
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceComprehensive characterization of polyproline tri-helix macrocyclic nanoscaffolds for predictive ligand positioning†
Chia-Lung
Tsai
a,
Je-Wei
Chang
b,
Kum-Yi
Cheng
c,
Yu-Jing
Lan
 a,
Yi-Cheng
Hsu
a,
Qun-Da
Lin
a,
Tzu-Yuan
Chen
a,
Orion
Shih
b,
Chih-Hsun
Lin
c,
Po-Hsun
Chiang
a,
Mantas
Simenas
d,
Vidmantas
Kalendra
d,
Yun-Wei
Chiang
*a,
Chun-hsien
Chen
*c,
U-Ser
Jeng
*bef and
Sheng-Kai
Wang
*ag
a,
Yi-Cheng
Hsu
a,
Qun-Da
Lin
a,
Tzu-Yuan
Chen
a,
Orion
Shih
b,
Chih-Hsun
Lin
c,
Po-Hsun
Chiang
a,
Mantas
Simenas
d,
Vidmantas
Kalendra
d,
Yun-Wei
Chiang
*a,
Chun-hsien
Chen
*c,
U-Ser
Jeng
*bef and
Sheng-Kai
Wang
*ag
aDepartment of Chemistry, National Tsing Hua University, Hsinchu 300044, Taiwan. E-mail: skwang@mx.nthu.edu.tw; ywchiang@mx.nthu.edu.tw
bNational Synchrotron Radiation Research Center, Hsinchu 300092, Taiwan. E-mail: usjeng@nsrrc.org.tw
cDepartment of Chemistry and Centre for Emerging Materials and Advanced Devices, National Taiwan University, Taipei 106319, Taiwan. E-mail: chhchen@ntu.edu.tw
dFaculty of Physics, Vilnius University, Sauletekio 3, LT-10257 Vilnius, Lithuania
eDepartment of Chemical Engineering, National Tsing Hua University, Hsinchu 300044, Taiwan
fCollege of Semiconductor Research, National Tsing Hua University, Hsinchu, 300044, Taiwan
gFrontier Research Center on Fundamental and Applied Sciences of Matters, National Tsing Hua University, Hsinchu 300044, Taiwan
First published on 1st January 2024
Abstract
Multivalent ligands hold promise for enhancing avidity and selectivity to simultaneously target multimeric proteins, as well as potentially modulating receptor signaling in pharmaceutical applications. Essential for these manipulations are nanosized scaffolds that precisely control ligand display patterns, which can be achieved by using polyproline oligo-helix macrocyclic nanoscaffolds via selective binding to protein oligomers and cell surface receptors. This work focuses on synthesis and structural characterization of different-sized polyproline tri-helix macrocyclic (PP3M) scaffolds. Through combined analysis of circular dichroism (CD), small- and wide-angle X-ray scattering (SWAXS), electron spin resonance (ESR) spectroscopy, and molecular modeling, a non-coplanar tri-helix loop structure with partially crossover helix ends is elucidated. This structural model aligns well with scanning tunneling microscopy (STM) imaging. The present work enhances the precision of nanoscale organic synthesis, offering prospects for controlled ligand positioning on scaffolds. This advancement paves the way for further applications in nanomedicine through selective protein interaction, manipulation of cell surface receptor functions, and developments of more complex polyproline-based nanostructures.
1. Introduction
Multivalent interactions are involved in many essential biological processes including recognition1 and signal transduction.2 These interactions between multiple ligands and receptors are commonly observed in carbohydrate-lectin binding, where the protein forms oligomers with a fixed arrangement such that the matching glycan ligand pattern leads to strong and selective multivalent binding.3 In contrast, mobile neighboring protein receptors on the cell surface can change their organizational arrangement to give alternative signaling by binding to the pre-organized multiple ligands.4 In both scenarios, precise presentation of multiple ligands results in alternative protein responses over the conventional monovalent ligands and therefore represents a promising strategy to manipulate biological processes.As the separation among the proteins involved in multivalent interactions is typically at the nanoscale, nano-sized objects are usually considered for application as scaffolds to support multiple ligands in nanomedicine. Nevertheless, most of these objects are self-assembled or fabricated with a specific growing process with no precise control on individual ligand positions. Instead, some families of macrocyclic compounds, such as cyclodextrins5 and calixarenes,6 have been used as multivalent scaffolds due to their well-defined structures and available sites for ligand conjugation. However, even these widely used scaffolds are not ideal because of their limited available sizes and, more importantly, the difficulty of controlling ligand patterns through selective conjugation, especially when asymmetric ligand patterns are required. In contrast to the shortcomings of these popular scaffolds, cyclic peptide scaffolds provide versatile scaffold designs.7 We exploited polyproline peptides to create a new cyclic scaffold system which allows precise control of ligand positions via the peptide sequence.
Polyproline peptides favor a stable and rigid polyproline helix II (PPII) conformation in aqueous solution and have been widely exploited as a molecular ruler.8 Structural investigations have indicated the PPII structure is highly symmetrical, with helical periodicity of 3 residues per turn and a pitch of about 0.9 nm (Fig. 1).9,10 The combination of its unique structure and biocompatibility has allowed researchers to control the directions and spacing of the ligands attached to the proline ring under aqueous conditions for a variety of biomedical-related applications.11–17
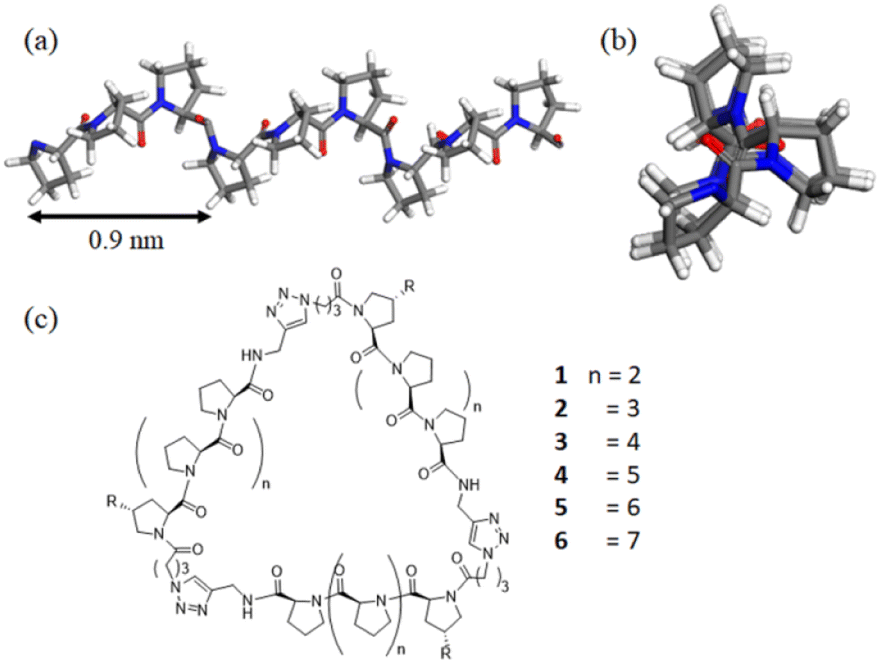 | ||
| Fig. 1 (a) Side view of the left-handed PPII helix showing the pitch; (b) axial view of the PPII helix showing the symmetry of the residues; (c) the chemical structures of the nanoscaffolds involved in this work. | ||
Previously, our group took advantage of these features to create polyproline tri-helix macrocyclic (PP3M) scaffolds to control ligand patterns at the nanoscale. This design has several advantages over other popular cyclic scaffolds. One is that the size of the PP3M scaffold can be adjusted over a broader range by using different lengths of the polyproline helix. More importantly, as the ligand conjugation site can be precisely controlled by using the sequence of the peptide and the order of peptide assembly, either symmetric or asymmetric ligand patterns on the scaffold can be tailored to have desired ligand numbers. These breakthroughs have enabled investigations on how ligand patterns affect the avidities to protein oligomers and cell surface receptors.17
The knowledge of the structure of the polyproline cyclic nanoscaffolds would greatly facilitate the prediction of the relative locations of the ligands in the design of the ligand pattern for a protein target. However, structural studies are challenging, as polyproline peptides are well-known for not crystalizing easily,10 NMR studies are hampered by the nature of these peptides. In contrast, other techniques, such as small-angle X-ray scattering (SAXS), can be used to probe the global conformation of biomolecules in a solution,18 including intrinsically disordered or unfolded proteins.19,20 Recently, combined analysis of SAXS and wide-angle X-ray scattering (SWAXS) was shown to improve the structural probing resolution for interfacial/local structures of proteins. Electron spin resonance (ESR) spectroscopy, another powerful tool, provides spatial information within a biomolecule. In particular, a widely used ESR spectroscopy method is double electron–electron resonance (DEER), which allows one to obtain distance distributions between pairs of chemically modified nitroxide spin labels within a biomolecule, thereby facilitating examination of the structure and conformational flexibility of a protein or peptide.21–23
In this study, we report the synthesis of PP3M scaffolds of tri-helix cyclic polyproline peptides, with different peptide lengths of Pro4 and Pro5 helices (Fig. 1). The smallest PP3M to date might be used for targeting smaller protein oligomers. Using the recent new synchrotron BioSWAXS beamline of the Taiwan Photon Source (TPS) featuring simultaneous SAXS and WAXS measurements and an online size exclusion column (SEC)24 for biomolecular solutions,25,26 we are able to reveal the correlated global structures of PP3M in solutions as the helix length of PP3M changes. Furthermore, with integrated analysis of circular dichroism (CD), small- and wide-angle X-ray scattering (SAXS and WAXS), ESR spectroscopy and scanning tunneling microscopy (STM), we propose possible solution structures of the PP3M scaffolds to elucidate the global structural features of the three PPII helices.
2. Experimental section
2.1 Synthetic procedures
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, final concentration 0.1 M), and then the solution was added to the resins that had already swelled and been gently shaken at room temperature overnight. Next, the resins were washed with DMF and CH2Cl2 repeatedly. A solution of CH2Cl2/MeOH/i-Pr2NEt (17:2:1) was added and shaken for 1 h to block the unreacted sites on the resins. The resultant resins were washed with DMF and CH2Cl2 and underwent a quantitative Fmoc test to determine the amino acid loading. The resins were further used in iterative peptide synthesis. The Fmoc protecting group was removed by treating the resins in a reaction vessel with 10% piperidine in DMF for 10 min and washed with DMF and CH2Cl2 repeatedly. Then the coupling solution was prepared by dissolving Fmoc-Pro-OH (4.0 equiv.) or trans-N-Alloc-4-amino prolines, PyBOP (4.0 equiv., for Fmoc-Pro-OH) or hexafluorophosphate azabenzotriazole tetramethyl uronium (HATU) (4.0 equiv., for trans-N-Alloc-4-amino prolines) in DMF, followed by the addition of N-methylmorpholine (NMM, 4.0 equiv.) to give a final concentration of 0.05 M. The coupling solution was added to resins in the reaction vessel, gently shaken at room temperature for 1 h, and then washed with DMF and CH2Cl2 repeatedly. After each coupling step, the resins were treated with Ac2O (10 vol% in pyridine, 5.0 mL) for 10 min to block the unreacted amino groups and then washed with DMF and CH2Cl2. These procedures were repeated until the desired sequence was synthesized. To cleave the product peptide, the resins 7, 8 and 19 were washed with CH2Cl2 and treated with CH2Cl2/TFA/(i-Pr)3SiH (90:5:5) under shaking at room temperature for 1 h. The filtrate was collected and the remaining resins were treated with the cleaving agent again for 30 min. The combined filtrates were collected and all of the volatiles were removed under reduced pressure. The residues were dissolved in water (1–2 mL) and centrifuged before the supernatant was purified by HPLC (Agilent Technology, 1260 Infinity) with a Vydac C18 column (218TP510 10 mm × 250 mm). 0.1% TFA in water (solvent A) and acetonitrile (solvent B) served as the mobile phase for peptide chromatography.
:1, peptide concentration 0.02 M), propargylamine (3.0 equiv.) and Et3N (5.0 equiv.) were added. After stirring for 1 h, CH2Cl2 was removed from the reaction mixture under reduced pressure. The remaining mixture was purified by HPLC with a Vydac C18 column.
:1, peptide concentration 0.6 M) in a vial, acetic acid (540 equiv.) and Bu3SnH (120 equiv.) were added, and the solution was stirred for 2 h before quenching with water. After the removal of CH2Cl2 under reduced pressure, the deprotected peptide was purified by HPLC with a Vydac C18 column.
:1, final concentration 0.1 M), and then the solution was added to the resins that had already swelled and been gently shaken at room temperature overnight. Next, the resins were washed with DMF and CH2Cl2 repeatedly. A solution of CH2Cl2/MeOH/i-Pr2NEt (17:2:1) was added and shaken for 1 h to block the unreacted sites on the resins. The resultant resins were washed with DMF and CH2Cl2 and underwent a quantitative Fmoc test to determine the amino acid loading. The resins were further used in iterative peptide synthesis. The Fmoc protecting group was removed by treating the resins in a reaction vessel with 10% piperidine in DMF for 10 min and washed with DMF and CH2Cl2 repeatedly. Then the coupling solution was prepared by dissolving Fmoc-Pro-OH (4.0 equiv.) or trans-N-Alloc-4-amino prolines, PyBOP (4.0 equiv., for Fmoc-Pro-OH) or hexafluorophosphate azabenzotriazole tetramethyl uronium (HATU) (4.0 equiv., for trans-N-Alloc-4-amino prolines) in DMF, followed by the addition of N-methylmorpholine (NMM, 4.0 equiv.) to give a final concentration of 0.05 M. The coupling solution was added to resins in the reaction vessel, gently shaken at room temperature for 1 h, and then washed with DMF and CH2Cl2 repeatedly. After each coupling step, the resins were treated with Ac2O (10 vol% in pyridine, 5.0 mL) for 10 min to block the unreacted amino groups and then washed with DMF and CH2Cl2. These procedures were repeated until the desired sequence was synthesized. To cleave the product peptide, the resins 7, 8 and 19 were washed with CH2Cl2 and treated with CH2Cl2/TFA/(i-Pr)3SiH (90:5:5) under shaking at room temperature for 1 h. The filtrate was collected and the remaining resins were treated with the cleaving agent again for 30 min. The combined filtrates were collected and all of the volatiles were removed under reduced pressure. The residues were dissolved in water (1–2 mL) and centrifuged before the supernatant was purified by HPLC (Agilent Technology, 1260 Infinity) with a Vydac C18 column (218TP510 10 mm × 250 mm). 0.1% TFA in water (solvent A) and acetonitrile (solvent B) served as the mobile phase for peptide chromatography.
:1, peptide concentration 0.02 M), propargylamine (3.0 equiv.) and Et3N (5.0 equiv.) were added. After stirring for 1 h, CH2Cl2 was removed from the reaction mixture under reduced pressure. The remaining mixture was purified by HPLC with a Vydac C18 column.
:1, peptide concentration 0.6 M) in a vial, acetic acid (540 equiv.) and Bu3SnH (120 equiv.) were added, and the solution was stirred for 2 h before quenching with water. After the removal of CH2Cl2 under reduced pressure, the deprotected peptide was purified by HPLC with a Vydac C18 column.
2.2 Small- and wide-angle X-ray scattering (SWAXS)
Aqueous sample solutions of PP3M nanoscaffolds 1–6 were prepared for simultaneous SAXS-WAXS at the Taiwan Photon Source (TPS) 13A BioSWAXS beamline of the National Synchrotron Radiation Research Center (NSRRC), Hsinchu.25,26 The beamline is featured with two in-vacuum pixel detectors of Eiger X 9M and Eiger X 1M, and an online high-performance liquid chromatography (HPLC) system (Agilent chromatographic system 1260 series) for size-exclusion-column (SEC) SAXS-WAXS. However, the column-bypass mode with a sample flow (with 10–50 μL sample of 10–50 mg mL−1) range of 0.1–0.3 mL min−1 was26 selected to avoid unwanted sample interactions with the silica-gel-based size exclusion column used. A 15 keV X-ray beam was irradiated on a thin-wall (ca. 25 μm) quartz capillary cell (2.0 mm diameter) for SAXS-WAXS measurements at 288 K, together with simultaneous UV-vis absorption and differential refractive index measurements. The SAXS and WAXS (SWAXS) data were combined to cover a wider scattering vector q-range, where q = 4πλ−1sinθ is defined by the scattering angle 2θ and X-ray wavelength λ (0.8266 Å) used. The radii of gyration Rg were extracted using the ATSAS package.28 Initial molecular models of nanoscaffolds 1–6 were built using Material Studio (Accelrys Software, Inc.); the models were then refined through fitting the theoretical SWAXS profiles to the corresponding SWAXS data for minimized χ2 values (ESI†) using CRYSOL software.28
2.3 DEER measurements and analysis
The pulsed ESR experiments were conducted at 80 K using a Bruker ELEXSYS E580-400 CW/pulsed spectrometer, with a split-ring resonator (EN4118X-MS3) and a helium gas flow system (4118CF). The DEER experiments were performed using the typical four-pulse constant-time DEER sequence as previously described.29 For enhanced sensitivity, we employed an EPR cryoprobe of a similar design as reported previously.30Data analysis for DEER was performed using the model-based analysis method as detailed below. Inter-spin distance distributions can be modeled as a sum of Gaussians, as previously demonstrated. The Gaussian-based analysis and error estimations for DEER were then performed utilizing the program DD (version 7).30,31 In DD, the errors in distance distribution are quantified in terms of a confidence band about the best-fit solution. This study presents the results in a confidence band of 2σ (95.4%), which reflects the influence of both the noise in the measured DEER data and the uncertainty in the estimate of the background correction. The confidence band is calculated from the full covariance matrix using the delta method.
2.4 Molecular model establishment
The PPII helix structure was obtained from the Cambridge Crystallographic Data Centre (CCDC).10 Subsequently, the helices were organized into a triangular arrangement. Each of the helices was altered in position (helices were moved in/out to the centroid) (d) and incline angle (θ), generating a number of arrangements. These arrangements were then connected by connector moieties. Following the constraint of the PPII structure, each arrangement underwent geometry optimization using a universal forcefield (Both electrostatic and van der Waals terms employ atom-based summation methods with a cubic spline truncation method. The cutoff distance is 15.5 Å, and the spline width and buffer width are set to 1 Å and 0.5 Å, respectively.) in the Forcite module of Materials Studio (Accelrys Software, Inc.) to optimize the structure of the added connector moieties.The two favorable structure models for 6 were then subjected to alterations in the helix rotation angle along the axis (ϕ), resulting in multiple arrangements. For simplicity, we use the nitrogen atom on Pro-6 (sixth proline residue from the N-terminus) to define the helix rotation angle as it is close to the center of mass for scaffold 6. The helix rotation angle that allowed the smallest distance between this nitrogen atom and the center of mass was defined as 0°. These arrangements were added linkers and spin labels. Following the constraint of the PPII structure, the linker and spin label moieties of each arrangement underwent geometry optimization in the Forcite module of Materials Studio to optimize the structures. Subsequently, with constraint of the PPII structure, the arrangements were subjected to annealing and further geometry optimization using a universal forcefield. An initially energy-minimized structure was subjected to 50 annealing cycles with the initial and midcycle temperatures of 300 and 1400 K, respectively, twenty heating ramps per cycle, one thousand dynamic steps per ramp, and one dynamic step per femtosecond. For each arrangement, 50 conformers of the linker and spin label moieties after annealing were generated.
The structure of Helix pomatia agglutinin complexed with GalNAc was obtained from the RCSB PDB database.32 Subsequently, the polyproline tri-helix macrocycles were superimposed and connected to the GalNAcs using linkers as in 28. With the polyproline tri-helix macrocycles and GalNAcs constrained, the model underwent geometry optimization using a universal forcefield in the Forcite module of Materials Studio to optimize the structure of the linkers.
2.5 STM imaging
To prepare the sample for STM imaging, the substrate, HOPG (highly oriented pyrolytic graphite, Bruker, ZYB-grade), was pre-modified with a porous monolayer of TMA (trimesic acid, Sigma-Aldrich, 98%). Detailed preparation and characterization of the TMA adlayer are reported elsewhere.33 Briefly, TMA was dissolved in PO (1-phenyloctane, TCI, 98%). After dropcasting of a 2.0 μL aliquot of saturated TMA on HOPG, the excess solvent was removed by using a piece of Kimwipes placed against the edge of the HOPG. A trace amount of scaffold 27 was dissolved in acetonitrile (aencore, >99.9%) and a 2.0 μL aliquot was placed on the TMA-modified HOPG. After most of the acetonitrile was evaporated, PO (<1 μL) was added on HOPG, and STM images were acquired by using a MultiMode NanoScopeIIIa (Bruker). The STM tips were home-made mechanically cut Pt/Ir wires (80%/20%, diameter 0.25 mm, California Fine Wires).3. Results and discussion
3.1 Synthesis of PP3M scaffolds
We exploited efficient solid-phase peptide synthesis/ligation to prepare PP3M scaffolds.17 This modular strategy not only allowed the ligand pattern to be fully controlled but also facilitated convenient pattern change by swapping the peptide units during assembly.17 To achieve the smallest PP3M scaffold to date, we prepared Pro4 and Pro5-PP3M with an approach similar to that for synthesizing the Pro6-based counterpart.34 2-Chlorotrityl chloride resins were exploited to prepare Pro4 and Pro5 peptides to allow further modifications on the C-terminal acid groups after cleaving from resins 7 and 8 (Scheme 1). A propargyl group was installed on the peptide C-terminal through amide coupling to give alkynyl Pro4 and Pro5 peptides 9 and 10. The remaining resins 7 and 8 were modified at the N-terminus by Fmoc deprotection, followed by the coupling of connector 11 to install azido groups (13 and 14) for modular assembly. Next, the robust Cu(I) catalyzed alkyne–azide cycloaddition (CuAAC) reaction35 was utilized to efficiently ligate alkynyl peptides 9 or 10 to the resin-bound peptides 13 and 14. The resulting di-helix intermediate underwent Fmoc deprotection and azido connector installation at the N-terminus to prepare for the next peptide ligation. The three-step cycle, including CuAAC ligation, Fmoc deprotection and azido connector installation, was repeated again to give the tri-helix intermediates 15 and 16 on the solid supports. Then the tri-helix peptides were cleaved from the resins and the propargyl groups were installed at their C-termini through amide coupling reactions. The resultant linear tri-helix peptides 17 and 18 then underwent the intramolecular CuAAC reaction for cyclization, which was performed at low concentration with no other oligomer products observed. Due to the smaller sizes compared with the other reported PP3M scaffolds, it was interesting to determine whether the cyclic strain from 17 and 18 could prevent this process. As the cyclization products 1 and 2 have the same molecular weights as the corresponding linear tri-helix peptides 17 and 18, we used excess small molecular alkyne compound to react with 1 and 2 under CuAAC conditions to confirm the completion of cyclization by mass spectrometry. While the linear peptide 17 conjugated to the added alkyne compound through its free azide group, scaffolds 1 and 2 did not react with the alkyne under the same CuAAC conditions (ESI Fig. S13 and S14†). This observation confirmed no degradation of the functional groups (no mass change) and that no free azide was available on scaffolds 1 and 2 showing that the scaffolds were cyclized. These results indicated that the CuAAC reaction is strong enough to cyclize the tri-helix ring despite the strains from the smaller PP3M nanoscaffolds. | ||
| Scheme 1 Synthesis of polyproline tri-helix macrocycle scaffolds. Synthesis of 3–6 has been reported previously in ref. 34. | ||
In addition to Pro4 and Pro5 PP3M scaffolds 1 and 2, previous Pro6- to Pro9-based scaffolds 3–6 (ref. 34) (Fig. 1) were also included in the structural investigation in this work. Pro9-PP3M 6 is particularly interesting as the ligand pattern created on this scaffold has been demonstrated to selectivity bind protein oligomers.17 To aid further investigation on the Pro9-based PP3M structure, we decided to install functional moieties on the scaffold. The Pro9 peptide was synthesized on the same resins, but the N-terminal proline residue was replaced by an Alloc-protected trans-4-aminoproline (19, Scheme 1). The alkyne modification on the C-terminus (20) allowed solid-phase peptide ligation and the following deprotection/coupling to be repeated to give linear tri-helix intermediate 22. The cyclization by the CuAAC reaction went smoothly to give scaffold 24 with three Alloc-protected amino groups located at (1,1′,1′′) positions. These Alloc groups were then deprotected to give the free amine for coupling to pyrroline nitroxide-based ESR spin label 25 resulting in a triply labeled nanoscaffold 26 for measurement of the distance between labels. In addition, as the previous attempt for AFM/STM analysis of unmodified PP3M has failed, the alkylation of the PP3M scaffold was also performed through Alloc group deprotection of 24 followed by the Cu(II)-catalyzed diazo-transfer reaction27 to have three azido groups installed onto the scaffold. Then the CuAAC conjugation with N-propargytridecanamide gave the desired trialkylated scaffold 27, which was subjected to further structural analysis.
3.2 Helical feature revealed by CD analysis
CD spectroscopy is an essential tool for investigating the polyproline peptide conformations, which may also reflect the ring strains in the corresponding cyclic scaffolds. The CD spectra measured for the new PP3M members 1 and 2 in aqueous solution (Fig. 2a), showed typical PPII CD spectra with a strong valley at ca. 205 nm and weak positive peak at around 228 nm.36,37 The aqueous CD spectra indicated Pro4, Pro5 and Pro9 were still in the PPII conformation, which allowed us to construct structural models based on PPII helices.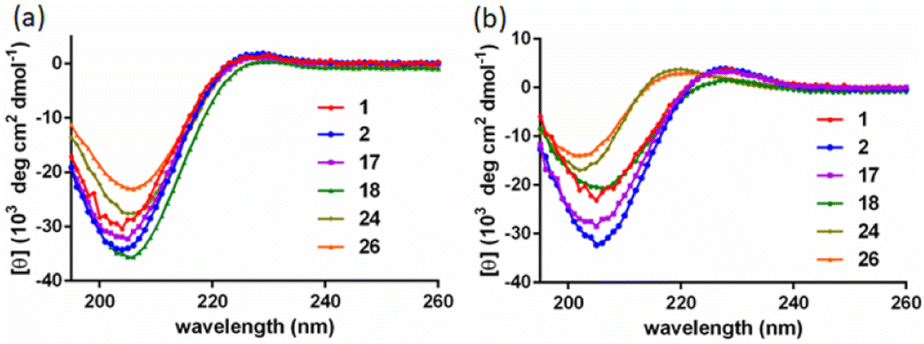 | ||
| Fig. 2 The CD spectra of linear and cyclized scaffolds in (a) water and (b) 1-propanol. All samples were measured at 100 μM. CD spectra of 3–6 were reported in ref. 17 and 34. | ||
Previous reports have suggested that polyproline peptides might favor polyproline helix I (PPI) in an environment of a less-polar solvent such as 1-propanol, leading to an opposite helix handedness and a different pitch and number of residues per turn compared to those in PPII.38 The preference between PPI and PPII conformations involves many factors including the charges at the peptide termini,39 length of the polyproline peptide,40 modification of the peptides41–44 and solvent.38,45,46 In our previous investigation, we observed that the strain which arose from cyclization of the polyproline tri-helix peptide to the PP3M scaffold promoted the PPI conformation for some sizes of the scaffolds in 1-propanol.17,34 Herein, as shown in Fig. 2b, we found that the CD spectra measured for scaffolds 1 and 2, as well as the linear tri-helix counterparts 17 and 18, in 1-propanol had the characteristics of the PPII helix (Fig. 2b). These results conform to the previous observation that PP3M scaffolds with a helix length below that of Pro8 exhibit PPII conformation.34 This weak tendency for the PPI conformation in 1-propanol for shorter polyproline peptides has also been reported.40 Therefore, this similarity in CD spectra for the linear and cyclized peptides due to helix length has prevented us from revealing the cyclic strain through CD spectroscopy. In addition to the smaller scaffolds, the CD analysis of larger Pro9 PP3M derivatives 24 and 26 showed the PPII conformation in water and slight PPI propensity in 1-propanol as reflected by the λmax and ratio of the positive and negative peak intensity (Rpn)47 (Table 1). The lipid-derived scaffold 27 has a lower solubility and is not compared for the mean residue ellipticity as shown in Fig. 2. However, it also has a PPII conformation in aqueous solution and exhibits more PPI propensity in 1-propanol (ESI Fig. S12†).
3.3 Combined analysis of SWAXS and molecular modeling
The SWAXS data for scaffolds 1–6 in aqueous solution are presented in Fig. 3. As the CD spectra have shown that the polyproline peptides are in the PPII conformation for these scaffolds under aqueous conditions (Fig. 2a and ref. 34), it is possible to optimize PP3M molecular models to fit the measured SWAXS data. The models can be refined against SWAXS data with the two structural parameters: the common distance d (from the center of each helix to the center of the horizontal plane defined by the helix centers, Fig. 4a) and the common inclined angle θ of each helix (with respect to the horizontal plane, Fig. 4a). A third parameter, the common rotation angle ϕ (of each polyproline helix along its axis), was found to be insensitive to the SWAXS data fitting and thus was neglected; however, this parameter can be better determined by ESR, as shown below. | ||
| Fig. 3 The SWAXS data of nanoscaffolds 1–6 in water. The predicted SWAXS curves from structural models for data fitting are indicated in red. | ||
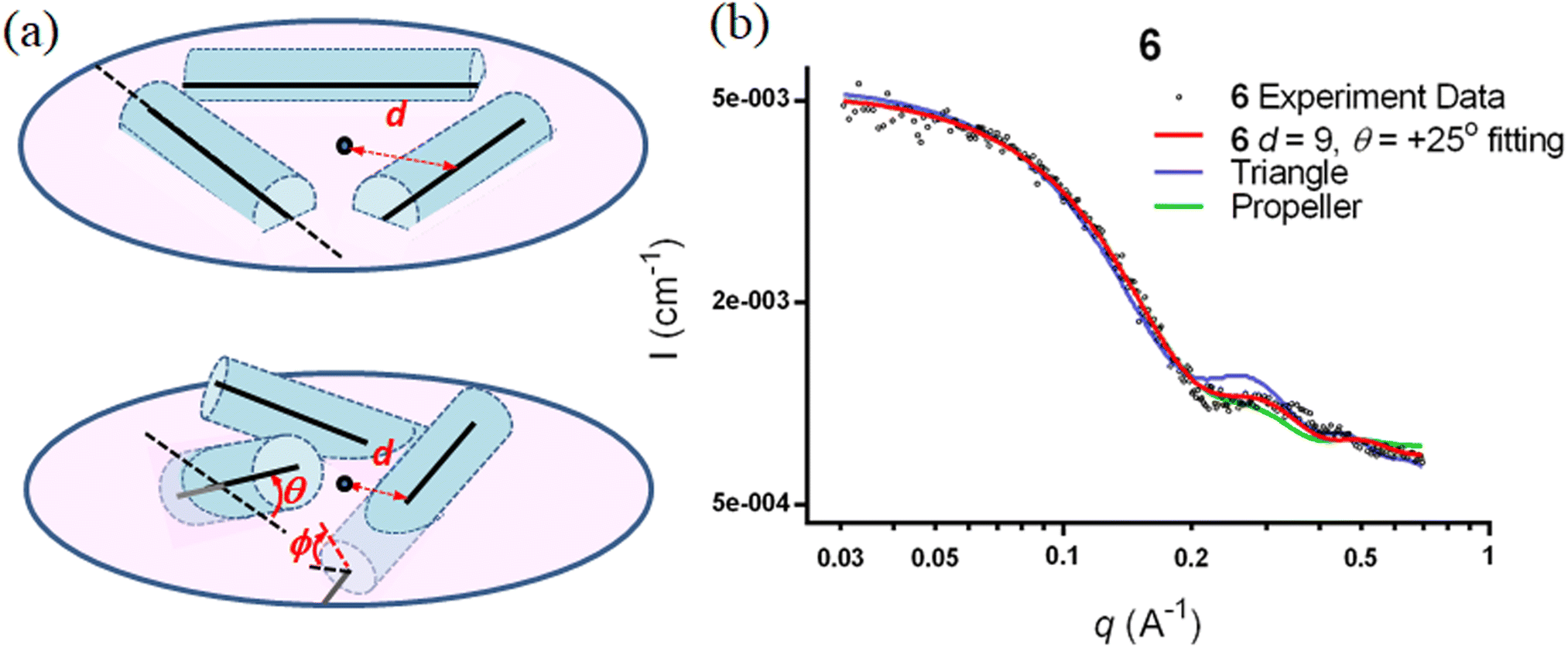 | ||
| Fig. 4 Optimization of the nanoscaffold structure with a systematic arrangement of polyproline helices to fit the SWAXS curve. (a) Parameters for helix arrangement: d is distance from the center of each helix axis to the center of the plane formed by the helices in both flat (top) and tilted (bottom) arrangements. θ is the incline angle for the helix against the plane formed by the center of the helices. ϕ is the helix rotation angle along the axis. The curved arrows for θ and ϕ indicate the directions of positive helix tilt and rotation. (b) The predicted SWAXS curves for extreme triangle (blue curve) and propeller structural models (green curve), as well as the refined structural model (red curve) for Pro9 PP3M scaffold 6, are plotted with the experimental data (black dots). | ||
Previously, we exploited the Pro9-based PP3M scaffold 6 to control ligand patterns for selective protein binding. Herein, we use the SWAXS data of scaffold 6 to illustrate the model refining process of three structural parameters for a more accurately defined structural model. First, we excluded d values smaller than 8 Å to avoid collisions between helices and increased d by 1 Å per step until it reached 10 Å, which is the maximum scaffold size that can be held by the connectors (11). Then, among the varied macrocycle radii designated by the d values, we further gradually changed the θ in 5° steps from −25° to +25°, which are the limits for the connectors to remain intact. These systematic adjustments of the helices resulted in the possible conformations that could fit all the SWAXS profiles (Fig. 3) with the radius of gyration (Rg) shown in Table S1 (ESI).† To evaluate the quality of fit to the experimental data, the fitting χ2 values (Table S1, ESI†) obtained from deviations of the model curves from the experimental data (Fig. 4b) were used to represent the goodness of fit. To better present the χ2 changes with the fitting parameters of d and θ, 2D χ2 maps for scaffolds 1–6 are shown in Fig. 5, and the best fitting parameters and Rg are listed in Table 2.
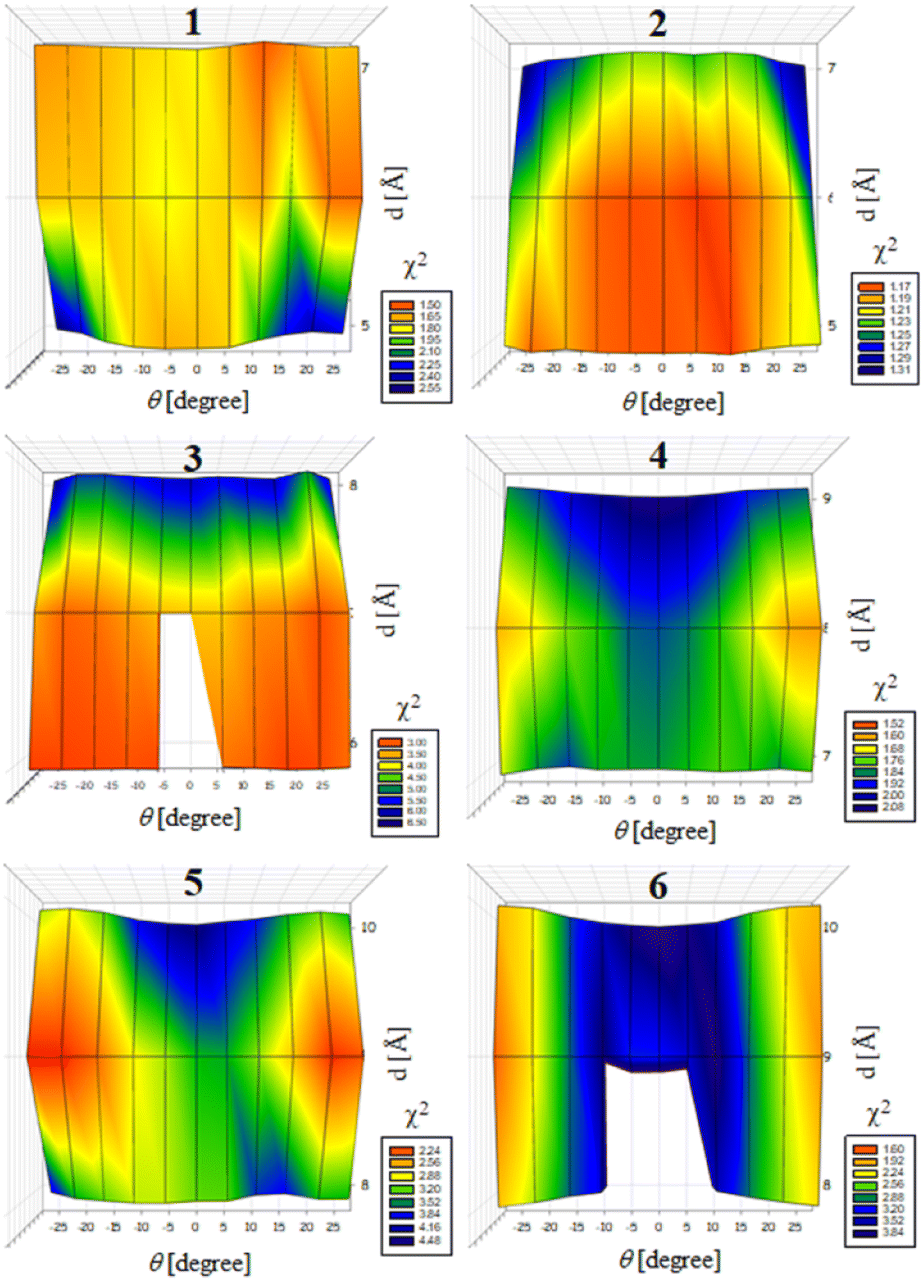 | ||
| Fig. 5 2D maps of χ2 as a function of the two parameters d and θ for evaluating the likeliness of the scaffold models that can fit well the SWAXS data of scaffolds 1–6. The blank regions indicate inappropriate parameters that cannot maintain an intact structure. | ||
| Nanoscaffold | 1 | 2 | 3 | 4 | 5 | 6 | |||
|---|---|---|---|---|---|---|---|---|---|
| d [Å] | Insensitive | Insensitive | 6 | 8 | 9 | 9 | |||
| θ [°] | Insensitive | Insensitive | Insensitive | +25 | −25 | +20 | −25 | +25 | −25 |
| R g [Å] | — | — | — | 12.4 ± 0.1 | 12.3 ± 0.1 | 13.6 ± 0.1 | 13.5 ± 0.1 | 14.4 ± 0.1 | 14.0 ± 0.1 |
As shown in Fig. 5 (also in Table S1 in the ESI†), the structural models of 6 with −25° or +25° incline angles have smaller χ2 values compared to those with θ close to 0° (also the triangle model curve in Fig. 4), suggesting that these tilted helix arrangements are favored over the flat-positioned helices. In addition, the data also indicate that d = 9 Å is favorable over the other sets to provide a suitable diameter for the model. The overall best fitting models for scaffold 6 are therefore identified as having d = 9 Å, with equally possible incline angles of θ = −25° or +25°. Similar processes were also performed for PP3M scaffolds 1–5 consisting of Pro4–Pro8 helices (Fig. 5 and Tables S2–S6 in ESI†). Although scaffolds 4 and 5 have very similar trends in favoring a tilted helix arrangement resembling that in scaffold 6, it is relatively difficult to determine the incline angle unambiguously for the smaller nanoscaffolds 1–3. The insignificant changes in χ2 values related to the change in the incline angles may be attributed to their relatively smaller size, as compared with scaffolds 4–6; consequently, similar changes in the incline angle θ result in smaller structural differences in space that cannot be differentiated sensitively enough with the SWAXS data of a similar q-range.
We also compared these structures to those in a previous investigation, in which the PP3M scaffolds had the drift time measured with ion mobility spectrometry (IMS) to calculate the molecular collision cross section (CCS).34 In the previous work, we built two extreme models having either the largest d with a flat helix arrangement (θ = 0°, the triangular model) or tilted helices with the smallest d (the propeller model). Although IMS analysis favored the propeller model over the triangular model, it is unlikely that further structural optimization could be provided based on only the measured drift time. In contrast, the SWAXS data covering the q-range up to 0.7 Å−1 could be used to validate the model structure parameterized by θ and d with the designed step resolutions. To further check the quality of the previous extreme models, we also plotted the predicted SWAXS curves of the triangle (blue) and propeller (green) models in Fig. 4b. The results indicate that the models with the refined parameters (red curve) indeed matched the experimental data better than the two previous models did. An additional advantage with the SWAXS analysis is that the scaffolds were measured in aqueous solution, faithfully reflecting the structures in potential application environments, as compared with those under the dry conditions of the IMS measurement. The SWAXS data-refined models of scaffolds 4–6 are shown in Fig. S17 in the ESI.†
3.4 ESR characterization
The above work provided 2 possible refined helix arrangements having positive or negative incline angles for PP3M scaffold 6. To determine which is a likely structural configuration in our study and determine the helix rotation angle ϕ of the scaffold (Fig. 4a), we designed spin labels on the side chain of specific proline residues to monitor the helix rotation angle by using the probe positions and performed DEER measurements. We chose pyrroline-carboxylic acid-based nitroxide 25 (Scheme 1) as the spin label for its convenient amide conjugation to our scaffold system. Therefore, we prepared an Alloc protected amine group at C-4 of proline and installed this residue at the N-termini of Pro9 helices in PP3M scaffold 24 (Scheme 1). The deprotection17 and spin label conjugation successfully gave the triply labelled PP3M scaffold 26 as described in the previous section.The DEER measurement for 26 (Fig. 6a) indicated three symmetrically arranged spin probes distributed 30 Å apart. This information led us to improve the PP3M structural model of 6 for the possible helix rotation angles by calculating the spin probe distance. We found that the possible helix rotation angles ϕ were limited by the connector between two polyproline helices ranging from −20° to +40° for both the favored arrangements of 6 with incline angles θ = +25° and −25°. By changing ϕ at intervals of 10°, we calculated the distance between spin labels in 26 (Table 3). Interestingly, the distances varied differently for the two models: when θ = +25° (Fig. 6b), the distance significantly varied with ϕ, whereas when θ = −25° (Fig. 6c), the distance responded with less ϕ change. Combining the experimental measurement of 30 Å and the calculated distances, the results favor the θ = +25° structural model and suggest a helix rotation angle ϕ of around +30° (Fig. 6c). This structural model is designated as the resulting structural model of scaffold 6.
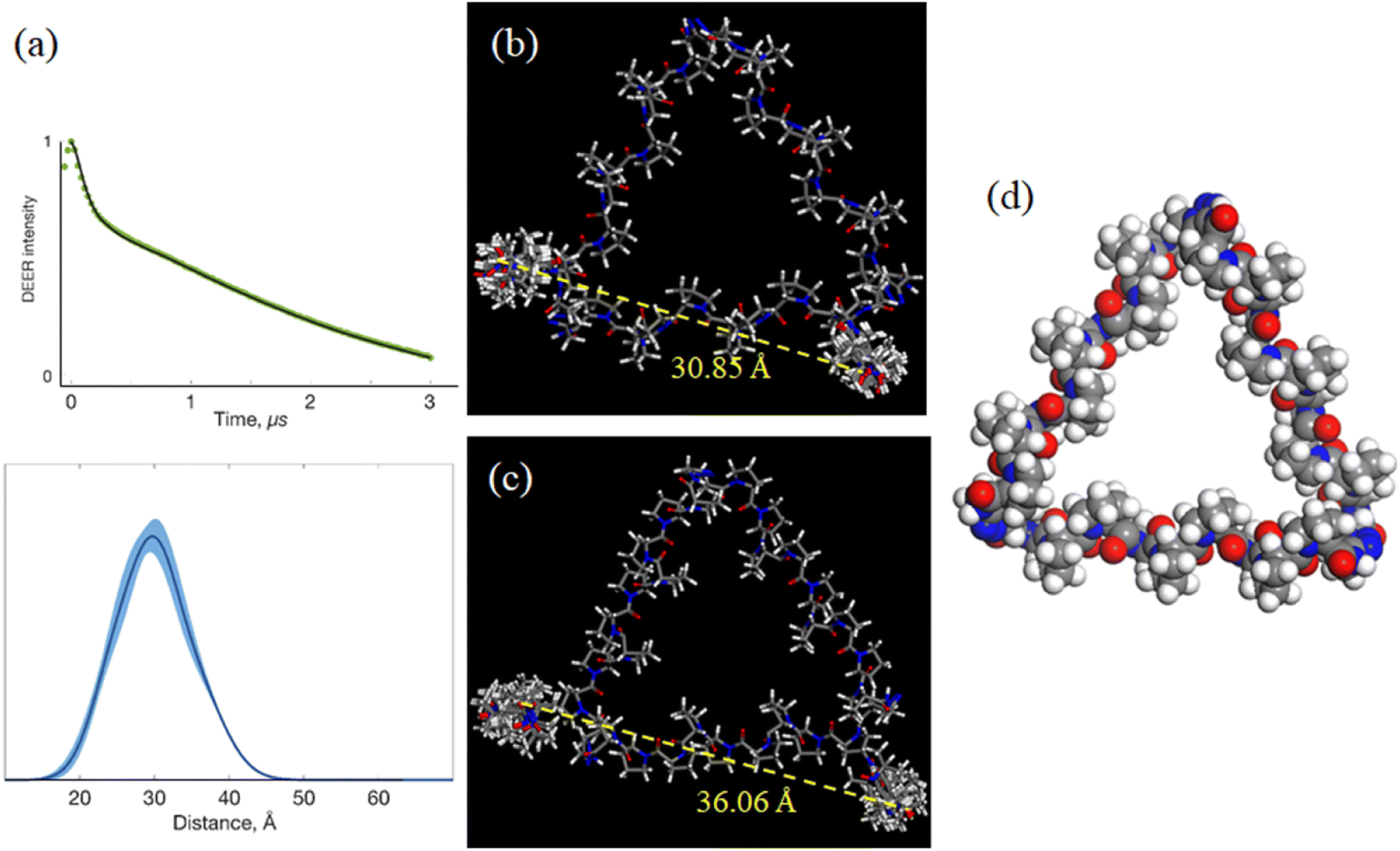 | ||
| Fig. 6 Double electron–electron resonance (DEER) analysis and models for scaffold 6. (a) Top: Raw experimental DEER trace (green dots) and the model fits (black line) to the data. Bottom: Interspin distance distribution obtained from the model fits. Shading around the line indicates uncertainty bounds (2σ confidence bands, where σ represents a standard deviation). (b) Structure and calculated probe distance for the best matched conformation (d = 9 Å, θ = +25°, and ϕ = +30°). (c) Structure and calculated probe distance for another conformation with an opposite helix stacking (d = 9 Å, θ = −25°, and ϕ = +10°). (d) Top view for the resulting structural model of Pro9 PP3M scaffold 6. | ||
| θ = +25° | θ = −25° | ||
|---|---|---|---|
| Φ | Probe–probe distance [Å] | Φ | Probe–probe distance [Å] |
| −20° | 39.41 ± 0.48 | −20° | 39.91 ± 0.71 |
| −10° | 38.21 ± 0.35 | −10° | 39.64 ± 0.93 |
| 0° | 36.63 ± 0.40 | 0° | 38.55 ± 2.32 |
| +10° | 34.94 ± 0.54 | +10° | 36.06 ± 1.97 |
| +20° | 33.20 ± 0.56 | +20° | 39.16 ± 0.91 |
| +30° | 30.85 ± 0.46 | +30° | 38.58 ± 0.34 |
| +40° | 28.86 ± 0.69 | +40° | 37.19 ± 0.49 |
3.5 STM characterization
In addition to SAXS and DEER analysis, we employed complementary approaches to investigate the structures of the PP3M scaffolds. Scanning tunneling microscopy (STM) is powerful in unveiling structural information, particularly for those lacking crystallinity.48–53 As aforementioned, our first STM attempts for underivatized scaffold 6 on graphite (highly oriented pyrolytic graphite, HOPG) found the feature of HOPG only, indicative of weak adsorbate–substrate interactions. To acquire STM images, the rule of thumb is to maintain spatial steadiness of the target compounds. Straight-chain alkyls that increase interactions with the substrate and between the nearest neighbors are typically derivatized to molecules of interest. However, unlike those with planar aromatic moieties or alkanes, which bear significant adsorbate-substrate contact areas and strong intermolecular interactions, our scaffolds are organized such that only a few points are in contact with the planar graphite substrate (e.g., the lower panel of Fig. 7d). To strengthen the contact, we prepared a 2-dimensional porous monolayer on HOPG so that the non-coplanar scaffolds could become lodged against and trapped by the pores. The porous layer can be clearly discerned in Fig. 7a and appears as the hexagonal underlayer. It was formed by trimesic acid (TMA), in which the meta positions have a total of 3 carboxylic acid functional groups. The center-to-center spacing between neighboring pores is about 1.7 nm; each pore is composed of 6 TMA molecules held together by H-bonded carboxylic acid dimers. We previously developed this approach to resolve the details of isolated fullerenes33 and an H-bonded azaindole trimer54 under ambient conditions. For the TMA porous layer on HOPG, high resolution STM images at the sub-molecular level are available elsewhere.33,55–59 | ||
| Fig. 7 STM images (a–c) and models on HOPG (d and e) of scaffold 27. Panel a exhibits a 50 nm × 50 nm image of scaffold 27 features with 3 lobes. Panels b and c show top views of the remnants after successively slicing away the upper portion to reveal possible segments with direct contact with the substrate. Representative models of top- and side-views are illustrated in panels d and e. For the images of scaffold 27, the portions in contact with the substrate presumably have a higher tunneling probability than those with void gaps.52,53,60 Panels b and c were magnified from areas denoted as b and c in panel a. The samples were prepared by drop-casting scaffold 27 (in acetonitrile) on HOPG pre-modified with a 2-dimensional porous monolayer of TMA. After acetonitrile was evaporated, a small drop of octanoic acid was placed on the sample where the imaging was performed. Imaging conditions: itunneling, 30 pA; Vbias, −0.9 V (tip-grounded). | ||
Fig. 7a shows a 50 nm × 50 nm image, in which the hexagonally arranged pattern in the background is the TMA layer. There were large and small adsorbed species. The features, such as those indicated in boxes, were consistent with the expected size of scaffold 27. Although the relative intensity and size varied significantly, they appeared to comprise 3 lobes. The center-to-center distances between lobes are about 3 nm, consistent with the results of DEER and the structural model proposed (vide infra).
In Fig. 7, the brighter regimes indicate higher tunneling probabilities. Fig. 7e illustrates that Scaffold 27 is not planar and, therefore, some regimes directly came into contact with the substrate while some segments did not. The efficiency of electron tunneling for the latter is inferior because it would involve substrate-to-scaffold and then scaffold-to-STM tip pathways. Two tunneling gaps resulted in lower tunneling currents and thus dimmer signals than those of the direct contact regimes (additional STM images and interpretations can be found in the ESI†).52,53,60 To help visualize the contact regimes of scaffold 27, STM images in Fig. 7b and c are overlaid with the resulting scaffold 6 model structure. Parts of the models are removed horizontally (also see Fig. 7d and e), manifesting the parts proximal to the substrate. The similarity between the images and the models (Fig. 7b and c) supports the success of synthesizing cyclized scaffold 27. The C3-symmetry of the Pro9-based PP3M scaffold was also confirmed by 1H-NMR (Fig. S15 in the ESI†).
3.6 Structural model for Helix pomatia agglutinin binding
With the SWAXS and DEER data contributing to the optimization of the PP3M structure, which was further supported by the STM observations, we further checked whether the resulting scaffold model structure could help to interpret the multivalent ligand binding to protein oligomers. In the previous investigation, we adjusted the ligand patterns on a Pro9-based PP3M scaffold to selectively bind to hexameric (as a symmetric trimer on either face) lectin Helix pomatia agglutinin (HPA).17 One active glycocluster 28 (Fig. 8a) had three α-GalNAc ligands symmetrically conjugated to the N-terminal residue on each polyproline helix of the scaffold to provide strong binding by matching the 3-fold symmetric binding sites of HPA.17 As the glycocluster used the same Pro9 tri-helix scaffold 6 to present α-GalNAc ligands, we tested whether the resulting scaffold 6 model structure would allow all three α-GalNAc ligands to reach the binding sites. We superimposed the structural model of scaffold 6 with the α-GalNAc-complexed structure of HPA (PDB:2CCV),32 connected the α-GalNAc to the scaffold 6 structure by using the linkers in glycocluster 28,17 and adjusted the relative position of the PP3M scaffold. The outcome indicated that the resulting scaffold model structure allowed positioning of the ligands to protein binding sites without structural clash or distortion (Fig. 8b–d). From the complexed structure of HPA, the oxygen atoms of the anomeric OH on three GalNAc ligands are 26 Å apart, whereas a distance of 24 Å was estimated for the gaps between nitrogen atoms attached to the C-4 of the (1, 1′, 1′′) proline residues on the resulting scaffold 6 model structure. This scaffold dimension favored the linkage to the GalNAc, which has its α anomeric linkage pointing outward from the HPA complex (Fig. 8c). These findings further support the resulting structural model of scaffold 6 agreeing well with the reported HPA-GalNAc complex structure.32 Based on this approach, other ligand patterns from PP3M scaffolds may be designed to target protein oligomers with known structures.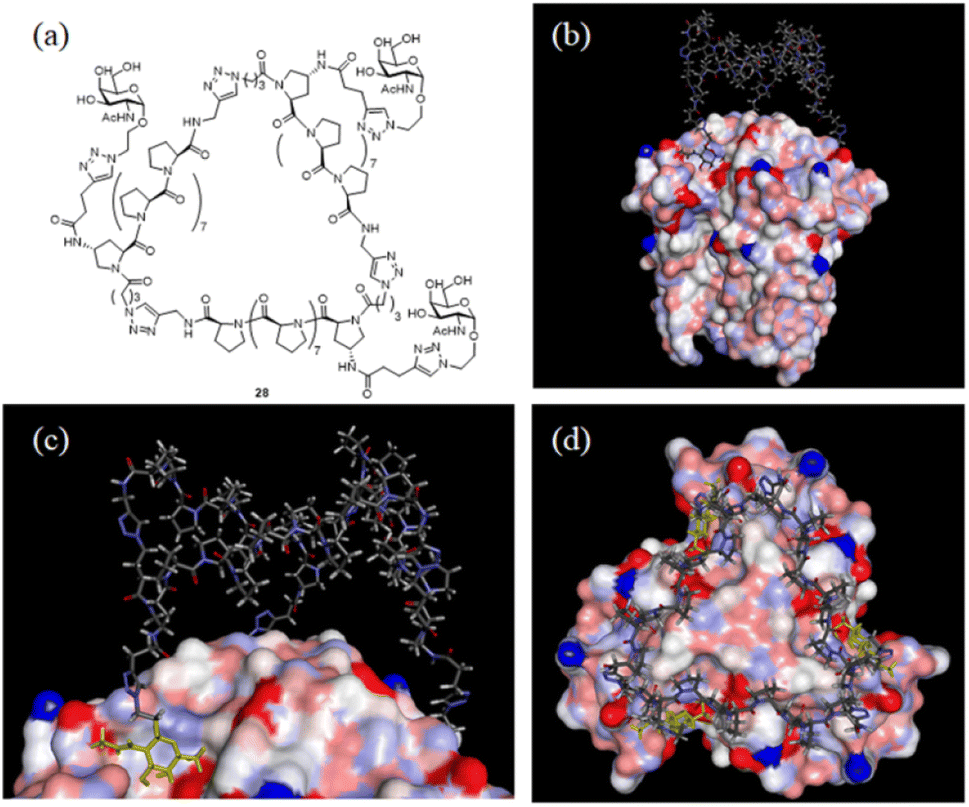 | ||
| Fig. 8 Superimposed structures of GalNAc glycocluster 28 to the HPA-GalNAc complex. (a) The chemical structure of glycocluster 28 (ref. 17). (b–d) Superimposed structures using the resulting scaffold 6 model structure as the core. 3 GalNAcs of the HPA complex structure are shown in yellow. | ||
4. Conclusions
It is very challenging to fabricate nano-objects with good size and shape control. It is even more difficult to control the conjugation locations of small molecules to such nano-objects. Without the capability for these precise manipulations, many applications of nanotechnology, such as controlling ligand patterns for nanomedicine, may be limited. Organic synthesis provides delicate tools to precisely build a molecule. When this advantage is applied to biopolymers with controllable sequences, it is possible to extend the precision of organic synthesis to the nanoscale, as long as the structure of the biopolymer can be predicted. In this study, we have made progress towards this end. By merging the capabilities of CD spectroscopy, SWAXS, DEER, and STM, we have devised a robust model for Pro9 helix-based macrocyclic nanostructures. The established strategy would further allow the prediction of structural models for other PP3M nanoscaffold analogs. We anticipate that this breakthrough will open up many opportunities in nanomedicine, including selective interaction with proteins, modulation of the functions of cell surface receptors, and construction of more complex polyproline-based nanostructures. All these exciting prospects are now within reach, signaling a promising future for molecular controls with nanoscale precision.Author contributions
SKW and CLT designed the experiments (conceptualization). CLT, YCH, QDL, TYC and PHC conducted chemical synthesis (investigation). JWC, CLT, OS and USJ performed SWAXS measurement and data analysis (investigation). YJL and YWC performed ESR experiments and data analysis (investigation). KYC, CHL and ChC performed the STM experiment and data analysis (investigation). CLT performed structural model calculation (investigation). MS and VK enhanced the sensitivity of ESR spectroscopy (resources). SKW, USJ, ChC, YWC and CLT wrote the manuscript (writing).Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the financial support from the National Science and Technology Council, Taiwan (111-2113-M-007-023, 112-2113-M-007-027, 111-2112-M-213-014-MY3, 112-2811-M-002-002, 112-2123-M-002-004 and 112-2923-M-007-003-MY3). We thank the National Center for High-performance Computing (NCHC) for providing computational and storage resources. The research reported in this publication was supported by funding from the European Union HORIZON-MSCA-2021-PF-01 Marie Skłodowska-Curie Fellowship (Project ID: 101064200; SPECTR, https://epr.ff.vu.lt/spectr) (M. S.), and the Research Council of Lithuania (LMTLT), agreement no. S-LLT-23-1 (V. K.).Notes and references
- M. Mammen, S. K. Choi and G. M. Whitesides, Polyvalent Interactions in Biological Systems: Implications for Design and Use of Multivalent Ligands and Inhibitors, Angew. Chem., Int. Ed., 1998, 37, 2754–2794 CrossRef PubMed.
- L. L. Kiessling, J. E. Gestwicki and L. E. Strong, Synthetic Multivalent Ligands as Probes of Signal Transduction, Angew. Chem., Int. Ed., 2006, 45, 2348–2368 CrossRef CAS PubMed.
- C. Fasting, C. A. Schalley, M. Weber, O. Seitz, S. Hecht, B. Koksch, J. Dernedde, C. Graf, E. W. Knapp and R. Haag, Multivalency as a Chemical Organization and Action Principle, Angew. Chem., Int. Ed., 2012, 51, 10472–10498 CrossRef CAS PubMed.
- D. Morzy and M. Bastings, Significance of Receptor Mobility in Multivalent Binding on Lipid Membranes, Angew. Chem., Int. Ed., 2022, 61, e202114167 CrossRef CAS PubMed.
- Á. Martínez, C. Ortiz Mellet and J. M. García Fernández, Cyclodextrin-based Multivalent Glycodisplays: Covalent and Supramolecular Conjugates to Assess Carbohydrate-Protein Interactions, Chem. Soc. Rev., 2013, 42, 4746–4773 RSC.
- F. Sansone and A. Casnati, Multivalent Glycocalixarenes for Recognition of Biological Macromolecules: Glycocalyx Mimics Capable of Multitasking, Chem. Soc. Rev., 2013, 42, 4623–4639 RSC.
- D. Goyard, A. M. Ortiz, D. Boturyn and O. Renaudet, Multivalent Glycocyclopeptides: Conjugation Methods and Biological Applications, Chem. Soc. Rev., 2022, 51, 8756–8783 RSC.
- B. Schuler, E. A. Lipman, P. J. Steinbach, M. Kumke and W. A. Eaton, Polyproline and the "Spectroscopic Ruler" Revisited with Single-Molecule Fluorescence, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 2754–2759 CrossRef CAS PubMed.
- P. M. Cowan and S. McGavin, Structure of Poly-L-proline, Nature, 1955, 176, 501–503 CrossRef CAS.
- P. Wilhelm, B. Lewandowski, N. Trapp and H. Wennemers, A Crystal Structure of an Oligoproline PPII-Helix, at Last, J. Am. Chem. Soc., 2014, 136, 15829–15832 CrossRef CAS PubMed.
- C. Kroll, R. Mansi, F. Braun, S. Dobitz, H. R. Maecke and H. Wennemers, Hybrid Bombesin Analogues: Combining an Agonist and an Antagonist in Defined Distances for Optimized Tumor Targeting, J. Am. Chem. Soc., 2013, 135, 16793–16796 CrossRef CAS PubMed.
- J. Kuriakose, V. Hernandez-Gordillo, M. Nepal, A. Brezden, V. Pozzi, M. N. Seleem and J. Chmielewski, Targeting Intracellular Pathogenic Bacteria with Unnatural Proline-Rich Peptides: Coupling Antibacterial Activity with Macrophage Penetration, Angew. Chem., Int. Ed., 2013, 52, 9664–9667 CrossRef CAS PubMed.
- L. Corcilius, G. Santhakumar, R. S. Stone, C. J. Capicciotti, S. Joseph, J. M. Matthews, R. N. Ben and R. J. Payne, Synthesis of Peptides and Glycopeptides with Polyproline II Helical Topology as Potential Antifreeze Molecules, Bioorg. Med. Chem., 2013, 21, 3569–3581 CrossRef CAS PubMed.
- P. Liu, L. Chen, J. K. C. Toh, Y. L. Ang, J. E. Jee, J. Lim, S. S. Lee and S. G. Lee, Tailored Chondroitin Sulfate Glycomimetics via a Tunable Multivalent Scaffold for Potentiating NGF/TrkA-Induced Neurogenesis, Chem. Sci., 2015, 6, 450–456 RSC.
- T. H. Lin, C. H. Lin, Y. J. Liu, C. Y. Huang, Y. C. Lin and S. K. Wang, Controlling Ligand Spacing on Surface: Polyproline-Based Fluorous Microarray as a Tool in Spatial Specificity Analysis and Inhibitor Development for Carbohydrate-Protein Interactions, ACS Appl. Mater. Interfaces, 2017, 9, 41691–41699 CrossRef CAS PubMed.
- Y. A. Nagel, P. S. Raschle and H. Wennemers, Effect of Preorganized Charge-Display on the Cell-Penetrating Properties of Cationic Peptides, Angew. Chem., Int. Ed., 2017, 56, 122–126 CrossRef CAS PubMed.
- C. H. Lin, H. C. Wen, C. C. Chiang, J. S. Huang, Y. Chen and S. K. Wang, Polyproline Tri-Helix Macrocycles as Nanosized Scaffolds to Control Ligand Patterns for Selective Protein Oligomer Interactions, Small, 2019, 15, e1900561 CrossRef PubMed.
- M. A. Schroer and D. I. Svergun, Recent Developments in Small-Angle X-ray Scattering and Hybrid Method Approaches for Biomacromolecular Solutions, Emerging Top. Life Sci., 2018, 2, 69–79 CrossRef CAS PubMed.
- Y. Q. Yeh, K. F. Liao, O. Shih, Y. J. Shiu, W. R. Wu, C. J. Su, P. C. Lin and U. S. Jeng, Probing the Acid-Induced Packing Structure Changes of the Molten Globule Domains of a Protein near Equilibrium Unfolding, J. Phys. Chem. Lett., 2017, 8, 470–477 CrossRef CAS PubMed.
- J. Trewhella, A. P. Duff, D. Durand, F. Gabel, J. M. Guss, W. A. Hendrickson, G. L. Hura, D. A. Jacques, N. M. Kirby, A. H. Kwan, J. Pérez, L. Pollack, T. M. Ryan, A. Sali, D. Schneidman-Duhovny, T. Schwede, D. I. Svergun, M. Sugiyama, J. A. Tainer, P. Vachette, J. Westbrook and A. E. Whitten, Publication Guidelines for Structural Modelling of Small-Angle Scattering Data from Biomolecules in Solution: an Update, Acta Crystallogr., Sect. D: Struct. Biol., 2017, 73(9), 710–728 CrossRef CAS PubMed.
- C. C. Li, T. Y. Kao, C. C. Cheng and Y. W. Chiang, Structure and Regulation of the BsYetJ Calcium Channel in Lipid Nanodiscs, Proc. Natl. Acad. Sci. U.S.A., 2020, 117, 30126–30134 CrossRef CAS PubMed.
- C. J. Tsai and Y. W. Chiang, Effects of Anisotropic Nanoconfinement on Rotational Dynamics of Biomolecules: an Electron Spin Resonance Study, Phys. Chem. C, 2012, 116, 19798–19806 CrossRef CAS.
- R. F. Tsai, N. C. Lin, T. Y. Kao, Y. H. Kuo, F. C. Lo, W. F. Liaw and Y. W. Chiang, Nitrosylation of the Diiron Core Mediated by the N Domain of YtfE, J. Phys. Chem. Lett., 2020, 11, 8538–8542 CrossRef CAS PubMed.
- T. Bizien, D. Durand, P. Roblina, A. Thureau, P. Vachette and J. Pérez, A Brief Survey of State-of-the-Art BioSAXS, Protein Pept. Lett., 2016, 23, 217–231 CrossRef CAS PubMed.
- D. G. Liu, C. H. Chang, L. C. Chiang, M. H. Lee, C. F. Chang, C. Y. Lin, C. C. Liang, T. H. Lee, S. W. Lin, C. Y. Liu, C. S. Hwang, J. C. Huang, C. K. Kuan, H. S. Wang, Y. C. Liu, F. H. Tseng, J. Y. Chuang, W. R. Liao, H. C. Li, C. J. Su, K. F. Liao, Y. Q. Yeh, O. Shih, W. R. Wu, C. A. Wang and U. Jeng, Optical Design and Performance of the Biological Small-Angle X-ray Scattering Beamline at the Taiwan Photon Source, J. Synchrotron Radiat., 2021, 28, 1954–1965 CrossRef CAS PubMed.
- O. Shih, K. F. Liao, Y. Q. Yeh, C. J. Su, C. A. Wang, J. W. Chang, W. R. Wu, C. C. Liang, C. Y. Lin, T. H. Lee, C. H. Chang, L. C. Chiang, C. F. Chang, D. G. Liu, M. H. Lee, C. Y. Liu, T. W. Hsu, B. Mansel, M. C. Ho, C. Y. Shu, F. Lee, E. Yen, T. C. Lind and U. Jeng, Performance of the New Biological Small- and Wide Angle X-ray Scattering Beamline 13A at the Taiwan Photon Source, J. Appl. Crystallogr., 2022, 55, 340–352 CrossRef CAS PubMed.
- P. T. Nyffeler, C. H. Liang, K. M. Koeller and C. H. Wong, The Chemistry of Amine-Azide Interconversion: Catalytic Diazotransfer and Regioselective Azide Reduction, J. Am. Chem. Soc., 2002, 124, 10773–10778 CrossRef CAS PubMed.
- K. Manalastas-Cantos, P. V. Konarev, N. R. Hajizadeh, A. G. Kikhney, M. V. Petoukhov, D. S. Molodenskiy, A. Panjkovich, H. D. T. Mertens, A. Gruzinov, C. Borges, C. M. Jeffries, D. I. Svergun and D. Franke, ATSAS 3.0: Expanded Functionality and New Tools for Small-Angle Scattering Data Analysis, J. Appl. Crystallogr., 2021, 54, 343–355 CrossRef CAS PubMed.
- G. Jeschke, DEER Distance Measurements on Proteins, Annu. Rev. Phys. Chem., 2012, 63, 419–446 CrossRef CAS PubMed.
- M. Šimėnas, J. O’Sullivan, C. W. Zollitsch, O. Kennedy, M. Seif-Eddine, I. Ritsch, M. Hülsmann, M. Qi, A. Godt, M. M. Roessler, G. Jeschke and J. J. L. Morton, A Sensitivity Leap for X-band EPR Using a Probehead with a Cryogenic Preamplifier, J. Magn. Reson., 2021, 322, 106876 CrossRef PubMed.
- E. J. Hustedt, F. Marinelli, R. A. Stein, J. D. Faraldo-Gómez and H. S. Mchaourab, Confidence Analysis of DEER Data and Its Structural Interpretation with Ensemble-Biased Metadynamics, Biophys. J., 2018, 115, 1200–1216 CrossRef CAS PubMed.
- J. F. Sanchez, J. Lescar, V. Chazalet, A. Audfray, J. Gagnon, R. Alvarez, C. Breton, A. Imberty and E. P. Mitchell, Biochemical and Structural Analysis of Helix pomatia Agglutinin. A Hexameric Lectin with a Novel Fold, J. Biol. Chem., 2006, 281, 20171–20180 CrossRef CAS PubMed.
- K. Y. Cheng, S. L. Lee, T. Y. Kuo, C. H. Lin, Y. C. Chen, T. H. Kuo, C. C. Hsu and C. H. Chen, Template-Assisted Proximity for Oligomerization of Fullerenes, Langmuir, 2018, 34, 5416–5421 CrossRef CAS PubMed.
- C. L. Tsai, S. Y. Wu, H. K. Hsu, S. B. Huang, C. H. Lin, Y. T. Chan and S. K. Wang, Preparation and Conformational Analysis of Polyproline Tri-Helix Macrocycle Nanoscaffolds of Varied Sizes, Nanoscale, 2021, 13, 4592–4601 RSC.
- V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, A Stepwise Huisgen Cycloaddition Process: Copper(I)-Catalyzed Regioselective "Ligation" of Azides and Terminal Alkynes, Angew. Chem., Int. Ed., 2002, 41, 2596–2599 CrossRef CAS PubMed.
- A. L. Rucker and T. P. Creamer, Polyproline II Helical Structure in Protein Unfolded States: Lysine Peptides Revisited, Protein Sci., 2002, 11, 980–985 CrossRef CAS PubMed.
- E. W. Ronish and S. Krimm, The Calculated Circular Dichroism of Polyproline II in the Polarizability Approximation, Biopolymers, 1974, 13, 1635–1651 CrossRef CAS PubMed.
- W. Traub and U. Shmueli, Structure of Poly-L-proline I, Nature, 1963, 198, 1165–1166 CrossRef.
- M. Kuemin, S. Schweizer, C. Ochsenfeld and H. Wennemers, Effects of Terminal Functional Groups on the Stability of the Polyproline II Structure: a Combined Experimental and Theoretical Study, J. Am. Chem. Soc., 2009, 131, 15474–15482 CrossRef CAS PubMed.
- S. Kakinoki, Y. Hirano and M. Oka, On the Stability of Polyproline-I and II Structures of Proline Oligopeptides, Polym. Bull., 2005, 53, 109–115 CrossRef CAS.
- J. C. Horng and R. T. Raines, Stereoelectronic Effects on Polyproline Conformation, Protein Sci., 2006, 15, 74–83 CrossRef CAS PubMed.
- A. K. Pandey, D. Naduthambi, K. M. Thomas and N. J. Zondlo, Proline Editing: a General and Practical Approach to the Synthesis of Functionally and Structurally Diverse Peptides. Analysis of Steric versus Stereoelectronic Effects of 4-substituted Prolines on Conformation within Peptides, J. Am. Chem. Soc., 2013, 135, 4333–4363 CrossRef CAS PubMed.
- W. H. Tseng, M. C. Li, J. C. Horng and S. K. Wang, Strategy and Effects of Polyproline Peptide Stapling by Copper(I)-Catalyzed Alkyne-Azide Cycloaddition Reaction, ChemBioChem, 2019, 20, 153–158 CrossRef CAS PubMed.
- M. C. Li, Y. J. Liu, K. C. Hsu, T. H. Lin, C. W. Lin, J. C. Horng and S. K. Wang, Design and Synthesis of Fluorinated Peptides for Analysis of Fluorous Effects on the Interconversion of Polyproline Helices, Bioorg. Chem., 2022, 119, 105491 CrossRef CAS PubMed.
- F. Gornick, L. Mandelkern, A. F. Diorio and D. E. Roberts, Evidence for a Cooperative Intramolecular Transition in Poly-L-proline, J. Am. Chem. Soc., 1964, 86, 2549–2555 CrossRef CAS.
- J. Engel and G. Schwarz, Cooperative Conformational Transitions of Linear Biopolymers, Angew. Chem., Int. Ed., 1970, 9, 389–400 CrossRef CAS PubMed.
- Y. Feng, G. Melacini, J. P. Taulane and M. Goodman, Acetyl-Terminated and Template-Assembled Collagen-Based Polypeptides Composed of Gly-Pro-Hyp Sequences. 2. Synthesis and Conformational Analysis by Circular Dichroism, Ultraviolet Absorbance, and Optical Rotation, J. Am. Chem. Soc., 1996, 118, 10351–10358 CrossRef CAS.
- Y. Zheng, W. Luo, L. Yu, S. Chen, K. Mao, Q. Fang, Y. Yang, C. Wang, H. Zhu and B. Tu, Heterochirality-Mediated Cross-Strand Nested Hydrophobic Interaction Effects Manifested in Surface-Bound Peptide Assembly Structures, J. Phys. Chem. B, 2022, 126, 723–733 CrossRef CAS PubMed.
- Y. Guo, L. Jiang, A. P. Seitsonen, B. Zhang, J. Reichert, A. C. Papageorgiou and J. V. Barth, Interaction of Cyclosporin A Molecules with Alkali and Transition Metal Atoms on Cu(111), Chem. Commun., 2021, 57, 2923–2926 RSC.
- N. Naruse, H. Satooka, K. Todo, A. Nakanishi, H. Taguchi and Y. Mera, Oligomers Imaging of Amyloid-β1-42 by Scanning Tunneling Microscopy, Jpn. J. Appl. Phys., 2019, 58, SIIB30 CrossRef CAS.
- S. L. Lee, N. T. Lin, W. C. Liao, C. H. Chen, H. C. Yang and T. Y. Luh, Oligomeric Tectonics: Supramolecular Assembly of Double-Stranded Oligobisnorbornene through pi-pi Stacking, Chem.–Eur. J., 2009, 15, 11594–11600 CrossRef CAS PubMed.
- C. M. Chou, S. L. Lee, C. H. Chen, A. T. Biju, H. W. Wang, Y. L. Wu, G. F. Zhang, K. W. Yang, T. S. Lim, M. J. Huang, P. Y. Tsai, K. C. Lin, S. L. Huang, C. H. Chen and T. Y. Luh, Polymeric Ladderphanes, J. Am. Chem. Soc., 2009, 131, 12579–12585 CrossRef CAS PubMed.
- H. C. Yang, S. L. Lee, C. H. Chen, N. T. Lin, H. C. Yang, B. Y. Jin and T. Y. Luh, One-Handed Helical Double Stranded Polybisnorbornenes, Chem. Commun., 2008, 44, 6158–6160 RSC.
- T. H. Tu, Y. T. Chen, Y. A. Chen, Y. C. Wei, Y. H. Chen, C. L. Chen, J. Y. Shen, Y. H. Chen, S. Y. Ho, K. Y. Cheng, S. L. Lee, C. H. Chen and P. T. Chou, The Cyclic Hydrogen-Bonded 6-Azaindole Trimer and its Prominent Excited-State Triple-Proton-Transfer Reaction, Angew. Chem., Int. Ed., 2018, 57, 5020–5024 CrossRef CAS PubMed.
- X. Zeng, Y. Hu, R. Xie, S. B. Khan and S. L. Lee, Monolayer and Bilayer Formation of Molecular 2D Networks Assembled at the Liquid/Solid Interfaces by Solution-Based Drop-Cast Method, Molecules, 2021, 26, 7707 CrossRef CAS PubMed.
- J. Ubink, M. Enache and M. Stöhra, Bias-Induced Conformational Switching of Supramolecular Networks of Trimesic Acid at the Solid-Liquid Interface, J. Chem. Phys., 2018, 148, 174703 CrossRef CAS PubMed.
- D. C. Y. Nguyen, L. Smykalla, T. N. H. Nguyen, T. Rüffer and M. Hietschold, Deposition-Temperature- and Solvent-Dependent 2D Supramolecular Assemblies of Trimesic Acid at the Liquid–Graphite Interface Revealed by Scanning Tunneling Microscopy, J. Phys. Chem. C, 2016, 120, 11027–11036 CrossRef CAS.
- W. Li, J. Jin, X. Leng, Y. Lu, X. Liu and L. Wang, Modulation of Coordinate Bonds in Hydrogen-Bonded Trimesic Acid Molecular Networks on Highly Ordered Pyrolytic Graphite Surface, J. Phys. Chem. C, 2016, 120, 12605–12610 CrossRef CAS.
- S. J. H. Griessl, M. Lackinger, F. Jamitzky, T. Markert, M. Hietschold and W. M. Heckl, Room-Temperature Scanning Tunneling Microscopy Manipulation of Single C60 Molecules at the Liquid–Solid Interface: Playing Nanosoccer, J. Phys. Chem. B, 2004, 108, 11556–11560 CrossRef CAS.
- P. Samorí, A. Fechtenkötter, F. Jäckel, T. Böhme, K. Müllen and J. P. Rabe, Supramolecular Staircase via Self-Assembly of Disklike Molecules at the Solid-Liquid Interface, J. Am. Chem. Soc., 2001, 123, 11462–11467 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Additional characterization of nanoscaffolds and peptide intermediates; full SWAXS data and additional STM images are available. See DOI: https://doi.org/10.1039/d3na00945a |
| This journal is © The Royal Society of Chemistry 2024 |