DOI:
10.1039/D4MR00056K
(Communication)
RSC Mechanochem., 2024,
1, 437-446
Mechanical approach for creating different molecular adducts and regulating salt polymorphs: a case study of the anti-inflammatory medication ensifentrine†
Received
27th May 2024
, Accepted 20th September 2024
First published on 20th September 2024
Abstract
An intriguing technique for crystal engineering is mechanochemistry, which frequently yields various solid forms (salts, cocrystals, polymorphs, etc.) that are challenging to acquire using traditional solution-based approaches. However, generating new and potentially beneficial solid forms remains an ongoing task in this field. Moving forward in this demanding arena, several molecular adducts (salts and salt polymorphs) of the model drug ensifentrine (ENSE) with different GRAS (generally recognized as safe) co-former were synthesised for the first time using a mechanochemical technique, followed by a slow evaporation crystallisation procedure. All the newly obtained solid forms were characterized by employing single crystal X-ray diffraction (SCXRD), powder X-ray diffraction (PXRD), thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC). Crystal structure analysis verified salt generation, revealing proton transfer from the carboxylic acid group of salt formers to the mesitylimino nitrogen atom of ENSE. Additionally, the phase transition behaviour of the produced salt polymorphs was examined through variable temperature PXRD (VT-PXRD) analysis. Furthermore, a detailed investigation of the physicochemical features of these recently produced entities was carried out, and their solubility in pH 1.2 and pH 7 environments was examined. Results demonstrate that, as compared to the parent drug, the binary adduct's solubility rate significantly increased at pH 7. Moreover, a thorough examination of the residue recovered after solubility confirmed that the majority of the molecular adducts were stable at pH 7 and did not show any phase change or dissociation, whereas at pH 1.2, the majority of the adducts were stable, except for those generated with malonic acid, which moved to a new stable form—a comprehensive study revealed that it was converted into ENSE·Cl salt. To the best of our knowledge, this is the first study to investigate various forms of ENSE, demonstrating that mechanical energy can be employed as a powerful control parameter to produce novel solid forms with superior physicochemical features. We hope that the current discovery will offer a valuable outlook prior to ENSE drug formulation.
1. Introduction
The metabolism and effectiveness of pharmacological substances largely depend on their solubility and dissolution rate. Thus, one of the most difficult tasks in the pharmaceutical business is to create a cost-effective way to increase the solubility and dissolution rate of active pharmaceutical ingredients (APIs).1–5 The discovery of novel polymorphs, amorphous forms, solid dispersions, cocrystals, salt forms, and inclusion complexes, among others, is now being investigated.6–15 Cocrystals and salt forms are gaining popularity in the pharmaceutical sector owing to their ability to enhance the physicochemical characteristics of APIs.16 Controlling cocrystal and salt characteristics is achievable considering the wide variety of (generally recognized as safe) GRAS co-former properties and potential interactions in the solid state.17–19 There are several approaches and procedures for creating cocrystals.20–24 Nowadays, solid-state grinding—also referred to as mechanochemical synthesis—has emerged as a fascinating substitute process for cocrystal formation.25–30 It is a more appealing and environmentally friendly method for creating a wide range of molecular adducts. Since it is considered an environmentally benign process that can be carried out in the absence of solvent or with very small amounts of solvent (catalytic amounts), it has grown in importance as a synthetic tool across a variety of industries. It can replace traditional solution reactions, which frequently require large volumes of solvents, in several situations. Numerous areas of chemistry, including catalysis, organic synthesis, the creation of metal complexes, and the fabrication of multicomponent medicinal solid forms, have greatly benefited from these sustainable procedures.31–40 More crucially, preparation from solution may be the sole workable method in situations when identifying the structure demands proper single crystals. However, due to the time-consuming solvent selection method, the likelihood of multi-component crystallisation, the formation of solvates, and the overall lack of environmental friendliness, mechanochemical synthesis is the only alternative.
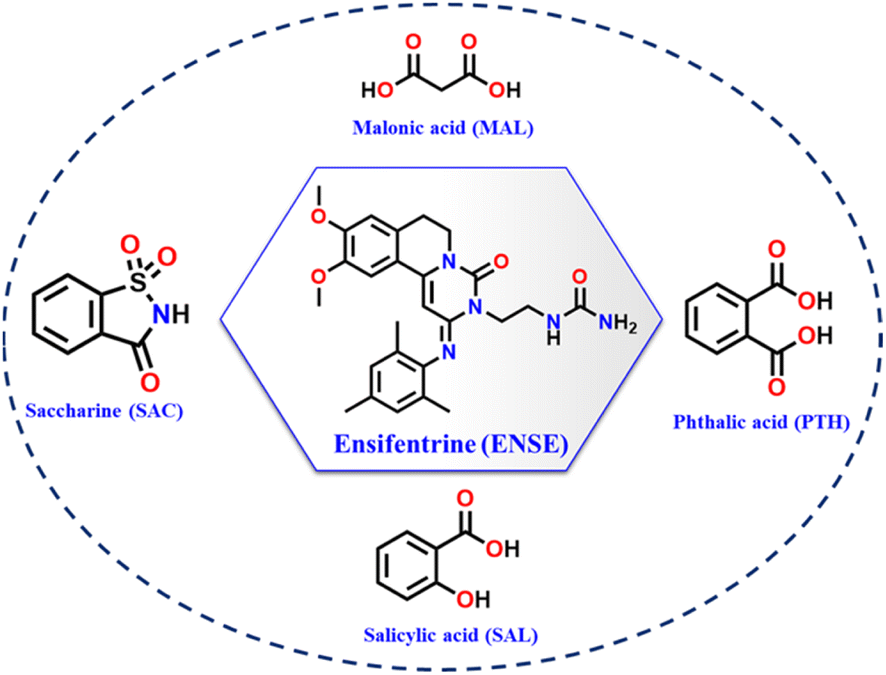
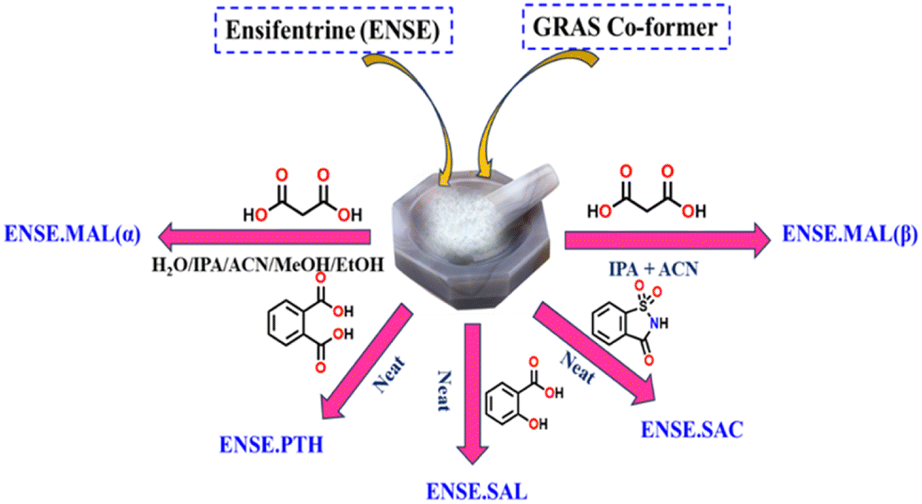
Ensifentrine (ENSE) is a bifunctional dual phosphodiesterase 3/4 inhibitor with bronchodilatory and anti-inflammatory properties.41 It is currently undergoing clinical trials to treat respiratory inflammatory diseases, such as chronic obstructive pulmonary disease (COPD) and other related conditions.42,43 Due to ENSE's potential advantages in the treatment of respiratory issues, inhalation is the preferred method of administration. However, ENSE cannot fulfill its intended therapeutic use because of its poor water solubility (0.001 mg mL−1).44 Even though there are currently very few articles and patents focusing on the creation of different solid forms of ENSE, the field is still in its infancy.44,45 Therefore, our goal was to enhance ENSE solubility by crystal engineering utilising several GRAS co-formers (Scheme 1). We predicted that ENSE and GRAS former would interact via hydrogen bonds, with proton transfer dependent on ΔpKa values (Table S1†). The current study used both NEAT and LAG (Liquid Assisted Grinding) methods depending on the solvent required to generate the molecular adducts. Interestingly, in most cases, molecular adducts were acquired by neat grinding, but in the case of MAL, LAG with certain selected solvents imparted salt polymorphs (Fig. 2). A schematic illustration of the obtained salt and salt polymorphs of ENSE is presented in Fig. 1. In addition, the grounded mixture was exposed to slow evaporation to produce high-quality crystals that aid in gaining a comprehensive understanding of the many interactions involved in stabilizing the structure and altering its properties. Tables 1 and S2† provide details on the geometrical characteristics of hydrogen bonding and crystallographic data. In addition to the aforementioned GRAS co-former (Scheme 1), we tested different aliphatic and aromatic carboxylic acid-based GRAS co-formers, but in most cases, we obtained a mixture of the corresponding compounds. The screening of an array of GRAS co-formers and the associated acquired PXRD data are outlined in Fig. S1 and Table S1.†
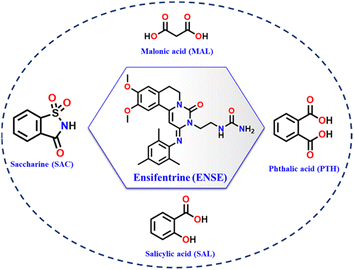 |
| | Scheme 1 Chemical structure of (a) ENSE and salt formers used in this study. | |
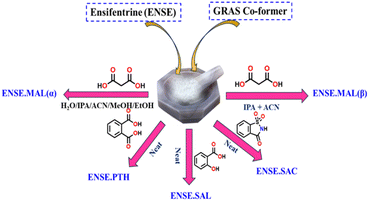 |
| | Fig. 1 An illustration of the obtained salt and salt polymorphs of ENSE. | |
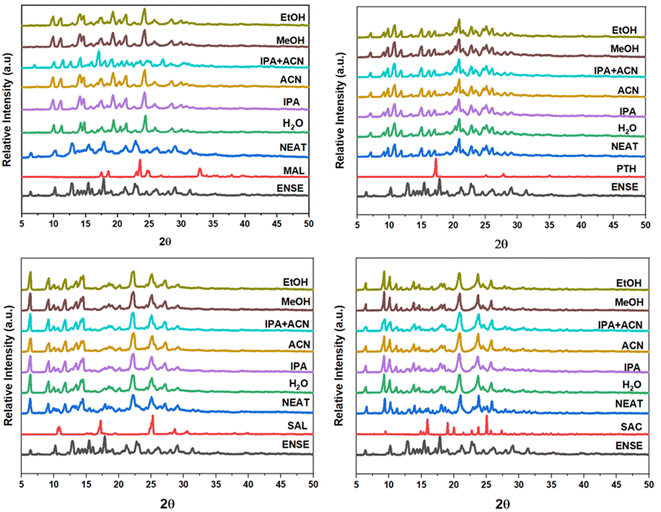 |
| | Fig. 2 PXRD screening method to evaluate the probable production of binary adducts between ENSE and the GRAS co-former. | |
Table 1 Crystallographic parameters of ENSE solid forms
| Parameters |
ENSE·MAL(α) |
ENSE·MAL(β) |
ENSE·PTH |
ENSE·SAL |
ENSE·SAC |
| Formula |
C26H32N5O4:C3H3O4 |
C26H32N5O4:C3H3O4 |
C26H32N5O4:C8H4O4 |
C26H32N5O4:C7H5O3 |
C26H32N5O4:C7H5NO3S |
|
M
r
|
581.62 |
581.62 |
642.68 |
615.68 |
660.74 |
| Crystal shape |
Block |
Plate |
Plate |
Plate |
Plate |
| Crystal colour |
Colourless |
Pale yellow |
Pale yellow |
Colourless |
Colourless |
| Crystal system |
Monoclinic |
Monoclinic |
Monoclinic |
Monoclinic |
Monoclinic |
| Space group |
P21/n |
P21/n |
P21/n |
P21/c |
P21/n |
|
T, K |
273(2) |
273(2) |
273(2) |
273(2) |
273(2) |
|
λ (Mo-Kα)/Å |
0.71073 |
0.71073 |
0.71073 |
0.71073 |
0.71073 |
|
a/Å |
15.925(3) |
8.8751(3) |
10.3617(12) |
8.3541(8) |
8.8693(3) |
|
b/Å |
10.5056(19) |
15.2902(6) |
19.203(2) |
19.1514(17) |
19.0166(7) |
|
c/Å |
17.711(4) |
21.2672(8) |
16.1287(17) |
20.0769(18) |
19.9781(8) |
|
α/° |
90 |
90 |
90 |
90 |
90 |
|
β/° |
90.685(6) |
98.005(1) |
93.901(4) |
100.231(3) |
101.451(1) |
|
γ/° |
90 |
90 |
90 |
90 |
90 |
|
V/Å3 |
2962.9(10) |
2857.75(18) |
3201.8(6) |
3161.1(5) |
3302.5(2) |
|
Z
|
4 |
4 |
4 |
4 |
4 |
|
D
c/g cm−3 |
1.304 |
1.352 |
1.331 |
1.292 |
1.329 |
|
μ, mm−1 |
0.096 |
0.100 |
0.096 |
0.092 |
0.155 |
| 2θ range [°] |
2.25–27.52 |
2.34–27.52 |
2.41–30.52 |
2.32–27.62 |
2.36–27.51 |
| Limiting indices |
−20 ≤ h ≤ 20 |
−11 ≤ h ≤ 11 |
−14 ≤ h ≤ 14 |
−10 ≤ h ≤ 10 |
−11 ≤ h ≤ 11 |
| −13 ≤ k ≤ 13 |
−19 ≤ k ≤ 19 |
−27 ≤ k ≤ 27 |
−24 ≤ k ≤ 24 |
−24 ≤ k ≤ 24 |
| −23 ≤ l ≤ 23 |
−26 ≤ l ≤ 26 |
−23 ≤ l ≤ 23 |
−26 ≤ l ≤ 26 |
−25 ≤ l ≤ 25 |
|
F(000) |
1232 |
1232 |
1352 |
1300 |
1392 |
| Total reflections |
47![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 388 388 |
41406 |
60755 |
57687 |
49152 |
| Unique reflections |
6817 |
6582 |
9795 |
7344 |
7599 |
| Reflection at I > 2σ (I) |
2938 |
3694 |
4723 |
3446 |
4510 |
| No. of parameters |
402 |
394 |
430 |
417 |
438 |
|
R
1, I > 2σ (I) |
0.0571 |
0.0512 |
0.0750 |
0.0631 |
0.0545 |
| wR2I > 2σ (I) |
0.1710 |
0.1477 |
0.2404 |
0.2316 |
0.1819 |
| GoF on F2 |
1.008 |
1.026 |
0.983 |
1.059 |
1.058 |
| CCDC no. |
2358076
|
2358075
|
2358078
|
2358077
|
2358074
|
2. Experimental section
2.1. Materials
All the chemicals were purchased from Sigma-Aldrich with >99% purity and were used without additional purification. The solvents used in the crystallisation investigations were of the spectroscopy grade of the highest available quality. Ensifentrine drug was synthesised in the lab according to the patent WO 2018/020249 A1 and US6794391 B2.46,47
2.2. Solid form screening
ENSE (47.75 mg, 0.1 mmol) and an equivalent amount of co-formers (Scheme 1) were crushed in a mortar pestle for around 30 minutes, while the grinding was done with two drops of water in the case of ENSE·MAL(α) and the mixture of isopropanol (IPA) and acetonitrile (ACN) in the case of ENSE·MAL(β), respectively. Furthermore, the powdered mixture was heated in a mixture of IPA and ACN solution in a water bath and allowed to evaporate slowly at ambient temperatures. Within 48 hours, good-quality single crystals appropriate for X-ray diffraction analysis were produced.
2.3. Single crystal X-ray diffraction (SCXRD)
The single-crystal data were collected using a Bruker diffractometer. The diffractometer is equipped with an APEX CCD area detector using graphite-monochromated MoKα radiation (λ = 0.71073 Å). The data were collected in φ and ω scan modes at room temperature. In all the cases, the process was smooth, and the crystals were found to be stable throughout the experiment. Bruker suite of data processing programme (SAINT) (SAINT; Bruker AXS, I. Analytical X-ray Systems 2000, 5373) was used to process the intensity data, and absorption corrections were applied using SADABS (SADABS; Siemens Industrial Automation, I. 1996). The structure solution for all the complexes was carried out by applying direct methods, and refinements were performed by full matrix least squares on F2 using the SHELXTL-PLUS suite of programs.48 All the non-hydrogen atoms were refined anisotropically, and all the hydrogen atoms were fixed using the AFIX constraint.
2.4. Powder X-ray diffraction (PXRD)
Powder X-ray diffraction (PXRD) data were collected using a Bruker (D8 Advance) diffractometer with Cu-Kα radiation (λ = 1.54060 Å). An X-ray generator set up at 40 kV and 30 mA was used to collect intensity data with a step size of 0.0170 (2θ) in a continuous scanning mode. Diffraction patterns were collected in the 2θ range of 5–50° at room temperature.
2.5. Thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC) measurements
The thermal analysis was performed using thermogravimetric analysis (TGA) (Model 4000, PerkinElmer) and differential scanning calorimetry (DSC) (Model 6000, PerkinElmer). The samples were put into Al2O3 crucible plates for TG measurements and sealed in aluminium plates for DSC ones. Powder samples (∼5.0–10.0 mg) were heated from 35 to 500 °C with a heating rate of 10 °C min−1, and a 20 mL min−1 N2 flow was performed as the purge gas.
2.6. Hirshfeld surface (HS) analyses
The Crystal Explorer package ver. 3.1.49 was used to calculate the Hirshfeld surface (HS) and 2D fingerprint (2D FP). A color scale of −0.407–1.424 Å was used to map the dnorm surface.
2.7. Solubility measurements using UV-visible spectroscopy
The equilibrium solubility of ENSE and all generated molecular salts were assessed at room temperature at 298 K in a solution of 0.1 N HCl (pH 1.2) and Millipore water (pH 7). UV visible spectroscopy was used to determine equilibrium solubility. Beer–Lambert's rule, A = εCl, was utilised to estimate the extinguishment coefficients (ε) for each of the solid samples by producing standard solutions and charting the absorbance (A) against the concentration (C) curve. Using the shake-flask method, which involves shaking an excess amount of the solid sample in 5 mL of pH 1.2 and pH 7 solution for 24 hours at room temperature, equilibrium solubility values were estimated. After that, the saturated solutions were diluted and passed through a syringe micron filter (0.45 μm). Subsequently, the saturated solutions underwent filtration using a syringe micron filter (0.45 μm) and were diluted to obtain the desired absorbance value. By multiplying the concentration of the diluted solution by the dilution factor, the equilibrium solubility of the resulting saturated solution was computed. After filtering, the residue was dried in the open, and PXRD was used to confirm the stability of the powdered components.
3. Results and discussion
All molecular salts generated anhydrous complexes and had a 1:1 stoichiometric ratio of ENSE and the corresponding salt former (Fig. S3†). Crystal structure analysis verified salt generation, revealing proton transfer from the carboxylic acid group of salt formers to the mesitylimino nitrogen atom of ENSE. Furthermore, all the molecular adducts of ENSE crystallized in a monoclinic system with space group, P21/n, while ENSE·SAL crystallized in the same crystal system with a different space group (P21/c). The analysis of the crystal packing is comparable and isostructural, which is strongly correlated with their similar crystallographic parameters.
3.1. Structural investigation of salt polymorphs of ENSE (ENSE·MAL(α) and ENSE·MAL(β))
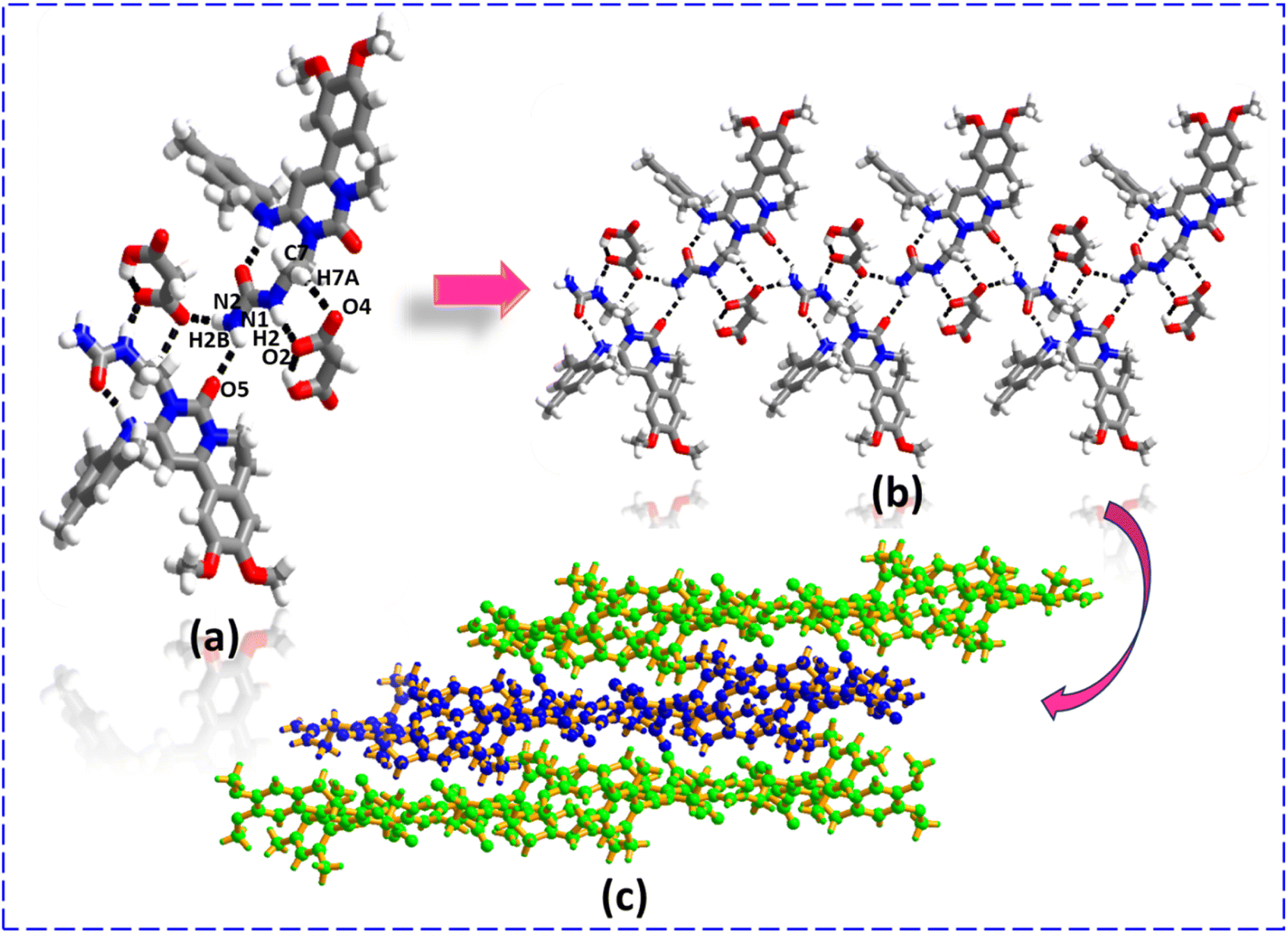
Utilising a selective solvent, the LAG technique yields two salt polymorphs of ENSE with MAL. ENSE·MAL(α) was produced using H2O, while ENSE·MAL(β) was produced with a 1:1 combination of IPA and ACN. In both molecular complexes, molecular recognition and packing are completely different. In ENSE·MAL(α), the MAL interacts with ENSE via N1–H1B⋯O2 (H1B⋯O2, 2.06 Å), N2–H100⋯O1− (H100⋯O1−, 2.01 Å) hydrogen bonds (formation of R22 (8) ring motif) and C15–H15⋯O3 (H15⋯O3, 2.30 Å) hydrogen bonds to form a trimeric network (Fig. 3(a)), extending along the crystallographic axis to form a cyclic entity (Fig. 3(b)). Furthermore, these cyclic entities interact with each other via C9–H101⋯O2 (H101⋯O2, 2.42 Å) hydrogen bonds, forming a honeycomb network structure (Fig. 3(c)). In the case of ENSE·MAL(β), ENSE interacts with MAL via dimeric N1–H2⋯O2− (H2⋯O2−, 1.98 Å), C7–H7A⋯O4 (H7A⋯O4, 2.57 Å) hydrogen bonds (formation of R22 (9) ring motif) and N2–H2B⋯O5 (H2B⋯O5, 2.50 Å) monomeric hydrogen bonds. These interactions resulted in a tetrameric network (Fig. 4(a)), which was further extended along a crystallographic axis to form a molecular tape (Fig. 4(b)). Additionally, these molecular tapes form a three-dimensional stacked layer structure by stacking on top of one another (Fig. 4(c)).
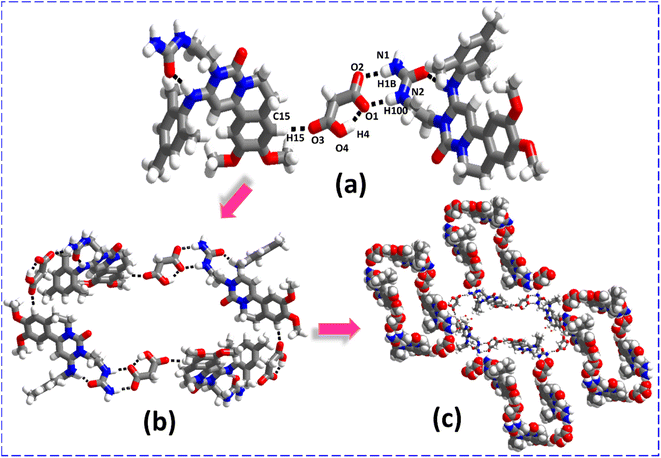 |
| | Fig. 3 (a) Molecular recognition among the binary adducts of the ENSE·MAL(α) form. (b) Cyclic network structure of the binary adduct. (c) Honeycomb network architecture of the binary adduct. | |
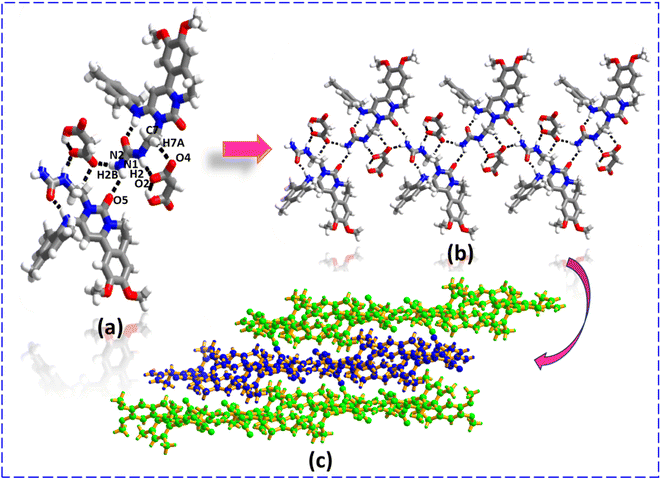 |
| | Fig. 4 (a) Molecular recognition among the binary adducts of the ENSE·MAL(β) form. (b) Formation of the molecular tape. (c) 3D stacked layer structure. | |
3.2. Structural analysis of ENSE·PTH
Furthermore, as demonstrated by earlier complexes, ENSE with PTH has a completely distinct recognition pattern. The ENSE molecule maintains its homomeric interaction via C2–H2E⋯O2 (H2E⋯O2, 2.57 Å) hydrogen bonds, while the interaction between ENSE and salt former occurs via N1–H1A⋯O8 (H1A⋯O8, 2.18 Å) and N2–H2B⋯O7− (H2B⋯O7−, 2.12 Å) hydrogen bonds through R22 (9) ring motif and forms a molecular tape (Fig. 5(a)). Fig. 5(b) illustrates the cyclic network created by continued interaction in a twisting fashion. Furthermore, N2–H2A⋯O3 (H2A⋯O3, 2.26 Å) hydrogen bonds allow these cyclic networks to expand throughout the crystal lattice and create cyclic molecular chains (Fig. 5(c)).
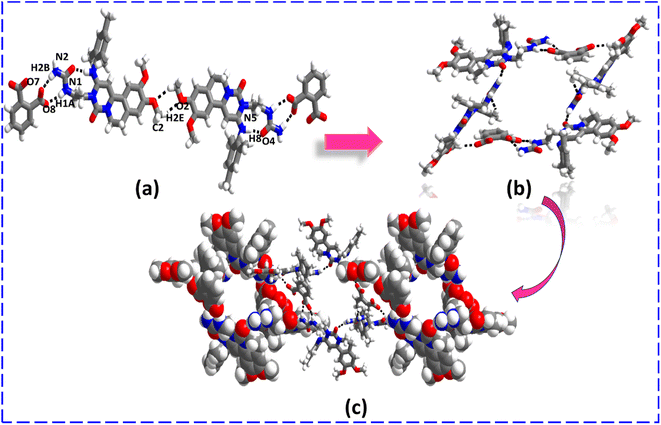 |
| | Fig. 5 (a) Molecular recognition among the binary adducts (ENSE and PTH). (b) Cyclic network structure of the binary adduct. (c) Formation of the cyclic molecular chain. | |
3.3. Structural analysis of ENSE·SAL and ENSE·SAC
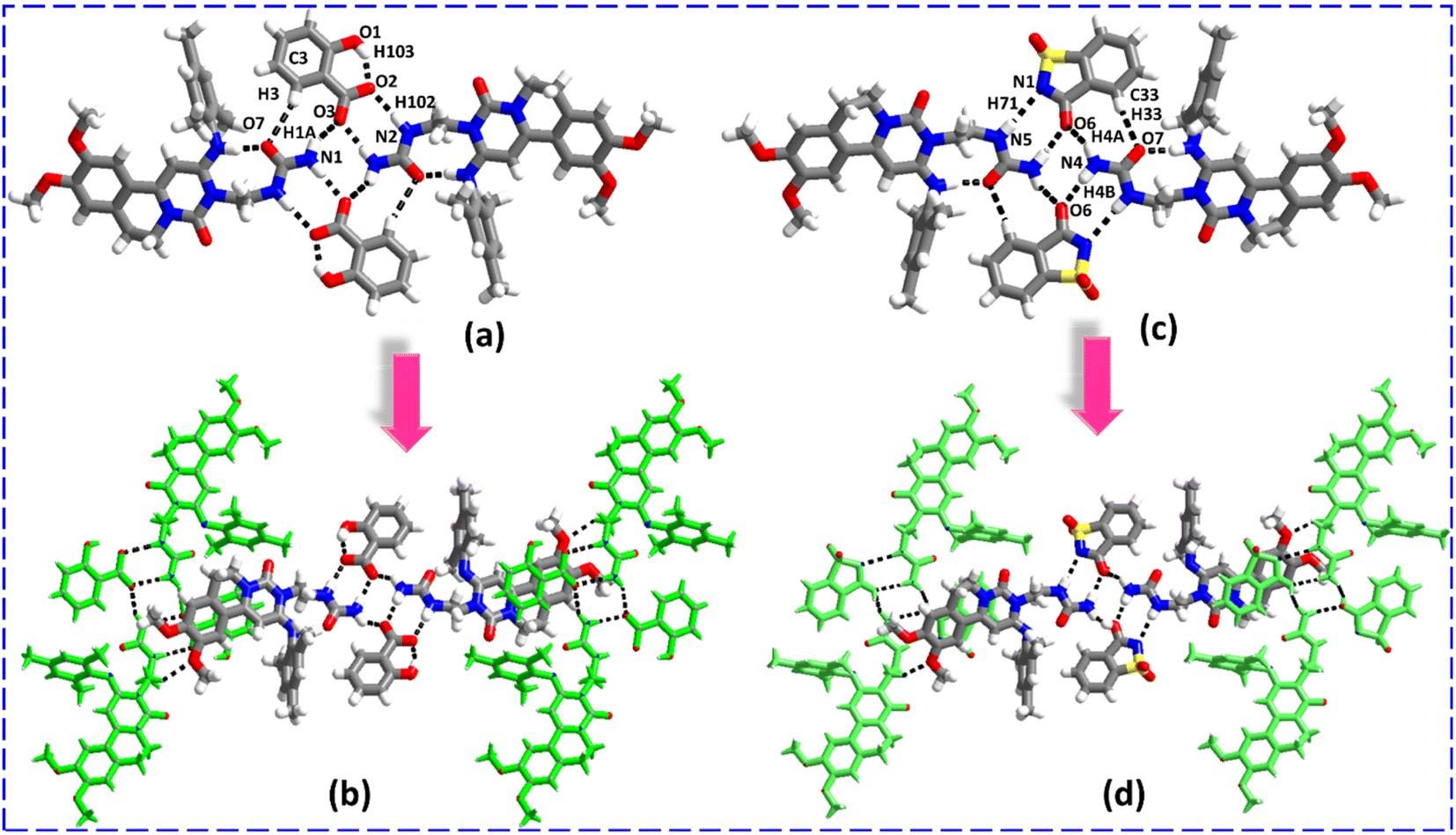
Analysis of crystal packing and interactions reveals that the recognition patterns of ENSE·SAL and ENSE·SAC are identical and isostructural although with different salt formers. In both molecular complexes, the urea moiety of ENSE established heteromeric interactions with SAL and SAC through C3–H3⋯O7 (H3⋯O7, 2.90 Å), N1–H1A⋯O3 (H1A⋯O3, 2.20 Å), N2–H102⋯O2− (H102⋯O2−, 2.04 Å), N1–H1B⋯O3 (H1B⋯O3, 1.99 Å) hydrogen bonds (in the case of ENSE·SAL), N1–H71⋯N5− (H71⋯N5−, 2.44 Å), N4–H4B⋯O6 (H4B⋯O6, 2.02 Å), N4–H4A⋯O6 (H4A⋯O6, 2.25 Å), and C33–H33⋯O7 (H33⋯O7, 2.62 Å) hydrogen bonds (in case of ENSE·SAC) with the formation tetrameric network via R22 (9), R22 (8), R22 (8), and R22 (9) ring motif, respectively (Fig. 6(a) and (c)). Furthermore, these tetrameric networks interact with each other via C19–H19B⋯O4 (H19B⋯O4, 2.47 Å) (in the case of ENSE·SAL) and C15–H15B⋯O2 (H15B⋯O2, 2.40 Å) (in the case of ENSE·SAC) hydrogen bonds produce a molecular tape structure (Fig. 6(b) and (d)).
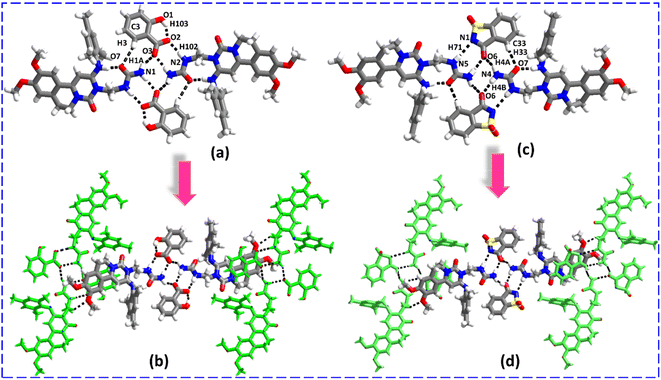 |
| | Fig. 6 (a and c) Molecular recognition among the binary adducts (ENSE and SAL; ENSE and SAC). (b and d) Molecular tape structure of the binary adduct. | |
3.4. Thermal analysis
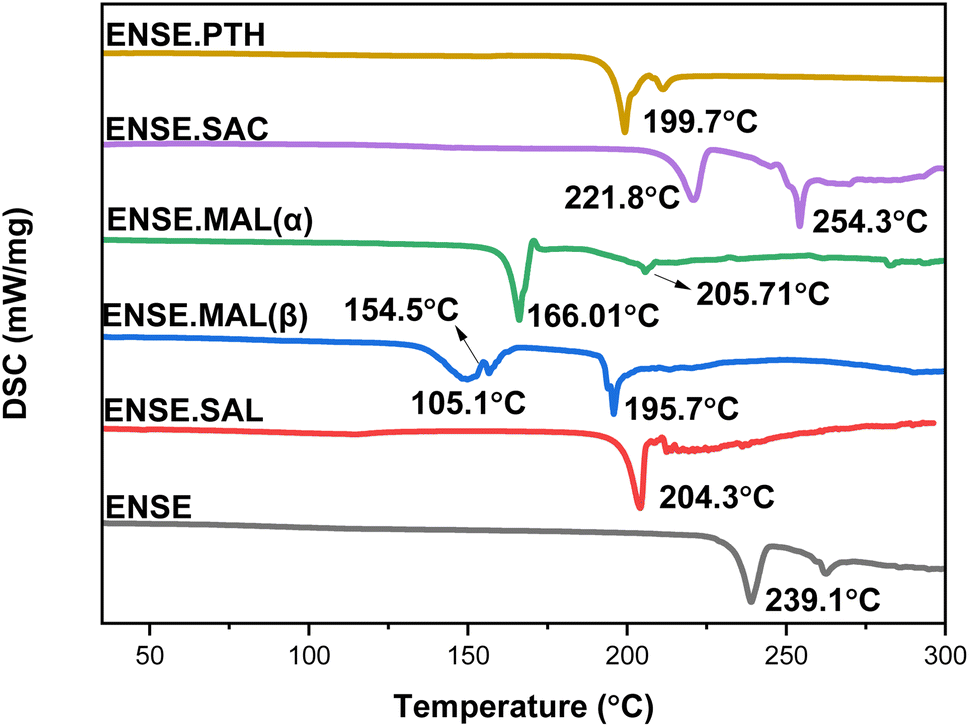
The thermal behaviour of these recently obtained molecular salts was investigated using TGA and DSC. Fig. 7 and S4† illustrate an overview of the DSC and TGA profiles. The TGA plots (Fig. S5†) for Form I–III demonstrate that there is no weight loss prior to 150 °C, indicating the absence of solvent residue in these adducts. Furthermore, in the instance of ENSE·PTH, a sudden weight drop was observed from 210 °C to 228 °C, indicating that the molecule disintegrated. Similarly, to assess the nature of a phase transition, the associated DSC profiles were analysed. There was no phase transition in ENSE·SAL, ENSE·MAL(α), and ENSE·PTH, as evidenced by the absence of endothermic or exothermic peaks in the DSC profiles of all forms prior to the melting point. In contrast, ENSE·MAL(β) showed a broad endothermic peak at 150.1 °C, followed by a tiny exotherm transition at 154.5 °C, followed by melting at 195.7 °C. Similarly, in the case of ENSE·SAC, two endothermic peaks at 221.8 °C and 254.3 °C were observed. To delve deeper into the phase transition, variable-temperature PXRD (VT-PXRD) was used.
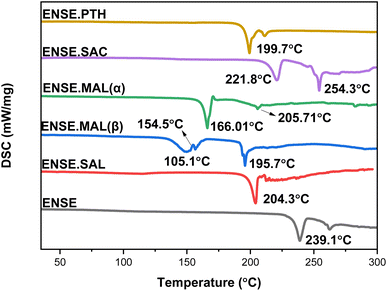 |
| | Fig. 7 DSC profiles of various molecular adducts of ENSE. | |
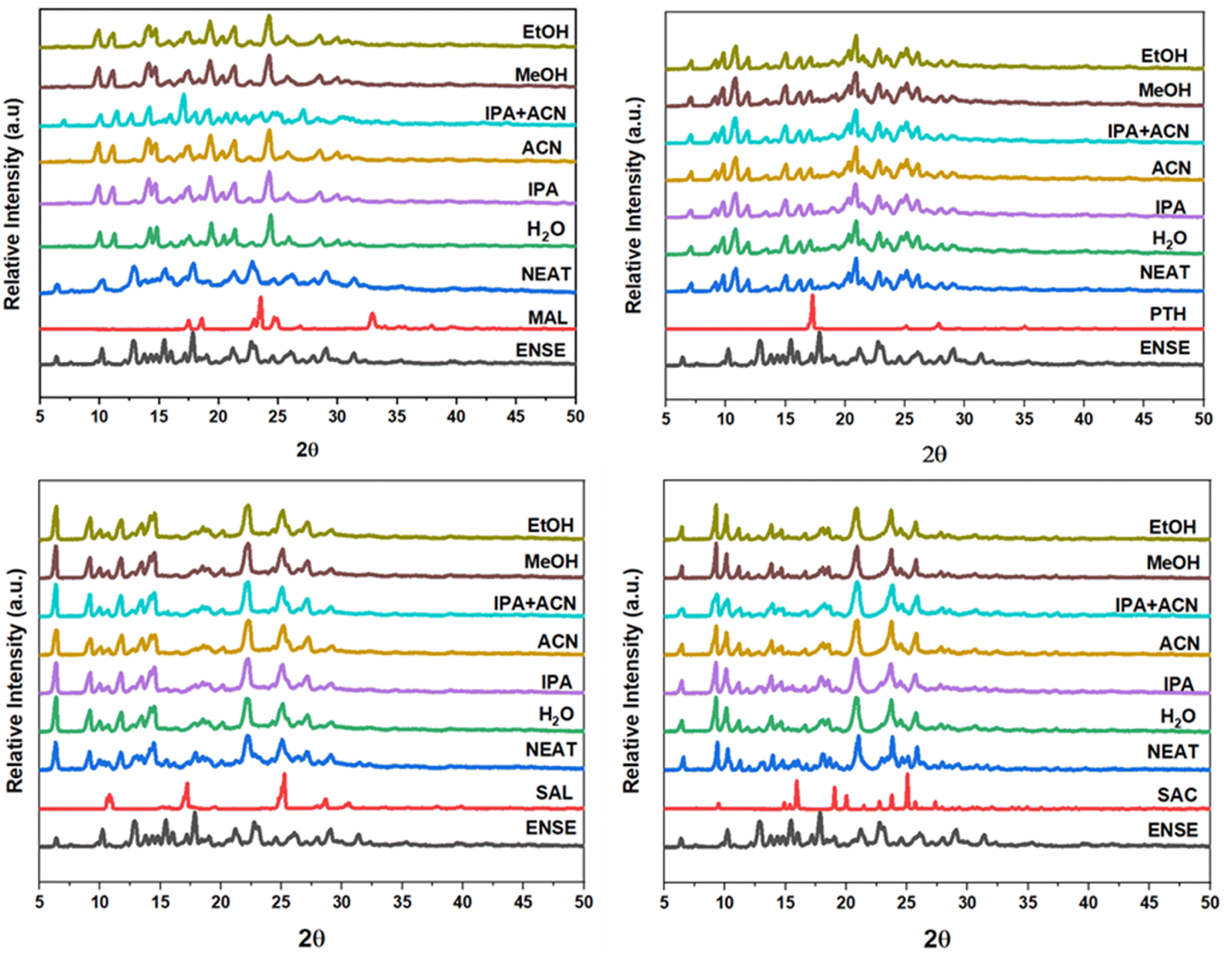
3.5. Analysis of form transitions by variable-temperature PXRD (VT-PXRD)
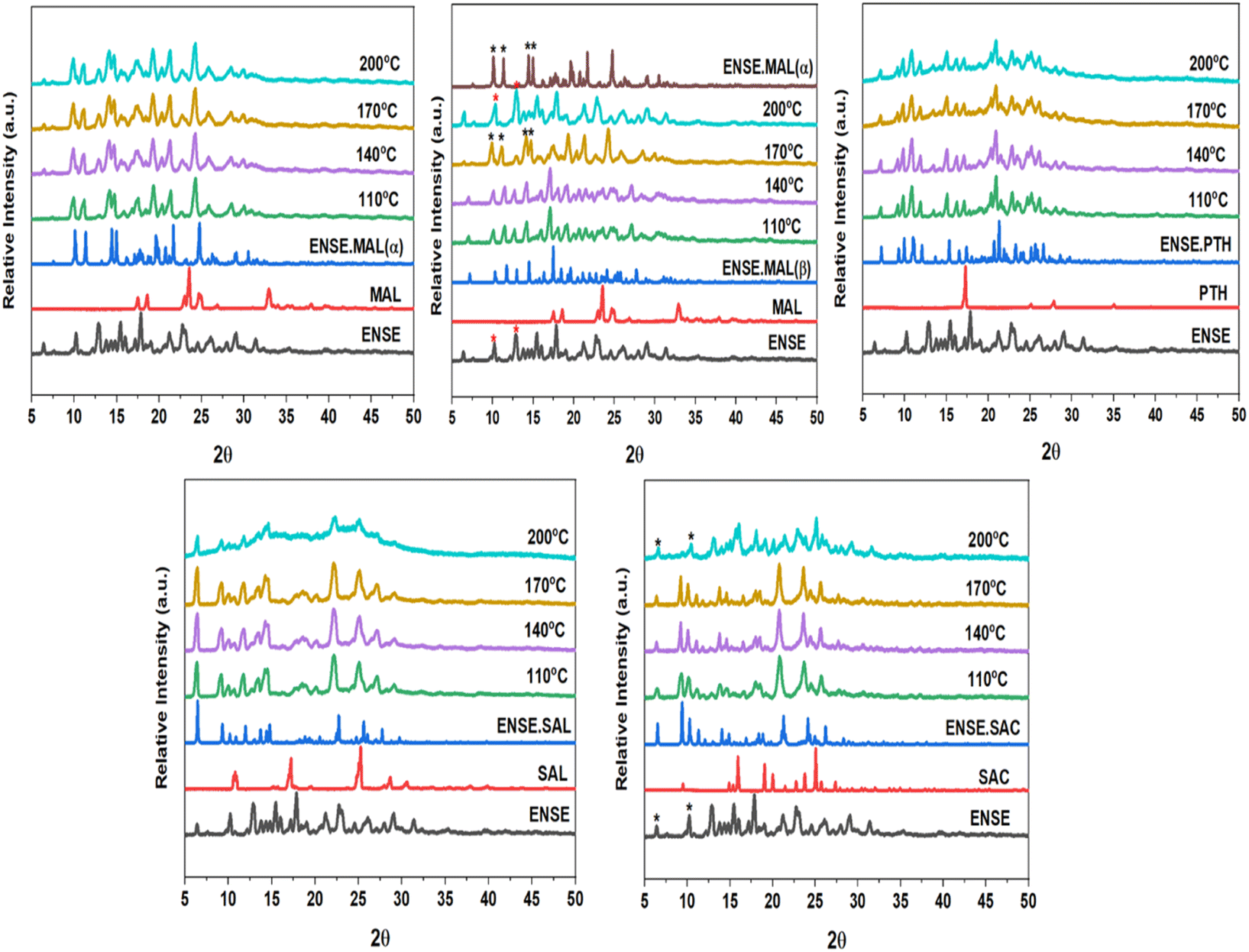
Precise kinetic and thermodynamic processes govern the phase transition of a material from one crystal form to another in a crystalline molecular adduct. These alterations might occur spontaneously as a result of changes in temperature or pressure, or they can be induced by external stimuli, such as solvent interactions or mechanical force. Variable temperature PXRD (VT-PXRD) plots were taken between 110 and 200 °C to gain a better understanding of the thermal stability of the unique genetic solid forms of ENSE. The majority of the adduct's PXRD patterns exhibited no apparent alteration. However, it is noteworthy to note that at 170 °C, ENSE·MAL(β) was converted into ENSE·MAL(α), while at 200 °C, it shifted to another form. Furthermore, the PXRD stacking pattern of the newly acquired form is closely related to the pure drug ENSE. Similarly, ENSE·SAC is stable at 170 °C, but beyond 170 °C, it takes on a new shape, and the PXRD pattern corresponds to that of the pure drug ENSE. Therefore, we may assume that the substance begins to disintegrate above 170 °C. Fig. 8 illustrates a schematic representation of the distinct phase transitions and stability observed in the various types of ENSE molecular salts.
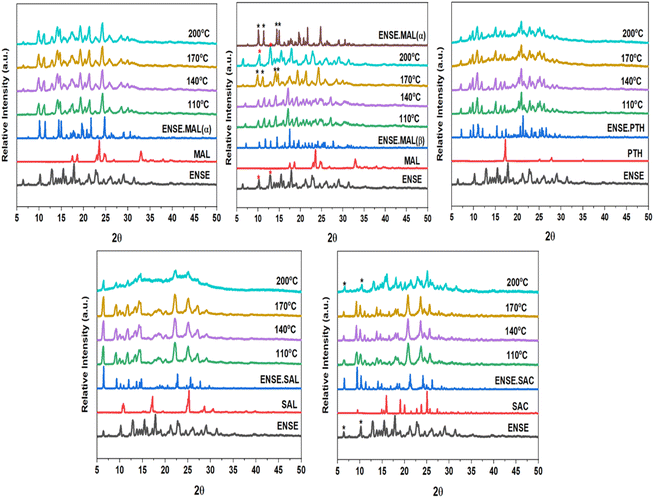 |
| | Fig. 8 VT-PXRD overlay of different adducts of ENSE. | |
3.6. Hirshfeld (HS) analysis
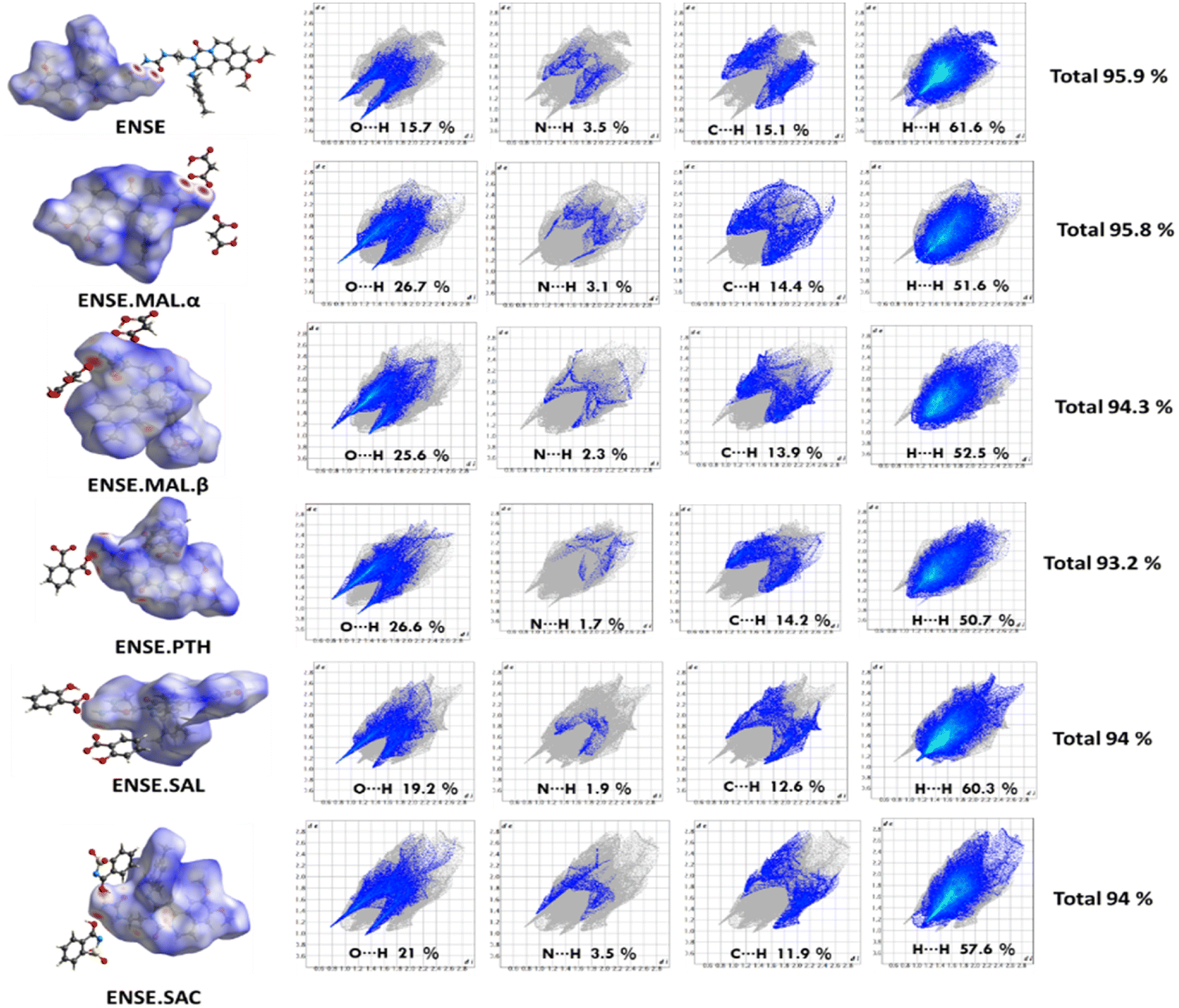
The Hirshfeld surface is frequently peculiar to a certain crystal structure, which raises the possibility of obtaining novel insights into various molecular crystal forms. It is also useful for developing a thorough comprehension of the hydrogen-bonding interactions between molecules that occur in patterns of crystal stacking. All the crystalline materials in this investigation had their HS analyses completed using the CrystalExplorer programme, and the resulting 2D fingerprint plots and 3D maps are shown in Fig. 9. A fingerprint plot represents the HS surface in two dimensions, with de and di representing the separations between the nearest atoms within and outside, respectively. Three colours (red, blue, and white) are used to map the HS surface to the 3D dnorm values: contacts with red regions are closer together and have a negative dnorm value; contacts with blue regions are farther apart and have a positive dnorm value; and contacts with white regions are separated by van der Waals radii and have a zero dnorm value. Fig. 9 displays the overall percentages of strong intermolecular interactions (O⋯H, N⋯H, C⋯H and H⋯H). This indicates that between ENSE·SAL and ENSE·SAC the total percentage contribution is equal, indicating that both are isostructural and that their involvement in total molecular interaction is the same. This is also well consistent with the crystal molecular packing pattern. Similarly, the produced salt polymorphs also demonstrated the participation of various interaction patterns.
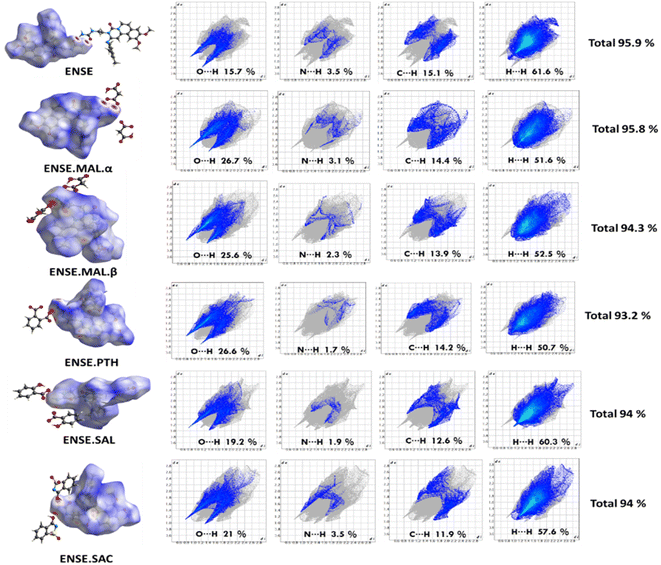 |
| | Fig. 9 HS 3D maps and 2D fingerprints; the total% of the contact population involved in ENSE and its molecular salts. | |
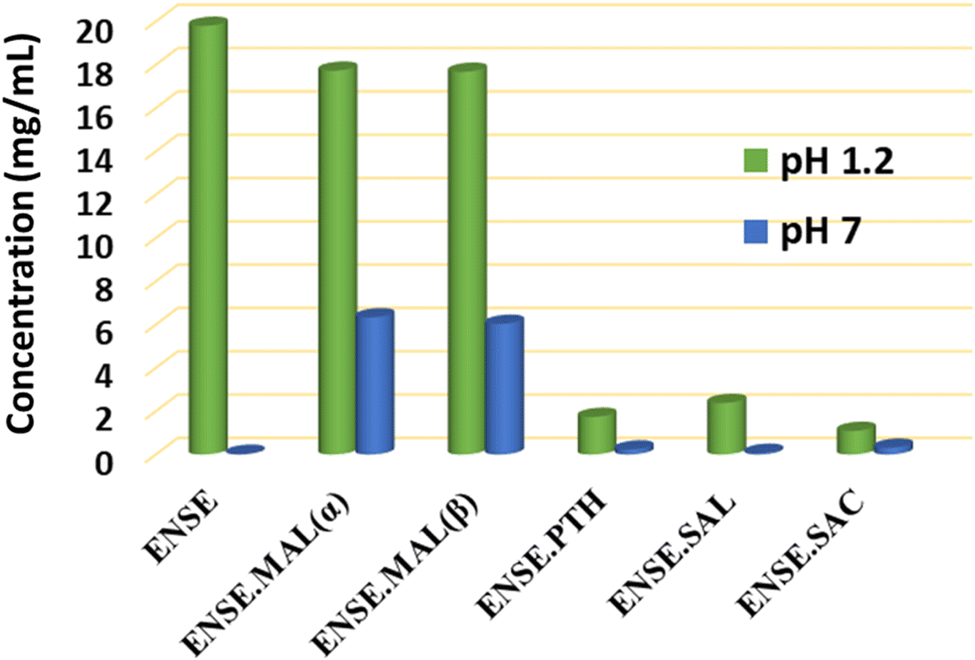
3.7. Solubility study
Despite having various therapeutic effects, ENSE's low solubility (∼0.001 mg mL−1) limits its clinical application due to limited bioavailability. The solubility investigation was conducted in Millipore water (pH = 7) and 0.1 N HCl (pH = 1.2). Prior to the solubility test, the overall purity of the powder materials was confirmed by PXRD measurement (Fig. S4†). A λmax value of 356 nm was used to calculate the absorbance of all the solid adducts of ENSE in both media (see ESI†). The collected data are illustrated in Table 2 and Fig. 10. The results demonstrated that at pH 7, all adducts are more soluble than the pure drug ENSE, with the maximum solubility in the case of ENSE·MAL(α) and ENSE·MAL(β), which is eighteen times more than the original term ENSE. However, three solid forms (ENSE·PTH, ENSE·SAL and ENSE·SAC) displayed a decrease in solubility at pH 1.2. As we know, many variables impacting the solubility of drugs include pH levels, polarity of drug and solvent, particle size, solution process, participation of different intermolecular interactions, and molecular packing. The solubility of medicine can alter with the pH of the solution; consequently, various solubility trends are observed in the two different pH mediums and the maximum solubility of ENSE·MAL might be attributable to a lower melting point than other molecular adducts or to the engagement of fewer intermolecular contacts than other adducts. Similarly, molecular recognition and intermolecular interactions play an important role in solubility. Therefore, the molecular recognition pattern of the resulting adducts was compared to that of the original drug (Fig. S7†). It is noteworthy that, in most cases, the dimeric contacts between ENSE molecules present in the parent drug are lacking. This might be one of the reasons the parent medication has a higher melting point at pH 7 and is less soluble than other drugs. To further understand the causes of the solubility discrepancy at both pH levels, PXRD data (Fig. S6†) for solid residues were acquired. The PXRD data show that while the majority of the solid forms remain stable at pH 7, ENSE·MAL and parent drug ENSE transformed into a new form of ENSE at pH 1.2. To obtain a deeper comprehension of the novel form, the solid residue was recrystallized, and crystal data were collected using SCXRD. According to the findings, it created an ENSE·Cl salt (Fig. S8 and S9†). The crystal structure and crystallographic details of the newly discovered ENSE·Cl salt are included in ESI.†
Table 2 Solubility parameters of ENSE and its molecular adducts
|
|
pH 1.2 |
pH 7 |
| Absorbance |
Equilibrium solubility (24 h, mg mL−1) |
Final solid phase |
Absorbance |
Equilibrium solubility (24 h, mg mL−1) |
Final solid phase |
| ENSE |
1.434 |
19.795 |
SALT |
1.884 |
0.029 |
ENSE |
| ENSE·MAL(α) |
1.284 |
17.722 |
SALT |
0.402 |
6.329 |
ENSE·MAL(α) |
| ENSE·MAL(β) |
1.281 |
17.671 |
SALT |
0.383 |
6.029 |
ENSE·MAL(β) |
| ENSE·PTH |
0.126 |
1.737 |
ENSE·PTH |
0.361 |
0.228 |
ENSE·PTH |
| ENSE·SAL |
0.172 |
2.375 |
ENSE·SAL |
0.116 |
0.073 |
ENSE·SAL |
| ENSE·SAC |
0.079 |
1.086 |
ENSE·SAC |
0.309 |
0.309 |
ENSE·SAC |
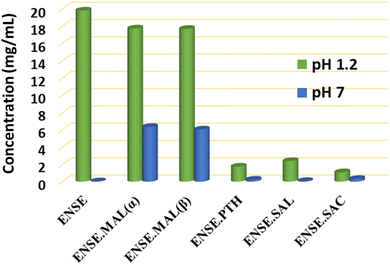 |
| | Fig. 10 Solubility profile of ENSE and its molecular adducts at pH 1.2 and pH 7. | |
4. Conclusions
For the first time, multiple molecular adducts of ensifentrine, an anti-inflammatory medicine, have been successfully synthesised using a mechanochemical approach and characterised using various analytical techniques. The transition from molecular salt to salt polymorphs highlights the importance of the mechanical approach, solvent selection and temperature. Furthermore, solubility tests on synthesised molecular adducts were conducted at various physiological pH levels to investigate the impact of molecular adducts on in vitro release behaviours. In most circumstances, the solubility of molecular adducts exceeds that of the parent medicine. As far as we know, this is the first study to develop different salt forms of ENSE, and mechanical energy may be used as a potent control parameter to create new solid forms with better physicochemical properties. We believe that this new discovery may provide some important insight prior to the ENSE medication development.
Data availability
The authors confirm that the data supporting the findings of this study are available within the article and its ESI.†
Author contributions
The authors acknowledge ICT-IOC, Bhubaneswar for providing necessary support. Rambabu Dandela thanks DST-SERB for Ramanujan fellowship (SB/S2/RJN-075/2016), Core research grant (CRG/2018/000782), and ICT-IOC startup grant.
Conflicts of interest
There are no conflicts to declare.
Notes and references
- J. M. M. Salmani, H. Lv, S. Asghar and J. Zhou, Pharm. Dev. Technol., 2015, 20, 465–472 CrossRef CAS PubMed.
- C. M. Bento, A. T. Silva, B. Mansano, L. Aguiar, C. Teixeira, M. S. Gomes, P. Gomes, T. Silva and R. Ferraz, Int. J. Mol. Sci., 2023, 24, 1402 CrossRef CAS PubMed.
- L. Liu, J.-R. Wang and X. Mei, CrystEngComm, 2022, 24, 2002–2022 RSC.
- D. N. Ridhurkar, K. A. Ansari, D. Kumar, N. S. Kaul, T. Krishnamurthy, S. Dhawan and R. Pillai, Drug Dev. Ind. Pharm., 2013, 39, 1783–1792 CrossRef CAS.
- T. Kipping and H. Rein, Int. J. Pharm., 2016, 497, 36–53 CrossRef CAS.
- X. Shi, Y. Deng, Z. Wang, X. Liu, Q. Chen, J. Peng, X. Xing and W. Su, J. Drug Delivery Sci. Technol., 2023, 84, 104511 CrossRef CAS.
- A. Salem, A. Hagymási, B. Vörös-Horváth, T. Šafarik, T. Balić, P. Szabó, F. Gősi, S. Nagy, S. Pál, S. Kunsági-Máté and A. Széchenyi, Eur. J. Pharm. Sci., 2021, 156, 105599 CrossRef CAS PubMed.
- A. Mathers, M. Pechar, F. Hassouna and M. Fulem, Int. J. Pharm., 2022, 623, 121855 CrossRef CAS.
- S. Khan, M. Rahman, H. M. Marwani, R. H. Althomali and M. M. Rahman, Z. Phys. Chem., 2024, 238, 291–311 CrossRef CAS.
- A. Kar, L. Giri, G. Kenguva, S. R. Rout and R. Dandela, CrystEngComm, 2023, 25, 1874–1883 RSC.
- S. R. Rout, G. Kenguva, L. Giri, A. Kar and R. Dandela, Chem. Commun., 2023, 59, 4640–4643 RSC.
- L. Giri, S. R. Rout, A. Kar, G. Kenguva and R. Dandela, J. Mol. Struct., 2022, 133746 CrossRef CAS.
- J. Y. Chen, H. Wu, C. Y. Guo, B. Zhu and G. Bin Ren, Int. J. Pharm., 2019, 572, 118776 CrossRef CAS.
- M. Banerjee and B. Brettmann, Pharm., 2020, 12, 995 CAS.
- M. Nowak, A. J. Dyba, J. Janczak, A. Morritt, L. Fábián, B. Karolewicz, Y. Z. Khimyak, D. E. Braun and K. P. Nartowski, Mol. Pharm., 2022, 19, 456–471 CrossRef CAS.
- G. Bolla and A. Nangia, Chem. Commun., 2016, 52, 8342–8360 RSC.
- A. Rehman, A. Delori, D. S. Hughesa and W. Jones, IUCrJ, 2018, 5, 309–324 CrossRef CAS PubMed.
- F. Leng, K. Robeyns and T. Leyssens, Pharmaceutics, 2021, 13, 671 CrossRef CAS PubMed.
- M. K. Gautam, M. Besan, D. Pandit, S. Mandal and R. Chadha, AAPS PharmSciTech, 2019, 20, 1–17 CrossRef CAS PubMed.
- S. Aher, R. Dhumal, K. Mahadik, J. Ketolainen and A. Paradkar, Pharm. Dev. Technol., 2013, 18, 55–60 CrossRef CAS.
- V. S. Vuppuluri, P. J. Samuels, K. C. Caflin, I. E. Gunduz and S. F. Son, Propellants, Explos., Pyrotech., 2018, 43, 38–47 CrossRef CAS.
- H. Zhang, Y. Zhu, N. Qiao, Y. Chen and L. Gao, Pharmaceutics, 2017, 9, 54 CrossRef.
- C. Neurohr, A. Erriguible, S. Laugier and P. Subra-Paternault, Chem. Eng. J., 2016, 303, 238–251 CrossRef CAS.
- S. Hiendrawan, B. Veriansyah, E. Widjojokusumo, S. N. Soewandhi, S. Wikarsa and R. R. Tjandrawinata, Int. J. Pharm., 2016, 497, 106–113 CrossRef CAS PubMed.
- S. Li, T. Yu, Y. Tian, C. P. McCoy, D. S. Jones and G. P. Andrews, Mol. Pharm., 2016, 13, 3054–3068 CrossRef CAS PubMed.
- M. Karimi-Jafari, A. Ziaee, E. O'Reilly, D. Croker and G. Walker, Pharm., 2022, 14, 634 CAS.
- H. Kulla, A. A. L. Michalchuk and F. Emmerling, Chem. Commun., 2019, 55, 9793–9796 RSC.
- Y. Xiao, C. Wu, X. Hu, K. Chen, L. Qi, P. Cui, L. Zhou and Q. Yin, Cryst. Growth Des., 2023, 23, 4680–4700 CrossRef CAS.
- S. Parakatawella, D. Gogoi, P. Deka, Y. Xu, C. Sandaruwan, A. C. A. Jayasundera, M. Arhangelskis, R. Thakuria and N. M. Adassooriya, ChemSusChem, 2022, 15, e202102445 CrossRef CAS.
- C. C. P. da Silva, C. C. de Melo, M. S. Souza, L. F. Diniz, R. L. Carneiro and J. Ellena, J. Pharm. Innov., 2019, 14, 50–56 CrossRef.
- D. Chen, J. Zhao, P. Zhang and S. Dai, Polyhedron, 2019, 162, 59–64 CrossRef CAS.
- S. Nakagawa, Y. Xin, H. Nishikawa, R. Yoneyama, T. Nakagawa, A. Yoshikawa and T. Shirai, Catal. Lett., 2024, 154, 582–592 CrossRef CAS.
- Y. Shi, B. Liang, A. Alsalme, R. B. Lin and B. Chen, J. Solid State Chem., 2020, 287, 121321 CrossRef CAS.
- R. Dubadi, S. D. Huang and M. Jaroniec, Mater., 2023, 16, 1460 CrossRef CAS PubMed.
- P. Zhang and S. Dai, J. Mater. Chem. A, 2017, 5, 16118–16127 RSC.
- C. Hu, P. van Bonn, D. E. Demco, C. Bolm and A. Pich, Angew. Chem., 2023, 135, e202305783 CrossRef.
- M. J. Cliffe, C. Mottillo, R. S. Stein, D. K. Bučar and T. Friščić, Chem. Sci., 2012, 3, 2495–2500 RSC.
- J. G. Hernández, I. S. Butler and T. Friščić, Chem. Sci., 2014, 5, 3576–3582 RSC.
- A. D. Katsenis, A. Puškarić, V. Štrukil, C. Mottillo, P. A. Julien, K. Užarević, M. H. Pham, T. O. Do, S. A. J. Kimber, P. Lazić, O. Magdysyuk, R. E. Dinnebier, I. Halasz and T. Friščić, Nat. Commun., 2015, 61(6), 1–8 Search PubMed.
- L. S. Germann, M. Arhangelskis, M. Etter, R. E. Dinnebier and T. Friščić, Chem. Sci., 2020, 11, 10092–10100 RSC.
- M. Cazzola, C. Page, L. Calzetta and M. G. Matera, Pharm. Pat. Anal., 2018, 7, 249–257 CrossRef CAS PubMed.
- M. Cazzola, C. Page and M. G. Matera, US Respir. Pulm. Dis., 2022, 7, 48 Search PubMed.
- J. F. Donohue, T. Rheault, M. Macdonald-Berko, T. Bengtsson and K. Rickard, Int. J. Chronic Obstruct. Pulm. Dis., 2023, 18, 1611 CrossRef CAS.
-
M. J. A. Walker, B. M. C. Plouvier, J. S. Northen and P. Fernandes, US Pat. Application, US 9062047, 2015.
- A. Kar, L. Giri, G. Kenguva, S. R. Rout and R. Dandela, CrystEngComm, 2024, 26, 3783–3790 RSC.
-
P. Spargo, WO Pat. 2018/020249 A1, 2018.
-
A. W. Oxford, and D. Jack, US Pat. Application, US 6794391, 2004.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- P. R. Spackman, M. J. Turner, J. J. McKinnon, S. K. Wolff, D. J. Grimwood, D. Jayatilaka and M. A. Spackman, J. Appl. Crystallogr., 2021, 54, 1006–1011 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence