 Degradation properties of a series of poly-Schiff base coatings before and after deionized water immersion (DPC-0.2 and DPC refer to freshly prepared coatings). (A) XPS spectra, (B) and (D) C 1s XPS spectra, and (C) and (E) N 1s XPS spectra. (F) and (G) Contact angle and Surface morphology of the coatings after immersion in deionized water for 5 days in the dark. (H) Contact angle of g-C and g-C-NS. (I) Contact angle and (J) typical shape of the contact angle of the coatings at degradation equilibrium. | ||
In addition, the surface morphology and water contact angles of the coatings were characterized in a simulated application environment to further understand their degradation behavior. As depicted in Fig. 5G, g-C-NS and g-C were not detected on the coating surfaces. Following immersion for 5 days, some concave holes with an average diameter of 4.5 μm were formed on the DPC surface (Fig. S6, ESI†), attributed to the hydrolysis of the imine bonds in the polymer chains. Hydrolysis gradually broke down and renewed the surface polymers of the composite coatings, exposing the internal g-C-NS or g-C. The density of holes correspondingly increased with the increasing g-C-NS content. Notably, the DPC-0.2 surface has the largest number of exposed g-C-NS with clear edge morphology. The anchored g-C-NS were uniformly distributed on the coating surface, while g-C was not sufficiently exposed on the DPC-g-C-0.2 surface. Additionally, cross-sectional SEM images indicated that g-C-NS had better dispersibility and compatibility with the poly-Schiff base polymer interface than g-C (Fig. S7, ESI†). g-C particles were significantly agglomerated in DPC-g-C-0.2, resulting in increased defects and porosity. The enhanced interfacial compatibility may stem from the stronger hydrogen bonding interactions between g-C-NS and the poly-Schiff base polymer matrix. In summary, it was shown that g-C-NS were more suitable than g-C for enhancing the photocatalytic and degradation performance of the poly-Schiff base coating.
The water contact angles of the coatings before and after degradation are shown in Fig. 5F. The contact angles of EP coating were 88.16° and 87.53° before and after immersion, respectively, showing a stable surface wettability. In contrast, the contact angle of DPC reduced from 77.35° to 70.48°. This was due to the increase in surface hydrophilic –CHO and –NH2 terminals (Fig. 5D and E), which readily formed hydrogen bonds with water molecules. Before degradation, the contact angle of the composite coatings was significantly increased due to the wrinkled structure of the surface (Fig. S6, ESI†). These inter-nanostructure gaps diminished the contact area between water droplets and the polymer. After degradation, internal g-C-NS were gradually exposed on the surface of DPC-x. These nanosheets popped up like small buds, forming concave–convex structures and further reducing the contact angle. Moreover, the average contact angle of g-C-NS (61.9°) was larger than that of g-C (37.9°), which indicated that the nano-flake structure conferred greater hydrophobicity than the micro-particle structure (Fig. 5H). After 50 days of degradation, the contact angle of DPC-0.2 rapidly decreased from 97.82° to 58.4° over time, whereas that of DPC only altered slightly (Fig. 5I and J). At this stage, the contact angle values were lower than the original value at 5 days of immersion. It showed that the number of aldehyde and amino terminals on the surface increased with the duration of immersion. Water molecules initially encountered the surface created by g-C-NS, and then gradually interacted with a large number of amino and aldehyde groups on the surface, leading to a rapid decrease in the contact angle to form a hydrophilic surface. It was conducive for the formation of a hydration layer to prevent the microorganism adhesion.60
| ln(C0/Ct) = kt |
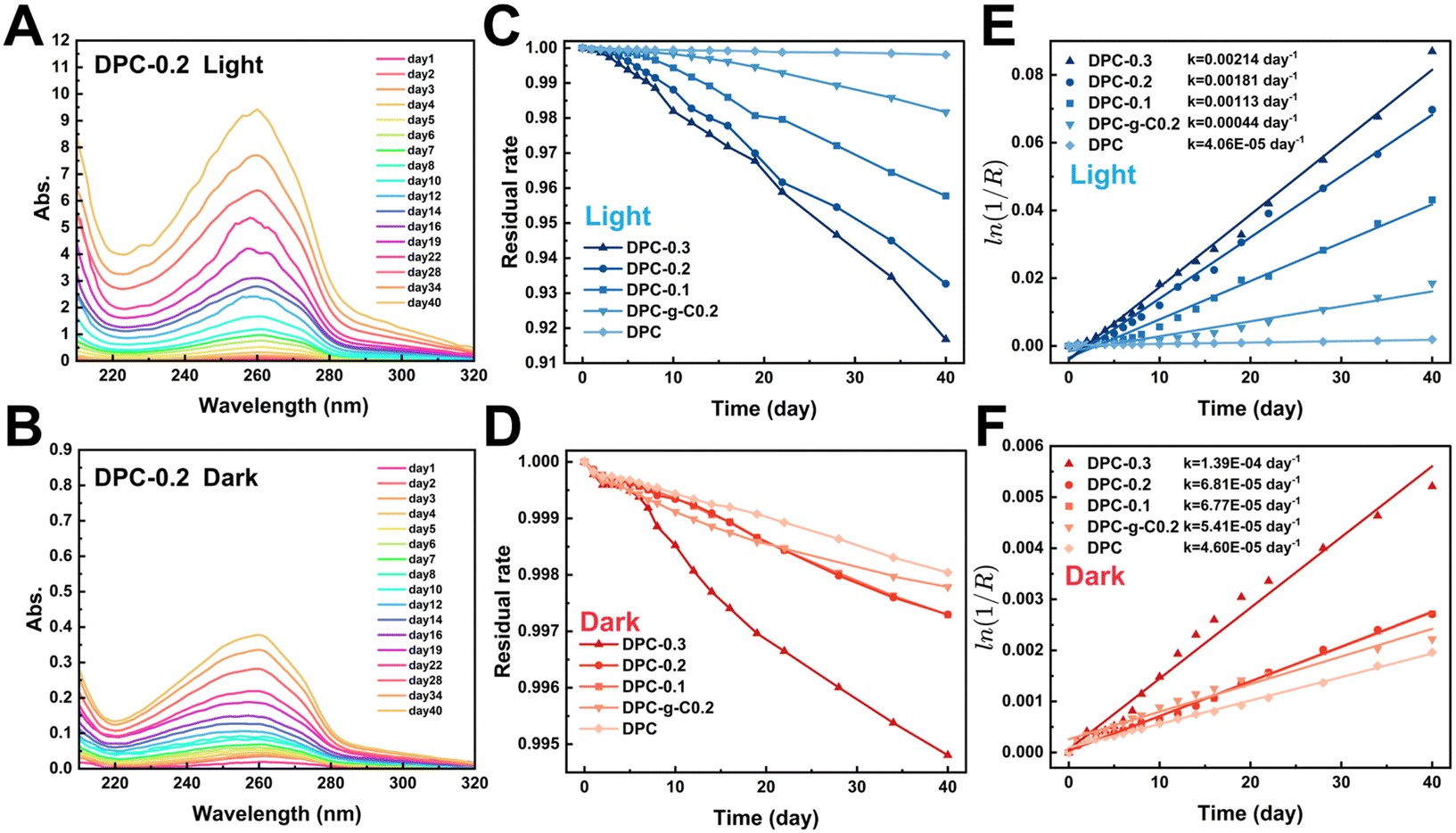 | ||
| Fig. 6 (A) and (B) Absorption spectra of DPC-0.2 degradation solution under light/dark conditions at 293.15 K. (C) and (D) Residual rate of composite coatings under light/dark conditions. (E) and (F) Linear fittings of the pseudo-first-order kinetic model for degradation of the coating under light/dark conditions at 293.15 K. | ||
Notably, the characteristic absorbance of TPA in DPC-x was significantly enhanced in the light (Fig. S9, ESI†). The change in absorbance indirectly reflected the extent of the degradation. Fig. 6C and D illustrate that the mass loss rate of the composite coatings increased significantly in the light. After 20 days, the residual rate profiles approached linearity, indicating stable degradation rates. There were 1–2 orders of magnitude differences in degradation reaction rate constant values for the DPC-x in the dark/light, while that of DPC was almost the same (Fig. 6E and F). For instance, the absorption peak of the DPC-0.3 degradation solution was 16 times higher in the light than in the dark, and the rate constant values were also larger in the light (2.14/103 day−1) than in the dark (1.39/104 day−1). The rate of the fracture of CN bonds of the poly-Schiff base polymer was accelerated by the photocatalytic effect of g-C/g-C-NS, leading to a significant increase in degradation product TPA. Moreover, the degradation reaction rate of DPC-0.2 notably exceeded that of DPC-g-C0.2 in the light, which were 1.81/103 day−1 and 4.4/104 day−1, respectively. Specifically, the degradation reaction rate of DPC-0.2 was 40 times that of DPC. This showed that the photocatalytic effect of g-C-NS accelerated dynamic self-renewal processes of poly-Schiff base polymer more than g-C, and this promotion was greatly pronounced with the increase of the g-C-NS content.
With the increased exposure of g-C-NS on the DPC-0.2 surface, the holey structures were larger in diameter and more numerous compared to the pre-light (Fig. S6 and S10, ESI†). In addition, the holes formed by DPC-0.2 were denser and more homogeneous than those of DPC-g-C0.2. From the cross-sectional morphology, g-C-NS was also more uniformly disseminated in the coating than g-C, which was consistent with before degradation (Fig. S7 and S10, ESI†). The rough surface increased the degradation sites of the polymer and accelerated the interfacial self-renewal. We considered that the active species (ROS, e−, and h+) generated by the g-C-NS interacted with the poly-Schiff base polymer framework, which rapidly initiated interfacial reactions between the g-C-NS, water, and the polymer in light. The exposure of g-C-NS to the coating surface improved the photocatalytic efficiency. With the accelerated degradation of the polymer matrix surrounding g-C-NS, the exposed area of g-C-NS in water was increasing until it was released. A hole centered on g-C-NS was formed on the DPC-x surface. This degradation mechanism ensured the continuous exposure of g-C-NS from the DPC-x, which not only prompted an efficient photocatalytic reaction but also made 100% utilization of g-C-NS possible.
3.4. Photocatalytic antibacterial activity of composite coating
Bacteria preferred to adhere to glass and EP surfaces, suggesting that non-degradable solid surfaces were prone to biofilm formation for bacterial growth and adhesion (Fig. 7A). The number of E. coli adhered to the DPC and composite coatings was dramatically reduced compared to glass and epoxy coatings under light/dark conditions (Fig. 7B and Fig. S12, ESI†). Surprisingly, the DPC exhibited an excellent antibacterial ability under both light and dark conditions, with antibacterial rates of 98.2% and 94.58%, respectively, highlighting the inherent fouling-release capabilities (Fig. 7C). In visible light, the antibacterial rate of the DPC-x reached nearly 100%. In the dark, the DPC-x also maintained great antimicrobial resistance, with DPC-0.2 achieving 99.31%, followed by DPC-0.3 and DPC-0.1, both with 97.36%. The antibacterial efficacy of the DPC-x was enhanced with increasing g-C-NS content, responsible for the improved photocatalytic antifouling and self-renewal properties. There was an overwhelming number of E. coli on the DPC-g-C0.2 surface, which was attributed to a conducive environment for bacterial adhesion created by the bulk g-C particles. In addition, in light for 12 h, the g-C/g-C-NS and degradation products released via the self-renewal of the DPC-g-C0.2 and DPC-0.2 surface killed 94.18% and 85.2% of the bacterial in the solution, while the number of bacteria in the degraded solution of DPC remained the same under light/dark conditions (Fig. 7B). It was suggested that the g-C/g-C-NS released by the composite coating yielded a notable photocatalytic antibacterial effect, although the released small molecules with aldehyde and amine groups had a slight inhibitory effect on E. coli.61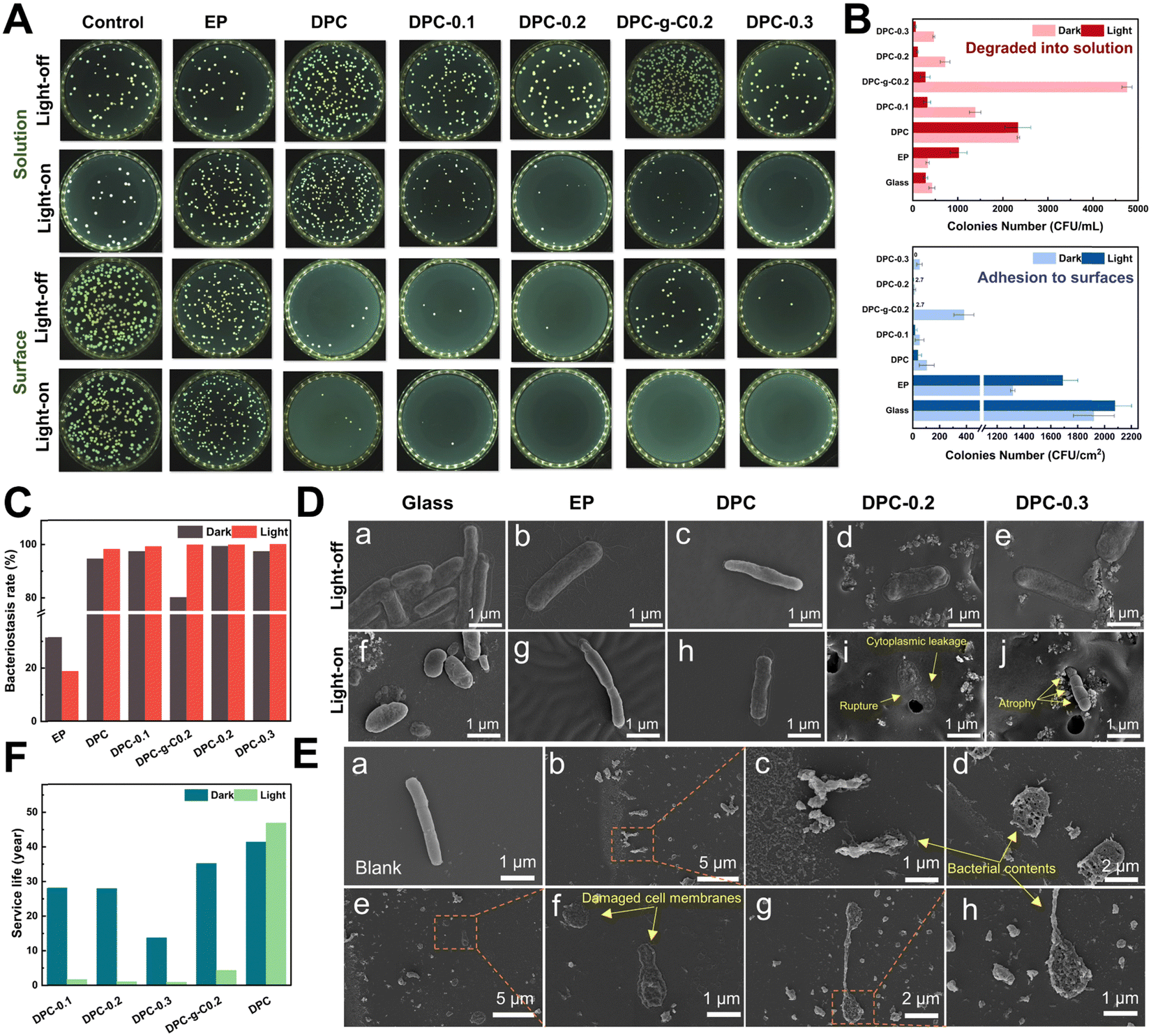 | ||
| Fig. 7 (A) Photographs of colonies from antibacterial tests under light/dark conditions. (B) Concentration of surviving bacteria attached to the surface of the coating/in the solution after the coatings were immersed in PBS solution for 12 h. (C) Bacteriostasis rate of coatings. (D) Morphological changes of bacteria on the glass and coating surfaces. (E) Morphology of bacteria in the blank control group (a) and in g-C-NS suspensions (b)–(h) (0.04 mg mL−1) after 24 h illumination (λ > 400 nm). (F) Service life of the degradable coatings calculated by the half-life formula. | ||
It was impressive to find that the g-C/g-C-NS and degradation products released by the composite coating effectively suppressed the growth of bacteria in the solution, even with a degradation time of only 12 h. The outstanding antibacterial performance was due to the reactive oxygen species (ROS) generated by the g-C-NS in light, which were known to damage bacterial cell walls and membranes, thus inhibiting or killing the bacteria.63 In addition, an antibacterial test with different concentrations of photocatalysts was conducted. All the synthesized g-C3N4-based catalysts suppressed at least 80% of bacterial proliferation at an exceedingly low concentration of 0.01 mg mL−1 (Fig. S11, ESI†). These findings showed that the DPC-x and g-C-NS possessed excellent photocatalytic antimicrobial properties in visible light.
Rod-shaped E. coli, approximately 2–3 μm in length, were detected on glass, EP surfaces, and PBS solutions with full cytoplasm, intact cell membranes, and no signs of damage (Fig. 7D(a), (b), (f), (g) and E(a)). On the DPC surface, the hydrolyzed surface of the poly-Schiff base polymer had little disruptive effect on the integrity of the E. coli (Fig. 7D(c) and (h)). Conversely, bacteria on DPC-x showed cell membrane rupture and cytoplasmic leakage, while those that had not yet ruptured appeared to have membrane atrophy (Fig. 7D(i) and (j)). Besides, numerous leaked contents of dead bacteria, including damaged cell membranes and part of the cytoplasm, were observed in the g-C-NS suspensions (Fig. 7E(b)–(h)). g-C-NS impaired the integrity of the bacterial cells and oxidized organic substances, leading to bacterial demise. These tests emphasized that the photocatalytic effect of g-C-NS on bacterial damage was reliable and effective, whether exposed to the coating surface or present in the suspension.
Antifouling efficacy and service life are two crucial references in the practical application of anti-biofouling coatings, which directly affect the maintenance and economy of equipment. Theoretically, based on the half-life equation t1/2 = ln2/k of pseudo-first-order reaction, the half-life (t1/2) of the degradable coatings serves as a reference index for their service life (Fig. 7F). Accordingly, under continuous 24 h illumination per day, the estimated service life (in years/100 μm) of coating to reach t1/2 is: DPC (46.8 years) > DPC-g-C0.2 (4.32 years) > DPC-0.1 (1.68 years) > DPC-0.2 (1.05 years) > DPC-0.3 (0.887 years). Similarly, under continuous 24 h darkness per day: DPC (41.3 years) > DPC-g-C0.2 (35.1 years) > DPC-0.1 (28.1 years) > DPC-0.2 (27.9 years) > DPC-0.3 (13.7 years). Therefore, we conservatively predict the lifetime of antifouling coating (100 μm) under simulated actual diurnal cycling to be, in order, DPC (42.6 years) > DPC-g-C0.2 (12.6 years) > DPC-0.1 (5.7 years) > DPC-0.2 (3.8 years) > DPC-0.3 (3.0 years). Conventional degradable polymer coatings have been reported to achieve control of degradation rates of polymers by modifying the structure of the polymers with degradable main chains and hydrolyzable side chains.12,64 In this work, the desired self-renewal rate of the poly-Schiff base resin coating can be adjusted by regulating the content of g-C-NS and setting the light processes. Considering the service life and antibacterial efficacy of the DPC-x, 20 wt% of the g-C-NS seems to be the best choice.
3.5. Synergistic antibacterial mechanism of the DPC-x
Traditional photocatalytic antimicrobial coatings are predominantly effective in strong UV light, with diminished antibacterial efficacy in visible light or subpar illumination. Conversely, the DPC-x prepared in this study exhibit an antibacterial rate of at least 97.36% in the dark and nearly 100% under visible light irradiation. It greatly makes up for the shortcomings of existing photocatalytic coatings. The excellent inhibition may be attributed to synergistic antifouling mechanisms (Fig. 8). Firstly, the enhanced self-renewal of the DPC-x, which continuously hydrolyzed to release small molecules to form a dynamic self-renewal surface that removed adhering bacteria. The hydrolysis-driven degradation mechanism had applicability even under light-free conditions, effectively improving the fouling-release ability. Secondly, the wettability of the coating was rapidly shifted from hydrophobic to hydrophilic by the exposed g-C-NS on the DPC-x surface, which further facilitated the fouling-release and the formation of a hydration layer. g-C-NS also represented a negative zeta potential in water (Fig. S13, ESI†). Upon hydrolysis of the coating surface, the exposed g-C-NS helped to repel bacterial surface anions, creating a barrier that deterred bacteria from approaching. Thirdly, g-C-NS boosted the self-renewal of the DPC-x surface and exhibited exceptional photocatalytic antibacterial activity. Moreover, the released g-C-NS from the DPC-x were the most effective agents for killing bacteria in visible light, preventing the biofouling of the coating surroundings. Therefore, the DPC-x avoided the deactivation of the photocatalytic function on the active top layer of the coating via the dynamic self-renewal surface, so that the g-C-NS remained in contact with water, light, and bacteria all the time, which ensured that the photocatalytic reaction could be continued. Ultimately, the enhanced self-renewal and effective photocatalytic antimicrobial properties allowed DPC-x to exhibit robust, long-term, and all-weather biofouling resistance. | ||
| Fig. 8 Synergistic antimicrobial mechanism of the composite coating. | ||
4. Conclusions
In summary, we designed and prepared a photocatalytic and self-renewed g-C3N4 nanosheet/poly-Schiff base composite coating, which effectively eliminated microbial adhesion on the coating and reduced bacterial proliferation in the solution surrounding the coating. Ultrathin g-C-NS (1.15 nm) showed 4-time light absorption (200–800 nm) in 0.2 mg mL−1 suspensions compared to bulk g-C, and they also produced reactive oxygen species (˙OOH, ˙OH) more rapidly. g-C-NS were homogeneously dispersed in the degradable resin coating, significantly enhancing the self-renewal rate of the poly-Schiff base resin coating in visible light. Specifically, the degradation reaction rate of DPC-0.2 (20 wt% g-C-NS) was 40 times that of DPC, thus improving the surface self-renewal and fouling-release capabilities. In addition, g-C-NS at a concentration of 0.01 mg mL−1 inhibited about 80% of bacterial growth in visible light. The g-C-NS exposed from the DPC-x led to bacterial membrane rupture/atrophy and cytoplasmic leakage, showing exceptional photocatalytic antibacterial effects. Under light conditions for 12 h, the g-C-NS and degradation products released via the self-renewal of the DPC-0.2 surface killed 85.2% of the bacteria in the solution. The antimicrobial rates of DPC and DPC-0.2 were 94.58%, 99.31% in the dark and 98.2% and 99.87% in visible light. With the dual-functional antifouling mechanism of enhanced self-renewal and the highly efficient photocatalytic antibacterial activity, the DPC-x exhibited excellent antifouling effects under all-weather conditions. The self-renewal rate of DPC-x and the release of g-C-NS can be controlled by visible light, and the predicted service life of DPC-0.2 (thickness of 100 μm) is about 3.8 years. The dual-functional g-C3N4 nanosheet/poly-Schiff base composite coating is expected to achieve long-term biofouling resistance and address the limitations of conventional photocatalytic coatings in light absorption and photocatalyst utilization.Author contributions
Saijun Wu: conceptualization, methodology, investigation, data curation, formal analysis, visualization, and writing original draft. Minglong Yan: conceptualization, methodology, investigation, formal analysis, visualization, funding acquisition, project administration, and review and editing. Yinghao Wu, Yangmin Wu, Xijian Lan and Jianjun Cheng: investigation, resources and validation. Wenjie Zhao: supervision, validation, funding acquisition, visualization, and writing, review and editing.Data availability
All relevant data are within the manuscript and its ESI. And detailed data are available from the corresponding author upon reasonable request.Conflicts of interest
The authors declare no competing interests.Acknowledgements
This work was supported by the National Natural Science Foundation of China (52103133), the Science and Technology Program of Zhejiang Province (2023C03013), Zhejiang Province Key Technology Project (2022C01183), the Science and Technology Innovation 2025 Major Project of Ningbo (2020Z053), the National Natural Science Foundation of China (52105230), and “Pioneer” and “Leading Goose” R&D Program of Zhejiang (2023C01089).References
- P. Vuong, A. McKinley and P. Kaur, npj Mater. Degradation, 2023, 7, 50 CAS
.
- H. Qiu, K. Feng, A. Gapeeva, K. Meurisch, S. Kaps, X. Li, L. Yu, Y. K. Mishra, R. Adelung and M. Baum, Prog. Polym. Sci., 2022, 127, 101516 CAS
- J. Pan, X. Ai, C. Ma and G. Zhang, Acc. Chem. Res., 2022, 55, 1586–1598 CAS
- S. Pourhashem, A. Seif, F. Saba, E. G. Nezhad, X. Ji, Z. Zhou, X. Zhai, M. Mirzaee, J. Duan, A. Rashidi and B. Hou, J. Mater. Sci. Technol., 2022, 118, 73–113 CAS
- E. Seo, M. R. Seong, J. W. Lee, H. Lim, J. Park, H. Kim, H. Hwang, D. Lee, J. Kim, G. H. Kim, D. S. Hwang and S. J. Lee, ACS Omega, 2020, 5, 11515–11521 CAS
- S. Tian, D. Jiang, J. Pu, X. Sun, Z. Li, B. Wu, W. Zheng, W. Liu and Z. Liu, Chem. Eng. J., 2019, 370, 1–9 CAS
- W. Wang, Y. Lu, H. Zhu and Z. Cao, Adv. Mater., 2017, 29, 1606506 Search PubMed
- Z. Yu, X. Li, X. Li, B. Zheng, D. Li, D. Xu and F. Wang, Adv. Funct. Mater., 2023, 33, 2305995 CAS
- Z. Tong, F. Gao, S. Chen, L. Song, J. Hu, Y. Hou, J. Lu, M. K. H. Leung, X. Zhan and Q. Zhang, Adv. Mater., 2024, 36, 2308972 CAS
- Y. Guo, M. Yan and W. Zhao, J. Colloid Interface Sci., 2024, 653, 833–843 CAS
- H. Agarwal, L. D. J. Quinn, S. C. Walter, T. J. Polaske, D. H. Chang, S. P. Palecek, H. E. Blackwell and D. M. Lynn, ACS Appl. Mater. Interfaces, 2022, 14, 17940–17949 CAS
- G. Dai, X. Ai, L. Mei, C. Ma and G. Zhang, ACS Appl. Mater. Interfaces, 2021, 13, 13735–13743 CAS
- M. Yan, Y. Guo and W. Zhao, Adv. Mater. Interfaces, 2022, 9, 2101920 CAS
- W. Liu, M. Yan and W. Zhao, J. Colloid Interface Sci., 2023, 629, 496–507 CAS
- J. Ran, J. Zhang, J. Yu, M. Jaroniec and S. Z. Qiao, Chem. Soc. Rev., 2014, 43, 7787–7812 CAS
- H. Guo, L. Song, J. Hu, T. Lin, X. Li, H. Yu, D. Cheng, Y. Hou, X. Zhan and Q. Zhang, Chem. Eng. J., 2021, 420, 127676 CAS
- L. Marín-Caba, G. Bodelón, Y. Negrín-Montecelo and M. A. Correa-Duarte, Adv. Funct. Mater., 2021, 31, 2105807 Search PubMed
- W.-J. Ong, L.-L. Tan, Y. H. Ng, S.-T. Yong and S.-P. Chai, Chem. Rev., 2016, 116, 7159–7329 CAS
- Q. Cao, B. Kumru, M. Antonietti and B. V. K. J. Schmidt, Mater. Horiz., 2020, 7, 762–786 CAS
- J. Wang and S. Wang, Coord. Chem. Rev., 2022, 453, 214338 CAS
- Y. Wang, X. Wang and M. Antonietti, Angew. Chem., Int. Ed., 2012, 51, 68–89 CAS
- H. Dong, X. Guo, C. Yang and Z. Ouyang, Appl. Catal., B, 2018, 230, 65–76 CAS
- X. Sun, H. Huang, Q. Zhao, T. Ma and L. Wang, Adv. Funct. Mater., 2020, 30, 1910005 CAS
- Q. Lin, L. Li, S. Liang, M. Liu, J. Bi and L. Wu, Appl. Catal., B, 2015, 163, 135–142 CAS
- S. Yang, Y. Gong, J. Zhang, L. Zhan, L. Ma, Z. Fang, R. Vajtai, X. Wang and P. M. Ajayan, Adv. Mater., 2013, 25, 2452–2456 CAS
- Y. Shi, B. Wang, L. Duan, Y. Zhu, Z. Gui, R. K. K. Yuen and Y. Hu, Ind. Eng. Chem. Res., 2016, 55, 7646–7654 CAS
- X. Li, H. Liu, S. Meng, F. Wang, T. Mei, X. Cai and Y. Ran, Chem. Eng. J., 2024, 488, 150731 CAS
- S. Zhang, Y. Shen, J. Lu, Z. Chen, L. Li, F. Guo and W. Shi, Chem. Eng. J., 2024, 483, 149232 CAS
- X. Rao, L. Du, J. J. Zhao, X. D. Tan, Y. X. Fang, L. Q. Xu and Y. P. Zhang, J. Mater. Sci. Technol., 2022, 118, 35–43 CAS
- W. Jiang, W. Luo, R. Zong, W. Yao, Z. Li and Y. Zhu, Small, 2016, 12, 4370–4378 CAS
- J. Sun, B. V. K. J. Schmidt, X. Wang and M. Shalom, ACS Appl. Mater. Interfaces, 2017, 9, 2029–2034 CAS
- M. Saeed, M. Muneer, A. U. Haq and N. Akram, Environ. Sci. Pollution Res., 2022, 29, 293–311 CAS
- S. C. Yan, Z. S. Li and Z. G. Zou, Langmuir, 2009, 25, 10397–10401 CAS
- I. Papailias, N. Todorova, T. Giannakopoulou, N. Ioannidis, N. Boukos, C. P. Athanasekou, D. Dimotikali and C. Trapalis, Appl. Catal., B, 2018, 239, 16–26 CAS
- M. Wu, Y. Gong, T. Nie, J. Zhang, R. Wang, H. Wang and B. He, J. Mater. Chem. A, 2019, 7, 5324–5332 CAS
- H. Yan, Y. Chen and S. Xu, Int. J. Hydrogen Energy, 2012, 37, 125–133 CAS
- X. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, J. M. Carlsson, K. Domen and M. Antonietti, Nat. Mater., 2009, 8, 76–80 CAS
- A. Thomas, A. Fischer, F. Goettmann, M. Antonietti, J.-O. Müller, R. Schlögl and J. M. Carlsson, J. Mater. Chem., 2008, 18, 4893–4908 CAS
- X. Wang, X. Chen, A. Thomas, X. Fu and M. Antonietti, Adv. Mater., 2009, 21, 1609–1612 CAS
- K. Li, Y. Jiang, W. Rao, Y. Li, X. Liu, J. Zhang, X. Xu and K. Lin, Chem. Eng. J., 2022, 431, 134075 CAS
- H. Ou, L. Lin, Y. Zheng, P. Yang, Y. Fang and X. Wang, Adv. Mater., 2017, 29, 1700008 Search PubMed
- C. Zhang, D. Pan, Y. Zhang, L. Lin, Y. Wang, M. Zhou, Z. Li and S. Xu, J. Environ. Chem. Eng., 2024, 12, 111956 CAS
- I. Różalska, P. Kułyk and I. Kulszewicz-Bajer, New J. Chem., 2004, 28, 1235–1243 Search PubMed
- J. Wen, F. Hu, B. Liu, H. Chen, M. Yang, Y. Liu and H. Li, Eur. Polym. J., 2023, 201, 112596 CAS
- Z. Zhang, D. Jiang, D. Li, M. He and M. Chen, Appl. Catal., B, 2016, 183, 113–123 CAS
- X. Zhang, X. Xie, H. Wang, J. Zhang, B. Pan and Y. Xie, J. Am. Chem. Soc., 2013, 135, 18–21 CAS
- Q. Zheng, D. P. Durkin, J. E. Elenewski, Y. Sun, N. A. Banek, L. Hua, H. Chen, M. J. Wagner, W. Zhang and D. Shuai, Environ. Sci. Technol., 2016, 50, 12938–12948 CAS
- D. Zhao, C.-L. Dong, B. Wang, C. Chen, Y.-C. Huang, Z. Diao, S. Li, L. Guo and S. Shen, Adv. Mater., 2019, 31, 1903545 CAS
- Y. Yang, G. Zeng, D. Huang, C. Zhang, D. He, C. Zhou, W. Wang, W. Xiong, X. Li, B. Li, W. Dong and Y. Zhou, Appl. Catal., B, 2020, 272, 118970 CAS
- T. Wang, Z. Jiang, T. An, G. Li, H. Zhao and P. K. Wong, Environ. Sci. Technol., 2018, 52, 4774–4784 CAS
- E. G. A. Owusu, A. J. MacRobert, I. Naasani, I. P. Parkin, E. Allan and E. Yaghini, ACS Appl. Mater. Interfaces, 2019, 11, 12367–12378 CAS
- Y. Ichihashi, T. Sekiguchi, K. Hiramatsu, Y. Tokui, K. Kumagai, H. Matsuyama, K. Taniya and S. Nishiyama, Appl. Catal., B, 2023, 325, 122326 CAS
- L. Chen, J. Duan, P. Du, W. Sun, B. Lai and W. Liu, Water Res., 2022, 221, 118747 CAS
- K. Sobańska, P. Pietrzyk and Z. Sojka, ACS Catal., 2017, 7, 2935–2947 Search PubMed
- A. Fujishima and K. Honda, Nature, 1972, 238, 37–38 CAS
- Z. Xu, C. Shan, B. Xie, Y. Liu and B. Pan, Appl. Catal., B, 2017, 200, 439–447 CAS
- G. Li, W. Yang, S. Gao, Q. Shen, J. Xue, K. Chen and Q. Li, Chem. Eng. J., 2021, 404, 127115 CAS
- Y. Zhang, J. M. Lucas, P. Song, B. Beberwyck, Q. Fu, W. Xu and A. P. Alivisatos, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 8959–8964 CAS
- C. Zhou, C. Lai, P. Xu, G. Zeng, D. Huang, C. Zhang, M. Cheng, L. Hu, J. Wan, Y. Liu, W. Xiong, Y. Deng and M. Wen, ACS Sustainable Chem. Eng., 2018, 6, 4174–4184 CAS
- Y. Deng, L. Xia, G.-L. Song, Y. Zhao, Y. Zhang, Y. Xu and D. Zheng, Composites, Part B, 2021, 225, 109263 CAS
- M. Yan, X. Lan and W. Zhao, Prog. Org. Coat., 2024, 186, 108075 CAS
- M. Gómez, M. D. Murcia, E. Gómez, J. L. Gómez and N. Christofi, Desalination, 2011, 274, 156–163 Search PubMed
- Y. Li, W. Zhang, J. Niu and Y. Chen, ACS Nano, 2012, 6, 5164–5173 CAS
- W. Xu, C. Ma, J. Ma, T. Gan and G. Zhang, ACS Appl. Mater. Interfaces, 2014, 6, 4017–4024 CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4mh00550c |
| This journal is © The Royal Society of Chemistry 2024 |