
Controlling nano- and microfilament morphology by strategically placing chiral centers in the side chains of bent-core molecules†
Ashwathanarayana
Gowda
 a,
Gourab
Acharjee
b,
Suraj Kumar
Pathak
a,
Grace A. R.
Rohaley
ac,
Asmita
Shah
a,
Robert P.
Lemieux
d,
Marianne E.
Prévôt
*abc and
Torsten
Hegmann
*abce
a,
Gourab
Acharjee
b,
Suraj Kumar
Pathak
a,
Grace A. R.
Rohaley
ac,
Asmita
Shah
a,
Robert P.
Lemieux
d,
Marianne E.
Prévôt
*abc and
Torsten
Hegmann
*abce
aAdvanced Materials and Liquid Crystal Institute, Kent State University, Kent, OH 44242, USA. E-mail: thegmann@kent.edu; mprevot1@kent.edu
bDepartment of Chemistry and Biochemistry, Kent State University, Kent, OH 44242, USA
cMaterials Science Graduate Program, Kent State University, Kent, OH 44242, USA
dDepartment of Chemistry, University of Waterloo, Waterloo, Ontario N2L 3G1, Canada
eBrain Health Research Institute, Kent State University, Kent, OH 44242, USA
First published on 8th October 2024
Abstract
Self-assembled lamellar nano- and microfilaments formed by select types of bent-core molecules are prime examples of the interplay between molecular conformation and morphological chirality. Here, we demonstrate how the strategic placement of chiral centers at C-1 and/or C-3 in the terminal alkyloxy side chains, largely based on a priori calculations of molecular conformation, leads to the predictable formation of increasingly complex nano- and microfilament morphologies. Adding to the previously described diversity of twisted and writhed filament types, we here demonstrate and explain the formation and coexistence of flat nanoribbons, nanocylinders, or nano- as well as microfilaments where the morphology spontaneously changes along the filament long axis. For some these more exotic types of filament morphology, helical multilayer filaments suddenly unwind to form flat nanoribbons that also twist again under preservation (not perversion) of the helical twist sense. Moreover, the morphologies formed by this series of molecules now allows us to demonstrate the complete transformation from flat multilayer ribbons over microfilaments and helical-wrapped nanocylinders to helical nanofilaments depending on the number and position of chiral centers in the aliphatic side chains.
New conceptsWe demonstrate that the shape or morphology of self-assembled organic nano- and microfilaments can be preconceived by logical molecular design. For a series of molecules with a pronounced bent molecular shape, these design concepts rely on a priori molecular conformational analysis coupled with the rational placement of chiral centers at the junctions between the rigid aromatic cores and the flexible aliphatic side chains. The complete series now illuminates the full morphological transition from flat microribbons to strongly twisted helical nanofilaments. In due course, we discovered complex new morphologies including, for example, flat multilayer microribbons that gradually decrease in width—in analogy to down feathers or split hair ends—to ultimately twist at the ends as well as nanofilaments that untwist at their ends or mid filament, notably without any perversions. Altogether, combined with existing morphologies formed by tris-biphenyl bent-core molecules, these new morphologies serve as snapshots to illuminate the entire possible transformation, from flat multilayer ribbons over writhed nanocylinders and microfilaments mimicking bladed worm corkscrews to helical nanofilaments with negative Gaussian curvature. Relevant for numerous applications, these nano- and microfilaments can serve as templates for nanomaterials or emitters, among other materials, and display structural coloration that varies with the dimensions of the ultimate filament. |
1. Introduction
Chiral filament morphologies are omnipresent building blocks in nature, found equally in flora and fauna at length scales ranging in length or width from a few nanometers to multi-meter-long macroscopic objects.1–9 In materials science, chiral filaments with nano- or microscale dimensions are increasingly generated using a wide array of materials classes and studied for promising uses in optics, photonics, sensing, and many other purposes.10The most common filament morphologies comprise single rod- or flat ribbon subfilaments or several individual subfilaments stacked on top of or twisting around one another with the cross-sections of the final multi-layer filaments being flat, elliptical, or round.10
The flat (lamellar) ribbons, as basic building blocks for the family of B4 filaments formed by molecules with a pronounced bent molecular shape, have been shown to assemble into multilayer helicoids, helical ribbons (cylindrical helices), or flat as well as occasionally twisted nanoribbons depending on the number and position of chiral centers in either one or both aliphatic side chains.11 Here, continuous twisting along a straight centerline leads to twisted multilayer filaments with Gaussian saddle-like curvature, continuous bending around the width of the ribbons to cylindrical helices with a helical centerline.10
The original helical nanofilament or HNF phase, appropriately named after the discovery of its internal structure and overall morphology by Clark et al., is a perfect example of self-assembly directed by molecular conformation.12 Multilayer helicoids form due to an intralayer mismatch between the two molecular halves that can only be relieved by local saddle splay. This, in turn, leads to twisted filaments composed of a limited number of layer stacks (about 5–7 layers). An intralayer mismatch is here the inherent misfit (in an anisotropic multi-layer configuration) that leads to twist.13 The two molecular halves tilt in orthogonal directions from the layer mid-plane (i.e., each at a subfilament angle of θ ∼ 45°) that generates a continuous twist of the flat ribbon along a straight center-line of about 2° nm−1 forming a helicoid. When formed by achiral molecules, HNFs exist as a chiral conglomerate composed of macroscopic chiral domains with opposite handedness.12 Within each domain, HNFs grow, chirality preserving (left-handed in one domain and right-handed in an adjacent domain),14 with a preset bulk secondary twist among HNFs governed by excluded volume interactions of strongly twisted filaments (i.e., right-handed HNFs form a right-handed secondary twist and vice versa).15–17 Early portrayal of this phase, historically named the B4 phase, was that of a crystalline solid phase.18 However, solid-state magic-angle spinning (MAS)-NMR experiments clarified that the HNF phase is neither a traditional crystalline solid nor a conventional liquid crystal19 that is also characterized by crystal or hexatic in-layer ordering.12
Morphological variants of the classical HNFs now include helical nano- and microfilaments with varying dimensions20–24 (width, height, and helical pitch) and intra- or interlayer modulation (A–C, Fig. 1a), heliconical nanocrystallites,25 heliconical-layered nanocylinders26,27 (D, Fig. 1a), and flat or occasionally twisted flat nanoribbons28 (E and F, Fig. 1a). The majority of these flat, twisted, or cylindrical nano- or microfilament morphologies were realized by strategically incorporating chiral centers into the aliphatic side chains of tris-biphenyl aromatic core bent-core molecules.10,11 Noteworthy, all these morphologies display characteristic blue structural color in reflection mode, whose origin is not fully understood but surely linked to diffraction, scattering, or interference via the periodic nano- to microscale layering and packing of these filaments in thin films.29–31
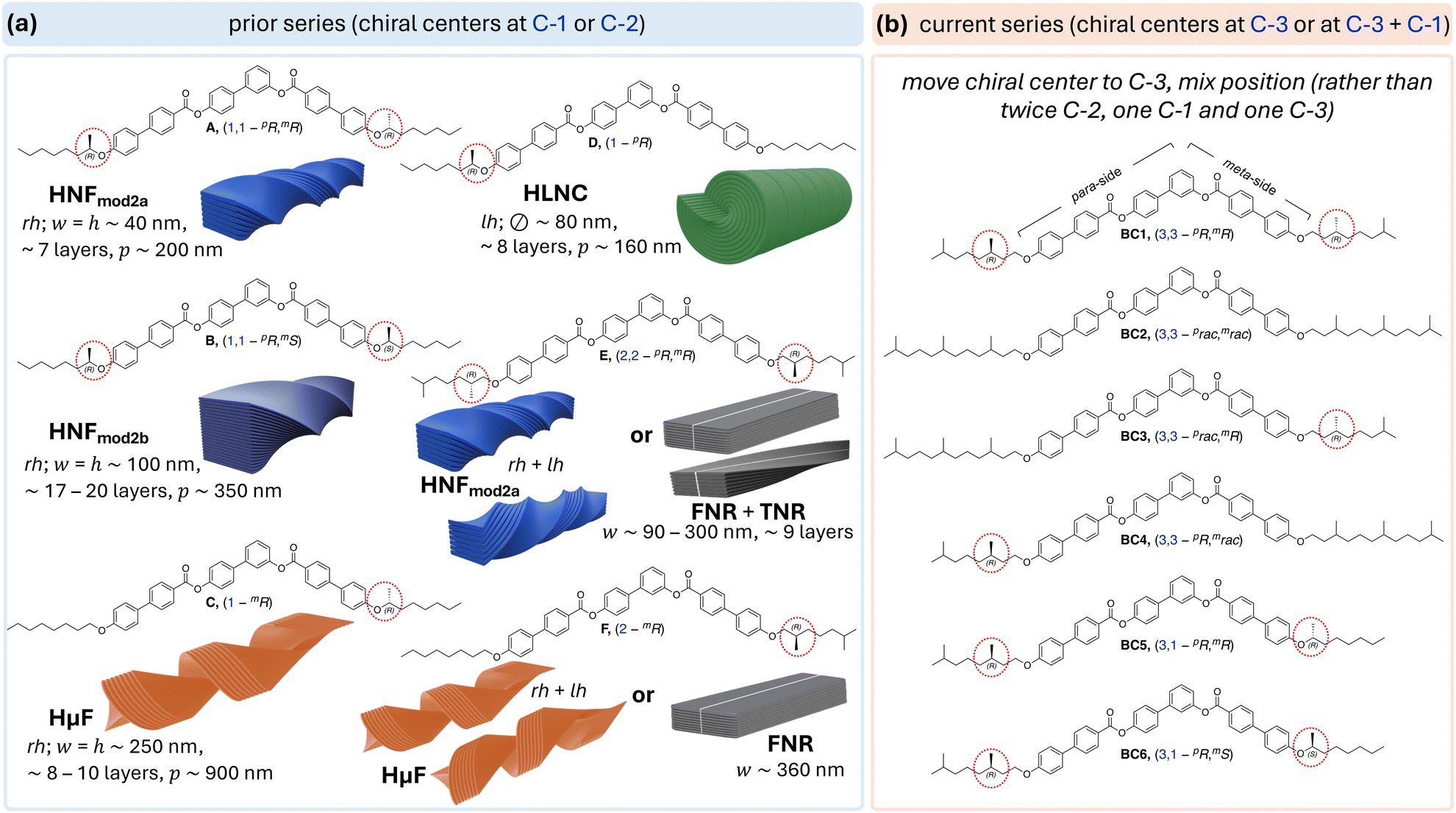 | ||
| Fig. 1 (a) Structures of tris-biphenyl bent-core molecules A–F20–22,28 with chiral centers at C-1 or C-2 in the aliphatic side chains and type (incl. handedness, dimensions, and number of layers) of nano- or microfilament formed depending on the rate of cooling from the isotropic liquid (rh = right-handed, lh = left-handed, w = width, h = height, p = pitch, and = diameter). (b) Chemical structures of the new series with chiral centers at C-3 (BC1–BC4) or at C-1 in the shorter meta-side and C-3 in the longer para-side (BC5 and BC6). | ||
Single-molecule stochastic dynamics (SD) atomistic simulations as well as density functional theory (DFT) calculations (B3LYP/6-31G*) revealed that the position of the chiral center at or near the core-chain junctions of the bent-core molecules in either one or both the shorter meta- or the longer para-side of these asymmetric bent-shaped molecules imparts significant changes in the core-chain angle distributions and thus overall molecular conformation.26 Basically, a chiral center residing at C-1 in the longer para-side, such as in compound D, leads to a significant shift to lower bend angles at the core-chain junction (resulting in the formation of heliconical-layered nanocylinders or HLNCs) compared to the larger bend angle at the core-chain junction calculated for compound C (forming helical microfilaments or HμFs), where the chiral center at C-1 is situated in the shorter meta-side (Fig. 1a). Even then, as long as both core-chain junctions in the molecule feature a chiral center (and branching point) at C-1, dual-modulated HNFs are generated; with lower overall dimensions for the homochiral derivative A (HNFmod2a) in comparison to B (HNFmod2b) with alternating configuration of the chiral centers (Fig. 1a).20,21
Considering the established links between filament morphology and lowest-energy conformations affected by the distribution of dihedral angles in the core-chain junction,26 the formation of flat or weakly twisted flat nanoribbons (FNRs or TNRs) when the chiral center(s) were relocated to C-2 (E and F, Fig. 2a) seem a logical consequence. In line with earlier established trends for helical pitch (p) in chiral nematic (N*) phases or spontaneous polarization values in chiral smectic-C (SmC*) phases as a chiral center is moved further away from a core-chain juncture,32 the formation of non-twisted, flat filaments, at least upon slow cooling from the isotropic liquid phase (at ∼5 °C min−1), is the result of a reduction of the restraint of rotation about the chiral center that effectively reduces the effects of chirality in the side chain. This is additionally supported by the fact that upon rapid cooling (at a rate ≥50 °C min−1) conglomerates of left- and right-handed HNFmod2a and HμF morphologies were seen for E and F (Fig. 1a) despite the homochiral nature of the molecules.28
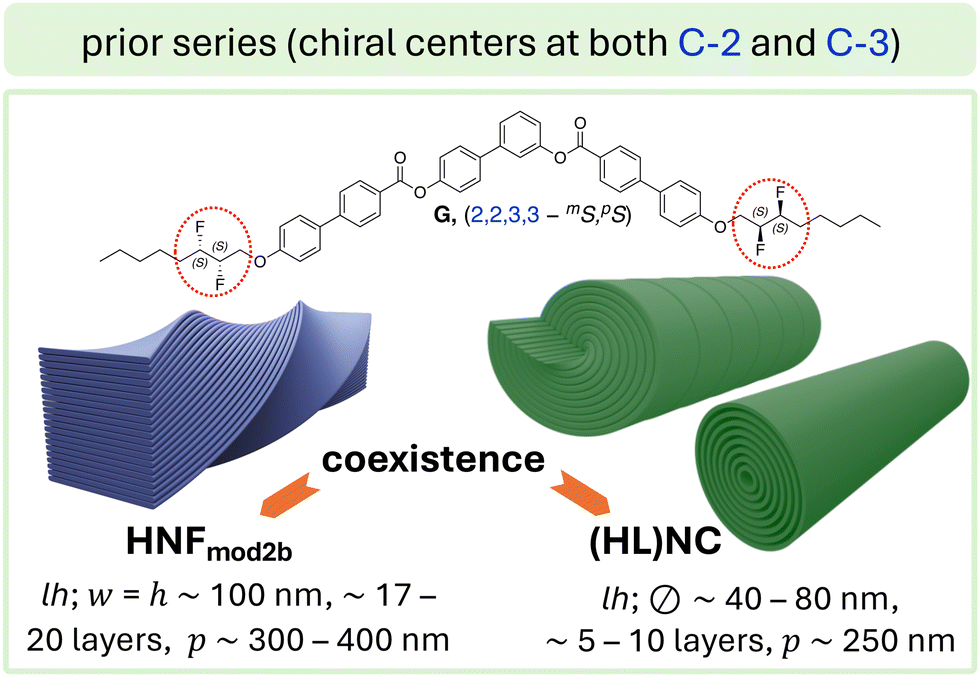 | ||
| Fig. 2 Bent-core molecule with chiral centers at C-2 and C-3 via the introduction of (S,S)-2,3-difluorooctyloxy side chains (compound G):27 upon slow as well as rapid cooling, G showed a coexistence of two morphologies (HNFmod2b) and (HL)NC; heliconical layering was here only observed for some of nanocylinders (unlike for compound D; Fig. 1, which showed heliconical layering depending on the configuration of the chiral center). | ||
To further confirm that this trend holds up, the current work focuses on a new set of bent-core tris-biphenyl materials for which the chiral centers were further removed from the core-chain juncture, now to C-3. BC1–BC4 have chiral centers at C-3 in both the meta- and para-arm. BC1 is homochiral, featuring two (R)-3,7-dimethyloctan-1-olyl chiral side chains with a further branching point at the chain terminus—in analogy to E and F. BC2–BC4 carry hexahydrofarnesolyl33 groups in the side chains (two racemic side chains for BC2 and one each in either the longer para-arm for BC3 or in the shorter meta-arm for BC4 with the other side chain analogous to BC1). Finally, to probe the effect of mixed chiral center positions, BC5 and BC6 feature chiral centers at C-3 in the para-arm and C-1 in the meta-arm; homochiral (R,R)- for BC5 and heterochiral (R,S)- for BC6 (Fig. 1b). Examining the self-assembly of these two derivatives will allow for a direct comparison with compound E featuring chiral centers at C-2 in either side chain.
Further insight into the role of low-energy conformations at the core-chain juncture will occur when we ultimately compare the final filament morphologies formed by BC1, BC5, and BC6 (each with chiral centers at C-3) with those formed by compounds E (chiral centers at C-2; Fig. 1a) and G (chiral centers at C-2 and C-3; Fig. 2). In the case of G,27 calculated conformational energy profiles revealed a greater conformational rigidity about the C-2–C-3 bond axis due to repulsive interactions of the two fluoro-substituents and the “gauche effect”34 associated with this structural unit (i.e., hyperconjugative interactions between vicinal C–H and C–F bonds in the lowest energy conformation). As a result, no flat or weakly twisted nanoribbons were seen in this case, rather, a coexistence of HNFmod2b and HLNC (Fig. 2).
To clarify our expectations upfront for filament morphology due to the relocation of chiral centers in the core-chain junctures to C-3, we first proceeded to calculate the conformational energy profile about the C-2–C-3 bond along O–C-1–C-2–C-3 using DFT calculations (B3LYP/6-31G*).35–37 Comparing the obtained energy profile with the ones previously calculated for related substructures with chiral centers either at C-1 or C-2 (Fig. 3a) shows that chiral centers at C-3 impart similar conformational rigidity as chiral centers at C-1. The key difference appears to be that the residual aliphatic hydrocarbon chain now point in the opposite directions28 for the same configuration of the chiral centers at C-1 and C-3 (see (R)-configuration plots and Newman projections in Fig. 3b). In contrast, a chiral center at C-2 does not show preferential energy minimum for the two syn-clinical or the antiperiplanar lower-energy conformations (Fig. 3b). Global energy minimum models of the core-chain substructure conformation with chiral center at C-3 nevertheless appear to show that this substructure is overall relatively linear with respect to the bent-core arm molecular substructure (Fig. 3c) as was seen earlier for the substructure with the chiral center at C-2 (compounds E and F in Fig. 1). Thus, the new derivatives with chiral centers exclusively at C-3 (BC1–BC4) should show a tendency for the formation of flat ribbon filaments due to the diminished effect of chirality when the chiral center is relocated even farther away from the core-chain juncture, the additional branching point at the chain terminus as for E and F (Fig. 1), and the overall linear substructure conformation. However, the situation is more subtle for BC5 and BC6. A chiral center at C-1 in the shorter meta-side should favor the formation of HμFs (as compounds C and F, Fig. 1), but an additional chiral center in the longer para-side would perhaps lead to the formation of one of the other chiral morphologies (HNF or HLNC), in analogy to all other prior-series compounds (Fig. 1 and 2).
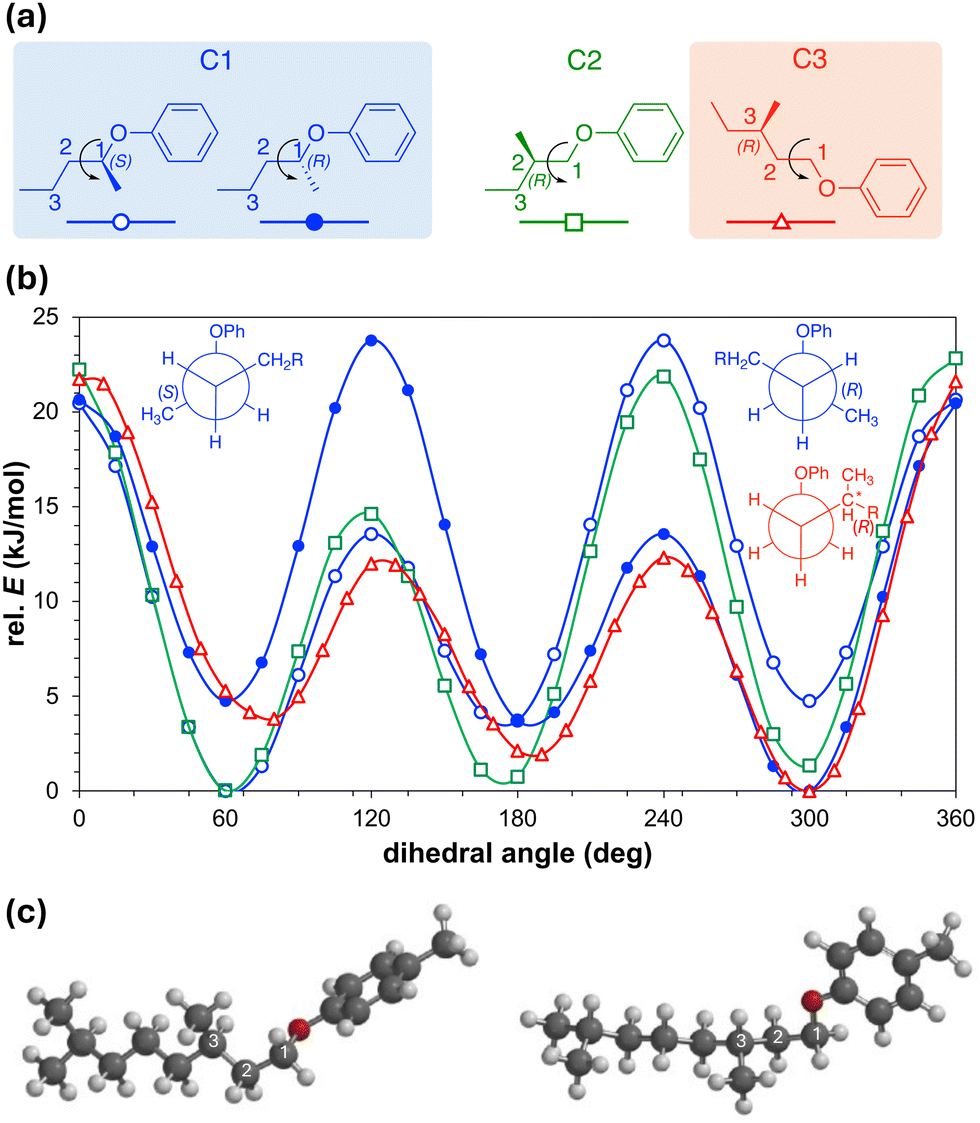 | ||
| Fig. 3 (a) Substructures of branched chiral side chains with chiral centers at C-1, C-2, or C-3 used for the calculation of the relative conformational energy profiles shown in (b). Newman projections for the two enantiomeric structures with chiral center at C-1 and for the (R)-enantiomer with the chiral center at C-3 are shown. (c) Two projections of the lowest energy conformation for the substructure with the chiral center at C-3. | ||
Thus, to further advance our understanding of the role of chiral center relocation (away from the core-chain juncture) in these tris-biphenyl bent-core molecules and the ensuing molecular conformation on final filament morphology, all new-series compounds with chiral centers at C-3 or C-1 and C-3, BC1 – BC6, were fully characterized by polarized optical microscopy (POM), differential scanning calorimetry (DSC), thin film circular dichroism (CD) spectropolarimetry, scanning and transmission electron microscopy (SEM, TEM), and X-ray diffraction (XRD) both after slow and rapid cooling from the isotropic liquid.
2. Experimental
2.1 Materials
General and analytical methods as well as synthesis details and analytical data for intermediates and bent-core compounds BC1–BC6 are given in the ESI† (Section S1, Scheme S1, Fig. S1–S6).2.2 Optical and thermal characterization
POM analysis and imaging were carried out using an Olympus BX-53 polarizing microscope equipped with a Linkam LTS420E heating/cooling stage. Samples were heated above the clearing point as indicated by DSC and then cooled at either <5 °C min−1 (commonly at 2 °C min−1; slow cooling) or ≥50 °C min−1 (rapid cooling or thermal quench). The phase transition temperatures and enthalpies were measured using either a PerkinElmer Pyris 1 DSC or a PerkinElmer DSC-7 at two heating/cooling rates (either at 5 °C min−1 or at 50 °C min−1), reporting data from the second heating/cooling runs, respectively. The temperature was calibrated with indium and zinc standards.2.3 CD spectropolarimetry
For thin film CD measurements, to eliminate contributions from linear dichroism and birefringence, samples and sample areas were investigated at 45° interval sample rotation angles. The spectra were then summed up or averaged to provide genuine CD signals of the sample area.20,38 The area interrogated by the light beam, given by instrumental slit size and shape, is ∼0.4 cm in diameter and larger than most individual domains seen by POM of the samples sandwiched between two untreated quartz substrates using nominal spacers of 10 μm.2.4 Scanning and transmission electron microscopy
SEM analysis was performed using a Quanta 450 FEG SEM commonly without prior metal deposition. TEM was carried out on an FEI Tecnai F20 microscope, operating at 200 kV, and equipped with a Schottky field emission gun and a twin-blade anticontaminator. All images were recorded using a Gatan 4 K Ultra Scan charge-coupled device camera. Since these organic material films are sensitive to electron beam irradiation, the films were normally previewed rapidly at a dose of 20 e− nm−2. Selected areas were then imaged at a dose level of 200 e− nm−2, which did not cause radiation damage. A drop of a solution of each compound in an organic solvent (CH2Cl2 or n-hexane) was placed either on indium tin oxide substrates for SEM or carbon-coated Cu grids for TEM imaging. After complete evaporation of the solvent in vacuum, samples were heated above the clearing point and cooled as described for the POM experiments, and then imaged. Image analysis was done using ImageJ®.392.5 X-ray diffraction
Variable-angle, temperature-controlled XRD experiments were performed with an in-house Xenocs Xeuss 3.0 using a Cu Kα source (λ = 1.54 Å) equipped with a Linkam HFSX350 heating–cooling stage. Samples were sealed into X-ray diffraction glass capillaries (Charles Supper Co.).3. Results and discussion
3.1 Optical and thermal analysis
DSC data (ESI,† Section S2, Fig. S7, and Table S1) show that all compounds, except for BC2, show only one significant phase transition from the isotropic liquid phase on slow cooling (at a rate of 5 °C min−1). BC1 and BC3, assuming quasi-crystalline B4-type structures, show either one or several crystal-crystal phase transitions on cooling, respectively. BC2, in contrast, shows a phase transition on cooling at higher temperature characterized by a significantly lower phase transition enthalpy. Considering phase sequences frequently observed for prior series of tris-biphenyl bent-core derivatives, this could be a columnar phase, that, here, forms over a narrow (2 °C) temperature interval. On rapid cooling at 50 °C min−1, BC1–BC6 show only one obvious phase transition (some with a discernible weak shoulder) and no glass transitions, often seen for B4-type bent-core materials, near room temperature.POM studies performed next (see Fig. 4) may provide further evidence about phase type or B4 morphology. Homochiral BC1, on slow cooling, displays a feather-like texture resembling, at least in overall appearance, the texture previously recorded for compound E on slow cooling.28 Upon rapid thermal quenching to room temperature, though, the texture observed only shows smaller, grainy birefringent domains. Nearly identical textures were seen for BC2 and BC4 with racemic hexahydrofarnesolyl side chains either in both the meta- and the para-side or only in the shorter meta-side, respectively (BC4 features a chiral center at C-3 in the longer para-side). The texture of the 2 °C wide, high temperature phase of BC2 (ESI,† Section S3 and Fig. S8) is best described as a broken spherulitic-type texture previously seen for columnar B1 phase, either neat or in coexistence with a B4-phase morphology.
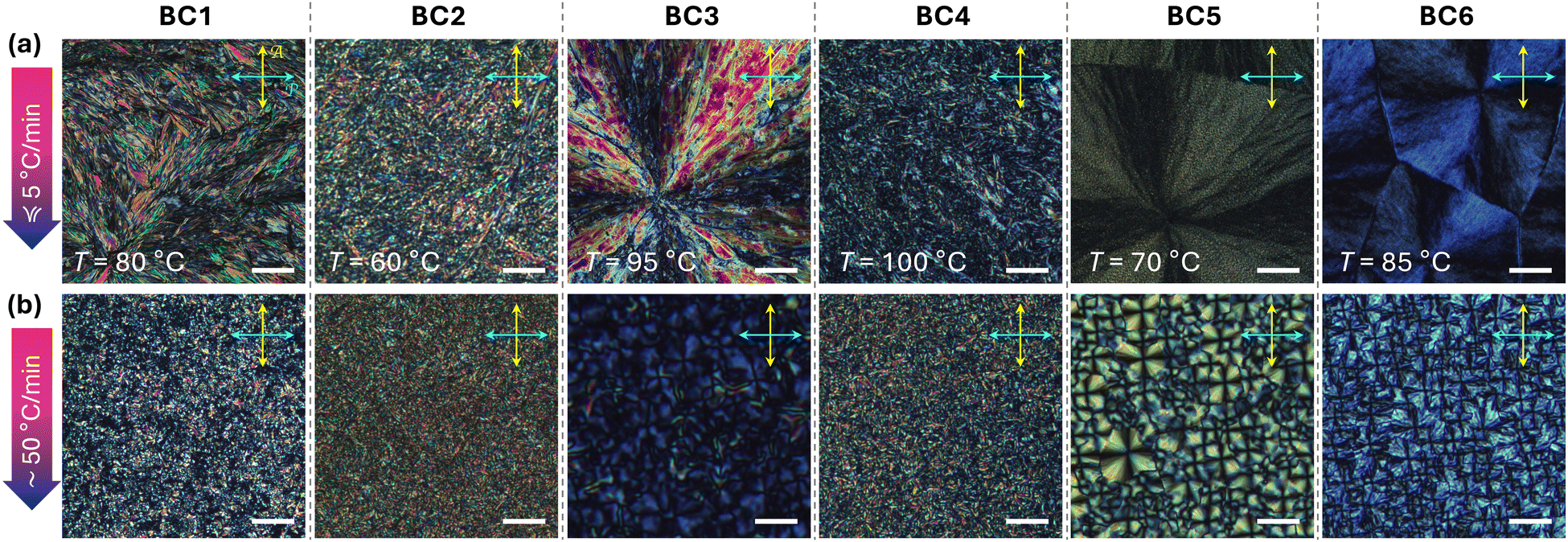 | ||
| Fig. 4 Polarized optical photomicrographs of BC1–BC6 (crossed polarizer and analyzer) after heating to the isotropic liquid phase (i.e., >125–153 °C depending on clearing point of each compound) and subsequent: (a) slow cooling to room temperature at the cooling rate of <5 °C min−1 (temperature indicated) and (b) rapid cooling to room temperature (to T ∼ 20 °C via rapid thermal quench) at a rate ≥50 °C min−1 (scale bars = 200 μm). | ||
In contrast, BC3, BC5, and BC6 show textures on slow as well as rapid cooling that closely resemble the textures earlier reported for the various nano- and microfilament phases;20–22,28 bluish in appearance and with the usual focal conic- or spherulitic-like domains. Since these materials are non-racemic, uncrossing of polarizer and analyzer clock- or anticlockwise did not generate the darker and brighter domains that would reverse upon uncrossing the analyzer in the opposite direction12 as for the chiral HNF conglomerates of achiral or racemic compounds (ESI,† Section S3 and Fig. S9). Another typical feature seen in all sets of POM photomicrographs for these compounds are the smaller domains observed under the rapid cooling regime. Since no polymer alignment layers were used in these POM experiments (only plain, pre-cleaned glass slides), identical textures were seen for preparations between untreated quartz substrates used next for thin film CD spectropolarimetry.
3.2 Thin film CD spectropolarimetry
For practically all the previous nano- and microfilaments formed by these tris-biphenyl bent-core materials, sign and wavelength of the major bands in the thin film CD spectra provided a clear indication of the handedness and in many cases even the type of filament morphology. An analysis of the CD spectra measured for BC1–BC6 (Fig. 5) was significantly less straightforward. BC1 on slow cooling showed individual CD spectra at the various sample rotation angles, all with weak positive bands centered around λ = 325 nm and a stronger negative band around λ = 350 nm. The sum CD signal is then similar with a positive as well as a negative band with maxima at these wavelengths; the latter one tailing off over a broad wavelength range (Fig. 5a). The sum spectrum measured upon rapid cooling is similar. Here, only the intensities of the two sum maxima are reversed (Fig. 5b). Both preparations with a nominal thickness of 10 μm, while weakly, show the distinctive bluish hue of the structural color commonly seen for B4 filament phases. The absence of a single clear maximum in these sum CD spectra already hints at a more complex, different, or the coexistence of more than just one morphology. The CD spectra of BC2 and BC3 (Fig. 5c–f) are then even more puzzling and less clear. On both slow and rapid cooling from the isotropic liquid state, all individual 45° sample rotation spectra randomly show either positive or negative CD bands over the entire spectral region ranging from λ = 250–750 nm without any noteworthy sum-CD signal, which is why the average of these individual spectra is reported in these cases. Together with the lack of even a hue of blue structural color (see insets), these CD data would indicate that BC2 and BC3 do not form a chiral morphology under either cooling regime. For BC4, however, the situation reverses again, and sum-CD spectra like those collected for BC1 (especially the sum-CD found on rapid cooling); a positive band centered near λ = 325 nm and a further negative band now centered at a longer wavelength around λ = 375 nm (Fig. 5g and h). The relative similarity of the CD data for BC4 and BC1 may already indicate the presence of similar filament morphology for these two materials.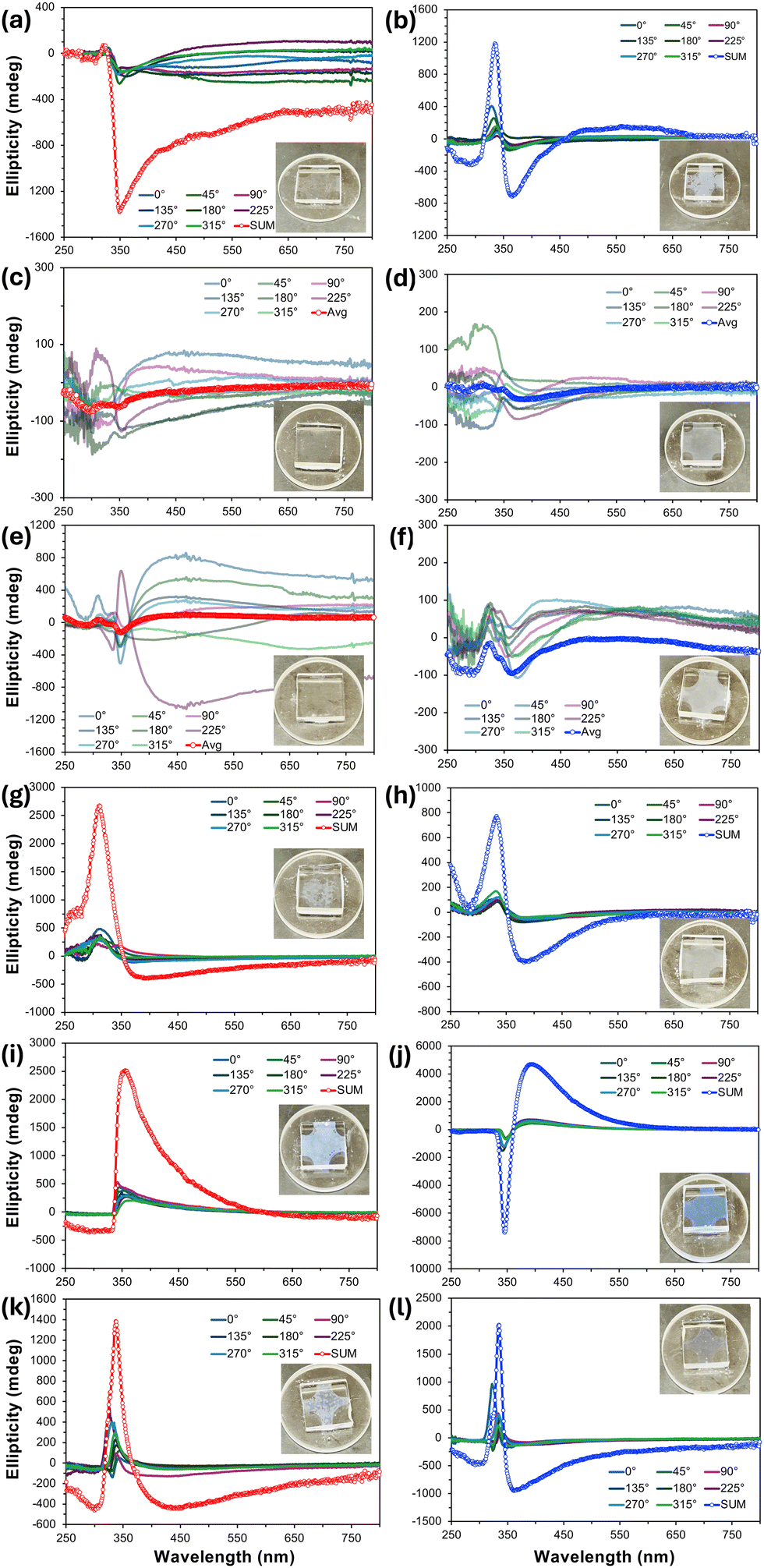 | ||
| Fig. 5 Thin-film CD spectra taken at 45° sample rotation angles (Δε [mdeg] vs. λ [nm]) of 10-μm films between untreated quartz substrates after cooling at a different rate from the isotropic liquid phase to room temperature (T ∼ 20 °C) at different sample rotation angles as indicated in the legends (red SUM or average (Avg) datasets to cancel out linear dichroism and birefringence are for slow cooling (<5 °C min−1); blue SUM or Avg datasets for rapid cooling (≥50 °C min−1)): (a) and (b) BC1, (c) and (d) BC2, (e) and (f) BC3, (g) and (h) BC4, (i) and (j) BC5, and (k) and (l) BC6. Insets in each graph are the sample photographs either showing or lacking blue structural color. Average rather than SUM spectra were generated from the individual 45° sample rotation datasets when the resulting graphs showed randomly positive or negative weaker signals over broader or specific spectral ranges (i.e., for BC2 and BC3). | ||
Finally, taking the comparatively intense blue structural color into account, it was not surprising that the CD spectra for BC5 and BC6 showed the most intense individual and sum-CD bands. On slow cooling, both compounds show intense positive sum-CD signals; centered around λ = 370 nm for BC5 and around λ = 340 nm for BC6 (Fig. 5i and k). The identical positive sign of these CD bands on slow cooling is surprising considering the enantiomeric relationship of the involved chiral centers at C-1 in the shorter meta-side of these molecules, especially since the CD spectra taken on rapid cooling display the expected mirror relationship for enantiomeric species (Fig. 5j and l), albeit at slightly different wavelength maxima. Given what we learned from CD data of prior series with diminished chirality due to chiral center relocation away from core-chain junctures, these CD datasets already indicate that we might see filaments of either handedness by electron microscopy.
3.3 SEM and TEM imaging
Initial observation of the filament morphologies formed upon slow or rapid cooling from the isotropic liquid by SEM revealed very similar filament types for BC1–BC3 (Fig. 6). Feather-like in appearance flat-looking filaments that curve almost look like split hair ends becoming finer, decreasing and further deceasing in width from w ∼ 10 μm to less than 1 μm toward the filament ends. Moreover, the apparent curvature of these flat filaments appears to result in cracks, as indicated by white lines that run parallel to the filament widths. As the filaments become finer and finer towards their ends (e.g., highlighted areas with green boundary for BC1 in Fig. 6), SEM images were not able to resolve if any of the thinner flat filament endings twist. Twist would be particularly favorable for thinner flat filaments (here the ends), and potentially expected given that the constituent molecules have chiral side chains. Considering the formation of twisted nanoribbons (TNRs) upon slow cooling and both left- and right-handed filaments upon rapid cooling by the closest relative in prior series, i.e., compound E with chiral centers at C-2 (Fig. 1), we expected to see a similar behavior at least for BC1, given: (1) that the thin film CD spectra (Fig. 5a and b) show clear negative (slow cooling) as well as negative and positive sum-CD bands (rapid cooling), (2) the faint but present blue structural color (insets, Fig. 5a and b), and (3) the lowest energy conformation of the BC molecules with a chiral center at C-3 that seems more closely related to those with a chiral center at C-1 (forming HNFs)20 than at C-2 (forming FNRs28 among other morphologies).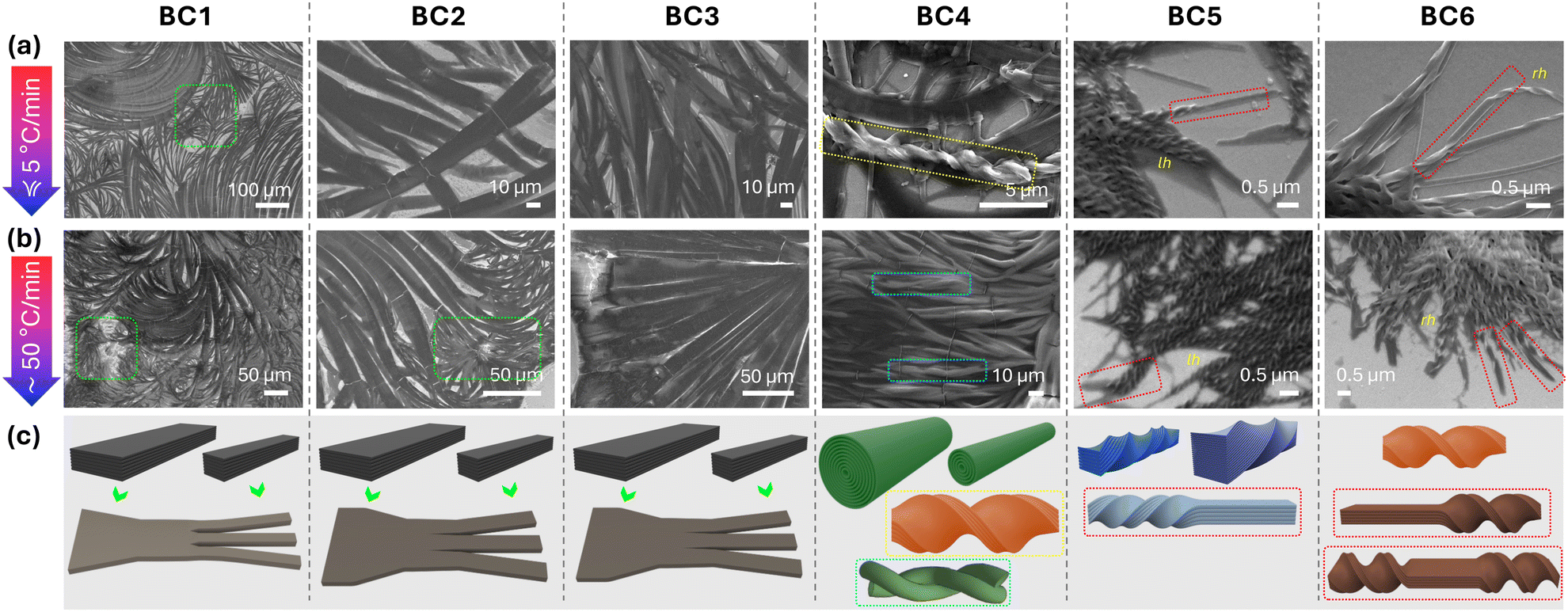 | ||
| Fig. 6 Representative SEM images obtained for BC1–BC6: (a) after slow cooling from the isotropic liquid state at a rate of <5 °C min−1 and (b) after rapid cooling (thermal quench) at a rate of ≥50 °C min−1. (c) Schematic 3-D renderings depicting the morphologies seen in the SEM images. For additional SEM images of BC4 see ESI,† Section S4, Fig. S10. | ||
BC4, despite all its structural similarity to BC1–BC3, however, forms two rather different morphologies, as can be seen in the SEM images (Fig. 6). One of the two morphologies, with similar features and overall dimensions on both slow and rapid cooling, and again splitting increasingly finer filaments, appears rather cylindrical with a spherical cross-section (and a diameter ranging from ∼200 nm–2 μm) rather than flat as for BC1–BC3. Assuming a nanocylinder-like morphology, no helicoidal wrapping of the outer layer is seen, as first reported for D (Fig. 1)26 with a chiral center at C-1 in the longer para-side. A similar lack of helicoidal wrapping, however, was reported for a homologue of D with longer aliphatic chains and a chiral center at para-C-128 and G with chiral centers at C-2 and C-327 (Fig. 2). As seen before for D, these cylinder-like morphologies braid (wrap around one another) as revealed in the highlighted area with turquoise boundary upon rapid cooling. The other clearly discernible morphology formed by BC4 exclusively upon slow cooling is those of twisted microfilaments with dimensions of w ∼ 900 nm and p ∼ 3.5 to 4 μm (highlighted area with yellow boundary in Fig. 6 and ESI,† Section S4 and Fig. S10), i.e., just about fourfold larger than the HμFs formed by C and F (Fig. 1).22,28 In line with the positive sign of the sum-CD band (Fig. 5g), these somewhat isolated HμFs are all right-handed.
Finally, once the chiral center in the shorter meta-side relocates back from C-3 to C-1, the morphology observed by SEM changes again. However, before stressing the differences between the filaments formed by BC5 and BC6 regarding type, handedness, and dimensions, a common filament feature not seen before is a change in the morphology within one and the same filament. Seen for both BC5 and BC6 upon slow as well as rapid cooling, twisted filaments suddenly unwind to form flat ribbons. Most of these twisted filaments unwind at the ends; some show shorter unwound flat segments, which then twist again with the identical twist sense (see highlighted areas in Fig. 6 with red boundaries). Twisted sections of the filaments formed by BC5 show smaller width and pitch (w ∼ 100–180 nm, p ∼ 300–450 nm) than those formed by BC6 (w ∼ 300 nm, p ∼ 900 nm). Thus, twisted sections of BC5 should be considered HNFs in analogy to the HNFmod2bs formed by B (Fig. 1);21 those of BC6 HμFs in analogy to C (Fig. 1).22 The filaments with larger width clearly visible for BC5 in the SEM images are predominantly left-handed—in contrast to the positive sum-CD band recorded by CD on slow cooling (Fig. 5i) but in line with the sign of the first sum-CD signal on rapid cooling (Fig. 5j). The handedness of the HμFs formed by BC6 (right-handed) is in line with the sum-CD bands seen upon slow and rapid cooling (Fig. 5k and l) but in contrast to the configuration of the chiral center at C-1 in the shorter meta-side of the molecule compared to C (Fig. 1). C featuring a chiral center at C-1 with (R)-configuration formed solely right-handed HμFs.22 So, assuming that a chiral center at C-1 should be the more dominating factor in deciding filament handedness, given that chirality diminishes as the chiral center is relocated farther away from the core-chain juncture, BC6 having an (S)-configuration chiral center at C-1 in the meta-side should form left-handed HμFs. Considering the images shown in Fig. 7, however, TEM data for BC1–BC6 provided far more clarity on the types and specific filament shapes.
 | ||
| Fig. 7 Representative TEM images obtained for BC1–BC6: (a) after slow cooling from the isotropic liquid state at a rate of <5 °C min−1 and (b) after rapid cooling (thermal quench) at a rate of ≥50 °C min−1. (c) Schematic 3-D renderings depicting the various morphologies seen in the TEM images. For additional TEM images of BC1, BC5, and BC6 see ESI,† Section S4, Fig. S11–S16. | ||
Significantly, TEM imaging revealed that among the smaller flat filaments formed by BC1, which could not be clearly resolved by SEM, several of those with smaller widths do eventually twist at the ends. As predicted by theory,40–42 multilayer filament twist is energetically only favorable for lower filament heights and more narrow widths, which is exactly what is observed for BC1. Wider flat ribbons branch into narrower ones, which twist at the ends (Fig. 7c). Some, as seen for BC5 and BC6 by SEM imaging, also show flat (untwisted) segments between two twisted ones (area highlighted with a red boundary for BC1 in Fig. 7b). Most of the filaments, however, as already seen in the SEM images, are FNRs with many of the narrow filaments showing values of w ∼ 450 nm (since those could now be imaged by TEM). Further supporting the just-made argument about the correlation between width and the probability for twist, dimensions of the twisted filament branches, almost identical under either cooling regime, are consistently smaller, with w ∼ 400 nm and p ∼ 2 μm. Thus, the twisted filament ends are HμFs with dimensions about twofold larger than those initially reported for C (Fig. 1), likely a consequence of the diminished chirality when the chiral centers are moved farther away from the core-chain juncture, as predicted earlier. After slow cooling, filaments are primarily left-handed, as shown in Fig. 7 and the ESI† (Section S4 and Fig. S11), in line with the negative sum-CD band centered around λ ∼ 350 nm for BC1 (Fig. 5a). After rapid cooling, mostly left- but also isolated right-handed filaments can be seen by TEM (ESI,† Section S4, Fig. S12), again in line with the thin film CD data (Fig. 5b).
For BC2 and BC3, TEM confirmed the flat ribbon nature, simply adding that the width can be as low as a few hundred nm (as low as w ∼ 300 nm), however, without any visible twist even for the narrowest filaments.
For BC4, TEM allowed for a better visualization of nanocylinders (NCs) with smaller diameters, now as low as ∅∼130–180 nm (Fig. 7a and b), which often bundle into three-dimensional constructs (Fig. 7a), quite like those reported previously for D (Fig. 1).26 While the nanocylinder-type nature is more challenging to recognize in TEM images, several do show cross-sectional electron density profiles (i.e., darker-brighter-darker) comparable to those we reported earlier for D,26 possibly even suggesting a hollow inner core.
Finally, for BC5 and BC6, both types and dimensions of filaments imaged by TEM drastically changes in comparison to BC1–BC4. The first obvious difference is the overall much smaller filament dimensions seen for BC5 and BC6, as already seen by SEM. The second obvious difference is that the TEM images now show a more intricate mix of filament types and dimensions. Moreover, many filaments display a change in morphology along one and the same filament, hinted at by SEM. For BC5, both left- and right-handed twisted filaments can be seen, some with dimensions of w ∼ 40 nm and p ∼ 240 nm as HNFmod2a formed by A and E (Fig. 1), some with dimensions of w ∼ 70–120 nm and p ∼ 320–550 nm more similar to the HNFmod2b formed by B (Fig. 7 and ESI,† Section S4, Fig. S13, S14). The potential presence or absence of further intra- or interlayer modulations will be clarified by XRD in the next section. Notably, HNFs with smaller widths form upon slow cooling from the isotropic liquid. Under each cooling regime, many twisted filaments (in some areas almost all) are unwound, forming FNRs at the ends, while several twisted filaments flatten for sections as long as 500 nm, only to twist again with the same twist sense (Fig. 7 and ESI,† Section S4, Fig. S13), i.e., without perversions. The filaments formed by BC6 share almost all these traits with the one exception that these filaments are larger. With an average width of w ∼ 240–280 nm and a pitch of p ∼ 950 nm (Fig. 7 and ESI,† Section S4, Fig. S15 and S16), these filament should be considered HμFs in analogy to those formed by compounds C and F (Fig. 1). The evident random change of handedness in the TEM images also makes a meaningful correlation with the thin film CD data challenging (Fig. 5i–l). The (R)-configuration at C-1 for BC5 (assuming this to be the ‘dominant’ chiral center) should lead to right-handed HNFs and positive CD bands, the (S)-configuration at C-1 for BC6 to left-handed HNFs and thus negative CD bands considering a comparison to compound A.20 However, prior studies on compounds with mixed configuration on either side at C-1 suggested that the opposite configuration of the two chiral centers leads to larger filament dimensions (as for BC6 in comparison to BC5 in the current series). To confirm the crystalline lamellar nature (i.e., B4-type) and any potential intra- or interlayer modulation within the filaments formed by BC1–BC6, we performed XRD measurements both after slow and rapid cooling from the isotropic liquid state.
3.4 X-ray diffraction data
All B4 morphologies discovered so far are based upon lamellar structures—layers that are either flat, twisted, or writhed. Yet, the global molecular conformation of the constituent bent-core molecules governs local packing and thus internal structure. Accordingly, each B4 morphology discovered to date has been characterized by, or better, originated from a distinct molecular packing (incl. in-layer tilt and intra- or interlayer modulations), ultimately giving rise to distinct X-ray diffraction patterns.20–22,27,28 Thus, given the coexistence of several morphologies or morphological evolutions detected by electron microscopy we should expect reasonably complex XRD patterns for BC1–BC6. Fig. 8 shows all medium-angle XRD (MAXD) patterns (q ∼ 0.1–0.7 Å−1) for BC1–BC6; diffraction patterns covering also the wide-angle (WAXD) region as well as peak listings, relative intensities, peak deconvolution, peak fitting, and Miller indices are provided in the ESI† (Section S5 and Fig. S17–S30).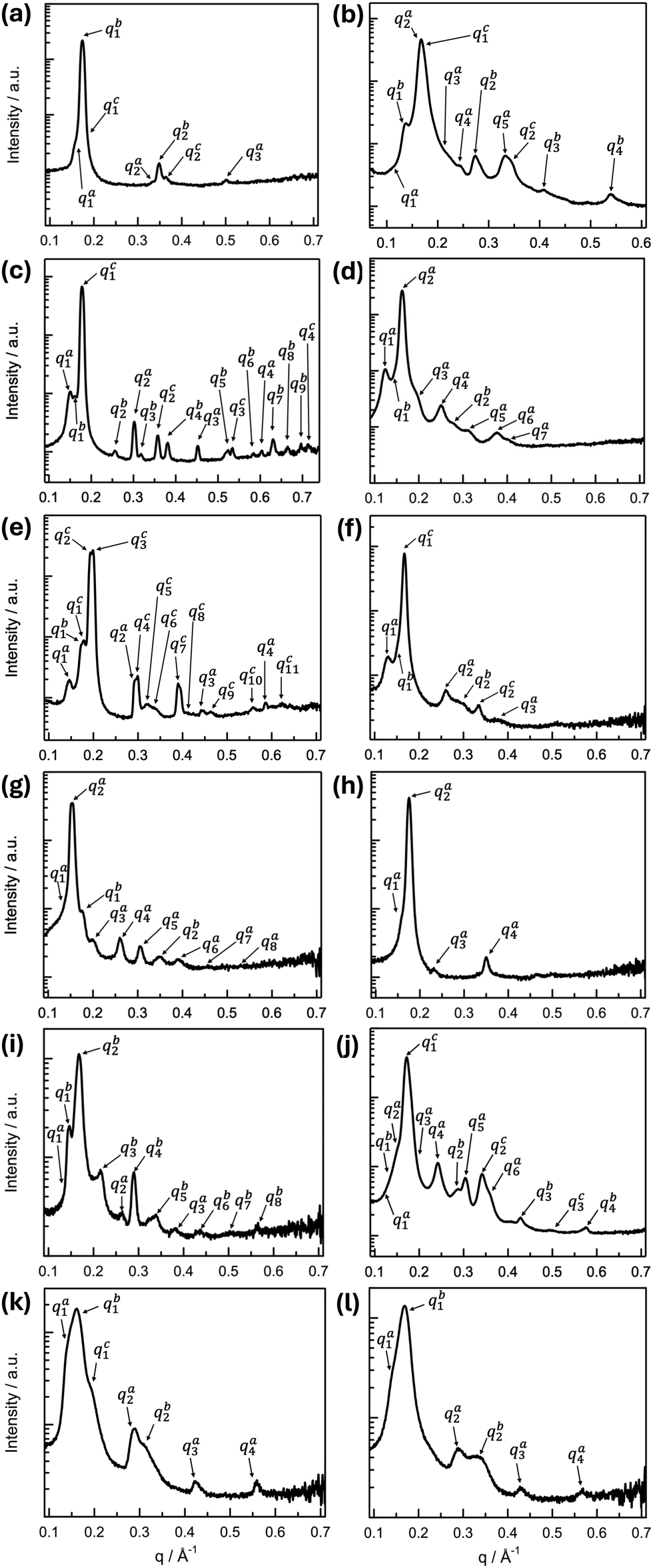 | ||
| Fig. 8 XRD analysis data showing intensity (a.u.) vs. wave vector q (Å−1) of the MAXD region: (left column) slow cooling and (right column) fast cooling. (a) and (b) BC1, (c) and (d) BC2, (e) and (f) BC3, (g) and (h) BC4, (i) and (j) BC5, and (k) and (l) BC6. Peak positions, relative intensity values, and Miller indices are provided in the ESI,† Section S5, Fig. S17–S30 and Tables S2–S15. | ||
BC1 showed the coexistence of FNRs either varying in width or splitting into finer and finer FNRs with gradually lower widths as well as HμFs that occasionally unwind at the ends. BC1, as a result thereof, then produces an XRD pattern where upon slow (Fig. 8a) as well as upon rapid cooling to room temperature (Fig. 8b) three sets of (00l) diffraction peaks with l = 1, 2, … can be indexed (qa1 = 0.165 Å−1, qb1 = 0.174 Å−1, and qc1 = 0.181 Å−1 on slow cooling and qa2 = 0.167 Å−1, qb1 = 0.137 Å−1 and qc1 = 0.169 Å−1 on rapid cooling; for the corresponding higher-order reflections, qxn with n = 2, 3 and x = a − c see ESI,† Section S5, Fig. S17, S18 and Tables S2, S3). One of the two structures seen upon rapid cooling is clearly a modulated layer structure with qa3 = (qa21 + qa22)1/2. The q values for the (001) reflections then indicate variable layer spacings for each morphology, ranging from d = 3.81–3.47 nm upon slow cooling and d = 4.58 nm and d = 3.72 nm upon rapid cooling, driven by variations in molecular tilt within the layers from θ ∼ 13° for one of the structures seen upon rapid cooling to 42° for one detected upon slow cooling, given a calculated molecular length of l = 4.7 nm. Given the spatial heterogeneity of coexisting morphologies seen by SEM and TEM, it seems not surprising that interrogating another sample location by XRD (realized by moving the X-ray capillary within the hot stage) revealed coexistence with yet another structure (ESI,† Section S5, Fig. S19 and Table S4) on rapid cooling. This subset of diffraction maxima qi (i = 1, …, 10) is best indexed as (001), (100), (010), (101), (111), (002), (200), (11![[2 with combining macron]](https://www.rsc.org/images/entities/char_0032_0304.gif) ), (020), and (120) with an oblique lattice and monoclinic space group P2 (3D), as shown in the ESI† (Section S5, Fig. S19, Table S4). The calculated lattice parameters of this 3D Colob-P2 phase are a = 40.2 Å, b = 34.9 Å, c = 42.4 Å, and β = 102°. Similar phase coexistences as seen here for BC1 upon rapid cooling (B1 Colob-P2 phase; structure shown in ESI,† Fig. S19) have previously been reported for other compounds in prior reported tris-biphenyl series.28
), (020), and (120) with an oblique lattice and monoclinic space group P2 (3D), as shown in the ESI† (Section S5, Fig. S19, Table S4). The calculated lattice parameters of this 3D Colob-P2 phase are a = 40.2 Å, b = 34.9 Å, c = 42.4 Å, and β = 102°. Similar phase coexistences as seen here for BC1 upon rapid cooling (B1 Colob-P2 phase; structure shown in ESI,† Fig. S19) have previously been reported for other compounds in prior reported tris-biphenyl series.28
For BC2, upon slow cooling, a phase coexistence was observed between two non-modulated B4 structures and a B1 Colr–c2mm phase (Fig. 8c). The two B4 structure sets of (00l) diffraction peaks with l = 1, …, 4 were identified with qa1 = 0.151 Å−1 and qc1 = 0.178 Å−1. The corresponding higher-order reflections, qnx with n = 2–4 and x = a, c are given in the ESI,† Section S5, Fig. S20 and Table S5. The q values for these 001 reflections indicate layer spacings for the two morphologies, likely the wider and narrower FNRs, of d = 4.16 nm (qa1 series) and d = 3.53 nm (qc1 series), again associated with variations in molecular tilt within the layers of θ ∼ 39° or 49°, respectively, taking into account a calculated molecular length of BC2 of l = 5.4 nm. A third set of diffraction peaks labeled qb1 − qb9 were indexed as (001), (101), (002), (102), (103), (301), (203), (104), and (302) signifying a rectangular lattice with c2mm plane group (2D) and lattice parameters of a = 31.4 Å and c = 39.0 Å, in which the molecules with highly branched side chains tilt significantly—a carry-over from a phase transition seen by DSC (ESI,† Fig. S7c). On rapid cooling, BC2 shows only two B4 structures (Fig. 8d). One is a modulated layer structure with qa3 = (qa21 + qa22)1/2 and maxima at qa1 = 0.123 Å−1, qa2 = 0.161 Å−1, and qa3 = 0.202 Å−1; values for the other reflections indexed as (200), (002), (300), and (202) are given in the ESI† (Section S5, Fig. S21 and Table S6). The second structure is non-modulated with qb1 = 0.139 Å−1 and qb2 = 0.278 Å−1. The layer spacing of the modulated phase amounts to d = 5.11 nm (indicating a tilt of the molecules within the layer of θ ∼ 19°); the non-modulated phase layer spacing is d = 4.52 nm from which a molecular tilt of θ ∼ 33° can be calculated.
Relatively similar phase coexistences were again found for BC3, not unpredicted given the similarity in coexisting morphologies seen by electron microscopy. After slow cooling, BC3 exhibits diffraction maxima (Fig. 8e) best assumed to be from two non-modulated B4 structures with layer spacings of d = 4.33 nm (qa1 series) and d = 3.65 nm (qb1 series)—assuming a calculated molecular length of l = 5.1 nm and tilt angles θ ∼ 32° and 44°, respectively, in addition to a coexisting B1 Colob-P2 (3D) phase (similar to BC2 upon rapid cooling) with somewhat similar lattice parameters of a = 32.7 Å, b = 31.6 Å, c = 35.3 Å, and β = 102° (ESI,† Section S5, Fig. S22, Table S7). Upon rapid cooling, a somewhat simpler diffraction pattern indicates the formation of three non-modulated filament structures for BC3 indexed with three sets of (00l) reflections (l = 1–3), i.e., lamellar structures with layer spacings of d = 4.87, 4.16 and 3.76 nm and tilt angles ranging from θ ∼ 17° to 42° (Fig. 8f and ESI,† Section S5, Fig. S23, Table S8). BC3 is the only compound in this series that forms a high temperature phase above the B4 phase (see DSC data in the ESI,† Fig. S7e). Considering the coexisting B1 Colob-P2 phase seen upon slow cooling, unsurprisingly, this high-temperature phase is also a B1 Colob-P2 (3D) phase with lattice parameters of a = 37.1 Å, b = 33.2 Å, c = 41.8 Å, and β = 102° that are slightly larger as a result of this phase being observed at a temperature range between T = 139 to 142 °C on heating (ESI,† Fig. S24, Table S9).
For BC4, considering that we only observed two morphologies upon slow cooling (NCs and HμFs) and just NCs on rapid cooling, expectations were that the X-ray diffraction pattern will confirm this. Indeed, upon slow cooling, BC4 shows the coexistence of a modulated and a non-modulated B4 structure (Fig. 8g). The modulated layer structure features qa3 = (qa21 + qa22)½ and maxima at qa1 = 0.130 Å−1 (100), qa2 = 0.153 Å−1 (001), and qa3 = 0.200 Å−1 (101). The values for the other reflections indexed as (200), (002), (300), (003), and (400) are given in the ESI† (Section S5, Fig. S25, Table S10). The layer spacing of this morphology is d = 4.10 nm and the tilt θ ∼ 36° considering the calculated molecular length of l = 5.1 nm. The non-modulated structure shows (00l) reflections with l = 1, 2, and 4 with qa1 = 0.172 Å−1 (001) and thus a layer spacing of d = 3.65 nm and a larger tilt of θ ∼ 44° (ESI,† Section S5, Fig. S25, Table S10). Given that the single structure seen upon rapid cooling is also modulated, we assume that the NC morphology seen by electron microscopy after both cooling regimes is the one with the modulated structure. The layer spacing is d = 3.57 nm and the tilt θ ∼ 45° (Fig. 8h and ESI,† Section S5, Fig. S26, Table S11).
BC5, which forms smaller HNFmod2as and larger HNFmod2bs that intermittently or at the ends untwist, as seen in the electron micrographs, shows complex diffraction patterns that confirm the presence of multiple coexisting morphologies as well. Upon slow cooling, a modulated and a non-modulated B4 structure were assigned with qa1 = 0.134 Å−1 and qb2 = 0.166 Å−1 (Fig. 8i). The corresponding higher-order reflections, qxn with n = 3 or 8 and x = a, b are given in the ESI† (Section S5, Fig. S27, Table S12). The modulated layer structure according to qb3 = (qb21 + qb22)1/2 features a layer spacing of d = 3.79 nm and a molecular tilt of θ ∼ 34° considering a calculated molecular length of l = 4.6 nm. The non-modulated B4 structure is quasi non-tilted with a layer spacing of d = 4.67 nm that near matches the molecular length of BC5. Upon rapid cooling of BC5, three coexisting B4 structures were identified in the diffraction pattern (Fig. 8j). Again, one is modulated, and the other two non-modulated. The (001) reflections for the two non-modulated structures at qb1 = 0.143 Å−1 and qc1 = 0.172 Å−1 provide values for the layer spacing of d = 4.39 and d = 3.65 nm and thus tilt angles of θ ∼ 17° and θ ∼ 37°, similar to the values calculated for other morphologies formed by BC1–BC4. The modulated structure with qa3 = (qa21 + qa22)½ has a layer spacing of d = 4.11 nm and a tilt of θ ∼ 27° (values for higher order reflections are given in the ESI,† Fig. S28, Table S13).
Finally, BC6's diffraction pattern upon slow cooling revealed three non-modulated B4 structures with qa1 = 0.143 Å−1, qb1 = 0.159 Å−1, and qc1 = 0.185 Å−1 (layers spacings ranging from d = 4.39–3.40 nm and tilt angles of θ ∼ 17°, 31°, and 42°), and upon rapid cooling two non-modulated B4 structures with qa1 = 0.146 Å−1 and qb1 = 0.169 Å−1, and thus, each with very similar values for d and θ to two seen on slow cooling (d = 4.30 nm, θ ∼ 21° and d = 3.72 nm, θ ∼ 36°). For values of higher order reflections, see ESI† (Fig. S29, S30 and Tables S14, S15).
Overall, analysis of the complex XRD patterns further supports what was already suggested by the thin film CD data and clearly seen by EM imaging. The local packing of the molecules in layers affected by the overall molecular conformation via the presence of strategically placed chiral centers and their configuration, and further, additional branching points in the side chains, leads to frustration that globally manifests itself in the formation of coexisting nano- and microscale filament morphologies. While we cannot assign sets of specific internal structural features to any particular morphology, differences in local packing clearly control what overall morphologies are formed by BC1–BC6.
Fig. 9 and Table 1 summarize the overall findings, highlighting trends with respect to filament type and dimensions as well as dimensions (d, θ) of the internal structure.
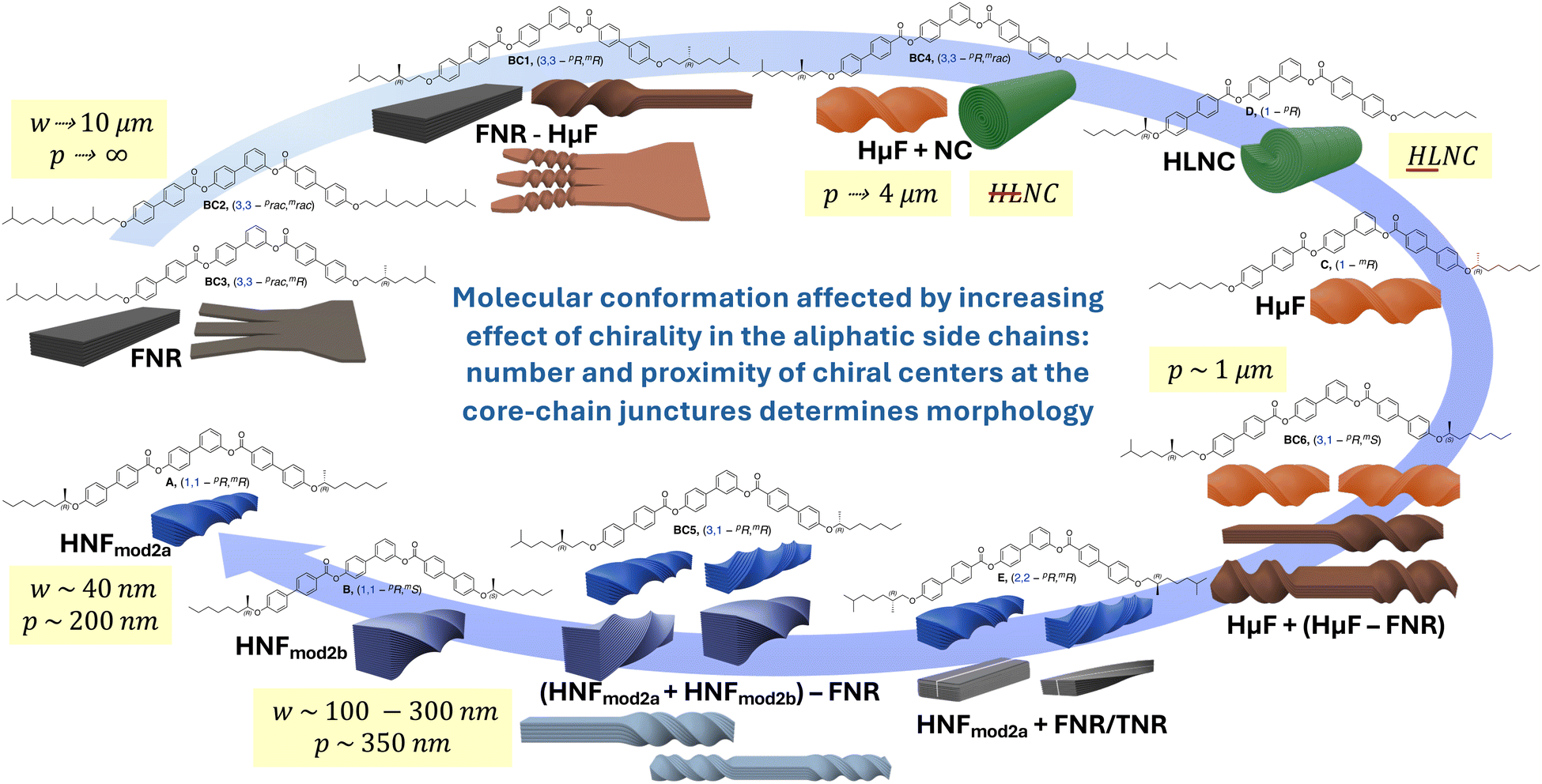 | ||
| Fig. 9 Evolution of B4 morphologies discovered to date depending on the increasing effect of chirality in the side chains near or at the core-chain juncture(s)—precisely the number and position of the chiral centers—from flat nanoribbons (FNRs) formed by BC1–BC3 with branching points and chiral centers at C-3 over microfilaments and nanocylinder morphologies for single chiral centers at C-1 to the helical nanofilaments formed by BC5, A,20 and B,21 with chiral centers at C-1. | ||
| # | Cooling regime | Morphologies | Dimensions | Handedness | Layer spacing d (nm) | Tilt angle θ (°) | Structural color | |
|---|---|---|---|---|---|---|---|---|
| Abbreviations/symbols: SC = slow cooling (5 °C min−1), FC = fast cooling (≥50 °C min−1), lh = left-handed, rh = right-handed, + sign = coexisting morphologies, morphology—morphology = progressive changes in morphology, mod = modulated (i.e., intra-layer modulation), (mod) = modulation observed for some but not all the coexisting morphologies, N/A = not applicable. | ||||||||
| BC1 | SC | (FNRwide–FNRnarrow–HμF) | FNR—: w = h < 1–10 μm | lh | 3.81–3.47 | 13–42 | No | |
| FC | (FNRwide–FNRnarrow–HμF)(mod) + Colob-P2 | HμF: w ∼ 400 nm | 4.58–3.72 | Yes | ||||
| p ∼ 2 μm | ||||||||
| BC2 | SC | (FNRwide–FNRnarrow) + Colr-c2mm | w ∼ <1–10 μm | N/A | 4.16–3.53 | 39–49 | No | |
| FC | (FNRwide–FNRnarrow)(mod) | 5.11–4.52 | 19–33 | No | ||||
| BC3 | SC | (FNRwide–FNRnarrow) + Colob-P2 | w ∼ <1–10 μm | N/A | 4.33–3.65 | 32–44 | No | |
| FC | FNRwide–FNRnarrow | 4.87–3.76 | 17–42 | No | ||||
| BC4 | SC | NCmod + HμF | NCmod | ⊘ ∼ 0.13–2 μm | rh | 4.10 | 36 | Yes, weak |
| HμF | w ∼ 900 nm | 3.65 | 44 | |||||
| p ∼ 3.5–4.0 μm | ||||||||
| FC | NCmod | ⊘ ∼ 0.13–2 μm | 3.57 | 45 | Yes, weak | |||
| BC5 | SC | [HNFa + HNFb + HNFa–FNR–(HNFa) + HNFb-FNR-(HNFb)](mod) | HNFa: w ∼ 40 nm | lh & rh | 4.67–3.79 | 0–34 | Yes | |
| p ∼ 240 nm | ||||||||
| HNFb: w ∼ 70–120 nm | ||||||||
| FC | p ∼ 320–550 nm | 4.39–3.65 | 17–37 | Yes | ||||
| —FNR: w ∼ 40–120 nm | ||||||||
| BC6 | SC | HμF + HμF–FNR–(HμF) | HμF—: w ∼ 240–280 nm | lh & rh | 4.39–3.40 | 17–42 | Yes | |
| p ∼ 950 nm | ||||||||
| FC | —FNR: w ∼ 240–280 nm | 4.30–3.72 | 21–36 | Yes | ||||
4. Conclusions
As graphically recapitulated in Fig. 9, the set of asymmetric tris-biphenyl bent-core compounds discussed here, BC1–BC6, fully closes the circle from flat non-twisted to helical multilayer ribbons with the smallest possible physical dimensions (width, height, and helical pitch). As reviewed recently in this journal,10 inter- or intralayer misfits caused by anisotropic multi-layer configurations, templating, packing frustration, amphiphilicity, intrinsic chirality, or specific molecular conformations imposing supramolecular chirality ultimately generate twisted or writhed flat ribbons. Combined with our earlier work, we here show that intralayer misfits and thus the ensuing nano- or microfilament shapes and dimensions can be predicted and engineered by strategically incorporating chiral centers/branching points into the aliphatic side chains. The most critical structural parameters to adjust for such predictable morphology engineering are the number and configuration of chiral centers as well as their exact position within the core-chain juncture (now altered from C-1 to C-3 in the alkyloxy side chains). All morphologies previously reported, even in cases of coexisting morphologies, adopted a specific morphology, either separately or along with another morphology. In the present series, BC2 and BC4 are examples of this type of behavior. In some instances, the cooling rate, either slow or rapid, decided which morphology (phase) was formed or coexisted (e.g., compounds C–F, Fig. 1). BC3 in the current series is another example of this cooling rate dependence.In stark contrast, BC1, BC5, and BC6 exhibit clear morphological transformations within one and the same filament. BC1 forms flat multilayer ribbons that initially and progressively lessen in width to ultimately twist at the ends when a lower threshold of width is reached. Accordingly, BC1 serves as a unique example and visual glimpse (quasi as a still image of a nonexistent movie) in support of the earlier discussed relationship between limited filament width and height that lends itself to energetically favorable twist. Comparable gradual shape evolutions have previously been reported only for very few materials such as peptide amphiphiles.43 The reduced effect of chirality when the chiral centers are relocated from C-2 (as for compound E, Fig. 1) to C-3, according to Gray and McDonnell rules,44 is visually apparent by comparing the dimensions of the twisted filaments (HNFs formed by E exclusively upon rapid cooling and HμFs by BC1). Yet, the more pronounced conformational bias toward one conformation when the chiral center resides at C-3 (Fig. 3b) leads to the formation of only one handedness for the HμFs at the filament ends seen for BC1.
BC5 and BC6 with mixed positions of the chiral centers near the core-chain junctures (C-1 and C-3) show another type of shape evolution. Here, twisted filaments (varying in width/height) are untwisted either somewhere along the filament and/or at their ends. In each case, and in analogy to E with chiral centers at C-2, both left- and right-handed filaments form simultaneously. Surprisingly then, that not a single one of the filaments showed a perversion45 (i.e., a change in handedness before and after a flat ribbon section). We assume that the intrinsic chirality of the molecules and their asymmetric overall structure prevent the conservation of zero overall twist. Lastly, again in analogy to other compounds in the series, BC5, with identical (homochiral) configuration of the two chiral centers, forms filaments with smaller dimensions (HNFa and HNFb), while BC6 with alternating chiral center configuration forms HμFs (see Table 1). The same behavior was earlier reported for compounds A and B (Fig. 1 and Fig. 9) forming smaller HNFmod2a and larger HNFmod2b, respectively.20,21 Overall, our expansion of the series discussed here highlights the intricate interplay between the position of the chiral center(s) and the ensuing effects of overall molecular conformation on the self-assembly into these fascinating and now even more complex B4 morphologies. As an example of the now realized predictability regarding filament type, dimensions and handedness, let us compare the nanofilaments formed by A, B, E, and BC5 by using the simplified molecular conformations shown in Fig. 10. BC5 is a de facto mix of the concepts used for A (or B) and E with the average distance of the chiral centers two carbon atoms away from the core-chain juncture. Thus, the observation of FNRs was not a surprise for BC5. The bent of the aliphatic side chain in the same direction (see Fig. 3b) for BC5 (3,1-pR,mR) mimics the conformation of B forming the larger HNFmod2bs, which are observed for BC5. Similarly, conformation and configuration of and caused by the chiral center at C-3 for BC5 lead, as expected, to the formation of filaments with either handedness. Similar predictability arguments related to overall conformation that is clearly affected by chiral center positions and configuration can be made for the other filaments formed by this new series of bent-core molecules.
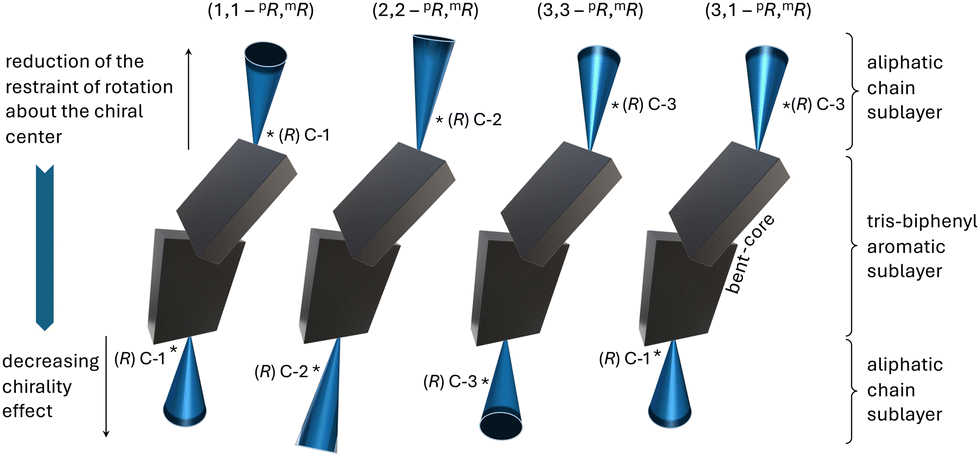 | ||
| Fig. 10 Summary of the interplay between the position(s) of the chiral center(s) and overall conformation of the tris-biphenyl bent-core molecules. Both parameters play key roles in the ensuing formation of the various B4 morphologies. Contributions from both structural effects (configuration of chiral center(s) and overall molecular conformation) can be prota- or antagonistic, variable in the strength of contribution, depending on the position within the molecule (meta- vs. para-side). | ||
In closing, while further structural modifications with respect to the placement of chiral centers in the aliphatic side chains are certainly possible (and about to be pursued), the set of currently available tris-biphenyl derivatives provides some very powerful insights into the nature of nano- and microfilament formation, and how to best manipulate chemical structures to achieve a specific shape, twist sense, coexistence, and overall filament dimension outcome that can potentially be applied to a range of related systems. B4 nano- and microfilaments are excellent and even dynamic templates for emissive dyes, nanomaterials, and various liquid crystal phases, among other guests,46–55 and the now more straightforwardly obtainable, ever-increasing number of nano- and microfilament polymorphs will continue to provide geometrically distinct templates for an increasing number of materials and potential applications, and thus access to exciting overall materials properties.29,30,50,56,57 For example, alignment of the new, more complex nano- and microfilament types could allow for spatially varying the efficacy of generating circular polarized luminescence (CPL) if nanomaterial or organic molecule emitters are templated by these filaments. Logical blending of nano- and microfilament morphologies (i.e., generating a desired morphology by mixing two others in the melt), as demonstrated by us recently,58 could further allow the finetuning of such emissive properties that are sought after in encryption technology applications such as physical unclonable functions (PUFs),59 especially if further coupled with the tunable structural color provided by these nano- and microfilaments.
Author contributions
T. H., M. E. P., and A. G. conceived the idea and the experiments. A. G. with help from G. A. accomplished the syntheses and full analytical characterization of the materials. A. G. performed the POM and CD studies, A. G., G. A., and G. A. A. R. SEM and TEM imaging. M. E. P., S. K. P., and A. S. did the XRD experiments, M. E. P. with some input from T. H. analyzed the XRD data. R. P. L. calculated the conformational energy profiles. G. A. R. R. designed the 3D filament models. T. H. directed the research and wrote the manuscript with contributions from all coauthors.Data availability
All data needed to evaluate the conclusions discussed in this article are either present in the paper itself or available via the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Science Foundation (NSF; DMR-1904091), the Ohio Third Frontier program for Ohio Research Scholars “Research Cluster on Surfaces in Advanced Materials” (T. H.) that supports the Materials Characterization and Imaging Facility at the Advanced Materials and Liquid Crystal Institute (AMLCI) at Kent State University, where current SEM and TEM data were acquired. R. P. L. acknowledges funding from the Natural Sciences and Engineering Research Council of Canada (NSERC). The authors further acknowledge access to the X-ray scattering facility at the AMLCI, which was financially supported by the National Science Foundation (NSF; DMR-2017845), the State of Ohio (The Ohio Department of Higher Education Action Fund), and Kent State University. Finally, we would like to acknowledge the mass spectrometry laboratory at Indiana University for providing all the high-resolution mass spectrometry data.References
- A. W. P. Fitzpatrick, B. Falcon, S. He, A. G. Murzin, G. Murshudov, H. J. Garringer, R. A. Crowther, B. Ghetti, M. Goedert and S. H. W. Scheres, Nature, 2017, 547, 185–190 CrossRef CAS PubMed.
- D. Kurouski, X. F. Lu, L. Popova, W. Wan, M. Shanmugasundaram, G. Stubbs, R. K. Dukor, I. K. Lednev and L. A. Nafie, J. Am. Chem. Soc., 2014, 136, 2302–2312 CrossRef CAS PubMed.
- T. Izoré, D. Kureisaite-Ciziene, S. H. McLaughlin and J. Löwe, e-Life, 2016, 5, e21600 Search PubMed.
- D. M. Bassen, Y. Hou, S. S. Bowser and N. K. Banavali, Sci. Rep., 2016, 6, 31723 CrossRef CAS PubMed.
- M. A. Constantino, M. Jabbarzadeh, H. C. Fu and R. Bansil, Sci. Adv., 2016, 2, e1601661 CrossRef PubMed.
- I. Gutsche, A. Desfosses, G. Effantin, W. L. Ling, M. Haupt, R. W. H. Ruigrok, C. Sachse and G. Schoehn, Science, 2015, 348, 704–707 CrossRef CAS PubMed.
- M. C. S. Kingsley and M. A. Ramsay, Arctic, 1988, 41, 236–238 Search PubMed.
- H. L. Kui, X. Y. Liu, J. Liu, W. Liang, S. W. Zhang, Z. H. Qian and L. Ren, Front. Bioeng. Biotechnol., 2020, 8, 363 CrossRef PubMed.
- J. S. Wang, G. Wang, X. Q. Feng, T. Kitamura, Y. L. Kang, S. W. Yu and Q. H. Qin, Sci. Rep., 2013, 3, 3102 CrossRef PubMed.
- A. Gowda, S. K. Pathak, G. A. R. Rohaley, G. Acharjee, A. Oprandi, R. Williams, M. E. Prévot and T. Hegmann, Mater. Horiz., 2024, 11, 316–340 RSC.
- D. P. N. Gonçalves, M. E. Prévôt, S. Üstünel, T. Ogolla, A. Nemati, S. Shadpour and T. Hegmann, Liq. Cryst. Rev., 2021, 9, 1–34 CrossRef.
- L. E. Hough, H. T. Jung, D. Kruerke, M. S. Heberling, M. Nakata, C. D. Jones, D. Chen, D. R. Link, J. Zasadzinski, G. Heppke, J. P. Rabe, W. Stocker, E. Korblova, D. M. Walba, M. A. Glaser and N. A. Clark, Science, 2009, 325, 456–460 CrossRef CAS PubMed.
- R. V. Kohn and E. O'Brien, J. Elasticity, 2018, 130, 115–143 CrossRef.
- D. Chen, J. E. Maclennan, R. Shao, D. K. Yoon, H. T. Wang, E. Korblova, D. M. Walba, M. A. Glaser and N. A. Clark, J. Am. Chem. Soc., 2011, 133, 12656–12663 CrossRef CAS PubMed.
- M. M. C. Tortora, G. Mishra, D. Presern and J. P. K. Doye, Sci. Adv., 2020, 6, eaaw8331 CrossRef CAS PubMed.
- E. Grelet and M. M. C. Tortora, Nat. Mater., 2024, 23, 1276–1282 CrossRef CAS PubMed.
- G. A. R. Rohaley and T. Hegmann, Nat. Mater., 2024, 23, 1161–1163 CrossRef CAS PubMed.
- G. Pelzl, S. Diele and W. Weissflog, Adv. Mater., 1999, 11, 707–724 CrossRef CAS.
- D. M. Walba, L. Eshdat, E. Korblova and R. K. Shoemaker, Cryst. Growth Des., 2005, 5, 2091–2099 CrossRef CAS.
- L. Li, M. Salamonczyk, A. Jákli and T. Hegmann, Small, 2016, 12, 3944–3955 CrossRef CAS PubMed.
- S. Shadpour, A. Nemati, M. Salamonczyk, M. E. Prévôt, J. Liu, N. J. Boyd, M. R. Wilson, C. H. Zhu, E. Hegmann, A. I. Jákli and T. Hegmann, Small, 2020, 16, 1905591 CrossRef CAS PubMed.
- L. Li, M. Salamonczyk, S. Shadpour, C. H. Zhu, A. Jákli and T. Hegmann, Nat. Commun., 2018, 9, 714 CrossRef PubMed.
- E. Tsai, J. M. Richardson, E. Korblova, M. Nakata, D. Chen, Y. Q. Shen, R. F. Shao, N. A. Clark and D. M. Walba, Angew. Chem., Int. Ed., 2013, 52, 5254–5257 CrossRef CAS PubMed.
- M. Castillo-Vallés, M. Cano, A. Bermejo-Sanz, N. Gimeno and M. B. Ros, J. Mater. Chem. C, 2020, 8, 1998–2007 RSC.
- M. Alaasar, M. Prehm and C. Tschierske, Chem. – Eur. J., 2016, 22, 6583–6597 CrossRef CAS PubMed.
- S. Shadpour, A. Nemati, N. J. Boyd, L. Li, M. E. Prévôt, S. L. Wakerlin, J. P. Vanegas, M. Salamonczyk, E. Hegmann, C. H. Zhu, M. R. Wilson, A. I. Jákli and T. Hegmann, Mater. Horiz., 2019, 6, 959–968 RSC.
- B. Sezgin, J. Liu, D. P. N. Gonçalves, C. H. Zhu, T. Tilki, M. E. Prévôt and T. Hegmann, ACS Nanosci Au, 2023, 3, 295–309 CrossRef CAS PubMed.
- J. Liu, S. Shadpour, M. E. Prévôt, M. Chirgwin, A. Nemati, E. Hegmann, R. P. Lemieux and T. Hegmann, ACS Nano, 2021, 15, 7249–7270 CrossRef CAS PubMed.
- W. Park, T. Ha, T. T. Kim, A. Zep, H. Ahn, T. J. Shin, K. I. Sim, T. S. Jung, J. H. Kim, D. Pociecha, E. Gorecka and D. K. Yoon, NPG Asia Mater., 2019, 11, 45 CrossRef.
- W. Park, J. M. Wolska, D. Pociecha, E. Gorecka and D. K. Yoon, Adv. Opt. Mater., 2019, 7, 1901399 CrossRef CAS.
- J. Y. Sun, B. Bhushan and J. Tong, RSC Adv., 2013, 3, 14862–14889 RSC.
- J. W. Goodby, A. J. Slaney, C. J. Booth, I. Nishiyama, J. D. Vuijk, P. Styring and K. J. Toyne, Mol. Cryst. Liq. Cryst., 1994, 243, 231–298 CrossRef CAS.
- G. Lemahieu, J. Aguilhon, H. Strub, V. Molinier, J. F. Ontiveros and J. M. Aubry, RSC Adv., 2020, 10, 16377–16389 RSC.
- C. Thiehoff, Y. P. Rey and R. Gilmour, Isr. J. Chem., 2017, 57, 92–100 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1993, 98, 1372–1377 CrossRef CAS.
- R. Ditchfield, W. J. Hehre and J. A. Pople, J. Chem. Phys., 1971, 54, 724–728 CrossRef CAS.
- W. J. Hehre and W. A. Lathan, J. Chem. Phys., 1972, 56, 5255–5257 CrossRef CAS.
- H. Qi, J. D. O'Neil and T. Hegmann, J. Mater. Chem., 2008, 18, 374–380 RSC.
- C. A. Schneider, W. S. Rasband and K. W. Eliceiri, Nat. Methods, 2012, 9, 671–675 CrossRef CAS PubMed.
- G. M. Grason, Soft Matter, 2020, 16, 1102–1116 RSC.
- D. M. Hall and G. M. Grason, Interface Focus, 2017, 7, 20160140 CrossRef PubMed.
- G. M. Grason, J. Chem. Phys., 2016, 145, 110901 CrossRef.
- H. Cui, T. Muraoka, A. G. Cheetham and S. I. Stupp, Nano Lett., 2009, 9, 945–951 CrossRef CAS PubMed.
- G. W. Gray and D. G. McDonnell, Mol. Cryst. Liq. Cryst., 1976, 34, 211–217 CrossRef.
- P. E. S. Silva, J. L. Trigueiros, A. C. Trindade, R. Simoes, R. G. Dias, M. H. Godinho and F. V. de Abreu, Sci. Rep., 2016, 6, 23413 CrossRef CAS PubMed.
- Y. Cao, T. Y. Tan, D. M. Walba, N. A. Clark, G. Ungar, C. H. Zhu, L. Zhang and F. Liu, Nano Lett., 2022, 22, 4569–4575 CrossRef CAS PubMed.
- H. Kim, S. H. Ryu, M. Tuchband, T. J. Shin, E. Korblova, D. M. Walba, N. A. Clark and D. K. Yoon, Sci. Adv., 2017, 3, e1602102 CrossRef PubMed.
- C. H. Zhu, C. Wang, A. Young, F. Liu, I. Gunkel, D. Chen, D. Walba, J. Maclennan, N. Clark and A. Hexemer, Nano Lett., 2015, 15, 3420–3424 CrossRef CAS PubMed.
- D. Chen, M. R. Tuchband, B. Horanyi, E. Korblova, D. M. Walba, M. A. Glaser, J. E. Maclennan and N. A. Clark, Nat. Commun., 2015, 6, 7763 CrossRef PubMed.
- R. A. Callahan, D. C. Coffey, D. Chen, N. A. Clark, G. Rumbles and D. M. Walba, ACS Appl. Mater. Interfaces, 2014, 6, 4823–4830 CrossRef CAS PubMed.
- D. Chen, C. H. Zhu, H. T. Wang, J. E. Maclennan, M. A. Glaser, E. Korblova, D. M. Walba, J. A. Rego, E. A. Soto-Bustamante and N. A. Clark, Soft Matter, 2013, 9, 462–471 RSC.
- J. Liu, Y. Molard, M. E. Prévôt and T. Hegmann, ACS Appl. Mater. Interfaces, 2022, 14, 29398–29411 CrossRef CAS PubMed.
- W. Lewandowski, N. Vaupotic, D. Pociecha, E. Gorecka and L. M. Liz-Marzán, Adv. Mater., 2020, 32, 1905591 CrossRef CAS PubMed.
- M. Baginski, M. Tupikowska, G. Gonzalez-Rubio, M. Wojcik and W. Lewandowski, Adv. Mater., 2020, 32, 1904581 CrossRef CAS PubMed.
- S. Parzyszek, J. Tessarolo, A. Pedrazo-Tardaos, A. M. Ortuño, M. Bagiñski, S. Bals, G. H. Clever and W. Lewandowski, ACS Nano, 2022, 16, 18472–18482 CrossRef CAS PubMed.
- H. Kim, Y. Yi, D. Chen, E. Korblova, D. M. Walba, N. A. Clark and D. K. Yoon, Soft Matter, 2013, 9, 2793–2797 RSC.
- G. Park, H. Park, J. M. Wolska, J. G. Park and D. K. Yoon, Mater. Horiz., 2022, 9, 2542–2550 RSC.
- J. Liu, S. Shadpour, A. Nemati, M. E. Prévôt, E. Hegmann, C. Zhu and T. Hegmann, Liq. Cryst., 2021, 48, 1129–1139 CrossRef CAS.
- X. Lin, Q. Li, Y. Tang, Z. Chen, R. Chen, Y. Sun, W. Lin, G. Yi and Q. Li, Adv. Sci., 2024, 11, 2401983 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis details; analytical data; DSC data; additional SEM and TEM images. See DOI: https://doi.org/10.1039/d4mh01243g |
| This journal is © The Royal Society of Chemistry 2024 |