
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhotoluminescence quantum yield of carbon dots: emission due to multiple centers versus excitonic emission†
Oleg
Dimitriev
 abc,
Dmytro
Kysil
*a,
Alexander
Zaderko
d,
Oksana
Isaieva
ae,
Andrii
Vasin
af,
Yuri
Piryatinski
g,
Mats
Fahlman
bc and
Alexei
Nazarov
af
abc,
Dmytro
Kysil
*a,
Alexander
Zaderko
d,
Oksana
Isaieva
ae,
Andrii
Vasin
af,
Yuri
Piryatinski
g,
Mats
Fahlman
bc and
Alexei
Nazarov
af
aV. Lashkaryov Institute of Semiconductor Physics, NAS of Ukraine, Pr. Nauky 41, Kyiv 03028, Ukraine. E-mail: kdmitr93@gmail.com
bLaboratory of Organic Electronics, Linköping University, Norrköping 60174, Sweden
cWallenberg Wood Science Center, Laboratory of Organic Electronics, Linköping University, Norrköping 60174, Sweden
dInstitute of High Technologies, Taras Shevchenko National University, Kyiv 01033, Ukraine
eNational University “Kyiv-Mohyla Academy”, Skovorody, 2, Kyiv 04070, Ukraine
fNational Technical University “Igor Sikorsky Kyiv Polytechnic Institute”, 37, Peremohy Ave., Kyiv 03056, Ukraine
gInstitute of Physics, NAS of Ukraine, Pr. Nauki 46, Kyiv 03028, Ukraine
First published on 27th March 2024
Abstract
Carbon dots (CDs) are recognized as promising fluorescent nanomaterials with bright emission and large variations of photoluminescence quantum yield (PLQY). However, there is still no unique approach for explanation of mechanisms and recipes for synthetic procedures/chemical composition of CDs responsible for the enhancement of PLQY. Here, we compare photophysical behavior and PLQY of two types of CDs synthesized by different routes, leading to the different extent of oxidation and composition. The first type of CDs represents a conjugated carbon system oxidized by F, N and O heteroatoms, whereas the second type represents a non-conjugated carbon system oxidized by oxygen. Photophysical data, photoemission spectroscopy and microscopy data yield the suggestion that in the first case, a structure with a distinct carbon core and highly oxidized electron-accepting shell is formed. This leads to the excitonic type non-tunable emission with single-exponent decay and high PLQY with a strong dependence on the solvent polarity, being as high as 93% in dioxane and as low as 30% in aqueous medium, but which is vulnerable to photobleaching. In the second case, the oxidized CDs do not indicate a clear core–shell structure and show poor solvatochromism, negligible photobleaching, low PLQY varying in the range of 0.7–2.3% depending on the solvent used, and tunable emission with multi-exponent decay, which can be described by the model of multiple emission centers acting through a clustering-triggered emission mechanism. The obtained results lead to a strategy that allows one to design carbon nanomaterials with principally different PLQYs that differ by orders of magnitude.
1. Introduction
Carbon dots (CDs) are recognized as promising fluorescent nanomaterials that are relatively stable and possess tuned photoluminescence (PL), excellent biocompatibility, nontoxic properties and low cost of production. These advantages of CDs enable their use in various applications such as light-emitting diodes, bio-imaging, solar cells, sensor probes, photo-catalysis, phototherapy, information encryption, etc.1–6 Photoluminescence quantum yield (PLQY) of CDs is an important parameter in the above applications, which can vary largely. The PLQY is particularly dependent on the synthetic procedures/chemical composition of the CDs, yielding values as low as less than one percent to more than 90%.7 A record PLQY of 94.5% has been obtained for CDs in water,8 but there is still no unique approach for explaining the mechanisms leading to and responsible for the enhancement of PLQY in CDs as there are many internal and external factors that influence their light emission. The internal factors include conjugation effects, surface states, degree of surface oxidation, presence of surface functional groups and doping atoms, structural defects, etc., and the external ones include environmental conditions such as solvent medium, temperature, pressure effects, and also molecular states due to molecules physicochemically adsorbed from solution on the CD surface.9By exploring factors that lead to the enhancement of PLQY in CDs, a solid understanding of the intrinsic mechanisms of CD light emission needs to be established. At least three different models have been proposed for description of the PL mechanism in CDs due to independent chromophores present in the particle core or particle surface.10 First, a bandgap transition model that assumes that CDs are quantum dots that experience a quantum confinement effect of conjugated π-domains due to graphene fragments constituting the CD core and yielding emission wavelengths dependent on the CD core confinement. This model is supported by experiments performed by Yuan et al. who demonstrated size-dependent blue-to-red emission shift in N-doped CDs with sizes between 1.95 and 6.68 nm.11 Second, a surface state emission model where surface oxidation, doping, or other surface defects that create traps are responsible for radiative recombination of excited electron–hole pairs. The surface states were proposed to arise not due to a single chemical group on the surface but as a result of synergetic hybridization of the surface chemical groups and the carbon core, e.g. attributed to the special molecular conformations of carboxyl-based groups and several carbon atoms on the edge of the carbon backbone.12 This model is supported by observation of a dual-band emission in CDs, where the high-energy band is attributed to the core and the low-energy band to surface state emission.13 Third, the molecular state model that forms a PL center solely due to some organic fluorophores which are located either on the surface or in the interior of the carbon core and which can provide independent PL emissions due to the molecular surface state or the carbon core state. Emission of the surface molecular state usually obeys the Vavilov rule, whereas the carbon core state emission is excitation dependent since it represents superposition of different chromophores that emit incoherently. Separation of the molecular surface and carbon core states can be performed, for example, by applying high temperatures that “kill” surface chromophores.14
Less often description of the PL mechanism in CDs is presented due to collective excitation of chromophores constituting the CDs (Fig. 1). On one hand, resonance interaction of different chromophores can result in formation of excitons, self-trapped excitons, or excimers, which have broad and structureless emission.15 On the other hand, cross-linking of non-conjugated carbon units containing heteroatoms can result in crosslink enhanced emission or cluster-triggering emission mechanism observed in the visible.16,17 The latter mechanisms are less specific with respect to chromophore requirements which can be present in CDs and therefore are more universal.
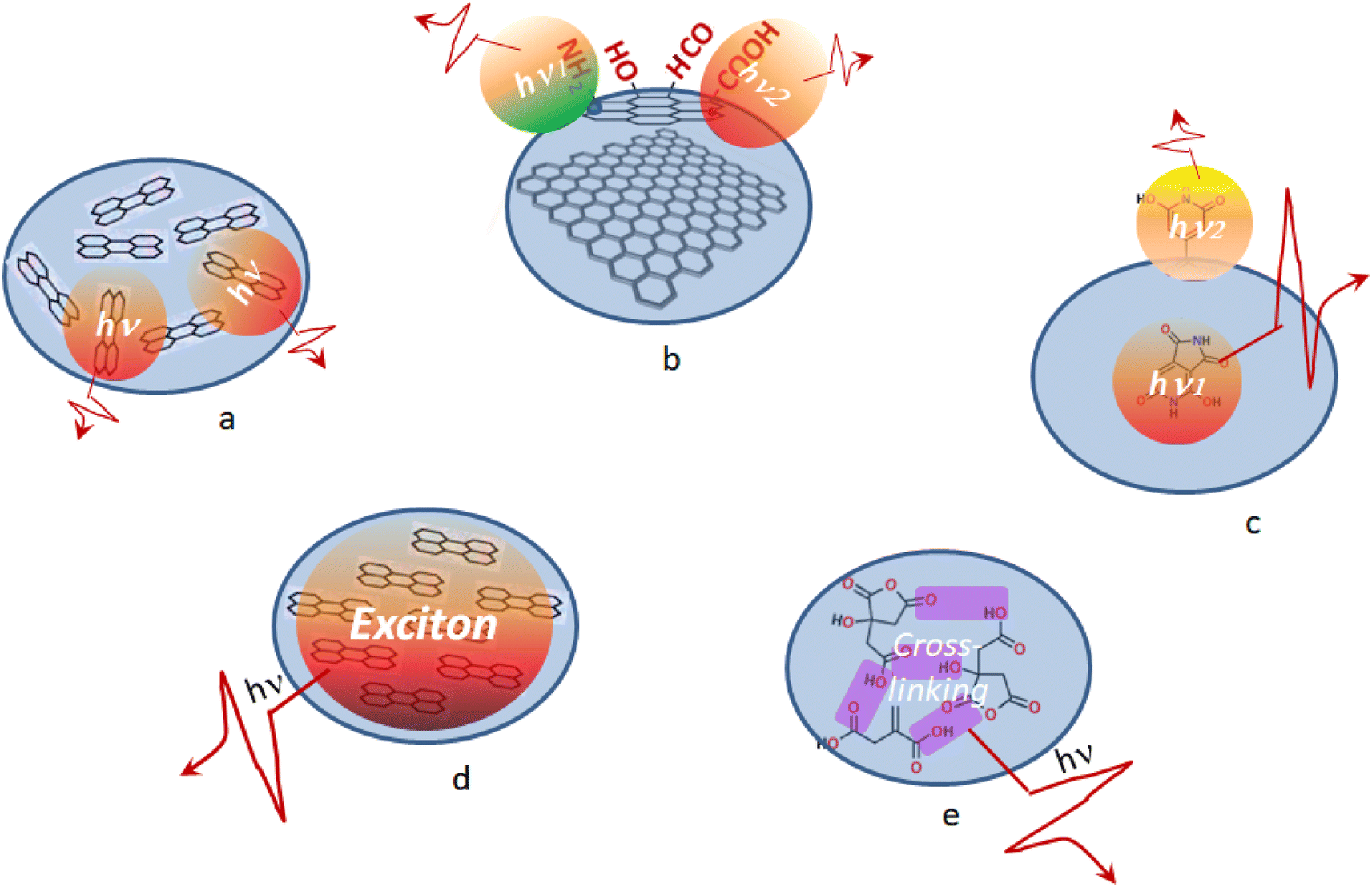 | ||
| Fig. 1 Schematic illustration of PL emission mechanisms in CDs as a result of (a–c) individual and (d and e) collective excitation of chromophores: (a) bandgap transition emission; (b) surface state emission; (c) molecular state emission; (d) excitonic emission; (e) crosslink enhanced emission. | ||
However, the lack of a unique model able to describe the light emission mechanism of CDs can be related to the large variety of structural units and composition of CDs. For example, several recent studies demonstrated that heterogeneity of surface states and charge traps,13 or a cocktail of different fluorophores coexisting in a given CD sample18,19 can be responsible for the fluorescence tunability of CDs depending on the excitation wavelength. On the other hand, the significant role of molecular units responsible for the fluorescence of CDs has been established as well.20
In this work, we compare photophysical properties of CDs synthesized by different methods that result in different compositions and structures of the CDs, namely, CDs with conjugated carbon moieties in the CD core and a highly oxidized shell containing fluorine, nitrogen and oxygen heteroatoms, on one hand, and moderately but comprehensively oxidized CDs by oxygen with no conjugated carbon units. These two types of CDs show different photophysical behavior expressed through the different light emission mechanisms. The first type of CDs features the excitonic type non-tunable emission with single-exponent decay, remarkable solvatochromism, significant photoinduced bleaching, and yields a high PLQY with a strong dependence on the solvent polarity: as high as 93% in dioxane and as low as 30% in aqueous medium. The second type of CDs shows near negligible solvatochromism and photobleaching, low PLQY between 0.7 and 2.3%, and a tunable light emission with multi-exponent decay, best described by the model of multiple emission centers. Thus, our findings provide ways for understanding how to link the CD structure to light emission mechanisms and PLQY.
2. Experimental
2.1. Synthesis of CDs
CD dispersions were prepared by two different methods, resulting in two significantly different types of CDs. Fluoralkylated CDs (Fluocar® Nano,21 referred to as CD1), containing F, N, and O heteroatoms, were synthesized via a solvothermal method described elsewhere.21,22 In brief, a mixture of urea (Pro Analysis, 5 g), citric acid (Anh. Pharm., 8 g), and m-trifluoromethyl aniline (Merck, Pro Synth., 3.35 g) was autoclaved at 135 °C for 2 hours, with the working temperature reached in about 15 minutes. As a result of the heat treatment, a dark yellow product was formed, which showed a faintly blue emission under UV illumination. After cooling down the reactor to 90 °C the heating cycle was repeated at 165 °C for 2 hours with the working temperature reached in about 10 minutes. After first 30 minutes of heating, the mixture increased in density and began to emit smoke. After another 90 minutes, the mixture visibly charred. At the end of the process, the mixture turned brown-black. The obtained product was cooled down, dissolved in an ethanol–water mixture and filtered through a 0.45 μm nylon acrodisk.Synthesis of CDs by thermal decomposition of sucrose (referred to as CD2) was performed following the procedure described in our previous work.23 The advantage of this method is the use of a solvent with a high boiling point, i.e., dimethyl sulfoxide (DMSO, Tb = 189 °C), which allow us to avoid autoclaving. In brief, a solution of sucrose (C12H22O11) in DMSO with a sucrose concentration of 200 mmol l−1 was thermally treated in an open quartz vessel on a laboratory furnace with a ceramic coating at a temperature of 170–180 °C for 5 minutes. The volume of the initial solution (before the heat treatment procedure) was about 7 ml. The heating time of the solution to a temperature of 170 °C was about 10 minutes. The cooling time of the solution to a temperature of 35 °C was about 12 minutes. The temperature of the solution was controlled using a Benetech GM333A pyrometer.
2.2. Measurements
Dioxane (Diox), isopropyl alcohol (IPA), methanol (MeOH), and dimethyl sulphoxide (DMSO) were obtained from Sigma-Aldrich. CD dispersions for the measurements were prepared by adding a small amount of the CD stock solution to a cuvette with the corresponding solvent. Absorption and photoluminescence (PL) spectra of the samples were recorded using an AvaSpec-2048 spectrophotometer. Solutions were measured using a 10 mm quartz cuvette; pure solvent was used as a reference. For PL emission, two excitation sources were used. For CW excitation, a solid-state diode laser operating at a wavelength of 405 nm with a power of 50 mW was used as an excitation source. For impulse excitation, a Xe lamp with a fixed pulse duration of 5 μs but a controlled pulse frequency per second (10 to 100 Hz) was used. The lamp was additionally equipped with a 405 nm interference filter. Solutions were diluted to minimize the reabsorption effect. The emission light from the sample was collected into a 600 μm fiber mounted at a right angle to the excitation light beam and was registered using a Peltier-cooled CCD detector. The relative quantum yield of the PL emission ΦS was determined by comparison with the fluorescence standard with the known quantum yield, i.e., 0.1 M NaOH aqueous solution of fluorescein (Φref = 79%),24 and calculated according to eqn (1):25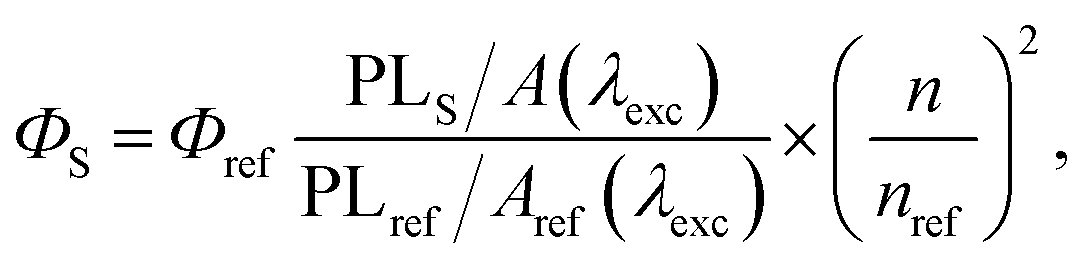 | (1) |
Light emission lifetimes were measured in a single photon counting regime using a Life Spec-II spectrometer (Edinburgh Instruments Ltd.) with ≈100 ps time resolution. A picosecond pulsed diode laser with a specific wavelength of 405 nm was used as an excitation source. The pulse repetition rate was up to 20 MHz and the pulse width was 50 ps. Before and after each fluorescence lifetime measurement, the instrument response function (IRF) was measured using scattered laser light from a diluted suspension of microparticles. The width of the resulting IRF was determined to be ∼200 ps (FWHM). The measured sample light emission was detected with a delayed gate pulse, and then the signal was dispersed in a monochromator and focused onto a single photon avalanche photodiode (iD Quantique). Measurements were performed at room temperature under ambient conditions.
Transmission electron microscopy (TEM) images of the samples deposited on a microgrid were obtained using an FEI Titan3 60-300 instrument with image and probe Cs correctors and a monochromated high brightness XFEG gun with an acceleration voltage of 300 kV.
Photoelectron emission (XPS and UPS) spectra were measured using the home-built Moses photoelectron spectrometer at a base pressure of 10−10 mbar. Monochromatic Al (Kα) X-rays at hν = 1486.6 eV were used for XPS and non-monochromatized He I radiation at hν = 21.22 eV for UPS, respectively. The binding energies were calibrated using the Fermi edge and 4f7/2 peak of gold at 0 and 84.0 eV, respectively. Additionally, the work function WF was determined from the UPS measurements as the difference between the excitation energy hν (21.22 eV) and the spectrum edge of the secondary electrons Ecutoff, WF = hν − Ecutoff. For XPS/UPS studies, the CD samples were cast from solution onto conductive ITO substrates.
3. Results
3.1. Morphology and composition of the samples
TEM studies revealed that the samples have different average sizes of the nanoparticles depending on the synthesis route, being about 4–5 nm for CD1 and ∼1 nm for CD2 (Fig. 2a and b). The diffraction patterns obtained by fast Fourier transform (FFT) of the samples (see insets in Fig. 2a and b) lead to the conclusion that both samples are amorphous, albeit the CD1 sample indicated slightly different features, which can be related to the presence of graphene fragments in the particle core.26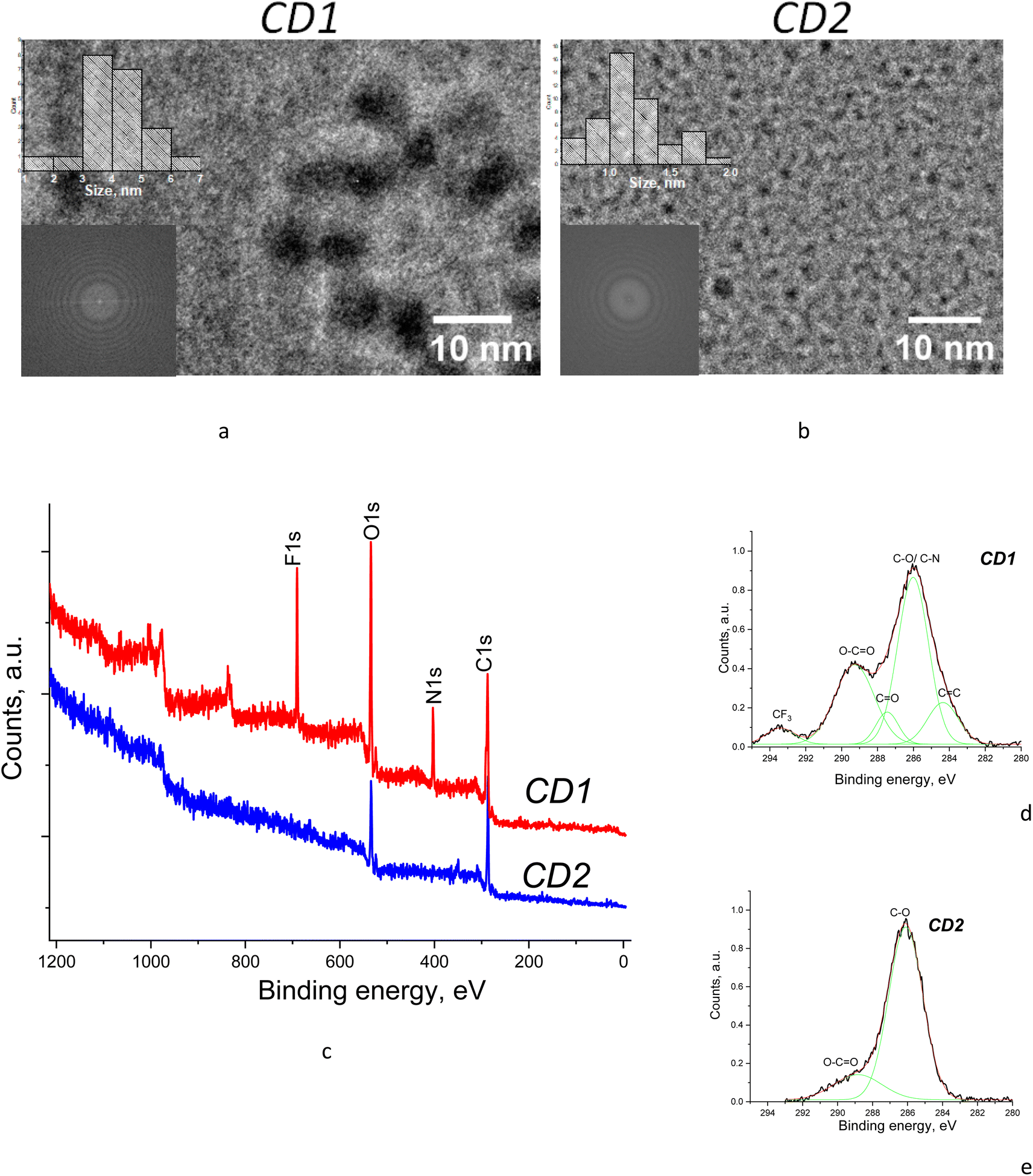 | ||
| Fig. 2 (a and b) TEM images and (c–e) XPS spectra of the samples with (d and e) high-resolution C 1s core level spectra fitted with Gaussian components. | ||
XPS measurements were performed to determine the composition of the samples depending on their synthesis routes. The CD2 XPS survey spectrum features the contribution of only two components, i.e., carbon and oxygen, whereas the CD1 XPS survey spectrum reveals the presence of fluorine and nitrogen in addition to carbon and oxygen (Fig. 2c). Detailed analysis of C 1s core level spectra for both CD1 and CD2 shows a significant contribution from oxidized carbon in different forms, i.e., C–O, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, and O–CO, which can be identified due to specific peak positions in the spectrum,27,28 whereas the presence of a graphene/graphitic component (due to CC peak) is found only in the CD1 sample (Fig. 2d and e). Moreover, the oxidation level of the samples is different. While the CD2 sample shows the dominant C–O peak, usually attributed to ethers and peroxides, the CD1 sample shows additionally developed peaks corresponding to the CO and O–CO oxides which are characteristic of carbonyl groups (Fig. 2c–e). This is consistent with the ratio of oxygen to carbon present in the samples as can be seen from the survey XPS spectra (Fig. 2c). Namely, the ratio of intensities of the O 1s to the C 1s peak is about 2.0 in CD1, whereas it is only 0.9 in the CD2 samples. In addition, the CD1 sample is oxidized due to the presence of fluorine which is a strong electron acceptor.
O, and O–CO, which can be identified due to specific peak positions in the spectrum,27,28 whereas the presence of a graphene/graphitic component (due to CC peak) is found only in the CD1 sample (Fig. 2d and e). Moreover, the oxidation level of the samples is different. While the CD2 sample shows the dominant C–O peak, usually attributed to ethers and peroxides, the CD1 sample shows additionally developed peaks corresponding to the CO and O–CO oxides which are characteristic of carbonyl groups (Fig. 2c–e). This is consistent with the ratio of oxygen to carbon present in the samples as can be seen from the survey XPS spectra (Fig. 2c). Namely, the ratio of intensities of the O 1s to the C 1s peak is about 2.0 in CD1, whereas it is only 0.9 in the CD2 samples. In addition, the CD1 sample is oxidized due to the presence of fluorine which is a strong electron acceptor.
The above details of the component distribution in XPS spectra allow us to make certain suggestions about structural features of the samples. Namely, the presence of the significant features corresponding to CC and carbon oxides in CD1 evidences that the sample composition includes both pure carbon with conjugated bonds and oxidized carbon, which is consistent with the core–shell structure with a carbon core, probably in the form of graphene nanoflakes, and the highly oxidized shell owing to the presence of specific groups such as C–O, CO, O–CO and C–F. The CD2 samples, on the other hand, do not reveal a sizeable contribution from CC but only the features related to carbon oxides (Fig. 2e). Therefore, no distinct core–shell structure can be suggested for CD2 as both the particle surface and the particle core are oxidized. The absence of the carbon core in CD2 is consistent with the previous reports that the carbogenic core starts forming at higher pyrolysis temperatures (>180 °C), while at low pyrolysis temperatures formation of molecular fluorophores predominates.29
3.2. Specific photophysical behavior of CDs
Synthesis conditions created a great impact on photophysical behavior of the CD dispersions. Specifically, the absorption maximum of CD2 in DMSO is located at ∼365 nm, while the same absorption feature appears only as a shoulder in the spectrum of CD1 where the absorption maximum is found at 415 nm (Fig. 3a). The absorption feature at 365 nm is assigned to n–π* transitions related to CO moieties located in the core15 and the feature at 415 nm to the electronic transition from the valence band of the carbon core to the manifold of midgap (π*) empty states localized on the particle surface containing carboxylic or amide groups,30 respectively. Separation of the absorption features in CD1 into those relevant to the core and the surface and the lack of such separation in CD2 support the conclusion from the XPS and TEM results that CD1 particles possess a core–shell structure, whereas in CD2 the whole particle itself is oxidized with no distinctly oxidized shell formed.
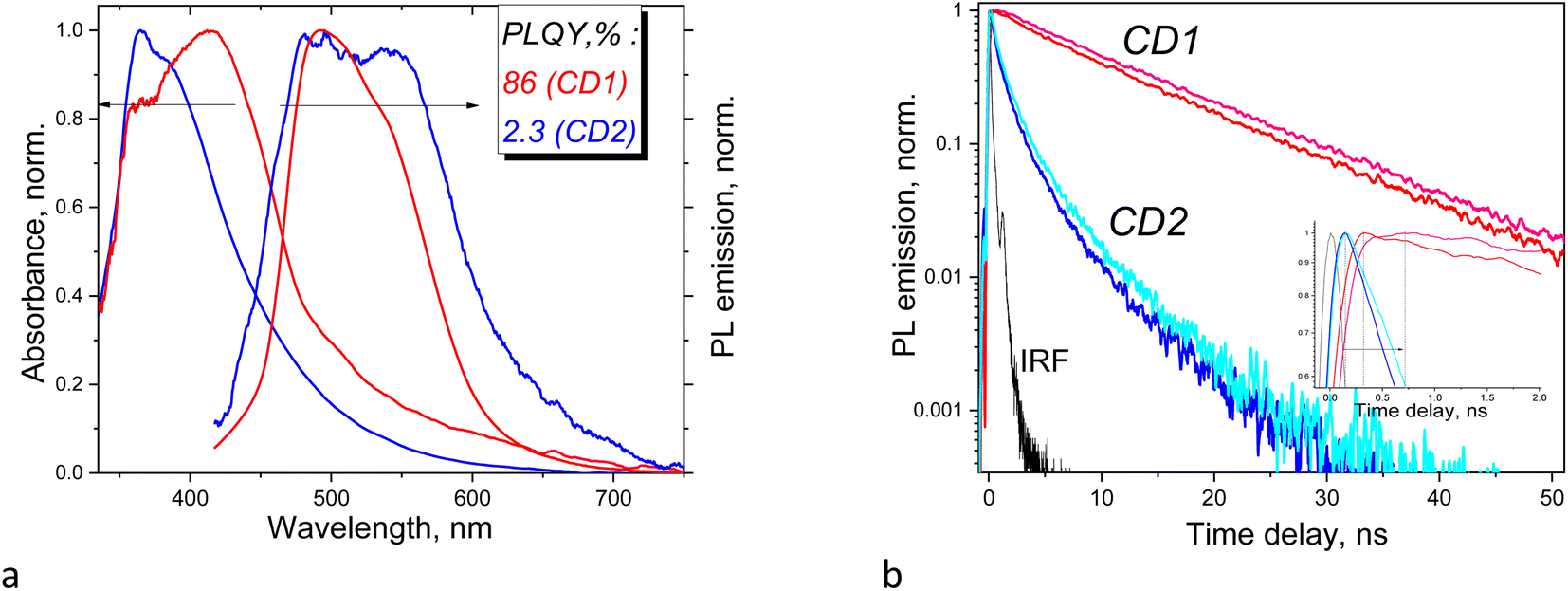 | ||
| Fig. 3 (a) Normalized absorption and PL emission spectra of DMSO dispersions of CD1 (red), CD2 (blue), and (b) corresponding decay kinetics of PL emission of the samples measured at 480 nm (blue and red) and 525 nm (light-blue and pink) in IPA; inset shows kinetics during the first 2 ns. | ||
The PL emission profiles of the samples also reveal significant differences. The PL spectrum of CD1 has an emission maximum at 493 nm followed by a shoulder at ∼540 nm. The PL emission spectrum of CD2 contains two features of approximately equal intensity located at 480 and 540 nm (Fig. 3a). The Stokes shift is 115 nm (6564 cm−1) and 78 nm (3812 cm−1) for CD2 and CD1, respectively. The PL emission kinetics of the samples also indicates different behavior, featuring single exponent (CD1) and multi-exponent (CD2) emission decay (Fig. 3b), respectively, suggesting that in the latter case, the sample combines two or more light emitters, which are affected by each other. The fastest emission decay is found for the CD2 sample, with a time constant as small as 540 ps, whereas CD1 shows time constants about one order of magnitude larger (Table 1). Moreover, the kinetic curves of CD1 show a rise during the first ∼0.5 ns after the laser excitation, indicating gradual population of the emission centers, whereas CD2 features an immediate decay from the very beginning (inset in Fig. 3b). Monitoring the emission at two different wavelengths allows us to conclude that the above particular behavior originates from solvent relaxation, i.e., it is accompanied by reorientation of molecular dipoles of the environment with respect to the emitter dipole which depends both on the polarity of the CD surface and the solvent polarity so that the emission occurs from a relaxed sub-state of the CD that becomes populated as a result of and depending on the extent of the solvent relaxation.31 When the excited state lifetime of the emitter is long enough and longer than the solvent relaxation time then the light emission maximum experiences a redshift as a function of time; therefore, monitoring the PL emission at a longer wavelength will result in a more delayed maximum formation, which is observed in our experiment (inset in Fig. 3b). The PL maximum is observed at a ∼0.3 ns delay when monitoring at 480 nm, and at a ∼0.7 ns delay when monitoring at 525 nm (Fig. 3b). It should be noted that the above sub-nanosecond formation period of the light emission maximum corresponds to the slow component of the solvent dipolar reorientation time constant which also lies in the sub-nanosecond range as reported by Khan et al.31 and by Cushing et al.,32 while the lifetime of the excited state of CD1 is one order of magnitude longer. The much longer excitation lifetime of CD1 means that light emission in this sample occurs from the lowest sub-state when the solvent relaxation processes have finished. The lifetime of the excited state of CD2 is much shorter and therefore CD2 does not demonstrate delay of the maximum light emission after excitation.
| Composition | Registration wavelength | τ 1 (ns) | A 1 (%) | τ 2 (ns) | A 2 (%) | τ ave (ns) |
|---|---|---|---|---|---|---|
| CD1 | 480 nm | 10.97 | 100 | — | — | 10.97 |
| 525 nm | 11.91 | 100 | — | — | 11.91 | |
| CD2 | 480 nm | 0.54 | 68 | 2.51 | 32 | 1.17 |
| 525 nm | 0.75 | 70 | 3.06 | 30 | 1.44 |
PLQYs of CDs estimated relative to the reference sample with a known quantum yield are found to be 86% and 2.3% for CD1 and CD2 in DMSO dispersions, respectively. The large difference in PLQY of the samples correlates well with their different composition, structure, and photophysical behavior described above. The relatively low PLQY of CD2 is associated with the distribution of oxygen and oxidized carbon throughout the CD structure and also with the relatively short emission time constants with multi-exponent decay. The latter can be due to significant contribution of nonradiative recombination processes originating from excitation quenching of interacting chromophores. The impressively high PLQY of CD1 is associated with long excitation lifetimes and single-exponent decay and the formation of a well-defined carbon core and a strongly oxidized shell. It was reported that specific surface passivation of CDs by nitrogen, fluorine, etc., leads to increased PLQY.33–38 Therefore, a possible mechanism of PLQY enhancement can be associated here with the presence of strong electron accepting groups at the surface of CD1, such as O–CO, CN, and CF3. A mechanism was suggested by Xu et al. where an increase of a strong electron-withdrawing CN group content on the surface of CDs produces push and pull electrons that improve intramolecular charge transfer efficiency leading to PLQY enhancement by almost a factor of five.39
3.3. Effect of solvent polarity on the emission of CDs
Both the shape and intensity of the PL emission of CDs are dependent on the solvent used but to different extents. For CD1, the relative quantum yield of light emission changes by about three times between aqueous and dioxane dispersions. On the other hand, for CD2, the PL intensity significantly drops only for the aqueous dispersion, while the emission shape almost does not change at all (Fig. 4). This suggests different mechanisms for light emission between CD1 and CD2 as well.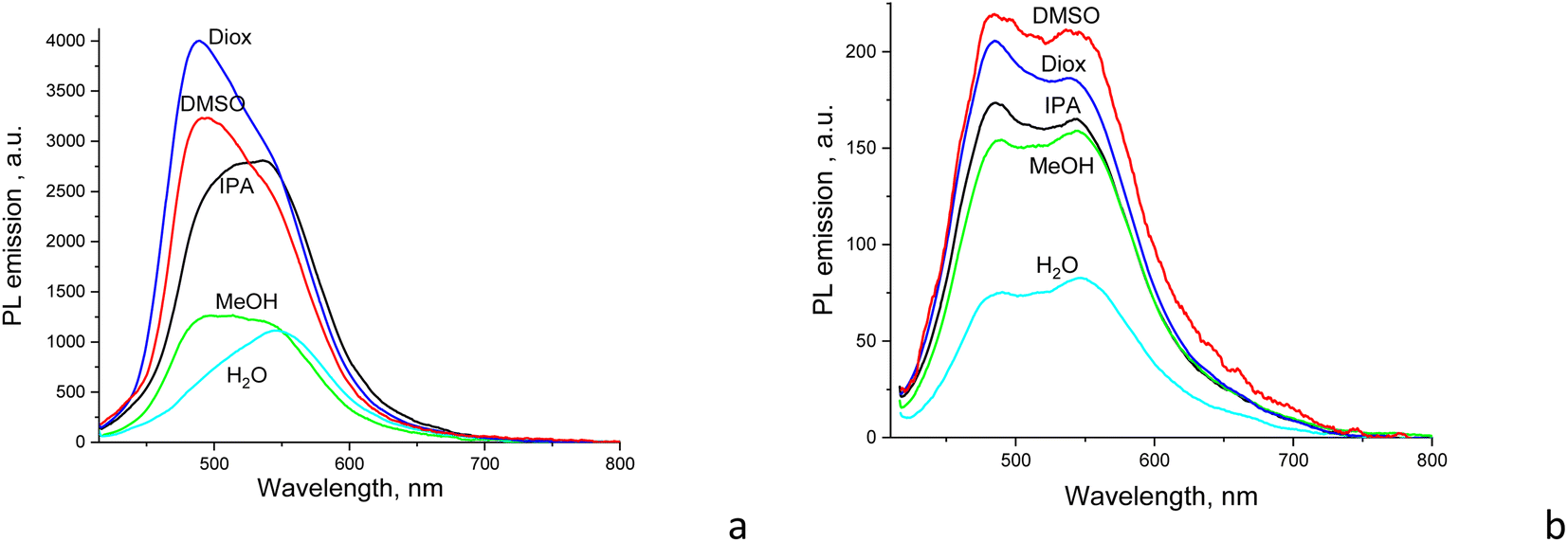 | ||
| Fig. 4 PL emission of (a) CD1 and (b) CD2 dispersions in different solvents used (λexc = 405 nm). | ||
The CD1 and CD2 dispersions both show a bright blue-green emission that can be separated into at least two components, with the first component near 480 nm for CD2 and 500 nm for CD1, respectively, with a second emission component in the form of either a clear peak or a shoulder at ∼525–540 nm (Fig. 3a and 4). This indicates the presence of at least two emission centers in both CD1 and CD2, where the spectral profiles can be fitted with two or more Gaussians (Fig. 5 and S2†). For CDs with dual emission, it was suggested that carbon-core states largely contribute to the short-wavelength blue emission. This emission originates from radiative recombination of surface electrons in surface energy traps (attributed to various single and double bonded oxygen functional groups on the surface of the carbon dots) and holes located in the carbon core. The green PL feature can be assigned to emission from intrinsic molecular states (double bonded oxygen functional groups (CO and COOH) on the surface of carbon dots).40–43
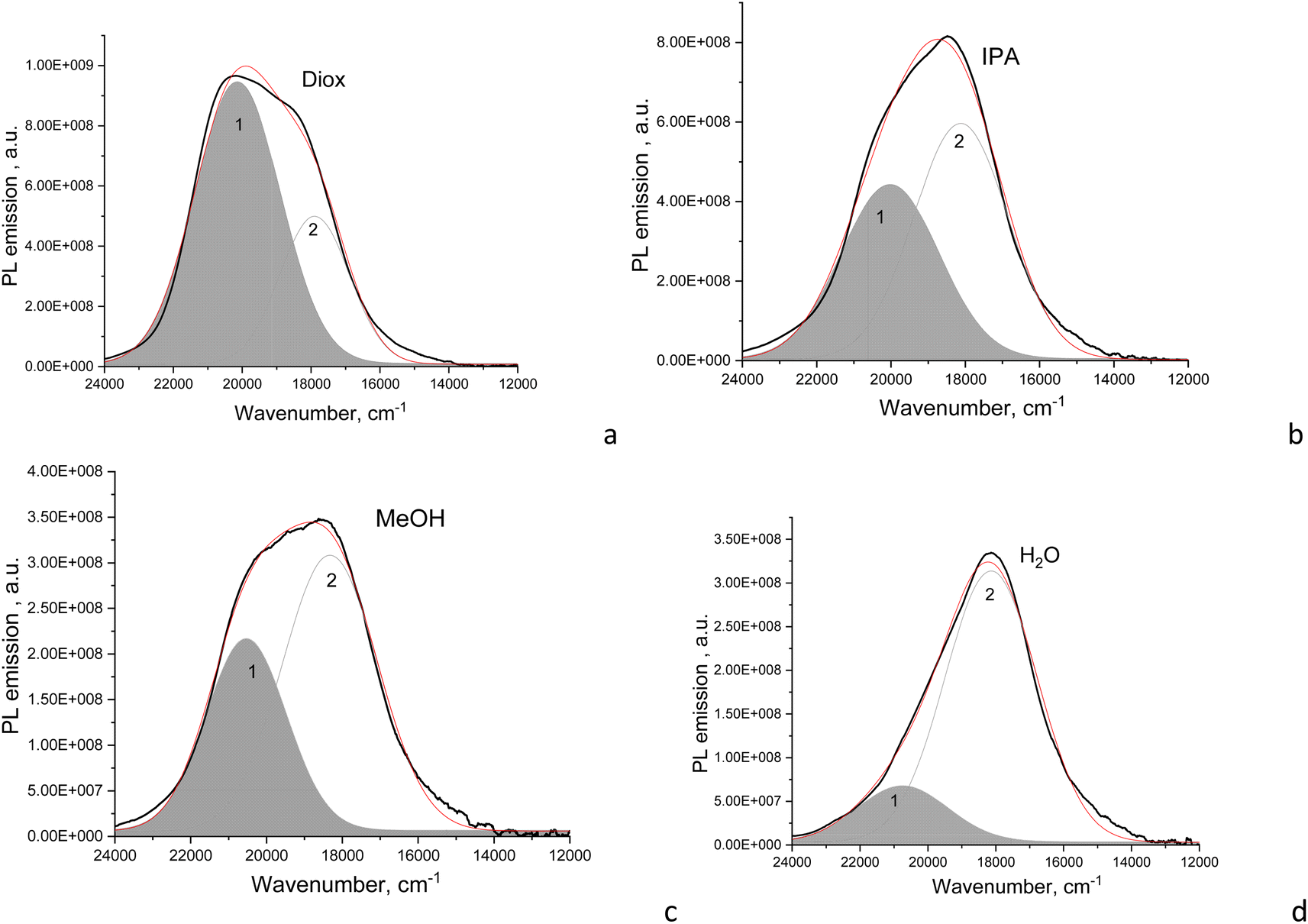 | ||
| Fig. 5 PL emission of CD1 dispersions (black curves) in (a) dioxane, (b) IPA, (c) methanol, and (d) water, and deconvolution of the PL bands into Gaussians (grey curves, the red one is their superposition). | ||
The fitted Gaussian emission components are dependent differently on the solvent polarity for the CD2 and CD1 samples (Fig. 5 and S2†). In CD1, when the solvent polarity increases from dioxane to water, the contribution of the high-energy PL component gradually decreases from the maximum (70%) in dioxane to the minimum (17%) in the aqueous medium (Fig. 5 and Table 2). Just this component mostly determines the strong dependence of PLQY on solvent polarity, while the low-energy emission component almost does not change (Fig. 6a). However, the latter mainly contributes to PLQY of CD1 in highly polar solvents (Fig. 6a). In contrast, PLQY of CD2 indicates only a slight dependence on solvent polarity, with the PL band shape being practically unchanged and the relative contributions of the two PL emission components varying only slightly with the solvent used, sharing a contribution of about 45% and 55%, respectively (Table 2).
| Diox | DMSO | IPA | MeOH | H2O | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Peak 1 | Peak 2 | Peak 1 | Peak 2 | Peak 1 | Peak 2 | Peak 1 | Peak 2 | Peak 1 | Peak 2 | |
| CD1 | ||||||||||
| Maximum position, cm−1 | 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 130 130 |
17896 |
20070 |
17971 |
20010 |
18126 |
20520 |
18325 |
20738 |
18140 |
| Contribution ratio | 70% | 30% | 67% | 33% | 44% | 56% | 37% | 63% | 17% | 83% |
| Relative PLQY, % | 65 | 28 | 58 | 28 | 35 | 45 | 16 | 27 | 5 | 25 |
|
||||||||||
| CD2 | ||||||||||
| Maximum position, cm−1 | 20600 |
17867 |
20600 |
17845 |
20585 |
17852 |
20539 |
17873 |
20375 |
17752 |
| Contribution ratio | 48% | 52% | 42% | 58% | 46% | 54% | 41% | 59% | 45% | 55% |
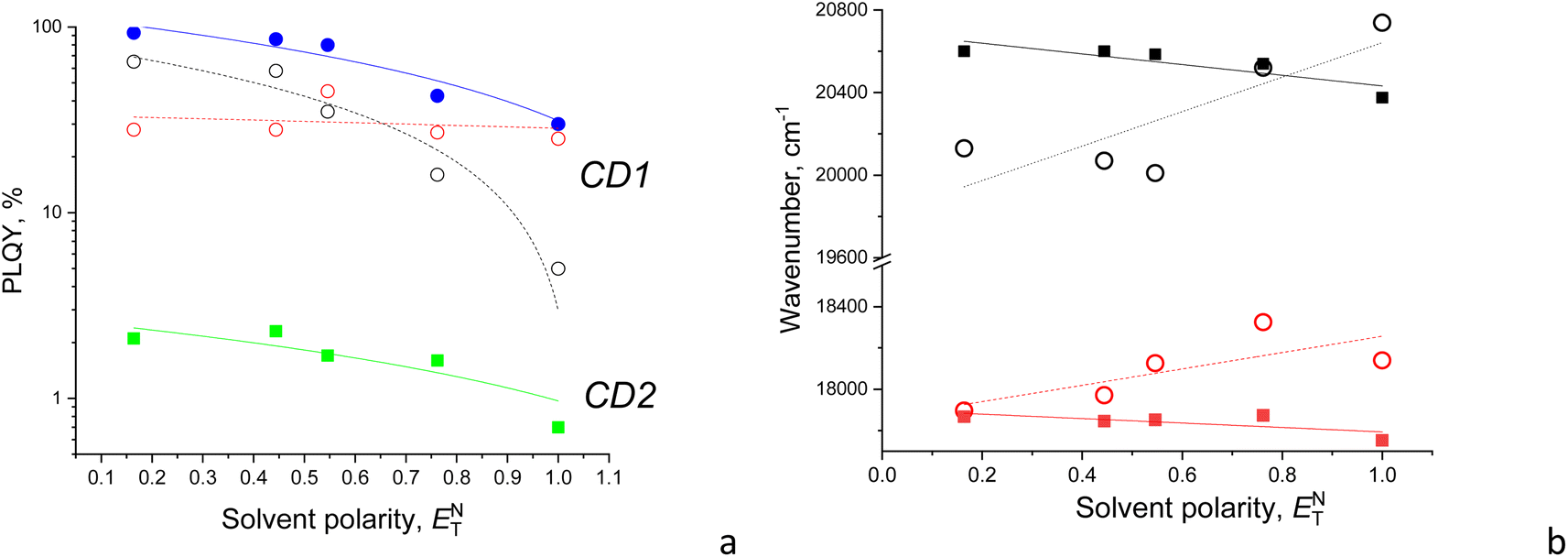 | ||
| Fig. 6 (a) PLQY (in log scale) and (b) maximum position of PL emission components as a function of the normalized solvent polarity. Color code: (a) CD1 (blue circles) with high-energy (black open circles) and low-energy (red open circles) emission components, and CD2 (green squares); (b) high-energy (black symbols) and low-energy (red symbols) emission components of CD1 (open circles) and CD2 (solid squares). | ||
Also, a solvatochromic shift of the emission of CD1 and CD2 is observed to be different, resulting in blue and red shifts of their PL components with increasing solvent polarity, respectively (Fig. 6b). The different signs of solvatochromism observed in CD1 and CD2 are associated with the different nature of their ground states, being polar and neutral, respectively. The polar ground state in CD1 is well consistent with its structure composed of the carbon core and a polar shell as discussed above, resulting in lowering the HOMO and increasing the bandgap, respectively, in the polar medium (Table 3).
| Solvent | Dioxane | DMSO | IPA | MeOH | H2O |
|---|---|---|---|---|---|
| Refractive index | 1.4224 | 1.4793 | 1.3776 | 1.3284 | 1.3330 |
| Normalized solvent polarity | 0.164 | 0.444 | 0.546 | 0.762 | 1 |
| PLQY of CD2, % | 2.1 | 2.3 | 1.7 | 1.6 | 0.7 |
| PLQY of CD1, % | 93 | 86 | 80 | 43 | 30 |
3.4. Evolution of the emission spectra due to photobleaching
The samples reveal gradual suppression of PL emission to some saturation level under CW excitation within the first minute of illumination. However, the above suppression or photobleaching occurs differently for each sample and is dependent on the solvent environment. The emission is stable in a strongly polar aqueous medium; however, in solvents with relatively low polarity, such as dioxane and DMSO, the emission significantly decreases during the first minute of CW irradiation at 405 nm with a 50 mW power. Specifically, the photobleaching is significant only for CD1 samples, showing almost a twofold drop of the emission intensity to saturation level during CW excitation, whereas the CD2 dispersion reveals only some minor changes (Fig. 7 and S6†). The shape of the emission band also changes as a result of photobleaching of the CD1 sample, where the behavior of the fitted Gaussians of the PL band shows that photobleaching of the emission components occurs differently. The high-energy component decreases by a factor of ∼1.7, while the low-energy component decreased by a factor of ∼1.5 (Fig. 7b). The photo-bleaching quantum yield ϕB is estimated as the ratio of the photons NB spent for bleaching with respect to the total amount of the absorbed photons Nabs,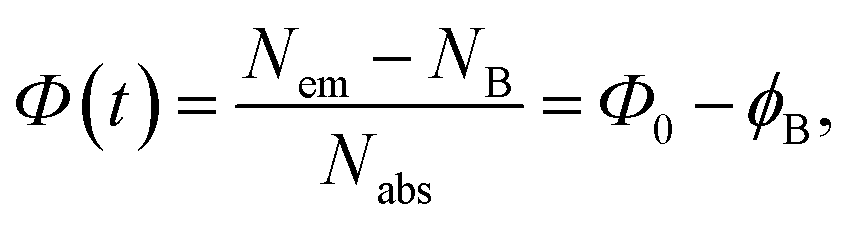 | (2) |
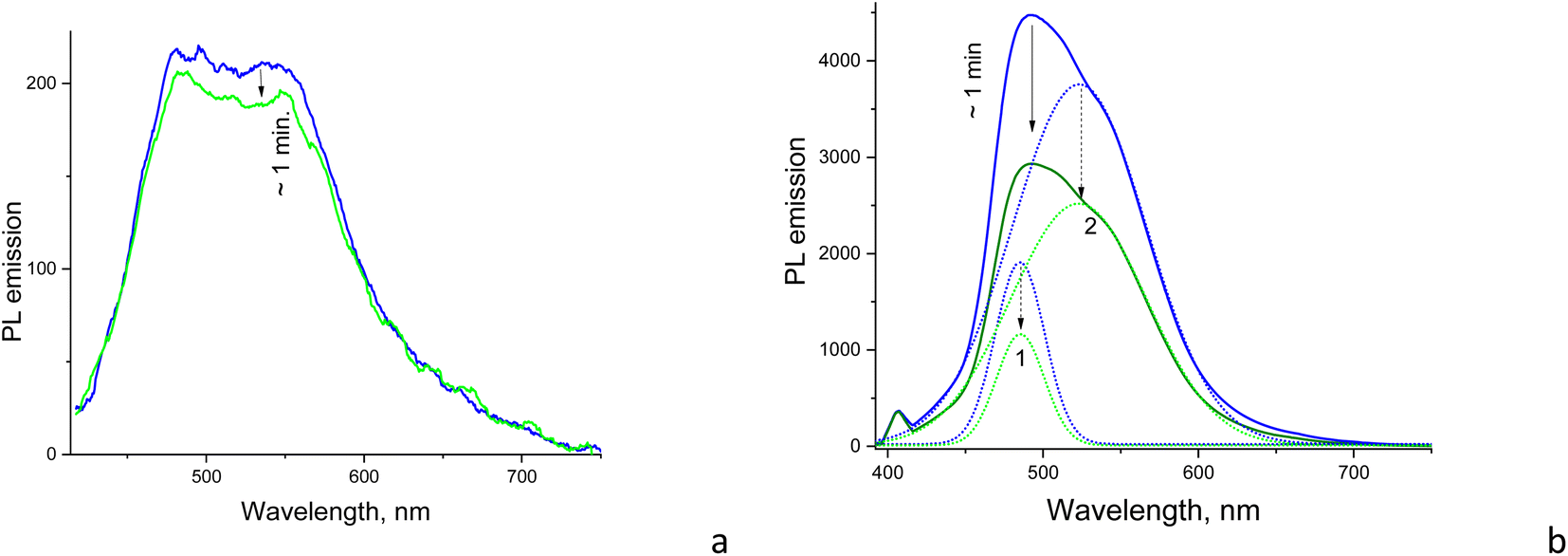 | ||
| Fig. 7 Photobleaching of the PL emission of DMSO solutions of (a) CD2 and (b) CD1 with the Gaussian components (dotted curves) shown. Blue curves correspond to the spectrum taken in the first 50 ms and green curves to the photobleached spectrum after 1 min of illumination by CW excitation at 405 nm (50 mW) until the “saturation” spectrum was reached. The integration time of each measurement was 50 ms. | ||
The above phenomenon is confirmed in experiments with the pulsed excitation of the samples, where the PL spectrum reveals dependence on the on–off time ratio or the duty cycle of the chain of excitation impulses. Specifically, the excitation pulse duration was constant (∼5 μs) but the pulse frequency per second was changed between 10 and 100 Hz (Fig. 8a). Here, the higher the frequency, the shorter the time for the sample recovery between excitation impulses and a more pronounced photobleaching effect, respectively. This effect is observed in the form of the delayed increase of the PL intensity as a function of the increasing frequency or effective duty cycle, where the latter is equivalent to the increasing acquisition time which should yield a proportional increase in the registering signal. In case of no delay caused by the photobleaching, the integral PL intensity should be strictly proportional to the excitation frequency. However, the plot of integral PL intensity versus excitation frequency fitted as a linear dependence shows that the slope is smaller than unity, which indicates that the increase in PL intensity with pulse excitation slows down and declines from the above strict proportionality (Fig. 8b). Moreover, the increase in PL spectrum components occurs disproportionally, i.e., the low-energy component at ∼550 nm increases more slowly compared to the high-energy emission component at ∼480 nm (Fig. 8a). That means that the low-energy emission component is subjected to stronger photobleaching than the high-energy counterpart.
 | ||
| Fig. 8 (a) PL emission spectra (λexc = 405 nm) and (b) PL integral emission intensity of CD1 in DMSO as a function of the frequency or effective duty cycle (pulse amount per second with the fixed pulse duration of 5 μs) of the pulsed excitation at 405 nm with pulse frequencies of 10, 20, 50, and 100 Hz. In (a), Gaussian components of the spectrum at 10 Hz are shown. Note that the frequency as high as 200 kHz (duty cycle = 100%) is needed to be equivalent here to the CW excitation conditions. | ||
It has been reported that carbon nanomaterials, including carbon nanodots44 and carbon nanotubes,45 undergo photoinduced bleaching, demonstrating temporal evolution of PL with similar time constants in the range of 20–60 s, which was assigned to photooxidation processes. Zhang et al. reported photoinduced bleaching of carbon dots due to the photoinduced dehydration of the deprotonated surface of CDs in dimethyl sulfoxide.46 However, we observed that emission in our case is completely recovered after a few minutes of interruption of CW irradiation; therefore, it cannot be assigned due to photoinduced chemical reactions. Instead, the observed photoinduced bleaching in CD1 can be related to population of the surface states in the core–shell structure as a result of dissociation of part of the excitons. This is consistent with the fact that the carboxyl group and trifluoromethyl group located at the particle surface possess high electron affinity and therefore good electron accepting properties47 in the CD1 samples.
4. Discussion
4.1. Intrinsic mechanism of PL emission
Based on our results, different intrinsic mechanisms of PL emission in CD1 and CD2 can be inferred. At the first glance, it appears that the photophysical behavior of CD2 is most consistent with the model of multiple PL centers that feature a wide distribution of energy levels assigned to polycyclic molecules in the carbon core such as anthracene, pyrene, perylene, etc.48 This model predicts tunability of the PL emission spectrum dependent on the excitation wavelength as well as tunability of the PL excitation spectrum dependent on the wavelength at which the spectrum is detected, which is observed for CD2 indeed (Fig. S3†). The other feature of this model is the multi-exponent excitation decay, which is typical for multichromophoric systems where the emission is accompanied by excitation quenching due to interaction of the different emitters,49 which is consistent with the low PLQY observed in CD2. The behavior of multiple emission centers can be accompanied by large Stokes shifts of over 100 nm (6350 cm−1) which was attributed by Fu et al. to formation of self-trapped excitons,48 whose mobility is largely impeded due to the existence of a strong local potential field.50 For CD2, the observed Stokes shift is over 115 nm (6564 cm−1) (Fig. 3), but it results from contributions of different chromophores in the multichromophoric system. To disentangle the contribution of the separate components, different emission wavelengths of the complex PL spectrum should be compared with their counterparts in the excitation spectrum (Fig. S4†); this gives a real Stokes shift of the disentangled components, being in the range of 50–100 nm (∼2600–5900 cm−1). Given this lower value for the Stokes shift, formation of self-trapped excitons within this model is unlikely to occur. The alternative model featuring an ensemble of emissive states due to spectral migration following relaxation processes31 also is not valid here, since this model implies a single excitation center which is not consistent with the CD2 PL excitation spectra (Fig. S4†). However, applicability of the model of multiple PL centers assigned to polycyclic molecules is also arguable here, since no CC moieties and therefore no π-conjugated carbon units attributed to such molecules are found in the XPS spectra of CD2 (see Fig. 2e). Instead, the PL emission mechanism can be related here to a crosslink enhanced or clustering-triggered emission, where multiple non-conventional emission centers act together through formation of crosslinked bonding.51 The idea of clustering-triggered emission extended to non-conventional chromophores, i.e., non-conjugated molecules with hydroxyl (–OH), ester (–COOR), anhydride (–COOCO–), amide (–NHCO–), amine (–NH2), etc., functional groups, explains fluorescence of the system that is inaccessible by the isolated molecules as a result of formation of clusters by through-space conjugation, which results in extended electron delocalization and a rigid conformation of the whole cluster.52 Clustering-triggered emission usually features emission in the blue to green region, a weak quantum yield, a tunable PL spectrum dependent on the excitation wavelength, and a multi-exponent decay kinetics of PL;51–54 all these features are attributed to photophysical behavior of CD2. It was shown that polysaccharides based on glucose monomers, i.e., the same material taken for synthesis of CD2, possess clustering-triggered emission as well.55 All this provides a strong argument in favor of the clustering-triggered emission mechanism of CD2 nanoparticles.
The photophysical behavior of CD1 is associated with both core and surface state transitions. As discussed, the excitation observed as a shoulder at 366 nm in the UV-Vis spectrum is attributed to the n–π* transition due to the CO moiety in the carbon core.15 The red shifted maximum absorption at 414 nm and extended tail towards 700 nm are attributed to surface transitions due to CO/CN moieties and graphitic nitrogen centers of the carbon shell which can inject excess electrons into the unoccupied π* orbital and thus reduce the HOMO–LUMO gap and the energies of the corresponding optical transitions.15 The above transitions give rise to two major emission components, with emission maxima at ∼485 and ∼525 nm, respectively (Fig. 5). The first emission component originates predominantly from excitation at ∼360 nm, corresponding to the n–π* transition in the carbon core, whereas the second emission component shares its origin from both core and surface excitations that involve CO moieties (see the PL excitation spectrum, Fig. S4†), suggesting a collective or resonance character of the above excitations. Again, a relatively small Stokes shift of 78 nm (3810 cm−1) as observed in the spectra (Fig. 3a) features an intermixture of spectral components of the different emitters. Separation of the spectra into components yields a more real Stokes shift for each component in the range of 100–120 nm (∼4680–5400 cm−1), which is larger than the apparent one and can be relevant to the excitonic model of excitation that can evolve to self-trapped states. Indeed, the absence of tunability of both emission components as a function of the excitation wavelength (Fig. S4†), relatively long excitation lifetimes and single-exponent excitation decay, as well as high PLQY, leads to inapplicability of the model of multiple emission centers coexisting in the same core for this type of CD and favors the conclusion that the entire CD1 particle operates as a single emitter, producing an exciton which localizes either within the particle core or at the particle surface, giving rise to the two main emitting components at ∼480 and 525 nm, respectively. The broad and structureless emission band rules out potential suggestions on the molecular origin of the emission since the observed PL band is not accompanied by vibronic sidebands normally present in the single molecule fluorescence spectrum.
The excitonic model implies collective excitations generated by resonant electronic interactions among different chromophore units within the nanoparticle. As applied to CDs, the excitonic model is considered to be due to H-aggregate-type quantum coherence spreading over the whole nanoparticle as a result of strong coupling among the transition dipoles of adjacent chromophores arranged in a co-facial stacking and exciton transport to emissive traps within the carbon particle.56,57
The conclusion concerning formation of excitons that are localized or self-trapped within the core in CD1 is supported by observation of a blue shift of the emission attributed to the first component as a result of dilution of the dispersion of CD1 that yields smaller particles, whereas the second component related to the surface did not change the spectral position (Fig. S5†). In this sense, CD1 particles behave as true quantum dots, changing the bandgap when changing the particle size. The conclusion concerning formation of localized excitons at the particle surface giving rise to the second emission component is consistent with its larger photoinduced bleaching, as discussed above. We speculate that the surprisingly different contribution of the components to the overall emission and PLQY can be due to specific geometries of the excitons, where the first one has an electron located on the shell and a hole located in the core, which might easily result in exciton dissociation due to the higher electron affinity of the shell, while the second one has an electron–hole pair located at the surface which can easily yield radiative recombination.
The excitonic model is also consistent with the observed dependence of CD1 emission on solvent polarity, leading to substantial solvatochromism. Generally, the effect of solvent polarity on PL emission depends both on the dipole of solvent molecules and the dipole of the emitter. Excitation leads to solvent dipole reorientation or relaxation around the dipole of the emitter, which lowers the energy of the excited state, but solvent dipole reorientation is more dramatic when the excited emitter dipole reaches higher magnitude. The higher dipole is expected just for the excitonic excitation, since the entire CD1 particle operates as a single emitter, producing an exciton. On the other hand, excitation due to multiple emission centers coexisting in the same core results in reduced total dipole since these represent superposition of independent chromophores that emit incoherently and do not provide collinear dipoles.
4.2. Outlook into potential optoelectronic application
The potential application of highly emitting CD particles is associated with fabrication of thin-film devices that possess specific energetic parameters and interface properties of the CD films, which should be adjusted for the relevant optoelectronic applications. For that, we reconstructed the energy diagram of thin films of the samples based on the UPS data (Fig. S7†) and compared these with another widely used carbon material, C60. For UPS studies, C60 was cast from chlorobenzene solution on the ITO substrate, i.e., similar to the preparation method of the CD samples. The band gap of C60 films was taken from literature data as 1.6 eV.58The found work function (WF) of the CD2 film (4.65 eV) is very close to that of the C60 film (4.6 eV), and also to other carbon materials such as diamond (WF = 4.5 eV (ref. 59)) and pyrolytic graphite (WF = 4.7 eV (ref. 60)), whereas the CD1 samples containing fluorine, oxygen and nitrogen show a noticeably larger work function of 4.9 eV (Fig. 9). The ionization potential (IP) of the CD1 sample (6.0 eV) is very close to that of the C60 films (6.1 eV), whereas the IP of the CD2 films is lower (5.85 eV). Also, the electron affinity (EA) of both CD samples is lower compared to C60 films by more than 1 eV (Fig. 9). The latter indicates that the CD samples are poor electron acceptors compared to C60. This leads to the different position of the Fermi level within the band gap: in the C60 films, the Fermi level position corresponds to n-type conductivity whereas for the CD films, the Fermi level lies a little bit below the mid-gap, showing a tendency of changing the intrinsic conductivity to p-type.
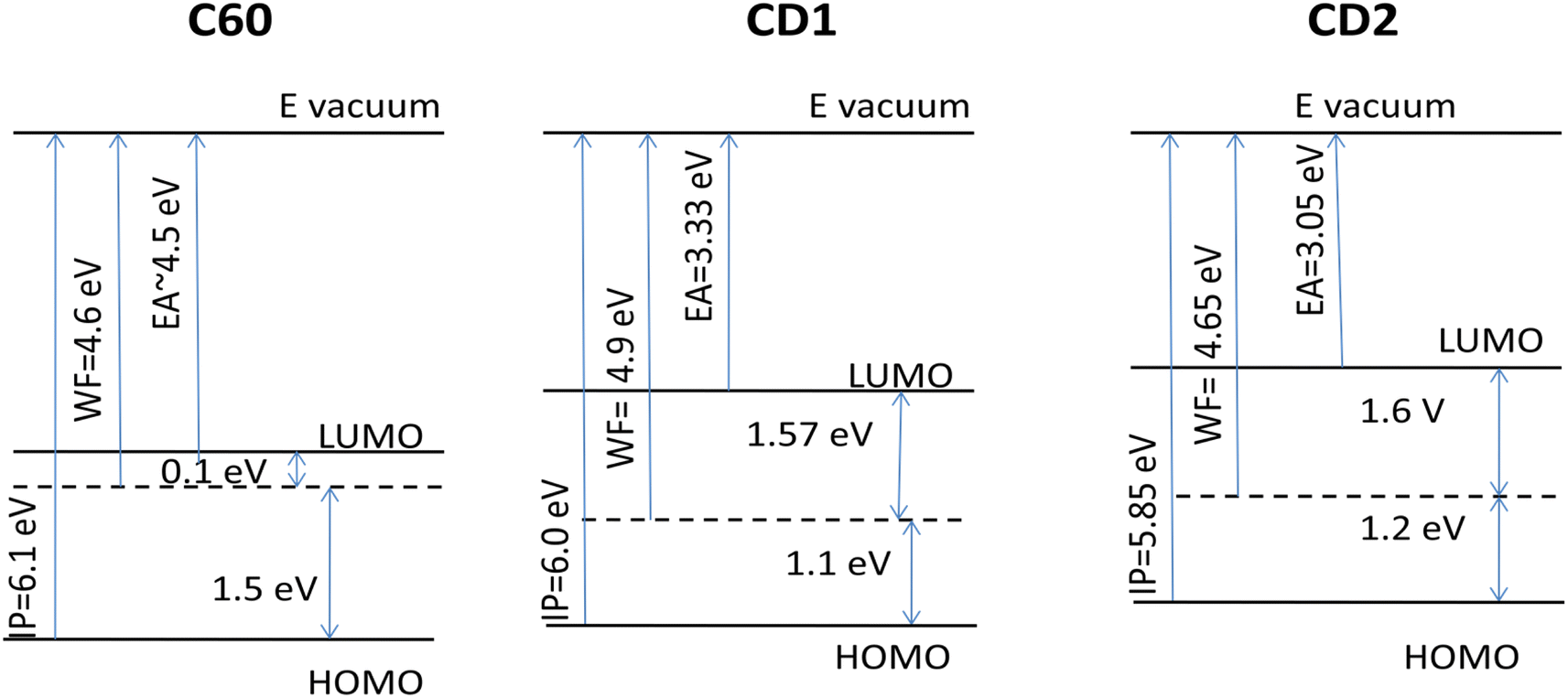 | ||
| Fig. 9 Reconstructed energy diagram of thin films of CD1 and CD2 samples compared to film of C60. | ||
Based on the obtained energetic parameters it can be concluded that the CD materials cannot be considered as good electron acceptors, but rather as electron donors. However, their IP and work functions are still high, which limits their practical applications as electron donors. They can be considered as overlayers for the anode material. For example, incorporation of carbon quantum dots within PEDOT:PSS for high-performance inverted organic solar cells has been recently demonstrated.61 But most interestingly the WF and IP of CD samples can be controllably varied using electronegative elements such as F, O, and N, which results in significant lowering of energy levels with respect to vacuum and increasing both ionization potential and work function.
5. Conclusions
In this work, we have compared the PLQY and photophysical behavior of CDs synthesized by different methods that yield samples with different structures, compositions and oxidation. Based on the obtained results, the intrinsic mechanism of PL emission in oxidized CDs obtained by thermal decomposition of sucrose is consistent with the model of the multiple PL centers which act together in the carbon core through a clustering-triggered emission mechanism. These CD particles possess tunable emission depending on the excitation wavelength, relatively short excitation lifetimes with multi-exponent decay, and poor sensitivity to solvent polarity as well as poor photoinduced bleaching. The PL mechanism in CDs heavily doped by fluorine, nitrogen and oxygen is relevant to excitonic emission, assigned due to recombination of the core hole and electron located either in the core or on the surface of CDs. This type of CD particle possesses non-tunable emission, relatively long excitation lifetimes with single-exponent decay, solvatochromism and dependence of PLQY on solvent polarity. Independent of the emission mechanisms, the above results indicate that PLQY of CDs can be sensitive not only to the environmental conditions but is also time-dependent within a relatively short time (∼one minute) from the beginning of CW excitation. Taking into account that steady-state PLQY, i.e., PLQY measured when the PL spectrum is “saturated”, of CD1 in DMSO and dioxane is high enough (86% and 93%, respectively), PLQY measured during the first seconds of excitation may even exceed these values. This leads to the concept of the dynamic PLQY in specific carbon nanoparticles where careful determination of PLQY requires references not only with respect to the environmental conditions but also with respect to the beginning of acquisition time when starting CW excitation, where the transient period can be as long as ∼102 seconds. The obtained results provide a means to separate mechanisms of emission in CDs that lead to principally different PLQYs that differ by orders of magnitude. Thus, our findings lead to a strategy for design of CDs based on the revealed link between their emission mechanism and PLQY.Author contributions
O. D.: conceptualization, investigation, writing, review & editing; D. K.: synthesis, investigation, review; A. Z.: synthesis, investigation; O. I. and Y. P.: investigation; A. V.: review & investigation; A. N.: review, supervision & project administration; M. F.: review & editing, project administration.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
O. D. and M. F. are grateful for financial support from the Knut and Alice Wallenberg Foundation (KAW) through the Wallenberg Wood Science Center. A. Z. acknowledges partial financial support of this work by EU Horizon 2020 Research and Innovation Staff Exchange Program (RISE) under Marie Skłodowska-Curie Action (project “UNAT”, no. 101008159) and Visegrad Scholarship Program ID 52310672. O. D. appreciates the help of Anna Elsukova for measurements and discussion of TEM images. Swedish Research Council and SSF for access to ARTEMI, the Swedish National Infrastructure in Advanced Electron Microscopy (2021-00171 and RIF21-0026) is gratefully acknowledged.References
- J. Liu, R. Li and B. Yang, ACS Cent. Sci., 2020, 6(12), 2179–2195 CrossRef CAS PubMed.
- A. S. Rasal, S. Yadav, A. Yadav, A. A. Kashale, S. T. Manjunatha, A. Altaee and J. Y. Chang, ACS Appl. Nano Mater., 2021, 4(7), 6515–6541 CrossRef CAS.
- A. Cadranel, J. T. Margraf, V. Strauss, T. Clark and D. M. Guldi, Acc. Chem. Res., 2019, 52(4), 955–963 CrossRef CAS PubMed.
- B. Vercelli, Coatings, 2021, 11(2), 232 CrossRef CAS.
- M. Semeniuk, Z. Yi, V. Poursorkhabi, J. Tjong, S. Jaffer, Z. H. Lu and M. Sain, ACS Nano, 2019, 13(6), 6224–6255 CrossRef CAS PubMed.
- B. Gayen, S. Palchoudhury and J. Chowdhury, J. Nanomater., 2019, 2, 1–19 Search PubMed.
- K. Hagiwara, S. Horikoshi and N. Serpone, Chem.–Eur. J., 2021, 27(37), 9466–9481 CrossRef CAS PubMed.
- H. Liu, Z. Li, Y. Sun, X. Geng, Y. Hu, H. Meng, J. Ge and L. Qu, Sci. Rep., 2018, 8(1), 1086 CrossRef PubMed.
- L. Ai, Y. Yang, B. Wang, J. Chang, Z. Tang, B. Yang and S. Lu, Sci. Bull., 2021, 66(8), 839–856 CrossRef CAS PubMed.
- S. Zhu, Y. Song, X. Zhao, J. Shao, J. Zhang and B. Yang, Nano Res., 2015, 8, 355–381 CrossRef CAS.
- F. Yuan, Z. Wang, X. Li, Y. Li, Z. Tan, L. Fan and S. Yang, Bright multicolor bandgap fluorescent carbon quantum dots for electroluminescent light-emitting diodes, Adv. Mater., 2017, 29, 1604436 CrossRef PubMed.
- L. Wang, S. J. Zhu, H. Y. Wang, S. N. Qu, Y. L. Zhang, J. H. Zhang, Q. D. Chen, H. L. Xu, W. Han, B. Yang and H. B. Sun, ACS Nano, 2014, 8, 2541–2547 CrossRef CAS PubMed.
- P. Yu, X. Wen, Y.-R. Toh and J. Tang, J. Phys. Chem. C, 2012, 116, 25552–25557 CrossRef CAS.
- S. Zhu, Q. Meng, L. Wang, J. Zhang, Y. Song, H. Jin, K. Zhang, H. Sun, H. Wang and B. Yang, Angew. Chem., Int. Ed., 2013, 52, 3953–3957 CrossRef CAS PubMed.
- M. Liu, Nanoarchitectonics, 2020, 1(1), 1–12 CrossRef.
- S. Zhu, Y. Song, J. Shao, X. Zhao and B. Yang, Angew. Chem., Int. Ed., 2015, 54(49), 14626–14637 CrossRef CAS PubMed.
- S. Tao, C. Zhou, C. Kang, S. Zhu, T. Feng, S. T. Zhang, Z. Ding, C. Zheng, C. Xia and B. Yang, Light: Sci. Appl., 2022, 11(1), 56 CrossRef CAS PubMed.
- S. Zhu, J. Zhang, X. Liu, B. Li, X. Wang, S. Tang, Q. Meng, Y. Li, C. Shi, R. Hu and B. Yang, RSC Adv., 2012, 2, 2717–2720 RSC.
- L. Wang, S. J. Zhu, H. Y. Wang, S. N. Qu, Y. L. Zhang, J. H. Zhang, Q. D. Chen, H. L. Xu, W. Han, B. Yang and H. B. Sun, ACS Nano, 2014, 8, 2541–2547 CrossRef CAS PubMed.
- Y. Xiong, J. Schneider, E. V. Ushakova and A. L. Rogach, Nano Today, 2018, 23, 124–139 CrossRef CAS.
- A. Zaderko, The Process for Obtaining of Fluoralkylated Carbon Quantum Dots, WO2020121119, 2020, https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2020121119, accessed 25 Jan 2021.
- V. V. Lisnyak, A. N. Zaderko, R. Mariychuk, V. Lysenko, O. Y. Boldyrieva, V. A. Skryshevsky, G. Mussabek, Y. Taurbayev, N. Zhylkybayeva and O. Y. Tananiko, Appl. Nanosci., 2022, 12(3), 795–803 CrossRef CAS.
- A. Vasin, D. Kysil, S. Sevostianov, O. Isaieva, G. Rudko, B. Capoen, M. Bouazaoui, H. El Hamzaoui, V. Tertykh, S. Starik and A. Nazarov, Phys. Status Solidi A, 2021, 218(15), 2000817 CrossRef CAS.
- R. E. Kellogg and R. G. Bennett, J. Chem. Phys., 1964, 41, 3042–3045 CrossRef CAS.
- J. N. Demas and G. A. Crosby, J. Phys. Chem., 1971, 75, 991–1024 CrossRef.
- D. Li, Y. Lu and C. Zhang, Nanomaterials, 2022, 12(24), 4387 CrossRef CAS PubMed.
- J. V. Rojas, M. Toro-Gonzalez, M. C. Molina-Higgins and C. E. Castano, Mater. Sci. Eng., B, 2016, 205, 28–35 CrossRef CAS.
- X. Chen, X. Wang and D. Fang, Fullerenes, Nanotub. Carbon Nanostruct., 2020, 28(12), 1048–1058 CrossRef CAS.
- M. J. Krysmann, A. Kelarakis, P. Dallas and E. P. Giannelis, J. Am. Chem. Soc., 2012, 134(2), 747–750 CrossRef CAS PubMed.
- A. Sciortino, E. Marino, B. V. Dam, P. Schall, M. Cannas and F. Messina, J. Phys. Chem. Lett., 2016, 7(17), 3419–3423 CrossRef CAS PubMed.
- S. Khan, A. Gupta, N. C. Verma and C. K. Nandi, Nano Lett., 2015, 15(12), 8300–8305 CrossRef CAS PubMed.
- S. K. Cushing, M. Li, F. Huang and N. Wu, ACS Nano, 2014, 8(1), 1002–1013 CrossRef CAS PubMed.
- Y. H. Yuan, Z. X. Liu, R. S. Li, H. Y. Zou, M. Lin, H. Liu and C. Z. Huang, Nanoscale, 2016, 8(12), 6770–6776 RSC.
- X. Sun, C. Brückner and Y. Lei, Nanoscale, 2015, 7(41), 17278–17282 RSC.
- J. Zhu, H. Chu, J. Shen, C. Wang and Y. Wei, J. Colloid Interface Sci., 2021, 586, 683–691 CrossRef CAS PubMed.
- X. Miao, X. Yan, D. Qu, D. Li, F. F. Tao and Z. Sun, ACS Appl. Mater. Interfaces, 2017, 9(22), 18549–18556 CrossRef CAS PubMed.
- K. Luo, Y. Wen and X. Kang, Molecules, 2022, 27(14), 4620 CrossRef CAS PubMed.
- L. Sciortino, A. Sciortino, R. Popescu, R. Schneider, D. Gerthsen, S. Agnello, M. Cannas and F. Messina, J. Phys. Chem. C, 2018, 122(34), 19897–19903 CrossRef CAS.
- X. Xu, L. Mo, Y. Li, X. Pan, G. Hu, B. Lei, X. Zhang, M. Zheng, J. Zhuang, Y. Liu and C. Hu, Adv. Mater., 2021, 33(49), 2104872 CrossRef CAS PubMed.
- W. Zhou, J. Zhuang, W. Li, C. Hu, B. Lei and Y. Liu, J. Mater. Chem. C, 2017, 5(32), 8014–8021 RSC.
- R. Mohan, J. Drbohlavova and J. Hubalek, Chem. Phys. Lett., 2018, 692, 196–201 CrossRef CAS.
- J. R. Macairan, T. V. de Medeiros, M. Gazzetto, F. Y. Villanueva, A. Cannizzo and R. Naccache, J. Colloid Interface Sci., 2022, 606, 67–76 CrossRef CAS PubMed.
- J. Bai, G. Yuan, Y. Zhu, Z. Huang, L. Zhang, X. Wang, S. Wu and L. Ren, J. Phys. Chem. C, 2021, 125(33), 18543–18551 CrossRef CAS.
- A. Terracina, A. Armano, M. Meloni, A. Panniello, G. Minervini, A. Madonia, M. Cannas, M. Striccoli, L. Malfatti and F. Messina, ACS Appl. Mater. Interfaces, 2022, 14(31), 36038–36051 CrossRef CAS PubMed.
- C. Georgi, N. Hartmann, T. Gokus, A. A. Green, M. C. Hersam and A. Hartschuh, ChemPhysChem, 2008, 9(10), 1460–1464 CrossRef CAS PubMed.
- B. Zhang, B. Wang, E. V. Ushakova, B. He, G. Xing, Z. Tang, A. L. Rogach and S. Qu, Small, 2022, 2204158 Search PubMed.
- J. E. True, T. D. Thomas, R. W. Winter and G. L. Gard, Inorg. Chem., 2003, 42(14), 4437–4441 CrossRef CAS PubMed.
- M. Fu, F. Ehrat, Y. Wang, K. Z. Milowska, C. Reckmeier, A. L. Rogach, J. K. Stolarczyk, A. S. Urban and J. Feldmann, Nano Lett., 2015, 15(9), 6030–6035 CrossRef CAS PubMed.
- O. P. Dimitriev, Y. P. Piryatinski and Y. L. Slominskii, J. Phys. Chem. C, 2019, 123, 28611–28619 CrossRef CAS.
- L. Xiao, Y. Wang, Y. Huang, T. Wong and H. Sun, Nanoscale, 2017, 9(34), 12637–12646 RSC.
- P. Liao, J. Huang, Y. Yan and B. Z. Tang, Mater. Chem. Front., 2021, 5(18), 6693–6717 RSC.
- C. Xiaohong, W. Yunzhong, Z. Yongming and Y. Wangzhang, Prog. Chem., 2019, 31(11), 1560 Search PubMed.
- L. Xu, X. Liang, S. Zhong, Y. Gao and X. Cui, ACS Sustain. Chem. Eng., 2020, 8(51), 18816–18823 CrossRef CAS.
- Q. Li, X. Wang, Q. Huang, Z. Li, B. Z. Tang and S. Mao, Nat. Commun., 2023, 14(1), 409 CrossRef CAS PubMed.
- W. Z. Yuan and Y. Zhang, J. Polym. Sci., Part A: Polym. Chem., 2017, 55(4), 560–574 CrossRef.
- A. P. Demchenko, C, 2019, 5(4), 71 CAS.
- A. P. Demchenko and M. O. Dekaliuk, Nanoscale, 2016, 8(29), 14057–14069 RSC.
- A. Skumanich, Chem. Phys. Lett., 1991, 182(5), 486–490 CrossRef CAS.
- N. Sato, Y. Saito and H. Shinohara, Chem. Phys., 1992, 162(2–3), 433–438 CrossRef CAS.
- R. F. Willis, B. Feuerbacher and B. Fitton, Phys. Rev. B: Solid State, 1971, 4(8), 2441 CrossRef.
- D. C. T. Nguyen, B. S. Kim, G. H. Oh, V. P. Vu, S. Kim and S. H. Lee, Synth. Met., 2023, 298, 117430 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4na00033a |
| This journal is © The Royal Society of Chemistry 2024 |