
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDirect restoration of photocurable cross-linkers for repeated light-based 3D printing of covalent adaptable networks†
Loc Tan
Nguyen
 and
Filip E.
Du Prez
*
and
Filip E.
Du Prez
*
A Polymer Chemistry Research Group, Centre of Macromolecular Chemistry (CMaC), Department of Organic and Macromolecular Chemistry, Faculty of Sciences, Ghent University, Krijgslaan 281 S4, 9000 Ghent, Belgium. E-mail: Filip.DuPrez@UGent.be
First published on 1st October 2024
Abstract
Light-based processing of thermosets has gained increasing attention because of its broad application field including its use in digital light processing (DLP) 3D printing. This technique offers efficient design and fabrication of complex structures but typically results in non-recyclable thermoset-based products. To address this issue, we describe here a photocurable, dynamic β-amino ester (BAE) based cross-linker that is not only suitable for DLP printing but can also be chemically degraded via transesterification upon the addition of 2-hydroxyethyl methacrylate (HEMA) as a decross-linker. This conceptually new protocol allows for efficient depolymerization with the direct restoration of curable monomers in a single step without the addition of external catalysts or solvents. By implementing this protocol, we have established a chemical recycling loop for multiple cycles of photo-cross-linking and restoration of cross-linkers, facilitating repeatable DLP 3D printing without generating any waste. The recycled materials exhibit full recovery of thermal properties and Young's modulus while maintaining 75% of their tensile strength for at least three cycles. Simultaneously, the presence of BAE moieties enables the (re)processability of these materials through compression molding.
New conceptsFirst of all, not only the broadly applicable dynamic β-amino ester chemistry has been applied for the first time on digital light-based processing (DLP 3D printing) of covalent adaptable networks (CANs) but the necessary photocurable cross-linkers have also been synthesized via a catalyst-free and up-scalable procedure from cost-effective building blocks. Although the first printable CANs to establish recyclable 3D printing have been reported recently, the repeated printing typically necessitates chemical recycling processes with costly catalysts or monomers and/or complex procedures that undermine the sustainability of this technology. To address these concerns, we established in this study a novel approach for closed-loop chemical recycling, enabling the direct regeneration of photocurable cross-linkers from cross-linked networks without any post-functionalization that is typically required in most of the current state-of-the-art protocols. In summary, the implementation of this pioneering protocol facilitates a repeatable DLP 3D printing process in a solvent- and catalyst-free way, and thus contributes to its potential industrial breakthrough. |
Introduction
The escalating accumulation of plastic waste, especially of non-recyclable thermosets, has become a global concern that requires substantial efforts from the scientific community to address.1–3 Thermosets are covalently cross-linked polymeric materials that are well known for their exceptional mechanical performance and thermal stability, which makes them suitable for advanced applications in a wide range of industrial sectors.4–7 However, due to their stable cross-links, such materials or derived composites are generally not recyclable, thus typically resulting in incineration or landfilling after use.8–10 With advancements in polymer and material science, covalent adaptable networks (CANs), also referred to as dynamic polymer networks, have emerged as a promising solution for reducing this thermoset-related pollution, and the first products have been commercialized.11–15 More specifically, this new class of polymeric materials contains bonds that are reversible by applying activated stimuli, and thus enabling the material to be recyclable while retaining its excellent performance at service conditions.16–19 A wide range of dynamic chemistry platforms has been explored over the past decades, which finally enabled the recycling via thermo-mechanical (or physical) processing such as injection molding or hot pressing.20–24 Despite some limitations related to energy consumption and thermal degradation, these recycling approaches could potentially be used with conventional thermoplastic processing techniques, which are widely used in plastics manufacturing.4,25,26Besides the well-established physical processing techniques for polymeric materials, significant attention has been recently directed towards photo-induced cross-linking, owing to its key advantages such as rapid and efficient processing. This is particularly beneficial in the context of 3D printing applications, where speed and precision are crucial.27–30 Light-based 3D printing techniques, especially digital light processing (DLP), also referred to as additive manufacturing, potentially enable the design and fabrication of complex structures that are applicable in for example robotics, medicine, and aviation.31–35 However, conventional printable polymeric resins typically result in permanent networks that possess limited capabilities for (re)processing and repairing, particularly in scenarios of printing failure or when end-of-use product modifications are desired.36,37 Unfortunately, these limitations render the majority of existing 3D printing technology unsustainable in the current mainstream of plastic waste management approaches, for which circularity is targeted.
To address this environmental issue, several dynamic chemistry platforms making use of thiourethanes, carbamates, disulfides, boronate esters, and Diels–Alder chemistry, have been utilized in photochemistry over the past years to synthesize photo-cross-linkable CANs that are potentially applicable for 3D printing.38–48 In 2018, Zhang et al. developed a UV-curable resin with dynamic properties based on transesterification, accelerated by Lewis acid, that enabled successful fabrication of healable/recyclable dynamic polymer networks via DLP 3D printing.49 Inspired by this work, Li et al. subsequently introduced a method to recycle such networks by grinding them into a powder and mixing them with a UV-curable resin, establishing a re-printable recycling process.50 While this mechanical recycling approach enables circular 3D printing, achieving high printing resolution and full recovery of properties necessitate fine grinding into microparticles and a post-treatment at elevated temperature (180 °C), which may cause undesirable degradation. Chemical recycling offers a promising alternative by recovering monomers or oligomers that can be reused as fresh reagents to (re)produce products, ideally, without sacrificing performance.4,7,51,52 In light-based processing, chemical recycling is especially valuable because it preserves the photo-responsive chemical functionalities and therefore enables repeatable photocuring, which recently emerged as an attractive strategy.
In 2023, Bowman et al. demonstrated a method to degrade printed thioester-based CANs by adding tetrathiol and triethylamine as a catalyst, yielding thiol-functionalized solutions that could be photo-cured and thus (re)printed via thiol–ene reactions.53 Later in the same year, Zhang et al. introduced a β-carbonyl ester that enables transesterification without the need for external catalysts, and successfully implemented it in photocurable cross-linkers, which can be 3D printed.54 Moreover, the resulting CANs could be reverted into photocurable resins through a two-step process, which involves the dissolution of the networks in ethylene glycol via transesterification and subsequent acrylate functionalization, allowing for repeated 3D printing processability. Very recently, Dove and coworkers reported circular resins for additive manufacturing through the synthesis of photoreactive cyclic disulfide monomers, which could undergo reversible (de)polymerization. The disulfide groups enabled the dissolution of the photocured networks to regenerate monomers with up to 98% yield, thus successfully establishing one of the most efficient light-based processing to date.55 Despite significant progress over the past decade, closed-loop photo-processing of CANs is still challenging and mostly requires costly catalysts or monomers, solvents and/or complex procedures that potentially undermine the sustainability of CANs.38,56
Taking advantage of the catalyst-free and easily upscalable dynamic β-amino ester (BAE) platform, which allows the fabrication of highly creep resistant CANs (up to 100 °C), initially developed by our research group in 2021 from acrylates and primary amines as bulk chemicals (see Fig. 1A),57–60 one of the objectives of this research is to develop dynamic cross-linkers containing BAEs that can be photocured and are applicable for 3D printing. Furthermore, based on the dynamic nature of BAE moieties, we aimed in this study for the straightforward chemical recovery of the obtained dynamic networks to the initial cross-linkers, thus offering closed-loop photocuring processing.
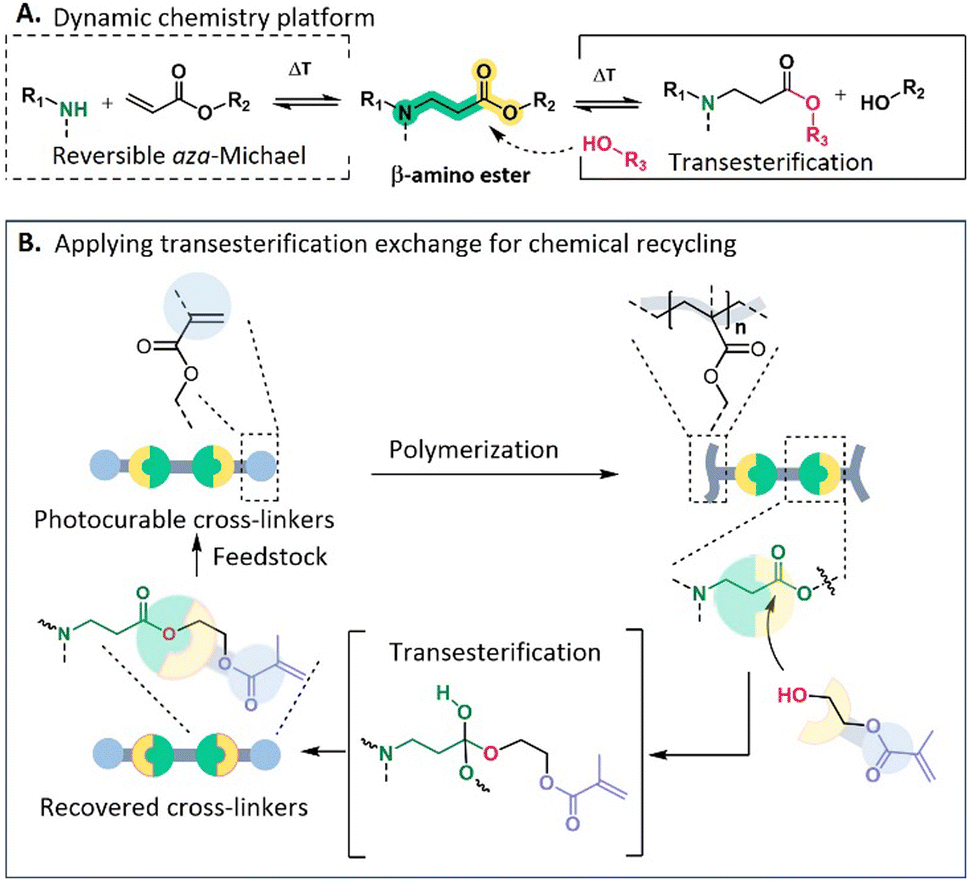 | ||
| Fig. 1 Schematic overview of: (A) exchange mechanism of β-amino ester chemistry that can be thermally activated, enabling dynamic exchange via two pathways including (retro) aza-Michael addition and transesterification (in the presence of hydroxyl groups); (B) the implementation of β-amino esters to design a circular chemical recycling protocol. HEMA is applied as a decross-linker for the regeneration of photocurable cross-linkers. | ||
In particular, we will first introduce the one-step design of a difunctional (meth)acrylate terminated cross-linker containing BAE as dynamic moieties for its use in vat photopolymerization, which is well established in light-based 3D printing.61,62 Subsequently, it will be demonstrated how 2-hydroxylethyl methacrylate (HEMA) will be used as a reactive compound that we will further refer to as a functional decross-linker, in which its hydroxyl group can exchange with the activated ester cross-links to degrade the network while simultaneously introducing new methacrylate functionalities. This newly established procedure allows for the direct regeneration of photocurable cross-linkers, thus paving the way for repeatable DLP 3D printing (Fig. 1B). Moreover, based on the presence of dynamic BAEs, these networks can also be thermo-mechanically (re)processed through compression molding, thus providing an additional recycling option.
Results and discussions
Our study started with the synthesis of an acrylate-terminated β-amino ester-containing cross-linker to examine the feasibility of chemical recycling of poly(β-amino ester)s (vide infra). For this purpose, a low molar mass poly(β-amino ester) cross-linker (BAEC) was synthesized via a one-step, upscalable and quantitative aza-Michael addition between commercially available building blocks, i.e. 1,6-hexanediol diacrylate (1, 1.25 eq.) and 4,4′-trimethylenedipiperidine (2, 1.0 eq.) (Fig. 2A and Fig. S1 and S2, ESI†). The cyclic secondary bisamine 2 was specifically chosen because of its commercial availability and cost-effectiveness, compared to linear bifunctional secondary amines.63,64 | ||
| Fig. 2 (A) The synthesis of poly(β-amino ester) macromolecular cross-linker (BAEC) from 1,6-hexanediol diacrylate (1, 1.25 eq.) and 4,4′-trimethylenedipiperidine (2, 1.0 eq.); (B) general representation of the depolymerization process of linear BAEC in the presence of HEMA; (C) kinetics of the depolymerization using varying HEMA content (i.e. 10, 20 and 30 eq. with respect to BAE moieties) at 80 and 100 °C, followed via SEC; (D) time sweep result and (E) corresponding visualization of the decross-linking of BAE-based network (BEN) in HEMA (20 eq.) at 100 °C. | ||
The depolymerization of the poly(β-amino ester)s was examined by adding various amounts of HEMA as a functional decross-linker, as schematically depicted in Fig. 2B. HEMA was chosen because of the lower propagation rate of methacrylate compared to acrylate monomers, which helps to minimize undesirable homopolymerization at elevated temperature.63,64 The reaction kinetics, followed by SEC-analysis, indicated a temperature-dependent transesterification, in which the depolymerization of BAEC was significantly faster at 100 °C, compared to the same experiment at 80 °C (Fig. 2C). It should be noted that the reaction temperatures were limited to 100 °C in order to minimize undesired self-polymerization of the presenting (meth)acrylates. Increasing the excess of HEMA to BAE (nOH/nBAE) from 10 to 20 eq. considerably accelerated the reaction while a further increase to 30 eq. did not lead to any significant changes in the depolymerization rate. As observed in the SEC analysis, the depolymerization of BAEC at 100 °C reached a plateau after around 10 h, yielding a mixture with number average molecular weight (Mn) of 700 g mol−1. Additionally, electrospray ionization mass spectrometry (ESI-MS) confirmed that the depolymerized mixture consists of methacrylate-terminated compounds, thus providing experimental proof for the proposed approach (Fig. S3, ESI†).
To apply this concept at the material level, BAEC was cross-linked through free radical polymerization by exposure to UV light in the presence of 2,2-dimethoxy-2-phenylacetophenon as photoinitiator (DMPA, 1 wt%), yielding a BAE-based network (BEN). Chemical recycling of BEN was attempted by immersing the material in 10 eq. of HEMA (with respect to BAEs in the material) at 100 °C for 1 h, resulting in incomplete dissolution of BEN, which is ascribed to insufficient amount of the decross-linker. On the other hand, clear solutions were obtained when increasing the amount of HEMA to 20 or 30 eq., indicating successful degradation (see Fig. S4, ESI†). These visual observations were further confirmed by a rheological time sweep experiment. In fact, while SEC analysis indicated that the depolymerization of the linear BAEC reached equilibrium after approximately 10 h, the time sweep experiment performed on the cross-linked material showed a much faster response. A gradual decrease in complex viscosity, associated with decross-linking, was observed within the first hour (Fig. 2D). This rapid decross-linking accounts for the visual network dissolution as seen in Fig. 2E and the time-lapse ESI-movie,† even though full depolymerization proceeds over a longer time frame (as shown by SEC analysis). In addition, a reference polyacrylate network prepared from the photocuring of 1,6-hexanediol diacrylate, maintained its structure in comparable conditions, hence demonstrating the contribution of BAE moieties to the dynamic nature of BEN (Fig. S5, ESI†).
In order to reduce the viscosity of the photocurable resin (PCR) to a range that is typically applied for DLP 3D printing (≤4 Pa s−1), mixtures of BAEC and HEMA were prepared in varying amounts. With increasing HEMA concentration, a significant decrease in viscosity (from 7 to less than 1 Pa s−1) was observed, while gel times remained below 10 s (up to 50 wt% of HEMA) (Fig. S6, ESI†). This characteristic supports the potential use of the resin in DLP 3D printing, which typically requires a low-viscous PCR and a quick sol-to-gel transition. Besides, the addition of HEMA improved the hydrolytic resistance of the networks since those containing HEMA all showed slower hydrolysis rates compared to BEN without additional monomer (Fig. S7, ESI†). However, when more than 20 wt% of HEMA was added (BEN-30 to BEN-80), the gel fraction significantly decreased during the hydrolysis test, indicating that exceeding this limit could compromise the network's stability and should therefore be avoided. Based on the viscosity and hydrolytic stability studies, BEN-20 was thus selected for further investigations.
Besides the primary objective of establishing a chemical recycling methodology, the dynamic nature of BAEs provides the photocured materials also with the potential for a thermo-mechanical (re)processability. Therefore, we investigated the feasibility of (re)processing the photocured BEN-20 network by compression molding. The results showed that the dynamic network BEN-20 could indeed undergo compression molding (at 150 °C under 2 tons of pressure for 30 min) yielding a homogeneous sample (Fig. 3A). In contrast, the reference network without BAE bonds did not exhibit any (re)processability (Fig. S8, ESI†). Further rheological analysis through frequency sweep measurements shows a drop in storage modulus with increased temperatures, which corresponds to a loss of cross-link density and reflects the contribution of dissociative exchange via (retro) aza-Michael addition (Fig. S9, ESI†). Finally, stress–relaxation measurements demonstrated the ability to relax the applied stress in the BEN-20 network, resulting in an activation energy of approximately 105 kJ mol−1 (Fig. 3B and C). Although the shear modulus and activation energy of the three-times remolded sample (BEN-20R3) slightly increased (Fig. S10, ESI†), no notable change in thermal properties and chemical character was observed via DSC and FT-IR, which proves the effective (re)processability of this photocured material (Fig. S11 and S12, ESI†).
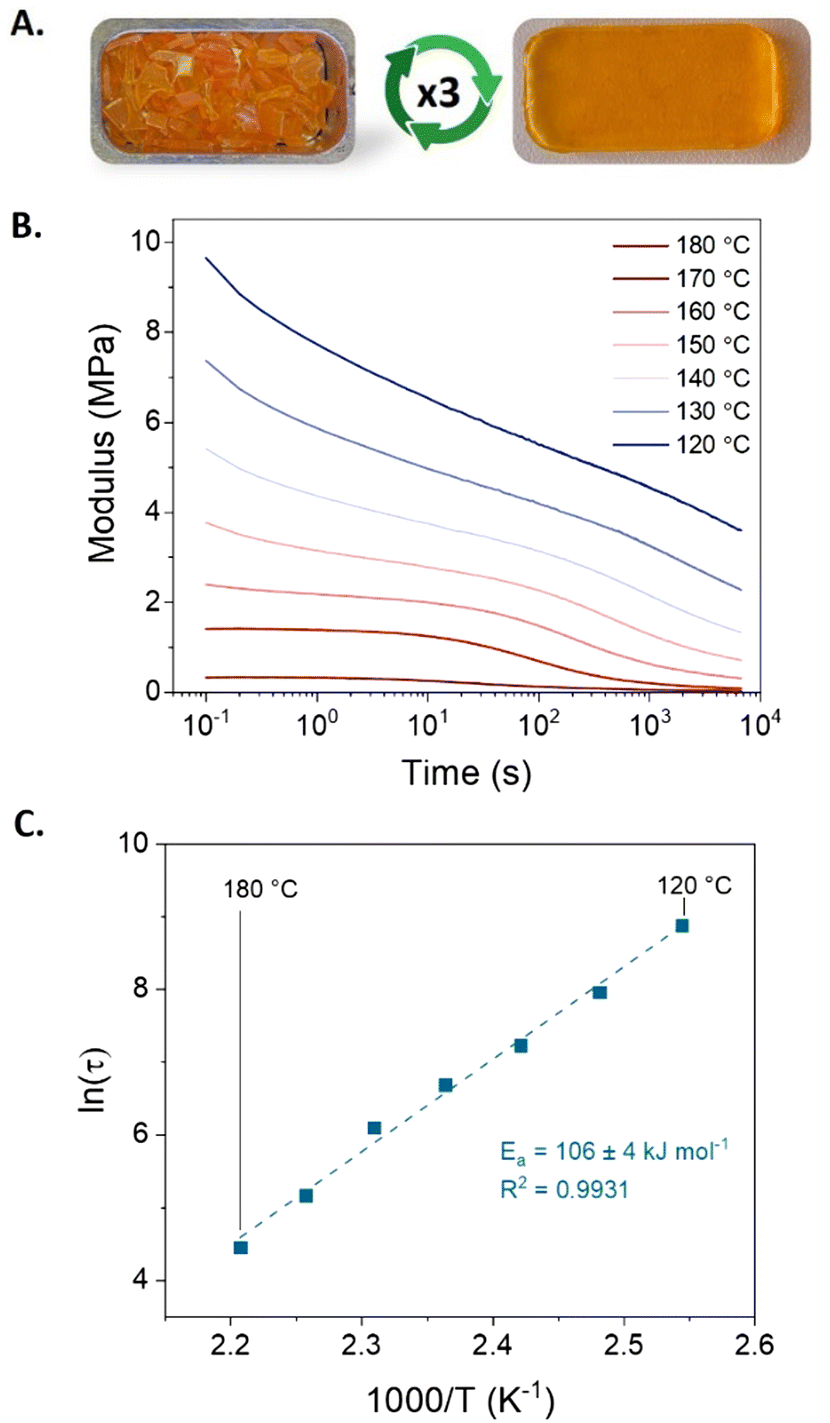 | ||
| Fig. 3 (A) Thermo-mechanical recycling of photocured BEN-20 through compression molding at 150 °C under 2 tons of pressure for 30 min. (B) Stress–relaxation curves and (C) Arrhenius plots of BEN-20 after the first recycling via compression molding. | ||
Returning to the primary focus of this study, the conceptual process for chemical recycling is displayed in Fig. 4. After freshly preparing BEN-20 (Fig. 4A), the network was immersed in HEMA (BEN-20/HEMA = 1/9 wt/wt, vide supra) to perform the depolymerization at 100 °C for 16 h (Fig. 4B). The obtained depolymerized mixture was directly reused as a photocurable resin, with an estimated concentration of BAEC of 10 wt% in HEMA. Technically, the excess of HEMA could be extracted by distillation to recover the reagent, or to modify the photocurable resin composition. However, from a proof-of-concept perspective, fresh BAEC was introduced to adjust the composition to 20 wt% of HEMA. The reformulated PCR was then cured under UV light, resulting in BEN-20U1 (name refers to a chemically recycled network).
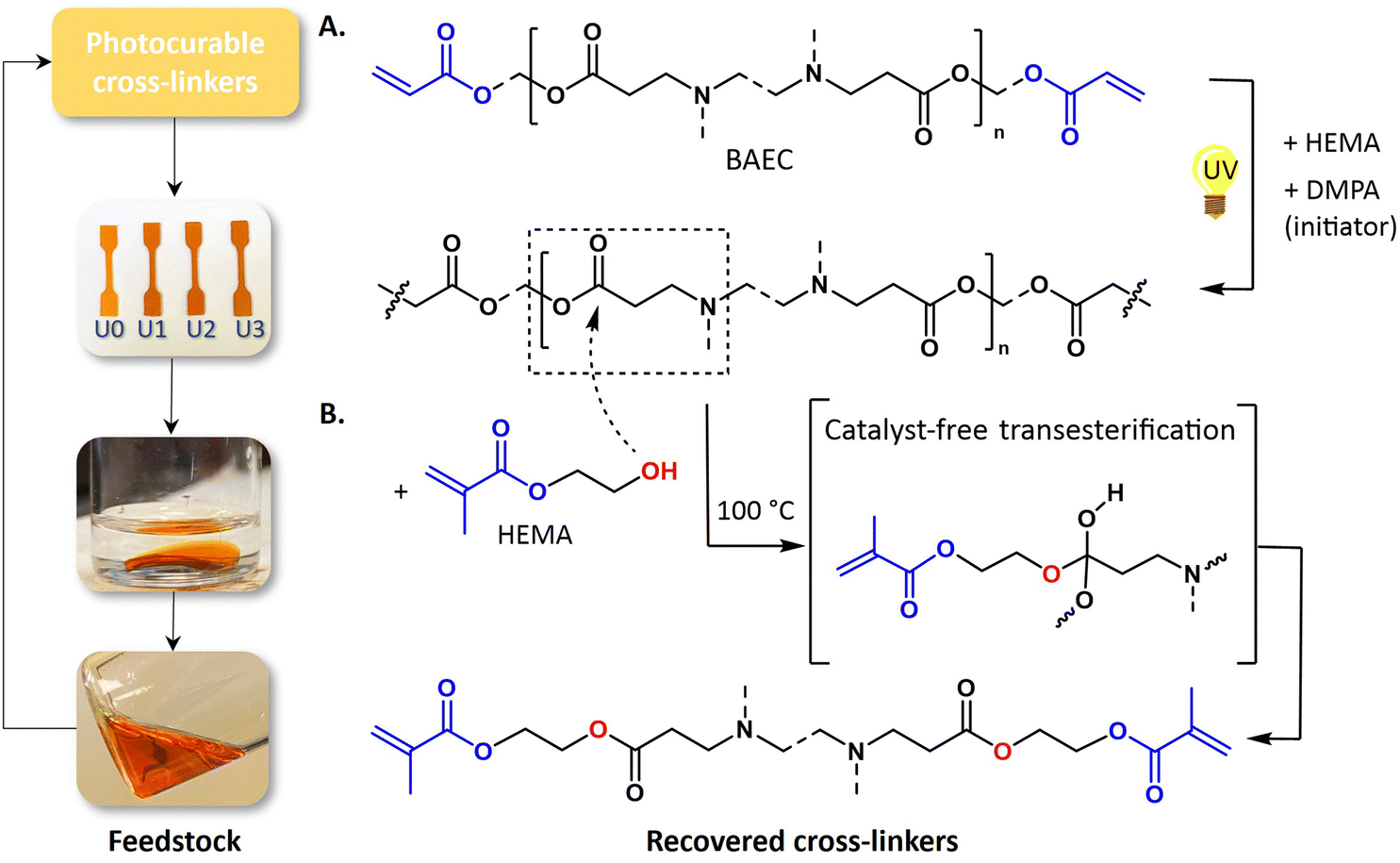 | ||
| Fig. 4 Overview of the recycling process. (A) β-Amino ester-based networks were prepared by exposing a photocurable resin comprising BAEC, HEMA and photoinitiator to UV light (λ-range: 320–400 nm). (B) HEMA was added as a functional decross-linker, wherein hydroxyl groups de-cross-linked the network via transesterification and simultaneously introduced methacrylate groups, resulting in the recovery of dynamic crosslinkers. | ||
The recycling efficiency of the resins was assessed through mechanical characterization. In the tensile test results, a significant loss in mechanical strength was observed with a drop in ultimate stress from 14 MPa of the original BEN-20 to 7 MPa of the recycled BEN-20U1 (neat) network (Fig. 5A).
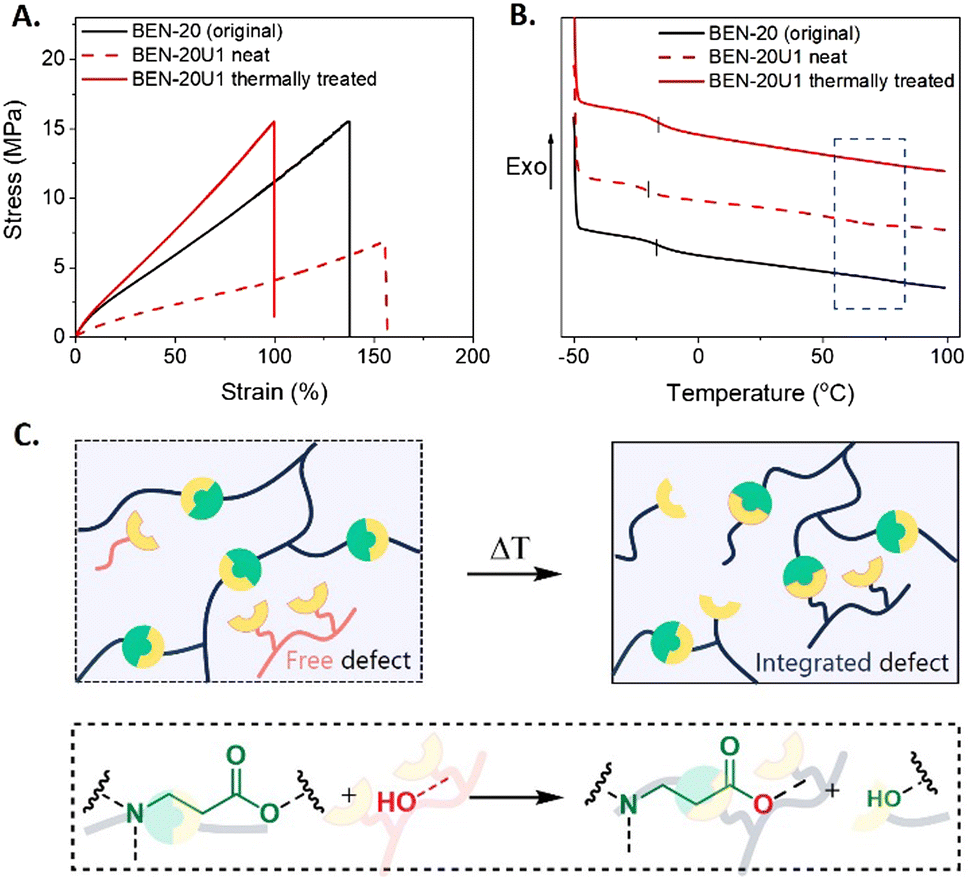 | ||
| Fig. 5 (A) Stress–strain curves of the initial BEN-20 network (black line) and the recycled network before and after thermal treatment (red dashed and solid lines, respectively). (B) DSC thermogram of the recycled network BEN-20U1 with and without thermal treatment (red solid and dashed lines, respectively) compared to the original network BEN-20 (black line). (C) Schematic representation of the integration of hydroxyl-containing uncross-linked molecules into the network (top), promoted by transesterification (bottom). | ||
A potential reason for that behavior is the fact that not only photocurable methacrylate compounds were obtained in the depolymerized solution, but also a fraction of unfunctionalized, hydroxyl containing molecules that act as defects, thus impacting the recycled material properties (Fig. S13, ESI†). The presence of small compounds caused a slight decrease in Tg (from −15 to −20 °C) of BEN-20U1 (neat) as a result of the plasticizing effect. On the other hand, larger uncross-linked compounds seem to introduce another small Tg transition at around 65 °C ascribed to phase separation (Fig. 5B). At that point, we hypothesized that these defects could be integrated into the networks as their hydroxyl groups might undergo transesterification with the BAE moieties, potentially restoring the mechanical strength (Fig. 5C). Indeed, a time sweep experiment conducted at 120 °C showed an increase in storage modulus, suggesting the recovery of mechanical properties based on the covalent integration of the compounds into the network (Fig. S14, ESI†). For this reason, an additional thermal treatment of the recycled network was performed. As anticipated, BEN-20U1 could restore its ultimate stress and unique Tg after 2 h of thermal treatment at 120 °C, revealing the effective recovery of its properties (Fig. 5A and B).
The material properties of the original material and the recycled ones (after thermal treatment) in Table 1 indicate that the recycled network BEN-20U1 could indeed recover its thermal properties (Tg and Td), compared to the original BEN-20 with a low soluble fraction (<5%).
| Network | T g (°C) | T d-5% (°C) | Swelling ratioc (%) | Soluble fractionc (%) | Ultimate stressd (Pa) | Total elongationd (%) | Modulusd (MPa) |
|---|---|---|---|---|---|---|---|
| a Determined from DSC analysis (10 °C min−1). b TGA onset temperatures after 5% weight loss. c Obtained after swelling in chloroform at room temperature for 24 h. d Determined from tensile testing (0.02 N preload and 4 mm min−1). | |||||||
| BEN-20U0 | −14 | 251 | 649 ± 46 | 4.7 ± 0.3 | 16.3 ± 1.1 | 136 ± 9 | 29.7 ± 9.3 |
| BEN-20U1 | −17 | 253 | 465 ± 85 | 4.4 ± 0.3 | 15.8 ± 1.9 | 122 ± 1 | 29.6 ± 4.0 |
| BEN-20U2 | −21 | 248 | 483 ± 19 | 4.0 ± 0.9 | 12.0 ± 0.8 | 95 ± 5 | 21.8 ± 3.3 |
| BEN-20U3 | −16 | 253 | 438 ± 15 | 3.3 ± 0.5 | 12.6 ± 2.0 | 80 ± 12 | 27.1 ± 5.1 |
Besides the lower swelling degree and elongation at break, attributed to the change in cross-link density of BAE-20U1, comparable ultimate tensile strength and Young's modulus were obtained, indicating an almost full recovery of properties after recycling. The chemical recycling protocol has been repeated 3 times, showcasing fully restored thermal properties and retaining up to 75% of the tensile strength, while preserving photocurable capability as well as chemical characteristics for at least three cycles (Table 1 and Fig. S15 and S16, ESI†).
To take this protocol a step further into a 3D printing application, we conducted DLP 3D printing on the dynamic photocurable resin BEN-20 (BAEC solution with 20 wt% of HEMA). This technique allows to project digitalized UV patterns onto photocurable resins, locally curing them to build up 3D structures layer-by-layer. The successful fabrication of simple cube and heart shape structures in this proof-of-concept experiment is shown in Fig. 6, highlighting the suitability of this particular resin for 3D printing.
 | ||
| Fig. 6 Illustration of repeatable DLP 3D printing using a dynamic photocurable resin. | ||
Additionally, multiple printing steps were performed with recycled PCR to assess the repeatability of this procedure. The resulting samples exhibited consistent shapes with no significant change in appearance for at least three cycles, thus demonstrating the repeatable printability after chemical recycling (Fig. S17, ESI†).
Conclusions
In this study, a photocurable dynamic cross-linker, based on poly(β-amino ester)s, has been successfully synthesized from readily available bulk chemicals. By using HEMA as a functional decross-linker, the photo-cured networks can be degraded via transesterification within 1 h at 100 °C. Kinetic analysis using SEC indicated that the catalyst-free depolymerization of BAEC yields methacrylate-comprising oligomers (characterized by ESI-MS analysis), which can be reused as cross-linkers, thus establishing a closed-loop chemical recycling protocol. By combining BAEC with the bulk monomer HEMA, photocurable resins with low viscosity (below 4 Pa s−1) and short gel time (<10 s), applicable for DLP 3D printing, have been obtained. The implementation of such protocol has allowed full recovery of the thermal properties and Young's modulus of the recycled materials, while maintaining 75% of the tensile strength and the photocurable function for at least three cycles. Subsequently, repeated DLP 3D printing has been demonstrated to fabricate 3D structures. In addition to the primary focus on chemical recycling, the obtained dynamic network also exhibits a potential for efficient thermo-mechanical recycling. (Re)processability was demonstrated by compression molding, showing an activation energy of approximately 110 kJ mol−1 and no significant change in properties of the (re)molded materials for at least three times.Author contributions
Loc Tan Nguyen: conceptualization, data curation, formal analysis, investigation, validation, writing original draft – review & editing. Filip E. Du Prez: conceptualization, supervision, resources, review & editing.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
The authors declare that they have no conflict of interest.Acknowledgements
This project received funding from the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation programme 101021081 (ERC-AdG-2020, CiMaC-project). Loc Tan Nguyen would like to thank Bernhard De Meyer for technical support and Dr Nezha Badi, Dr Tapas Debsharma, Dr Susanne Fischer, Dr Filip Van Lijsebetten, Dr Matthieu Soete, and Stephan Maes for fruitful discussion. The authors would like to thank the Polymer Chemistry & Biomaterials Group (PBM research group, Ghent University), especially Anna Szabó and Astrid Quaak for their assistance with DLP 3D printing.Notes and references
- A. R. Rahimi and J. M. Garciá, Nat. Rev. Chem., 2017, 1, 0046 CrossRef.
- R. Geyer, J. R. Jambeck and K. L. Law, Sci. Adv., 2017, 3, e1700782 CrossRef PubMed.
- X. Wu, P. Hartmann, D. Berne, M. De Bruyn, F. Cuminet, Z. Wang, J. M. Zechner, A. D. Boese, V. Placet, S. Caillol and K. Barta, Science, 2024, 384, eadj9989 CrossRef PubMed.
- N. Zheng, Y. Xu, Q. Zhao and T. Xie, Chem. Rev., 2021, 121, 1716–1745 CrossRef CAS PubMed.
- W. Denissen, J. M. Winne and F. E. Du Prez, Chem. Sci., 2016, 7, 30–38 RSC.
- N. J. Van Zee and R. Nicolaÿ, Prog. Polym. Sci., 2020, 104, 101233 CrossRef CAS.
- L. Matt, R. Sedrik, O. Bonjour, M. Vasiliauskaité, P. Jannasch and L. Vares, ACS Sustainable Chem. Eng., 2023, 11, 8294–8307 CrossRef CAS PubMed.
- D. Montarnal, M. Capelot, F. Tournilhac and L. Leibler, Science, 2011, 334, 965–968 CrossRef CAS PubMed.
- M. A. Bin Rusayyis and J. M. Torkelson, ACS Macro. Lett., 2022, 11, 568–574 CrossRef CAS PubMed.
- J. Zheng, Z. M. Png, S. H. Ng, G. X. Tham, E. Ye, S. S. Goh, X. J. Loh and Z. Li, Mater. Today, 2021, 51, 586–625 CrossRef CAS.
- C. J. Kloxin, T. F. Scott, B. J. Adzima and C. N. Bowman, Macromolecules, 2010, 43, 2643–2653 CrossRef CAS PubMed.
- Mallinda, VITRIMAX, https://mallinda.com/, (accessed 10 June 2024).
- ASTP Innovations, Aromatic Thermosetting coPolyesters (ATSP) EsthermTM resins, https://www.atspinnovations.com/, (accessed 10 June 2024) Search PubMed.
- V. Schenk, K. Labastie, M. Destarac, P. Olivier and M. Guerre, Mater. Adv., 2022, 3, 8012–8029 RSC.
- Z. Lei, H. Chen, S. Huang, L. J. Wayment, Q. Xu and W. Zhang, Chem. Rev., 2024, 124, 7829–7906 CrossRef CAS PubMed.
- J. M. Winne, L. Leibler and F. E. Du Prez, Polym. Chem., 2019, 10, 6091–6108 RSC.
- C. Bowman, F. Du Prez and J. Kalow, Polym. Chem., 2020, 11, 5295–5296 RSC.
- L. T. Nguyen, H. Q. Pham, D. T. Thi Phung, T. T. Truong, H. T. Nguyen, T. Chanh Duc Doan, C. M. Dang, H. Le Tran, P. T. Mai, D. T. Tran, T. Q. Nguyen, N. Q. Ho and L. T. T. Nguyen, Polymer, 2020, 188, 122144 CrossRef CAS.
- D. Reisinger, S. Kaiser, E. Rossegger, W. Alabiso, B. Rieger and S. Schlögl, Angew. Chem., Int. Ed., 2021, 60, 14302–14306 CrossRef CAS PubMed.
- L. T. Nguyen, F. Portone and F. E. Du Prez, Polym. Chem., 2024, 15, 11–16 RSC.
- C. Cui, F. Wang, X. Chen, T. Xu, Z. Li, K. Chen, Y. Guo, Y. Cheng, Z. Ge and Y. Zhang, Adv. Funct. Mater., 2024, 34, 2315469 CrossRef CAS.
- X. Li, T. Zhang, B. Song, K. Yang, X. Hao and J. Ma, J. Appl. Polym. Sci., 2024, 141, e54762 CrossRef CAS.
- C. Pronoitis, M. Hakkarainen and K. Odelius, Polym. Chem., 2021, 12, 5668–5678 RSC.
- C. Taplan, M. Guerre, J. M. Winne and F. E. Du Prez, Mater. Horiz., 2020, 7, 104–110 RSC.
- I. Vollmer, M. J. F. Jenks, M. C. P. Roelands, R. J. White, T. van Harmelen, P. de Wild, G. P. van der Laan, F. Meirer, J. T. F. Keurentjes and B. M. Weckhuysen, Angew. Chem., Int. Ed., 2020, 59, 15402–15423 CrossRef CAS PubMed.
- Z. O. G. Schyns and M. P. Shaver, Macromol. Rapid Commun., 2021, 42, 2000415 CrossRef CAS PubMed.
- G. Zhu, H. A. Houck, C. A. Spiegel, C. Selhuber-Unkel, Y. Hou and E. Blasco, Adv. Funct. Mater., 2024, 34, 2300456 CrossRef CAS.
- Y. Jia, H. Xie, J. Qian, Y. Zhang, H. Zheng, F. Wei, Y. Li and Z. Zhao, Adv. Funct. Mater., 2024, 34, 2307279 CrossRef CAS.
- E. Rossegger, R. Höller, K. Hrbinič, M. Sangermano, T. Griesser and S. Schlögl, Adv. Eng. Mater., 2023, 25, 2200749 CrossRef CAS.
- G. Zhu, N. von Coelln, Y. Hou, C. Vazquez-Martel, C. A. Spiegel, P. Tegeder and E. Blasco, Adv. Mater., 2024, 36, 2401561 CrossRef CAS PubMed.
- H. Cui, D. Yao, R. Hensleigh, H. Lu, A. Calderon, Z. Xu, S. Davaria, Z. Wang, P. Mercier, P. Tarazaga and X. (Rayne) Zheng, Science, 2022, 376, 1287–1293 CrossRef CAS PubMed.
- J. G. Jardine, T. Ota, D. Sok, M. Pauthner, D. W. Kulp, O. Kalyuzhniy, P. D. Skog, T. C. Thinnes, D. Bhullar, B. Briney, S. Menis, M. Jones, M. Kubitz, S. Spencer, Y. Adachi, D. R. Burton, W. R. Schief and D. Nemazee, Science, 2015, 349, 156–161 CrossRef CAS PubMed.
- S. Gantenbein, K. Masania, W. Woigk, J. P. W. Sesseg, T. A. Tervoort and A. R. Studart, Nature, 2018, 561, 226–230 CrossRef CAS PubMed.
- A. Vedrtnam, P. Ghabezi, D. Gunwant, Y. Jiang, O. Sam-Daliri, N. Harrison, J. Goggins and W. Finnegan, Compos. C: Open Access, 2023, 12, 100418 CAS.
- L. Strohmeier, H. Frommwald and S. Schlögl, RSC Adv., 2020, 10, 23607–23614 RSC.
- V. S. D. Voet, J. Guit and K. Loos, Macromol. Rapid Commun., 2021, 42, 2000475 CrossRef CAS PubMed.
- Q. Shi, K. Yu, X. Kuang, X. Mu, C. K. Dunn, M. L. Dunn, T. Wang and H. Jerry Qi, Mater. Horiz., 2017, 4, 598–607 RSC.
- X. Lopez de Pariza, O. Varela, S. O. Catt, T. E. Long, E. Blasco and H. Sardon, Nat. Commun., 2023, 14, 5504 CrossRef PubMed.
- C. Cui, L. An, Z. Zhang, M. Ji, K. Chen, Y. Yang, Q. Su, F. Wang, Y. Cheng and Y. Zhang, Adv. Funct. Mater., 2022, 32, 2203720 CrossRef.
- L. S. Hamachi, D. A. Rau, C. B. Arrington, D. T. Sheppard, D. J. Fortman, T. E. Long, C. B. Williams and W. R. Dichtel, ACS Appl. Mater. Interfaces, 2021, 13, 38680–38687 CrossRef PubMed.
- S. Maes, V. Scholiers and F. E. Du Prez, Macromol. Chem. Phys., 2023, 224, 2100445 CrossRef.
- X. Li, R. Yu, Y. He, Y. Zhang, X. Yang, X. Zhao and W. Huang, ACS Macro Lett., 2019, 8, 1511–1516 CrossRef PubMed.
- Z. Chen, M. Yang, M. Ji, X. Kuang, H. J. Qi and T. Wang, Mater. Des., 2021, 197, 109189 CrossRef CAS.
- X. Li, R. Yu, Y. He, Y. Zhang, X. Yang, X. Zhao and W. Huang, Polymer, 2020, 200, 122532 CrossRef CAS.
- L. L. Robinson, J. L. Self, A. D. Fusi, M. W. Bates, J. Read De Alaniz, C. J. Hawker, C. M. Bates and C. S. Sample, ACS Macro Lett., 2021, 10, 857–863 CrossRef CAS PubMed.
- D. Kojic, K. Ehrmann, R. Wolff, Y. Mete, T. Koch, J. Stampfl, S. Baudis and R. Liska, Polym. Chem., 2023, 14, 4809–4818 RSC.
- K. P. Cortés-Guzmán, A. R. Parikh, M. L. Sparacin, A. K. Remy, L. Adegoke, C. Chitrakar, M. Ecker, W. E. Voit and R. A. Smaldone, ACS Sustainable Chem. Eng., 2022, 10, 13091–13099 CrossRef.
- E. Rossegger, R. Höller, D. Reisinger, J. Strasser, M. Fleisch, T. Griesser and S. Schlögl, Polym. Chem., 2021, 12, 638–644 Search PubMed.
- B. Zhang, K. Kowsari, A. Serjouei, M. L. Dunn and Q. Ge, Nat. Commun., 2018, 9, 1831 CrossRef PubMed.
- H. Li, B. Zhang, R. Wang, X. Yang, X. He, H. Ye, J. Cheng, C. Yuan, Y. F. Zhang and Q. Ge, Adv. Funct. Mater., 2022, 32, 2111030 CrossRef CAS.
- S. Huang, M. Podgórski, X. Han and C. N. Bowman, Polym. Chem., 2020, 11, 6879–6883 RSC.
- Y. Liu, Z. Yu, B. Wang, P. Li, J. Zhu and S. Ma, Green Chem., 2022, 24, 5691–5708 RSC.
- A. S. Kuenstler, J. J. Hernandez, M. Trujillo-Lemon, A. Osterbaan and C. N. Bowman, ACS Appl. Mater. Interfaces, 2023, 15, 11111–11121 CrossRef CAS PubMed.
- J. Cui, F. Liu, Z. Lu, S. Feng, C. Liang, Y. Sun, J. Cui and B. Zhang, Adv. Mater., 2023, 35, 2211417 CrossRef CAS PubMed.
- T. O. Machado, C. J. Stubbs, V. Chiaradia, M. A. Alraddadi, A. Brandolese, J. C. Worch and A. P. Dove, Nature, 2024, 629, 1069–1074 CrossRef CAS PubMed.
- P. S. Klee, C. Vazquez-Martel, L. Florido Martins and E. Blasco, ACS Appl. Polym. Mater., 2024, 6, 935–942 CrossRef CAS.
- C. Taplan, M. Guerre and F. E. Du Prez, J. Am. Chem. Soc., 2021, 143, 9140–9150 CrossRef CAS PubMed.
- J. O. Holloway, C. Taplan and F. E. Du Prez, Polym. Chem., 2022, 13, 2008–2018 RSC.
- D. Berne, G. Coste, R. Morales-Cerrada, M. Boursier, J. Pinaud, V. Ladmiral and S. Caillol, Polym. Chem., 2022, 13, 3806–3814 RSC.
- L. T. Nguyen, C. Mertens and F. E. Du Prez, Macromolecules, 2024, 57, 4817–4825 CrossRef CAS.
- W. H. Lee, Z. Wan, A. Shegiwal and D. Haddleton, Polym. Chem., 2024, 15, 2959–2969 RSC.
- V. Saygin, K. Snapp, A. E. Gongora, R. Kolaghassi and K. A. Brown, Adv. Mater. Technol., 2023, 8, 2202022 CrossRef CAS.
- M. Buback, A. Feldermann, C. Barner-Kowollik and I. Lacík, Macromolecules, 2001, 34, 5439–5448 CrossRef CAS.
- R. A. Hutchinson, S. Beuermann, D. A. Paquet and J. H. McMinn, Macromolecules, 1997, 30, 3490–3493 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4mh00823e |
| This journal is © The Royal Society of Chemistry 2024 |