
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePrecise and rapid point-of-care quantification of albumin levels in unspiked blood using organic field-effect transistors†
Ajoy
Mandal
a,
Suman
Mandal
a,
Samik
Mallik
b,
Sovanlal
Mondal
b,
Subhendu Sekhar
Bag
 c and
Dipak K.
Goswami
*ab
c and
Dipak K.
Goswami
*ab
aOrganic Electronics Laboratory, Department of Physics, Indian Institute of Technology Kharagpur, Kharagpur – 721302, India. E-mail: dipak@phy.iitkgp.ac.in
bSchool of Nanoscience and Technology, Indian Institute of Technology Kharagpur, Kharagpur – 721302, India
cBioorganic Chemistry Laboratory, Department of Chemistry, Indian Institute of Technology Guwahati, Guwahati –781039, India
First published on 13th December 2023
Abstract
Nanowire-based field-effect transistors (FETs) are widely used to detect biomolecules precisely. However, the fabrication of such devices involves complex integration procedures of nanowires into the device and most are not easily scalable. In this work, we report a straightforward fabrication approach that utilizes the grain boundaries of the semiconducting film of organic FETs to fabricate biosensors for the detection of human serum albumin (HSA) with an enhanced sensitivity and detection range. We used trichromophoric pentapeptide (TPyAlaDo-Leu-ArTAA-Leu-TPyAlaDo, TPP) as a receptor molecule to precisely estimate the concentration of HSA protein in human blood. Bi-layer semiconductors (pentacene and TPP) were used to fabricate the OFET, where the pentacene molecule acted as a conducting channel and TPP acted as a receptor molecule. This approach of engineering the diffusion of receptor molecules into the grain boundaries is crucial in developing OFET-based HSA protein sensors, which cover a considerable detection range from 1 pM to 1 mM in a single device. The point-of-care detection in unspiked blood samples was confirmed at 4.2 g dL−1, which is similar to 4.1 g dL−1 measured using a pathological procedure.
Introduction
Organic field-effect transistors (OFETs) have shown tremendous promise for the fabrication of biosensors by exploiting complex molecular interactions, such as hydrogen bonding, charge transfer, or simply π–π stacking interactions, between analytes and receptor molecules attached to the semiconducting channel.1,2 The incorporation of receptor molecules into the semiconducting channel to obtain better access to the charge-conducting pathway of the sensor while sensing the analyte molecules is crucial for the development of a precise sensor.3–6 Increasing the surface area of the active layer by introducing porosity into the conducting channel or by using nanomaterials without compromising the conducting pathway of the charge flow or the use of ultra-thin semiconducting films were some of the aspects exploited to achieve high sensitivity.7–9 The conductivity of most of the bio-receptor molecules is very poor.10 Therefore, using these molecules to fabricate electrical device-based sensors is very difficult. Recently, these bio-molecules have been attached to a conductive semiconductor to fabricate a good selective bio-sensor.11 However, the thickness and concentration of the bio-receptor are very crucial factors that determine the sensitivity of the device. Most of the researchers have ignored this aspect. In this work, we have used trichromophoric pentapeptide (TPyAlaDo-Leu-ArTAA-Leu-TPyAlaDo, TPP) as a receptor molecule for the precise estimation of HSA protein concentration in human blood using the OFET as a sensor platform.12 The molecular structure of TPP is shown in Fig. S1.† Peptides are dielectric materials and can be used only as a gate dielectric layer for the fabrication of OFET under top contact/bottom-gate (TCBG) configuration. However, we used a electrolyte-based gate dielectric material in the bottom contact/top gate (BCTG) configuration for the fabrication of OFET-based biosensors. Electrolyte gate-based OFET exhibit design complexity with limited portability in the development of POC sensor systems. In this work, we report the fabrication methodology of OFET-based HSA protein sensors with peptides as the sensing material in the TCBG configuration. This approach has advantages owing to the easy fabrication method suitable for the large-scale fabrication over the devices fabricated using 2D materials or single nanowires-based sensors. We have systematically incorporated peptide material within the pentacene semiconducting channel. Therefore, a bilayer combination of the peptide with pentacene is used as the sensing layer. The pentacene film defines the conducting channel at the semiconductor/dielectric interface. However, the peptide molecules diffuse through the grain boundaries of the dendrite structures of the pentacene film to obtain access to the interface. As the peptide molecules interact with the HSA protein present in the buffer solution, it changes the electronic structure of the interface, and the device current is changed. Therefore, one can easily estimate the concentration of HSA molecules from the relative changes in the device current. There are reports for the fabrication of ambipolar OFETs using a semiconducting bilayer channel consisting of n-and p-type materials.13 Conducting channels consisting of 2D material or nanowires functionalized with receptor molecules are mostly used for the fabrication of FET-based biosensors.14–17 However, such devices have fabrication complexity in large-scale fabrication for commercialization. In this work, we used a bilayer channel consisting of a semiconducting material, whereas the other was a dielectric sensing material. The two layers were grown separately with ease and low-cost fabrication capabilities. Our approach for HSA sensing relies on electrostatic π–π interaction between HSA and ArTAA_Py-Py pentapeptide (TPP).The quantitative determination of HSA is crucial in clinical diagnosis to predict various health parameters. HSA is the most abundant protein that takes up about 50% of the total plasma protein produced in the liver.18 It plays several pivotal roles in the human body, such as maintaining the oncotic pressure and carrying hormones, fatty acids, thyroxin, and metabolites.19 The standard reference level of HSA is about 3.5–5.5 g dL−1 in blood plasma.18–20 However, this concentration can be much lower in urine (∼2.2–35 × 10−4 g dL−1) and in saliva (∼0.05 g dL−1).21–27 An unusual concentration of HSA may lead to various diseases, such as hyperalbuminemia, hypoalbuminemia, liver diseases, nephrosis, cardiovascular diseases, and even type-2 diabetes mellitus.23,24 Therefore, the estimation of HSA level with high accuracy and precision covering the range from ultra-low (∼10−9 g dL−1) to few g dL−1 is essential for various clinical diagnoses at point-of-care (POC). Several methods have been applied to quantitatively estimate HSA levels from blood samples or different buffer media. Such methods include high-performance liquid chromatography (HPLC),28 fluorescence-based sensors,29–33 colorimetric methods,34 and electrochemical methods.35,36 However, many of these techniques have a limited detection range are mostly used in clinical laboratories, and are not suitable for POC application. Therefore, real-time, low-cost, yet low-voltage consuming biosensors will be necessary for such diagnostics. OFETs have drawn much attention to the development of biosensors due to the decisive advantage of tailoring the organic material to adjust its properties to sense a specific biological analyte.37 Nevertheless, it is easier to miniaturize OFET-based sensors to fit into measuring electronics by enabling easy scalability. Being transistors, OFETs can also amplify the sensing signal within the device to enhance detection sensitivity. However, enhancing sensitivity and detection range in OFET-based sensors is also challenging. We have demonstrated a new methodology for the fabrication of low-voltage, ultra-precession OFET-based biosensors to detect HSA protein in the blood samples.
In this article, we report the fabrication of OFET-based low-voltage, ultra-precession biosensors for the detection of HSA protein from blood samples covering a detection range from ultra-low (∼1 pM, 6.60 × 10−9 g dL−1) to a high concentration (∼1 mM, 6.6 g dL−1). We synthesized a TPP with selective binding with HSA protein as a sensing material. We achieved exceptional sensitivity to detect 1 pM by covering a range of up to 1 mM in a single device.
Results and discussion
In the first part of our work, we optimized the thickness of the pentacene films for OFET fabrication. We used a bilayer dielectric system, which includes thin Al2O3 and BaTiO3 (BTO) layers. The Al2O3 layer was grown by the anodization of a part of gate (Al) contact. This layer reduces the gate leakage current. h-BTO is a high dielectric constant material and has been used to reduce the operating voltage. The thickness of pentacene films was controlled by the deposition time in the OMBD growth chamber. We deposited 10 nm, 20 nm, 25 nm, 44 nm, and 60 nm pentacene films to fabricate OFETs. The UV-Vis spectra of these pentacene films are shown in Fig. S1a.† The surface morphology of the 20 nm pentacene film is shown in Fig. 1a. The schematic design of the pentacene-based OFET is shown in Fig. 1b. The field-effect mobility extracted from the transfer curve of different thicknesses of pentacene OFET is represented in Fig. 1d. The mobility of the pentacene film increases from 10 nm to 20 nm. There is a sharp decrease for 24 nm pentacene film and is saturated. The surface roughness of the pentacene film increases with the thickness, as shown in Fig. S2.† However, the crystalline quality of the film improves with the thickness leading to enhanced carrier mobility. Therefore, initially, the charge transport increases with the thickness of up to 20 nm films. The roughness also induces grain boundary density, which acts like a trap. Therefore, we observed a significant decrease in carrier mobility in the devices fabricated with thicker films. For the sensor fabrication, we used 20 nm pentacene films owing to high mobility. The transfer and output characteristic curve of the 20 nm pentacene film are shown in Fig. 1c and S1b,† respectively. | ||
| Fig. 1 (a) Surface morphology of the pentacene film showing the grain boundaries of the dendrite structures. (b) Schematic diagram of the OFET. (c) Transfer characteristic of the 20 nm pentacene film-based OFET. (d) Variation of mobility of the pentacene film grown on BaTiO3/Al2O3/Al/Glass. A 20 nm pentacene film was selected for the sensing experiment due to higher field-effect mobility. (e) Surface morphology of the peptide film (5000 rpm coated) on a pentacene film. | ||
In the second step, we optimized the TPP film growth on a 20 nm pentacene-based OFET. We spin-coated different films of TPP molecules on a glass substrate by varying the RPM (rotation per minute) of the spin-coater system. The surface morphology of the TPP film (5000 rpm) on the pentacene film is shown in Fig. 1e. The UV-vis spectra of different TPP films (5000 rpm, 4000 rpm, 3000 rpm, 1500 rpm, and 500 rpm) are shown in Fig. S4a.† The optical band gap extracted using a Tauc plot is around 3.12 eV (Fig. S4b†). Due to the high optical band gap, the TPP molecule showed poor conductivity. Although we tried to fabricate transistors only using this molecule, it did not work. Due to its high bio-recognizing properties to albumin protein, the TPP molecule was spin-coated on the standard pentacene-based OFET and the device was used for the bio-sensing application. Maintaining a better crystallinity of the pentacene film coated with the TPP film is essential for a higher device current. Charge transport through OFETs works in the accumulation mode at the semiconductor/dielectric interface. Therefore, the device current flows through the interface of the pentacene and dielectric (h-BTO) layers. The degradation of crystallinity of the pentacene film is limited to the thin film phase for the TPP film grown at higher RPM. Therefore, thin TPP film grown at a higher RPM would be better to achieve a higher device current. We developed OFETs with various TPP-coated pentacene channels. We observed that the field-effect carrier mobility for the OFETs decreases sharply up to the 4000 rpm-coated TPP film, and after that, it gets saturated (Fig. S4c†).
The reduction in carrier mobility is due to the structural degradation of the pentacene film caused by the penetration of TPP molecules in the grain boundaries. However, the diffusion of TPP molecules gets saturated in the 4000 RPM-coated film. As a result, the carrier mobility is saturated in the film coated at lower RPM. Nevertheless, 5000 RPM-coated TPP film-based devices showed better device performances with carrier mobility of 0.055 cm2 V−1 s−1 and a threshold voltage of −0.75 V. As the TPP molecules are not soluble in water, it is expected that the penetration of the water molecules through the TPP film into the device is not favourable. Therefore, TPP molecules present in the film may protect the device from possible damage from water.
Fig. 2a represents the typical design of the OFET-based sensors used to detect HSA protein. Besides, we used a bilayer dielectric system, which included 15 nm Al2O3 and 60 nm hexagonal BaTiO3 (h-BTO) layers. h-BTO is a high dielectric constant material and has been used to reduce the operating voltage of OFETs to less than 2V.38 TPP molecules get easy access to the pentacene/BTO interface by diffusing through grain boundaries. Therefore, TPP molecules can efficiently influence the drain current of OFETs during the sensing of HSA protein. Consequently, it is expected that the sensitivity of detection of HSA protein using TTP molecules will be much higher in this design of sensor fabrication.
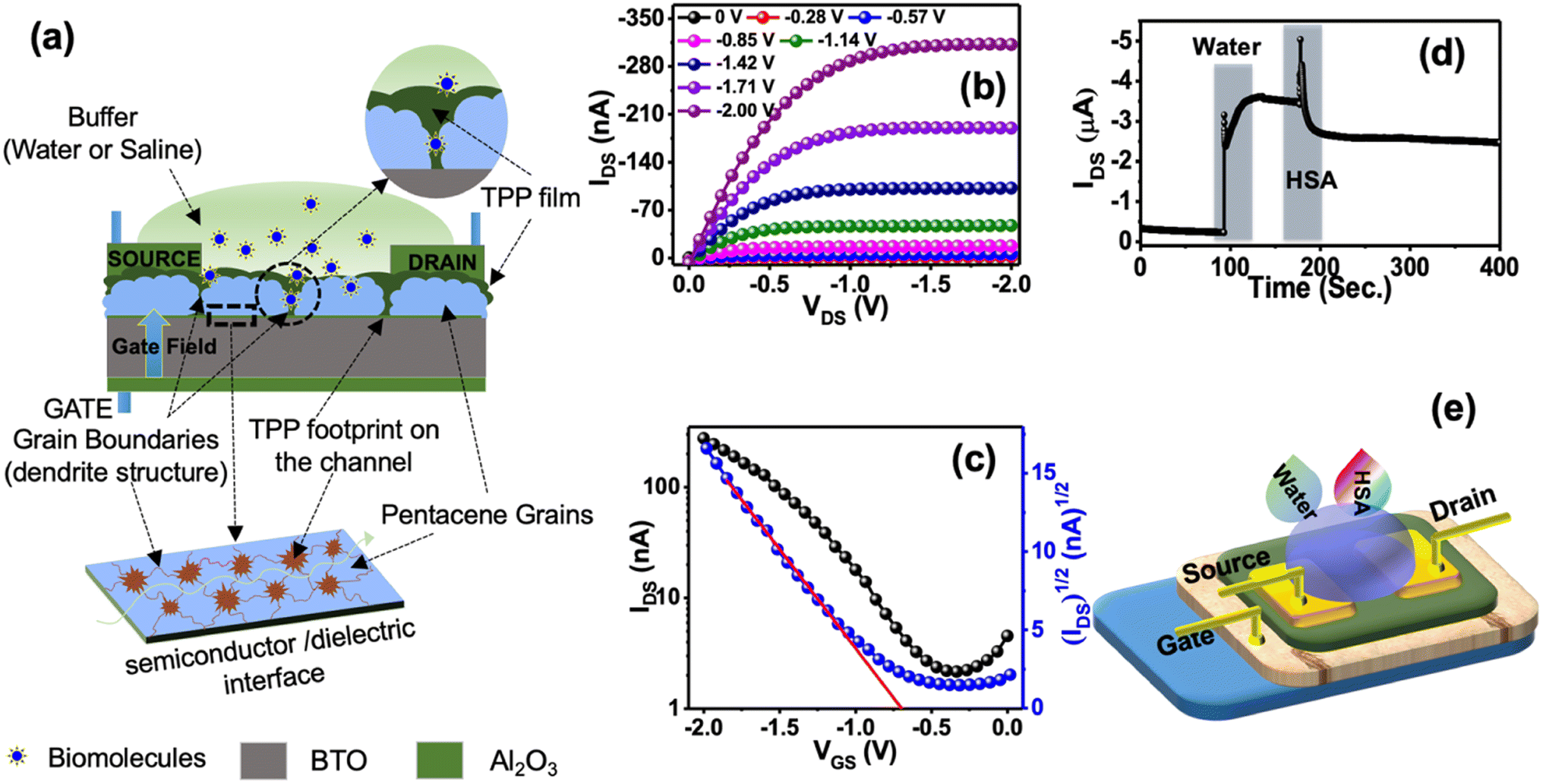 | ||
| Fig. 2 Device structure and OFET performances: (a) schematic 2D design of TPP and pentacene-based bi-layer OFETs. The footprint of TPP films is due to diffusion through the grain boundaries at the pentacene/h-BTO interfaces. (b) A conducting pathway at the interface is also marked. (c) Transfer and output characteristics of the bi-layer OFETs. (d) A typical response of the OFET drain current (ISD) profile during the sensing of HSA protein. The response to DI water followed by HSA protein is marked. (e) The schematic 3D diagram shows the DI water and HSA protein solution droplets on the sensors during measurement. | ||
The typical output and transfer characteristic curves of bilayer OFET are shown in Fig. 2b and c. Charge injection from the source to the semiconductor channel may be affected due to the presence of TPP molecules at the interface. However, we did not observe any kink or double slope in the transfer characteristics, confirming a better charge injection from the contact into the channel.36 The OFETs are operated at less than 2 V bias voltage. To test the performance of the sensors, we continuously monitored the drain current of the device biased at a constant voltage with VGS = VDS = –2 V after dropping 10 μl of buffer DI water in the channel region. A sharp increase in the drain current was observed, as shown in Fig. 2d. Such a sudden increase in drain current is explained in terms of surface conductivity due to the electrolysis of water.37 The amount of current depends on the volume of the water placed.39 However, soon the drain current gets saturated at a higher current. Once the current is stable, the second drop of HSA protein is added to the water droplet. The drain current further shoots up once the second drop is added. However, when TPP starts binding with HSA molecules present in the solution, the drain current is modified depending on the concentration of HSA protein present in the solution.
The stability of the sensors was tested under different conditions. As the sensors work under a buffer medium, we checked the device current in the buffer medium. There are two components of the device current under the buffer medium. Besides current due to the surface conductivity, the second part of the device current is the drain current flowing from the source to the drain via the semiconductor/dielectric interfaces. However, the drain current is dominated over the surface current in the presence of water or saline as we observed that the relative changes in transfer curves under these conditions were minimal, as shown in Fig. 3a and b. These results confirm that sensors were still working under the buffer media. We have also checked the stability of the device for one year. Fig. S5a† represents the transfer curve of the fresh device and the estimated mobility was 0.055 cm2 V−1 s−1. Fig. S5b† represents the transfer curve of the same device after one year and the estimated mobility was 0.013 cm2 V−1 s−1. The observed change in carrier mobility could be due to the degradation of TPP molecules over time. However, the reported sensing results are from freshly fabricated devices.
 | ||
| Fig. 3 Transfer characteristic curve of the bi-layer OFET under different buffer media. (a) Comparison of transfer curve under normal conditions and deionized water. (b) Comparison of transfer curve in normal condition and saline. It is confirmed from the above figure that the device is still working in buffer environments. There are two components of the device current, such as surface current and device current. In the presence of water/saline, the drain current was dominating over the surface current. | ||
There are more than 20 proteins existing in human plasma. However, the most abundant proteins consisting of 99% of total plasma protein in human serum are albumin (HSA), immunoglobulin G (IgG), and fibrinogen. In this study, we demonstrated the sensing of albumin and IgG from human serum. Besides, we also checked the sensing responses of lysozyme and RNase as non-blood proteins to confirm the sensors' selectivity. TPP was used to sense bovine serum albumin (BSA) protein by the fluorescence method.12 We used two different buffer media (e.g., DI water and saline), where the analyte molecules were dispersed before dropping onto the sensors. Initially, we dropped 10 μl of DI water on the sensing region of the OFET, which was under constant bias with VGS = VDS = −2 V. As we added various concentrations of HSA solutions ranging from 1 pM to 1 mM to the existing water droplets, we monitored the relative changes in the normalised device current. We used different devices for sensing each concentration of HSA. To quantify the relative changes in the response current, we considered the normalised device current (IP/IW), where IW and IP are the saturated currents after adding the water drop and HSA protein to the water drop, respectively. The normalized current responses for different concentrations of HSA are shown in Fig. 4a. It is observed that the normalized saturated device currents decrease with the increase of the HSA concentration.
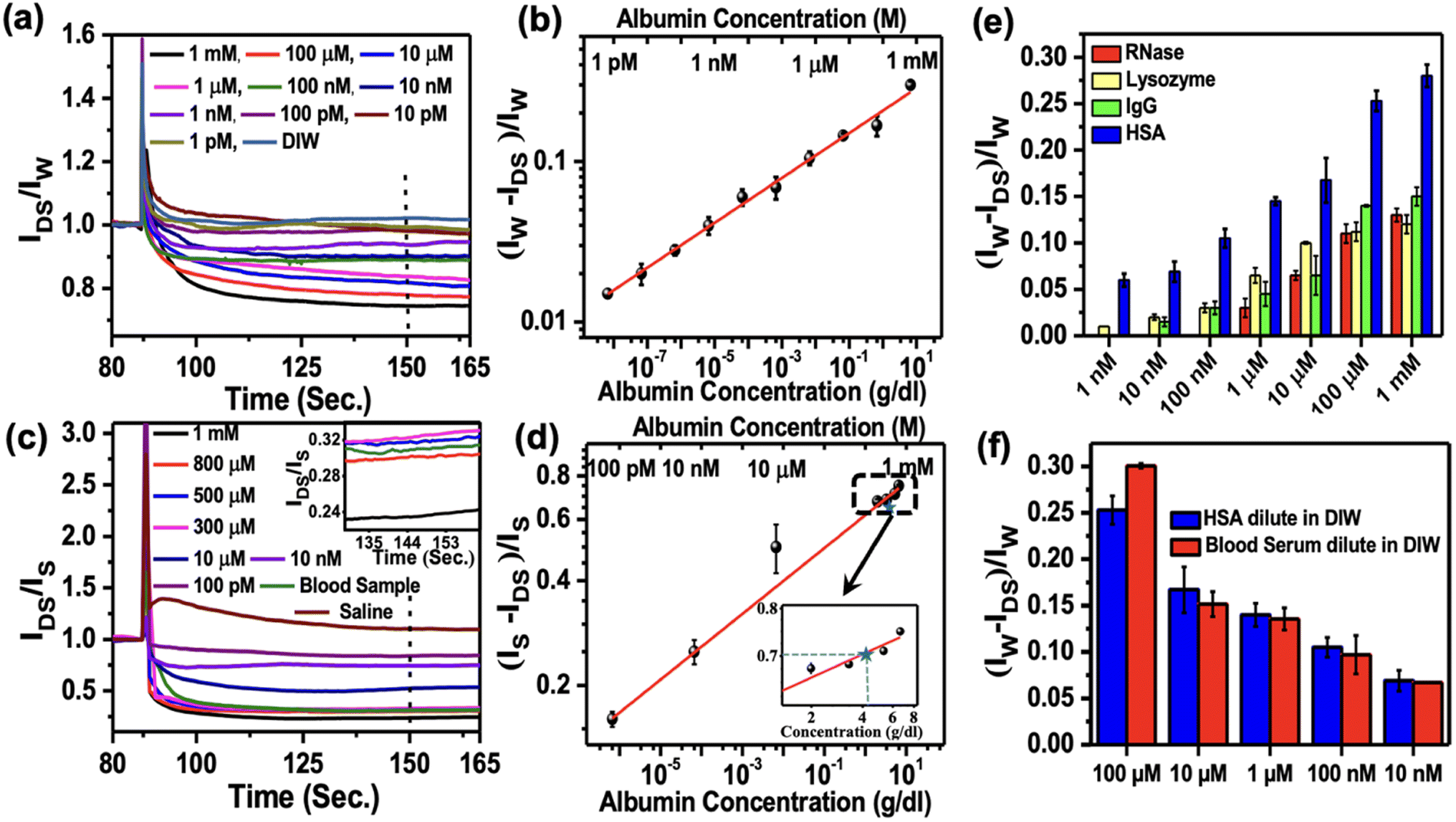 | ||
| Fig. 4 Normalized device current after exposing different albumin protein concentrations and buffer. IP, IW and IS are devices current in the presence of the albumin protein, DI water, and saline. (a) A normalized current of the device after exposure to different concentrations of HSA protein in DI water. (b) Change of the normalized current with varying concentrations of HSA protein (1 mM to 1 pM) in DI water. (c) A normalized current of the device after exposure to different concentrations of HSA protein in saline. (d) Change of the normalized current with varying concentrations of HSA protein (1 mM to 100 pM) in saline. (e) Dynamic change in the device current at different protein (HSA, IgG, lysozyme, and RNase) concentrations (1 mM to 1 nM). (f) Comparison of normalized current with varying concentrations of HSA protein of blood plasma and only HSA protein. | ||
The probe TPP is an aromatic triazolo amino acid scaffolded trichromophoric fluorescent pentapetide adopted β-sheet conformation wherein the scaffold (ArTAA, Fig. S3†) itself is a chromophore. The fluorescent triazolylpyrene (TPy) unnatural amino acids (TPyAlaDo) are other two chromophores attached to the N-and C-terminus, respectively, of the scaffold via an intervening natural amino acid, leucine BocNH-TPyAlaDo-Leu-ArTAADo-Leu-TPyAlaDo-CONMe(OMe). TPP is known as a “dual door entry to excimer emission” probe exhibiting a Förster Resonance Energy Transfer (FRET) mediated excimer emission at 470 nm via the π-stacked excited state complex formation among the two-terminal TPyAlaDo when it absorbs light of 290 nm. The FRET occurs from the scaffold amino acid ArTAA to TPyAlaDo to afford the excimer emission along with a monomer emission at 410 nm. The peptide was found to interact strongly with BSA, placing it in the hydrophobic pocket involved in hydrophobic and π–π-stacking interactions.12 Most effectively, the TPy moieties of the terminal amino acids TPyAlaDo got accommodated in the hydrophobic pocket of BSA, forming excimer and leaving aside the peptide chain on the surface. As the amount of BSA increased, π–π-stacked excited state complex formation between two TPy was hindered. The isothermal titration calorimetry indicated mostly an exothermic binding process for site II with a free energy change of −5.6 kcal mol−1. Spectroscopic evidence and molecular docking calculation suggested the close proximity of TPy of TPP and tryptophan-134 of BSA. They remained surrounded by other hydrophobic amino acids of the hydrophobic pocket of subdomain IB of site I of BSA. All the results suggested that both the hydrophobic as well as electrostatic interactions played an important role in the BSA–TPP interaction process. BSA and HSA proteins are 76% similar proteins. In this work, the detection of HSA protein was studied by considering similar interactions of BSA and TPP molecules in the devices.
With this experimental report and the 76% sequence homology between HSA and BSA, it is quite obvious that the similar interaction between TPP and HSA plays a crucial role in reducing the mobile charge density and hence, decreasing the current with the increasing amount of added HSA. We defined the responsivity of the sensors as ΔRN = (IW − IP)/IW typically calculated after the 60 s of dropping the HSA solution, as marked by the dashed line in Fig. 4a. The log–log variation of ΔRN with the concentration of HSA protein ranging from 1 pM to 1 mM is plotted in Fig. 4b and was used as a calibration curve. This shows a power law variation with the exponent 0.15 ± 0.02. The limit of detection (LOD) for the sensors is 1 pM, which is the lowest concentration we could measure. We also carried out similar experiments with saline as a buffer in place of DI water to detect HSA. The results are shown in Fig. 4c. The corresponding responsivity is defined as ΔRN = (IS − IP)/IS, with IS as saturated device current in the saline buffer. The results are shown in Fig. 4d. We again observed power law variation with the exponent 0.09 ± 0.03. The limit of detection in the case of saline buffer was around 100 pM. The inset of Fig. 4d displays the enlarged part of the calibration curve around mM concentrations. A comparison of various reported sensors for the detection of HSA is listed in Table S1.† It confirmed that the sensitivity of detection of HSA protein is much higher in water than in saline. This is also reflected in the lower value of the exponent as measured in the case of saline.
We studied the sensing response of IgG, lysozyme, and RNase with varying concentrations from 1 mM to 1 nM, as shown in Fig. 4e. We followed the same sensing procedure as used in the case of HSA protein sensing. Similar responses were observed for lysozyme, IgG, and RNase. However, we observed over 50% more responsivity for HSA in the higher concentration range than the other proteins. As the protein concentration decreases, the responsivity for all other proteins decreases significantly than HSA up to 1 nM concentration. This observation revealed the enhanced selectivity of TPP as a sensing material for the detection of HSA protein. To emphasize the role of the TPP film on sensing proteins, we also checked the responses of only pentacene-based OFETs without TPP films for sensing proteins and observed no significant responses (Fig. S7†). In order to check if there is any migration of Au materials from contacts to the channel in the presence of water or saline during sensing measurement, we carried out an EDAX experiment on the channel. No significant Au signal was observed on the channel confirming no damage to the contact during the measurement (Fig. S7†).
In order to explore the point-of-face usability of the sensors, we measured the HSA concentration directly from human blood plasma. Various concentrations of HSA from 100 μM to 10 nM were prepared by diluting blood plasma and the concentration of albumin in each solution was tested by the standard method used in the pathological laboratory. Additionally, we also measured the concentration by these sensors. To compare the responses, we carried out the testing of the same concentration of only HSA protein samples prepared by diluting HSA protein in DI water. A comparison of these two sets of results is shown in Fig. 4f. It is interesting to observe that the results collected from these sensors are comparable within the error bar with the results obtained using the standard pathological laboratory. The observations confirmed that the performance of the OFETs-based sensors developed by engineering the grain boundaries is on par with that of the standard methodology used for HSA detection in the pathological laboratory. Our method of developing biosensors will be attractive for commercial applications at POC.
We tested these sensors for the detection of HSA concentration directly from real blood samples without further processing. Blood samples were collected from a healthy person and the blood albumin level was tested using our OFET-based sensors three times. In these sensing experiments, we used saline as the buffer medium in place of DI water and followed the same experimental procedure as described above. We observed that the normalized average responses (ΔRN = (IS − IP)/IS) was around 0.714 (taken 60 seconds after the addition of blood), which corresponded to 4.2 ± 0.02 g dL−1 of the albumin concentration according to the calibration curve shown in Fig. 4d. The normalized device current for the blood samples tested in three different devices is shown in the supplementary Fig. S8.† The albumin concentration of the same blood sample was also measured from the pathological laboratory and the value obtained was around 4.1 g dL−1. The albumin concentration calculated using our protocol is about 2.5% higher than the value obtained from the standard clinical methods. The relatively higher value obtained in our method may be due to the presence of IgG (the second most abundant plasma protein in the blood) as we observed a non-zero response from IgG up to 10 nM concentration, as shown in Fig. 4e. Nevertheless, we typically have about ±5% error in the detection carried out using clinical methods used in the pathological laboratories. This confirmed that the sensors are suitable for the detection of HSA protein concentration directly from the blood samples, and it does not require any further sample preparation for post-blood collection.
Understanding the Debye length is essential for optimizing the design and performance of the electrochemical sensors, especially those that operate in solutions with varying ionic strengths. For 10 μL of human blood dissolved in 10 μL saline at 27 °C with an ionic concentration of 150 mM, the device has a Debye length, λD = 0.69 nm; and the same for saline at 27 °C with an ionic concentration of 30 mM, Debye length, λD = 1.5 nm.
Conclusions
We report the development of highly sensitive HSA protein sensors using OFETs as a detection platform. TPP molecules are used as sensing materials, which are incorporated into pentacene grain boundaries of the conducting channel of the devices. The diffused TPP molecules have easy accessibility to the conducting pathway of the drain current across the interface of the pentacene and BTO dielectric layer. As a result, the sensitivity of the detection of HSA protein was increased significantly. We observed high selectivity towards albumin protein detection in blood plasma and unprocessed blood samples. The measured LOD was found to be 1 pM. The sensors are capable of covering a huge concentration ranging from 1 pM to 1 mM concentration. The responses for the detection of different components present in the blood plasma showed high selectivity towards HSA protein. The results confirmed the suitability of these sensors for POC applications.Experimental section
Materials
Pentacene was purchased from TCI with a purity of 99.9%. Hexagonal barium titanate (h-BTO), used as gate dielectric material, was synthesized using the sol–gel method.35 HSA, lysozyme, IgG, and RNase molecules were purchased from Sigma Aldrich and used without further modification. TPP molecules were synthesized following the method described in literature.12Device fabrication and characterization
The top contact bottom gate pentacene-TPP-based bilayer OFET devices were fabricated on the glass substrate. Substrates were ultrasonically cleaned stepwise using DI water, acetone, isopropanol, and again DI water for 20 minutes in each solution. Thereafter, the substrates were taken out of the solution and purged with dry N2 Gas. An aluminum (Al) film (as gate electrode) of 120 (±10) nm was deposited on a clean glass substrate using the thermal evaporation technique with chamber pressure 2 × 10−6 mbar. A part of Al film was converted into alumina (Al2O3) of 15 nm thickness using an anodization technique, where a glass substrate with the aluminium strip (working electrode) and platinum wire (counter electrode) was immersed in an electrolyte solution, prepared by diluting citric acid in DI water. The h-BTO sol was spin-coated on Al2O3 at 3000 RPM for 1 min to grow the second dielectric layer. The film was annealed at 80 °C for 80 minutes after spin coating. A thin pentacene film of 20 nm was vacuum sublimed at 240 °C on the h-BTO layer using an organic molecular beam deposition (OMBD) chamber at a substrate temperature of 80 °C at 1 × 10−6 mbar chamber pressure. TPP solutions were prepared by diluting TPP molecules in ethanol and vigorously stirring for 1 hour at room temperature. Then, TPP molecules were spin-coated by varying the spinning speeds from 500 rpm to 5000 rpm for 1 minute on pentacene surfaces. In order to dry the excess solvent, TPP films were kept in a vacuum desiccator for 1 hour. Gold (Au) films were grown using a shadow mask for the source and drain contacts in a thermal evaporation chamber at a pressure of 2 × 10−6 mbar. The typical channel length and width for the devices were 30 μm and 2 mm, respectively. All the electrical measurements were carried out using Keithley 2450 source meters inside a probe station. Crystalline structure and surface morphology of TPP and pentacene films were characterized by X-ray diffraction (XRD) using the Bruker instrument, atomic force microscopy (AFM) in tapping mode (Agilent Technologies, model 5100), and Scanning Electron Microscopy (SEM) (Zeiss supra 40).Sensing experiment
All the protein sensing experiments were performed in a buffer medium of either DI water or Saline under ambient conditions. Protein molecules were diluted in DI water at different concentrations. At constant bias voltage (–2 V), 0.5 μL of DI water or saline was dropped on the active area of OFET, followed by adding a drop of 0.5 μL protein solution in the same area to let the interaction between HSP and TPP occur in the liquid environments.HSA detection using the bromocresol green (BCG) method in pathology laboratory
Quantitative estimation of HSA was carried out by EM 360 automated clinical chemistry analyser using BCG assay kits. The measuring range of this system was 0.1–7.2 g dL−1 with an error of ± 0.25 g dL−1. In the BCG method, due to the binding between the bromocresol green dye and human serum albumin, a chromophore that can be detected at wavelength 630 nm is formed. The intensity of the absorbance is linearly proportional to the amount of albumin present in serum. Blood samples were procured from the clinic for testing purposes. BCG measurements were carried out by the clinicians.Conflicts of interest
The authors declare no competing interests.Acknowledgements
This work was partially supported by (MeitY) 5(1)/2017-NANO, (DST) DST/NM/NNetRA/2018(G)-IIT-KGP. We would like to thank Mr Rajib Das (Pathologist at the B. C. Roy Technology Hospital, IIT Kharagpur) for helping with the measurements of the albumin samples using an EM 360 automated clinical chemistry analyser.References
- L. Torsi, M. Magliulo, K. Manoli and G. Palazzo, Chem. Soc. Rev., 2013, 42, 8612–8628 RSC.
- Y. H. Lee, O. Y. Kweon, H. Kim, J. H. Yoo, S. G. Han and J. H. Oh, J. Mater. Chem. C, 2018, 6, 8569–8612 RSC.
- Y. J. Wang, Q. Gong and Q. Miao, Mater. Chem. Front., 2020, 4, 3505–3520 RSC.
- M. Jang, H. Kim, S. Lee, H. W. Kim, J. K. Khedkar, Y. M. Rhee, I. Hwang, K. Kim and J. H. Oh, Adv. Funct. Mater., 2015, 25, 4882–4888 CrossRef CAS.
- A. Mandal, S. Mandal, S. P. Verma, S. Mallik, S. S. Bag and D. K. Goswami, Adv. Mater. Interfaces, 2023, 10, 2202293 CrossRef CAS.
- A. Mandal, S. Mallik, S. Mondal, S. Subhadarshini, R. Sadhukhan, T. Ghoshal, S. Mitra, M. Manna, S. Mandal and D. K. Goswami, ACS Sens., 2022, 7, 3006–3013 CrossRef CAS PubMed.
- J. J. Lu, D. P. Liu, J. C. Zhou, Y. L. Chu, Y. T. Chen, X. H. Wu and J. Huang, Adv. Funct. Mater., 2017, 27, 1700018 CrossRef.
- B. Kang, M. Jang, Y. Chung, H. Kim, S. K. Kwak, J. H. Oh and K. Cho, Nat. Commun., 2014, 5, 4752 CrossRef CAS.
- F. J. Zhang, G. Qu, E. Mohammadi, J. G. Mei and Y. Diao, Adv. Funct. Mater., 2017, 27, 1701117 CrossRef.
- D. Xu, G. D. Watt, J. N. Harb and R. C. Davis, Nano Lett., 2005, 5, 571–577 CrossRef CAS.
- C.-A. Vu and W.-Y. Chen, Sensors, 2019, 19, 4214 CrossRef CAS PubMed.
- S. S. Bag, S. Jana, M. K. Pradhan and S. Pal, RSC Adv., 2016, 6, 72654–72658 RSC.
- S. Nam, H. Han, J. Seo, M. Song, H. Kim, T. D. Anthopoulos, I. McCulloch, D. D. C. Bradley and Y. Kim, Adv. Electron. Mater., 2016, 2, 1600264 CrossRef.
- M. M. Azmi, Z. Tehrani, R. Lewis, K.-A. Walker, D. Jones, D. Daniels, S. Doak and O. Guy, Biosens. Bioelectron., 2014, 52, 216–224 CrossRef.
- S. Xu, S. Jiang, C. Zhang, W. Yue, Y. Zou, G. Wang, H. Liu, X. Zhang, M. Li, Z. Zhu and J. Wang, Appl. Surf. Sci., 2018, 427, 1114–1119 CrossRef CAS.
- M. Tian, M. Qiao, C. Shen, F. Meng, L. A. Frank, V. V. Krasitskaya, T. Wang, X. Zhang, R. Song, Y. Li and J. Liu, Appl. Surf. Sci., 2020, 527, 146839 CrossRef CAS.
- Y. Zhao, R. J. Tong, M. Q. Chen and F. Xia, Sens. Actuators, B, 2019, 284, 96–102 CrossRef CAS.
- G. J. Quinlan, G. S. Martin and T. W. Evans, Hepatology, 2005, 41, 1211–1219 CrossRef CAS.
- M. Taverna, A. L. Marie, J. P. Mira and B. Guidet, Ann. Intensive Care, 2013, 3, 4 CrossRef PubMed.
- X. M. He and D. C. Carter, Nature, 1992, 358, 209–215 CrossRef CAS PubMed.
- D. Barchel, D. Almoznino-Sarafian, M. Shteinshnaider, I. Tzur, N. Cohen and O. Gorelik, Eur. J. Intern. Med., 2013, 24, 772–778 CrossRef CAS PubMed.
- E. A. Mutlu, A. Keshavarzian and G. M. Mutlu, Scand. J. Gastroenterol., 2006, 41, 759–760 CrossRef CAS PubMed.
- S. Arques, Eur. J. Intern. Med., 2018, 52, 8–12 CrossRef CAS PubMed.
- T. B. Horwich, K. Kalantar-Zadeh, R. W. MacLellan and G. C. Fonarow, Am. Heart. J., 2008, 155, 883–889 CrossRef CAS PubMed.
- H. Martin, Clin. Biochem. Rev., 2011, 32, 97–102 Search PubMed.
- J. L. Zhang, R. Zhang, Y. T. Wang, H. Y. Li, Q. Q. Han, Y. C. Wu, T. L. Wang and F. Liu, J. Diabetes Res., 2019, 7825804 CAS.
- P. B. Vaziri, M. Vahedi, S. H. Abdollahzadeh, H. R. Abdolsamadi, M. Hajilooi and S. H. Kasraee, Iran. J. Public Health, 2009, 38, 54–59 Search PubMed.
- J. H. Contois, C. Hartigan, L. V. Rao, L. M. Snyder and M. J. Thompson, Clin. Chim. Acta, 2006, 367, 150–155 CrossRef CAS.
- R. S. E. Wang, L. Tian and Y. H. Chang, J. Pharm. Biomed. Anal., 2012, 63, 165–169 CrossRef CAS PubMed.
- J. Qian, M. M. Sun, M. Liu and W. Gu, ACS Omega, 2019, 4, 11949–11959 CrossRef CAS PubMed.
- G. Liu, J. Wei, X. Li, M. Tian, Z. Wang, C. Shen, W. Sun, C. Li, X. Li, E. Lv and S. Tian, Adv. Sci., 2022, 26, 2202505 CrossRef PubMed.
- M. De, S. Rana, H. Akpinar, O. R. Miranda, R. R. Arvizo, U. H. F. Bunz and V. M. Rotello, Nat. Chem., 2009, 1, 461–465 CrossRef CAS PubMed.
- P. Anees, S. Sreejith and A. Ajayaghosh, J. Am. Chem. Soc., 2014, 136, 13233–13239 CrossRef CAS.
- B. T. Dumas, W. A. Watson and H. G. Biggs, Clin. Chim. Acta, 1997, 258, 21–30 CrossRef CAS.
- Z. Stojanovic, J. Erdossy, K. Keltai, F. W. Scheller and R. E. Gyurcsanyi, Anal. Chim. Acta, 2017, 977, 1–9 CrossRef CAS.
- E. Arkana, R. Saber, Z. Karimi, A. Mostafaie and M. Shamsipur, J. Pharm. Biomed. Anal., 2014, 92, 74–81 CrossRef.
- H. Li, W. Shi, J. Song, H. J. Jang, J. Dailey, J. S. Yu and H. E. Katz, Chem. Rev., 2019, 119, 3–35 CrossRef CAS PubMed.
- S. Mandal, M. Banerjee, S. Roy, A. Mandal, A. Ghosh, B. Satpati and D. K. Goswami, ACS Appl. Mater. Interfaces, 2019, 11, 4193–4202 CrossRef CAS PubMed.
- P. A. Bobbert, A. Sharma, S. G. J. Matheijssen, M. Kemerink and D. M. Leeuw, Adv. Mater., 2012, 24, 1146–1158 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3na00564j |
| This journal is © The Royal Society of Chemistry 2024 |