
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Peptide nucleic acid-clicked Ti3C2Tx MXene for ultrasensitive enzyme-free electrochemical detection of microRNA biomarkers†
Muhsin
Ali
a,
Erol
Hasan
 a,
Sharat Chandra
Barman
a,
Mohamed Nejib
Hedhili
b,
Husam N.
Alshareef
a and
Dana
Alsulaiman
*a
a,
Sharat Chandra
Barman
a,
Mohamed Nejib
Hedhili
b,
Husam N.
Alshareef
a and
Dana
Alsulaiman
*a
aMaterial Science and Engineering, Physical Science and Engineering Division, King Abdullah University of Science & Technology (KAUST), Thuwal 23955-6900, Saudi Arabia. E-mail: dana.alsulaiman@kaust.edu.sa
bImaging and Characterization Core Lab, King Abdullah University of Science and Technology, Thuwal 23955-6900, Saudi Arabia
First published on 3rd August 2024
Abstract
We report the engineering and synthesis of peptide nucleic acid-functionalized Ti3C2Tx MXene nanosheets as a novel transducing material for amplification-free, nanoparticle-free, and isothermal electrochemical detection of microRNA biomarkers. Through bio-orthogonal copper-free click chemistry, azido-modified MXene nanosheets are covalently functionalized with clickable peptide nucleic acid probes targeting prostate cancer biomarker hsa-miR-141. The platform demonstrates a wide dynamic range, single-nucleotide specificity, and 40 aM detection limit outperforming more complex, amplification-based methods. Its versatility, analytical performance, and stability under serum exposure highlight the immense potential of this first example of click-conjugated MXene in the next generation of amplification-free biosensors.
New conceptsEngineering robust, rapid, and ultrasensitive biosensors to detect disease-specific biomarkers, like microRNA, in liquid biopsy holds significant promise in minimally-invasive diagnostics. While electrochemical biosensors have shown potential in this regard, existing strategies generally rely on complex enzyme-based amplification or nanocomposite materials to achieve sufficient sensitivity. The advent of two-dimensional (2D) nanomaterials has enabled notable advancements; however, the functionalization of their surfaces with bioreceptors often compromises their electrical conductivity, thereby limiting performance. Remarkably, MXenes have emerged as a family of 2D materials that bypass this compromise, exhibiting both metallic conductivity and abundant surface functional groups conducive to covalent immobilization. Exploiting bio-orthogonal copper-free click chemistry, we demonstrate the design and synthesis of the first bespoke peptide nucleic acid probe-clicked Ti3C2Tx MXene as a highly efficient transducing material for the electrochemical detection of microRNA. Implementing the prostate cancer biomarker, hsa-miR-141, as a proof-of-concept, our biosensor outperforms more complex approaches in terms of sensitivity (40 aM calculated limit-of-detection), dynamic range, and specificity, while circumventing the need for enzymes and nanomaterial composites. With its versatility, analytical performance, and stability even under serum exposure, bioreceptor-clicked MXene emerges as a robust and efficient transducing material poised to drive the next generation of rapid, amplification-free biosensors enabling precision medicine. |
Introduction
There is a pressing need to shift from traditional invasive approaches in cancer diagnostics to minimally invasive and cost-effective approaches based on the detection of clinically relevant biomarkers within biofluid samples like serum or urine. MicroRNA (miRNA) represents a class of short (∼22–25 nt) non-coding RNA1 that have gained prominence as liquid biopsy-based clinical biomarkers owing to their gene regulatory roles and dysregulated patterns in diseases, including cancer and neurodegenerative diseases.2,3 Nevertheless, their short lengths, low concentrations (femtomolar to picomolar range),4 and high sequence homology among family members pose challenges to their detection even with gold-standard methods such as reverse-transcription (RT)-qPCR and hybridization-based microarrays. Moreover, these conventional approaches suffer from being complex, semiquantitative, time-consuming, and costly.5,6 To this end, electrochemical biosensors have emerged as promising platforms to drive precision medicine by enabling efficient and ultrasensitive detection of disease biomarkers. However, to achieve the sensitivity required to detect low-abundance biomarkers like clinically relevant miRNA (whose concentrations vary across several orders of magnitude, often present at sub-picomolar concentrations), electrochemical biosensors often necessitate complex multistep approaches involving target or signal amplification and the use of expensive reagents like enzymes and nanomaterial composites.7Two-dimensional (2D) nanomaterials such as graphene8 and black phosphorus9 have enabled transformative advancements in electrochemical biosensing.10,11 However, most 2D nanomaterials suffer a major trade-off between functionalization and electrical conductivity: introducing functionalities on their surfaces considerably degrades their electrical conductivity and increases noise. Remarkably, MXenes have emerged as a new family of 2D transition metal carbides, nitrides, and carbonitrides that can circumvent this trade-off.12 They exhibit metallic conductivity and contain abundant surface functional groups including –OH, which render them hydrophilic and facilitate the covalent immobilization of bioreceptors or biosensing probes.10,13,14 Among the MXene families, Ti3C2Tx offers exceptional intrinsic properties such as metallic conductivity, strength, stability, catalytic properties, and hydrophilicity.15–17 These features, in addition to their redox characteristics,18 have prompted substantial research into leveraging them as charge-transfer promoters and transducing materials in the electrochemical detection of proteins,19 DNA,20 and more recently, miRNA.21,22 For example, Mohammadniaei et al. developed nanocomposites of MXene and ssDNA-functionalized gold nanoparticles for miRNA detection based on duplex-specific nuclease amplification.23 Duan et al. combined 2D MXene nanosheets with iron phthalocyanine quantum dots for the impedimetric detection of miR-155.24 Despite their sensitivity, most reported approaches for electrochemical biosensing involve the use of nanocomposites (typically with zero-dimensional nanomaterials25) often in combination with complex enzyme-based amplification strategies to achieve sufficient analytical performance (ESI,† Table S1).
This work presents the first demonstration of a robust electrochemical biosensor for miRNA detection which circumvents the use of enzymes, amplification strategies, and nanoparticles by covalently functionalizing MXene nanosheets with bespoke peptide nucleic acid (PNA) probes. PNAs are synthetic oligonucleotides characterized by a neutral pseudopeptide backbone, superior chemical stability, resistance to nucleases and proteases,26,27 as well as higher binding affinity and specificity to target nucleic acids compared with equivalent DNA/DNA or DNA/RNA duplexes.28 Employing bio-orthogonal copper-free click chemistry, clickable PNA probes are designed and covalently attached onto ultrathin Ti3C2Tx nanosheets enabling a highly stable and uniform immobilization as well as high loading density under ambient conditions (Fig. 1A). These PNA-functionalized MXene nanosheets serve as highly efficient transducers of electron transfer between the biorecognition events and the electrode, herein, glassy carbon electrode (GCE). The presented biosensing strategy is based on the sequence-specific hybridization of the target miRNA to the immobilized PNA, facilitating the intercalation of redox-active methylene blue (MB) dye within the PNA:miRNA duplex. This design yields a rapid, highly specific, and quantitative electrochemical turn-on signal proportional to the amount of captured miRNA, without requiring expensive enzymes, amplification strategies, or other nanomaterial-based labels (Fig. 1B). Comprehensive materials characterization including X-ray photoelectron spectroscopy (XPS) and Raman spectroscopy is conducted to validate the successful functionalization of the MXene nanosheets, laying the groundwork for subsequent electrochemical characterization and biosensing evaluations of the platform's analytical performance with miRNA in buffer and serum samples.
 | ||
| Fig. 1 (A) Schematic illustration of the PNA-MXene electrochemical biosensor for miRNA detection based on chemical functionalization of MXene nanosheets via silanization to introduce azido functionalities followed by bio-orthogonal copper-free click chemistry with a DBCO–PNA probe, creating the PNA-MXene nanosheets which are drop-casted on a GCE working electrode. (B) The biosensing strategy is based on adding the serum sample containing miRNA on the working electrode followed by washing non-specifically bound molecules, incubation with methylene blue (MB) to enable intercalation within the duplex and acquiring electrochemical measurements following another wash step. (C) XRD patterns of the Ti3AlC2 MAX phase (blue) and Ti3C2Tx MXene (black) indicating the large interlayer spacing of Ti3C2Tx. (D) AFM micrographs to visualize a monolayer of Ti3C2Tx nanosheets, with inset depicting thickness profile traced along the white line (scale bar = 2 μm). (E) Transmission electron microscopy (TEM) image of single Ti3C2Tx flakes with a zoom-in (F) showing the delaminated flake is a high-quality ultrathin nanosheet. | ||
Results and discussion
Ti3C2Tx MXene nanosheets were synthesized from the Ti3AlC2 MAX phase based on our previous protocols as detailed in the methods section below. The quality of the synthesized Ti3C2Tx MXene nanosheets was investigated using X-ray diffraction (XRD) analysis. XRD was conducted using a powder X-ray diffractometer (Bruker D8 Advance) with Cu Kα radiation at a wavelength of 1.5418 Å. The scanning rate was 0.5 s per step at a step size of 0.02° in the 2θ range of 5°–60°. Fig. 1C presents the typical XRD patterns of Ti3C2Tx MXene and its parent Ti3AlC2 MAX phase with their distinguishing (002) peaks. As deduced from the XRD data, the interlayer spacing (d-spacing), which signifies the interlayer distance characteristic (002) XRD peak of Ti3C2Tx emerged at ∼8.3° rather than ∼9.4°. The shift and broadening in the characteristic (002) peak of Ti3C2Tx are accredited to the substitution of the Al layers with the surface-terminating groups (Tx) during the exfoliation step.29To confirm the successful synthesis of a few layers of MXene, Atomic force microscopy (AFM, Bruker, Dimension Icon SPM) was used to depict the surface morphology and quantify the thickness of the synthesized MXene flakes. Fig. 1D shows a single sheet with a 1.7 nm thickness, validating the successful synthesis of high-quality delaminated MXene sheets. Furthermore, TEM was used to obtain an image of the Ti3C2Tx flakes as depicted in Fig. 1E along with the selected area electron diffraction (SAED) pattern of the nanosheet as an inset image. The SAED pattern shows that the delaminated flake is of high quality and ultrathin, best evidenced by the absence of oxide particles or apparent defects as depicted in Fig. 1F.
As a more robust, stable, and sequence-specific alternative to ssDNA and RNA-based oligonucleotide hybridization probes, PNA-based bioreceptor probes were designed and applied for miRNA detection. Owing largely to their neutral backbone, PNAs exhibit higher chemical and thermal stability and greater binding affinity to target nucleic acids even at low ionic strength conditions; therefore, they have been widely exploited as promising alternatives to natural nucleic acid probes.30,31 Two PNA probes with different lengths (7-mer or 17-mer) were prepared in this work and designed to be complementary to a promising cancer biomarker, hsa-miR-141, which is upregulated in prostate and colorectal cancer patients.32 To enable the covalent immobilization of the probe on the MXene nanosheets, the PNA was functionalized at the N-terminus with a dibenzocyclooctyne (DBCO) moiety, which is suitable for bio-orthogonal copper-free click chemistry conjugation.33 Furthermore, two arginine amino acid residues were also introduced at the N-terminus of the probes to facilitate solubility in aqueous solutions.34 PNA synthesis, based on solid-phase peptide synthesis using Fmoc chemistry,35 was performed on an automated peptide synthesizer (Purepep Chorus, Gyros Protein Technologies), while purification was performed via reverse-phase preparative high-performance liquid chromatography (HPLC). The successful synthesis of the PNA probes was verified using liquid chromatography-mass spectroscopy (LCMS), as shown in the ESI† (Fig. S1).
Before clicking the PNA probes onto the MXene surfaces, bare Ti3C2 MXene nanosheets (produced in our lab following previously published protocols)36 were silanized with 3-azidopropyl triethoxysilane (AzPTES) to introduce the azido functionality. After decorating the MXene surfaces with these clickable azide (–N3) functional groups, DBCO–PNA was added to the azido-MXene nanosheets, and the copper-free click chemistry reaction was conducted in aqueous media, at room temperature overnight. After multiple wash steps to remove unreacted groups, the PNA-MXene nanosheets were stored in a −20 °C freezer until further use. Further details of the synthesis procedures and a workflow diagram can be found in the Methods and Fig. S2 (ESI†), respectively.
Successful silanization and click chemistry-based functionalization of the MXene nanosheets were validated using several characterization techniques. Firstly, XPS was used to analyse the surface chemical composition of the bare Ti3C2Tx MXene compared to that of the MXene after silanization (azido-MXene) and subsequent click chemistry (PNA-MXene) (Kratos Analytical, AMICUS/ESCA 3400) (Fig. 2). The overlayed survey spectra of the bare MXene, azido-MXene, and PNA-MXene are shown in Fig. 2A(i). Compared to the spectrum of bare MXene (red), the azido-MXene spectrum (black) showed the appearance of Si, accompanied by a drastic increase in the N content from 1% in the bare MXene to 13% in the azido-MXene sample (and an increase in the N/C ratio from 0.04 to 1.62), confirming successful silanization. Similarly, the PNA-MXene (blue) spectrum showed both additional peaks but with higher C and N contents, attributed to the successful clicking of the PNA probe. Details of the atomic percentages are summarized in the ESI,† Table S2.
 | ||
| Fig. 2 (A) (i) XPS spectra of the bare MXene, azido-MXene, and PNA-MXene with the peaks for Si and N highlighted in green. (ii) C 1s and Ti 2p core-level spectra of the bare MXene sample. (B) (i) N 1s core-level spectrum of AzPTES and (ii) N 1s and Ti 2p core-level spectra of the azido-MXene sample. (C) (i) N 1s core-level spectrum of the PNA probe and (ii) N 1s and C 1s core-level spectra of the PNA-MXene sample. | ||
Next, high-resolution Ti 2p and C 1s core-level spectra of the bare MXene sample were acquired and analysed (Fig. 2A(ii)). Notably, the Ti 2p spectrum was fitted with six doublets (Ti 2p 2p3/2–Ti 2p1/2). The Ti 2p3/2 component centered at 454.8 eV was attributed to Ti bound to C. The Ti 2p3/2 components centered at 455.7 and 457.0 eV were attributed to Ti ions with valence states of 2+ and 3+ respectively,37,38 indicative of the presence of surface termination species including –O2− and –OH−. Of note is the presence of a small peak at 458.4 eV which is attributed to TiO2 in the bare MXene. The low percentage of TiO2 (making up only 5% of the total Ti content) signifies the synthesis of high-quality MXene with minimal oxidation during synthesis, storage, and analysis. The C 1s core-level spectrum of the bare MXene sample was also fitted with seven components and analyzed as detailed in Fig. S3 (ESI†).
Before characterization of the azido-MXene sample, a high-resolution N 1s core-level spectrum of the starting material, AzPTES, was acquired (Fig. 2B-i). The spectrum was fitted with three components located at 400.8 eV, 401.2 eV, and 404.7 eV, attributed to N−, N0, and N+ of the azide group, respectively.39,40 Additional high-resolution XPS spectra of AzPTES and azido-MXene are shown in Fig. S3 (ESI†). Importantly, the high-resolution N 1s of azido-MXene showed the same three abovementioned components, confirming successful functionalization with AzPTES (Fig. 2B-ii). The high-resolution Ti 2p spectrum of the azido-MXene sample was also acquired and deconvoluted. The major difference between the Ti 2p spectra of the azido-MXene compared to the bare MXene was the increased intensity of the Ti component at a binding energy (BE) of 458.4 eV binding energy. Coincidentally, this BE is the same for Ti 2p electrons from a TiO2 or Ti–O–Si bond. A control experiment was next conducted to investigate which bond contributed to the high-intensity peak observed after silanization and to (importantly) ensure that this peak is not a result of MXene oxidation during the silanization experiment. Specifically, bare MXene was exposed to the same reaction conditions as the silanization experiment; however, without the presence of the AzPTES material. Briefly, the bare MXene was incubated in ethanol and mixed under magnetic stirring at room temperature for 24 h before a high-resolution Ti 2p XPS spectrum was acquired. As shown in Fig. S4(A and B) (ESI†), this spectrum is similar to the bare untreated MXene with the TiO2 peak making up 5% of the total Ti content, signifying that no additional oxidation of titanium occurs. This consequently confirms that the high-intensity peak making up 28% of the total Ti signal post-silanization can be assigned mainly to the formation of new Ti–O–Si bonds. An XPS analysis table shown in the ESI† (Table S3) displays the atomic percentage of the Ti species in the four samples: bare MXene, MXene treated with ethanol, azido-MXene, and PNA-MXene.
Next, the high-resolution N 1s spectrum of the starting material, 17-mer DBCO–PNA probe, was acquired and characterized prior to PNA-MXene characterization (Fig. 2C). The N 1s core-level spectrum of PNA was fitted with three components located at 399.0 eV, 400.2 eV, and 400.9 eV corresponding to –N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) , –NH–CO–/tertiary nitrogen, and –NH2, respectively (Fig. 2C-i).41 Finally, for the PNA-MXene, the N 1s and C 1s core-level spectra were acquired (Fig. 2C-ii). Notably, comparing the N 1s spectra of the PNA starting material and PNA-MXene sample, similar peaks were observed, including the presence of amine and amide bonds, confirming successful clicking of PNA. Due to the difficulties of verifying the triazole bond formation via XPS (the theoretical triazole BE overlaps with that of nitrogen groups from the azido functionality at BE 400.8–401.2 eV and the PNA backbone), a control experiment was performed to verify that the PNA is indeed chemically rather than physically adsorbed on the MXene surface (Fig. S4C, ESI†). In this supplementary experiment, the click chemistry reaction was performed between DBCO–PNA and bare MXene (without the azido functionality). The presence of the same N 1s peaks after this reaction would indicate that the functionalization strategy proposed is based solely on physical adsorption of the PNA on the MXene rather than chemical functionalization. The N 1s XPS spectra of this MXene after the control click reaction was acquired and plotted against control spectra of the bare MXene (red), MXene without AzPTES (pink), PNA-MXene without AzPTES (black), and PNA-MXene (blue) in the ESI† (Fig. S4C). Notably, no N peaks were observed in this MXene sample, which confirms the absence of adsorbed PNA. This is expected as the developed click reaction protocol involves extensive washing of the MXene to remove any unreacted or physically adsorbed PNA from the MXene surfaces. These results further validate that the click reaction indeed enables covalent functionalization of the MXene surface with PNA probes as consistent with previous conclusions.
, –NH–CO–/tertiary nitrogen, and –NH2, respectively (Fig. 2C-i).41 Finally, for the PNA-MXene, the N 1s and C 1s core-level spectra were acquired (Fig. 2C-ii). Notably, comparing the N 1s spectra of the PNA starting material and PNA-MXene sample, similar peaks were observed, including the presence of amine and amide bonds, confirming successful clicking of PNA. Due to the difficulties of verifying the triazole bond formation via XPS (the theoretical triazole BE overlaps with that of nitrogen groups from the azido functionality at BE 400.8–401.2 eV and the PNA backbone), a control experiment was performed to verify that the PNA is indeed chemically rather than physically adsorbed on the MXene surface (Fig. S4C, ESI†). In this supplementary experiment, the click chemistry reaction was performed between DBCO–PNA and bare MXene (without the azido functionality). The presence of the same N 1s peaks after this reaction would indicate that the functionalization strategy proposed is based solely on physical adsorption of the PNA on the MXene rather than chemical functionalization. The N 1s XPS spectra of this MXene after the control click reaction was acquired and plotted against control spectra of the bare MXene (red), MXene without AzPTES (pink), PNA-MXene without AzPTES (black), and PNA-MXene (blue) in the ESI† (Fig. S4C). Notably, no N peaks were observed in this MXene sample, which confirms the absence of adsorbed PNA. This is expected as the developed click reaction protocol involves extensive washing of the MXene to remove any unreacted or physically adsorbed PNA from the MXene surfaces. These results further validate that the click reaction indeed enables covalent functionalization of the MXene surface with PNA probes as consistent with previous conclusions.
Next, the colloid stability and surface charge of the bare and azido-MXene nanosheets were investigated via characterizing their zeta potential using a Zetasizer (Nano ZL) instrument (ESI,† Fig. S5A). Owing to the negatively charged surface functional groups, bare MXene exhibited a surface potential of −32.5 mV in water, indicating good colloidal stability.42 After azide functionalization, the zeta potential of the MXene nanosheets increased to −18.5 mV, which can be attributed to the successful reaction of the OH− groups during the silanization step. Furthermore, the overall size of the individual MXene nanosheets was studied via dynamic light scattering, revealing an average lateral size of 1.4 ± 0.08 μm (ESI,† Fig. S5B). The zeta potential of MXene-PNA sample was not measured due to the large sample volume requirements, which exceed our synthesis scale.
Raman spectroscopy was next performed to study the molecular vibrations and chemical structure of the bare MXene and azido-MXene nanosheets (LabRam Aramis instrument equipped with a green laser at a wavelength of 532 nm) (Fig. 3A). The spectrum for bare MXene exhibited characteristic wave numbers at ∼205, 270, 385, 575, 620, and 720 cm−1 which can be correlated to the expected Raman vibration modes of Ti3C2Tx MXene observed in the literature.43 Notably, the peaks at ∼205 cm−1 and 720 cm−1 correspond to A1g out-of-plane vibrations from the surface functional groups and carbon atoms, respectively. The peaks at 280 cm−1 and 620 cm−1 correspond to Eg group vibrations, including the in-plane (shear) vibration modes of Ti, C, and surface functional groups (–O, –F, and –OH).44 Compared to the bare MXene spectrum, the azido-MXene spectrum displayed shifted peaks and the appearance of new peaks, indicative of surface functionalization (Fig. 3A, red trace).45 The spectral range spanning 230 cm−1 to 470 cm−1 is associated with vibrations related to surface atoms and thus can be used to probe the surface chemistry of MXenes and discern alterations arising from chemical or electrochemical transformations.46 Notably, compared to the spectrum for bare MXene, the azido-MXene spectrum exhibited additional peaks within this region at ∼273 cm−1 and 328 cm−1, which further validates the successful silanization reaction (and the formation of new Ti–O–Si bonds).47 Consistent with the literature, the peak at ∼156 cm−1 was assigned to the formation of oxidized Ti3C2 due to the high laser power during spectral acquisition.48
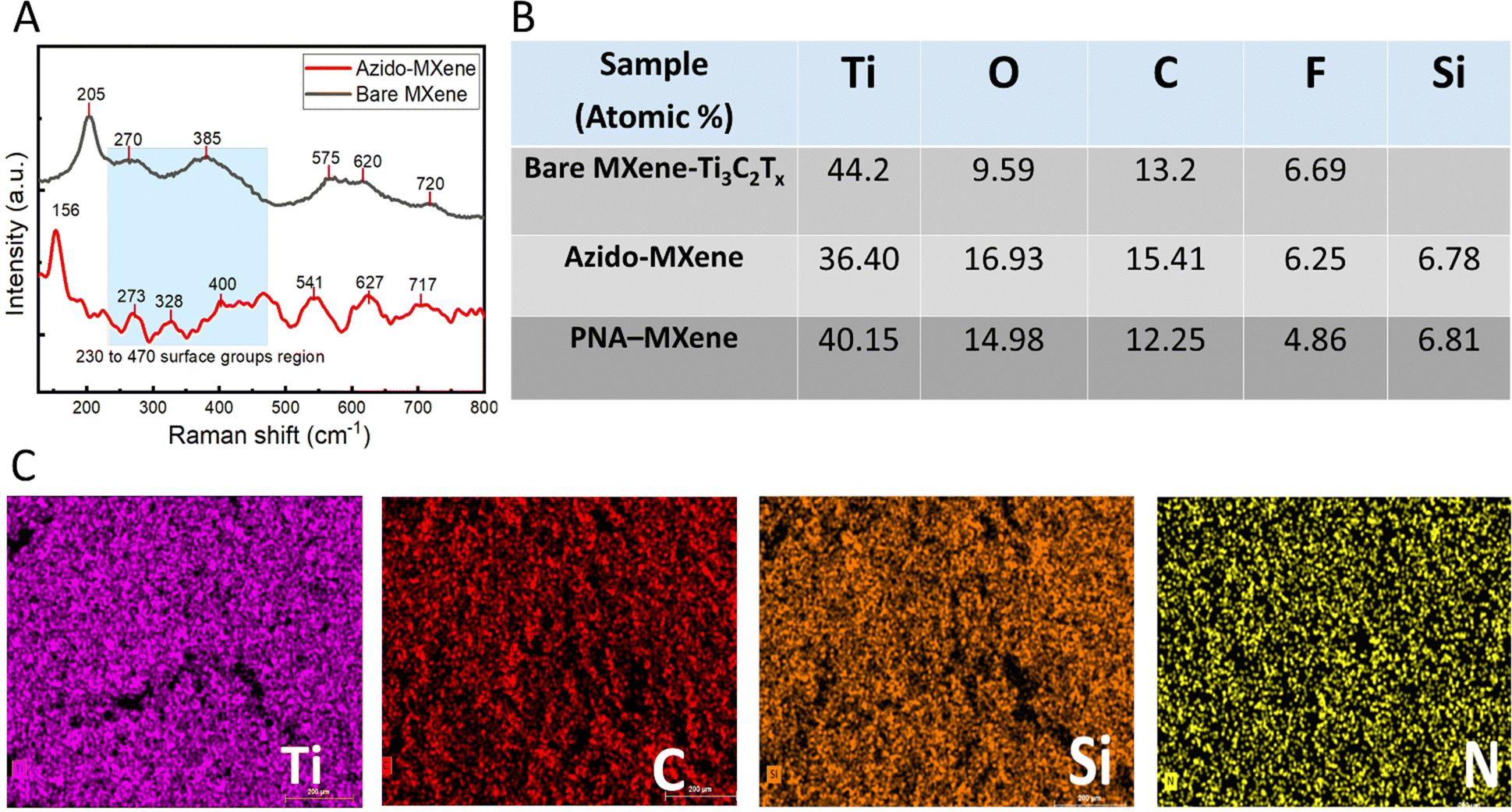 | ||
| Fig. 3 (A) Raman spectra of the bare MXene (black) and azido-MXene (red). (B) Bare MXene, azido-MXene and PNA-MXene atomic percentages. (C) The elemental mapping of the PNA-MXene was obtained via EDX spectroscopy. Scale bar shows 200 μm. | ||
To investigate the spatial distribution of various elements on the nanosheet surfaces, EDX analysis of the bare MXene, azido-MXene, and PNA-MXene was conducted (Fig. S6, ESI†). Ti, C, Si, and N elements were identified by combining EDX (20 keV) with EMAX energy spectroscopy. The elemental composition of the three samples is shown in Fig. 3B, demonstrating consistent results with XPS analysis. Ti and C were uniformly distributed on the nanosheet surfaces as shown in the elemental mapping in Fig. 3C. Similarly, after PNA functionalization, Si and N appeared to be uniformly distributed on the nanosheets, indicating the successful silanization and click chemistry reaction with the PNA probes and their homogeneous distribution on the nanosheet surfaces. Moreover, AFM analysis was conducted to characterize the surface topography and quantify the thickness of individual PNA-MXene nanosheets compared to the bare MXene. The AFM scans, height profiles, and 3D AFM images of the nanoconstruct are shown in the ESI† (Fig. S7). Notably, the line profile of the PNA-MXene sample was distinct from that of pristine MXene: it showed a larger overall sheet thickness and a higher, yet uniform surface roughness associated with the top layer of PNA.
To investigate the electrochemical properties of the MXene nanosheets before and after each functionalization step, a three-electrode electrochemical cell setup comprising GCE (2 mm2 working area) as the working electrode, Pt wire as the auxiliary electrode, and Ag/AgCl as the reference electrode was adopted. Notably, all electrochemical experiments were performed on a portable commercial potentiostat (Palmsens4, Palmsens), to demonstrate the amenability of the platform for future point-of-care applications. Firstly, cyclic voltammetry was performed to investigate the electrochemical processes at the electrode–solution interface in the presence of hexaammineruthenium(III) chloride ([Ru(NH3)6]Cl3, 5 mM) (RuHex) solution in the potential window of −0.6 to 0.2 V at a scan rate of 100 mV s−1. RuHex was used as the redox probe due to its ability to be oxidized and reduced at lower potential windows (i.e., more cathodic potentials) to minimize the probability of MXene oxidation.11
Using only GCE, two well-defined reversible redox peaks corresponding to the oxidation and reduction of the redox probe were observed at the working electrode (Fig. 4A, black). When the GCE was coated with bare MXene, the oxidation peak current of the redox probe increased by 40-fold from 2.24 μA to 89 μA, as shown in Fig. 4A (ii, blue), which can be attributed to both capacitive and faradaic currents. The increase in faradaic current (∼15.5 μA) is indicative of the enhanced efficiency of electron transfer within the MXene nanosheets, which can be attributed to the unique morphology (high specific surface area) and intrinsic electronic conductivity of the transducing material. These results agree with previous research on nanostructured and nanoporous electrodes. Notably, previous studies have demonstrated the ability to achieve accelerated electron transport and reduced charge screening using gold nanostructured electrodes.49 When the working GCE was coated with PNA-MXene, a decreased oxidation peak current compared to that of MXene-coated GCE was observed (Fig. 4A, red). This decreased oxidation peak current can be attributed to the presence of PNA probes on the MXene surfaces which may impede electron transfer kinetics.
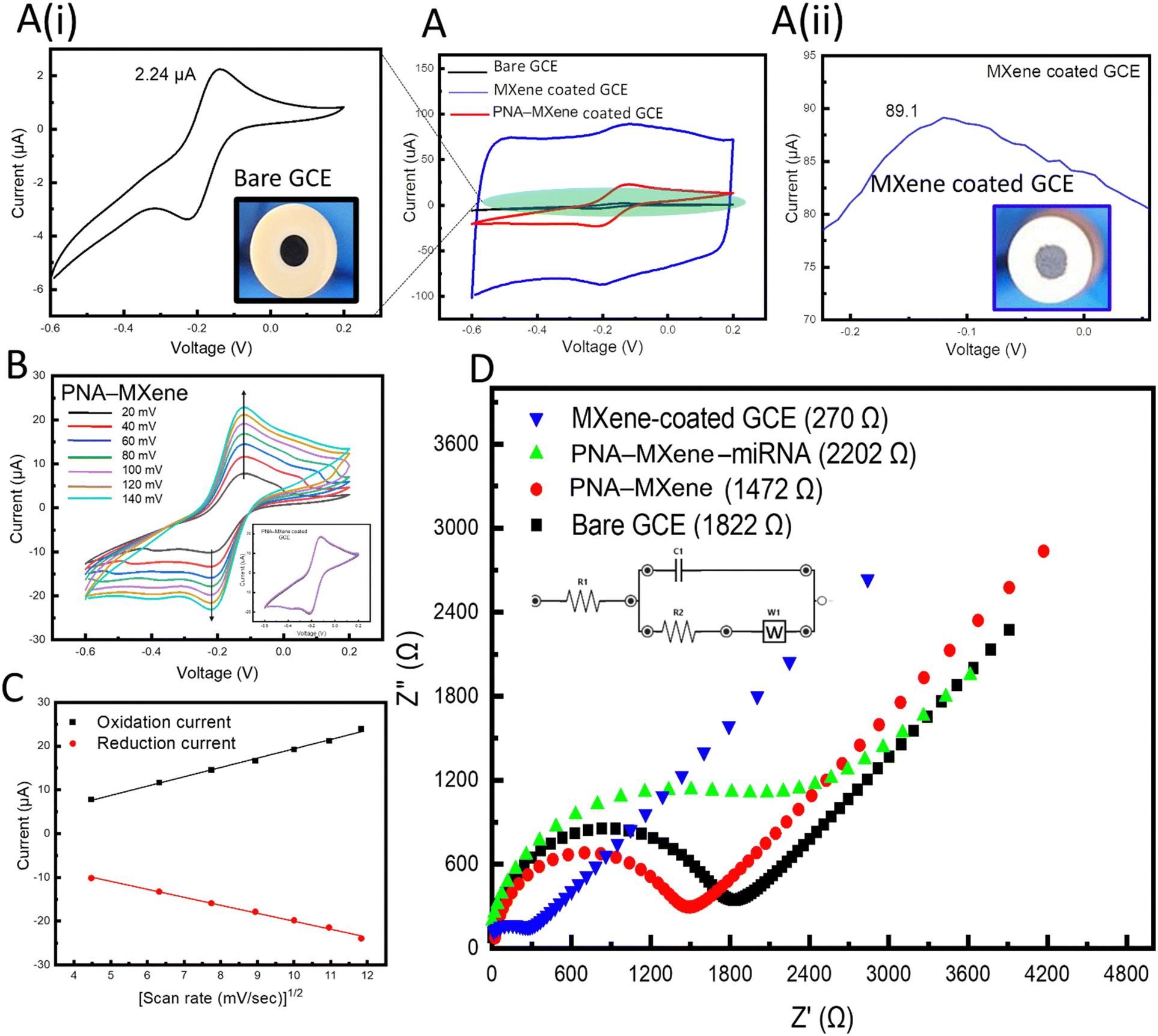 | ||
| Fig. 4 (A) CVs of bare GCE (black, i), MXene-coated GCE (blue, ii), and PNA-MXene-coated GCE (red) in 5 mM RuHex at a scan rate of 100 mV s−1. (B) CVs of PNA-MXene-coated GCE at different scan rates from 20 mV s−1 to 140 mV s−1. Inset shows stability after repeating five CV cycles. (C) Plots of oxidation (black) and reduction (red) currents versus square root of the scan rate, showing a linear relationship. (D) EIS of the bare GCE (black), MXene-coated GCE (blue), and PNA-MXene-coated GCE before (red) and after (green) hybridization with target miRNA. Inset shows the equivalent circuit diagram. | ||
Cyclic voltammograms were acquired to quantify the electrochemically active surface area of MXene-coated and PNA-MXene-coated GCEs compared to that of bare GCE. This area was calculated using the Randles–Sevcik equation,50Ip = 2.69 × 105AD1/2cv1/2, where Ip represents the peak current (in amps), A represents the electrochemically active surface area (in cm2), D represents the diffusion coefficient of the redox probe (i.e., 6.0 × 10−6 cm2 s−1), c represents the concentration of RuHex (i.e., 5 mM), and v represents the scan rate (in V s−1). The electrochemically active surface areas of the bare GCE and MXene-coated GCE were determined to be 7.6 × 10−4 cm2 and 50 × 10−4 cm2, respectively. This 7-fold increase in the electrochemically active surface area can be attributed to two main properties of the MXene nanosheets when drop-casted on the GCEs: their distinct nanoporous morphology and their intrinsically high metallic conductivity.51 Both these features render the MXene (and specifically probe-functionalized MXene) as a highly suitable transducing matrix for electrochemical biosensing applications.
Electrochemical kinetics of the bare MXene-coated GCE and PNA-MXene-coated GCE were studied at scan rates ranging from 20 mV s−1 to 120 mV s−1 (Fig. S8A, ESI†). For the bare GCE, the cathodic and anodic currents increased linearly with the square root of the scan rate, indicating a reversible and diffusion-controlled redox reaction (Fig. S8B, ESI†).52 Similarly, the electrochemical kinetics of the PNA-MXene-coated GCE were studied at scan rates ranging from 20 mV s−1 to 140 mV s−1, as shown in Fig. 4B. The cathodic and anodic currents also increased linearly with the square root of the scan rate (Fig. 4C). This indicates that the PNA probes do not affect the electrochemical behavior of the redox species generated at the electrode surface, i.e., the mechanism is also diffusion-controlled, and the electron transfer is reversible. The electrochemical stability of the MXene-coated GCE was investigated by acquiring multiple scans of cyclic voltammograms and observing the overlap of the CVs as shown in the ESI† (Fig. S8C).
Electrochemical impedance spectroscopy (EIS) was performed in Tris–EDTA (TE) buffer (50 mM, pH 7.4) with 5 mM RuHex to characterize the GCE modifications and monitor the changes in impedance induced by the bioreceptor functionalization of the MXene transducer. The charge-transfer resistance (Rct) was monitored across the frequency range of 0.1–100 kHz. As shown in Fig. 4D, coating the GCE with bare MXene nanosheets resulted in a seven-fold reduction in impedance from an Rct of 1.82 kΩ for the bare GCE down to 270 Ω for the MXene-coated GCE. This demonstrates that the modification of the GCE with the 2D nanomaterial enables robust and highly efficient electron transfer at the electrode surface. Moreover, modification of the GCE with PNA-MXene resulted in an increase in the impedance to 1.47 kΩ, verifying the functionalization and presence of bioreceptors, which hinder charge transfer. To confirm that this impedance is caused by the presence of our specific bioreceptor (PNA probe), the target miRNA was added to the electrode and allowed to hybridize at room temperature (∼25 °C) for 30 min. After extensive washing of the surface to remove non-specifically bound molecules, the corresponding EIS spectrum was acquired, revealing a further increase in impedance to 2.2 kΩ. This increase can be attributed to the reduced charge transfer at the electrode surface owing to the formation of the bulky PNA:miRNA duplex on the MXene nanosheets. Overall, the electrochemical characterization studies (both CV and EIS) consistently confirmed the enhanced electron transfer of the MXene nanosheets and high (covalent) loading of the bioreceptors on the MXene surfaces, favourable for electrochemical biosensing applications.
The biosensing performance of the PNA-MXene-coated GCE platform was initially investigated by performing a series of control experiments. Briefly, the protocol involved adding 5 μL of 10 pM target miRNAs onto the PNA-MXene-coated electrode and incubating for 30 min at RT for hybridization. After washing non-specifically bound molecules thrice with TE buffer, 5 μL of 10 mM MB was added and incubated for 5 min. After washing thrice, oxidation peak currents were recorded by cyclic voltammetry in the potential range of −0.6 to 0.2 V at a scan rate of 0.1 V s−1.
Firstly, in the absence of target miRNA and after MB incubation, no distinct oxidation peak was observed in the CV (Fig. 5A, red). However, after adding the target miRNA, a distinct oxidation peak current was observed (Fig. 5A, black). Since no other component in the system is redox-active, the observed peak was attributed to the oxidation of the MB present at the electrode surface due to binding and/or intercalation within the formed PNA:miRNA duplex.53 The mechanism of binding between the MB and PNA:miRNA duplex can be attributed to a combination of physical interactions including intercalation, electrostatic interactions, and groove binding.54,55
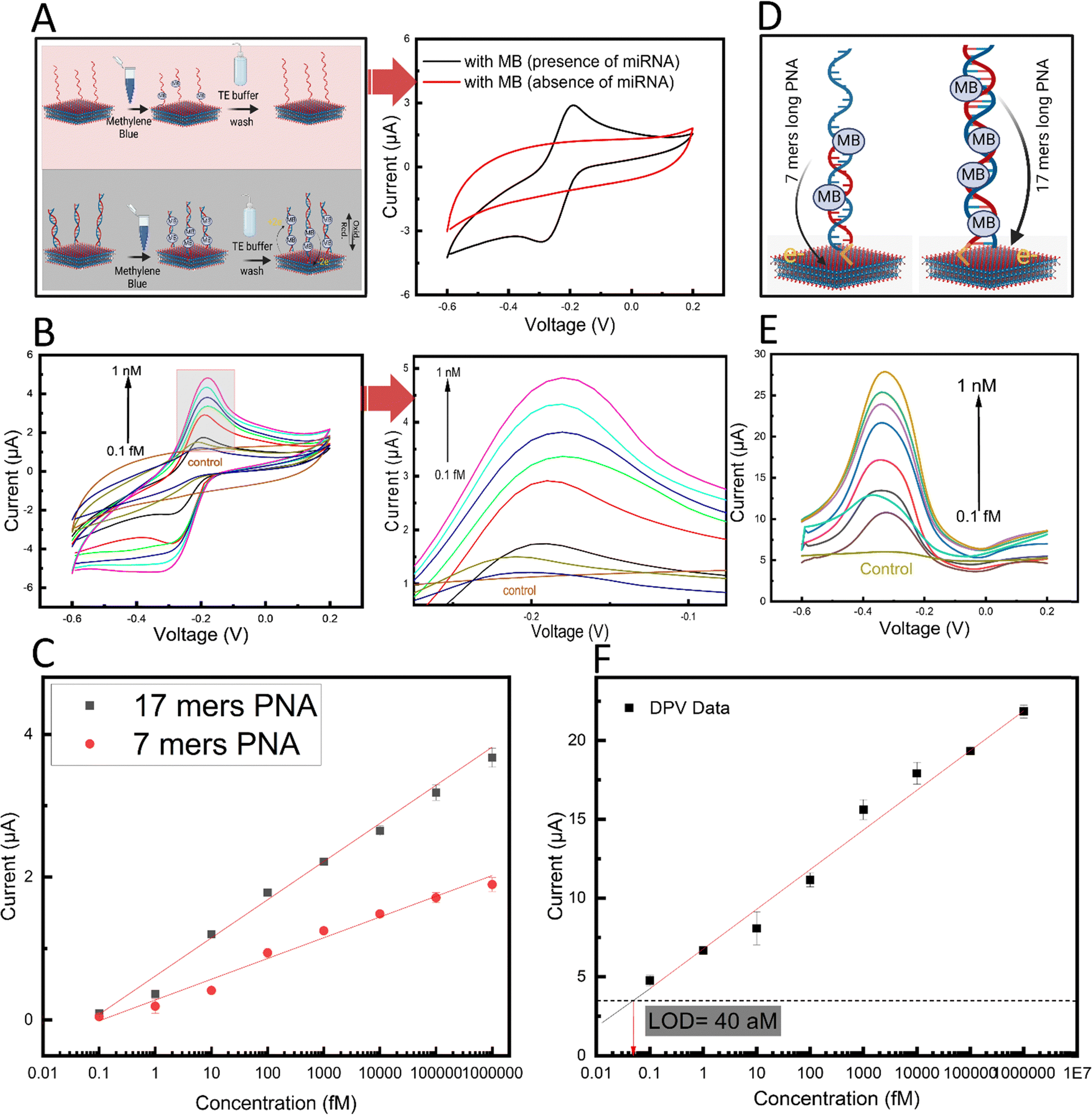 | ||
| Fig. 5 (A) Schematic representation of control experiments conducted in the absence (red) and presence (black) of the target miRNA along with the corresponding CVs acquired at a scan rate of 100 mV s−1. (B) Cyclic voltammograms in the presence of increasing concentrations of the target miRNA ranging from 0.1 fM to 1 nM along with the corresponding oxidation peaks (arrow upwards indicates trend direction). (C) Calibration curve for the detection of miR-141 using the short 7-mer PNA (red dots) and long 17-mer PNA (black squares). (D) Schematic illustration representing the MB-bound PNA:miRNA duplex formed by the 7-mer (left) and 17-mer (right) PNA probe. (E) DPV response of the 17-mer PNA-MXene biosensor with increasing concentrations of the target miRNA. (F) Calibration curve showing the dependence of the DPV peak currents on the target miRNA at concentrations ranging from 0.1 fM to 1 nM. Dashed black line indicates three times the standard deviation of the control (i.e., no miRNA) condition, used to calculate the LOD. Error bars in panels (C) and (F) indicate standard deviations from n = 6 data points collected from two individual biosensors with three CV or DPV scans acquired per concentration. | ||
Next, to characterize the biosensor sensitivity, calibration curves were plotted from the oxidation peak currents of the CVs in the presence of increasing concentrations of target miRNA from 0.1 fM to 1 nM (Fig. 5B). A linear regression line was fitted to the data and the limit of detection (LOD; calculated as three times the standard deviation of the control condition in the absence of miRNA) was calculated. Interestingly, a linear response (R = 0.98), wide dynamic range (over seven orders of magnitude), and low calculated LOD of 180 aM were observed (Fig. 5C, red). This remarkable analytical performance can be attributed to the efficient electron transfer between the intercalated MB redox label and the MXene transducing surface on the electrode. Apart from the unique morphology and conductivity of the MXene nanosheets, the direct covalent functionalization of the PNA bioreceptor in close proximity to the nanosheet surfaces may also contribute to this highly efficient redox process.
Considering the transduction mechanism in our biosensor, we hypothesized that using a longer PNA would result in an increase in the number of MB molecules within the PNA:miRNA duplex, ultimately leading to higher biosensor sensitivity (Fig. 5D). To this end, we designed and prepared a longer 17-mer PNA probe and assessed our biosensing performance. The longer probe was based on the same design as the original 7-mer PNA probe but with 10 more nucleotides complementary to hsa-miR-141 (chemical structure in Fig. S1, ESI†). Importantly, compared with standard hybridization probes based on negatively charged natural nucleic acid probes such as ssDNA, our design using PNA probes minimizes non-specific electrostatic interactions between positively charged MB and the probe in the absence of the target miRNA. With the longer PNA probe immobilized onto MXene nanosheets based on the developed methods, a second calibration curve was acquired by varying the target miRNA concentration from 0.1 fM to 1 nM (Fig. 5C, black). Interestingly, a distinct increase in the slope of the linear regression line by roughly 2-fold was observed, which is indicative of an enhanced biosensor sensitivity. This enhancement effect also contributed to a 56% decrease and thus substantial improvement in the calculated LOD to 78 aM. Next, owing to the low capacitive currents in differential pulse voltammetry measurements, this approach was used to conduct a calibration curve to further improve the sensitivity (Fig. 5E). The oxidation peak current versus miRNA concentration from 0.1 fM to 1 nM was plotted in a calibration curve, and a linear regression line was fitted as previously described (Fig. 5F). A further enhancement in sensitivity was achieved using DPV analysis, reaching a calculated LOD of 40 aM and a wide linear dynamic range (covering over seven orders of magnitude). It is noteworthy that while calculated LOD was 40 aM, the lowest concentration tested was 100 aM. With this ultralow (attomolar regime) limit of detection, the sensitivity of our proposed biosensor outperforms that of recently reported electrochemical biosensors for nucleic acid detection (ESI,† Table S1). Importantly, compared to most reported approaches which depend on enzymes and amplification strategies to achieve sufficient sensitivity, this study achieves attomolar sensitivity in an isothermal, enzyme-free, and amplification-free manner through advances in nanomaterials, marking a significant development in electrochemical biosensing.
The superior analytical performance of our biosensor achieving attomolar sensitivity in the absence of enzymes, amplification mechanisms, or additional 0D nanomaterials, can be attributed to the interplay between two synergistic effects, afforded by our novel PNA-MXene transducing material, which together increase the probability of hybridization events and the efficiency of electron transfer between the redox probe-intercalated duplex and the electrode surface. Firstly, the efficiency and bio-orthogonality of our click chemistry-based functionalization strategy (which exploits MXene's abundant terminal hydroxyl groups) ensures high probe grafting density which increases the probability of PNA:miRNA hybridization events within the material's porous structure.56 Secondly, the unique 3D morphology of our transducing material with its high electrochemically active surface area reduces Debye volume within the porous structure.57,58 This leads to more efficient electron transfer (by weakening charge screening) and an increase in the probability of faradaic electron transfer events for a given probe conformation. This effect has been shown to lower LOD by 4-fold in nanostructured electrodes compared to planar electrodes with the same footprint.59 The two aforementioned synergetic effects, illustrated in the ESI† (Fig. S9), provide insights into the mechanisms that provide our biosensor with such high sensitivity without enzymes or amplification strategies.
To study the specificity of the biosensing platform, the electrochemical response of the biosensor was investigated in the presence of the target miRNA sequence (hsa-miR-141) compared with a negative control miRNA (Cel-miR-39) in addition to three mutated hsa-miR-141 sequences designed with a different number of point mutations (Fig. 6A). The results were analyzed using an unpaired, one-tailed t-test (two groups) with equal variance not assumed (Welch correction), considering p < 0.05 as statistically significant. Specifically, the biosensor's response to one, two, or three single-nucleotide mutations (SNPs) along the miRNA sequence was investigated and quantified. All sequences tested are shown in the table displayed in Fig. 6B. Compared with the response from the target miRNA, which was normalized to 100%, the non-target sequences Cel-miR-39, miR-141-1SNP, miR-141-2SNPs, and miR-141-3SNPs caused 72%, 68%, 86%, and 99% reductions in the oxidation peak current, respectively. The high specificity of the sensor can be largely attributed to the choice of bioreceptor: the engineered PNA probes. Owing mainly to their neutral backbone, PNAs bind to target nucleic acids with greater affinity and thermal stability. For example, the melting temperature (Tm) of a 15-mer DNA:RNA duplex is 50.1 °C whereas that of an equivalent PNA:RNA duplex is 72.2 °C.28 This also means that nucleotide mismatches cause a greater destabilizing effect in the PNA:miRNA duplex compared to duplexes of natural nucleic acids.60 Another noteworthy factor that can contribute to the biosensor's high specificity is the sensitivity of MB, the hybridization redox indicator, to mismatches. Local disruptions such as mismatches within the duplex can hinder and potentially prevent the intercalation of MB.61 These results demonstrate the immense potential of our platform in nucleic acid sensing, especially when applied to miRNA biomarkers, which suffer from high sequence homology among family members.
 | ||
| Fig. 6 (A) Specificity test of the biosensor showing the current signal response to target miR-141 (normalized to 100%) (green) compared with non-target Cel-miR-39 (blue) and three mutated miR-141 sequences (orange) with one, two, or three individual single-nucleotide mutations within the sequence. *** indicates p-value <0.001 from a one-tailed t-test. Error bars indicate standard deviation from n = 6 data points collected from two individual biosensors with three DPV scans acquired per sample. (B) Table of oligonucleotide sequences used in this study including the synthesized PNA probes and commercial synthetic miRNAs. (C) The effect of time and storage conditions on biosensor stability was evaluated by quantifying the oxidation peak current of four sensors in the presence of 5 mM RuHex probe. Sensors 1 and 2 (grey) were tested on the fabrication day (i.e., fresh sensors), with the signal from sensor 1 normalized to 100%. Sensors 3 and 4 (blue) were examined after a 7-day storage at 4 °C. (D) Evaluation of sensor performance using DPV analysis with real biological samples (human serum) containing 5 mM RuHex probe. Compared to the response in buffer (normalized to 100%), a minimal 8% and 13% reduction in current was observed in the presence of 10% serum (yellow) and 100% serum (dark yellow) samples respectively. (E) Comparing the biosensor response to three concentrations of target miRNA prepared in TE buffer (grey) versus spiked into 100% serum (yellow). Control-subtracted current is calculated as the oxidation peak current in the presence of target miRNA minus the peak current in the absence of target (i.e., blank sample with MB control). Error bars in panels (C) to (E) indicate standard deviations from three consecutive DPV cycles. | ||
In addition to studying their analytical performance, developing robust point-of-care biosensors necessitates a thorough investigation of their reproducibility and stability over time. To this end, four sensors were prepared independently, and their current response in the presence of 5 mM RuHex probe was examined by DPV analysis. Sensors 1 and 2 were evaluated immediately after fabrication (termed ‘fresh sensors’), while sensors 3 and 4 were tested after seven days of storage at 4 °C. Fig. 6C illustrates the normalized data, considering sensor 1 as the reference, normalized to 100%. Notably, the results reveal two critical findings. Firstly, sensors prepared and stored under identical conditions exhibit remarkable reproducibility. Secondly, seven days of storage causes a minimal (roughly 11%) decrease in their performance.
Ensuring the stability of biosensors upon exposure to complex biological fluid samples, like serum, is of critical importance for the reliability and effectiveness of point-of-care platforms based on liquid biopsy. To this end, DPV analysis was employed to assess biosensor stability by evaluating the current response to 5 mM RuHex probe prepared in control buffer solutions (grey), 10% human serum (light yellow) and 100% human serum (yellow) samples (Fig. 6D). When compared to the signal obtained from buffer solutions (normalized to 100%), a minimal reduction in the signal of 8% and 13% was observed for the 10% serum and 100% serum samples, respectively. These findings offer valuable insights into the biostability of our biofunctionalized MXenes and highlight the presence of intrinsic anti-biofouling properties, which can be improved by additional strategies in future work.
Building upon these encouraging findings, we next sought to evaluate the biosensor's efficacy in detecting miRNA within model biological samples, specifically 100% human serum spike-in samples. As shown in Fig. 6E, compared to miRNA-in-buffer samples (normalized to 100%), the raw serum spike-in miRNA samples exhibited a nominal reduction in signal intensity, with decreases of 20%, 16%, and 22% observed for target miRNA concentrations of 1 pM, 10 pM, and 100 pM, respectively. This decrease in the analytical signal can be attributed to the biofouling effects stemming from serum proteins and other biological components on the MXene surface. While promising, our future work will focus on enhancing the anti-biofouling activity and biostability of MXenes to enable their use as key functional components and advanced materials in liquid biopsy-based point-of-care electrochemical biosensors.
Conclusions
To detect low-abundance nucleic acid biomarkers like miRNA, most approaches in electrochemical biosensing rely on the use of expensive enzymes for target or signal amplification or on the use of nanocomposites or nanomaterial-based probes. Despite their high sensitivity, these strategies suffer from high complexity and poor stability while being time-consuming and expensive, all of which hinder their application in point-of-care testing. This study develops a robust, enzyme-free, amplification-free, and nanoparticle-free electrochemical biosensor for miRNA detection, which is based on covalent functionalization of 2D MXene nanosheets with bespoke PNA hybridization probes using bio-orthogonal click chemistry strategies. Notably, this is the first demonstration of click-based bioconjugation of MXene nanosheets with a remarkable limit of detection of 40 aM, surpassing reported enzyme-based methods. The biosensing mechanism relies on the highly efficient electron transport between MB intercalators and the MXene-coated electrode, which is mediated by the formation of a PNA:miRNA duplex upon sequence-specific target detection. The outstanding sensitivity of the developed biosensor is attributed to a combination of intrinsic and engineered factors, namely, (i) the inherently high conductivity and electron transport of the MXene nanosheets, (ii) their unique morphology on the electrode, which enables accelerated and efficient electron transfer, and (iii) the stable and direct covalent bonding between the bioreceptor and the MXene surfaces. Single-nucleotide specificity was demonstrated using robust PNA probes as bioreceptors instead of natural nucleic acid probes. Importantly, extensive stability studies were performed demonstrating high sensor reproducibility as well as stability over time and upon exposure to raw biological samples, namely 100% human serum. Furthermore, the biosensor design can be tailored to detect any other nucleic acid biomarker and potentially other biomarker types by changing the immobilized bioreceptor type. This versatile and robust strategy demonstrates the immense potential of engineered PNA-MXene nanosheets to act as efficient and ultrasensitive electrochemical transducers in the next generation of advanced point-of-care liquid biopsy biosensors.Author contributions
Muhsin Ali: data curation, formal analysis, investigation, methodology, visualization, writing – original draft: lead; Sharat Chandra Barman: data curation and methodology: support; Erol Hasan, PhD: conceptualization, data curation, formal analysis: support, investigation, methodology, writing – review & editing: support; Mohamed Nejib Hedhili: data curation and formal analysis; support; Husam N. Alshareef, PhD: conceptualization, funding acquisition, supervision, writing – review & editing: support; Dana Alsulaiman, PhD: conceptualization, formal analysis, funding acquisition, investigation, supervision, writing – review & editing: lead, visualization, writing – original draft: support.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The research reported in this publication was supported by King Abdullah University of Science and Technology (KAUST). This work was also partially supported by a KAUST Smart Health Initiative grant (REI/1/5295-01-01). We also acknowledge Wildan Hanif for handling the serum samples, Yongjiu Lei for support with TEM, as well as Xiangming Xu and Somayah Qutub for their support with AFM.References
- D. P. Bartel, Cell, 2009, 136, 215–233 CrossRef CAS PubMed.
- J. Wang, J. Chen and S. Sen, J. Cell. Physiol., 2016, 231, 25–30 CrossRef CAS PubMed.
- H. Cheng, L. Zhang, D. E. Cogdell, H. Zheng, A. J. Schetter, M. Nykter, C. C. Harris, K. Chen, S. R. Hamilton and W. Zhang, PLoS One, 2011, 6, e17745 CrossRef CAS PubMed.
- M. de Planell-Saguer and M. C. Rodicio, Anal. Chim. Acta, 2011, 699, 134–152 CrossRef CAS PubMed.
- H. Dong, J. Lei, L. Ding, Y. Wen, H. Ju and X. Zhang, Chem. Rev., 2013, 113, 6207–6233 CrossRef CAS PubMed.
- C. De la Rosa and J. L. Reyes, Northern blot analysis of microRNAs and other small RNAs in plants, 2019, pp. 121–129 Search PubMed.
- A. Ramanavičius, A. Ramanavičienė and A. Malinauskas, Electrochim. Acta, 2006, 51, 6025–6037 CrossRef.
- A. T. Lawal, Biosens. Bioelectron., 2018, 106, 149–178 CrossRef CAS PubMed.
- H. Liu, H. Song, Y. Su and Y. Lv, Appl. Spectrosc. Rev., 2019, 54, 275–284 CrossRef CAS.
- L. Wu, X. Lu, Dhanjai, Z. S. Wu, Y. Dong, X. Wang, S. Zheng and J. Chen, Biosens. Bioelectron., 2018, 107, 69–75 CrossRef CAS PubMed.
- S. Kumar, Y. Lei, N. H. Alshareef, M. A. Quevedo-Lopez and K. N. Salama, Biosens. Bioelectron., 2018, 121, 243–249 CrossRef CAS PubMed.
- X. Cai, Y. Luo, B. Liu and H.-M. Cheng, Chem. Soc. Rev., 2018, 47, 6224–6266 RSC.
- J. Zheng, J. Diao, Y. Jin, A. Ding, B. Wang, L. Wu, B. Weng and J. Chen, J. Electrochem. Soc., 2018, 165, B227–B231 CrossRef CAS.
- P. A. Rasheed, R. P. Pandey, K. Rasool and K. A. Mahmoud, Sens. Actuators, B, 2018, 265, 652–659 CrossRef.
- M. Naguib, M. Kurtoglu, V. Presser, J. Lu, J. Niu, M. Heon, L. Hultman, Y. Gogotsi and M. W. Barsoum, Adv. Mater., 2011, 23, 4248–4253 CrossRef CAS PubMed.
- B. Akuzum, K. Maleski, B. Anasori, P. Lelyukh, N. J. Alvarez, E. C. Kumbur and Y. Gogotsi, ACS Nano, 2018, 12, 2685–2694 CrossRef CAS PubMed.
- G. Gao, A. P. O’Mullane and A. Du, ACS Catal., 2017, 7, 494–500 CrossRef CAS.
- R. A. Soomro, S. Jawaid, Q. Zhu, Z. Abbas and B. Xu, Chin. Chem. Lett., 2020, 31, 922–930 CrossRef CAS.
- Q. Wu, N. Li, Y. Wang, Y. liu, Y. Xu, S. Wei, J. Wu, G. Jia, X. Fang, F. Chen and X. Cui, Biosens. Bioelectron., 2019, 144, 111697 CrossRef CAS PubMed.
- P. Yadav, Z. Cao and A. Barati Farimani, ACS Nano, 2021, 15, 4861–4869 CrossRef CAS PubMed.
- X. Yang, M. Feng, J. Xia, F. Zhang and Z. Wang, J. Electroanal. Chem., 2020, 878, 114669 CrossRef CAS.
- Y.-H. Wang, L.-L. He, K.-J. Huang, Y.-X. Chen, S.-Y. Wang, Z.-H. Liu and D. Li, Analyst, 2019, 144, 2849–2866 RSC.
- M. Mohammadniaei, A. Koyappayil, Y. Sun, J. Min and M.-H. Lee, Biosens. Bioelectron., 2020, 159, 112208 CrossRef CAS PubMed.
- F. Duan, C. Guo, M. Hu, Y. Song, M. Wang, L. He, Z. Zhang, R. Pettinari and L. Zhou, Sens. Actuators, B, 2020, 310, 127844 CrossRef CAS.
- L. Liu, Y. Wei, S. Jiao, S. Zhu and X. Liu, Biosens. Bioelectron., 2019, 137, 45–51 CrossRef CAS PubMed.
- V. V. Demidov, V. N. Potaman, M. D. Frank-Kamenetskil, M. Egholm, O. Buchard, S. H. Sönnichsen and P. E. Nlelsen, Biochem. Pharmacol., 1994, 48, 1310–1313 CrossRef CAS PubMed.
- R. Corradini, S. Sforza, T. Tedeschi, F. Totsingan, A. Manicardi and R. Marchelli, Curr. Top. Med. Chem., 2011, 11, 1535–1554 CrossRef CAS PubMed.
- M. Egholm, O. Buchardt, L. Christensen, C. Behrens, S. M. Freier, D. A. Driver, R. H. Berg, S. K. Kim, B. Norden and P. E. Nielsen, Nature, 1993, 365, 566–568 CrossRef CAS PubMed.
- M. Shekhirev, C. E. Shuck, A. Sarycheva and Y. Gogotsi, Prog. Mater. Sci., 2021, 120, 100757 CrossRef CAS.
- J. Hu, K. Xiao, B. Jin, X. Zheng, F. Ji and D. Bai, Biotechnol. Bioeng., 2019, 116, 2764–2777 CrossRef CAS PubMed.
- S. Ruffin, I. A. Hung, U. M. Koniges and R. Levicky, ACS Sens., 2017, 2, 892–896 CrossRef CAS PubMed.
- Y. Wang, Z. Chen and W. Chen, Eur. J. Med. Res., 2017, 22, 51 CrossRef PubMed.
- J. M. Eeftens, J. van der Torre, D. R. Burnham and C. Dekker, BMC Biophys., 2015, 8, 9 CrossRef PubMed.
- M. J. Crawford, S. Rapireddy, R. Bahal, I. Sacui and D. H. Ly, J. Nucleic Acids, 2011, 2011, 1–10 CrossRef PubMed.
- Y. Wu and J.-C. Xu, Tetrahedron, 2001, 57, 8107–8113 CrossRef CAS.
- J. K. El-Demellawi, A. E. Mansour, A. M. El-Zohry, M. N. Hedhili, J. Yin, A.-H. M. Emwas, P. Maity, X. Xu, O. M. Bakr, O. F. Mohammed and H. N. Alshareef, ACS Mater Lett., 2022, 4, 2480–2490 CrossRef CAS.
- J. Zhu, Y. Tang, C. Yang, F. Wang and M. Cao, J. Electrochem. Soc., 2016, 163, A785–A791 CrossRef CAS.
- J. Halim, K. M. Cook, M. Naguib, P. Eklund, Y. Gogotsi, J. Rosen and M. W. Barsoum, Appl. Surf. Sci., 2016, 362, 406–417 CrossRef CAS.
- J. Boehmler, Well-controlled and well-described SAMs-based platforms for the study of material-bacteria interactions occuring at the molecular scale, PhD dissertation, Université de Haute Alsace-Mulhouse, 2012.
- T. Heinrich, C. H.-H. Traulsen, E. Darlatt, S. Richter, J. Poppenberg, N. L. Traulsen, I. Linder, A. Lippitz, P. M. Dietrich, B. Dib, W. E. S. Unger and C. A. Schalley, RSC Adv., 2014, 4, 17694–17702 RSC.
- A. Cattani-Scholz, D. Pedone, M. Dubey, S. Neppl, B. Nickel, P. Feulner, J. Schwartz, G. Abstreiter and M. Tornow, ACS Nano, 2008, 2, 1653–1660 CrossRef CAS PubMed.
- C. Sun, C. Wu, X. Gu, C. Wang and Q. Wang, Nanomicro Lett., 2021, 13, 89 CAS.
- M. Naguib, M. Kurtoglu, V. Presser, J. Lu, J. Niu, M. Heon, L. Hultman, Y. Gogotsi and M. W. Barsoum, Adv. Mater., 2011, 23, 4248–4253 CrossRef CAS PubMed.
- T. Hu, J. Wang, H. Zhang, Z. Li, M. Hu and X. Wang, Phys. Chem. Chem. Phys., 2015, 17, 9997–10003 RSC.
- S. Kumar, Y. Lei, N. H. Alshareef, M. A. Quevedo-Lopez and K. N. Salama, Biosens. Bioelectron., 2018, 121, 243–249 CrossRef CAS PubMed.
- A. Sarycheva and Y. Gogotsi, Chem. Mater., 2020, 32, 3480–3488 CrossRef CAS.
- F. Yang, Y. Huang, X. Han, S. Zhang, M. Yu, J. Zhang and X. Sun, ACS Appl. Nano Mater., 2023, 6, 3367–3377 CrossRef CAS.
- Y. Cao, Q. Deng, Z. Liu, D. Shen, T. Wang, Q. Huang, S. Du, N. Jiang, C.-T. Lin and J. Yu, RSC Adv., 2017, 7, 20494–20501 RSC.
- K. Fu, J. Seo, V. Kesler, N. Maganzini, B. D. Wilson, M. Eisenstein, B. Murmann and H. T. Soh, Adv. Sci., 2021, 8(23) DOI:10.1002/advs.202102495.
- T. R. L. C. Paixão, ChemElectroChem, 2020, 7, 3414–3415 CrossRef.
- A. Liu, X. Liang, X. Ren, W. Guan, M. Gao, Y. Yang, Q. Yang, L. Gao, Y. Li and T. Ma, Adv. Funct. Mater., 2020, 30(38) DOI:10.1002/adfm.202003437.
- N. Rohaizad, C. C. Mayorga-Martinez, M. Fojtů, N. M. Latiff and M. Pumera, Chem. Soc. Rev., 2021, 50, 619–657 RSC.
- B. J. Venton and D. J. DiScenza, Electrochemistry for Bioanalysis, Elsevier, 2020, pp. 27–50 Search PubMed.
- H.-A. Rafiee-Pour, M. Behpour and M. Keshavarz, Biosens. Bioelectron., 2016, 77, 202–207 CrossRef CAS PubMed.
- P. Paul, S. S. Mati and G. S. Kumar, J. Photochem. Photobiol., B, 2020, 204, 111804 CrossRef CAS PubMed.
- P. Daggumati, Z. Matharu and E. Seker, Anal. Chem., 2015, 87, 8149–8156 CrossRef CAS PubMed.
- K. Shoorideh and C. O. Chui, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 5111–5116 CrossRef CAS PubMed.
- V. Kesler, B. Murmann and H. T. Soh, ACS Nano, 2020, 14, 16194–16201 CrossRef CAS PubMed.
- K. Fu, J.-W. Seo, V. Kesler, N. Maganzini, B. D. Wilson, M. Eisenstein, B. Murmann and H. T. Soh, Adv. Sci., 2021, 8, e2102495 CrossRef PubMed.
- B. P. Belotserkovskii, R. Liu and P. C. Hanawalt, Mol. Carcinog., 2009, 48, 299–308 CrossRef CAS PubMed.
- E. Farjami, L. Clima, K. V. Gothelf and E. E. Ferapontova, Analyst, 2010, 135, 1443 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4mh00714j |
| This journal is © The Royal Society of Chemistry 2024 |