
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePolymorphism control of polyethylene terephthalate (PET) degradation product via mechanochemistry leads to accelerated microbial degradation†
Deepika
Shingwekar
 a,
Nicholas
Lutz
a,
Delbert S.
Botes
c,
Elani J.
Cabrera-Vega
d,
Gonzalo
Campillo-Alvarado
*a,
Jay L.
Mellies
*b and
Jesus Daniel
Loya
*a
a,
Nicholas
Lutz
a,
Delbert S.
Botes
c,
Elani J.
Cabrera-Vega
d,
Gonzalo
Campillo-Alvarado
*a,
Jay L.
Mellies
*b and
Jesus Daniel
Loya
*a
aDepartment of Chemistry, Reed College, Portland, OR 97202, USA. E-mail: gcampillo@reed.edu; jdloya@reed.edu
bDepartment of Biology, Reed College, Portland, OR 97202, USA. E-mail: melliesj@reed.edu
cDepartment of Chemistry, University of Missouri, Columbia, MO 65211, USA
dDepartment of Chemistry, Portland State University, Portland, OR 97201, USA
First published on 30th August 2024
Abstract
Widespread usage of single-use plastics such as polyethylene terephthalate (PET) has heavily contributed to a global plastic pollution crisis, necessitating the improvement and development of recycling methods. We previously established a chemo-microbial degradation process for post-consumer PET plastic, consisting of PET depolymerization to form bis(2-hydroxyethyl) terephthalate (BHET) followed by the complete degradation of BHET by a bacterial consortium found to synergistically degrade PET and BHET. The BHET produced during PET depolymerization consists of two polymorphic forms, the α and δ forms. This work investigates the effect of BHET polymorphism on microbial degradation to further optimize the chemo-microbial process. Reversible interconversion methods for BHET polymorphs were effectively developed using mechanochemistry, achieving pure α and δ forms by modulating milling conditions. When inoculated with the bacterial consortium, the α form was degraded faster than the δ form, indicating solid polymorphism is a significant factor for the biodegradation level. This work paves the way to optimize the chemo-microbial process for an increased degradation rate of post-consumer PET and furthers the effort for sustainable plastic recycling methods.
Introduction
Polyethylene Terephthalate (PET) is one the most widely used polyester-based plastics.1,2PET is used for packaging single-use beverage bottles, textiles, and more. While the discovery of plastic, like PET, has revolutionized our packaging needs, its environmental impact is extensive. The vast majority of plastic waste ends up in landfills or the environment.3–6 By 2050, it is estimated that 120 million tons of mismanaged plastic waste will be produced per year, demonstrating that the plastic pollution crisis will only slow down with appropriate intervention.7PET has gained considerable use in consumer products due to its attractive packaging properties such as UV resistance, transparency, and lightweight, leading to higher pollution rates.3 To combat the detrimental effects of plastic pollution, there is a pressing need to design new, efficient ways to recycle these materials.Traditional PET recycling is achieved through energy recovery, mechanical, and chemical recycling.8,9 In recent years, new techniques have emerged to degrade polymers via biological means, such as enzymatic and even microbial degradation.10–15 Depolymerization of PET produces four distinct molecules, terephthalic acid (TPA), ethylene glycol (EG), bis-2-(hydroxyethyl) terephthalate (BHET), and 2-hydroxyethyltherpethalic acid (MHET).16 The method used for depolymerization determines the specific monomer formed.
A main limitation of these biological processes is that the microbial and enzymatic degradation is affected by the degree of crystallinity of PET.17 Polymers, like PET, can be both amorphous and semi-crystalline depending on the thermal treatment properties during the synthesis of these materials.18,19 The biodegradation rate for semi-crystalline PET is slower than amorphous PET, due to the former having more intermolecular forces to overcome.16,20,21 To bypass this limitation, we previously reported a two-step chemo-microbial process for the complete degradation of PET.22 First, depolymerization of PET is achieved through orange peel ash (OPA) catalyzed glycolysis reaction, which forms BHET as the main product. Further degradation of BHET is achieved by using a microbial consortium. The bacterial consortium used for this chemo-microbial process has been previously reported to synergistically degrade PET and BHET as sole carbon sources.16,22–24 Biodegradation has the potential to become an alternative to conventional polymer recycling as it is inexpensive and has a comparatively low environmental impact. We sought to optimize our previously reported chemo-microbial degradation by investigating the relationship between the solid-state architecture (e.g., polymorphism) of BHET and the microbial consortium to evaluate its biodegradation speed. It has been noted in previous literature that optimizing the crystallization conditions (namely isolation of the different polymorphs) is necessary since it could restrict further processing of BHET due to impurities from the glycolysis product, and particle size optimization.25
BHET is known to exist in four different polymorphic forms.26–29 Polymorphism is the phenomenon of a chemical compound to crystallize in different solid-state architectures.30 The arrangement of molecules in the crystal structure can give rise to varied morphologies and physical properties between polymorphs.30,31 Only three out of the four structures have been reported in the crystallographic database.26–28 Polymorph interconversion has traditionally been achieved through modulation of temperature and pressure changes and more recently through mechanochemistry (e.g., co-crystal interconversion).31,32 Of the four reported structures, only two polymorphs are water-stable (i.e., the α and δ forms), which makes them suitable for bacterial degradation (Table S4†).27
To evaluate the effect of BHET polymorphism on microbial degradation, we report a mechanochemical method to interconvert between the two water-stable polymorphs of BHET (Scheme 1). Specifically, by carefully choosing the mechanochemical conditions, the α and δ forms can be selectively generated and interconverted in gram amounts. We demonstrate here that the solid-state architecture influences the biodegradation level, specifically the α form degrades at a faster rate than the δ form. To the best of our knowledge, our study demonstrates the connection between the polymorphism of a polymer degradation product and biodegradation for the first time. We envision our research will enable the development of improved and more efficient polymer degradation processes that account for solid-state packing architectures.
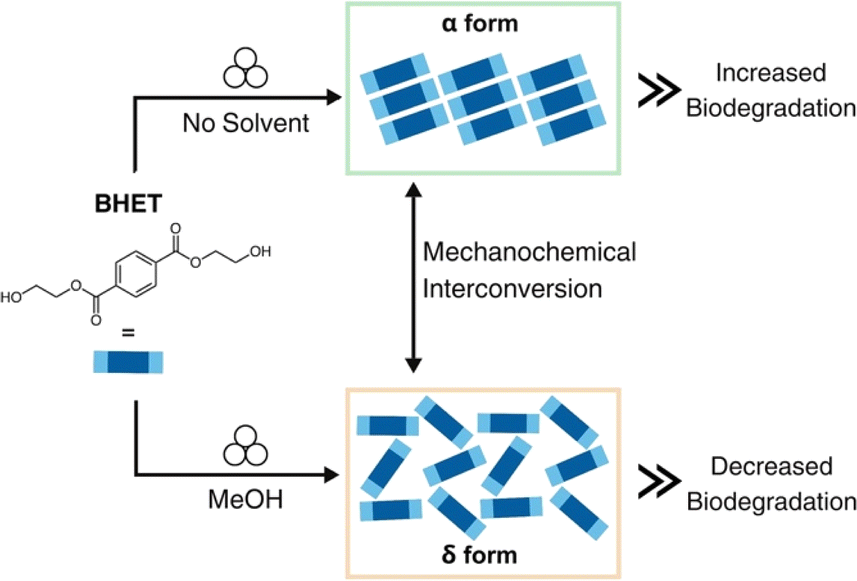 | ||
| Scheme 1 BHET polymorph interconversion through mechanochemistry and effect on biodegradation. | ||
Results and discussion
Structural differences in polymorph packing
The α form crystallizes in the monoclinic space group P21/a (CSD-HETPAL).26 The asymmetric unit contains two symmetry-independent half molecules of BHET. The structure is sustained through O–H⋯O hydrogen bonds between hydroxyl groups of different BHET molecules [O–H⋯O separation: 2.839 (7) Å], C–H⋯O hydrogen bonds between the carbonyl group of an ester from a BHET molecule and a hydroxyl group of a different BHET molecule [C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O⋯H–O separation: 2.839 (7) Å], and offset π–π stacking (4.29 Å) (Fig. 1).
O⋯H–O separation: 2.839 (7) Å], and offset π–π stacking (4.29 Å) (Fig. 1).
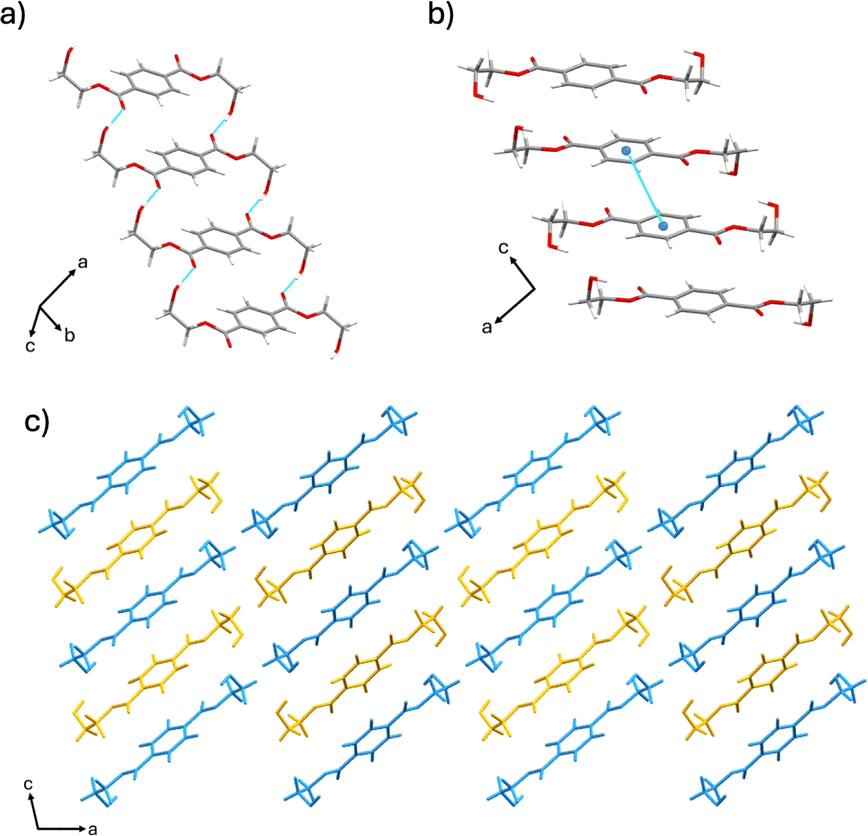 | ||
| Fig. 1 Crystal structure of the α form showing: (a) hydrogen bonding, (b) offset π–π stacking, and (c) extended crystal structure packing (molecules colored blue and yellow for visual clarity). | ||
The δ form crystallizes in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) (CSD-HETPAL02).28 The asymmetric unit contains four symmetry-independent half molecules of BHET. The δ form packs in a herringbone-like arrangement. The crystal structure is sustained through hydrogen bonds from the hydroxyl groups of neighboring BHET molecules [O–H⋯O separation: 2.734 (3) Å]. Additional offset π–π contacts (4.113 (15) Å) support the formation of ribbons along the a-axis (Fig. 2).
(CSD-HETPAL02).28 The asymmetric unit contains four symmetry-independent half molecules of BHET. The δ form packs in a herringbone-like arrangement. The crystal structure is sustained through hydrogen bonds from the hydroxyl groups of neighboring BHET molecules [O–H⋯O separation: 2.734 (3) Å]. Additional offset π–π contacts (4.113 (15) Å) support the formation of ribbons along the a-axis (Fig. 2).
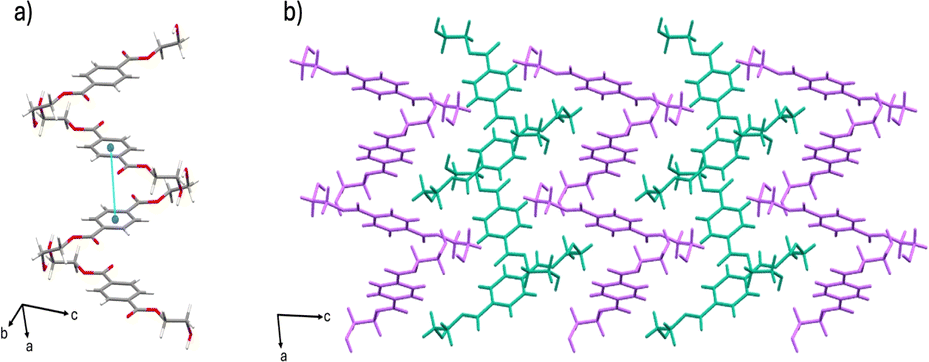 | ||
| Fig. 2 Crystal structure of the δ form showing: (a) hydrogen bonding and offset π–π stacking, and (b) extended crystal structure packing (molecules colored magenta and teal for visual clarity). | ||
Both polymorphs display parallel offset π-stacking and hydrogen bonding, but the orientation of molecules in the crystal structure gives rise to different extended packings. The hydrogen bonding of BHET molecules in the α form produces two unique hydrogen-bonded chains of BHET molecules. Both BHET chains lie along the crystallographic b-axis with the only difference being that one chain is slanted and the other is planar. The offset π–π stacking forms two hydrogen-bonded columns of BHET in the α form, where adjacent columns are linked with hydrogen bonds from the hydroxyl groups at the ends of the molecule and carbonyl oxygen atoms. Conversely, the herringbone pattern of the δ form is sustained through a hydrogen-bonded network between hydroxyl and carbonyl groups. Parallel offset π-stacking occurs between molecules in the δ form that, unlike the α form, do not have their ethylene groups in the same orientation.
Polymorph synthesis
Formation of BHET polymorphs is both solvent and temperature-dependent when growing suitable crystals for SCXRD analysis.26,27 Commercially available BHET was utilized to synthesize both polymorphs. The α form was readily made at room temperature in various solvents, including chloroform, methanol (MeOH), acetone, acetonitrile, and dichloromethane (DCM) (Table S1†). The δ form was only observed to grow at 4 °C in MeOH, indicating temperature dependence for the polymorph to form. When BHET was recrystallized in water at 4 °C, a mixture of both the α and δ forms was produced, confirming the results observed after the OPA-catalyzed glycolysis reaction workup. Although solution-based methods yielded successful syntheses of the α and δ forms, the time-consuming nature of the crystal growth, which took 3-4 days, could limit the scope and applicability of these methods.Mechanochemistry was employed to selectively form the α and δ polymorphs from the OPA-catalyzed PET depolymerization process (Table S2†). Specifically, neat milling of the reaction mixture for 90 minutes afforded the α form exclusively (Fig. S3†) as shown by powder X-ray diffraction (PXRD). The α form was also isolated via liquid-assisted grinding (LAG) with chloroform after 60 minutes (Fig. S5†). LAG of the reaction mixture at room temperature with MeOH for 120 minutes produced the δ form exclusively, demonstrating a reliable and efficient approach to control polymorphism at room temperature instead of 4 °C (Fig. S4†). In addition, the two polymorphs were also able to form via mechanochemistry when using commercial BHET as the starting material, with neat milling for 90 minutes producing the α form and LAG with MeOH for 210 minutes producing the δ form (Fig. S9†).
Polymorph interconversion via mechanochemistry
We next turned our attention to polymorph interconversion using mechanochemistry.32 The milled δ form powder was effectively converted to the α form using LAG, with chloroform, for 60 minutes (Fig. S6†). The milled α form powder was converted to the δ form using LAG, with methanol, for 210 minutes (Fig. S6†). 1H Nuclear Magnetic Resonance (NMR) spectroscopy showed no traces of the solvent used in LAG experiments (Fig. S1 and S2†). The results indicate the formation of the α and δ polymorphs is tunable with solvent and milling time. It had previously been reported that the α form could not be converted into any other form without melting.27 Our approach achieved reversible and ready interconversion between the α and δ forms through LAG.Polymorph conversion via heating and energetic considerations
Conversion of the δ form into the α form was observed using PXRD after heating the δ form at 107 °C for 30 minutes using a heating stage, showing a solid-state transition between the two polymorphs (Fig. S7†). This temperature-dependent conversion indicates the δ form has a lower thermal stability than the α form. When heating the milled α form powder, no change in crystalline phase was observed.To determine the energetically favored form, UNI intermolecular potential force field model was applied to both polymorphs to examine the interaction strength between molecules in the crystal lattice.33–35 The α form shows a stronger total packing energy (−186.4 kJ mol−1) than the δ form (−175.2 kJ mol−1). From the calculations and experimental results, we deemed the α form to be an energetically favored polymorph. We posit that the α and δ forms have a monotropic relationship, where one polymorph is always stable below the melting point of both polymorphs. In a monotropic system, heating the metastable form at any given temperature will cause that form to convert into the energetically stable form.36 In this system, the δ form is the metastable form, which can exist at room temperature, but will convert back into the α form once heat is introduced. The thermal stabilities of the α and δ polymorphs were assessed through differential scanning calorimetry (DSC) and thermogravimetric analysis (TGA) (Fig. S10–12†). Both polymorphs showed similar endotherms associated with melting points, at 110 °C (110.47 °C) and 110 °C (110.93 °C) for the α and δ forms respectively. While both had comparable endotherms, we observed a slight shoulder on the δ form endotherm, which could indicate a phase transition from the δ form to the α form. Attempts to resolve the peak by running the sample at a slower heating rate were unsuccessful. As seen on the TGA curves, the δ form has an onset decomposition temperature of 214 °C (214.93 °C) compared to the α form at 207 °C (207.05 °C). The overall mass loss for the δ form occurs at a faster rate than the α form, with approximately 20% and 46% mass left respectively at the ending temperature of 300 °C (Fig. S12†). We note that no interconversion between the α and δ forms was observed upon exposure to ambient conditions (25 °C) for 3 months (Fig. S8†).
Polymorph characterization
Solids of BHET were further analyzed via PXRD, Fourier Transform Infrared Spectroscopy (FT-IR), and dynamic light scattering (DLS). PXRD confirmed that the products of the mechanochemical reactions were indeed the α and δ forms (Fig. 3a). The PXRD pattern for the α form contains characteristic peaks at 6.98° and 16.58°, whereas the δ form has unique peaks at 10.86°, 14.90°, and 15.70°. The unmilled crystalline BHET glycolysis product had all of the aforementioned peaks, and through phase analysis of the PXRD pattern, contained 82.20% of the δ form and 17.80% of the α form. The phase analysis was conducted using the semi-quantitative Reference Intensity Ratio method using Match (Fig. 3c).37 In contrast, after mechanochemical interconversion, the v and δ products showed no trace of the other polymorph in their patterns. | ||
| Fig. 3 Polymorph characterization: (a) powder X-ray diffraction pattern of synthesized α and δ forms, and BHET depolymerization product. The top and bottom-most patterns are simulated diffraction patterns for the α and δ forms, respectively, generated using Mercury software version 2024.1.0.33 (b) FTIR spectra (4000 to 600 cm−1) of the δ form (top) and the α form (bottom). (c) Phase composition analysis of BHET depolymerization product from OPA-catalyzed glycolysis reaction. | ||
FT-IR spectra (Fig. 3b) confirmed the polymorphs' chemical bonding are unique to each solid structure. Hydrogen bonding decreases the vibrational frequency of the bonds involved in those interactions, as the non-covalent interaction weakens the covalent bond from groups participating.38 The O–H absorbance peak for the α form appears at 3440 cm−1 and is sharp. In contrast, the δ form O–H absorbance peak appears at 3280 cm−1 and is more broad-shaped, which is characteristic of hydrogen-bonded hydroxyl groups and suggests that the δ form has more H-bonding interactions involving the hydroxyl groups. Similarly, when looking at the carbonyl absorbance region, the α form has an absorbance band at 1686 cm−1 and 1712 cm−1 compared to an absorbance band at 1718 cm−1 for the δ form, indicating the molecule has free ester groups. The IR spectra indicate the α form has a mixture of hydrogen-bonded and free ester groups, whereas the δ form only has free ester groups.27 These results are in agreement with SCXRD data, which reveal that the packing structure of the α form primarily contains heterosynthons comprised of hydroxyl-carbonyl, and homosynthons of hydroxyl–hydroxyl hydrogen bonds between.
BHET molecules. Conversely, the packing of the δ form consists only of homosynthons between hydroxyl–hydroxyl hydrogen bonds.26,28 Particle size distribution analysis of powders of the α and δ forms was performed with DLS, showing mean hydrodynamic diameters of 5.53 ± 0.06 μm and 5.25 ± 0.46 μm, respectively (Fig. S14 and S15†), which are deemed comparable. The uneven particle size distribution of BHET has been observed previously.25 We note the particle sizing for particles above 1 μm is not accurate using DLS, the values are given here only as an indication for similar aggregation behavior of the α and δ forms.39 Further studies correlating particle size with microbial degradation are ongoing.
Chemo-microbial degradation
Both the α and δ polymorphs of BHET were inoculated with the full microbial consortium to determine whether polymorphism affects the level of biodegradation. To enable the hydrolysis of BHET, the bacterial cultures were grown in media primarily composed of water.Since the data was normalized against a mixture of LCFBM and BHET containing no bacteria, decreases in BHET concentration due to hydrolysis were removed from our measurements, allowing them to only consist of BHET biodegradation by the bacterial consortium. Both the α and δ forms were significantly degraded by the consortium, indicating that neither polymorph had a detrimental effect on microbial degradation (Fig. 4). Here, we found that the 4 weeks biodegradation level was 11.58% for the α form and 8.33% for the δ form. In addition, while weeks 1 and 2 did not show significance in the biodegradation level between the two polymorphs (p-value > 0.05; Dunnett's Test), the α and δ forms were statistically different from week 3 onwards in their biodegradation levels (week 3 p-value = 0.0289, week 4 p-value = 0.0138; Dunnett's Test), with the α form being degraded faster by the consortium. Our results confirm that BHET biodegradation is affected by the polymorph provided as a carbon source, despite having similar physical properties like the α and δ forms.
 | ||
| Fig. 4 Consortium biodegradation of the α and δ forms over time. The full consortium was grown on 0.6% (w/v) α form or δ form BHET and 0.05% (w/v) yeast extract in liquid carbon-free basal medium (LCFBM). Cultures were incubated statically at 30 °C. Degradation was measured weekly through HPLC, and concentration differences were measured using an internal standard compared to uninoculated negative controls. All treatments and time points were performed in triplicate. Error bars indicate the standard error of the mean. Statistical significance between the α form and δ form biodegradation levels was determined using a Dunnett's test, with one asterisk (*) indicating statistically significant results at the 95% confidence level. | ||
The 4 weeks biodegradation, hydrolysis, and total degradation rates for both polymorphs can be seen in Table S3.† The hydrolysis rates between both polymorphs were found to be statistically insignificant from one another (p-value = 0.3664), thus revealing that BHET hydrolysis does not contribute significantly to the biodegradation differences between the α and δ forms.
Looking at the structural differences between the polymorphs, it could be that a lack of symmetry increases the biodegradation level as there would be more gaps between molecules for enzymatic hydrolysis to occur. However, the α form exhibits higher biodegradation compared to the δ form. This is despite the α form having a higher degree of symmetry than the δ form. Further work is needed to determine why biodegradation differs between these two polymorphs and to understand how solid structure affects microbial degradation levels.
Conclusions
This work demonstrates that the α form of BHET is degraded faster by the bacterial consortium compared to the δ form, indicating that the solid structure of BHET does play a role in biodegradation. The present work discovered a novel way to cycle between the α and δ polymorphs of BHET using mechanochemistry, where the formation of δ polymorph had not yet been achieved at ambient room temperature. Polymorphism control of BHET can be utilized to improve the chemo-microbial process for an effective and sustainable way to combat PET pollution and mitigate the effects of environmental fallout without using costly or harmful reagents or procedures. We envisage emerging technologies for processing plastics considering solid-state polymorphism and particle size could be used to address the world's plastic pollution problem.Data availability
The data supporting these findings are within the article and the ESI.†Author contributions
D. S. contributed by conceptualization, carried out investigation, methodology, curation, validation, writing and reviewing of the original draft. N. L., D. S. B., and E. J. C.-V. performed formal analysis, data curation, and review of original draft. G. C.-A. contributed by conceptualization, performed formal analysis, review and editing of original draft. J. D. L. and J. L. M. contributed by conceptualization, project administration, and writing and reviewing of the original draft.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge Professor Kristin M. Hutchins for access to thermal characterization instruments and Professor Andrea M. Goforth for access to dynamic light scattering instrumentation. We are thankful to the Chemistry Department of Portland State University for access to the NMR facility and to Charlene Kupara for her assistance with sample collection. D. S. gratefully acknowledges support from the Ann and Don Pease Undergraduate Student Research Fund. Research in the laboratory of J. L. M. is supported by NSF RUI Collaborative grant #2246498. G. C.-A acknowledges support from the M. J. Murdock Charitable Trust (NS-202222588) and Reed College (start-up funds). J. D. L. appreciates support from Reed College and Consortium of Faculty Diversity Postdoctoral Fellowship.References
- C. B. Crawford and B. Quinn, in Microplastic Pollutants, Elsevier, 2017, pp. 57–100 Search PubMed.
- D. Danso, J. Chow and W. R. Streit, Appl. Environ. Microbiol., 2019, 85 DOI:10.1128/AEM.01095-19.
- R. Geyer, J. R. Jambeck and K. L. Law, Sci. Adv., 2017, 3, e1700782 CrossRef PubMed.
- M. Shams, I. Alam and M. S. Mahbub, Environ. Adv., 2021, 5, 100119 CrossRef CAS PubMed.
- Y. Peng, P. Wu, A. T. Schartup and Y. Zhang, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2111530118 CrossRef CAS PubMed.
- S. B. Borrelle, J. Ringma, K. L. Law, C. C. Monnahan, L. Lebreton, A. McGivern, E. Murphy, J. Jambeck, G. H. Leonard, M. A. Hilleary, M. Eriksen, H. P. Possingham, H. De Frond, L. R. Gerber, B. Polidoro, A. Tahir, M. Bernard, N. Mallos, M. Barnes and C. M. Rochman, Science, 2020, 369, 1515–1518 CrossRef CAS PubMed.
- W. W. Y. Lau, Y. Shiran, R. M. Bailey, E. Cook, M. R. Stuchtey, J. Koskella, C. A. Velis, L. Godfrey, J. Boucher, M. B. Murphy, R. C. Thompson, E. Jankowska, A. Castillo Castillo, T. D. Pilditch, B. Dixon, L. Koerselman, E. Kosior, E. Favoino, J. Gutberlet, S. Baulch, M. E. Atreya, D. Fischer, K. K. He, M. M. Petit, U. R. Sumaila, E. Neil, M. V. Bernhofen, K. Lawrence and J. E. Palardy, Science, 2020, 369, 1455–1461 CrossRef CAS PubMed.
- R. C. Thompson, C. J. Moore, F. S. Vom Saal and S. H. Swan, Philos. Trans. R. Soc., B, 2009, 364, 2153–2166 CrossRef CAS PubMed.
- S. C. Kosloski-Oh, Z. A. Wood, Y. Manjarrez, J. P. De Los Rios and M. E. Fieser, Mater. Horiz., 2021, 8, 1084–1129 RSC.
- S. Yoshida, K. Hiraga, T. Takehana, I. Taniguchi, H. Yamaji, Y. Maeda, K. Toyohara, K. Miyamoto, Y. Kimura and K. Oda, Science, 2016, 351, 1196–1199 CrossRef CAS PubMed.
- H. P. Austin, M. D. Allen, B. S. Donohoe, N. A. Rorrer, F. L. Kearns, R. L. Silveira, B. C. Pollard, G. Dominick, R. Duman, K. El Omari, V. Mykhaylyk, A. Wagner, W. E. Michener, A. Amore, M. S. Skaf, M. F. Crowley, A. W. Thorne, C. W. Johnson, H. L. Woodcock, J. E. McGeehan and G. T. Beckham, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, E4350–E4357 CrossRef CAS PubMed.
- B. Liu, L. He, L. Wang, T. Li, C. Li, H. Liu, Y. Luo and R. Bao, ChemBioChem, 2018, 19, 1471–1475 CrossRef CAS PubMed.
- Y.-H. V. Soong, M. J. Sobkowicz and D. Xie, Bioengineering, 2022, 9, 98 CrossRef CAS PubMed.
- S. Sulaiman, S. Yamato, E. Kanaya, J.-J. Kim, Y. Koga, K. Takano and S. Kanaya, Appl. Environ. Microbiol., 2012, 78, 1556–1562 CrossRef CAS PubMed.
- S. Tanasupawat, T. Takehana, S. Yoshida, K. Hiraga and K. Oda, Int. J. Syst. Evol. Microbiol., 2016, 66, 2813–2818 CrossRef CAS PubMed.
- C. Roberts, S. Edwards, M. Vague, R. León-Zayas, H. Scheffer, G. Chan, N. A. Swartz and J. L. Mellies, mSphere, 2020, 5, 1–20 CrossRef PubMed.
- C. Bach, X. Dauchy and S. Etienne, IOP Conf. Ser.: Mater. Sci. Eng., 2009, 5, 012005 Search PubMed.
- J. M. G. Cowie and V. Arrighi, Polymers: Chemistry and Physics of Modern Materials, CRC Press, Boca Raton, 3rd edn, 2008 Search PubMed.
- P. Benyathiar, P. Kumar, G. Carpenter, J. Brace and D. K. Mishra, Polymers, 2022, 14, 2366 CrossRef CAS PubMed.
- Å. M. Ronkvist, W. Xie, W. Lu and R. A. Gross, Macromolecules, 2009, 42, 5128–5138 CrossRef.
- F. Kawai, M. Oda, T. Tamashiro, T. Waku, N. Tanaka, M. Yamamoto, H. Mizushima, T. Miyakawa and M. Tanokura, Appl. Microbiol. Biotechnol., 2014, 98, 10053–10064 CrossRef CAS PubMed.
- D. Shingwekar, H. Laster, H. Kemp and J. L. Mellies, Bioengineering, 2023, 10, 1253 CrossRef CAS PubMed.
- R. León-Zayas, C. Roberts, M. Vague and J. L. Mellies, Microbiol. Resour. Announce., 2019, 8 DOI:10.1128/MRA.00237-19.
- S. Edwards, R. León-Zayas, R. Ditter, H. Laster, G. Sheehan, O. Anderson, T. Beattie and J. L. Mellies, Int. J. Mol. Sci., 2022, 23, 5612 CrossRef CAS PubMed.
- P. Yuan, B. Liu, Q. Li and H. Sun, J. Chem. Eng. Data, 2022, 67, 2693–2705 CrossRef CAS.
- S. Kashino and M. Haisa, Acta Crystallogr., Sect. B: Struct. Sci., 1975, 31, 1819–1822 CrossRef.
- A. Miyake, Bull. Chem. Soc. Jpn., 1957, 30, 361–363 CrossRef CAS.
- F. Scé, I. Cano, C. Martin, G. Beobide, Ó. Castillo and I. De Pedro, New J. Chem., 2019, 43, 3476–3485 RSC.
- W. S. McDonald, E. L. V. Lewis and D. I. Bower, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1983, 39, 410–412 CrossRef.
- A. Hill, W. Kras, F. Theodosiou, M. Wanat, D. Lee and A. J. Cruz-Cabeza, J. Am. Chem. Soc., 2023, 145, 20562–20577 CrossRef CAS PubMed.
- L. S. Germann, M. Arhangelskis, M. Etter, R. E. Dinnebier and T. Friščić, Chem. Sci., 2020, 11, 10092–10100 RSC.
- L. Ma, Q. Zheng, D. K. Unruh and K. M. Hutchins, Chem. Commun., 2023, 59, 7779–7782 RSC.
- C. F. Macrae, I. Sovago, S. J. Cottrell, P. T. A. Galek, P. McCabe, E. Pidcock, M. Platings, G. P. Shields, J. S. Stevens, M. Towler and P. A. Wood, J. Appl. Crystallogr., 2020, 53, 226–235 CrossRef CAS PubMed.
- A. Gavezzotti, Acc. Chem. Res., 1994, 27, 309–314 CrossRef CAS.
- A. Gavezzotti and G. Filippini, J. Phys. Chem., 1994, 98, 4831–4837 CrossRef CAS.
- E. H. Lee, Asian J. Pharm. Sci., 2014, 9, 163–175 CrossRef.
- H. Putz and K. Brandenburg GbR, Match! - Phase Analysis Using Powder Diffraction (Version Version 3) Crystal Impact, Kreuzherrenstr, 102, 53227, Bonn, Germany Search PubMed.
- T. Fornaro, D. Burini, M. Biczysko and V. Barone, J. Phys. Chem. A, 2015, 119, 4224–4236 CrossRef CAS PubMed.
- S. Hornig, H. Bunjes and T. Heinze, J. Colloid Interface Sci., 338, 56–62 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4mr00060a |
| This journal is © The Royal Society of Chemistry 2024 |