
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Atomistic simulations of mechanically activated reactions for oxygen release from polymers†
José
Cobeña-Reyes
,
Fakhrul H.
Bhuiyan
 and
Ashlie
Martini
*
and
Ashlie
Martini
*
Department of Mechanical Engineering, University of California Merced, USA. E-mail: amartini@ucmerced.edu
First published on 7th June 2024
Abstract
Singlet oxygen molecules are useful in several therapeutic applications involving photo-activated release of oxygen from carrier molecules toward targeted cells. However, the drawbacks of existing photo-activated methods encourage the development of alternatives, particularly polymer mechanophores that act as oxygen carriers. Here, we present a reactive molecular dynamics simulation-based study of an endoperoxide-based polymer for which oxygen release can be activated either thermally or mechanochemically. Simulations of the polymers heated are compared to simulations of the polymers subject to compression and shear at room temperature. Results show that oxygen release is preceded by deformation of the anthracene ring in both thermal and mechanochemical reactions. However, in the mechanically activated reaction, this deformation is imposed directly by chemical bonding between the oxygen and atoms in the shearing surfaces, eliminating the need for high temperature to initiate the oxygen release. These results could be useful in the development of alternative therapeutic protocols that do not rely on photo-activated reactions.
Introduction
The release of singlet oxygen molecules from oxygen carriers is a desired process in certain medical treatments. For instance, some cancer therapies require the release of singlet oxygen molecules near the tumor to form hydroxyl radicals that will eventually cause apoptosis on the cancer cells.1–5 A common therapy is photo-activated oxygen release, where the oxygen molecule is released upon molecular excitation under ultra violet light. However, disadvantages of photo-activated methods include the persistence of the photosensitizer molecules in the body after treatment and low yield in the oxygen production.5 Hence, the development of molecules that can release oxygen by methods other than photo-activation is highly desired. A promising alternative is the mechanochemical activation of molecules, i.e., mechanophores, that undergo chemical transformation in response to mechanical force.6Polymeric mechanophores are of particular interest because such mechanophores have shown the potential to release oxygen under mechanical force. For instance, multi-functional polymer-based micelles that release oxygen by sonication have been used to increase the concentration of hydroxyl radicals next to malignant cells.1 Other examples include polymers that contain an anthracene-endoperoxide (EPO) functional group. EPO-based polymers contain oxygen atoms in the peroxide group (O–O) loosely attached to the anthracene functional group that is part of the polymer backbone.7 It has been shown that the O–O pairs can be released by the force applied to the polymers during ball milling.2
Polymeric mechanophores are usually chemically modified, where a moiety that has shown mechanochemical activity is added to the structure as one or more of the monomer units.8 However, the reaction mechanisms of such mechanophores are still not fully understood.9,10 Exploring mechanochemical reaction mechanisms is challenging because a polymer can go through different reaction pathways and form different products depending on the method used to trigger the reaction. For instance, EPO polymeric mechanophores go through a cycloreversion reaction that is not observed when the reaction is attempted using solvent and temperature.6,11 Specifically, in a ball mill, the cycloreversion reaction of EPO-based polymers generates a singlet oxygen molecule and another double bond in the original ring. It was suggested that this reaction is primarily driven by the shear force exerted by the ball mill.6 Additionally, even within mechanochemical methods, the instrument used potentially affects the kinetics of the reaction. For instance, the rate of the cycloreversion reaction of EPO-based polymers can be increased by switching from a ball mill to a ring-and-puck mill.12 Efforts to determine reaction pathways13 and the effects of different moieties on the mechanochemical activity in polymers are currently underway.14 Understanding the mechanisms by which a polymer can be mechanochemically activated to release singlet oxygen is important for the development of potential cancer therapies.15 However, understanding these effects is challenging using only experimental tools.
Computational techniques such as molecular dynamics (MD) simulations and density functional theory (DFT) have been used to understand the mechanochemically driven cycloreversion reaction mechanisms that can lead to the release of molecules from EPO polymers. MD simulations with reactive force fields have been used to study cycloreversion reactions in polymers, where the bond scission happens in one of the aromatic rings in the backbone chain.16 Reactive MD simulations have also shown that the bond scission in cycloreversion reactions greatly depends on the direction of pulling force on the targeted atoms in a polymer.16 DFT methods have been used to study the effect of compression on cross-linked polymers and the release of groups of atoms not in the polymer backbone by a covalent bond scission.17 More recently, DFT was used to study the force dependence of energetic barriers in aromatic molecules, such as anthracene cycloadducts, that undergo cycloreversion reactions when subject to shear force.18
Previous work has shown that computational tools can be useful for investigating the mechanisms of mechanochemical reactions of EPO polymers. However, the thermal and mechanical paths leading to the release of oxygen are not completely understood, which impedes the development of new polymers that might be used in medical applications. In this study, we used reactive MD simulations to study the mechanisms of oxygen release for a polymethacrylate-endoperoxide (PMA-EPO) polymer activated either thermally or mechanically. Two sets of dynamic simulations were run, one with the polymers subject to heating and another with the polymers sheared between two graphite walls. The rate of oxygen release was tracked in both sets of simulations and then individual reactions were analyzed to identify the corresponding reaction pathways. Guided by visual observation of the pathways that identified molecular deformation as a key step in the process, deformation was quantified and compared between the thermal and mechanochemical reactions. Results provide insight into how mechanical force, particularly shear force, can directly activate oxygen release reactions from EPO polymers.
Methods
Reactive MD simulations were performed with the ReaxFF19 potential, with a parameter set20 that was previously developed to add a low-gradient correction to the original ReaxFF19 for long-range London Dispersion. This force field has been used to model polymeric reactions in previous studies.21 Two sets of MD simulations of the PMA-EPO were conducted, heating and shearing. The first set was used to determine the onset temperature at which the oxygen release occurred in a thermal reaction; the second set was used to investigate the effects of pressure and shear on bond breaking in the polymer. The polymers and shearing surfaces were created in Schrödinger Maestro v. 2022–3,22 using the polymer builder and the nanosheets modules, respectively.Fig. 1 shows the PMA-EPO monomer, a representative polymer, and the model system in heating simulations. The heating model comprised a periodic box with five 5-monomer PMA-EPO chains. After a minimization step, the system was equilibrated for 500 ps in the NVT ensemble at 300 K followed by an additional 500 ps in the NPT ensemble at 1 atm. The Nosé-Hoover thermostat and barostat were used. The initial system density was 1.37 g cm−3 and the density after thermal equilibration was 1.23 g cm−3. Finally, the temperature was increased from 300 K to 1200 K at a rate of 0.01 K fs−1 in the NPT ensemble.
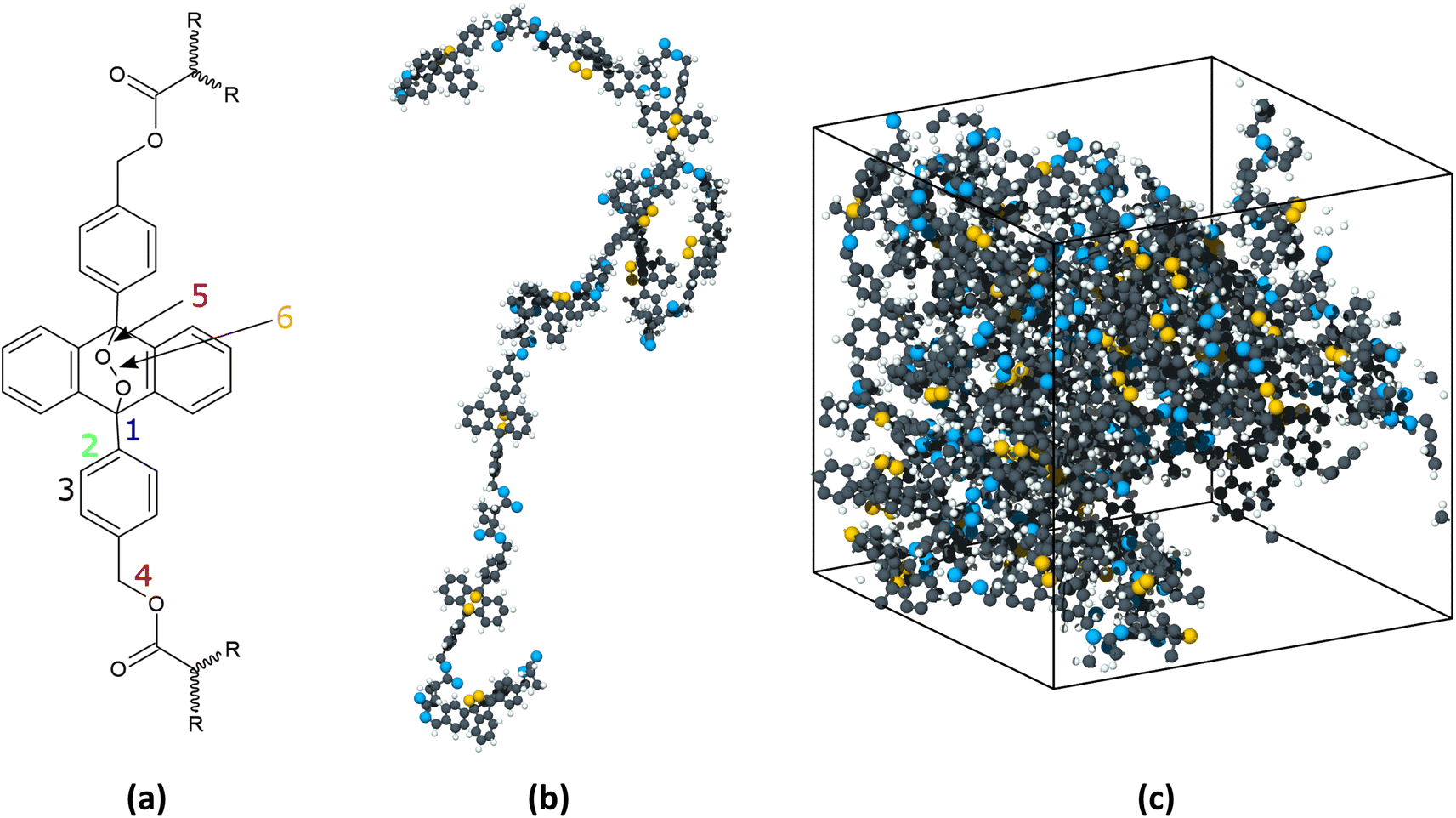 | ||
| Fig. 1 (a) Schematic of the PMA-EPO monomer where select bonds relevant to the reaction are numbered to facilitate discussion in the text. (b) Snapshot of one polymer used in the simulations. (c) Snapshot of the simulation box for the thermal reactions. The white and black spheres represent hydrogen and carbon atoms, respectively. The blue and gold spheres represent oxygen atoms, where the gold color is used to identify the atoms that will be released as oxygen molecules. | ||
For the shearing simulations, the model system consisted of five 5-monomer PMA-EPO chains confined between rigid graphene sheets (Fig. S1†). The choice of graphene was made due to its simplicity and relative inertness,23 while still being capable of bonding with polymers.24 The initial dimensions of the simulation box were 15 nm × 15 nm in the directions parallel to the graphene basal plane and 7 nm in the surface-normal direction. The top graphene sheet was treated as a rigid body and the positions of the atoms in the bottom sheets were fixed. The endmost monomer in each polymer chain was bonded to the upper of the two bottom graphene sheets. Specifically, the non-cyclic C atom from bond 4 and the non-peroxide O atom double bonded to the linear chain in one monomer (see Fig. 1a) were placed close enough to the lower graphene sheet such a chemical bond was formed per the ReaxFF potential.
Minimization and equilibration in the NVT ensemble followed the same settings as in the heating simulations. Next, a pressure of 1 GPa was applied to the topmost graphene sheet. Pressure was applied for 250 ps, during which time no bonds formed or broke. Then, the top graphene layer was moved laterally with a sliding speed of 10 m s−1. The high pressure and fast speed were selected to accelerate chemical processes such that a sufficient number of reactions occurred within the short timescale of the simulations. The sliding continued for 400 ps.
In all the simulations, the timestep was set to 0.25 fs. Simulations were run using the LAMMPS package,25 and the visualization of trajectories and structures was conducted using Ovito.26 Three realizations were conducted for both the heating and shearing simulations. The number of oxygen molecules was tracked during the simulations using the bond orders from the ReaxFF potential.
Results and discussion
Fig. 2 shows the number of oxygen molecules completely separated from the polymer chains in the heating and sliding simulations. Snapshots of the simulations highlighting the release of oxygen molecules are shown in Fig. S2.† In the heating simulation, the onset of oxygen release occurred at ∼800 K and then the number of oxygen molecules increased approximately linearly with the linear temperature increase, as shown by Fig. 2a. In contrast, the results from the sliding simulations in Fig. 2b show that oxygen release can occur at room temperature when mechanical force is present. Further, since no oxygen molecules were released until sliding began, these results show that shear stress is the key driver of the reactions, consistent with observations in previous studies.27–29 By the end of either the heating or sliding simulations, approximately 15 oxygen molecules (50%) were released. The reaction pathways leading to this release were then analyzed to understand the thermal and mechanochemical mechanisms.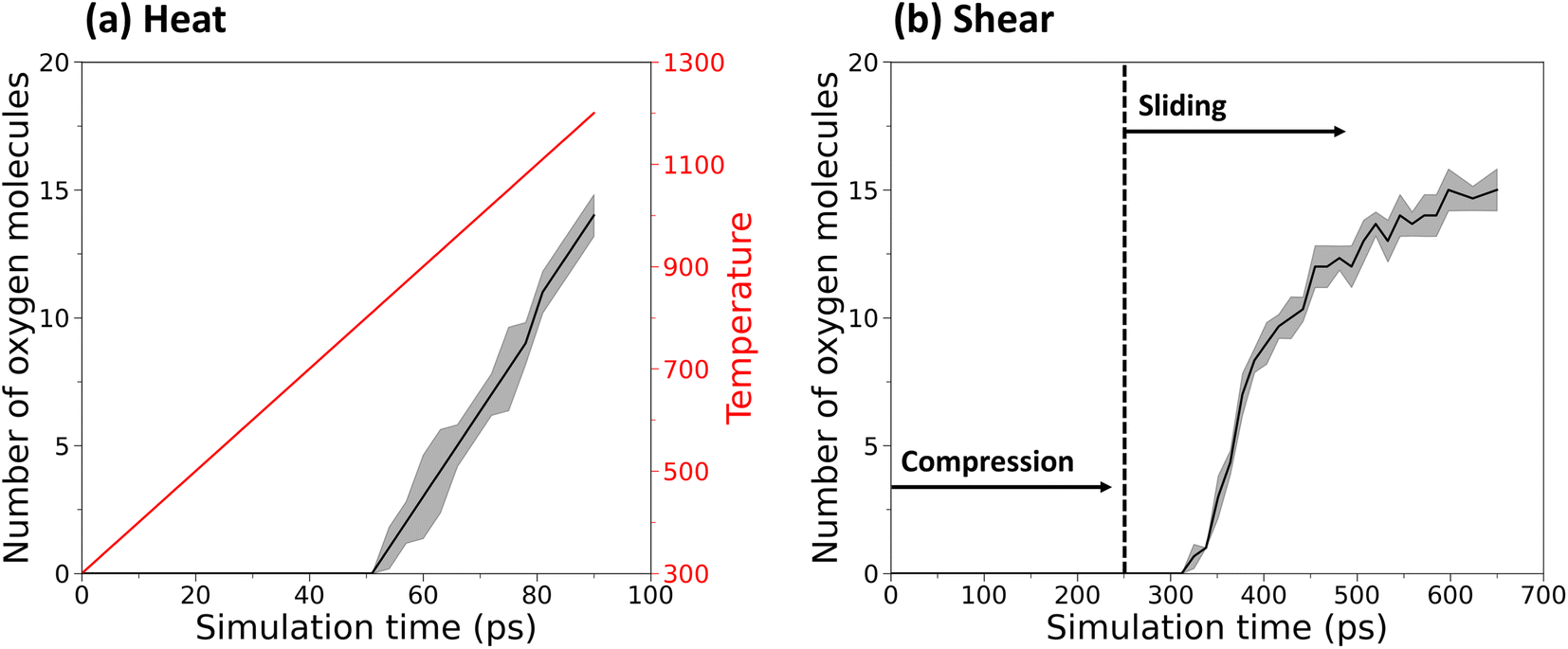 | ||
| Fig. 2 (a) Number of oxygen molecules (left y-axis, black) and system temperature (right y-axis, red) as a function of simulation time from the heating simulations. (b) Number of oxygen molecules as a function of simulation time from the compression and sliding simulations, where the onset of sliding is identified by a dashed vertical line. In both cases, the number is out of a total possible 30 molecules. The solid black lines in both plots represent the average of three independent simulations and the shaded gray regions reflect the standard deviation. | ||
The thermal pathway was tracked and found to involve three steps, depicted in Fig. 3a–c. First, the undeformed anthracene group of the monomer (Fig. 3a) received thermal energy during the heating process. With increasing thermal energy, the central ring of the anthracene group started to deform until one of the C–O bonds broke (Fig. 3b). This ring deformation continued as the temperature increased. Finally, the second C–O bond broke resulting in the formation of a double bond in the middle ring, producing a planar structure, and the oxygen molecule was completely released (Fig. 3c). Relative energies for these bonds from a DFT-based relaxed coordinate scan are shown in Fig. S3.† The double bond formation and structural changes observed in the central anthracene ring after the reaction are consistent with previous research on mechanochemical oxygen release from anthracene-endoperoxide-based mechanophore in ball milling.6
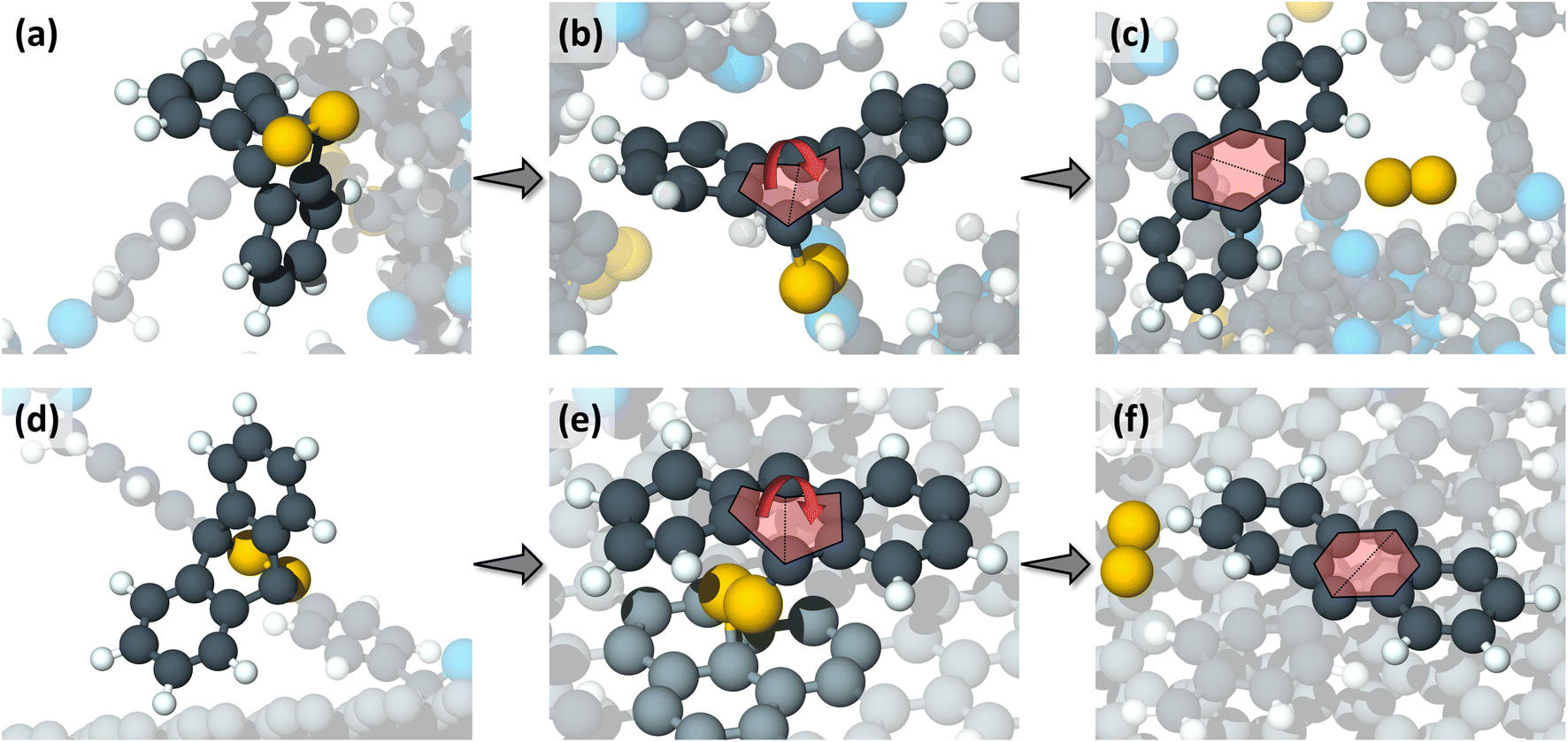 | ||
| Fig. 3 Close-up snapshots of representative reactions through the thermally driven pathway (a–c), and the shear-driven pathway (d–f). (a) The undeformed anthracene rings hold the peroxide oxygen atoms (in gold color) before undergoing any thermal reactions. (b) One of the two C–O bonds breaks due to thermally induced deformation in the anthracene rings. The deformation here was a flattening of the central anthracene ring as shown by the red arrow. (c) Finally, the second C–O bond breaks, the central anthracene ring becomes planar, and the oxygen molecule is released. (d) Undeformed anthracene rings before shear-driven reactions. (e) A graphite carbon atom (in light grey color) bonds with one of the oxygen causing the central anthracene ring to flatten and leading to the first anthracene C–O bond scission. (f) The second oxygen atom breaks free from the anthracene rings due to the sliding motion and a free oxygen molecule is released. The colors of the atoms are the same as in Fig. 1. | ||
The reaction mechanism observed in the shearing simulations also consisted of three steps. First, the undeformed anthracene group (Fig. 3d) approached the upper graphene layer. Then, as the sliding process continued, a C atom from the graphene layer bonded with one of the peroxide O atoms causing deformation of the central anthracene ring, eventually leading to the first C–O scission in the central anthracene ring (Fig. 3e). In the last step, the second O broke from the anthracene group due to the motion of the graphene layer and the two peroxide oxygen atoms were released as a free oxygen molecule (Fig. 3f). Similar to the thermal reaction pathway, a double bond formed at the central anthracene ring after the oxygen release, and the anthracene ring became planar (Fig. 3f).
The evolution of the reaction was tracked quantitatively for both thermally activated and shear-activated reactions in terms of the bond order of the C–O (bond 5) and O–O bonds (bond 6). Fig. 4 shows these bond orders averaged over all molecules that underwent the reaction. The data is plotted vs. reaction onset time where the reaction onset is identified as the point in time when the C–O bond is completely dissociated. Using reaction onset time for this analysis, as opposed to simulation time, is advantageous since the reactions proceeded at different times for the different molecules. Dissociation of the C–O bond in Fig. 4 is identified by the point where the C–O bond order becomes zero. The bond order is calculated by the ReaxFF simulation and is around one for single bonds and around two for double bonds. Therefore, as shown in Fig. 4, the order of the C–O bonds decreases from 0.8 to 0 as those bonds dissociate, and the order of the O–O bonds increases from 0.8 to about 1.6 as the reaction proceeds and an oxygen molecule is released. This occurs in both the heating and sliding simulations.
 | ||
| Fig. 4 Evolution of the average angle of the anthracene group (left y-axes) and the bond orders of the C–O and O–O bonds (right y-axes) in (a) heating simulations and (b) shearing simulations. The data is plotted vs. reaction coordinate, where reaction onset is defined as the coordinate at which the C–O bond order reaches zero for each molecule. The symbols and connecting solid lines in both plots represent the average of three independent simulations and the shaded regions reflect the standard deviation. | ||
Next, based on the visual observation of deformation prior to reaction as seen in Fig. 3, we tracked the angle at the central anthracene ring (angle definition in Fig. S4†). As shown in Fig. 4, prior to reaction, this angle is 120°. Then, as the C–O bond order decreases (the bond becomes weaker), the ring angle increases. Finally, at the end of the reaction, the angle is 180° such that the middle ring has a planar structure, and the oxygen molecule is released.
Fig. 4 confirms that the deformation of the monomer precedes the oxygen release reaction in both the heating and shearing simulations. The rate of change of the angle with reaction onset time cannot be compared between the two types of simulations since the temperature is linearly increasing during heating whereas the shearing simulation involves compression and then steady sliding. However, the order of events in the reaction appears to differ between the thermally and mechanochemically activated reactions. Specifically, in Fig. 4a, the C–O bond order starts to decrease before the angle has increased much from its initial value of 120°. In contrast, in Fig. 4b, the angle increases rapidly and is already larger than 160° when the C–O bond order begins decreasing. This suggests that molecular deformation is an effect of the thermally activated reaction but the initiator of the mechanochemical reaction.
Bond and molecule deformation has been observed in other mechanochemical reactions.10,30,31 Specifically with respect to reactions activated by shear force in a sliding interface, it has been reported that reactant species bond with the surfaces such mechanical energy is transmitted to the reactants as the surfaces move relative to one another.28,32,33 This is observed here as bond formation between the oxygen in the polymers and carbon atoms in the graphite (Fig. 3e) which directly causes deformation of the central anthracene ring (Fig. 4b). Importantly, the mechanochemical reaction proceeds at 300 K, in contrast with the thermal reaction that did not start until 800 K. These results demonstrate that molecular deformation and intermediate steps that involve the substrate enable the reaction to proceed at a lower temperature than the thermally driven reaction.
Conclusions
Reactive molecular dynamics simulations showed that oxygen release from a polymethacrylate-endoperoxide polymer can be activated either thermally or mechanochemically. Both reactions involved deformation of the anthracene holding the peroxide oxygen atoms. However, tracking the strength of the individual bonds and angles in the simulations suggested that deformation was a result of the thermal reaction and the cause of the mechanochemical reaction. This is consistent with the observation of chemical bonding between the reactant and the sliding surface, enabling transmission of shear force that directly deformed the monomer which led to bond dissociation. This direct activation by mechanical force enabled reactions that required high temperatures in thermal conditions to proceed at room temperature under shear. Although this study included just one type of polymer and simple graphite surfaces, the simulation methodology could be extended to other molecules and surfaces as a means to characterizing their mechanochemical susceptibility and understanding the role of deformation more generally. We hope these findings help in the design of polymeric mechanophores that can be used as oxygen carriers for therapeutic applications.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We thank Dr Sourabh Kumar for feedback on the DFT calculations. This work was supported by the National Science Foundation (NSF) Center for the Mechanical Control of Chemistry (CMCC), CHE-2303044. The CMCC is part of the NSF Centers for Chemical Innovation Program.References
- Y. Li, Y. Qin, Y. Shang, Y. Li, F. Liu, J. Luo, J. Zhu, X. Guo, Z. Wang and Y. Zhao, Mechano-Responsive Leapfrog Micelles Enable Interactive Apoptotic and Ferroptotic Cancer Therapy, Adv. Funct. Mater., 2022, 32, 2112000 Search PubMed.
- A. Turksoy, D. Yildiz, S. Aydonat, T. Beduk, M. Canyurt, B. Baytekin and E. U. Akkaya, Mechanochemical generation of singlet oxygen, RSC Adv., 2020, 10, 9182–9186 Search PubMed.
- T. Huang, M. Zhao, Q. Yu, Z. Feng, M. Xie, S. Liu, K. Y. Zhang, Q. Zhao and W. Huang, De Novo Design of Polymeric Carrier to Photothermally Release Singlet Oxygen for Hypoxic Tumor Treatment, Research, 2019, 9269081 Search PubMed.
- J. Liu, G. Yang, W. Zhu, Z. Dong, Y. Yang, Y. Chao and Z. Liu, Light-controlled drug release from singlet-oxygen sensitive nanoscale coordination polymers enabling cancer combination therapy, Biomaterials, 2017, 146, 40–48 Search PubMed.
- S. Martins, J. P. S. Farinha, C. Baleizão and M. N. Berberan-Santos, Controlled release of singlet oxygen using diphenylanthracene functionalized polymer nanoparticles, Chem. Commun., 2014, 50, 3317–3320 Search PubMed.
- J. G. Hernández, Polymer and small molecule mechanochemistry: closer than ever, Beilstein J. Org. Chem., 2022, 18, 1225–1235 Search PubMed.
- V. Brega, Y. Yan and S. W. Thomas, Acenes beyond organic electronics: sensing of singlet oxygen and stimuli-responsive materials, Org. Biomol. Chem., 2020, 18, 9191–9209 Search PubMed.
- N. Deneke, M. L. Rencheck and C. S. Davis, An engineer's introduction to mechanophores, Soft Matter, 2020, 16, 6230–6252 Search PubMed.
- G. Kim, Q. Wu, J. L. Chu, E. J. Smith, M. L. Oelze, J. S. Moore and K. C. Li, Ultrasound controlled mechanophore activation in hydrogels for cancer therapy, Proc. Natl. Acad. Sci., 2022, 119, e2109791119 Search PubMed.
- H. L. Adams, M. T. Garvey, U. S. Ramasamy, Z. Ye, A. Martini and W. T. Tysoe, Shear-induced mechanochemistry: pushing molecules around, J. Phys. Chem. C, 2015, 119, 7115–7123 Search PubMed.
- Y. Tian, X. Cao, X. Li, H. Zhang, C.-L. Sun, Y. Xu, W. Weng, W. Zhang and R. Boulatov, A polymer with mechanochemically active hidden length, J. Am. Chem. Soc., 2020, 142, 18687–18697 Search PubMed.
- B. A. Versaw, T. Zeng, X. Hu and M. J. Robb, Harnessing the power of force: development of mechanophores for molecular release, J. Am. Chem. Soc., 2021, 143, 21461–21473 Search PubMed.
- M. Stratigaki and R. Göstl, Methods for exerting and sensing force in polymer materials using mechanophores, ChemPlusChem, 2020, 85, 1095–1103 Search PubMed.
- D. E. Martínez-Tong, J. A. Pomposo and E. Verde-Sesto, Triggering Forces at the Nanoscale: Technologies for Single-Chain Mechanical Activation and Manipulation, Macromol. Rapid Commun., 2021, 42, 2000654 Search PubMed.
- S. A. Salim, T. A. Salaheldin, M. M. Elmazar, A. Abdel-Aziz and E. A. Kamoun, Smart biomaterials for enhancing cancer therapy by overcoming tumor hypoxia: a review, RSC Adv., 2022, 12, 33835–33851 Search PubMed.
- T. Stauch and A. Dreuw, Advances in quantum mechanochemistry: electronic structure methods and force analysis, Chem. Rev., 2016, 116, 14137–14180 Search PubMed.
- M. B. Larsen and A. J. Boydston, “Flex-activated” mechanophores: Using polymer mechanochemistry to direct bond bending activation, J. Am. Chem. Soc., 2013, 135, 8189–8192 Search PubMed.
- M. Walter, D. Linsler, T. König, C. Gäbert, S. Reinicke, M. Moseler and L. Mayrhofer, Mechanochemical activation of anthracene [4+ 4] cycloadducts, J. Phys. Chem. Lett., 2023, 14, 1445–1451 Search PubMed.
- A. C. Van Duin, S. Dasgupta, F. Lorant and W. A. Goddard, ReaxFF: a reactive force field for hydrocarbons, J. Phys. Chem. A, 2001, 105, 9396–9409 Search PubMed.
- L. Liu, Y. Liu, S. V. Zybin, H. Sun, I. I. I. Goddard and W. A. ReaxFF-lg, Correction of the ReaxFF reactive force field for London dispersion, with applications to the equations of state for energetic materials, J. Phys. Chem. A, 2011, 115, 11016–11022 Search PubMed.
- F. Avilés, Thermoresistivity of Carbon Nanostructures and their Polymeric Nanocomposites, Adv. Mater. Interfaces, 2023, 2300218 Search PubMed.
- K. J. Bowers, E. Chow, H. Xu, R. O. Dror, M. P. Eastwood, B. A. Gregersen, J. L. Klepeis, I. Kolossvary, M. A. Moraes, F. D. Sacerdoti, et al., Scalable algorithms for molecular dynamics simulations on commodity clusters, Proceedings of the 2006 ACM/IEEE Conference on Supercomputing, 2006, p. 84 Search PubMed.
- L. Liao, H. Peng and Z. Liu, Chemistry makes graphene beyond graphene, J. Am. Chem. Soc., 2014, 136, 12194–12200 Search PubMed.
- G. Guo and Y. Zhu, Cohesive-shear-lag modeling of interfacial stress transfer between a monolayer graphene and a polymer substrate, J. Appl. Mech., 2015, 82, 031005 Search PubMed.
- A. P. Thompson, H. M. Aktulga, R. Berger, D. S. Bolintineanu, W. M. Brown, P. S. Crozier, P. J. in’t Veld, A. Kohlmeyer, S. G. Moore and T. D. Nguyen, et al., LAMMPS – a flexible simulation tool for particle-based materials modeling at the atomic, meso, and continuum scales, Comput. Phys. Commun., 2022, 271, 108171 Search PubMed.
- A. Stukowski, Visualization and analysis of atomistic simulation data with OVITO–the Open Visualization Tool, Modell. Simul. Mater. Sci. Eng., 2009, 18, 015012 Search PubMed.
- F. H. Bhuiyan, S. H. Kim and A. Martini, Reactive molecular dynamics simulations of thermal and shear-driven oligomerization, Appl. Surf. Sci., 2022, 591, 153209 Search PubMed.
- F. H. Bhuiyan, Y.-S. Li, S. H. Kim and A. Martini, Shear-activated chemisorption and association of cyclic organic molecules, Faraday Discuss., 2023, 241, 194–205 Search PubMed.
- J. Zhang and H. Spikes, On the mechanism of ZDDP antiwear film formation, Tribol. Lett., 2016, 63, 1–15 Search PubMed.
- A. Vashisth, S. Khatri, S. H. Hahn, W. Zhang, A. C. Van Duin and M. Naraghi, Mechanical size effects of amorphous polymer-derived ceramics at the nanoscale: experiments and ReaxFF simulations, Nanoscale, 2019, 11, 7447–7456 Search PubMed.
- Y. S. Zholdassov, L. Yuan, S. R. Garcia, R. W. Kwok, A. Boscoboinik, D. J. Valles, M. Marianski, A. Martini, R. W. Carpick and A. B. Braunschweig, Acceleration of Diels-Alder reactions by mechanical distortion, Science, 2023, 380, 1053–1058 Search PubMed.
- A. Khajeh, X. He, J. Yeon, S. H. Kim and A. Martini, Mechanochemical association reaction of interfacial molecules driven by shear, Langmuir, 2018, 34, 5971–5977 Search PubMed.
- A. Martini and S. H. Kim, Activation volume in shear-driven chemical reactions, Tribol. Lett., 2021, 69, 1–14 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4mr00004h |
| This journal is © The Royal Society of Chemistry 2024 |