
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMechanochemical synthesis of β-cyclodextrin urea derivatives under reactive CO2 atmosphere by Staudinger aza-Wittig reaction†
Sawssen
Nasri
 ,
Maxime
Lestoquoy
,
Anne
Ponchel
,
Eric
Monflier
and
Stéphane
Menuel
*
,
Maxime
Lestoquoy
,
Anne
Ponchel
,
Eric
Monflier
and
Stéphane
Menuel
*
Univ. Artois, CNRS, Centrale Lille, Univ. Lille, UMR 8181, Unité de Catalyse et Chimie du Solide (UCCS), Rue Jean Souvraz, SP 18, 62300 Lens, France. E-mail: stephane.menuel@univ-artois.fr
First published on 4th June 2024
Abstract
Various ureido β-cyclodextrins can be easily synthesised by mechanochemistry from azido-β-cyclodextrins, carbon dioxide and amino derivatives. The reaction was carried out with short reaction times, without solvents and without thermal activation, greatly reducing the environmental impact of the synthesis.
Introduction
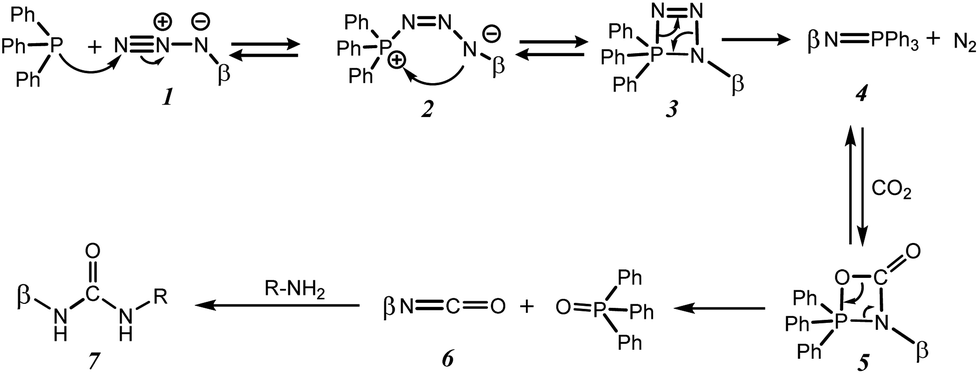
Staudinger aza-Wittig reactions are nowadays regularly employed in many fields of organic chemistry, as a step of a reaction sequence,1 in syntheses of poly-substituted cyclic derivatives,2–5 or for isotopic labelling.6 This sequential reaction involves the formation of iminophosphorane from phosphane and organic azide, known as the Staudinger reaction,7,8 and the reaction of the latter, known as the aza-Witting reaction, with aldehyde, CO2, CS2 or isocyanate to give imine, isocyanate, isothiocyanate or carbodiimide, respectively. The Staudinger aza-Wittig reaction has been widely used to obtain ureido-cyclodextrin derivatives since the pioneering work of Marsura, especially in solution in anhydrous media9,10 or in supercritical CO2.11 However, the use of solvents associated with long reaction times or harsh reaction conditions excludes this approach from the realm of green chemistry. To overcome this issue, we investigated this reaction using mechanochemical activation. Mechanochemistry consists of the study and use of mechanical energy for the activation of chemical reactions. The mechanical energy transferred to the reaction system can be provided by pestles, vibrating or planetary mills or extruders in order to carry out chemical reactions in the solid state.12 Mechanochemistry, by the reduction of reaction times, as well as by the important reduction of the quantity of solvent necessary to the process, allows an important reduction of the energy consumption compared to the traditional processes. Thus, the mechanochemical activation allows a more respectful approach of the environment and is fully in line with the sustainable development.Cyclodextrins are cyclic oligosaccharides; the most common of which are composed of 6, 7 and 8 glucose units for α, β and γ-cyclodextrins respectively. They form cone-shaped structures with non-polar cavities that can complex apolar guests.13 It is worth noting that cyclodextrins offer a versatile platform for chemical modification, enabling a wide array of tailored derivatives with diverse properties and functionalities for various applications including in textiles, flavor protection in food formulations, cholesterol removal in dairy products, drug delivery enhancement, agricultural chemical encapsulation, and separation in the chemical industry, owing to their complexation abilities, odor control, and molecular encapsulation properties.14
In this study, we employ functionalized cyclodextrins, specifically, monoazido-per-O-acetyl-β-cyclodextrin ((2,3-di-O-acetyl-6-deoxy-6-azido)-hexakis-(2,3,6-tri-O-acetyl)-cyclomaltoheptaose) 1A and monoazido-per-O-methyl-β-cyclodextrin ((2,3-di-O-acetyl-6-deoxy-6-azido)-hexakis-(2,3,6-tri-O-methyl)-cyclomaltoheptaose) 1B (Scheme 1). The reaction sequence involves a Staudinger reaction between the azido-cyclodextrin derivatives 1A or 1B and triphenylphosphine (TPP),15 followed by an aza-Wittig type transformation with CO2, and subsequently, ureation reactions with R–NH2 derivatives to form ureido-β-cyclodextrins products (Scheme 2).
 | ||
| Scheme 1 Cyclodextrin 1A and 1B. | ||
 | ||
| Scheme 2 Staudinger aza-Wittig ureation. | ||
During urea formation, it is essential to avoid any other nucleophilic species in the reaction environment to prevent adverse reactions with isocyanate 6. Therefore, the use of monoazido-β-cyclodextrin ((6-deoxy-6-azido)-cyclomaltoheptaose) with OH groups is not recommended as it may result in the formation of unwanted carbamate derivatives. However, the deprotection of cyclodextrin hydroxyl functions through ester function methanolysis using the Zemplen method is easily achievable,16 thereby enhancing the relevance of this study starting from 1A.
Results and discussion
Performance of grinding process vs. solution process
During this study, the mechanochemical processes were carried out in planetary ball milling with 20 mL grinding bowl and ten 10 mm balls in zirconium oxide. The reaction atmosphere can be introduced at pressures ranging from atmospheric pressure to 10 bars.We began by co-grinding 1A, TPP and 2-iodoaniline under 10 bars of CO2. After a 15 minute reaction, we obtained a yield of 47% for compound 7Ak, accompanied by the formation of 6% for coproduct in the form of a symmetrical bicyclodextrin (Scheme 3).
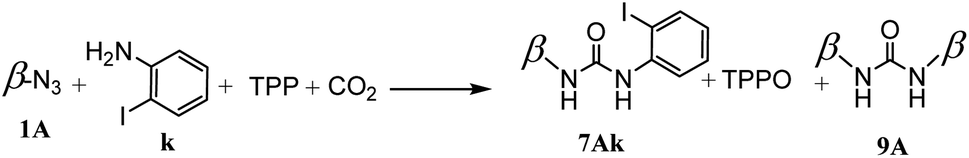 | ||
| Scheme 3 Ureation of 1A with 2-iodoaniline. | ||
During the Staudinger aza-Wittig urea reaction, a side reaction can occur between the iminophosphorane intermediates 4 and the isothiocyanate intermediates 6 to form carbodiimides 8, which will be hydrolyzed into symmetrical ureas 9 during analyses or purifications17 (Scheme 4). However, selectivity towards compound 7Ak was 80%.
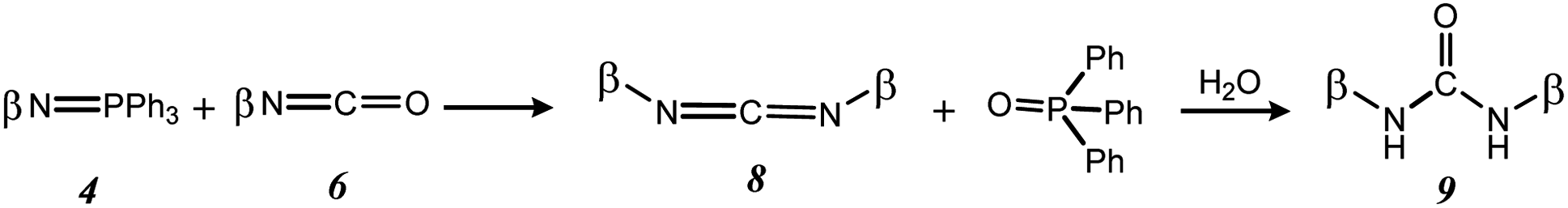 | ||
| Scheme 4 Side reaction during Staudinger aza-Wittig ureation. | ||
When we conducted the reaction involving 1A, TPP, and 2-iodoaniline under CO2 pressure directly in a chloroform solution for a duration of 15 minutes, the yield of 7Ak was found to be less than 1%, which makes the selectivity undefined (Table 1 entry 1). However, a yield of 48% could be obtained after a reaction time of 15 h, but the selectivity was lower than that obtained by grinding (Table 1 – entry 2). Intriguingly, upon pre-grinding 1A and TPP in the absence of additional reagents and solvents, followed by subsequent use of the pre-ground powder for a 15 minute reaction in a chloroform solution in the presence of CO2 and 2-iodoaniline, we achieved a significantly enhanced yield of 48% for compound 7Ak, characterized by an impressive 67% selectivity (Table 1 – entry 4). These findings suggest that the pre-milling of 1A and TPP can significantly enhance the Staudinger reaction step in this reaction sequence. Furthermore, the molar ratio obtained in chloroform solution at 20 °C closely resembles that achieved through ambient temperature milling.
| Entry | Method | Time | Yield (%) | Selectivity (%) |
|---|---|---|---|---|
| 7Ak | (7Ak/(7Ak + 2 × 9A)) × 100e | |||
| a 1A (1000 mg, 0.5 mmol, 1 equiv.), TPP (157 mg, 0.60 mmol, 1.2 equiv.), 2-iodoaniline (131 mg, 0.60 mmol, 1.2 equiv.), CHCl3 (15 mL), CO2 (10 bars), 1500 rpm, R.T. b 1A (1000 mg, 0.5 mmol, 1 equiv.), TPP (157 mg, 0.60 mmol, 1.2 equiv.), 2-iodoaniline (131 mg, 0.60 mmol, 1.2 equiv.), CO2 (10 bars), 20 mL zirconium oxide grinding bowl with ten 10 mm zirconium oxide grinding balls, 850 rpm. c 1A (1000 mg, 0.5 mmol, 1 equiv.), TPP (157 mg, 0.60 mmol, 1.2 equiv.), 20 mL zirconium oxide grinding bowl with ten 10 mm zirconium oxide grinding balls, 850 rpm, 15 min before introduction of resulting powder in solution of 2-iodoaniline (131 mg, 0.60 mmol, 1.2 equiv.) in CHCl3 (15 mL), CO2 (10 bars), 1500 rpm, R.T. d Isolated yield. e Calculated with isolated products quantity. f The reaction progress is too low to allow isolation of any product. | ||||
| 1 | Solutiona | 15 min | n.d.f | n.d. |
| 2 | Solutiona | 15 h | 48 | 67 |
| 3 | Grindingb | 15 min | 47 | 80 |
| 4 | Solutionc | 15 min | 48 | 67 |
Effect of substitution on performance in grinding process
This study was extended to a wide range of halogenated aniline, amino-pyridine derivatives or amino-adamantane to product unsymmetrical urea's cyclodextrin (7Ax or 7Bx) (Table 2).| a 1A or 1B (0.5 mmol, 1 equiv.), TPP (157 mg, 0.60 mmol, 1.2 equiv.), aniline derivative (0.60 mmol, 1.2 equiv.), CO2 (10 bars), 20 mL zirconium oxide grinding bowl with ten 10 mm zirconium oxide grinding balls, 850 rpm, 15 min – yield (Y%) and selectivity (S%) calculated with isolated products and S% = (7x/(7x + 2 × 9)) × 100. |
|---|
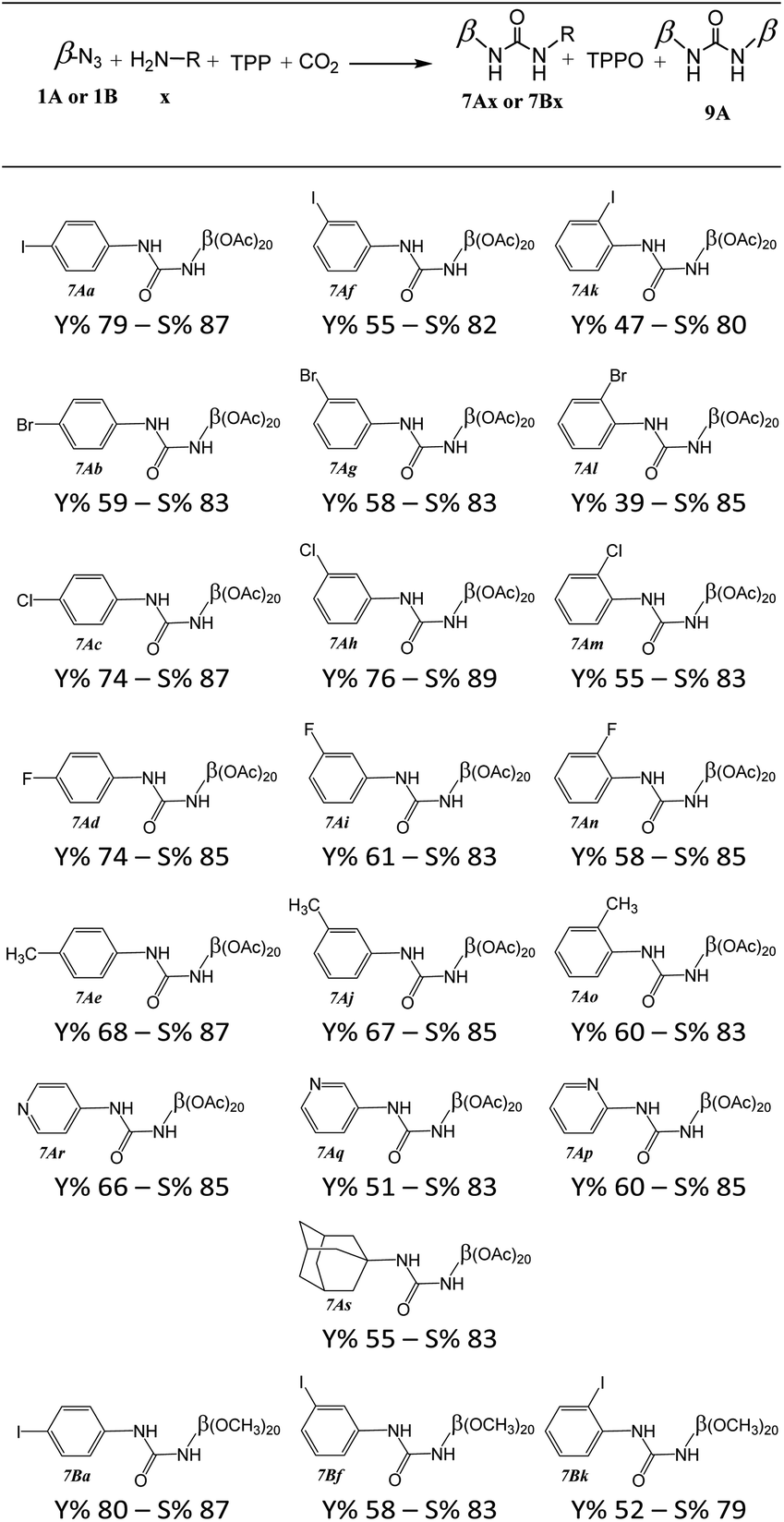
|
In the case of aniline derivatives, we first investigated the effect of halogenated substituents in the ortho, meta, and para positions on the yields with 1A. We did not observe significant electronic effects on the yields. However, we found that steric effects are predominant. Indeed, regardless of the presence of a halogenated substituent or a methyl substituent, the para-substituted series of aniline always gave the best yields. However, in the fluorinated series, although the absence of steric hindrance in the para position allows for the best yields, the differences in yields between the ortho and meta positions do not allow us to conclude a unique influence of steric effects. The fluorine may also induce electronic effects in addition to steric effects.
We also observed that the presence of a substituent in the ortho position of the NH2 function leads to significant steric hindrance resulting in low to moderate yields. However, by using aminopyridine derivatives, we were able to study the influence of electronic effects. We observed that the yields obtained are directly correlated to the nucleophilicity of the aminopyridine derivative.18 Thus, when steric effects are not present, the influences of electronic effects become predominant (Fig. 1). These results emphasize the importance of considering the steric and electronic effects of substituents when designing new derivatives for the synthesis of ureas by mechanoactivation.
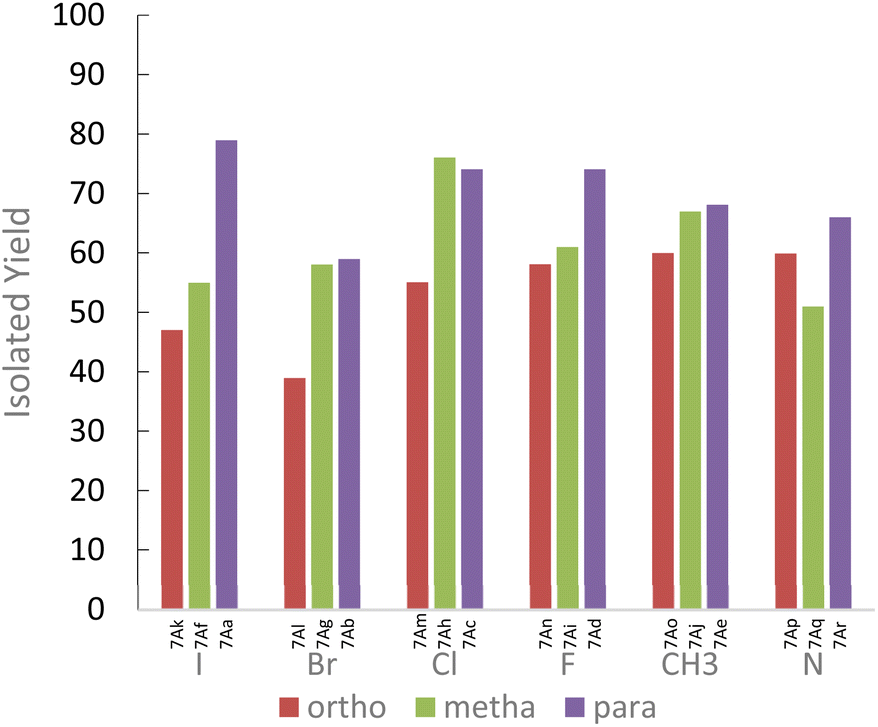 | ||
| Fig. 1 Effect of substrates on isolated yield of 7A type derivatives. | ||
Although 1-adamantanamine is a sterically hindered primary amine, it is perfectly recognized by cyclodextrin cavities,19 with which it forms complexes in solution with high association constants. We have demonstrated in previous articles that the complexes present in solution also exist in the solid phase,20,21 so it seems conceivable that this is also the case with 1-adamantanamine. Thus, it is likely that the obtained result can be explained by the spatial proximity of the reactive sites in the 1-adamantanamine/cyclodextrin complex. However, it is probable that complexes are not specifically oriented, and that a random distribution between the orientation of the amine function towards the secondary face (corresponding to positions 2 and 3 of the sugars) and the primary face (corresponding to positions 6 of the sugars) coexists.22 Thus, only complexes with an orientation of the amine function towards the primary face would have spatial proximity to the reactive function of cyclodextrins, which would explain yields of 47% to 80%.
Effect of CO2 pressure on performance in grinding process
We also investigated the effect of CO2 pressure (Table 3). We observed that the CO2 pressure did not significantly influence either the yield or the selectivity of the reaction. We attribute this finding to the high accessibility of the reactive sites to the gaseous reactants in the absence of solvent. In contrast to liquid-phase reactions, where mass transfer limitations are typically compensated by using high gas pressures to promote phase transfers, this limitation is absent in solid-phase reactions. Therefore, a high pressure of gaseous reactant is no longer necessary, and a nearly stoichiometric quantity of CO2 is sufficient to achieve high yields (Table 3). Recently, Bols and co.23 presented a compelling demonstration of the capacity of certain cyclodextrin derivatives to form complexes with CO2. This study was distinguished by the successful crystallization of specific complexes involving cyclodextrin derivatives and CO2, providing robust confirmation of the existence of these associations in the solid phase. Furthermore, this team elucidated a correlation between the heightened lipophilicity of cyclodextrin derivatives and a significant increase in association constants with CO2. Consequently, it becomes plausible that, in the solid phase, CO2 can indeed form inclusion complexes with cyclodextrins 1A and 2B, thereby enhancing chemical reactivity through proximity effects with reactive groups, without the necessity for high pressures.| Entry | CO2 (bar) | Yield (%) | Selectivity (%) |
|---|---|---|---|
| 7Ak | (7Ak/(7Ak + 2 × 9A)) × 100 | ||
| a 1A (1000 mg, 0.5 mmol, 1 equiv.), TPP (157 mg, 0.60 mmol, 1.2 equiv.), 2-iodoaniline (131 mg, 0.60 mmol, 1.2 equiv.), CO2, (10 bars), 20 mL zirconium oxide grinding bowl with ten 10 mm zirconium oxide grinding balls, 850 rpm, 15 min – yield and selectivity calculated with isolated products. | |||
| 1 | 10 | 47 | 81 |
| 2 | 7 | 48 | 80 |
| 3 | 2 | 47 | 80 |
Mechanism exploration
Our results suggest strongly that the Staudinger reduction constitutes a critical step. Many research teams have investigated this reaction using experimental techniques24–26 or computational methods.27 To scrutinize this critical phase, we used 31P NMR spectroscopy to follow the reaction progress. Experiments involved the co-grinding of TPP with per-methylated-β-cyclodextrin or per-acetylated-β-cyclodextrin cyclodextrins not functionalized by azide functions and without taking precautions on the atmosphere composition (air at atmospheric pressure), moreover, the atmosphere is renewed at each sampling every hour. Over a grinding time of 3 hours without or in the presence of per-methylated-β-cyclodextrin or per-acetylated-β-cyclodextrin, we didn't observe any oxidation of TPP. Similarly, a solution of TPP in CDCl3 at room temperature will only undergo an oxidation of less than 0.3% in 3 hours, the oxidation is about 3% after 46 h in solution at R.T. These observations allow us to use 31P-NMR for the analysis of our systems by not considering the oxidation during the short time of the analysis.We investigated the generation of iminophosphorane intermediates in both solution and under dry milling conditions. We used an equimolar mixture of azido-cyclodextrin derivatives 1A or 1B and TPP. Our analysis using 31P-NMR consistently revealed the presence of four distinct species in varying proportions. Specifically, TPP and TPPO exhibited signals at −5.0 ppm and 29.5 ppm, respectively. Additionally, we observed signals corresponding to triazaphosphadienes 2A at 41.4 ppm, 2B at 40.8 ppm, and iminophosphoranes 4A at 12.5 ppm, and 4B at 8.7 ppm (Fig. 2).
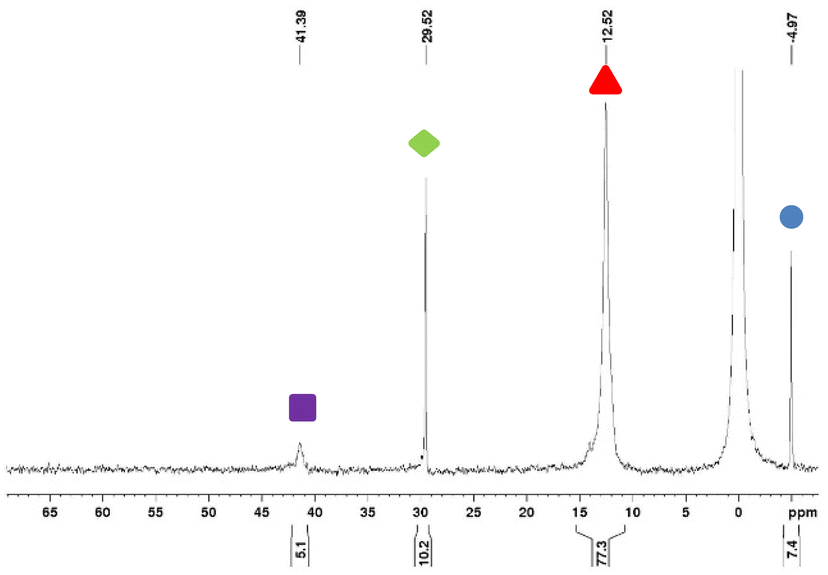 | ||
Fig. 2
31P NMR(CDCl3) spectra of the reaction of 1A with TPP in CDCl3 after 22 h at room temperature (H3PO4 is the outsourcing reference at 0 ppm).  TPP, TPP,  TPPO, TPPO,  2A, 2A,  4A. 4A. | ||
In solution at room temperature in CDCl3, we observe an evolution of the conversion of TPP over several hours in the presence of an equimolar quantity of azido-cyclodextrins 1A. The presence of intermediate 2A is always maintained at a proportion of about 5% (Fig. 3A).
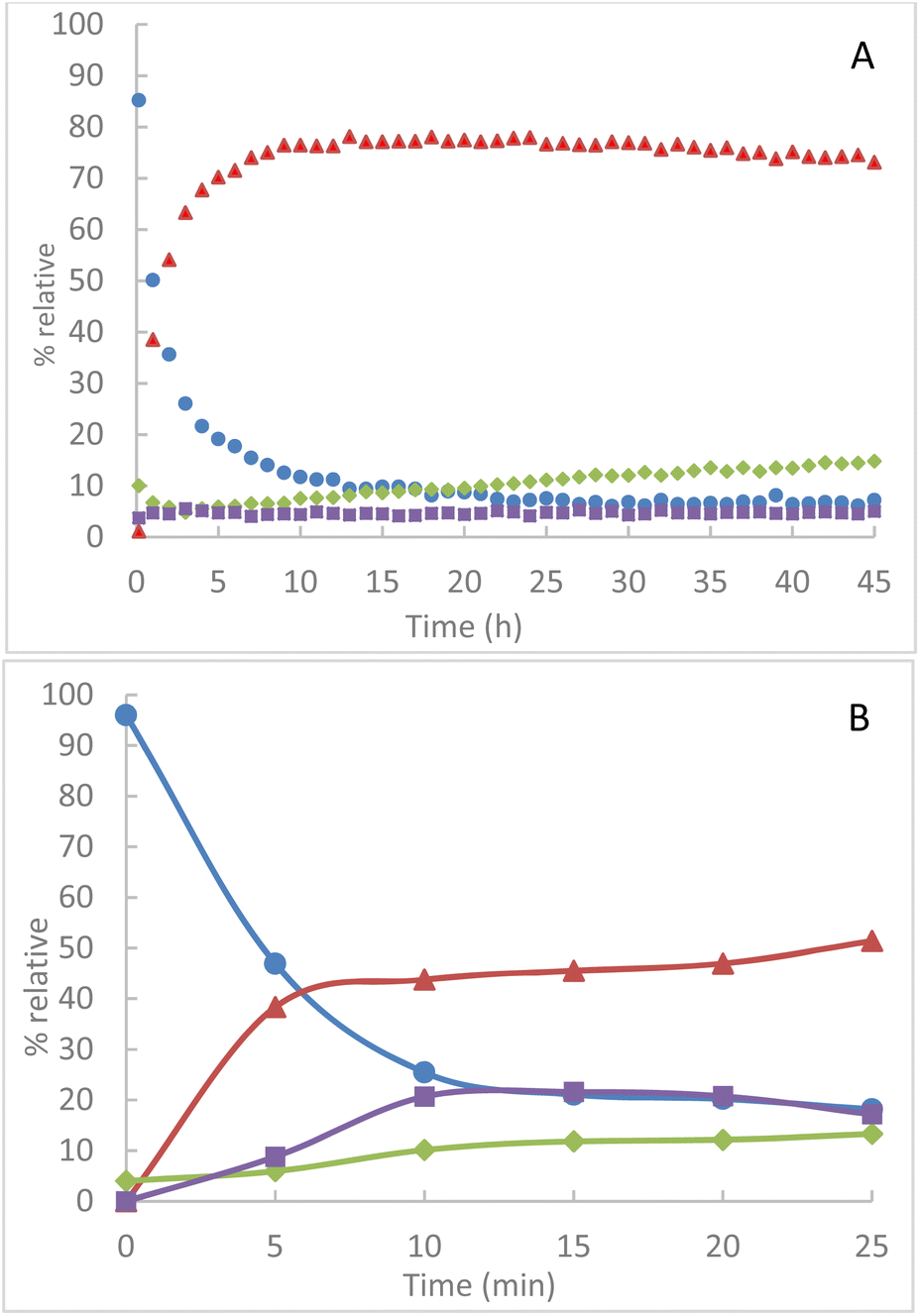 | ||
Fig. 3 Relative integrated intensity of phosphorus species by NMR 31P(CDCl3) (A) in solution at room temperature in CDCl3 (B) by grinding at 850 rpm  TPP, TPP,  TPPO, TPPO,  2A, 2A,  4A. 4A. | ||
The evolution of the proportion of TPPO in the medium is attributed to the greater sensitivity of iminophosphorane 4A to hydrolysis. By mechanochemical activation, we observe an extremely important increase in the conversion of TPP compared to reactions in solution at room temperature (Fig. 3B). Thus, milling of 1A and TPP leads to a conversion step of 80% of TPP in less than 20 min whereas in solution, a comparable conversion is obtained in more than 4 hours, i.e. a reduction of about 92% of the conversion time. However, the product distribution is very different.
Similarly, from an equimolar quantity of azido-cyclodextrins 1B and TPP in solution at room temperature in CDCl3, we observe an evolution of the conversion of TPP over several hours. The presence of intermediate 2B is always maintained at a small proportion of about 6% (Fig. 4A). The evolution of the proportion of TPPO in the medium is attributed to the greater sensitivity of iminophosphorane 4B to hydrolysis. From 1B, we also observe a much faster conversion of TPP by mechanochemical activation a plateau at 70% conversion in less than 15 min (Fig. 4B). Comparatively, a comparable conversion requires more than 13 hours in solution, i.e. a reduction of about 98% of the conversion time.
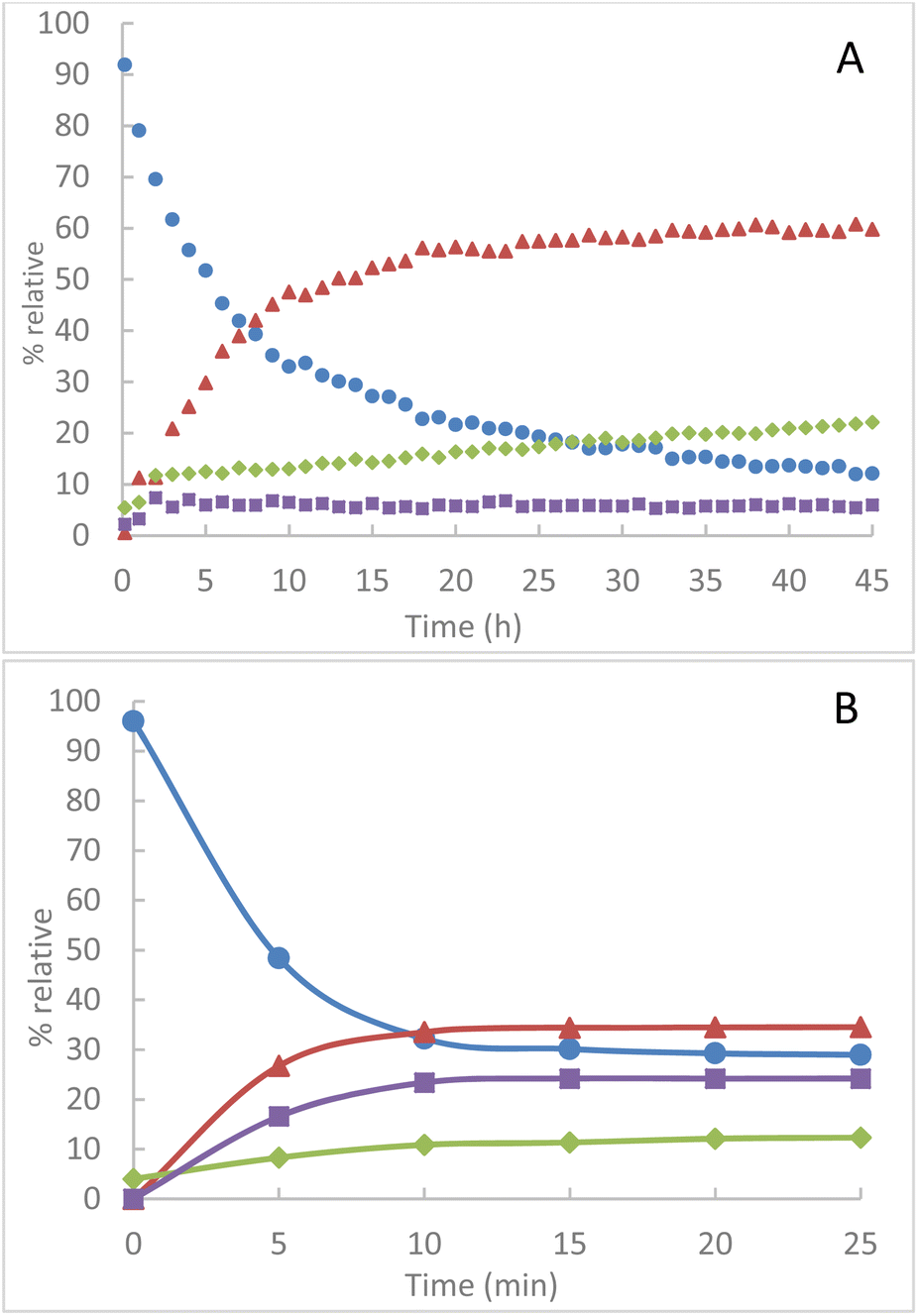 | ||
Fig. 4 Relative integrated intensity of phosphorus species by NMR 31P(CDCl3) (A) in solution at room temperature in CDCl3 (B) by grinding at 850 rpm  TPP, TPP,  TPPO, TPPO,  2B, 2B,  4B. 4B. | ||
Unexpectedly, in both solution and under dry milling conditions, we observe a faster conversion of TPP starting at 1A compared with 1B. We suggest attributing this result to the contribution of the acetate groups in shifting the equilibria in favor of iminophosphorane 2A formation through a transition by an 1,3,2-oxazaphosphetane 3A# (Scheme 5).28
 | ||
| Scheme 5 Proposed mechanism for assisting the ester function to shift equilibria towards the formation of iminophosphorane 4A. | ||
However, the diffusion dynamics of the reagents in the solids is fast when grinding from 1A or 1B. This diffusion allows a rapid reaction between the TPP and the azide functions, with the formation of the intermediate 2. Thus, the mechanical energy transferred to the powders allows intermolecular movement as well as reaction activation for the formation of the first intermediate 2. However, the intramolecular dynamics seems to be different in solid medium compared to the solution medium. Indeed, the reaction equilibria in solid phase lead to a distribution of reaction intermediates different from the distribution in solution. Thus, we assume that the intramolecular reaction allowing the formation of 4 from 2 intermediates is slower in solid phase than in the liquid phase. However, the intramolecular chemical equilibria remain displaceable by introducing another reactive molecule in the medium.
Green chemistry metrics of grinding process vs. solution process
The green chemistry metrics29 of the grinding process were compared with those of the solution process (Table 4).| Green chemistry metrics | In solutiona | By grindingb | Percentage off by grinding versus solution |
|---|---|---|---|
| a 1A (1000 mg, 0.5 mmol, 1 equiv.), TPP (157 mg, 0.60 mmol, 1.2 equiv.), 2-iodoaniline (131 mg, 0.60 mmol, 1.2 equiv.), CHCl3 (15 mL), CO2 (10 bars), 1500 rpm, R.T. b 1A (1000 mg, 0.5 mmol, 1 equiv.), TPP (157 mg, 0.60 mmol, 1.2 equiv.), 2-iodoaniline (131 mg, 0.60 mmol, 1.2 equiv.), CO2 (10 bars), 20 mL zirconium oxide grinding bowl with ten 10 mm zirconium oxide grinding balls, 850 rpm. | |||
| Atom Economy (AE) | 87.9% | # | |
| Reaction Mass Efficiently (RME) | 40.7% | 39.9% | Δ 0.8% |
| Process Mass Intensity (PMI) | 45 kg kg−1 | 2.6 kg kg−1 | 94% reduction |
| E-factor | 44 kg kg−1 | 1.6 kg kg−1 | 96% reduction |
| Time of reaction (min) | 900 | 15 | 98% reduction |
| Purification | Evaporation of organic solvents/column chromatography | Column chromatography | |
For Atom Economy (AE), both methods yield a similar result, with an atom economy of 87.9%, showcasing the efficiency of both approaches in utilizing reactants. Similarly, Reaction Mass Efficiency (RME) demonstrates congruence between the two methods, with both registering an RME of approximately 40%, implying the effective conversion of reactants into desired products.
In contrast, Process Mass Intensity (PMI) exhibits a substantial contrast between the two methods. While the solution-based approach yields a PMI of 45 kg kg−1, the grinding method remarkably reduces this metric to a mere 2.6 kg kg−1. This indicates a remarkable 94% reduction in process mass intensity when employing the grinding method.
Likewise, the E-factor illustrates a significant disparity between the two approaches. The solution-based method results in an E-factor of 44 kg kg−1, whereas the grinding method records an impressively lower E-factor of 1.6 kg kg−1, signifying a substantial 96% reduction in waste generation when grinding is employed. Furthermore, the time of reaction displays a substantial contrast, with the solution-based approach necessitating 900 minutes, whereas the grinding method remarkably reduces the reaction time to just 15 minutes, signifying a noteworthy 98% reduction.
Lastly, in terms of purification, the solution-based method involves the evaporation of organic solvents and column chromatography, while the grinding method employs just a column chromatography, which may be seen as a more simply purification strategy.
This comparison demonstrated that the grinding method offers significant advantages in terms of process mass intensity, E-factor, and reaction time when compared to the solution-based approach, thereby exemplifying its potential as a more environmentally sustainable and efficient synthetic route.
Conclusion
This study has clearly demonstrated that mechanochemistry under reactive atmosphere is a practical and effective method to functionalize cyclodextrins with urea functions. The formation of iminophosphorane has been identified as the kinetically limiting step in this reaction, whether in a solution or a solid phase. Additionally, our findings suggest that steric effects are generally more prevalent than electronic effects in the reaction. However, supramolecular effects facilitating complex formation can modulate steric effects by pre-organizing reactive functionalities, notably in the case of 1-adamantamine. Finally, it has been shown that the use of high-pressure reactive atmosphere is not necessary in the absence of solvent. These observations have contributed to a better understanding of the mechanisms involved in mechano-activated reactions in the absence of solvent and offer interesting perspectives for the optimization of this synthesis method. Similarly, this study demonstrates that mechanochemistry is an approach that makes it possible to reduce the environmental impact of reactions under a reactive atmosphere, in particular the ureation reaction by the Staudinger aza-Wittig reaction sequence under reactive atmosphere of CO2.Author contributions
Conceptualization SM; methodology, SM, AP, EM; validation SM, AP, EM; formal analysis, SN, ML, SM; investigation, SN, ML, AP, EM, SM; resources, SN, ML, SM; data curation, SN, ML, SM; writing—original draft preparation, SM; writing—review and editing, SM, EM, AP, SN; supervision, SM; project administration, SM, AP; funding acquisition, SM, AP.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The University of Artois is acknowledged for funding this work. Dr Jérémy Ternel is acknowledged for NMR analyses and Johan Hachani is acknowledged for MALDI analyses.Notes and references
- B. Mitasev, J. Yang, F. Gusovsky, D. Girish, A. S. Khile, V. S. Balla, V. Vikram, A. Vaddi, S. Bathula, S. R. Sugandham, C. Terli, V. Kalla, P. K. Rayaprolu, R. K. Talabhakthula, M. Gotoda, B. Melillo, S. L. Schreiber and F. G. Fang, Org. Process Res. Dev., 2022, 26(3), 817–831 CrossRef CAS.
- L. Zhao, M. L. Yang, M. Liu and M. W. Ding, Beilstein J. Org. Chem., 2022, 18, 286–292 CrossRef CAS PubMed.
- W. S. Zhang, Y. Li, H. Y. Zhou, X. L. Su, X. F. Zhang and W. Zhang, Tetrahedron Lett., 2021, 81, 153361 CrossRef CAS.
- H. Xie, Q. Q. Hu, X. T. Qin, J. M. Liang, L. Li, Y. L. Zhang and Z. Lu, Heterocycles, 2022, 104(3), 584–588 CrossRef.
- K. Pedrood, M. N. Montazer, B. Larijani and M. Mahdavi, Synthesis, 2021, 53(14), 2342–2366 CrossRef CAS.
- V. Babin, A. Sallustrau, O. Loreau, F. Caille, A. Goudet, H. Cahuzac, A. Del Vecchio, F. Taran and D. Audisio, Chem. Commun., 2021, 57(54), 6680–6683 RSC.
- H. Staudinger and J. Meyer, Helv. Chim. Acta, 1919, 2, 635–646 CrossRef CAS.
- J. C. Lopez, M. del Rio, A. Oliden, J. Banuelos, I. Lopez-Arbeloa, I. Garcia-Moreno and A. M. Gomez, Chem.–Eur. J., 2017, 23(69), 17511–17520 CrossRef PubMed.
- F. Sallas, A. Marsura, V. Petot, I. Pinter, J. Kovacs and L. Jicsinszky, Helv. Chim. Acta, 1998, 81(4), 632–645 CrossRef CAS.
- S. Menuel, R. E. Duval, D. Cuc, P. Mutzenhardt and A. Marsura, New J. Chem., 2007, 31(6), 995–1000 RSC.
- S. Menuel, M. Wagner, D. Barth and A. Marsura, Tetrahedron Lett., 2005, 46(19), 3307–3309 CrossRef CAS.
- A. A. L. Michalchuk, E. V. Boldyreva, A. M. Belenguer, F. Emmerling and V. V. Boldyrev, Front. Chem., 2021, 9, 685789 CrossRef CAS PubMed.
- G. Crini, Chem. Rev., 2014, 114, 10940–10975 CrossRef CAS PubMed.
- B. G. Poulson, Q. A. Alsulami, A. Sharfalddin, E. F. El Agammy, F. Mouffouk, A.-H. Emwas, L. Jaremko and M. Jaremko, Polysaccharides, 2022, 3(1), 1–31 CrossRef CAS.
- W. Q. Tian and Y. A. Wang, J. Org. Chem., 2004, 69, 4299–4308 CrossRef CAS PubMed.
- M. Mourer, F. Hapiot, E. Monflier and S. Menuel, Tetrahedron, 2008, 64(30–31), 7159–7163 CrossRef CAS.
- F. Charbonnier, A. Marsura, K. Roussel, J. Kovacs and I. Pinter, Helv. Chim. Acta, 2001, 84, 535–551 CrossRef CAS.
- N. A. Caballero, F. J. Melendez, C. Muñoz-Caro and A. Niño, Biophys. Chem., 2006, 155–160 CrossRef CAS PubMed.
- K. Sadrerafi, E. E. Moore and M. W. Lee Jr, J. Inclusion Phenom. Macrocyclic Chem., 1989, 7, 537–543 CrossRef.
- K. Cousin, S. Menuel, E. Monflier and F. Hapiot, Angew. Chem., Int. Ed., 2017, 56, 10564–10568 CrossRef CAS PubMed.
- S. Menuel, B. Leger, A. Addad, E. Monflier and F. Hapiot, Green Chem., 2016, 18, 5500–5509 RSC.
- J.-W. Wang, K.-X. Yu, X.-Y. Ji, H. Bai, W.-H. Zhang, X. Hu and G. Tang, Molecules, 2021, 26, 2412–2425 CrossRef CAS PubMed.
- C. H. Jessen, J. Bendix, T. B. Nannestad, H. Bordallo, M. J. Pedersen, C. M. Pedersen and M. Bols, Beilstein J. Org. Chem., 2023, 19, 1021–1027 CrossRef CAS PubMed.
- D. E. Shalev, S. M. Chiacchiera, A. E. Radkowsky and E. M. Kosower, J. Org. Chem., 1996, 61, 1689–1701 CrossRef CAS PubMed.
- S. Y. Corrigan and J. S. Poole, J. Phys. Org. Chem., 2023, 36, e442 CrossRef.
- T. Deb, J. Tu and R. M. Franzini, Chem. Rev., 2021, 121(12), 6850–6914 CrossRef CAS PubMed.
- W. Q. Tian and Y. A. Wang, J. Org. Chem., 2004, 69(13), 4299–4308 CrossRef CAS PubMed.
- N. Kano, X. J. Hua, S. Kawa and T. Kawashima, Tetrahedron Lett., 2000, 41(27), 5237–5241 CrossRef CAS.
- R. A. Sheldon, ACS Sustainable Chem. Eng., 2018, 6(1), 32–48 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4mr00020j |
| This journal is © The Royal Society of Chemistry 2024 |