
A self-organized sandwich structure of chromium nitride for ultra-long lifetime in liquid sodium†
Ming
Lou
a,
Ran
Chen
a,
Kai
Xu
a,
Jibin
Pu
 *a and
Keke
Chang
*ab
*a and
Keke
Chang
*ab
aKey Laboratory of Advanced Marine Materials, Ningbo Institute of Materials Technology and Engineering, Chinese Academy of Sciences, Ningbo 315201, China. E-mail: pujibin@nimte.ac.cn; changkeke@nimte.ac.cn
bCenter of Materials Science and Optoelectronics Engineering, University of Chinese Academy of Sciences, Beijing 100049, China
First published on 20th June 2024
Abstract
The development of fast neutron reactors with improved efficiency and sustainability, being a tangible solution to the large-scale utilization of nuclear energy, serves as a critical step prior to the commercialization of fusion energy. These reactors use liquid metal coolants, which can weaken the durability of metallic components. Conventional design of protective coatings counts upon thermodynamics, which often overlooks the kinetic factors such as structural evolutions, resulting in deteriorated coating properties. Herein, we present a novel interface-engineering strategy involving the control of the phase transformation direction and interface diffusion reaction. Through iterations of self-organization, desired surfaces and interfaces can be achieved for materials used in harsh environments. Specifically, a CrN-coated steel sample with an interfacial Cr layer was designed and fabricated. After ultra-long (up to 6000 h) immersion in liquid sodium, the CrN/Cr coating structure was converted into a sandwich Cr2N/CrN/Cr2N structure dynamically. As a consequence, the coating system exhibited enhanced properties, namely increased surface hardness (by ∼36%), reduced coefficient of friction (by ∼13%), and enhanced interfacial adhesion (by ∼37%). Thus, the proposed strategy can guide the future design of robust coatings with ultra-long service life in harsh environments.
New conceptsDevelopments of durable protective coatings for use in harsh environments have long been a pursuit, yet are extremely difficult to achieve. In this communication, we propose a novel interface-engineering strategy consisting of the deliberate control of the phase transformation direction and interface diffusion reaction, which differs from the conventional concept of materials design that mainly counts upon thermodynamics with the kinetic factors overlooked. Following such a strategy, we successfully attained self-organized surfaces and interfaces desired for components serving under harsh conditions, and the coating system exhibited time-dependent property reinforcements, i.e., a ∼36% rise in hardness, a ∼13% reduction in COF, and a ∼37% increase in adhesion strength, after ultra-long (over 6000 h) immersion in liquid sodium, in sharp contrast to the routinely observed property degradation upon high temperature exposure. Thus, this study provides a new thread for the design of self-adaptive coating systems serving in nuclear reactors, and the interface-engineering strategy proposed is expected to propel breakthroughs in the fields of coating development for harsh environments. |
Introduction
To reach net-zero emissions worldwide by 2050, the development of reliable fast neutron reactors has become a tangible solution to the large-scale utilization of nuclear energy. In this context, the European Union has recently launched a new act on climate change mitigation and adaptation covering certain nuclear activities, among which the promotion of advanced nuclear reactors with closed fuel cycles has been revitalized since 2023.1 Fast neutron reactors that use uranium-238 have the potential to solve the longstanding efficiency and sustainability issues associated with thermal reactors.2 For example, the current pressurized water reactors (PWRs) harness <1% of the total energetic power of uranium. In contrast, the next-generation sodium-cooled fast reactors (SFRs) have the potential to harness >60% while also featuring higher power density and reducing the radioactive waste lifetime. The SFR, which typically runs at 370–550 °C and utilizes liquid sodium as a coolant instead of water, requires reliable materials for a life expectancy of 60 years.3The in-core and out-of-core structures in the SFR, including fuel claddings, control rods, and their mechanical auxiliaries, are made of stainless steel (SS).2–4 Some of these SS components, such as the control rod drive mechanisms, are subjected to wear contact.5 A primary research focus has been laid on the compatibility between major types of SS materials and liquid sodium,6–8 and the austenitic SS was found to be less sensitive to liquid metal embrittlement than the ferritic and martensitic ones.7 Still, the dissolutions of iron, nickel, and chromium on the austenitic SS surface can be identified shortly after 250 h, with the resultant generations of NaCrO2 layers and discrete cavities underneath.8 Against this backdrop, proper coatings that are adaptive to the liquid sodium environment are deemed necessary to prolong the service life of SS. Relevant attempts included the thermally sprayed ceramic coatings (e.g., Y2Al4O9),9 the electroplated metallic coatings (e.g., Cr),10 and the nitrided-coated surfaces consisting of Fe3O4 and CrN;11 unfortunately, all these candidate coatings were unable to block the element diffusion at elevated temperatures. Recent endeavors with the construction of duplex nitride coatings,12,13 despite being more protective, encountered an inevitable decline of hardness at high temperatures, which raised additional concern on the tribological performance of coated SS.
Conventional strategies leverage thermodynamics to develop the coatings with anti-corrosion and anti-wear properties,14,15 which reckon on the phase equilibria to possibly acquire targeted phase compositions. Nevertheless, coatings are normally deposited via non-equilibrium processes where metastable phases can form.16,17 The evolution of the metastable phases towards their equilibrium states, albeit theoretically feasible, can hardly proceed in a controlled manner, accompanied with property deteriorations. For example, a variety of fuel cladding coatings, including the nitride (e.g., TiAlCrN) and metallic (e.g., Zr) ones,18,19 experienced phase decomposition or transformation upon high temperature exposure, resulting in a lessened barrier effect on oxygen diffusion; while some others, such as the CrN and TiO2 coatings,20,21 exhibited the formation of interfacial phases due to the dynamic diffusion of N (from CrN) and Cr (from SS substrates), producing internal stresses and defects as a consequence. These phase transitions and structural mutations at the surfaces and interfaces of coatings highlight the importance of kinetic factors in addition to the thermodynamic ones during the coating design.
Thus far, a cornerstone strategy for designing self-adaptive coating systems with long-term protectiveness under the stimuli of high temperature, corrosion, and stress in the SFR and other harsh environments is yet to be developed. Recent successes22,23 in the manipulation of segment migration and interface reaction in the functional coatings to endow strong interfacial adhesion in aquatic environments have offered some clues for designing robust self-adaptive coatings. In the current work, we propose an interface-engineering strategy (Fig. 1A) involving the control of phase transformation direction and interface diffusion reaction for realizing self-adaptive coating systems with long-term durability. The thermodynamic and kinetic aspects of this strategy were rationalized via the CALculation of PHAse Diagrams (CALPHAD) and the ab initio molecular dynamics (AIMD) approaches. Following such a strategy, we synthesized a CrN coating on austenitic 316L SS substrates with an interfacial Cr layer for the SFR application. As stable surface states of nuclear materials can be readily achieved within 1000–5000 h,24 we selected 6000 h as the total duration of liquid sodium exposure tests. After the immersion, the designed coating system exhibited enhanced properties (such as higher hardness, lower COF, and strengthened interfacial adhesion), as opposed to the routinely witnessed property degradations. Such property enhancements were attributable to the self-organized sandwich structure of Cr2N/CrN/Cr2N.
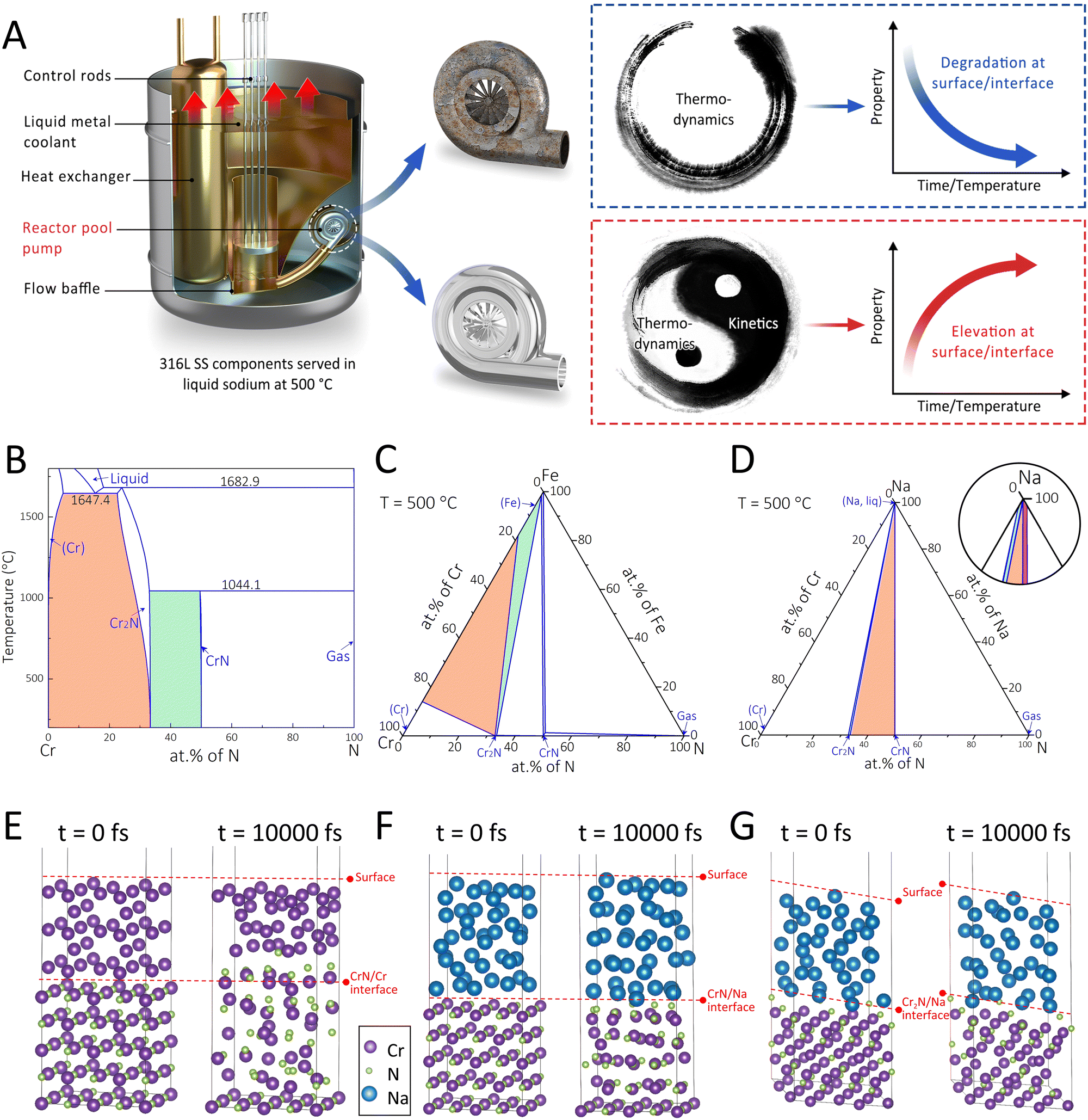 | ||
Fig. 1 (A) Illustration of the proposed interface-engineering strategy for developing a self-organized sandwich coating structure with enhanced properties for sodium-cooled fast reactors (SFRs). (B) Cr–N binary phase diagram. (C) Isothermal section of the Cr–Fe–N system at 500 °C. (D) Isothermal section of the Cr–Na–N system at 500 °C. Ab initio molecular dynamics (AIMD) results of (E) CrN/Cr, (F) CrN/Na, and (G) Cr2N/Na interface structures simulated at 500 °C for 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 fs. 000 fs. | ||
Results and discussion
The chromium nitride coating that has exhibited superior irradiation resistance over other ceramic and metallic coatings was selected as the model coating system.25 The purpose of the interface-engineering strategy adopted in this work is to fabricate the coating system enriched with the Cr2N phase that has been characterized by the high hardness and corrosion resistance yet a narrow processing window.26,27 Following such a strategy, the CrN/Cr duplex coatings on the SS substrates were considered, with the original and ultimate phase compositions assessed at three interfaces, namely (i) the CrN/Cr, (ii) the Cr/Fe, and (iii) the CrN/Na. The vertical section of the Cr–N binary phase diagram (Fig. 1B) was established to identify potential phase transitions at the CrN/Cr interface. Since the Cr2N single-phase region is located between the (Cr) and CrN single-phase regions, the equilibrium phase state at the CrN/Cr interface after the immersion in liquid sodium (at 500 °C) would most likely be (Cr) + Cr2N (marked orange) or CrN + Cr2N (marked green). As for the Cr/Fe interface, the isothermal section of the Cr–Fe–N ternary system at 500 °C (Fig. 1C) was evaluated. The equilibrium phase component would fall within the (Cr) + Cr2N + (Fe) three-phase region (marked orange) or the Cr2N + (Fe) two-phase region (marked green). Note that the (Cr) and (Fe) phases could not co-exist under this condition, unless the new phase (Cr2N) formed. For the CrN/Na interface, i.e., the coating top surface in contact with the corrosion medium, the isothermal section of the Cr–Na–N ternary system at 500 °C (Fig. 1D) shows that in addition to the coexistence of CrN and Na (marked red), Cr2N might also be present in the equilibrium composition as a Cr2N + CrN + Na three-phase region (marked orange) and a narrow Cr2N + Na two-phase region (marked green) can be noted as well.The kinetics of potential phase transformations towards the Cr2N phase in the model coating system were further investigated through AIMD. At the CrN/Cr interface (Fig. 1E), it is shown that both Cr and N atoms in the face-centered-cubic (fcc) CrN became irregularly arranged after the simulation at 500 °C for 10000 fs. In addition, with several N atoms diffusing into the body-centered-cubic (bcc) Cr, the original CrN/Cr interface collapsed. These kinetic results agree well with the calculated phase diagram (Fig. 1A). The potential phase transition at the CrN/Cr interface can be suggested as follows:
| (Cr) + CrN → Cr2N | (1) |
In terms of the coating top surface, the kinetic simulations considered the CrN (Fig. 1F) and Cr2N (Fig. 1G) in contact with liquid sodium at 500 °C, and the results reveal that both interfaces remained unchanged after 10000 fs. Nevertheless, the average radial distribution functions differed between the CrN and Cr2N structures (Fig. S1, ESI†). The Cr2N structure was well maintained after simulation, except for minor distortions. In contrast, the CrN structure was substantially destroyed, suggesting its instability when exposed to liquid sodium. These kinetic simulations together with the thermodynamic results (Fig. 1D) could indicate the high propensity of phase transformation from CrN to Cr2N at the coating top surface via the following reaction:
| (Cr) + CrN + (Na, liq) → Cr2N + (Na, liq) | (2) |
It is pertinent to note that the CrN phase should be more stable than the Cr2N phase at a temperature of 500 °C,28 while in the presence of liquid sodium the CrN structure became incapable of maintaining its integrity. Thus, the (Na, liq) may act as a catalyst to facilitate the phase transformation, and such inference may need further validation.
To validate the effectiveness of the proposed strategy, the designed coating system was fabricated on 316L SS substrates. The as-deposited coating was dark grey in color (Fig. S2, ESI†) and had a total thickness of ∼46 μm (Fig. S3, ESI†), which can be referred to as the CrN*/(Cr)* coating (where the asterisk denotes the main phase in each layer), as per the X-ray diffraction (XRD) and electron backscatter diffraction (EBSD) results. Fig. 2A shows that the as-deposited CrN* top layer was composed of CrN (space group: Fm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m) and minor Cr2N (space group: P1m), which, in addition, may incorporate marginal (Cr) phase (space group: Imm) at a level undetectable by XRD (Fig. 2B), as metallic macro-particles (droplets) can be commonly observed in arc evaporation-deposited coatings.29 In the (Cr)* transition layer, Fig. 2B, minor Cr2N coexisted with (Cr) phase, which originated from the phase transformation during coating deposition.
m) and minor Cr2N (space group: P1m), which, in addition, may incorporate marginal (Cr) phase (space group: Imm) at a level undetectable by XRD (Fig. 2B), as metallic macro-particles (droplets) can be commonly observed in arc evaporation-deposited coatings.29 In the (Cr)* transition layer, Fig. 2B, minor Cr2N coexisted with (Cr) phase, which originated from the phase transformation during coating deposition.
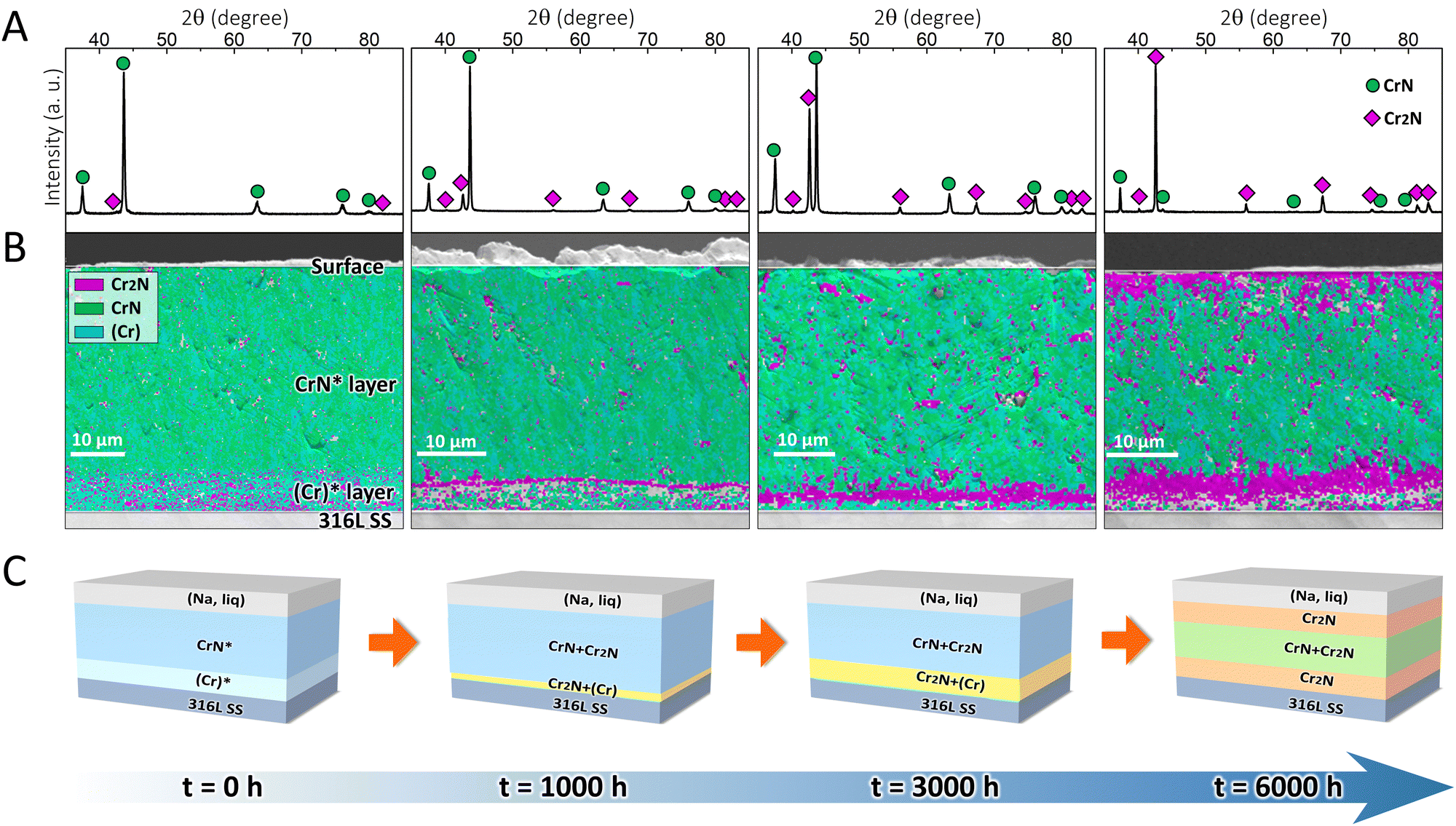 | ||
| Fig. 2 (A) X-ray diffraction (XRD) patterns and (B) electron backscatter diffraction (EBSD) phase maps of the as-deposited CrN*/(Cr)* coating and the coating immersed in liquid sodium at 500 °C for 1000, 3000, and 6000 h. (C) Schematic diagrams of the stepwise formation of a self-organized sandwich structure composed of Cr2N/CrN/Cr2N. The asterisk denotes the main phase in each layer. | ||
After the coating was immersed in 500 °C liquid sodium for different durations (1000, 3000, and 6000 h), its surface became light grey (Fig. S2, ESI†), which was owing to the exfoliation of oxide layers on the coating surface in liquid sodium.30 Simultaneously, the coating/substrate interface showed the gradual formation of a continuous layer, which was particularly prominent after 6000 h exposure in liquid sodium (Fig. S3, ESI†). According to the XRD (Fig. 2A) measurements, the coating surface that was immersed in liquid sodium for 1000 h exhibited sharper Cr2N peaks than the as-deposited one. The peak intensities of Cr2N continued to increase after 3000 h, which eventually became the dominant phase (instead of CrN) after 6000 h. The EBSD phase maps (Fig. 2B) further unveiled that the Cr2N phase was randomly distributed in the CrN* layer after 1000 h exposure, with a tendency to coalesce at the CrN*/(Cr)* interface. After 3000 h, the Cr2N phase was further assembled at the interface, while the top surface revealed only a trace amount of Cr2N formation. Eventually after 6000 h, the original (Cr)* transition layer was completely transformed into a Cr2N layer, while another Cr2N layer was concurrently generated at the top surface. Hitherto, a self-organized sandwich structure consisting of Cr2N/CrN/Cr2N was formed as designed after the ultra-long immersion in liquid sodium.
The schematic diagrams to illustrate the phase transformation-induced generation of the sandwich structure are shown in Fig. 2C. The original coating (t = 0 h) was in a metastable state, which finally became close to a phase-equilibrium state (t = 6000 h). The formation of the self-organized sandwich structure benefited from such metastable-to-stable phase transformation in a stepwise manner, during which intermediate states (t = 1000, 3000 h) were present. One should note that the phase transformation direction was well predicted by the thermodynamic calculations (Fig. 1B–D), while the earlier formation of the Cr2N layer at the interface compared with that at the top surface was also supported by the kinetic simulations (Fig. 1E–G).
The grain morphologies in the interfacial region before and after the phase transition were specifically investigated via transmission electron microscopy (TEM). According to Fig. 3A, both the CrN* layer and the (Cr)* layer exhibited lath-like grains with an average width of ∼70 nm. The transmission Kikuchi diffraction (TKD) and the energy dispersive X-ray spectroscopy (EDS) maps revealed that the original (Cr)* layer had a thickness of ∼500 nm. The selected area electron diffraction (SAED) pattern (Fig. 3A1) of the (Cr)* layer revealed its incorporation of nano-crystalline (Cr) grains. The nano-beam diffraction (NBD) pattern (Fig. 3A2) of the CrN* layer validated the presence of CrN grains with a cubic structure. The 6000 h immersion in liquid sodium resulted in remarkable microstructural variations in the interfacial region. Although rod-like grains with an average width of ∼110 nm were still observable adjacent to the SS substrate, a number of equiaxed grains with similar widths emerged right above them (Fig. 3B). In light of the TKD map, both rod-like and equiaxed grains belonged to the Cr2N phase. In addition, no element (Cr and N) segregation can be detected in this interfacial region, suggesting the thorough transformation from (Cr) to Cr2N. Further examination of the SAED pattern (Fig. 3B1) again confirmed the conversion of the original (Cr) into Cr2N, while the NBD pattern (Fig. 3B2) revealed the transformation of CrN into Cr2N as well, both of which validated the interface reaction proposed (eqn (1)). The dynamic phase transition thus eliminated the initial CrN*/(Cr)* interface, forming an integrated layer of Cr2N, which could be beneficial to the interfacial bonding, a point that will be examined in the following paragraph.
 | ||
| Fig. 3 (A) Transmission electron microscope (TEM) image with the transmission Kikuchi diffraction (TKD) map of the interfacial region of the as-deposited CrN*/(Cr)* coating system. The energy dispersive X-ray spectroscopy (EDS) maps of Cr, N, and Fe, as well as the selected area electron diffraction (SAED) and nano-beam diffraction (NBD) patterns are provided for the analyses of chemical and phase compositions. (B) TEM image with TKD map of the interfacial region of the coating system after 6000 h immersion in liquid sodium. The EDS maps of Cr, N, and Fe, as well as the SAED and NBD patterns are provided for the analyses of chemical and phase compositions. | ||
The effect of the stepwise evolution towards the Cr2N/CrN/Cr2N sandwich structure on the mechanical and tribological properties was examined periodically (after 1000, 3000, and 6000 h immersion). As shown in Fig. 4A, the benchmark adhesion force of the as-deposited coating to the SS substrate was measured as ∼117 N, which rose to ∼153 N after 1000 h and plateaued at ∼160 N after 3000 h and 6000 h. The average coefficient of friction (COF) value of the coated surface after 6000 h immersion in liquid sodium (∼0.47) became slightly lower than the pre-immersion one (∼0.54). The hardness of the coated surface, in addition, gradually increased over the exposure time, from an initial value of ∼15 GPa to a value of ∼21 GPa after 6000 h. Both the decrease in COF and the increase in hardness would indicate the dynamic improvement in the tribological properties of the engineered coating system. The temperature-induced changes in the hardness of typical nitride coatings and SS31–37 were further compared (Fig. 4B). Besides the TiAlN coating, which showed increased hardness (by ≤1%) after heat treatment,31 other types of nitride coatings exhibited prominent hardness decreases following the high temperature annealing (500 °C). For example, the TiN coating exhibited a ∼9% decline in hardness,31 and this downward trend was also supported by the theoretical results.37 Similarly, the SS materials encountered inevitable decreases in hardness (∼4% for 316 LN SS33 and ∼15% for 304 SS34) after high temperature exposure. In sharp contrast to these widely-used binary and ternary nitride coating systems (including their multilayer counterparts) and SS materials, the coating system engineered in this work showed a hardness increase rate of ∼37%, consistent with the hardness difference (∼36%) between CrN and Cr2N predicted using ab initio calculations. Thus, the coating system designed based on the interface-engineering strategy is expected to impart ultra-long surface protection to the SS components serving in SFR environments.
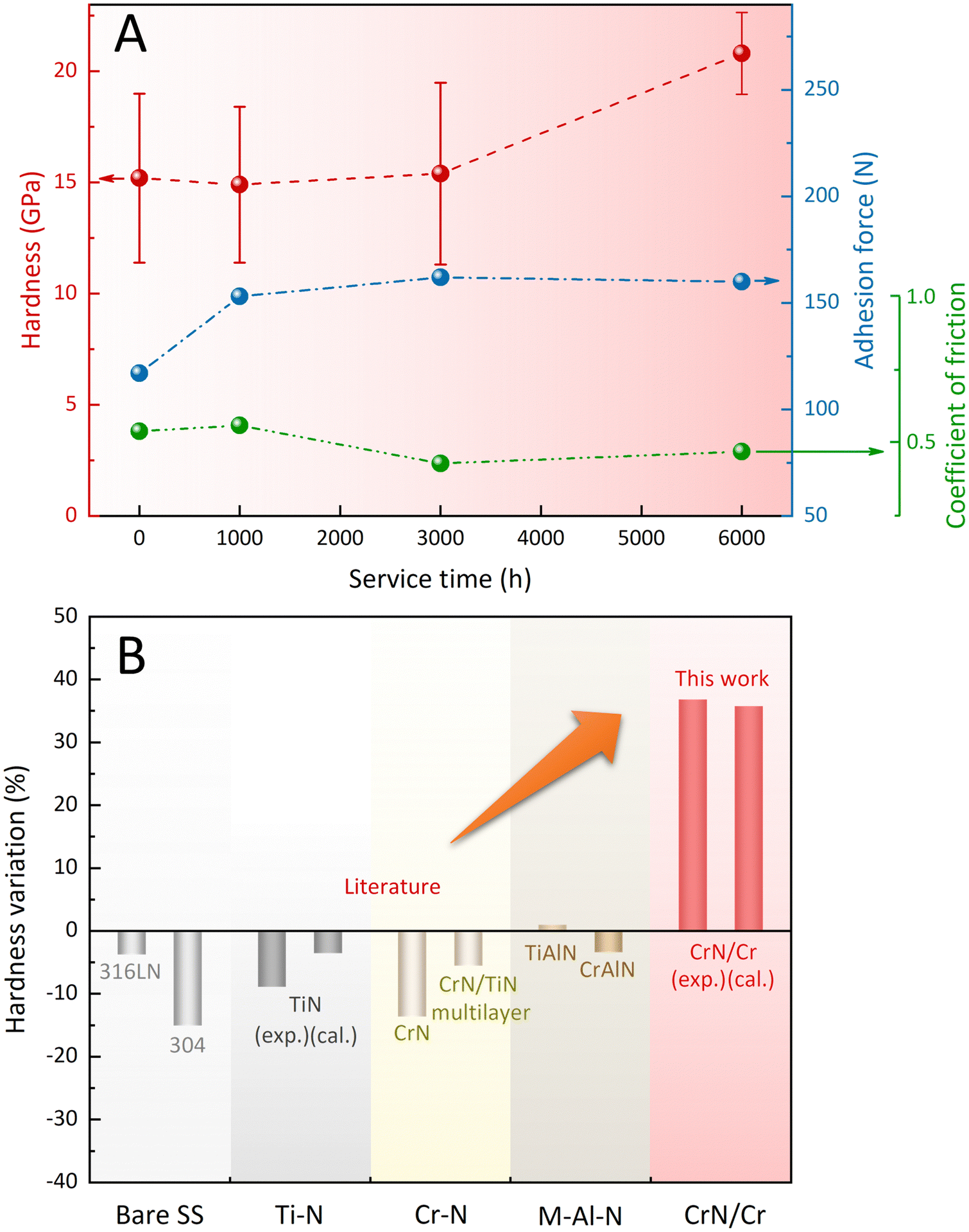 | ||
| Fig. 4 (A) Variations of hardness, adhesion force, and coefficient of friction (COF) with the increased duration of exposure to high temperature liquid sodium. (B) Temperature-induced hardness variations of the designed coating system in this work and the typical nitride coatings and SS materials after high temperature annealing at 500 °C reported in the literature.31–37 | ||
Conclusions
To avoid property degradation and prolong the service life of critical materials used in harsh environments, we propose a novel interface-engineering strategy that involves the subtle control of the phase transformation direction and interface diffusion reaction. The coating/substrate and coating/corrosion-medium interfaces became the areas of interests at which the thermodynamics and kinetics of phase transformations were considered. Based on such a strategy, a CrN/Cr coating system was designed, which could generate a self-organized sandwich structure of Cr2N/CrN/Cr2N after the ultra-long immersion (up to 6000 h) in high temperature liquid sodium. Along with the phase transitions, the coating system exhibited dynamic property reinforcements, namely an ∼37% rise in hardness, ∼13% reduction in COF, and ∼37% increase in adhesion strength. Thus, the proposed strategy may provide solutions superior to the conventional methods that relied solely on thermodynamics to amend the compositions and processing routines, and can usefully guide the material exploration for achieving ultra-long service life in nuclear power systems and other harsh-environment systems.Author contributions
J. P. and K. C. conceived the work. R. C. conducted the experiments. K. X. and K. C. performed the theoretical calculations. M. L. and K. C. contributed to the discussion of the results. M. L. drafted the manuscript and all authors contributed to the review & editing.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (52325503, 52222507, U23A6016). Prof. Jian Lu (City University of Hong Kong), Prof. Liping Wang (Ningbo Institute of Materials Technology and Engineering, Chinese Academy of Sciences), Prof. Yun-Tian Zhu (City University of Hong Kong), Prof. Xiang Chen (Nanjing University of Science and Technology), and Prof. Sida Liu (Xi’an Jiaotong University) are gratefully acknowledged for their helpful discussions.References
- D. Ferrie and A. Apostola, EU taxonomy: Complementary climate delegated act (europa.eu), 2022 Search PubMed.
- T. Allen, J. Busby, M. Meyer and D. Petti, Mater. Today, 2010, 13, 14–23 CrossRef CAS.
- S. J. Zinkle and G. S. Was, Acta Mater., 2013, 61, 735–758 CrossRef CAS.
- G. S. Was, D. Petti, S. Ukai and S. Zinkle, J. Nucl. Mater., 2019, 527, 151837 CrossRef CAS.
- E. Lemaire and M. Le Calvar, Wear, 2001, 249, 338–344 CrossRef CAS.
- P. Skeldon, J. P. Hilditch, J. R. Hurley and D. R. Tice, Corros. Sci., 1994, 36(4), 593–610 CrossRef CAS.
- S. Hemery, T. Auger, J. L. Courouau and F. Balbaud-Celerier, Corros. Sci., 2014, 83, 1–5 CrossRef CAS.
- M. Rivollier, J. L. Courouau, M. Tabarant, C. Blanc and M. L. Giorgi, J. Nucl. Mater., 2018, 500, 337–348 CrossRef CAS.
- P. Varghese, E. Vetrivendan, B. R. V. Krupa, P. K. Shukla, E. H. Rao and S. Ningshen, Surf. Coat. Technol., 2023, 458, 129353 CrossRef CAS.
- S. Yeo, J. H. Kim and S. H. Eom, J. Nucl. Mater., 2020, 530, 151980 CrossRef CAS.
- X. Chen, S. Feng, L. Wang, R. Tang, F. Zhang, S. Ming and Z. Cai, Surf. Coat. Technol., 2022, 562, 153583 CAS.
- Y. Chen, S. Wang, Y. Hao, J. Pu, X. Jiang, L.-F. Huang and L. Wang, Tribol. Int., 2020, 143, 106079 CrossRef CAS.
- S. Yeo, C. M. Lee, H. S. Yoon and J. H. Kim, Appl. Surf. Sci., 2022, 579, 152133 CrossRef CAS.
- K. Chang, F. Meng, F. Ge, G. Zhao, S. Du and F. Huang, J. Nucl. Mater., 2019, 516, 63–72 CrossRef CAS.
- H. Holleck, Surf. Eng., 1991, 7(2), 137–144 CrossRef CAS.
- S. Liu, K. Chang, S. Mráz, X. Chen, M. Hans, D. Music, D. Primetzhofer and J. M. Schneider, Acta Mater., 2019, 165, 615–625 CrossRef CAS.
- S. Liu, K. Chang, D. Music, X. Chen, S. Mráz, D. Bogdanovski, M. Hans, D. Primetzhofer and J. M. Schneider, Acta Mater., 2020, 196, 313–324 CrossRef CAS.
- X.-F. Ma, Y.-W. Wu, J. Tan, C.-Y. Meng, L. Yang, W.-A. Dang and X.-J. He, Surf. Coat. Technol., 2019, 358, 521–530 CrossRef CAS.
- H. C. Koh, P. Hosemann, A. M. Glaeser and C. Cionea, J. Nucl. Mater., 2017, 496, 367–378 CrossRef CAS.
- J. Liu, Z. Hao, Z. Cui, D. Ma, J. Lu, Y. Cui, C. Li, W. Liu, S. Xie, P. Hu, P. Huang, G. Bai and D. Yun, Corros. Sci., 2021, 185, 109416 CrossRef CAS.
- V. Firouzdor, J. Brechtl, B. Hauch, K. Sridharan and T. R. Allen, Appl. Surf. Sci., 2013, 282, 798–808 CrossRef CAS.
- X. Jiang, X. Xu, Z. Xia, D. Lin, Y. Chen, Y. Wang, D. Yu, X. Wu and H. Zeng, Adv. Funct. Mater., 2024, 34, 2314589 CrossRef CAS.
- D. Yu, J. Huang, Z. Zhang, J. Weng, X. Xu, G. Zhang, J. Zhang, X. Wu, M. Johnson, J. Lyu, H. Yang and W. Wang, Adv. Mater., 2022, 34, 2106908 CrossRef CAS PubMed.
- J. J. Park, D. P. Butt and C. A. Beard, Nucl. Eng. Des., 2000, 196, 315–325 CrossRef CAS.
- L. Song, B. Huang, J. Li, X. Ma, M. Liu, J. Jiang and Y. Hu, Ann. Nucl. Energy, 2021, 156, 108206 CrossRef CAS.
- P. Engel, G. Schwarz and G. K. Wolf, Surf. Coat. Technol., 1998, 98, 1002–1007 CrossRef CAS.
- Z. B. Zhao, Z. U. Rek, S. M. Yalisove and J. C. Bilello, Surf. Coat. Technol., 2004, 185, 329–339 CrossRef CAS.
- F. H. Lu and H. Y. Chen, Thin Solid Films, 2001, 398–399, 368–373 CrossRef CAS.
- L. Chen, K. K. Chang, Y. Du, J. R. Li and M. J. Wu, Thin Solid Films, 2011, 519, 3762–3767 CrossRef CAS.
- R. N. Johnson, S. L. Schrock and G. A. Whitlow, J. Vac. Sci. Technol., 1974, 11, 759–764 CrossRef CAS.
- P. H. Mayrhofer, A. Hörling, L. Karlsson, J. Sjölén, T. Larsson, C. Mitterer and L. Hultman, Appl. Phys. Lett., 2003, 83, 2049–2051 CrossRef CAS.
- R. Hahn, M. Bartosik, M. Arndt, P. Polcik and P. H. Mayrhofer, Int. J. Refract. Met. Hard Mater., 2018, 71, 352–356 CrossRef CAS.
- H. X. Pei, H. L. Zhang, L. X. Wang, S. L. Li, D. Z. Li and X. T. Wang, Mater. High Temp., 2014, 31, 198–203 CrossRef CAS.
- G. Sun, L. Du, J. Hu, B. Zhang and R. D. K. Misra, Mater. Sci. Eng., A, 2019, 746, 341–355 CrossRef CAS.
- L. Wang and X. Nie, J. Mater. Eng. Perform., 2014, 23, 560–571 CrossRef CAS.
- Z. Liu, L. Chen and Y. Xu, J. Am. Ceram. Soc., 2018, 101, 845–855 CrossRef CAS.
- A. Wang, S.-L. Shang, M. He, Y. Du, L. Chen, R. Zhang, D. Chen, B. Fan, F. Meng and Z.-K. Liu, J. Mater. Sci., 2014, 49, 424–432 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4mh00698d |
| This journal is © The Royal Society of Chemistry 2024 |