
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMagnetic anisotropy engineering in onion-structured metal oxide nanoparticles combining dual exchange coupling and proximity effects†
Kevin
Sartori
 ab,
Raul
Lopez-Martin
c,
Fadi
Choueikani
b,
Alexandre
Gloter
d,
Jean-Marc
Grenèche
e,
Sylvie
Begin-Colin
a,
Dario
Taverna
f,
Jose A.
De Toro
c and
Benoit P.
Pichon
*ag
ab,
Raul
Lopez-Martin
c,
Fadi
Choueikani
b,
Alexandre
Gloter
d,
Jean-Marc
Grenèche
e,
Sylvie
Begin-Colin
a,
Dario
Taverna
f,
Jose A.
De Toro
c and
Benoit P.
Pichon
*ag
aUniversité de Strasbourg, CNRS, Institut de Physique et Chimie des Matériaux de Strasbourg, UMR 7504, F-67000 Strasbourg, France. E-mail: benoit.pichon@unistra.fr
bSynchrotron SOLEIL, L'Orme des Merisiers, Saint Aubin – BP48, 91192 Gif-sur-Yvette, France
cInstituto Regional de Investigación Científica Aplicada (IRICA), Departamento de Física Aplicada, Universidad de Castilla-La Mancha, 13071 Ciudad Real, Spain
dLaboratoire de Physique des Solides, CNRS, Université Paris-Saclay, 91400 Orsay, France
eInstitut des Molécules et Matériaux du Mans, IMMM, UMR CNRS-6283, Le Mans Université, Avenue Olivier Messiaen, 72085 Le Mans Cedex 9, France
fInstitut de Minéralogie, de Physique des Matériaux et de Cosmochimie, UMR 7590, CNRS, Sorbonne Université, 75005 Paris, France
gInstitut Universitaire de France, 1 Rue Descartes, 75231 Paris Cedex 05, France
First published on 25th March 2024
Abstract
A series of exchange-coupled magnetic nanoparticles combining several magnetic phases in an onion-type structure were synthesized by performing a three-step seed-mediated growth process. Iron and cobalt precursors were alternatively decomposed in high-boiling-temperature solvents (288–310 °C) to successively grow CoO and Fe3−δO4 shells (the latter in three stages) on the surface of Fe3−δO4 seeds. The structure and chemical composition of these nanoparticles were investigated in depth by combining a wide panel of advanced techniques, such as scanning transmission electron microscopy (STEM), electron energy-loss spectroscopy–spectrum imaging (EELS-SI), 57Fe Mössbauer spectrometry, and X-ray circular magnetic dichroism (XMCD) techniques. The size of the nanoparticles increased progressively after each thermal decomposition step, but the crystal structure of core–shell nanoparticles was significantly modified during the growth of the second shell. Indeed, the antiferromagnetic CoO phase was progressively replaced by the CoFe2O4 ferrimagnet due to the concomitant processes of partial solubilization/crystallization and the interfacial cationic diffusion of iron. A much more complex chemical structure than that suggested by a simple size variation of the nanoparticles is thus proposed, namely Fe3−δO4@CoO-CoFe2O4@Fe3−δO4, where an intermediate Co-based layer was shown to progressively become a single, hybrid magnetic phase (attributed to proximity effects) with a reduction in the CoO amount. In turn, the dual exchange-coupling of this hybrid Co-based intermediate layer (with high anisotropy and ordering temperature) with the surrounding ferrite (core and outer shells) stabilized the particle moment well above room temperature. These effects allow for the production of Fe oxide-based magnetic nanoparticles with high effective anisotropy, thus revealing the potential of this strategy to design rare-earth-free permanent nanomagnets at room temperature.
Introduction
Permanent magnets are widely applied in most devices necessary for today's daily life applications, such as communication, transport, and renewable energies. However, their efficient magnetic properties require a high consumption of rare-earth (RE) and platinum group (PG) components, which are classified as critical raw materials by the European Union owing to potential supply risks.1 In this context, magnetic nanoparticles represent a true alternative to build high-performance technological devices.2 At the nanoscale, their physical properties can be efficiently modulated according to the size and shape of the objects. However, the transformation of soft RE- and PG-free magnetic nanomaterials into permanent magnets remains a huge challenge.Superparamagnetic iron oxide (Fe3O4) nanoparticles are probably the most well-known nanomagnets owing to their extensive investigations for biomedical applications, among other reasons.3,4 Their relatively low magnetic anisotropy can be significantly enhanced by growing a magnetically harder shell, which pins a softer core through interfacial exchange-coupling, thus increasing its effective anisotropy.5,6 Beyond the wide range of structural parameters (core size, shell thickness, chemical composition, defects, doping, interfacial roughness, etc.) significantly affecting exchange coupling,7–13 the selection of a hard phase is restricted by high crystal complementarity and large difference in anisotropy with a soft phase.6,14 Although CoO fulfils both criteria (the cell parameter of Fe3O4 (8.396 Å) is nearly twice that of CoO (4.26 Å) and magnetic anisotropy constants differ by two orders of magnitude, K(Fe3O4)15 = 2 × 104 J m−3 and K(CoO) = 5 × 106 J m−3),16 its antiferromagnetic order is lost at room temperature (Néel temperature, TN = 290 K).17 This drawback can be circumvented by embedding Co@CoO nanoparticles in a NiO matrix to exploit proximity effects.18 The doping of the Wüstite phase by divalent cations is also a potential route to enhance exchange-bias coupling with soft phases.19,20 Ultimately, the Wüstite phase can be replaced by a high-anisotropy spinel phase, such as in some ferrites, with a ferrimagnetic order far above room temperature.14,21,22
Synthetic processes can also favor the formation of ferrites, since high temperatures usually favor cation mobility through interfaces20,23 and partial solubilization.7,24 Therefore, Fe3O4@CoO nanoparticles can include an intermediate layer, such as a CoFe2O4 shell, and can be better described as a core@shell@shell structure.22,23 Such an onion-type structure offers the possibility to generate additional soft–hard interfaces to enhance exchange coupling. Nevertheless, they have been rarely reported because multi-step synthesis with fine control of the size and shape is certainly very difficult.25–27 Recently, we opened new perspectives into this research domain by reporting on the synthesis of Fe3O4@CoO@Fe3O4 nanoparticles displaying permanent magnetization at room temperature.28 However, their chemical structure turned out to be much more complex than expected, hampering the understanding of the magnetic properties (including the exchange coupling phenomenon). Indeed, we noticed that the formation of a second Fe3O4 shell resulted in a remarkable modification of the nanoparticle structure, which was dominated by the concomitant partial disappearance of the CoO Wüstite phase and the appearance of the CoFe2O4 spinel phase. Such a significant modification of the nanoparticle structure could certainly contribute to an increase in the magnetic anisotropy energy. However, given, the limited number of samples that were synthesized, we could not conclude on this point.
Herein, we report an in-depth study of the chemical structure of Fe3O4@CoO@Fe3O4 nanoparticles to better understand their magnetic properties, including their remanent magnetization at room temperature. Their synthesis consisted of a three-step seed-mediated synthesis that involved alternating thermal decompositions of iron and cobalt precursors. Crucially, the amount of iron precursor used in the third step was modified in order to investigate systematically the chemical structure of the onion nanoparticles and its relationship with the magnetic properties. A wide range of advanced characterization techniques, such as element-specific electron loss spectroscopy-spectral high-resolution imaging (EELS-SI), 57Fe Mössbauer spectrometry, and X-ray magnetic circular dichroism (XMCD), were used to accurately characterize the spatial distribution, site occupancy, and oxidation states of Fe and Co cations in the unexpectedly complex multilayer structures. This, in turn, enabled a greater understanding of the magnetic properties, in particular the unprecedented enhancement of the effective magnetic anisotropy in iron-rich oxide nanoparticles due to a combination of magnetic proximity and dual exchange-coupling effects.
Experimental section
Chemical compounds purchased
FeCl2·4H2O (99% Acros Organic), CoCl2·6H2O (reagent grade, Sigma), oleic acid (99%, Alfa Aesar), octyl ether (99%, Sigma), acetone (Pure, Carlo Erba), ethanol (Absolute, Carlo Erba), chloroform (pure stabilized with ethanol, Carlo Erba) were used as received.Synthesis of the metal precursors
Iron stearate (FeSt2) and cobalt stearate (CoSt2) were prepared by a home-made synthesis in order to control precisely the purity and the thermal decomposition process with high reproducibility of the nanoparticle structure.29Synthesis of the nanoparticles
For synthesis of the nanoparticles, a three-step synthesis process was followed. The first step consisted of the synthesis of iron oxide nanoparticles (denoted C) by the decomposition of iron(II) stearate (FeSt2) in dioFctyl ether (b. p. = 288 °C). The second step consisted of the decomposition of cobalt(II) stearate (CoSt2) in order to grow a CoO shell on the surface of the iron oxide nanoparticles, thus forming core@shell (CS) nanoparticles. Finally, the third step consisted of the thermal decomposition of FeSt2 in order to grow a second shell of iron oxide, e.g., core@shell@shell (CSS) nanoparticles. The amount of FeSt2 was set according to the molar ratio R = n(FeSt2 shell)/n(FeSt2 core) = 0.5, 1, 1.5 in order to modulate the thickness of the second shell, resulting in three samples named CSSa, CSSb, and CSSc, respectively.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm, 5 min). Finally, the obtained nanoparticles were stored as a colloidal suspension in chloroform.
000 rpm, 5 min). The obtained core@shell nanoparticles (CS) were finally stored as a colloidal suspension in chloroform.
000 rpm, 5 min). The obtained core@shell@shell nanoparticles (CSS) were finally stored as a colloidal suspension in chloroform.
000 rpm, 5 min). Finally, the obtained nanoparticles were stored as a colloidal suspension in chloroform.
000 rpm, 5 min). The obtained core@shell nanoparticles (CS) were finally stored as a colloidal suspension in chloroform.
000 rpm, 5 min). The obtained core@shell@shell nanoparticles (CSS) were finally stored as a colloidal suspension in chloroform.
| Sample | R ratio | m (iron(II) stearate) g | n (iron(II) stearate) mmol | m (oleic acid) g | n (oleic acid) mmol |
|---|---|---|---|---|---|
| CSSa | 0.5 | 0.156 | 0.25 | 0.141 | 0.50 |
| CSSb | 1 | 0.311 | 0.50 | 0.282 | 1.00 |
| CSSc | 1.5 | 0.468 | 0.75 | 0.423 | 1.50 |
Transmission electron microscopy (TEM) was performed using a JEOL 2100 LaB6 instrument with a 0.2 nm point-to-point resolution and a 200 kV acceleration voltage. EDX was performed with a JEOL Si(Li) detector. The average size of the nanoparticles was calculated by measuring at least 300 nanoparticles from the TEM micrographs using the ImageJ software. The average shell thickness was calculated as half of the difference between the size of the nanoparticles before and after the thermal decomposition step. The size distribution was fitted by a log–normal function.
High-resolution scanning transmission electron microscopy (STEM) and electron energy-loss spectroscopy (EELS) analyses were performed using Cs aberration-corrected STEM on a NION UltraSTEM200 system coupled with a high-sensitivity EEL spectrometer. The convergence and collection semi-angles in the EELS experiments were respectively 35 mrad and 50 mrad. CS, CSSa, and CSSb were analyzed with the STEM microscope operated at a 100 kV acceleration voltage, while the CSSc experiments were performed at 60 kV.
X-Ray diffraction (XRD) was performed using a Bruker D8 Advance instrument equipped with monochromatic copper radiation (Kα = 0.154056 nm) and a Sol-X detector in the 20°–80° 2θ range with a scan step of 0.02°. High purity silicon powder (a = 0.543082 nm) was systematically used as an internal standard. The crystal sizes were calculated by Scherrer's equation and the cell parameters by Debye's law.
Fourier transform infra-red (FT-IR) spectroscopy was performed using a PerkinElmer Spectrum spectrometer in the energy range 4000–400 cm−1 on samples diluted in KBr pellets.
Granulometry measurements were performed using a nano-sizer Malvern (nano ZS) zetasizer at a scattering angle of 173°. Each measurement corresponded to the average of 7 runs of 30 s.
Themogravimetry analyses (TGA) were performed using a SDTQ600 from TA instrument. Measurements were performed on dried powders under air in the temperature range of 20–600 °C at a heating rate of 5 °C min−1.
X-Ray absorption (XAS) and X-ray magnetic circular dichroism (XMCD) spectra were recorded at the L2,3 edges of Fe and Co, on the DEIMOS beamline at SOLEIL (Saclay, France).30 All the spectra were recorded at 4.2 K under UHV conditions (10−10 mbar) and using the total electron yield (TEY) recording mode. The measurement protocol was previously detailed by Daffé et al.31 An external parallel magnetic field H+ (antiparallel H−, respectively) was applied on the sample while a polarized σ+ (polarized σ−, respectively) perpendicular beam was directed on the sample. Isotropic XAS signals were obtained by taking the mean of the σ+ + σ− sum, where σ+ = [σL(H+) + σR(H−)]/2 and σ− = [σL(H−) + σR(H+)]/2, with σL and σR the absorption cross-sections measured respectively with left and right circularly polarized X-rays. XMCD spectra were obtained by taking the σ+–σ− dichroic signal with a ±6.5 T applied magnetic field.
At the DEIMOS beamline, the circularly polarized X-rays were provided by an Apple-II HU-52 undulator for both XAS and XMCD measurements while EMPHU65 with a polarization switching rate of 10 Hz was used to record hysteresis cycle at fixed energy.30 Measurements were performed between 700 and 740 eV at the iron edge and between 770 and 800 eV at the cobalt edge with a resolution of 100 MeV and a beam size of 800 × 800 μm. Both the XMCD and isotropic XAS signals presented here were normalized by dividing the raw signal by the edge jump of the isotropic XAS. The samples consisted of drop-casting suspensions of nanoparticles in chloroform onto a silicon substrate. The substrates were then affixed on a sample holder.
57Fe Mössbauer spectroscopy was performed at 77 K using a conventional constant acceleration transmission spectrometer with a 57Co(Rh) source and a bath cryostat. The samples consisted of 5 mg Fe per cm2 powder concentrated in a small surface due to the rather low quantities. The spectra were fitted by means of the MOSFIT program32 involving asymmetrical lines and lines with Lorentzian profiles, and α-Fe foil was used as the calibration sample. The values of isomer shifts are quoted relative to that of α-Fe at 300 K.
Magnetometry was performed using a superconducting quantum interference device (SQUID) magnetometer (Quantum Design MPMS-XL 5). Temperature-dependent zero-field-cooled (ZFC) and field-cooled (FC) magnetization curves were recorded as follows: powder samples were pressed into polycarbonate capsules and introduced in to the SQUID magnetometer at room temperature, then cooled down to 5 K with no applied magnetic field after a careful degaussing procedure. Then, a magnetic field of 7.5 mT was applied, and the ZFC magnetization curve was recorded upon heating from 5 to 400 K. The sample was then cooled down to 5 K under the same applied field, and the FC magnetization curve was recorded upon heating from 5 to 400 K. In-plane zero-field-cooled (ZFC) and field-cooled (FC, H = 70 kOe) hysteresis loops [M(H) curves] were measured at 10 and 300 K using a maximum field of 70 kOe. The coercive field (HC) and the MR/MS ratio were extracted from ZFC M(H) curves. The exchange bias field (HE) was measured in the FC M(H) curves. Magnetization saturation (MS) was measured from the hysteresis recorded at 5 K and was determined after correcting for the mass of organic ligands obtained from the TGA experiments.
Results
Multicomponent nanoparticles based on metal oxides were synthesized by a three-step seed-mediated growth process. Iron(II) stearate (FeSt2) and cobalt(II) stearate (CoSt2) were decomposed alternatively in high-boiling-temperature solvents (around 300 °C) in order to form successively C, CS, and CSS nanoparticles (Fig. 1).28 In the third step, the amount of FeSt2 was increased to synthesize CSSa, CSSb, and CSSc nanoparticles (see experimental part for details).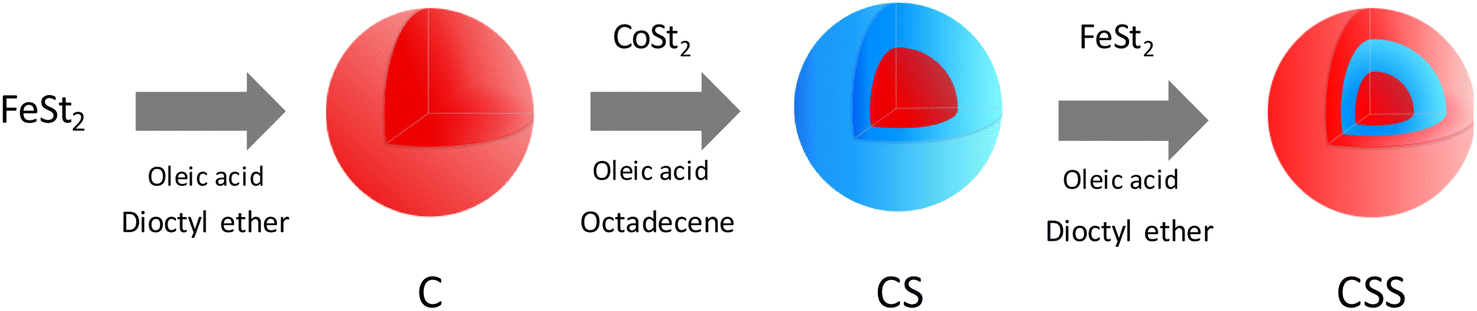 | ||
| Fig. 1 Schematic illustration of the synthesis of CSS nanoparticles by performing the three-step seed-mediated growth process based on the successive thermal decomposition of Fe(II) and Co(II) precursors. | ||
According to the TEM micrographs (Fig. 2), the shape of the nanoparticles was nearly spherical with narrow size distributions gradually shifting to larger average diameters after each thermal decomposition step from 10.1 to 15.6 nm (Table 2). Such size variations corresponded to an additional shell thickness of 2.0 nm for CS, while it was much thinner for CSSa (0.3 nm), CSSb (0.6 nm), and CSSc (0.8 nm). These values were smaller than the cell parameters of the iron oxide spinel, which would lead to an inhomogeneous growth (incomplete coating), as indicated by the shape of the nanoparticles slightly deviating from the spheres and by the broadening of their size distribution. Indeed, their surface was made up of facets, i.e., crystalline planes, with different surface energies yielding different kinetics for the growth of the components.33
 | ||
| Fig. 2 TEM images of (a) CS, (b) CSSa, (c) CSSb, and (d) CSSc and the corresponding size distributions (bottom). | ||
| C | CS | CSSa | CSSb | CSSc | |
|---|---|---|---|---|---|
| Diameter (nm) | 10.1 ± 1.1 | 14.0 ± 1.5 | 14.5 ± 1.5 | 15.1 ± 1.7 | 15.6 ± 2.3 |
| Additional layer thickness (nm) | — | 2.0 | 0.3 | 0.6 | 0.8 |
| Fe:Co at. ratio by EDX |
— | 45:55 |
57:43 |
68:32 |
73:27 |
| Hydrodynamic diameter (nm) | 12 | 16 | 18 | 21 | 21 |
| Cell parameter (Å) | 8.37(9) ± 0.01 | 8.40(9) ± 0.01 | 8.39(1) ± 0.01 | 8.40(1) ± 0.01 | 8.41(2) ± 0.01 |
| Crystal size (nm) | 8.0 ± 0.1 | 9.1 ± 0.1 | 11.4 ± 0.1 | 12.2 ± 0.1 | 12.7 ± 0.1 |
The chemical composition of the nanoparticles was also studied by energy dispersive X-ray spectrometry (EDX). The Fe:Co atomic ratio increased progressively from CS to CSSa, CCSb, and CSSc (Table 2), as expected by the growth of a thicker iron oxide shell. The spatial distribution of Fe, Co, and O atoms was further investigated by performing electron energy-loss spectroscopy – spectral imaging (EELS-SI). Although Co was distributed around the entire surface of the iron oxide core in CS, some areas corresponded to higher loadings (Fig. 3a and e), indicating the preferential growth of CoO as a thicker shell on specific facets. Although this was similar in CSSa, the distribution of CoO appeared to be progressively more uniform in CSSb and CSSc, indicating the redistribution of Co cations as the amount of the Fe precursor increased.
 | ||
| Fig. 3 Electron energy-loss spectroscopy – spectral imaging (EELS-SI) images at the Fe L-edge (top), Co L-edge (bottom). (a and e) CS, (b and f) CSSa, (c and g) CSSb and (d and h) CSSc nanoparticles. | ||
High-resolution STEM micrographs were recorded in dark-field mode to study the crystal structure of the nanoparticles (Fig. 4). Continuous and straight lattice fringes with no defects were observed across each nanoparticle, regardless of its composition (CS and all CSS particles). A doubling of the lattice fringes periodicity could be observed in some areas, which, according to the FFT, corresponded to the reflections of the spinel and Wüstite structures (Fig. 4). The interpenetration of the two lattice fringes was consistent with the epitaxial growth of the Wüstite shell (CoO) from the surface of the spinel core (Fe3−δO4), as expected from their similar cubic structures and low lattice mismatch. The cell parameter of Fe3O4 (8.396 Å, JCPDS card no 19-062) was almost twice that of CoO (4.26 Å, JCPDS card no 00-048-1719), giving a lattice mismatch of 1.5% between the two phases. This was also confirmed by the geometrical phase analysis (GPA), which evidenced a lattice difference of 1–2% for the CoO compared to the Fe3−δO4, indicating very low residual strains in the CS and CSSc nanoparticles (Fig. 4b and f).
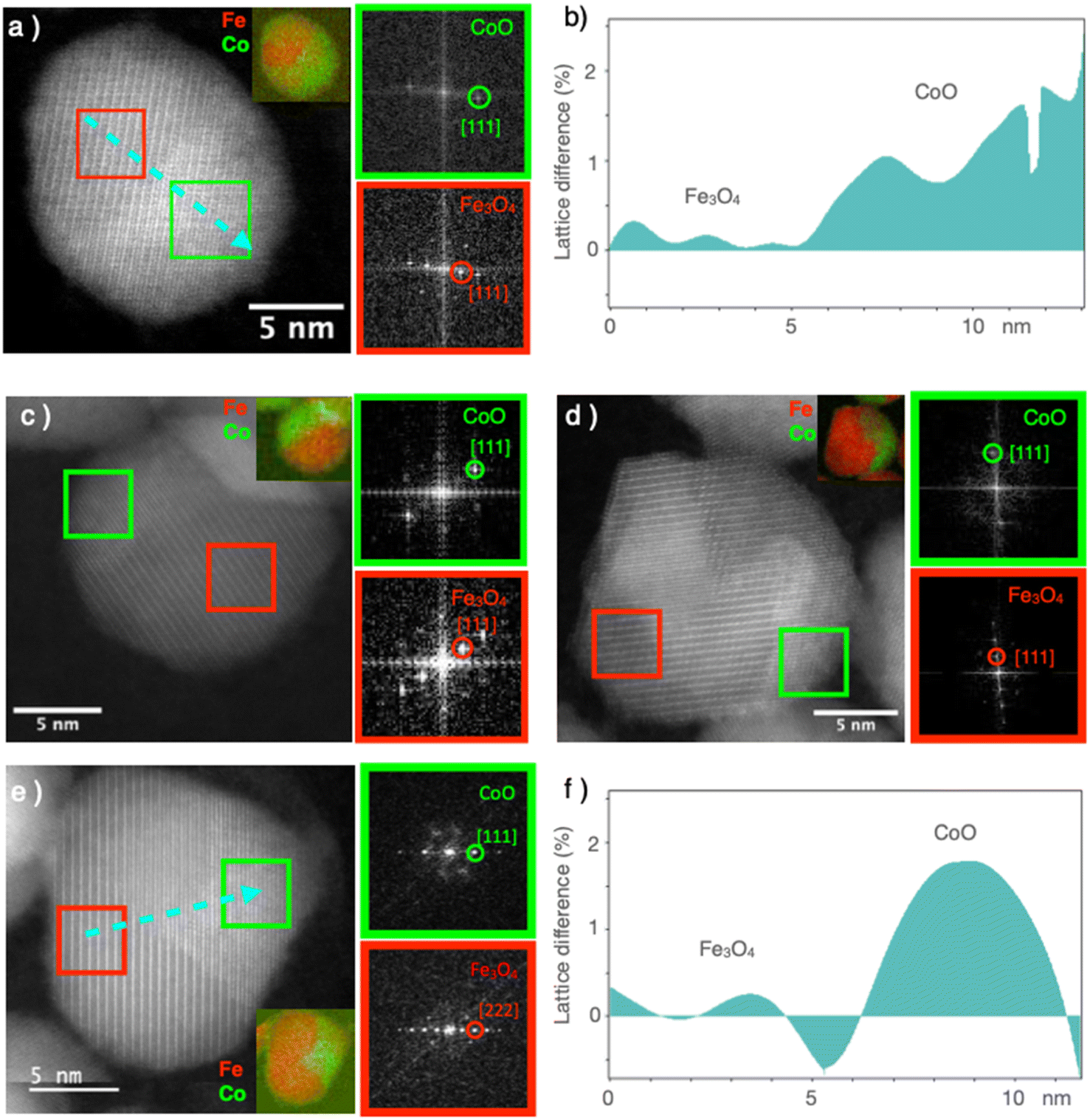 | ||
| Fig. 4 (a, c, d, and e) High-resolution dark-field STEM images with specific FFT corresponding to the selected zone in the HR STEM images. In the inset of the main panel: the corresponding EELS elemental maps of Fe (red) and Co (green) for: (a) CS, (c) CSSa, (d) CSSb, and (e) CSSc. (b and f) Geometrical phase analysis (GPA) showing the variation of the lattice mismatch of (a) and (b), respectively, along the direction of the dashed blue arrow. The periodicity of the spinel phase lattice fringes was taken as a reference for performing the GPA. | ||
The crystal structure of the nanoparticles was also studied by X-ray diffraction (XRD) (Fig. 5). The XRD patterns displayed peaks that could be indexed to the spinel structure. Additional peaks corresponding to the Wüstite phase were also observed, such as the one around 36.5°, in the CS and CSS nanoparticle patterns. The intensity of this peak was markedly reduced in CSSa and then gradually decreased in CSSb and CSSc, compared to the (311) reflection of the spinel structure. Concurrently, the spinel peaks became narrower, which was ascribed to the increase in the crystal sizes from 8.0 nm for C up to 9.1, 11.4, 12.2, and 12.7 nm for CS, CSSa, CSSb, and CSSc, respectively. This was consistent with the increase in the nanoparticle size observed in the TEM micrographs, while also indicating a high crystalline coherence of the spinel phase in the growth of the successive layers. The lower values of the crystal sizes compared to the TEM nanoparticle size may be also explained by the 2D projection of the faceted nanoparticles on the TEM micrographs, which tends to overestimate their size. While the unit cell parameter for C (8.379 Å) was in agreement with the partial oxidation of magnetite into maghemite, the values extracted for the other samples were higher than the lattice parameter of magnetite (a = 8.396 Å, JCPDS card no 19-062).34 This was ascribed to the strain induced by the lattice mismatch between the spinel and Wüstite phases (2aCoO = 8.52 Å, JCPDS card no 78-0431) and to the higher Fe2+ content, as previously observed in related studies.35,36
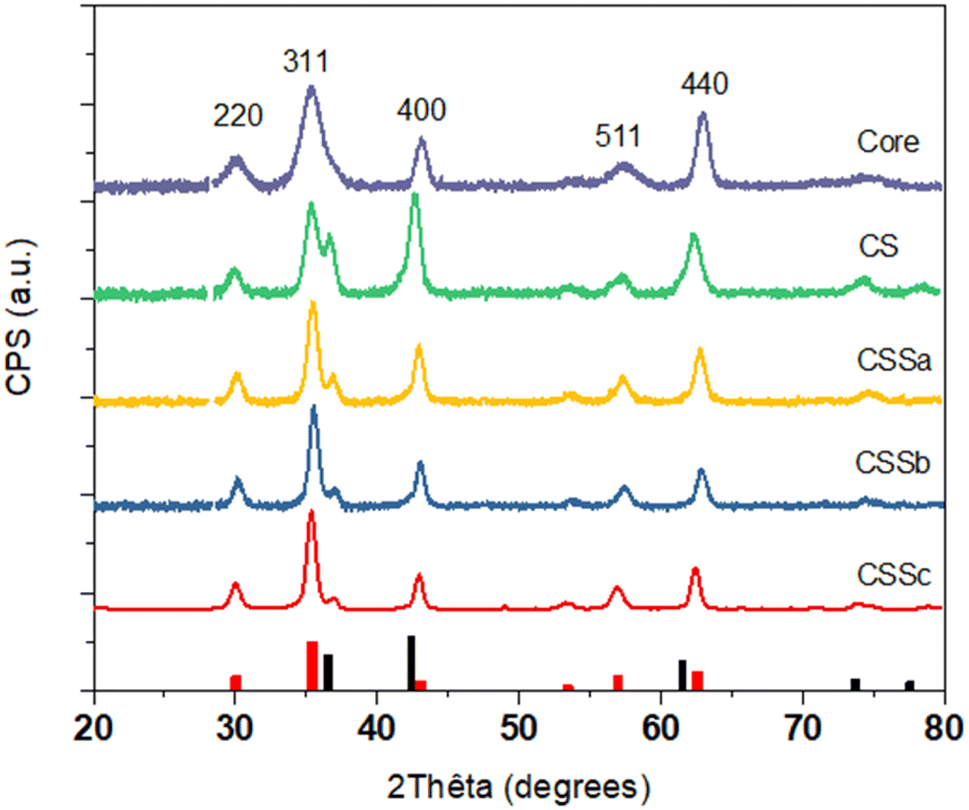 | ||
| Fig. 5 XRD patterns of C, CS, CSSa, CSSb, and CSSc nanoparticles. The red and black shapes at the bottom show the reference reflections of Fe3O4 (JCPDS card no 19-062) and CoO (JCPDS card no 78-0431) phases, respectively. hkl index refers to the spinel phase. | ||
These results were confirmed by FTIR (see ESI†), which shows that the band related to the M−O vibrational modes was shifted to shorter wavelengths (from 599 cm−1 for CS to 582 cm−1 for CSSc) when the amount of FeSt2 increased. Such a shift for the CSS samples, shorter than that of CoFe2O4 (591 cm−1), was in agreement with the increasing content of Fe3O4 (574 cm−1) as a second shell on the surface of the nanoparticles. The bands recorded for CSSb and CSSc mostly overlapped, which was consistent with a possible concomitant growth of the CoFe2O4 and Fe3−δO4 shells. Nevertheless, CSSb and CSSc did not display similar structures according to the above-mentioned results (longer coherence length of the spinel phase, higher Fe/Co ratio).
In order to obtain additional information on the environment and valence state of iron cations, 57Fe Mössbauer spectrometry was performed at 77 K. All the spectra consisted of magnetic sextets composed of asymmetrical and broad lines (Fig. 6). The C and CS spectra showed additional peaks corresponding to quadrupolar doublets ascribed to the presence of superparamagnetic fluctuations36 of the smallest particles in these systems. The spectral refinement led to several components associated with the hyperfine parameters that could well describe the hyperfine structure. The isomer shift (IS), hyperfine field (Bhf), and quadrupole shift (2ε) describe the oxidation state, magnetic environment, and local electronic structure of 57Fe atoms, respectively (see ESI†). Although spectra can be refined by different combinations of components, the mean values of each hyperfine parameter are invariant and independent on the fitting model.
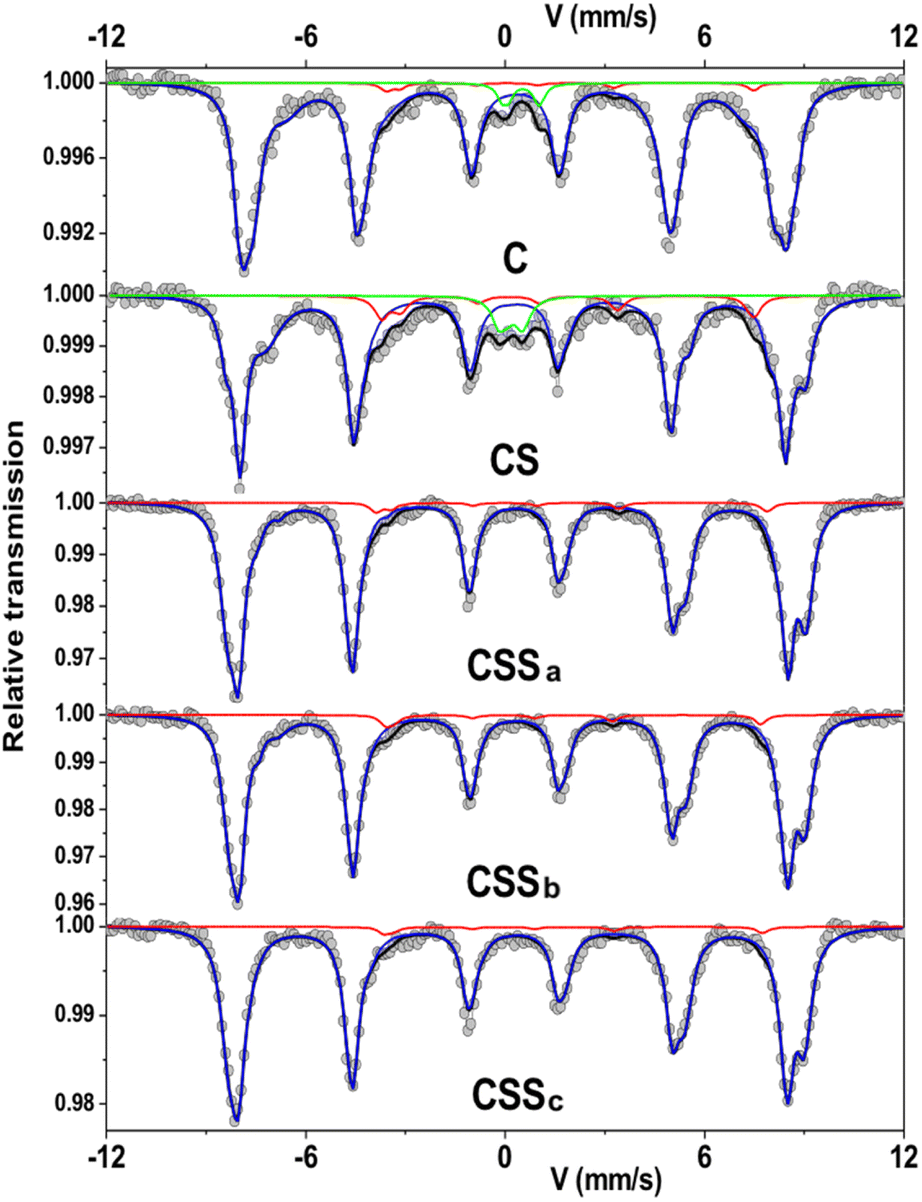 | ||
| Fig. 6 Mössbauer spectra recorded at 77 K in a zero-field condition. The red line represents the Fe2+ content. | ||
The mean isomer shift values at 77 K can be compared to the theoretical values of pure magnetite (0.61 mm s−1), maghemite (0.40 mm s−1), and cobalt ferrite (0.45 mm s−1) in order to determine the fraction of Fe3O4 in each type of nanoparticle.37–39 By considering a linear relationship between the experimental and the theoretical values, the Fe3O4 fraction vs. the Fe2+-deficient phases (γ-Fe2O3 and CoFe2O4) can be calculated. The increase in the mean isomer shift from C (0.504 mm s−1) to CS (0.528 mm s−1) indicated a higher content of Fe2+ in CS, as the CoO shell hindered the oxidation of Fe2+ when the nanoparticles were exposed to air. Thus, the Fe3O4 fraction increased from 34% in C to 49% in CS. The mean Bhf also increased from C (47.5 T) to CS (48.5 T), which was attributed to the presence of Co in the vicinity of Fe atoms,40,41i.e., due to the formation of an intermediate layer of Co-doped ferrite at the Fe3−δO4/CoO interface.7,23,24,42
For CSSa nanoparticles, the decrease in the mean isomer shift (0.489 mm s−1) indicated a lower relative content of Fe2+ compared to Fe3+, which was consistent with the larger fraction of CoFe2O4 indicated by FTIR and XRD. This confirmed the increase in the mean Bhf value above 51 T. However, the increase in the mean isomer shift of CSSb (0.510 mm s−1) corresponded to a larger amount of Fe2+. Finally, the isomer shift was decreased for CSSc (0.485 mm s−1), although the component tentatively ascribed to Fe2+ was stable (3% of the relative sub-spectral area) while that of Fe2-3+ increased from 5% to 8% (see ESI†). Considering these values, Fe3O4 accounted for 24% of CSSa, 38% of CSSb, and 22% of CSSc. The mean Bhf values of CSSb and CSSc remained rather constant above 51 T, showing the stability of CoFe2O4 regardless of the amount of Fe precursor used to grow the second shell.
Soft X-ray absorption spectroscopy (XAS) and X-ray magnetic circular dichroism (XMCD) experiments were performed to discriminate the site occupancies and oxidation states of Fe and Co cations (Fig. 7). The isotropic XAS spectra recorded at the Fe L2,3 and Co L2,3 edges were typical of the spinel structure, which contained both cations.43,44 The Fe2+/Fe3+ ratio in the octahedral (Oh) sites could be qualitatively determined from the intensity ratio I1/I2 of the peaks labeled in Fig. 7a, see Table 3,43 and markedly increased from C (0.56) to CS (0.82), indicating a higher content of Fe2+.23,36 The value of CS being higher than that of pure Fe3O4 was ascribed to Fe2+ in the Wüstite phase (FeO), as suggested by Mössbauer spectrometry. In contrast, the ratio calculated for CSSa (0.52) was lower than that measured in C. Then, it increased to 0.61 for CSSb and 0.65 for CSSc, which were indicative of the higher content of Fe2+.
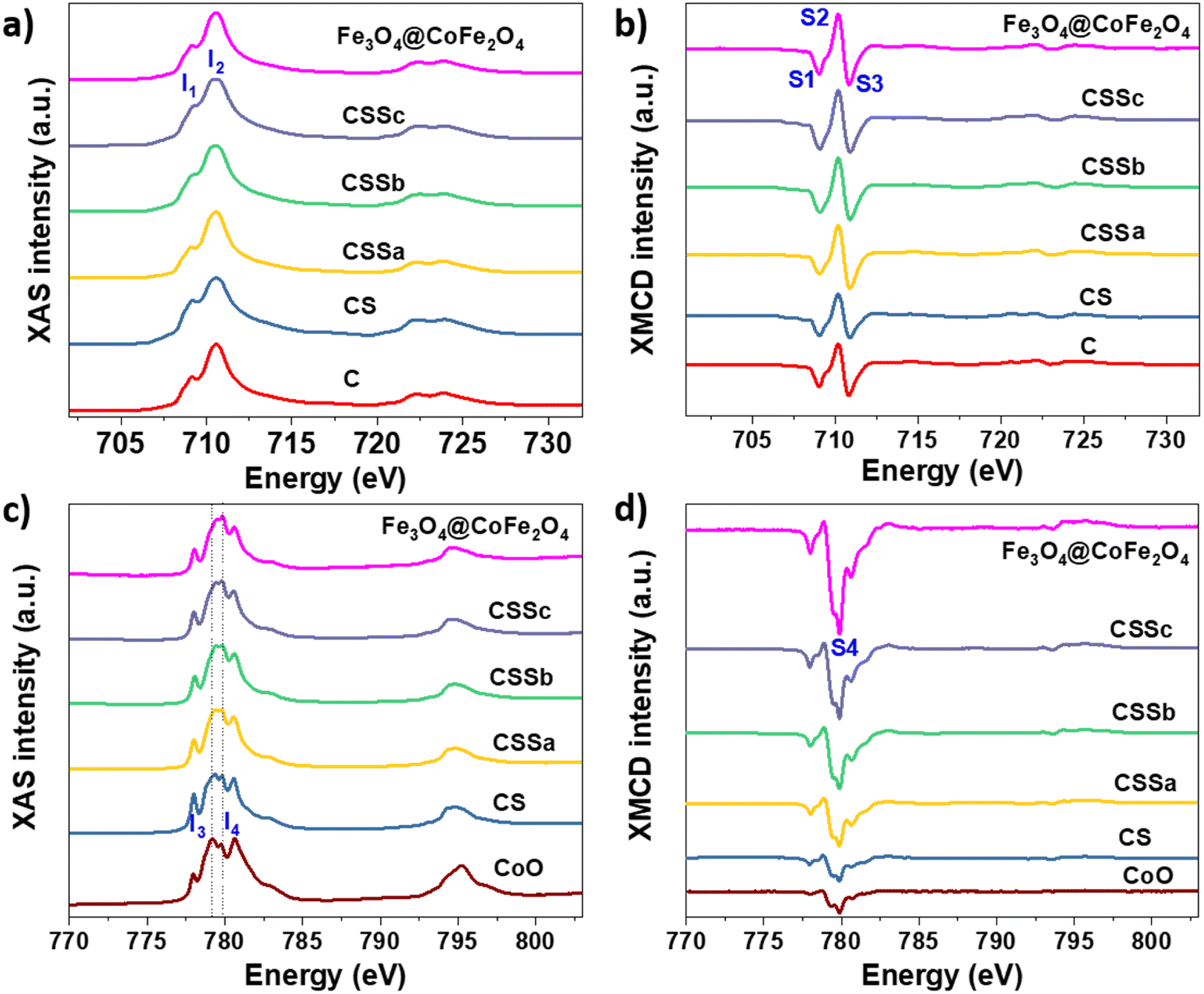 | ||
| Fig. 7 (a and c) XAS and (b and d) XMCD spectra recorded for C, CS, CSSa, CSSb, and CSSc nanoparticles at the (a and b) Fe L2,3 edges and (c and d) Co L2,3 edges. | ||
XMCD spectra were also recorded at the Fe L2,3 and were typical of the reverse spinel structure. The peaks indexed as S1, S2, and S3 corresponded to Fe2+ and Fe3+ in the Oh sites, Fe3+ in the tetrahedral sites (Td), and Fe3+ in the Oh sites, respectively. The ratio S = (S1 + S2)/(S2 + S3) displays a similar behavior to that of the I1/I2 ratio (Table 3).45 Considering the values ascribed to pure Fe3O4 (1.27)44 and γ-Fe2O3 (0.69),31 the extracted S values indicated an Fe2+ deficiency, ascribed to the formation of γ-Fe2O3 and CoFe2O4. In CS, we expect that the Fe2+ deficient fraction (57%) could be mostly ascribed to CoFe2O4, which resulted from the diffusion of Co2+ in the Oh vacancies on the surface of the Fe3−δO4 nanoparticles.7,23 Moreover, the significant decrease in the Fe2+ fraction in CSSa (12%) did not result from the oxidation of the core (protected by the CoO shell), but from a higher amount of CoFe2O4. In CSSb and CSSc, the thicker Fe3−δO4 shells resulted in a significant increase in the Fe2+ content (30% and 43%, respectively). These values were much higher than expected, because Fe2+ on the nanoparticle surface would be oxidized, as has been generally reported for Fe3−δO4 nanoparticles.46
It is worth noting that the I1/I2 ratio calculated from the XAS spectra of CS was higher than that for Fe3O4, which could be attributed to the presence of an additional fraction of Fe2+ in the Wüstite phase, as observed recently.24 Therefore, we expect the Wüstite shell to consist of Co1−xFexO resulting from the co-crystallization of Co2+ and Fe2+, with the latter resulting from partial solubilization at the early stages of the Wüstite shell formation. As the CoO shell is also partially solubilized during thermal annealing in a liquid medium, this resulted in a strong decrease in the I1/I2 for CSSa. In contrast, the S value was lower than that of pure Fe3O4 and C due to the formation of CoFe2O4. Therefore, the increase in the S ratio in CSSb and CSSc agreed with the presence of higher Fe2+ contents in the iron oxide shell. This is particularly true because 70% of the XMCD signal came from the first 2 nm of the nanoparticle surface.
In the XAS spectra recorded at the Co L2,3 edges, the intensities of peaks I3 and I4 refer to the distribution of Co2+ in the Wüstite and spinel phases, respectively.31,47 Therefore, the I4/I3 ratio evidenced the progressive conversion of CoO to CoFe2O4 from CSSa to CSSc.23,36 The XMCD spectra also recorded at the Co L2,3 edges showed a negative peak S4, corresponding to Co2+ in the Oh sites of the spinel structure.36,44 The intensity of this peak markedly increases from CS to CSSc, which also supported the increasing fraction of CoFe2O4, in agreement with the EDX, XRD, and FTIR data. Note that magnetically compensated spins in antiferromagnets do not contribute to the XMCD signal. Nevertheless, XMCD is sensitive to uncompensated Co spins of CoO, which are typically localized at the nanoparticle surface48 or coupled to Fe spins at the interface.23,24 The normalization of the XMCD spectra by the XAS signal allows determining the fraction of uncompensated spins. Here, the intensity of the S4 peak yielded 23% of the uncompensated Co spins in CS, which mainly consisted of a CoFe2O4 layer localized at the Fe3−δO4/CoO interface.24 In CSS nanoparticles, the S4 peak indicated there were much higher amounts of uncompensated Co spins (42%, 54%, and 68% in CSSa, CSSb, and CSSc, respectively). This result unambiguously confirmed the increasing fraction of CoFe2O4 in the nanoparticles at the expense of CoO, completing a consistent structural picture of the particles, as graphically summarized in Fig. 8.
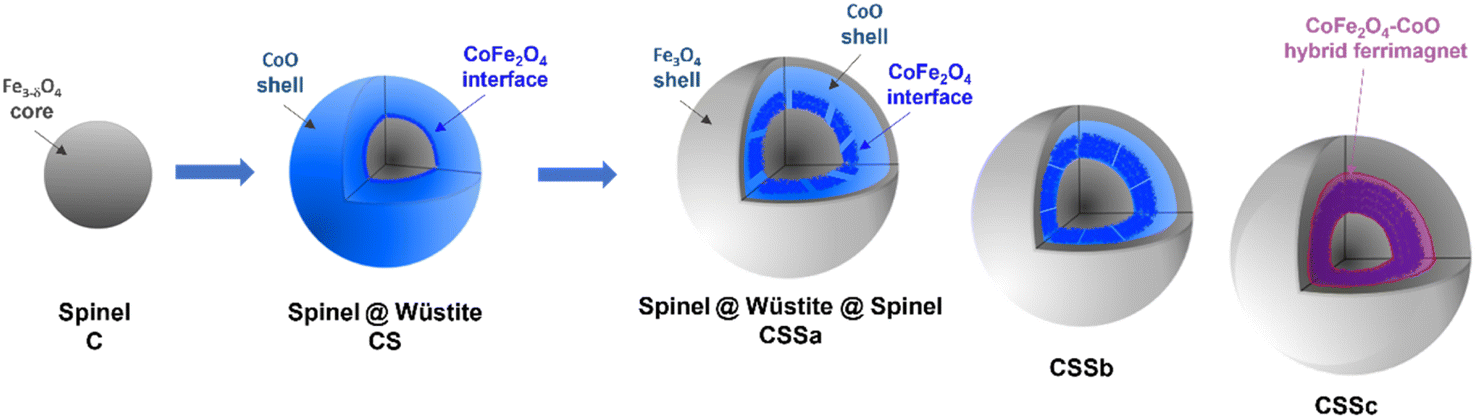 | ||
| Fig. 8 Schematic illustration of the chemical structure of CSS nanoparticles. The last (rightmost) cartoon depicts the suggested hybrid ferrimagnet, which develops progressively from the CSSa stage. | ||
Magnetic properties
As mentioned above, element-specific magnetization curves were registered to study the magnetic structure with respect to site occupancy and the oxidation states of Fe and Co cations (Fig. 9). The M(H) curves recorded at 4K displayed very similar coercive fields (HC) when measured at the different energy edges of Fe and Co (see ESI† for the detailed values), thus demonstrating the coherent reversal and, thus, the strong exchange coupling between the Fe and Co cations in all the spinel structures. The mean HC values were similar for all the CS and CSS nanoparticles (10.9, 10.8, 10.9, and 9.6 kOe for CS, CSSa, CSSb, and CSSc, respectively), that, at these low temperatures, the coercivity was mostly determined at the core–shell interface by exchange coupling of the core to similarly anisotropic Co-based phases across the series. Yet, the clear reduction in HC in CSSc was consistent with the larger fraction of magnetically soft Fe3−δO4 in the outer shell leading to a doubly exchange-coupled “soft core/hard layer/soft layer” system.36 However, the main factor behind this reduction was possibly the modification (with the progressive conversion of CoO into Co-ferrite) of the nature of the inner interface. These coercivity values were larger than those observed in many similar core/shell nanoparticles comprising magnetite and Co oxides; for instance, Fe3−δO4@CoO nanoaggregates of 9.9 nm (HC = 3 kOe),7 magnetite-doped cobalt ferrite nanoparticles of 40.3 nm (HC = 2.1 and 3.9 kOe at Fe and Co edges),44 and even exceed those we reported recently for optimized Fe3−δO4@CoFe2O4@Fe3−δO4 (9.6 kOe).36 There are few reports of similarly high coercivity in Co-ferrite-based nanoparticles of similar sizes.49 | ||
| Fig. 9 Element-specific magnetization curves recorded at 4K by XMCD between −6.5 T and +6.5 T at fixed energies corresponding to the maximum of S1, S2, S3 peaks (Fe L2,3 edges) and to the S4 peak (Co L2,3 edges) for (a) CS, (b) CSSa, (c) CSSb, and (d) CSSc. | ||
The magnetic properties were also investigated by SQUID magnetometry. Fig. 10a presents the low-field magnetothermal curves recorded after zero-field-cooling (ZFC) and field-cooling (FC). The peak temperature of the ZFC curve, Tmax, is often taken as the “blocking temperature” of the system in the measurement (magnetometry) timescale, which corresponds to the thermal energy comparable to the magnetic anisotropy energy barrier (kV ≈ 25 kBT). However, the blocking temperature (TB) is described more realistically as the median of a distribution of energy barriers, which can be extracted from the FC-ZFC difference as:50
| f(TB) = d(MFC − MZFC)/dT |
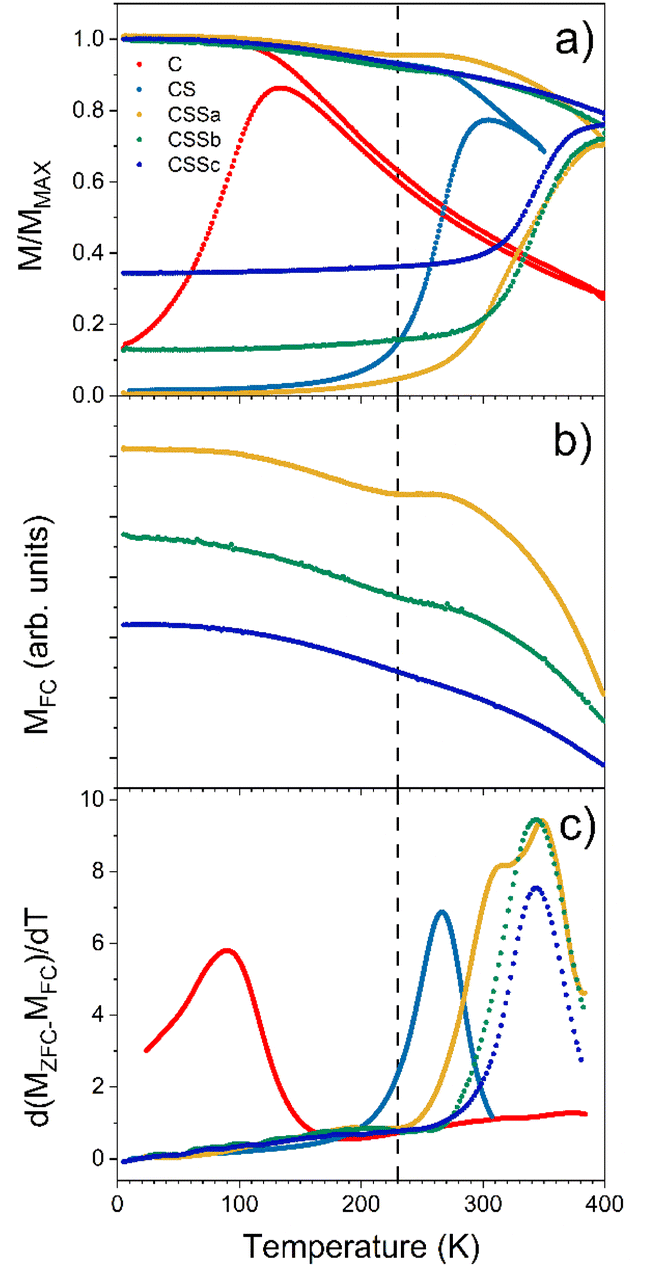 | ||
| Fig. 10 (a) Low-field magnetothermal field-cooled (FC) and zero-field-cooled (ZFC) curves recorded for CSSa, CSSb and CSSc. (b) Magnetization FC curves. (c) Distribution of blocking temperatures. The dashed line indicates the position of the MFC dip. | ||
This distribution was shifted to higher temperatures by interparticle dipolar interactions in the nanoparticle powders (Fig. 10c).51,52 However, since the interparticle interactions were roughly similar in all samples, the observed evolution of the median blocking temperature (or Tmax for the same reason) across the series mainly reflected variations in the average particle anisotropy barrier.53
The pristine Fe3−δO4 nanoparticles displayed a Tmax of 150 K and TB of 93 K, in agreement with the reported values for iron oxide nanoparticles of 10 nm.46 These values were strongly increased for CS (Tmax = 290 K, TB = 266 K), indicating an increase of the effective magnetic anisotropy energy (KefV), which unequivocally reflected the exchange coupling at the soft/hard interface.57 Remarkably, Tmax in CS was roughly equal to the Néel temperature of CoO (TN = 290 K), indicating that this shell was thick enough (at least 2 nm) to support thermally stable CoO grains up to the ordering temperature.18,54 Although some CoO grains in CS were thermally stable up to TN, a fraction of them started to fluctuate at ≈200 K (alternatively or concurrently, the AFM order in thinner shell regions may be lost due to size effects),54 below which all particles were pinned: note the flat shape of the ZFC curve below that temperature, as previously described in other biased NP systems.18,55 A similar behavior, modulated by the relative fraction of the remaining CoO phase, was also observed for CSSa. The ZFC curves of CSS remained flat for a wide range of low temperatures, which extended to higher temperatures for CSSa to CSSc. While the onset of the ZFC magnetization in CSSa was relatively gradual (the derivative in Fig. 10c shows that two overlapping mechanisms were at play), in CSSb and CSSc the onset was sharper and took place at about 290 K. The FC curves of these three samples are plotted separately in panel (b) to highlight the presence of a small but clearly visible dip for CSSa, signaling the loss of CoO-related exchange coupling at about 230 K. This feature vanished progressively with the decreasing amount of CoO in CSSb and CSSc. Yet, in these two systems the volume of CoO still amounted to nearly one-half and one-third, respectively, of the Co-containing species (see Table 3). This suggests that in CSSb and CSSc the remaining, possibly discontinuous, layer of CoO was so thin that it was effectively stabilized via proximity effects by the surrounding Co-ferrite layer, thus becoming a single magnetic object with properties in between those of CoO and Co-ferrite. This explains the vanishing dip in the FC curve (attributed to the “individual” magnetism of the CoO phase) while preserving, even enhancing, the median blocking temperature determined by the dual exchange coupling of the Fe oxide core and outer shell with the high-anisotropy hybrid Co–Fe–O shell in between them.
The magnetic response, M(H), measured at 300 K, for C and CS showed no hysteresis, thus confirming the room temperature superparamagnetism of these two samples (Fig. 11c). In contrast, CSS nanoparticles exhibited a remanent magnetization at 300 K, as expected from their higher blocking temperatures. Note that the single-loop aspect of all the hysteresis loops (no kinks, i.e., absence of a decoupled soft component, often observed as a low-field step)56,57 showed that the Fe3−δO4, CoO, and CoFe2O4 phases were all fully-coupled and rotated coherently upon reversal as required in applications of exchange-coupled soft–hard magnetic systems.13
 | ||
| Fig. 11 Hysteresis loops measured at (a) 10 K after zero field cooling, (b) 10 K after cooling under a field of 7 T, and (c) at 300 K. | ||
Hysteresis loops recorded at 10 K after cooling in zero-field showed a strong hysteresis in all the nanoparticles, except those measured in the simple C seeds (Fig. 11a). The coercive field of CS (16.4 kOe) was very high compared with similar Fe3−δO4@CoO nanoparticles reported in the literature.22,23,58 It increased to ≈17.3 kOe for CSSa and CSSb (due to the hard Co-ferrite contribution), and decreased down to ≈15.0 kOe for CSSc, consistently with the growth of a thicker soft iron oxide shell.14 As expected, cooling down in a magnetic field of 7 T resulted in yet higher HC values (Fig. 11b) due to the exchange coupling to uncompensated high-anisotropy (yet rotatable) spins at the soft–hard interfaces, an effect more apparent in the horizontal shift (exchange-bias field) due to the exchange-coupling with pinned uncompensated spins.5,25 In contrast with the ZFC loops, the coercivity was higher in CS (19.8 kOe) than in the CSS samples (18.5 to 15.8 kOe). In other words, the exchange-coupling-induced HC increase was significantly larger in the CS particles. This can be understood in terms of both the higher anisotropy of CoO (compared to CoFe2O4) and the large fraction of uncompensated spins (UCS) in the CoO phase. In any case, the HC values of our CSS particles were higher than those reported earlier at low temperatures for core@multi-shell magnetic nanoparticles, such as FeO@Fe3O4@MnO@Mn3O4 (1.2 kOe)25 or MnFe2O4@CoFe2O4@NiFe2O4 (7.7 kOe).27 The MR/MS ratio of the ZFC curves increased from 0.24 (C) to 0.38 (CS) and further up to about 0.60 in CSS. Therefore, the hysteresis curves became squarer upon the gradual replacement of CoO by CoFe2O4 and the increase in the iron oxide shell thickness, concurrently with an increase in saturation magnetization, thus leading to larger energy products (BH)max. The gradual removal of CoO (AFM) and substitution by Co-ferrite (FiM) were indeed clearly confirmed by the evolution of the saturation magnetization (MS), which markedly increased from 41 (CS) to 72 (CSSc) emu g−1 (Table 4), since the compensation of spins in the ordered fraction of the AFM structure resulted in a zero MS.22
| C | CS | CSSa | CSSb | CSSc | |
|---|---|---|---|---|---|
| Diameter (nm) | 10.1 ± 1.1 | 14.0 ± 1.5 | 14.5 ± 1.5 | 15.1 ± 1.7 | 15.6 ± 2.3 |
| Thickness (nm) | — | 2.0 | 0,3 | 0,6 | 0,8 |
| H C 300 K (ZFC) (kOe) | 0 | 0 | 0.5 | 0.3 | 0.5 |
| H C 10 K (ZFC) (kOe) | 0.4 | 16.4 | 17.5 | 17.2 | 15.0 |
| H C 10 K (FC) kOe | 0.4 | 19.8 | 18.5 | 17.9 | 15.8 |
| H E 10 K (kOe) | 0 | 5.3 | 2.8 | 0.9 | 0.5 |
| T max (K) | 150 | 290 | ≈400 | >400 | >400 |
| T B (K) | 93 | 266 | 315 | 343 | 343 |
| M S at 5 K (ZFC) (emu g−1) | 60 | 41 | 51 | 55 | 72 |
| M R/MS at 5 K (ZFC) | 0.24 | 0.38 | 0.59 | 0.68 | 0.66 |
As anticipated above, all the FC M(H) curves were shifted to negative magnetic fields as a result of the exchange-bias phenomenon, namely the pinning of the ferrimagnetic (FiM) phase magnetization by the uncompensated spins of the AFM phase, which aligned with the cooling field, establishing a unidirectional anisotropy. This exchange-bias field (HE), defined as the loop shift, was largest for CS (5.3 kOe) and decreased gradually down to 0.5 kOe for CSSc, consistently with the progressive substitution of the CoO antiferromagnet by CoFe2O4, as concluded from the structural characterization, together with a concurrent loss of magnetic anisotropy in CoO through proximity effects with the Co-ferrite in the CSSb and CSSc samples, as proposed above. Nevertheless, a fraction of CoO remained in each CSS nanoparticle, as observed by XRD and XAS/XMCD. In the CS nanoparticles, the significant vertical shift of the hysteresis loop evidenced a correspondingly large fraction of fully pinned uncompensated spins (which do not rotate with the core magnetization). These are the spins responsible for the strong biasing of the core magnetization reflected in the high HE value. In contrast with this FiM(soft)–CoO coupling, the FiM(soft)–FiM(hard) coupling did not introduce unidirectional anisotropy (the CoFe2O4 component essentially lacks such pinned UCS), as evidenced by the strong decrease in HE, but the large anisotropy of the CoFe2O4-CoO hybrid phase proposed above (majority in the CSS samples), combined with its high ordering temperature (in contrast with CoO), which stabilizes the overall moment beyond room temperature. Indeed, the HE(T) curves show that HE vanished at gradually lower Tonset values from CS (200 K) to CSSc (20 K) (Fig. 12). The pronounced reduction in both HE (10 K) and Tonset even for CSSa (for which we estimated above a modest decrease in CoO content from 63% to 51%, see Table 1) suggested that the conversion of CoO into CoFe2O4 was accompanied by the loss of UCS in the remaining CoO, a likely effect of the recently reported effective annealing provided by the third decomposition step.24 The same effects can be expected from a progressive hybridization of the shrinking CoO layer or interface islands with the Co-ferrite phase via proximity effects. Note that we have previously observed no HE values in core/shell Fe3O4/CoFe2O4 nanoparticles.36
 | ||
| Fig. 12 Temperature dependence of the coercive field (black squares) and exchange field (blue triangles) of (a) CS, (b) CSSa, (c) CSSb, and (d) CSSc. | ||
The decreasing number of CoO UCS (both pinned and rotatable) was confirmed by the difference between the HC values [HC(FC) − HC(ZFC)], which was highest for CS (3.4 kOe) and decreased markedly for the CSS particles (≤1 kOe). In short, the concurrent structural ordering of the CoO phase and its hybridization with Co-ferrite from CS to CSSc were consistent with the large reduction in HE across the CS–CSS series (roughly an order of magnitude) while essentially preserving the high coercivity (with a mere 20% decrease in the 10 K loops).
In the CSS samples, Fig. 12 shows that HE vanished at temperatures (Tonset) significantly lower than the FC feature signaled by the vertical line in Fig. 10, ascribed to the residual fraction of CoO, which still preserves its individual properties (c.f. the remaining CoO was hybridized with Co-ferrite via a proximity effect). Although the presence of such a residual fraction (decreasing from CSSa to CSSc) leaves a subtle fingerprint in the low-field M(T) curves (possibly at its Néel temperature), it was not capable of biasing the hysteresis loop above 100 K (less for CSSc). These relatively low Tonset values in the CSS particles indicated that the remarkable magnetic stabilization of the CSS particles up to (at least) 400 K was not due by exchange coupling to CoO (see also the strong coercivities measured well above Tonset), but was rather driven by exchange coupling to an intermediate CoO–CoFe2O4 hybrid shell stemming from a proximity effect from these two compounds, where, conveniently, CoO contributed a high anisotropy, and Co-ferrite a relatively large saturation magnetization. Then, the magnetic characterization of the samples gave rise to yet another scenario, as summarized in the last figure in Fig. 8.
Discussion
Although each thermal decomposition step resulted in the growth of additional layers onto the surface of the nanoparticles, a far more complex chemical structure than expected was revealed by a the results from a wide range of complementary techniques. Regarding the CS nanoparticles studied here, a number of new results were obtained with respect to previous studies on Fe3−δO4@CoO core–shell nanoparticles. First, we confirmed that the growth of the CoO shell partially preserved the Fe3−δO4 core from the oxidation of Fe2+ when the nanoparticles were exposed to air. Second, in the HRTEM micrographs the Wüstite phase was identified to grow epitaxially with low strain at the surface of the spinel phase, which may explain the efficient exchange coupling yielding unprecedentedly strong coercivities in this system. Third, EELS-SI showed that CoO did not grow uniformly at the surface of the Fe3−δO4 nanoparticles, which was attributed to the preferential growth on certain facets driven by the different surface energies. Fourth, Co2+ cations diffused at the surface of the Fe3−δO4 core to fill vacancies in the Oh sites, as shown by the Mössbauer and XMCD measurements. Therefore, in contrast with the simple core–shell scenario often assumed in the literature, i.e., well-defined core–shell interfaces, we demonstrated a complex structure involving three phases, namely Fe3−δO4@CoFe2O4@CoO with a discontinuous CoO shell.The structure of CSS nanoparticles is yet more complex. The Fe/Co atomic ratio measured by EDX indicated the formation of Fe species at the surface of CS nanoparticles. By considering a simple spherical core–shell–shell model, hypothetical Fe3−δO4 outer shells with mean thicknesses similar to those obtained from TEM micrographs were calculated. In CSSa, the small size increase resulted in an effective shell thickness of 0.3 nm, less than the Fe3O4 lattice parameter (8.396 Å). A non-uniform growth of Fe3O4 resulting in a discontinuous shell was expected, although a partial solubilization of the CoO shell followed by recrystallization certainly took place (see below), as we reported very recently.24 When the mean shell thickness increases, the shell is expected to be continuous. Although the EELS-SI micrographs showed that the spatial distribution of Fe and Co cations in CSSa was similar to that of CS, it became more homogeneous in CSSb and CSSc. These results suggest the progressive formation of CoFe2O4 at the expense of CoO. The Fe/Co signal ratio was also higher than for CSSa, confirming the formation of an Fe3−δO4 shell, which became thicker with increasing the amount of the Fe precursor. In fact, XRD confirmed the gradual disappearance of CoO and a concomitant increase in the spinel crystal size with increasing the amount of the Fe precursor used to grow the outer shell in the CSS system. The increase in the lattice parameter was consistent with the higher Fe2+ content and the strains resulting from the presence of CoO and a high content of defects in CoFe2O4.15 Nevertheless, CoFe2O4 resulted in lower strains, as shown for CSSc, in comparison with CS, which agreed with the better epitaxial growth expected between Fe3−δO4 and CoFe2O4. The shift in the FTIR band specific for metal oxides to shorter wavelengths consistently supported a significant increase in CoFe2O4 as well as a higher Fe2+ content in the outer shell. Although the shift was significant from CSSa to CSSb, the band of CSSc was centered at a very similar wavelength (582 cm−1), suggesting the concomitant growth of CoFe2O4 (591 cm−1) and Fe3O4 (574 cm−1), with the latter compensating the former, as otherwise it would be closer to the wavelength of pure magnetite (574 cm−1).
Although these results suggest the growth of CoFe2O4, it could not be clearly discriminated from Fe3−δO4 and CoO. Mössbauer spectrometry evidenced unequivocally the significant increase in CoFe2O4 at the expense of CoO in CSSa by the reduced mean isomer shift in comparison to CS. In contrast, the higher value calculated for CSSb indicated the formation of an Fe3−δO4 shell with a significant Fe2+ content. In CSSb, the formation of Fe3−δO4 predominated over that of CoFe2O4. The I1/I2 and S ratios calculated from the XAS and XMCD spectra showed the same evolution of Fe2+vs. Fe3+ as for the Mössbauer data, except for CSSc; whereby XAS and XMCD showed that the Fe2+ content was higher in CSSc than CSSb, while the Mössbauer results showed the opposite. This discrepancy can be explained by the higher surface sensitivity of soft X-rays, with 70% of the XAS and XMCD signal coming from a 2 nm surface layer (thereby, Fe2+ at the CSSc surface would be overestimated). The lower mean isomer shift in CSSc suggested a higher amount of Fe2+-deficient phases. According to the XAS and XMCD spectra, the increase in both the I1/I2 and S ratios rules out the oxidation of Fe2+ as a possible cause of the reduced mean isomer shift of CSSc. In CSSc, the replacement of CoO by CoFe2O4 predominated over the growth of the Fe3−δO4 outer shell. This result was confirmed by the M−O bands (FTIR spectra) of CSSc, which mostly overlapped with that of CSSb, with the increase in the Fe:Co ratio measured by EDX and the relatively homogeneous distribution of Co and Fe as shown by the EELS-SI micrographs. It is worth noting that in comparison to Fe3−δO4 nanoparticles, the surfaces of CSSb and CSSc seemed to contain a relatively high amount of Fe2+, although given the thickness of the outer shell, this should be mostly oxidized. The presence of CoFe2O4 in all CSS nanoparticles was also supported by their mean Bhf fields being significantly higher (about 51 T) than that of Fe3−δO4 nanoparticles (about 47 T). Finally, the XMCD spectra recorded at the Co L-edge unambiguously demonstrated the presence of CoFe2O4, which increased gradually up to 68% in CSSc at the expense of CoO. Although most of the CoO was converted into CoFe2O4 in CSSc, the CoO fraction still remained significant (32%), and has been shown to impact the magnetic properties of particles, even at room temperature.
As we reported elsewhere,23 the formation of CoO in the second thermal decomposition step favored the diffusion of Co cations into the vacancies at the surface of the Fe3−δO4 nanoparticles, i.e., the formation of an interlayer of Co-doped ferrite at the Fe3−δO4 @CoO interface. Due to the diffusion process, a concentration gradient of Co2+ was expected from the Fe3O4/CoO interface. The gradual increase in the CoFe2O4 fraction in CSS was directly related to the experimental conditions upon performing the third thermal decomposition process. The CoO shell is highly unstable in liquid medium at such high temperatures.24 Therefore, we expected that the CoFe2O4 phase in CSS nanoparticles resulted from the partial solubilization of the CoO shell during the third decomposition step, which was followed by the recrystallization of the solubilized Co species with the new Fe monomers (resulting from the decomposition of the Fe precursor). Here, CoO acted as a reservoir of Co monomers, enabling the growth of a CoFe2O4 shell before that of Fe3O4. This was clearer for low amounts of Fe precursor, e.g., CSSa. However, for larger amounts of Fe precursor, the increasing volume fraction of CoFe2O4 could not be caused solely by the solubilization of CoO, as this would imply that increasing the Fe monomers in the reaction medium enhanced the solubilization of CoO. Therefore, we expect that above a critical amount of Fe precursor, the Fe3−δO4 shell grows at the surface of nanoparticles. Considering the high temperatures involved (about 300 °C), Co2+ could be expected to diffuse at the CoO/Fe3−δO4 interface in order to partially replace CoO by CoFe2O4. In the case of CSSb and CSSc, the Fe precursor amount seemed to be sufficiently high to result in an Fe3−δO4 shell.
By combining the Mössbauer and XAS/XMCD results, we calculated the relative volume fraction of each phase in each nanoparticle (Table 5). To simplify our model, we considered that all the vacancies in Fe3−δO4 are filled by Co2+, resulting in a stable CoFe2O4 intermediate layer, given the self-limitation of cationic diffusion (Table 1). Considering the structure of CSS nanoparticles, we expect that the Fe3−δO4@CoFe2O4 structure would not be affected by the third thermal decomposition step, in contrast with the CoO shell.24 The CoO content decreased gradually from 63% in CS to 26% in CSSc, while that of CoFe2O4 increased from 19% to 55%. Finally, the Fe3O4 fraction remained stable (≈18%) due to the compensation in CSSb and CSSc between the formation of Fe3−δO4 at the outer shell and that of CoFe2O4 at the inner shell/outer shell interface. In CSSa, the extremely thin outer shell could not provide such compensation, explaining the lower fraction of Fe3−δO4 (12%).
| Compound | CS | CSSa | CSSb | CSSc |
|---|---|---|---|---|
| Fe3O4 | 18 | 12 | 18 | 19 |
| CoFe2O4 | 19 | 37 | 44 | 55 |
| CoO | 63 | 51 | 38 | 26 |
The magnetic properties were explained in correlation with the chemical composition and structure of the nanoparticles. The replacement of CoO by CoFe2O4 progressively removed the limitations in exchange coupling imposed by the relatively low AFM ordering temperature of CoO (the FiM order of CoFe2O4 remained well above room temperature, up to TC = 790 K). In fact, the ultrathin islands of CoO remaining in CSSb and CSSc enabled magnetic proximity effects with the neighboring Co-ferrite, rendering a hybrid Co-based phase with a high anisotropy and ordering temperature. Hence, the magnetic properties of the CSS nanoparticles, particularly at room temperature, were no longer driven by a simple FiM/AFM exchange-bias coupling, where TN (CoO) = 290 K, but by a “soft-FiM/hard–artificial FiM/soft-FiM” dual exchange-coupling in a core@hybrid–shell@shell structure. Indeed, the progressive replacement of CoO by CoFe2O4 in CSS resulted in blocking temperatures higher than room temperature. The concurrent increase in the saturation magnetization agreed with the estimated relative fractions of Fe3−δO4, CoO, and CoFe2O4. At low temperatures, the gradual reduction of the horizontal and vertical shifts measured in the FC hysteresis loops from CS to CSSc also reflected the progressive removal of the CoO phase. Only a small fraction of “independent” or interfacial spin disordered CoO remained, as shown by the residual exchange field observed at 10 K. Although the M(H) curves measured at 300 K showed moderate coercivity, they unambiguously verified that the magnetic moments of the CSS nanoparticles were blocked at room temperature in the timescale of the SQUID measurements.
It is worth noting that in the particular case of CSSa, where the CoO phase still corresponded to half of the nanoparticle volume but there was already a 37% content of CoFe2O4, two different exchange phenomena could be resolved from the thermal dependence of the magnetization (Fig. 10b and c), namely those between the core and the CoO phase (responsible for the still large exchange bias field at low temperatures) and between the core and the Co-ferrite (or initial stages of the hybridized CoO-Co-ferrite FiM), responsible for the increase in the blocking temperature up to at least 400 K.
Finally, the final magnetic scenario can be summarized as follows: (i) exchange coupling predominates in Fe3−δO4@CoO core–shell nanoparticles, even though a CoFe2O4 layer is already present at the core–shell interface; (ii) further increase in the CoFe2O4/CoO ratio in CSSa results in both Fe3−δO4-CoO and Fe3−δO4-CoFe2O4 couplings, thus two (un)blocking processes, as resolved in the double peak in d(Mfc-Mzfc)/dT curves; (iii) the significant reduction of the CoO fraction in CSSb and CSSc favors the proximity effects between CoO and CoFe2O4, leading us to consider the coupling between the Fe3−δO4 core and a hybrid Co shell; the latter combining both high anisotropy and ordering temperature, where CoO and Co-ferrite behave, as one as indicated by the fact that there was only one blocking process.
Conclusion
Small magnetic nanoparticles that were magnetically stable above 400 K were synthesized by a three-step synthesis growth process. Iron and cobalt precursors were alternately decomposed around 300 °C in order to form iron oxide particles first, onto which a CoO shell was successively grown, which then turned gradually into CoFe2O4 upon the further growth of a Fe3−δO4 outer shell. The thermal instability of the CoO shell during the third synthesis step led to its partial solubilization and Co diffusion at both the Fe3−δO4 core/CoO shell and CoO shell/Fe3−δO4 shell interfaces. The replacement of the AFM CoO phase by CoFe2O4 altered markedly the exchange coupling at the Fe3−δO4 core/CoO shell interface, although a significant but decreasing volume fraction of CoO remained present in the CSS series. The formation of a hard FiM CoFe2O4 inner shell resulted from the co-crystallization of the solubilized Co species and Fe monomers caused by the thermal decomposition of the Fe precursor and Co diffusion at the CoO/Fe3−δO4 interface. Two types of exchange coupling (core-AFM and core-FiM) were clearly observed (in the thermal dependence of the magnetization) in the only sample with a large content of both CoO and CoFe2O4 (CCSSa); whereas, for thinner layers of CoO (in CSSb and CSSc), a single blocking process is observed above room temperature, indicating the hybridization (magnetic proximity effects) of the two species into an artificial ferrimagnet with high anisotropy and ordering temperature, given that TC (CoFe2O4) ≫ TN (CoO). Besides growing a thicker Fe3−δO4 shell, increasing the amount of the Fe precursor accelerated the replacement of CoO by CoFe2O4. Furthermore, the concomitant formation of a CoFe2O4 inner shell and a thicker Fe3−δO4 outer shell by increasing the amount of Fe precursor resulted in dual interfacial coupling (soft core/hard shell/soft shell), preserving the coherent rotation of all the spins in the nanoparticles, thus allowing a further increase in the blocking temperature of CSSc above 400 K while increasing the saturation magnetization. This system constitutes one of the few examples of either single-phase or complex onion nanoparticles (as small as ∼15 nm) showing magnetic stability, i.e., blocked ferrimagnetism, well above room temperature. Hence, the complex structure of our iron oxide-based nanoparticles and the engineering of multiple exchange-couplings and magnetic proximity effects resulted in unprecedented magnetic stability. This strategy opens new perspectives for the design of rare-earth-free nanoparticles with tunable magnetic properties for a wide range of applications, including high density data storage, theranostics, and sensors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
K. S. was supported by a PhD grant from the French Agence Nationale de la Recherche (ANR) under the reference ANR-11-LABX-0058-NIE within the Investissement d'Avenir program ANR-10-IDEX-0002-02 and SOLEIL synchrotron/Laboratoire Léon Brillouin fellowship. The authors are grateful to SOLEIL synchrotron for providing the access to DEIMOS beamline. J. A. D. T. and R. L.-M. acknowledge support from Junta de Comunidades de Castilla-La Mancha [SPBLY/21/180501/000226] and Ministerio de Ciencia, Innovación y Universidades [PID2022-142267NB-I00]. R. L.-M. acknowledges his predoctoral research grant co-funded by the European Social Fund [2020-PREDUCLM-16730].References
- S. Massari and M. Ruberti, Resour. Policy, 2013, 38, 36 CrossRef
.
- O. Gutfleisch, M. A. Willard, E. Brück, C. H. Chen, S. G. Sankar and J. P. Liu, Adv. Mater., 2011, 23, 821 CrossRef CAS PubMed
- P. Tartaj, M. P. Morales, T. Gonzalez-Carreño, S. Veintemillas-Verdaguer and C. J. Serna, Adv. Mater., 2011, 23, 5243 CrossRef CAS PubMed
- S. Bedanta and W. Kleemann, J. Phys. D: Appl. Phys., 2008, 42, 013001 CrossRef
- J. Nogues and I. K. Schuller, J. Magn. Magn. Mater., 1999, 192, 203 CrossRef CAS
- J. Nogues, J. Sort, V. Langlais, V. Skumryev, S. Surinach, J. S. Munoz and M. D. Baro, Phys. Rep., 2005, 422, 65 CrossRef
- T. Gaudisson, R. Sayed-Hassan, N. Yaacoub, G. Franceschin, S. Nowak, J.-M. Grenèche, N. Menguy, Ph. Sainctavit and S. Ammar, CrystEngComm, 2016, 18, 3799 RSC
- W. Baaziz, B. P. Pichon, C. Lefevre, C. Ulhaq-Bouillet, J.-M. Greneche, M. Toumi, T. Mhiri and S. Begin-Colin, J. Phys. Chem. C, 2013, 117, 11436 CrossRef CAS
- A. López-Ortega, E. Lottini, G. Bertoni, C. de Julián Fernández and C. Sangregorio, Chem. Mater., 2017, 29, 1279 CrossRef
- G. C. Lavorato, E. L. Jr, D. Tobia, D. Fiorani, H. E. Troiani, R. D. Zysler and E. L. Winkler, Nanotechnology, 2014, 25, 355704 CrossRef PubMed
- R. F. L. Evans, D. Bate, R. W. Chantrell, R. Yanes and O. Chubykalo-Fesenko, Phys. Rev. B, 2011, 84, 092404 CrossRef
- F. G. Silva, J. Depeyrot, Yu. L. Raikher, V. I. Stepanov, I. S. Poperechny, R. Aquino, G. Ballon, J. Geshev, E. Dubois and R. Perzynski, Sci. Rep., 2021, 11, 5474 CrossRef CAS PubMed
- Q. Song and Z. J. Zhang, J. Am. Chem. Soc., 2012, 134, 10182 CrossRef CAS PubMed
- A. López-Ortega, M. Estrader, G. Salazar-Alvarez, A. G. Roca and J. Nogués, Phys. Rep., 2015, 553, 1 CrossRef
- M. Pauly, B. P. Pichon, P. Panissod, S. Fleutot, P. Rodriguez, M. Drillon and S. Begin-Colin, J. Mater. Chem., 2012, 22, 6343 RSC
- J. B. Tracy, D. N. Weiss, D. P. Dinega and M. G. Bawendi, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 72, 064404 CrossRef
-
C. N. R. Rao and B. Raveau, Transition Metal Oxides, Wiley-VCH, Germany, Weinheim, 2nd edn, 1995 Search PubMed
- J. A. De Toro, D. P. Marques, P. Muñiz, V. Skumryev, J. Sort, D. Givord and J. Nogués, Phys. Rev. Lett., 2015, 115, 057201 CrossRef PubMed
- B. Muzzi, M. Albino, M. Petrecca, C. Innocenti, C. de J. Fernández, G. Bertoni, C. Marquina, M. R. Ibarra and C. Sangregorio, Small, 2022, 18, 2107426 CrossRef CAS PubMed
- A. Lopez-Ortega, M. Estrader, G. Salazar-Alvarez, S. Estrade, I. V. Golosovsky, R. K. Dumas, D. J. Keavney, M. Vasilakaki, K. N. Trohidou, J. Sort, F. Peiro, S. Surinach, M. D. Baro and J. Nogues, Nanoscale, 2012, 4, 5138 RSC
- G. Salazar-Alvarez, J. Sort, A. Uheida, M. Muhammed, S. Surinach, M. D. Baro and J. Nogues, J. Mater. Chem., 2007, 17, 322 RSC
- X. Liu, B. P. Pichon, C. Ulhaq, C. Lefèvre, J.-M. Grenèche, D. Bégin and S. Bégin-Colin, Chem. Mater., 2015, 27, 4073 CrossRef CAS
- K. Sartori, G. Cotin, C. Bouillet, V. Halte, S. Begin-Colin, F. Choueikani and B. P. Pichon, Nanoscale, 2019, 11, 12946 RSC
- K. Sartori, D. Ihiawakrim, C. Lefèvre, S. Reguer, C. Mocuta, S. Bégin-Colin, F. Choueikani and B. P. Pichon, Mater. Adv., 2022, 8716–8728 RSC
- G. Salazar-Alvarez, H. Lidbaum, A. López-Ortega, M. Estrader, K. Leifer, J. Sort, S. Suriñach, M. D. Baró and J. Nogués, J. Am. Chem. Soc., 2011, 133, 16738 CrossRef CAS PubMed
- K. L. Krycka, J. A. Borchers, M. Laver, G. Salazar-Alvarez, A. Lopez-Ortega, M. Estrader, S. Surinach, M. D. Baro, J. Sort and J. Nogués, J. Appl. Phys., 2013, 113(17), 17B531 CrossRef
- V. Gavrilov-Isaac, S. Neveu, V. Dupuis, D. Taverna, A. Gloter and V. Cabuil, Small, 2015, 11, 2614 CrossRef CAS PubMed
- K. Sartori, F. Choueikani, A. Gloter, S. Begin-Colin, D. Taverna and B. P. Pichon, J. Am. Chem. Soc., 2019, 141, 9783 CrossRef CAS PubMed
- G. Cotin, C. Kiefer, F. Perton, M. Boero, B. Özdamar, A. Bouzid, G. Ori, C. Massobrio, D. Begin, B. P. Pichon, D. Mertz and S. Begin-Colin, ACS Appl. Nano Mater., 2018, 1, 4306 CrossRef CAS
- P. Ohresser, E. Otero, F. Choueikani, K. Chen, S. Stanescu, F. Deschamps, T. Moreno, F. Polack, B. Lagarde, J.-P. Daguerre, F. Marteau, F. Scheurer, L. Joly, J.-P. Kappler, B. Muller, O. Bunau and Ph. Sainctavit, Rev. Sci. Instrum., 2014, 85, 013106 CrossRef CAS PubMed
- N. Daffé, F. Choueikani, S. Neveu, M.-A. Arrio, A. Juhin, P. Ohresser, V. Dupuis and P. Sainctavit, J. Magn. Magn. Mater., 2018, 460, 243 CrossRef
-
J. Teillet and F. Varret, MOSFIT Software, Université du Maine, Le Mans, France Search PubMed
- E. Lima, E. L. Winkler, D. Tobia, H. E. Troiani, R. D. Zysler, E. Agostinelli and D. Fiorani, Chem. Mater., 2012, 24, 512 CrossRef CAS
- W. Baaziz, B. P. Pichon, Y. Liu, J.-M. Grenèche, C. Ulhaq-Bouillet, E. Terrier, N. Bergeard, V. Halté, C. Boeglin, F. Choueikani, M. Toumi, T. Mhiri and S. Begin-Colin, Chem. Mater., 2014, 26, 5063 CrossRef CAS
- A. López-Ortega, E. Lottini, C. de J. Fernández and C. Sangregorio, Chem. Mater., 2015, 27, 4048 CrossRef
- K. Sartori, A. Musat, F. Choueikani, J.-M. Grenèche, S. Hettler, P. Bencok, S. Begin-Colin, P. Steadman, R. Arenal and B. P. Pichon, ACS Appl. Mater. Interfaces, 2021, 13, 16784 CrossRef CAS PubMed
- P. M. A. de Bakker, E. De Grave, R. E. Vandenberghe and L. H. Bowen, Hyperfine Interact., 1990, 54, 493 CrossRef CAS
- T. J. Daou, G. Pourroy, S. Begin-Colin, J.-M. Greneche, C. Ulhaq-Bouillet, P. Legare, P. Bernhardt, C. Leuvrey and G. Rogez, Chem. Mater., 2006, 18, 4399 CrossRef CAS
- N. Yaacoub, H. Mortada, Z. Nehme and J.-M. Greneche, J. Nanosci. Nanotechnol., 2019, 19, 5014 CrossRef CAS PubMed
- F. L. Deepak, M. Bañobre-López, E. Carbó-Argibay, M. F. Cerqueira, Y. Piñeiro-Redondo, J. Rivas, C. M. Thompson, S. Kamali, C. Rodríguez-Abreu, K. Kovnir and Y. V. Kolen’ko, J. Phys. Chem. C, 2015, 119, 11947 CrossRef CAS
- M. Liu, M. Lu, L. Wang, S. Xu, J. Zhao and H. Li, J. Mater. Sci., 2016, 51, 5487 CrossRef CAS
- G. Franceschin, T. Gaudisson, N. Menguy, B. C. Dodrill, N. Yaacoub, J.-M. Grenèche, R. Valenzuela and S. Ammar, Part. Part. Syst. Charact., 2018, 35, 1800104 CrossRef
- E. Pellegrin, M. Hagelstein, S. Doyle, H. O. Moser, J. Fuchs, D. Vollath, S. Schuppler, M. A. James, S. S. Saxena, L. Niesen, O. Rogojanu, G. A. Sawatzky, C. Ferrero, M. Borowski, O. Tjernberg and N. B. Brookes, Phys. Status Solidi B, 1999, 215, 797 CrossRef CAS
- J. Li, N. Menguy, M.-A. Arrio, P. Sainctavit, A. Juhin, Y. Wang, H. Chen, O. Bunau, E. Otero, P. Ohresser and Y. Pan, J. R. Soc., Interface, 2016, 13, 20160355 CrossRef PubMed
- S. Brice-Profeta, M.-A. Arrio, E. Tronc, N. Menguy, I. Letard, C. Cartier dit Moulin, M. Noguès, C. Chanéac, J.-P. Jolivet and Ph. Sainctavit, J. Magn. Magn. Mater., 2005, 288, 354 CrossRef CAS
- W. Baaziz, B. P. Pichon, S. Fleutot, Y. Liu, C. Lefevre, J.-M. Greneche, M. Toumi, T. Mhiri and S. Begin-Colin, J. Phys. Chem. C, 2014, 118, 3795 CrossRef CAS
- M. W. Haverkort, arXiv, 2005, preprint, arXiv:cond-mat/0505214, DOI:10.48550/arXiv.cond-mat/0505214.
- A. G. Roca, I. V. Golosovsky, E. Winkler, A. López-Ortega, M. Estrader, R. D. Zysler, M. D. Baró and J. Nogués, Small, 2018, 14, 1703963 CrossRef PubMed
- P. Tancredi, P. C. Rivas-Rojas, O. Moscoso-Londoño, D. Muraca, M. Knobel and L. M. Socolovsky, J. Alloys Compd., 2022, 894, 162432 CrossRef CAS
- I. J. Bruvera, P. Mendoza Zélis, M. Pilar Calatayud, G. F. Goya and F. H. Sánchez, J. Appl. Phys., 2015, 118, 184304 CrossRef
- M. Pauly, B. P. Pichon, P.-A. Albouy, S. Fleutot, C. Leuvrey, M. Trassin, J.-L. Gallani and S. Begin-Colin, J. Mater. Chem., 2011, 21, 16018 RSC
- J. A. De Toro, P. S. Normile, S. S. Lee, D. Salazar, J. L. Cheong, P. Muñiz, J. M. Riveiro, M. Hillenkamp, F. Tournus, A. Tamion and P. Nordblad, J. Phys. Chem. C, 2013, 117, 10213 CrossRef CAS
- E. H. Sánchez, M. Vasilakaki, S. S. Lee, P. S. Normile, M. S. Andersson, R. Mathieu, A. López-Ortega, B. P. Pichon, D. Peddis, C. Binns, P. Nordblad, K. Trohidou, J. Nogués and J. A. De Toro, Small, 2022, 18, 2106762 CrossRef PubMed
- J. Nogues, V. Skumryev, J. Sort, S. Stoyanov and D. Givord, Phys. Rev. Lett., 2006, 97, 157203 CrossRef CAS PubMed
- P. S. Normile, J. A. De Toro, T. Muñoz, J. A. González, J. P. Andrés, P. Muñiz, R. E. Galindo and J. M. Riveiro, Phys. Rev. B, 2007, 76, 104430 CrossRef
- E. H. Sánchez, M. Vasilakaki, S. S. Lee, P. S. Normile, G. Muscas, M. Murgia, M. S. Andersson, G. Singh, R. Mathieu, P. Nordblad, P. C. Ricci, D. Peddis, K. N. Trohidou, J. Nogués and J. A. De Toro, Chem. Mater., 2020, 32, 969 CrossRef
- R. L. Antón, J. A. González, J. P. Andrés, P. S. Normile, J. Canales-Vázquez, P. Muñiz, J. M. Riveiro and J. A. De Toro, Nanomaterials, 2017, 7, 61 CrossRef PubMed
- E. Skoropata, R. D. Desautels, C.-C. Chi, H. Ouyang, J. W. Freeland and J. van Lierop, Phys. Rev. B: Condens. Matter Mater. Phys., 2014, 89, 024410 CrossRef
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3na01108a |
| This journal is © The Royal Society of Chemistry 2024 |