
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceProtecting-group-free mechanosynthesis of amides from hydroxycarboxylic acids: application to the synthesis of imatinib†
Tatsiana
Nikonovich
 a,
Tatsiana
Jarg
a,
Jevgenija
Martõnova
a,
Artjom
Kudrjašov
ab,
Danylo
Merzhyievskyi
ac,
Marina
Kudrjašova
a,
Fabrice
Gallou
d,
Riina
Aav
*a and
Dzmitry
Kananovich
*a
a,
Tatsiana
Jarg
a,
Jevgenija
Martõnova
a,
Artjom
Kudrjašov
ab,
Danylo
Merzhyievskyi
ac,
Marina
Kudrjašova
a,
Fabrice
Gallou
d,
Riina
Aav
*a and
Dzmitry
Kananovich
*a
aDepartment of Chemistry and Biotechnology, Tallinn University of Technology, Akadeemia tee 15, 12618, Tallinn, Estonia. E-mail: riina.aav@taltech.ee; dzmitry.kananovich@taltech.ee
bTallinna Tõnismäe Reaalkool, Pärnu mnt 50, Tallinn, Estonia
cDepartment of Chemistry of Bioactive Nitrogen-containing Heterocyclic Bases, V. P. Kukhar Institute of Bioorganic Chemistry and Petrochemistry, National Academy of Sciences of Ukraine, Academician Kukhar Str. 1, 02094 Kyiv, Ukraine
dNovartis Pharma AG, Novartis Campus, 4056 Basel, Switzerland
First published on 12th March 2024
Abstract
Despite considerable advancements in mechanochemical amide couplings, there is a paucity of studies addressing chemoselective issues in these transformations, such as the tolerance of unmasked hydroxyl groups. In view of the high practical significance of amide coupling reactions in the synthesis of active pharmaceutical ingredients (APIs), we aimed to investigate the tolerance of unprotected hydroxyls in carboxylic acids towards various reported mechanochemical amide coupling conditions. 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (EDC·HCl) in combination with ethyl acetate as a liquid-assisted grinding (LAG) additive was revealed as the most selective amide coupling system that delivers 76–94% yields of amides from a range of hydroxycarboxylic acids, including N-Boc-protected amino acids serine and tyrosine. The EDC-mediated amide coupling protocol was employed in the synthesis of imatinib, an anticancer drug included in the World Health Organization's List of Essential Medicines. The target API was synthesized in an overall 86% yield and 99% HPLC purity through a two-step mechanochemical C–N bond assembling reaction sequence starting from 4-(hydroxymethyl)benzoic acid.
Introduction
Mechanochemistry has shown great potential as a powerful tool for advancing the sustainability goals in the synthesis of active pharmaceutical ingredients (APIs).1–5 Focusing on the applications of mechanochemical C–N bond forming reactions,6,7 our objective was to develop a mechanochemical method for the synthesis of imatinib, an anti-cancer drug included in the World Health Organization's List of Essential Medicines.8 The planned synthetic route involved the amide coupling of 4-(hydroxymethyl)benzoic acid (1), followed by a subsequent nucleophilic substitution reaction at the benzylic hydroxyl (Scheme 1). In our previous work,7 we noticed that amide coupling of 1 with N-Boc piperazine mediated by the TCFH reagent (chloro-N,N,N′,N′-tetramethylformamidinium hexafluorophosphate) did not affect the hydroxyl group of 1. However, regarding the synthesis of imatinib, we were uncertain whether using a poorly nucleophilic aromatic amine would be equally successful, considering that competitive esterification reactions can be facilitated by common coupling reagents.9–15 While numerous mechanochemical amide coupling protocols have been reported,6,16–25 limited attention has been given to studying the chemoselectivity issues in these transformations. In particular, the amide bond formation in the presence of a free hydroxyl group in starting materials has not been systematically explored, with only sporadic instances of such reactions reported.7,26–30 As a prerequisite for the synthesis of imatinib, we aimed to perform a systematic screening of previously reported protocols6,16,17,21 on a model amide coupling reaction of 1. The goal was to identify a coupling reagent with the best tolerance to the unmasked hydroxyl functionality of 1 and to outline the limits of such tolerance by exploring amines with varying nucleophilicities. | ||
| Scheme 1 Outline of the work. | ||
The study identified 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide hydrochloride (EDC·HCl) as the most efficient amide coupler for 1 and other examples of hydroxycarboxylic acids. Subsequently, we demonstrate the application of the established chemoselective amide coupling methodology in the two-step synthesis of imatinib 2.
Results and discussion
The mechanochemical amidation protocols have been screened for the model coupling reaction of hydroxycarboxylic acid 1 with 4-bromo-3-methylaniline 3 as a poorly nucleophilic aromatic amine31 (Table 1).|
|
|||
|---|---|---|---|
| Entry | Coupling reagent/base | LAG additive, η (μL mg−1) | Yield of 4b, % |
| a General conditions: acid 1 (0.66 mmol, 100 mg), amine 3 (0.9–1 equiv.), coupling reagent (1–1.1 equiv.), base (0.85–3 equiv.), LAG additive (η = 0.19–0.25 μL mg−1), and ball milling at 30 Hz for 60 min (see the ESI for the details). b Yields are determined by 1H NMR with an internal standard (1,3,5-trimethoxybenzene). c Guanidinium derivative 3a was formed by the reaction of TCFH with 3. d Guanidinium derivative 3b was formed as a by-product. e Ester by-products resulting from self-condensation of 1 were observed. f The reaction was performed with ethyl ester of 1. g Almost no reaction: starting materials with a trace of unidentified by-products. h Anhydride 1a was formed in a reaction without amine 3. | |||
| 1 | TCFH/K2HPO4 | EtOAc (0.19) | 26c |
| 2 | COMU/K2HPO4 | EtOAc (0.19) | 83d,e |
| 3 | TCFH/NMI | Without | 74c |
| 4 | CDI | Without | 10e |
| 5 | t-BuOK | Without | 0f,g |
| 6 | EDC·HCl/DMAP | CH3NO2 (0.25) | 0g |
| 7 | EDC·HCl | EtOAc (0.25) | 90h |
| Structures of the reagents and identified by-products: | |||

|
|||
| NMI = 1-methylimidazole, CDI = carbonyldiimidazole. | |||

Initially, we expected that free hydroxyl of 1 could act as a competitive nucleophile,9–15 thereby leading to the formation of ester by-products. However, we observed a variety of side processes, the prevalence of which was influenced by the specific coupling conditions employed. The combination of TCFH with K2HPO4 (Table 1, entry 1)7 afforded amide 4 in low 26% yield due to a competitive reaction between TCFH and amine 3 with the formation of a guanidinium derivative 3a. 1,1′-Carbonyldiimidazole (CDI, entry 4)16 demonstrated a low 10% yield of 4 since the process was accompanied by self-condensation of 1 that delivered ester-type by-products. Several other reported methodologies17,21 failed to provide the amide product at all. The highest yield of 4 (90% by 1H NMR) was attained with the EDC·HCl reagent (entry 7) and ethyl acetate as a green LAG additive.32 The use of EDC·HCl for the mechanochemical amide coupling in combination with nitromethane as a LAG additive was first reported by Štrukil and co-workers,21 which was followed by several examples of peptide couplings, where nitromethane was replaced by ethyl acetate.22,33 In our case, ethyl acetate demonstrated comparable efficacy to nitromethane and sulfolane as LAG additives (Table S1, entries 1–3 in the ESI†). This is particularly important from a safety perspective, as it is essential to avoid the use of hazardous nitromethane32 under mechanochemical conditions. Pure amide 4 was isolated in 89% yield by following an operationally simple work-up protocol, which involved treatment with water, filtration and drying.
The results demonstrate that the amide coupling is generally more favoured with many of the tested reagent systems compared to the ester capping of the hydroxyl group. To further clarify the respective chemoselectivity concerns, certain amide coupling conditions were also tested for producing ester 5 through the reaction of 4 with 1 (Table 2). Both COMU/K2HPO4 and TCFH/NMI systems afforded 5 at moderate yields of 31% and 40%, respectively (Table 2, entries 1 and 2). However, using TCFH/K2HPO4 or EDC·HCl (entries 3 and 4) resulted in only trace amounts of 5, with the predominant process being the generation of anhydride 1a.
|
|
|||
|---|---|---|---|
| Entry | Coupling reagent/base | LAG additive, η (μL mg−1) | Yield of 5b, % |
| a General conditions: amide 4 (0.19 mmol, 60 mg), acid 1 (1 equiv.), coupling reagent (1–1.1 equiv.), base (3 equiv.), LAG additive (η = 0.19–0.25 μL mg−1), and ball milling at 30 Hz for 60 min. b Yields are determined by 1H NMR. c Anhydride 1a was formed in 70% yield. d Starting materials are left and anhydride 1a was formed in 30% yield. | |||
| 1 | COMU/K2HPO4 | EtOAc (0.19) | 31 |
| 2 | TCFH/NMI | Without | 40 |
| 3 | TCFH/K2HPO4 | EtOAc (0.19) | 5c |
| 4 | EDC·HCl | EtOAc (0.25) | <1d |

This suggests that COMU/K2HPO4 and TCFH/NMI systems are equally effective for synthesizing both esters and amides, thus requiring to incorporate protecting groups in the starting materials to achieve optimal yields. Conversely, EDC·HCl can be reliably used for amide synthesis in the presence of unprotected hydroxyls.
Then, the scope of the EDC-promoted coupling was briefly investigated (Scheme 2). First, we explored the effect of the nucleophilicity of the amine partner on the amide coupling of 1 (Scheme 2a). As a result, more nucleophilic N-Boc piperazine smoothly delivered amide 8 in a high 91% yield, while the least nucleophilic electron-deficient ethyl 4-aminobenzoate and sterically hindered 2,4,6-trimethylaniline formed the corresponding amides 6 and 7 in reduced 76% and 77% yields, respectively. These results confirm that poor nucleophilic amines are less reactive, but still amenable substrates. Besides the hydroxycarboxylic acid 1, the amino group in (4-(aminomethyl)phenyl)methanol was selectively acylated with benzoic acid, affording amide 9 in 85% yield, with no ester isomer of 9 being formed (Scheme 2b). The outcome complies with the analogous reactions in solution34,35 and by mechanochemistry.26–28 Next, we explored hydroxyl group containing amino acids, namely N-Boc-L-serine and N-Boc-L-tyrosine, in the reaction with aniline 3 and L-phenylalanine methyl ester (Scheme 2c). Notably, previous examples of mechanochemical peptide coupling reactions relied on the use of hydroxyl-protecting groups in serine and tyrosine.18,19 Amides 10 and 12 were prepared in good 82% and 90% yields in the reactions with 3. However, column chromatography purification was needed in these cases to separate unreacted amine 3. Dipeptides 11 and 13 were flawlessly prepared by coupling with L-phenylalanine methyl ester. Pure products were isolated in 84% and 83% yields after extractive workup. Importantly, dipeptide 13 was obtained with high diastereomeric purity (99![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 dr), indicating the absence of notable epimerization. Lithocholic acid in its coupling with 3 produced amide 14 in a high 93% yield, after simple treatment of the reaction mixture with water and drying in air. To compare the mechanochemical protocol with solution synthesis, dipeptide 11 was additionally prepared in CH2Cl2 solution. The solution-based reaction followed by extractive workup also afforded pure 11 in 90% yield, demonstrating that the chemoselectivity of the amide coupling is attributable to the coupling reagent rather than the mechanochemical conditions applied.
:1 dr), indicating the absence of notable epimerization. Lithocholic acid in its coupling with 3 produced amide 14 in a high 93% yield, after simple treatment of the reaction mixture with water and drying in air. To compare the mechanochemical protocol with solution synthesis, dipeptide 11 was additionally prepared in CH2Cl2 solution. The solution-based reaction followed by extractive workup also afforded pure 11 in 90% yield, demonstrating that the chemoselectivity of the amide coupling is attributable to the coupling reagent rather than the mechanochemical conditions applied.
 | ||
| Scheme 2 Scope of EDC-mediated mechanochemical amide coupling: (a) influence of amine's nucleophilicity; (b) amide coupling of aminoalcohol; (c) synthesis of amides and dipeptides from natural hydroxy (amino)acids. aYield of the isolated product after treatment with water, filtration and drying in air. bYield determined by 1H NMR with an internal standard. cYield of the isolated product after extraction work-up. dProduct isolated by silica gel column chromatography. | ||
Subsequently, the EDC-mediated amide coupling protocol was employed in the synthesis of anticancer drug imatinib 2 (the trade name Gleevec®) from the World Health Organization's List of Essential Medicines.8 The first synthetic route was patented in 1993 by Zimmermann,36 followed by a number of improved protocols,37–48 including flow-based approaches,49–52 and microwave-assisted solid-phase synthesis.53 However, mechanochemical preparations have not been reported yet.
Importantly, our planned route bypasses the formation of a chlorinated intermediate 17 with known genotoxic properties (Scheme 3), the content of which is strictly regulated (10 ppm)41 and which is commonly encountered in the mainstream synthetic strategies.38–40,44–47,49–51,53
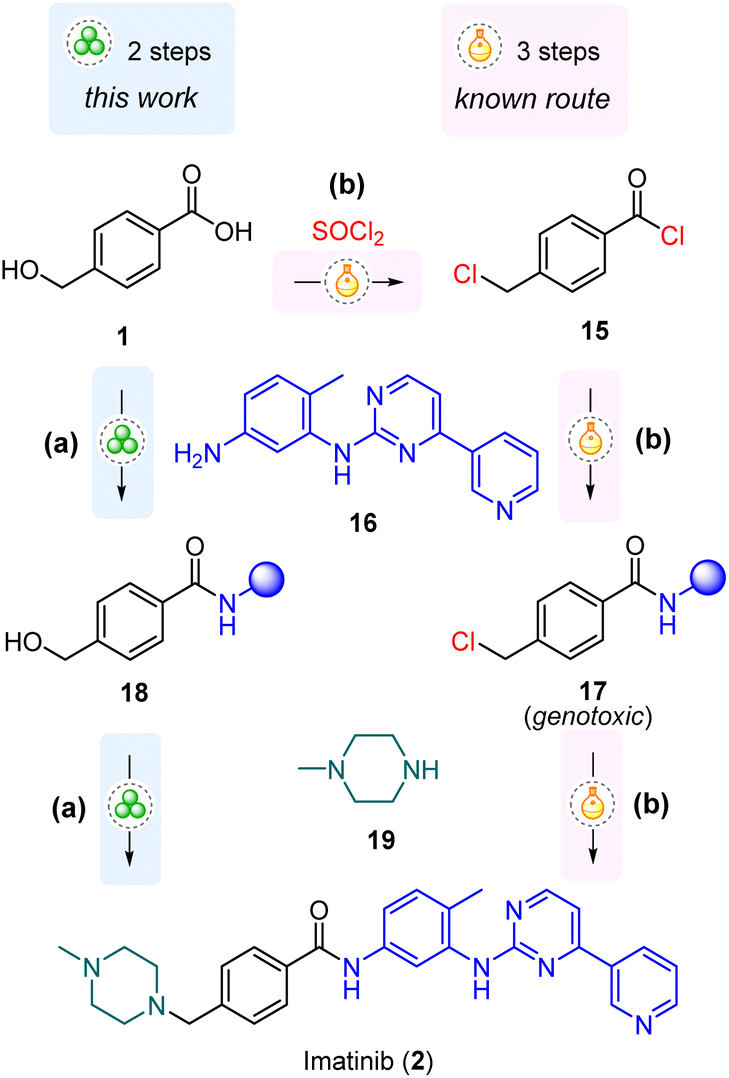 | ||
| Scheme 3 The developed mechanochemical route (a) and previously reported mainstream solution-based (b) approach for the synthesis of imatinib (2). | ||
After short optimization of the reaction conditions (see Tables S4 and S5 in the ESI†), imatinib 2 was prepared by EDC-mediated amide coupling of 1 with amine 16, followed by nucleophilic substitution of the hydroxyl group in the formed amide 18 with 1-methylpiperazine 19via generation of reactive isouronium intermediate 18a (Scheme 4a).7 The crude API was obtained in 86% overall yield, taking into account its 95% HPLC purity. The main impurities in 2 were identified as unreacted 18 (1.3%), quaternary salt 20 (2%) and 21 (1.4%; Scheme 4b). Purification of the crude 2 was achieved through recrystallization from a methanol-ethyl acetate mixture (1:1 ratio), yielding the target API with an HPLC purity of 99%. Crystals of intermediate 18 suitable for single-crystal X-ray diffraction analysis were obtained by crystallization from methanol solution and its solid-state structure was revealed, in a form of methanol solvate (CCDC 2287665, see Section 5 in the ESI†).
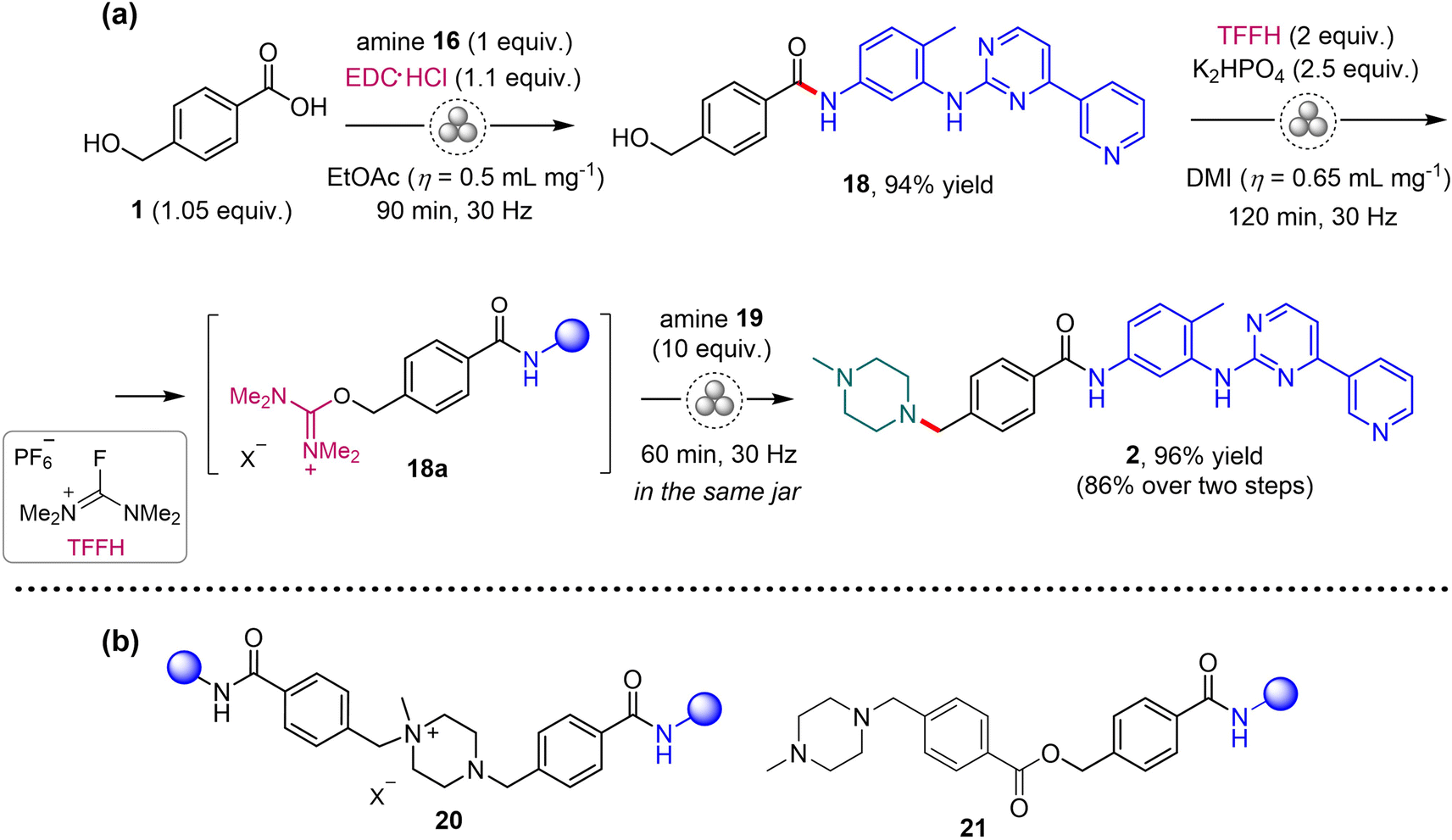 | ||
| Scheme 4 (a) Mechanochemical synthesis of imatinib (2). DMI = dimethyl isosorbide. (b) The main impurities identified in the crude 2. | ||
The genotoxic potential of intermediate 18 was checked by performing in silico assessment. Knowledge-based and statistical systems were used to predict potential mutagenicity following the recommendations of the ICH M7 (R1) (2018) guideline. As a result, 18 displayed no structural concern for mutagenicity. However, genotoxic amine 16 was still present in the product, in a content of ca. 560 ppm (see the ESI† for the details). This is similar to the content in the crude imatinib obtained by other methods and could be minimized during the downstream processing (see Section 6.2 in the ESI†).54
Next, we applied the CHEM21 toolkit55 to reveal the advantages of the developed mechanochemical synthesis in comparison to the similar early-stage development solution protocol described by Liu et al.39 (extended data and calculations for other similar routes are shown in Table S6 in the ESI†).
The main advantages of the mechanochemical protocol compared to the benchmarking solution-based approach included reduced process mass intensity (PMI, 221 vs. 564), the use of green and sustainable solvents (ethyl acetate, dimethyl isosorbide, and water) and room temperature operation (Fig. 1a). The exclusion of the genotoxic intermediate 17 (Scheme 3) was another important improve.
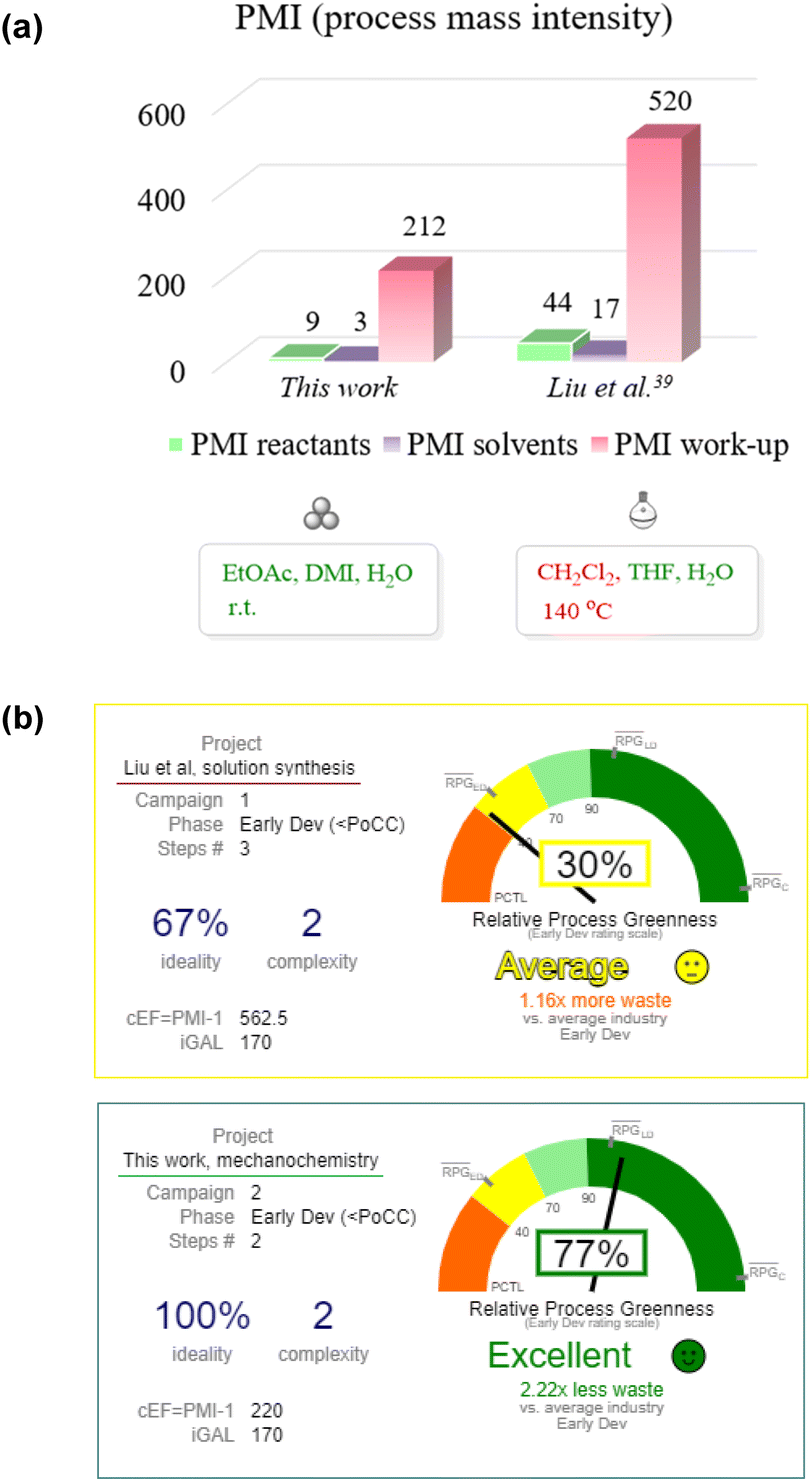 | ||
| Fig. 1 (a) Comparison of PMI values for the mechanochemical and a benchmarking solution-based method.39 (b) Graphical illustration of green chemistry innovation using the iGAL scorecard. | ||
In order to expose the green chemistry-related innovations of the developed method even more clearly, we applied the “innovation Green Aspiration Level” (iGAL) methodology56 by using the respective scorecard web calculator.57 The mechanochemical method demonstrated an excellent relative process greenness (RPG) of 77%, which is much higher when compared to the RPG (30%) of the solution-based method by Liu et al., used as a benchmark early-stage development process (Fig. 1b). Importantly, the mechanochemical approach exhibits a lower waste output of 2.22 times when compared to the average value at the early development stage. However, the developed methodology still relies on the use of stoichiometric coupling reagents (EDC·HCl and TFFH), which may pose environmental, safety or health hazards58 and reduce the atom economy of the reaction. Consequently, there is a need for further development of mechanochemical amidation methods with improved atom efficiency through the avoidance of stoichiometric amide coupling reagents.59
Conclusions
Here we demonstrated that mechanochemical amide coupling can be performed with unprotected hydroxycarboxylic acids by using EDC·HCl as a coupling reagent in combination with ethyl acetate as a LAG additive. High 76–94% yields of amides can be achieved, while the use of poor nucleophilic amines results in reduced coupling efficacy. The presence of an unmasked hydroxyl group in the obtained amide products allows their straightforward functionalization in a step-economical manner, such as nucleophilic substitution of the hydroxyl group. The application is illustrated by the first mechanochemical synthesis of imatinib, an anticancer drug included in the World Health Organization's List of Essential Medicines. The target API was prepared in 86% total yield and HPLC purity of 99% through a two-fold mechanochemical C–N bond construction sequence.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The research was supported by the Estonian Research Council grant PRG399 and COST Action CA18112 “Mechanochemistry for Sustainable Industry”. This project has received funding from the European Union's Horizon 2021–2027 research and innovation programme under grant agreement No. 101057286 (IMPACTIVE). Danylo Merzhyievskyi is grateful to the Education and Youth Board of Estonia for financial support of his research stay at the Tallinn University of Technology.References
- P. Ying, J. Yu and W. Su, Adv. Synth. Catal., 2021, 363, 1246–1271 CrossRef CAS.
- O. Bento, F. Luttringer, T. Mohy El Dine, N. Pétry, X. Bantreil and F. Lamaty, Eur. J. Org Chem., 2022, e202101516 CrossRef CAS.
- X. Yang, C. Wu, W. Su and J. Yu, Eur. J. Org Chem., 2022, e202101440 CrossRef CAS.
- M. Pérez-Venegas and E. Juaristi, ACS Sustainable Chem. Eng., 2020, 8, 8881–8893 CrossRef.
- O. Galant, G. Cerfeda, A. S. McCalmont, S. L. James, A. Porcheddu, F. Delogu, D. E. Crawford, E. Colacino and S. Spatari, ACS Sustainable Chem. Eng., 2022, 10, 1430–1439 CrossRef CAS.
- T. Dalidovich, K. A. Mishra, T. Shalima, M. Kudrjašova, D. G. Kananovich and R. Aav, ACS Sustainable Chem. Eng., 2020, 8, 15703–15715 CrossRef CAS.
- T. Dalidovich, J. V. Nallaparaju, T. Shalima, R. Aav and D. G. Kananovich, ChemSusChem, 2022, 15, e202102286 CrossRef CAS PubMed.
- World Health Organization, WHO Model List of Essential Medicines 23rd List, 2023 Search PubMed.
- M. Tsakos, E. S. Schaffert, L. L. Clement, N. L. Villadsen and T. B. Poulsen, Nat. Prod. Rep., 2015, 32, 605–632 RSC.
- B. Neises and W. Steglich, Angew. Chem., Int. Ed., 1978, 17, 522–524 CrossRef.
- A. Chighine, S. Crosignani, M.-C. Arnal, M. Bradley and B. Linclau, J. Org. Chem., 2009, 74, 4753–4762 CrossRef CAS PubMed.
- J. K. Twibanire and T. B. Grindley, Org. Lett., 2011, 13, 2988–2991 CrossRef CAS PubMed.
- M. Pittelkow, F. S. Kamounah, U. Boas, B. Pedersen and J. B. Christensen, Synthesis, 2004, 2485–2492 CAS.
- N. R. Luis, K. K. Chung, M. R. Hickey, Z. Lin, G. L. Beutner and D. A. Vosburg, Org. Lett., 2023 DOI:10.1021/acs.orglett.3c01611.
- H. Nishikawa, M. Kuwayama, A. Nihonyanagi, B. Dhara and F. Araoka, J. Mater. Chem. C, 2023, 11, 12525–12542 RSC.
- T.-X. Métro, J. Bonnamour, T. Reidon, J. Sarpoulet, J. Martinez and F. Lamaty, Chem. Commun., 2012, 48, 11781–11783 RSC.
- W. I. Nicholson, F. Barreteau, J. A. Leitch, R. Payne, I. Priestley, E. Godineau, C. Battilocchio and D. L. Browne, Angew. Chem., Int. Ed., 2021, 60, 21868–21874 CrossRef CAS PubMed.
- L. Gonnet, T. Tintillier, N. Venturini, L. Konnert, J.-F. Hernandez, F. Lamaty, G. Laconde, J. Martinez and E. Colacino, ACS Sustainable Chem. Eng., 2017, 5, 2936–2941 CrossRef CAS.
- J. Bonnamour, T.-X. Métro, J. Martinez and F. Lamaty, Green Chem., 2013, 15, 1116–1120 RSC.
- C. Bolm and J. G. Hernández, ChemSusChem, 2018, 11, 1410–1420 CrossRef CAS PubMed.
- V. Štrukil, B. Bartolec, T. Portada, I. Đilović, I. Halasz and D. Margetić, Chem. Commun., 2012, 48, 12100–12102 RSC.
- V. Porte, M. Thioloy, T. Pigoux, T.-X. Métro, J. Martinez and F. Lamaty, Eur. J. Org Chem., 2016, 3505–3508 CrossRef CAS.
- C. Duangkamol, S. Jaita, S. Wangngae, W. Phakhodee and M. Pattarawarapan, RSC Adv., 2015, 5, 52624–52628 RSC.
- T. Lainer, F. Czerny and M. Haas, Org. Biomol. Chem., 2022, 20, 3717–3720 RSC.
- J. G. Hernández, K. J. Ardila-Fierro, D. Crawford, S. L. James and C. Bolm, Green Chem., 2017, 19, 2620–2625 RSC.
- T. Portada, D. Margetić and V. Štrukil, Molecules, 2018, 23, 3163 CrossRef PubMed.
- Q. Cao, D. E. Crawford, C. Shi and S. L. James, Angew. Chem., Int. Ed., 2020, 59, 4478–4483 CrossRef CAS PubMed.
- F. Ravalico, S. L. James and J. S. Vyle, Green Chem., 2011, 13, 1778–1783 RSC.
- M. Anselmi, P. Stavole, E. Boanini, A. Bigi, E. Juaristi and L. Gentilucci, Future Med. Chem., 2020, 12, 479–491 CrossRef CAS PubMed.
- F. Santino, R. Petruzzelli, J. Zhao, E. Boanini and L. Gentilucci, Sustainable Chem. Pharm., 2021, 24, 100540 CrossRef CAS.
- T. Kanzian, T. A. Nigst, A. Maier, S. Pichl and H. Mayr, Eur. J. Org Chem., 2009, 6379–6385 CrossRef CAS.
- D. Prat, A. Wells, J. Hayler, H. Sneddon, C. R. McElroy, S. Abou-Shehada and P. J. Dunn, Green Chem., 2016, 18, 288–296 RSC.
- N. Pétry, H. Benakki, E. Clot, P. Retailleau, F. Guenoun, F. Asserar, C. Sekkat, T.-X. Métro, J. Martinez and F. Lamaty, Beilstein J. Org. Chem., 2017, 13, 2169–2178 CrossRef PubMed.
- D.-K. Kim, J. Y. Lee, J.-S. Kim, J.-H. Ryu, J.-Y. Choi, J. W. Lee, G.-J. Im, T.-K. Kim, J. W. Seo, H.-J. Park, J. Yoo, J.-H. Park, T.-Y. Kim and Y.-J. Bang, J. Med. Chem., 2003, 46, 5745–5751 CrossRef CAS PubMed.
- M. Parmentier, M. K. Wagner, K. Magra and F. Gallou, Org. Process Res. Dev., 2016, 20, 1104–1107 CrossRef CAS.
- J. Zimmermann, EP Pat., 0564409, 1993 Search PubMed.
- B. J. Deadman, M. D. Hopkin, I. R. Baxendale and S. V. Ley, Org. Biomol. Chem., 2013, 11, 1766–1800 RSC.
- Y. Heo, D. Hyun, M. R. Kumar, H. M. Jung and S. Lee, Tetrahedron Lett., 2012, 53, 6657–6661 CrossRef CAS.
- Y.-F. Liu, C.-L. Wang, Y.-J. Bai, N. Han, J.-P. Jiao and X.-L. Qi, Org. Process Res. Dev., 2008, 12, 490–495 CrossRef CAS.
- X. Zhang, J. Sun, T. Chen, C. Yang and L. Yu, Synlett, 2016, 27, 2233–2236 CrossRef CAS.
- A. Kompella, B. R. K. Adibhatla, P. R. Muddasani, S. Rachakonda, V. K. Gampa and P. K. Dubey, Org. Process Res. Dev., 2012, 16, 1794–1804 CrossRef CAS.
- K. C. Nicolaou, D. Vourloumis, S. Totokotsopoulos, A. Papakyriakou, H. Karsunky, H. Fernando, J. Gavrilyuk, D. Webb and A. F. Stepan, ChemMedChem, 2016, 11, 31–37 CrossRef CAS PubMed.
- C. Wang, X. Bai, R. Wang, X. Zheng, X. Ma, H. Chen, Y. Ai, Y. Bai and Y. Liu, Org. Process Res. Dev., 2019, 23, 1918–1925 CrossRef CAS.
- Z. Szakács, S. Béni, Z. Varga, L. Örfi, G. Kéri and B. Noszál, J. Med. Chem., 2005, 48, 249–255 CrossRef PubMed.
- W. Szczepek, W. Luniewski, L. Kaczmarek, B. Zagrodzki, D. Samson-Lazinska, W. Szelejewski and M. Skarzynski, World Pat., 2006071130A2, 2006 Search PubMed.
- A. Kompella, A. K. S. Bhujanga Rao, N. Venkaiah Chowdary and R. Srinivas, World Pat., 2004108699A1, 2004 Search PubMed.
- O. Loiseleur, D. Kaufmann, S. Abel, H. M. Buerger, M. Meisenbach, B. Schmitz and G. N. Sedelmeier, World Pat., 2003066613, 2003 Search PubMed.
- M. D. Khunt, N. S. Patil, H. S. Pagire and N. S. Pradhan, World Pat., 2011095835A1, 2011 Search PubMed.
- M. D. Hopkin, I. R. Baxendale and S. V. Ley, Chem. Commun., 2010, 46, 2450–2452 RSC.
- J. C. Yang, D. Niu, B. P. Karsten, F. Lima and S. L. Buchwald, Angew. Chem., Int. Ed., 2016, 55, 2531–2535 CrossRef CAS PubMed.
- N. Collins, D. Stout, J.-P. Lim, J. P. Malerich, J. D. White, P. B. Madrid, M. Latendresse, D. Krieger, J. Szeto, V.-A. Vu, K. Rucker, M. Deleo, Y. Gorfu, M. Krummenacker, L. A. Hokama, P. Karp and S. Mallya, Org. Process Res. Dev., 2020, 24, 2064–2077 CrossRef CAS.
- W. C. Fu and T. F. Jamison, Org. Lett., 2019, 21, 6112–6116 CrossRef CAS PubMed.
- F. Leonetti, C. Capaldi and A. Carotti, Tetrahedron Lett., 2007, 48, 3455–3458 CrossRef CAS.
- E. Grendele and A. Soldà, Eur. Pat., 2927223B1, 2015 Search PubMed.
- C. R. McElroy, A. Constantinou, L. C. Jones, L. Summerton and J. H. Clark, Green Chem., 2015, 17, 3111–3121 RSC.
- F. Roschangar, Y. Zhou, D. J. C. Constable, J. Colberg, D. P. Dickson, P. J. Dunn, M. D. Eastgate, F. Gallou, J. D. Hayler, S. G. Koenig, M. E. Kopach, D. K. Leahy, I. Mergelsberg, U. Scholz, A. G. Smith, M. Henry, J. Mulder, J. Brandenburg, J. R. Dehli, D. R. Fandrick, K. R. Fandrick, F. Gnad-Badouin, G. Zerban, K. Groll, P. T. Anastas, R. A. Sheldon and C. H. Senanayake, Green Chem., 2018, 20, 2206–2211 RSC.
- https://www.acs.org/green-chemistry-innovation-scorecard, 2023.
- J. C. Graham, A. Trejo-Martin, M. L. Chilton, J. Kostal, J. Bercu, G. L. Beutner, U. S. Bruen, D. G. Dolan, S. Gomez, J. Hillegass, J. Nicolette and M. Schmitz, Chem. Res. Toxicol., 2022, 35, 1011–1022 Search PubMed.
- T. Stolar, J. Alić, G. Talajić, N. Cindro, M. Rubčić, K. Molcanov, K. Užarević and J. G. Hernández, Chem. Commun., 2023, 59, 13490–13493 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2287665. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4mr00006d |
| This journal is © The Royal Society of Chemistry 2024 |