DOI:
10.1039/D4NA00223G
(Paper)
Nanoscale Adv., 2024,
6, 3911-3922
Functionalized graphene oxide by 4-amino-3-hydroxy-1-naphthalenesulfonic acid as a heterogeneous nanocatalyst for the one-pot synthesis of tetraketone and tetrahydrobenzo[b]pyran derivatives under green conditions†
Received
17th March 2024
, Accepted 23rd May 2024
First published on 24th May 2024
Abstract
4-Amino-3-hydroxy-1-naphthalenesulfonic acid-functionalized graphene oxide (GO-ANSA) was prepared and characterized using different spectroscopic, microscopic and analytical methods including energy-dispersive X-ray spectroscopy (EDS), EDS elemental mapping, Fourier-transform infrared (FT-IR) spectroscopy, field emission scanning electron microscopy (FESEM), X-ray diffraction (XRD), and thermogravimetry/differential thermogravimetry analysis (TGA/DTA). The obtained nanomaterial was used as a novel, highly efficient, and reusable solid acid carbocatalyst for the one-pot three-component synthesis of tetraketone, as well as tetrahydrobenzo[b]pyran derivatives via tandem Knoevenagel–Michael reactions under green conditions. All of the derivatives were prepared in EtOH, as a green solvent, under reflux conditions in high to excellent yields and very short reaction times. The nanocatalyst was recovered and reused at least five times without significant reduction in its activity. In addition, the absence of toxic transition metals, high to excellent yields, mild reaction conditions, simple procedure for the separation and purification of products, stability, and recycling of the catalyst are the most important advantages of this green procedure.
1. Introduction
Most recently, nanotechnology has found extensive application across many fields and become one of the attractive topics among different fields of science and technology, including chemistry, physics, biology, materials, medicinal, electronic,1 automotive,2 aerospace,3 and others.4–6 One of the most important applications of nanotechnology in the field of chemistry is the preparation of nanocatalysts, which have shown a significant impact on green chemistry.7–13 Indeed, a variety of inorganic and organic compounds have been reported as homogeneous catalytic species; however, they have disadvantages such as difficult separation and recycling, producing large quantities of hazardous waste, high cost, etc. To address these concerns, heterogeneous catalytic systems were introduced as alternatives for the homogeneous catalytic systems. In recent decades, nanocatalytic systems have emerged as a bridge between homogeneous and heterogeneous catalytic systems and demonstrate many advantages such as high activity, stability, and selectivity compared to the traditional catalytic systems. Among the catalyst supports, carbon materials are noteworthy due to their synergistic catalytic activation.14–19
Due to their unique properties, carbon-based nanomaterials have strongly influenced many research fields such as pollutant adsorption,20 drug delivery,21 biomedical imaging,22 sensing,23 and catalysis.24 Graphene oxide (GO) is attracting special attention in many research fields such as antimicrobial nanohybrids,25 batteries,26 nanomembranes,27 photocatalytic degradation of organic pollutants,28 and supported catalysts29,30 owing to its unique and exclusive properties including excellent chemical stability, high specific surface area, outstanding mechanical performance, and high thermal stability.31–33 Because of its oxygen-containing functional groups such as hydroxyl, epoxide, and carboxylic acid on both surface and edges, GO has resolved the non-dispersion of graphene sheets in aqueous media. Consequently, it has more active sites for the loading of catalytically active species than pristine graphene. These properties make GO an attractive candidate for a catalytic substrate in synthesizing organic materials, especially via MCRs.34–40
MCRs are synthetic processes that simultaneously afford products from several reactants in one-pot reactions. Recently, a wide range of MCRs have been developed using various catalysts.41–47 MCR procedures have many advantages such as reducing product purification processes, good atom economy, preventing the formation of side products, reducing the use of solvents, and mild reaction conditions. MCRs are very attractive processes for the synthesis of a variety of polymers, heterocyclic compounds, and materials with biological properties, and provide a wide range of products often having complex molecular structures in rapid and simple procedures.48–51 One of the most important parameters in the synthesis of organic compounds through MCRs and complying with the principles of green chemistry is choosing an eco-friendly solvent. In this context, performing the reactions for the synthesis of pharmaceutical intermediates or final active pharmaceutical ingredients (APIs) in EtOH, as a green solvent, is one of the best strategies.9,41,52,53
Recently, tetraketone and tetrahydrobenzo[b]pyran derivatives have attracted much attention due to their biological and medicinal properties. Tetraketones are a group of chemical compounds that exist in two forms (enol–keto tautomerism) and have antiviral,54 antibacterial, antioxidant55 as well as inhibitory properties of some enzymes such as lipoxygenase56 or tyrosinase (reduce overproducing skin melanoma).57 Also, these compounds are used in fabrication of laser dyes58 and considered the most popular precursors for the preparation of many important organic compounds such as acridindiones and xanthendiones.59 On the other hand, tetrahydrobenzo[b]pyrans are non-aromatic heterocyclic compounds that were first synthesized in 1962 through the thermal decomposition of 2-acetoxy-3,4-dihydro-2H-pyran. These compounds have many biological properties including antioxidant, antitumor or anticancer, antianaphylactic, antispasmodic, and antiinflammatory.35,60–63 Also, recent studies have shown that these heterocycles can be effective in the treatment of some diseases such as Alzheimer's, Down's syndrome, Schizophrenia, Parkinson's, etc. (Fig. 1).49,64,65
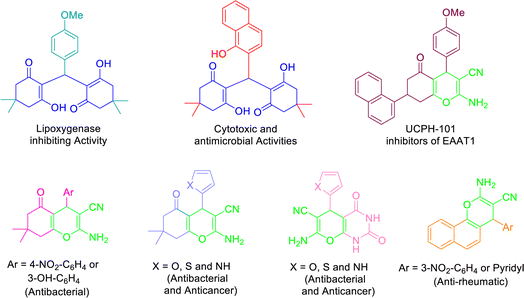 |
| | Fig. 1 Structure of selected examples of biologically- and pharmacologically-active tetraketones and 4H-pyrans. | |
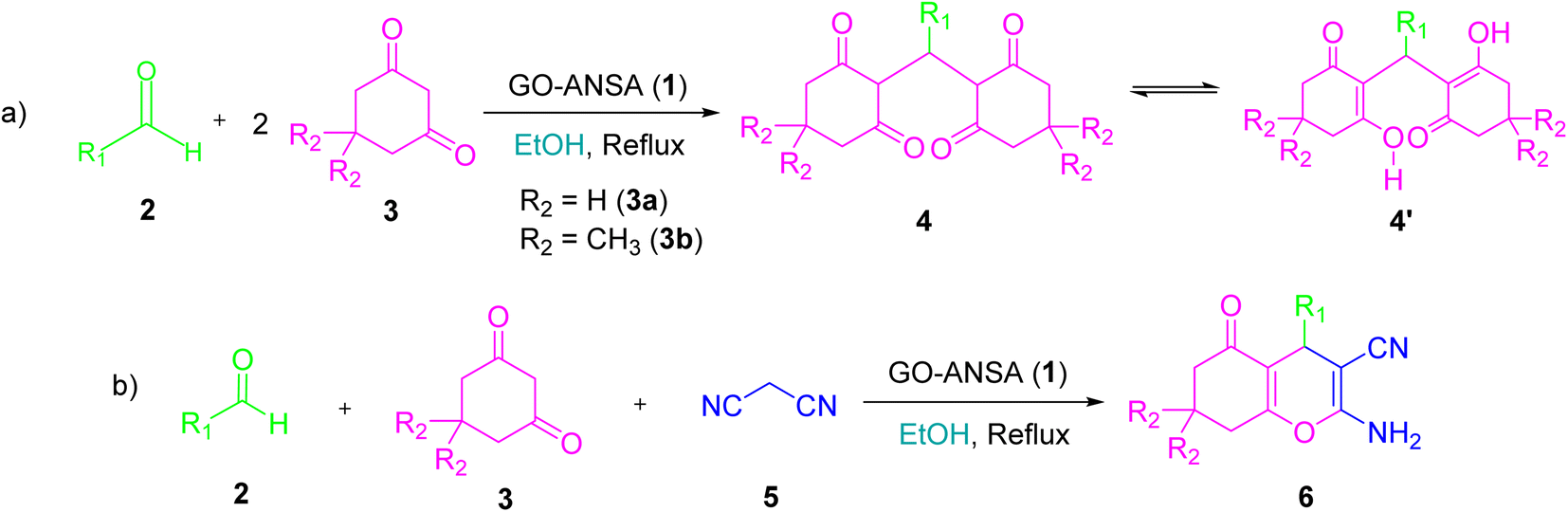
Different protocols have been introduced for the synthesis of tetraketone and tetrahydrobenzo[b]pyran derivatives by using a variety of catalysts in recent years.66,67 So far, numerous heterogeneous and homogeneous catalysts for the synthesis of tetraketones such as [H-Pyrr][HSO4],68L-histidine in ionic liquid,69 nano SiO2Cl,70 amino-appended β-cyclodextrin,71 Cu2(NH2-BDC)2(DABCO),72 Fe-zeolite,73 and 2-aminopyrazine74 have been reported. Similarly, a wide range of methods have been developed for the synthesis of tetrahydrobenzo[b]pyrans using different catalytic systems such as sodium alginate,75 piperazine–GO,76 SO3H-functionalized nano-MGO-D-NH2,77 Cs-EDTA-Cell network,78 nano α-Al2O3 supported ammonium dihydrogen phosphate (NH4H2PO4/Al2O3),79 proline- or propylamine-modified SBA-15,80 PMO-ICS,81 and FeNi3–SiO2 nanoparticles.82 To the best of our knowledge, there are no previous reports on the use of GO-ANSA for the synthesis of organic compounds. In continuation of our interest for exploring various solid acid nanocatalysts in different organic transformations, especially based on GO,83–88 we wish herein to report the catalytic activity of GO-ANSA (1) in the one-pot three-component reaction of aldehydes, enolizable compounds and/or malononitrile to afford the corresponding tetraketones or tetrahydrobenzo[b]pyrans (Scheme 1).
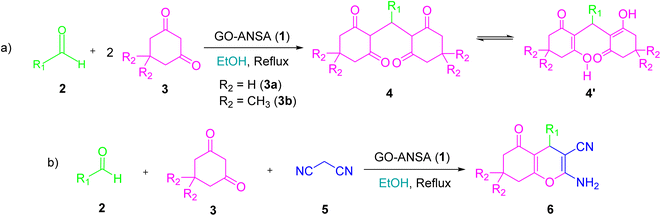 |
| | Scheme 1 One-pot three-component reaction of (a) aldehydes and enolizable compounds (2.0 equiv.), (b) aldehydes, enolizable compounds, and malononitrile catalyzed by the GO-ANSA nanomaterial. | |
2. Experimental
2.1 Materials and measurements
NaNO3 (98+%) was purchased from Sigma-Aldrich. Graphite flakes (99.8%), H2SO4 (98%), potassium permanganate (KMnO4, 98.5%), HCl (37%), H2O2 (30%), acetic anhydride, 4-amino-3-hydroxy-1-naphthalenesulfonic acid (96%), dimedone, 1,3-cyclohexanedione, benzaldehyde derivatives, malononitrile, DMF and EtOH (96%) were purchased from Merck. Distilled water was used in all required steps of reactions or purification. The XRD pattern was collected by a D8 advance Bruker with Cu Ka radiation (λ = 1.54050 Å). FESEM images, EDX and EDX mapping were taken by FESEM TESCAN-MIRA3. Atomic force microscopy (AFM, Full PLUS, Brisk, ARA Research) was used to visualize the surface morphology of the sample. FTIR spectroscopy was performed on a 1720-X PerkinElmer. TGA-DTA analysis was performed by SPA-503 BAHR. 1H NMR spectroscopy was carried out on a Bruker DRX-500 at 500 MHz using CDCl3 and DMSO, as solvent, and data are given in ppm relative to TMS at ambient temperature (CDCl3: 1H: δ = 7.26 ppm). All the products are known compounds and were identified by comparison of their physical, spectroscopic, and analytical data with the authentic samples.
2.2 Synthesis of GO
GO was prepared via the modified Hummers' method.89 The mixture of graphite (1.0 g) and NaNO3 (0.5 g) was added to the concentrated sulfuric acid solution (23.0 ml, 98%), and stirred while the reaction temperature was kept below 4 °C. Then, KMnO4 (3.0 g) was added at 20 °C and the stirring of the mixture was continued for 2 h until the reaction was complete. To avoid a sudden increase in the reaction temperature due to the exothermic nature of the reaction, KMnO4 was slowly added to the mixture. After that, distilled water (46.0 ml) was slowly added to the reaction mixture at a temperature of 96 °C. In the next step, distilled water (140.0 ml) was added and finally, hydrogen peroxide (10.0 ml, 30% v/v) was added until the color of the mixture turned to yellow. Then, the obtained mixture was allowed to rest for 24 h, and it was washed with HCl (5%) solution and distilled water, so that all the impurities were washed and removed. Finally, the obtained GO was dried in an oven at 60 °C.
2.3 Synthesis of 4-amino-3-hydroxy-1-naphthalenesulfonic acid-functionalized graphene oxide (GO-ANSA, 1)
DMF (20.0 ml) and GO (200.0 mg) were added to a round-bottom flask and dispersed with an ultrasonic probe to separate the GO sheets. Afterward, Ac2O (0.4 ml) was added to the sonicated solution and the obtained mixture was placed in an ultrasonic bath for 2.0 h. Then, ANSA (0.4 g) and Et3N (0.35 ml) were added, and the mixture was stirred at 80 °C for 48 h. At the end of the reaction, sulfuric acid (0.1 ml) was added to the mixture. Then, the obtained residue was filtered using filter paper, and washed with THF and EtOH. Finally, the obtained black powder was dried at ambient temperature for 4 h (Scheme 2).
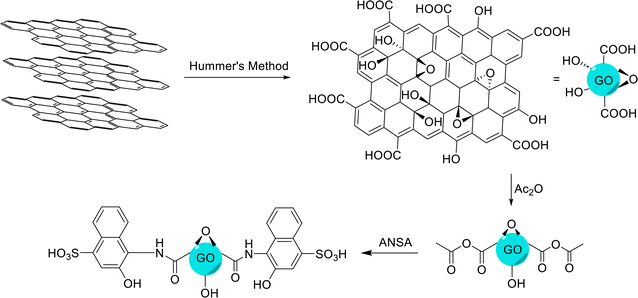 |
| | Scheme 2 Procedures for the preparation of GO, according to the modified Hummers' method, and subsequent preparation of GO-ANSA. | |
2.4 General procedure for the synthesis of tetraketone 4 and tetrahydrobenzo[b]pyran 6 derivatives catalysed by the GO-ANSA nanocatalyst
For the synthesis of the tetraketone derivatives 4, aldehyde derivative (2, 1.0 mmol), enolizable compound (3, 2.0 mmol), GO-ANSA (1, 15.0 mg), as a nanocatalyst, and EtOH (3.0 ml) were added to a 10 ml flask under stirring and reflux conditions. The progress of the reaction was followed by TLC (n-hexane![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :EtOAc, 1:3). After the completion of reaction, EtOH (2 ml) was added to the mixture and it was heated to remove the catalyst from crude reaction mixture by filtration. Then, the pure products 4 were recrystallized from the ethanolic filtrate. The products were identified by measurement of their melting point as well as FTIR and 1H NMR spectral data. The results are reported in Table 2.
:EtOAc, 1:3). After the completion of reaction, EtOH (2 ml) was added to the mixture and it was heated to remove the catalyst from crude reaction mixture by filtration. Then, the pure products 4 were recrystallized from the ethanolic filtrate. The products were identified by measurement of their melting point as well as FTIR and 1H NMR spectral data. The results are reported in Table 2.
For the synthesis of the tetrahydrobenzo[b]pyran derivatives 6, the aldehyde derivative (2, 1.0 mmol), malononitrile (5, 1.0 mmol), enolizable compound (3, 1.0 mmol), GO-ANSA (1, 10.0 mg) and EtOH (3.0 ml) were added to a 10 ml round-bottom flask equipped with a magnetic stirring and reflux condenser. The progress of the reaction was monitored by TLC (n-hexane:EtOAc, 1:3). After the completion of reaction, EtOH (2 ml) was added to the mixture and it was heated to remove the catalyst from crude reaction mixture by filteration. Then, the pure products 6 were recrystallized from the ethanolic filtrate. The products were identified by measurement of their melting point as well as FTIR and 1H NMR spectral data. The results are reported in Table 5.
3. Results and discussion
3.1 Characterization of GO and GO-ANSA
To determine the percentage of different atoms present in the structure of GO-ANSA (1), both energy-dispersive X-ray spectroscopy (EDS, Fig. 2a) and EDS elemental mapping were used (Fig. 2b). According to the data for the EDX spectra of the GO-ANSA, the atomic percentage of carbon, oxygen, nitrogen, and sulfur was 72.80%, 21.40%, 4.14%, and 1.66%, respectively, which indicates the presence of ANSA grafted mainly onto the edges of GO or even on its surface. To our delight, EDS elemental mapping showed that the amount of N element was significantly lower than that of other elements, especially the S element. In fact, both strong SO3H Brønsted acid centers and amidic NH basic centers exist simultaneously in the structure of GO-ANSA (1). This leads to a rapid proton transfer reaction from SO3H to the amidic NH. This proton transfer has two significant consequences: it renders the free electron pair of nitrogen unavailable and alters its electronic levels. Ultimately, the number of allowed electron transfers involving N is reduced compared to other elements. Interestingly, this observation aligns with findings reported in the literature.90
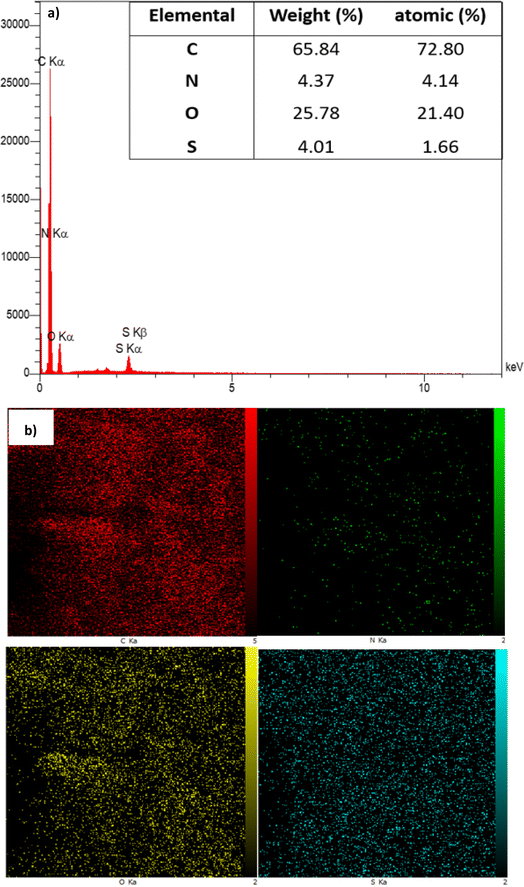 |
| | Fig. 2 (a) EDS spectrum and (b) EDS elemental mapping of GO-ANSA (1) for the distribution of C, N, O, and S atoms, respectively. | |
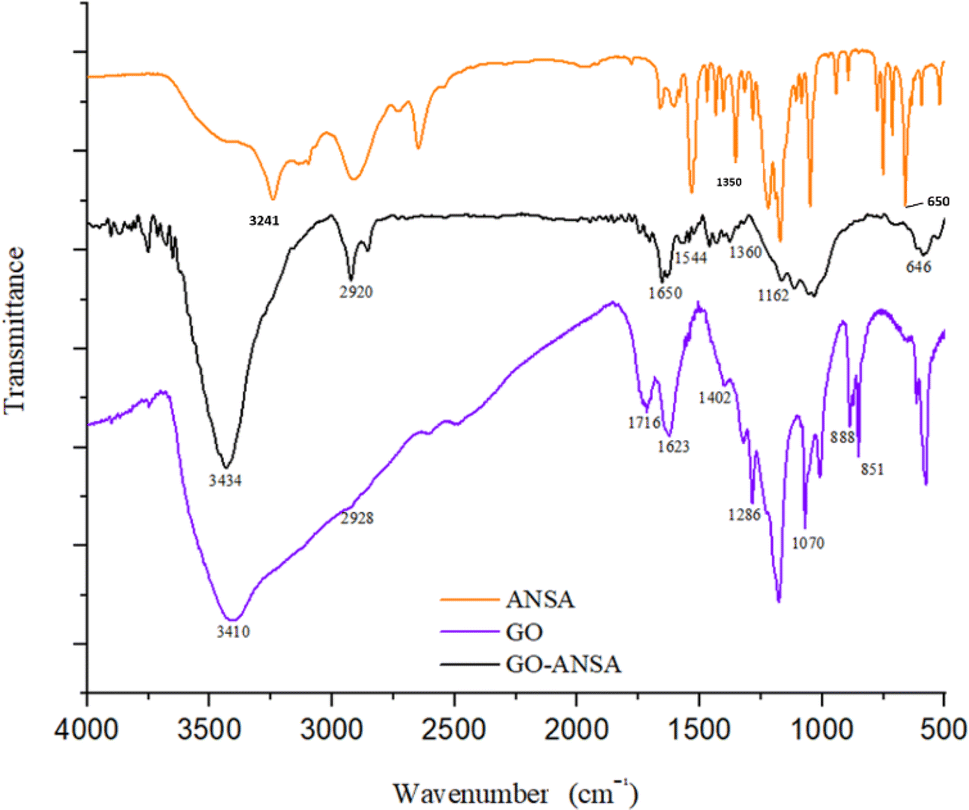
FTIR spectra of GO, ANSA, and GO-ANSA (1) are shown in Fig. 3. In the FTIR spectrum of GO, the broad bands at 2400 cm−1 to 3600 cm−1 are related to the O–H group of carboxylic, alcoholic or phenolic functional groups.91 The stretching vibrations at 1623 cm−1 and 1716 cm−1 are related to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds in aromatic rings and the CO bond in carboxylic acid, respectively.92 The bonds at 1178 cm−1 and 1070 cm−1 show the vibrational stretching of the C–O bond in the phenol and alcohol groups, respectively.93 In the FTIR spectrum of GO-ANSA (1), there was a broad peak at 3100 cm−1 to 3600 cm−1, which shows the vibrational stretching of O–H in sulfonic acid and hydroxyl groups as well as N–H in amide groups. Therefore, the carboxylic acid groups of GO were converted to the corresponding ones. The CH2 asymmetric stretching vibration was observed at 2920 cm−1.91 The band that appeared at 1650 cm−1 showed the stretching vibration of the carbonyl groups in amide and confirmed the existence of amide functional group in the GO-ANSA. Also, the absorption band at 1544 cm−1 can be attributed to a combination of two absorptions related to the stretching vibration of C–N bond and the bending vibration of N–H bond in the secondary amide structures.94
C bonds in aromatic rings and the CO bond in carboxylic acid, respectively.92 The bonds at 1178 cm−1 and 1070 cm−1 show the vibrational stretching of the C–O bond in the phenol and alcohol groups, respectively.93 In the FTIR spectrum of GO-ANSA (1), there was a broad peak at 3100 cm−1 to 3600 cm−1, which shows the vibrational stretching of O–H in sulfonic acid and hydroxyl groups as well as N–H in amide groups. Therefore, the carboxylic acid groups of GO were converted to the corresponding ones. The CH2 asymmetric stretching vibration was observed at 2920 cm−1.91 The band that appeared at 1650 cm−1 showed the stretching vibration of the carbonyl groups in amide and confirmed the existence of amide functional group in the GO-ANSA. Also, the absorption band at 1544 cm−1 can be attributed to a combination of two absorptions related to the stretching vibration of C–N bond and the bending vibration of N–H bond in the secondary amide structures.94
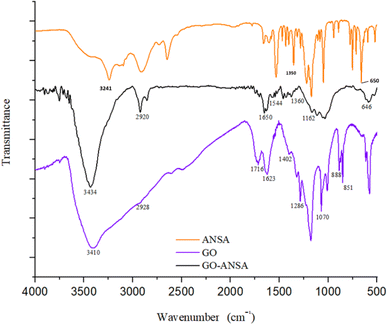 |
| | Fig. 3 FT-IR spectra of GO, ANSA, and GO-ANSA. | |
The XRD pattern for GO showed a peak with a very high intensity at 2θ = 9.3° (001) and another peak at about 2θ = 42.0° (100), which confirmed the successful formation of GO.95,96 After the functionalization of GO, the intensity of its corresponding peak was reduced in the XRD pattern of GO-ANSA (1). Both peaks were slightly shifted, which indicated the formation of a covalent bond between the functional groups of GO and ANSA. These observations are in agreement with the literature data on covalent-functionalization of GO by using different organic groups.97–102 GO and GO-ANSA interlayer d spacing is 0.69 and 0.95 nm, respectively. Also, a new peak appeared at 2θ = 25.0°, which was due to the addition of ANSA compound to GO (Fig. 4).
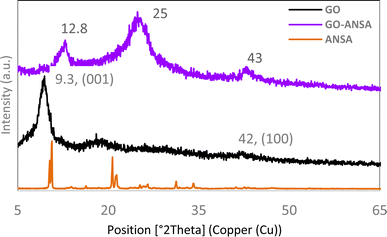 |
| | Fig. 4 XRD patterns of ANSA, GO and GO-ANSA (1) are shown from bottom to top. | |
Atomic force microscopy (AFM) was used to visualize the surface morphology of the sample and determine the maximum peak height—Rmax, which is the highest peak above the mean line in the profile. The scanned area was 2 × 2 μm2. The large-area GO's and GO-ANSA's AFM images and the corresponding deposited on a mica substrate are shown in Fig. 5. From the set of FE-SEM and AFM photographs taken, the most characteristic and repeatable images of the sample surface were selected for presentation. GO-ANSA's AFM images demonstrated that the morphology of GO changed after the catalyst preparation process and required calculations. The thickness of GO sheets was found to be 1.2 nm from the AFM images (Fig. 5e). However, the thickness increased to 2.1 nm in GO-ANSA, as shown in Fig. 5f.103 FESEM images were used to determine the morphology of GO and GO-ANSA (1) (Fig. 5). The FESEM images of prepared GO showed the formation of its layered structure. The two-dimensional and layered structure of GO is a consequence of utilizing of the Hummers' method by starting from graphite with a three-dimensional structure (Fig. 5a and b). GO-ANSA's FESEM images showed the morphology of GO changed after the catalyst preparation process (Fig. 5c and d).
 |
| | Fig. 5 FE-SEM images (a and b) for GO and (c and d) for GO-ANSA; atomic force microscopy measurement of (e) GO and (f) GO-ANSA (scanned area: 2 μm × 2 μm). | |
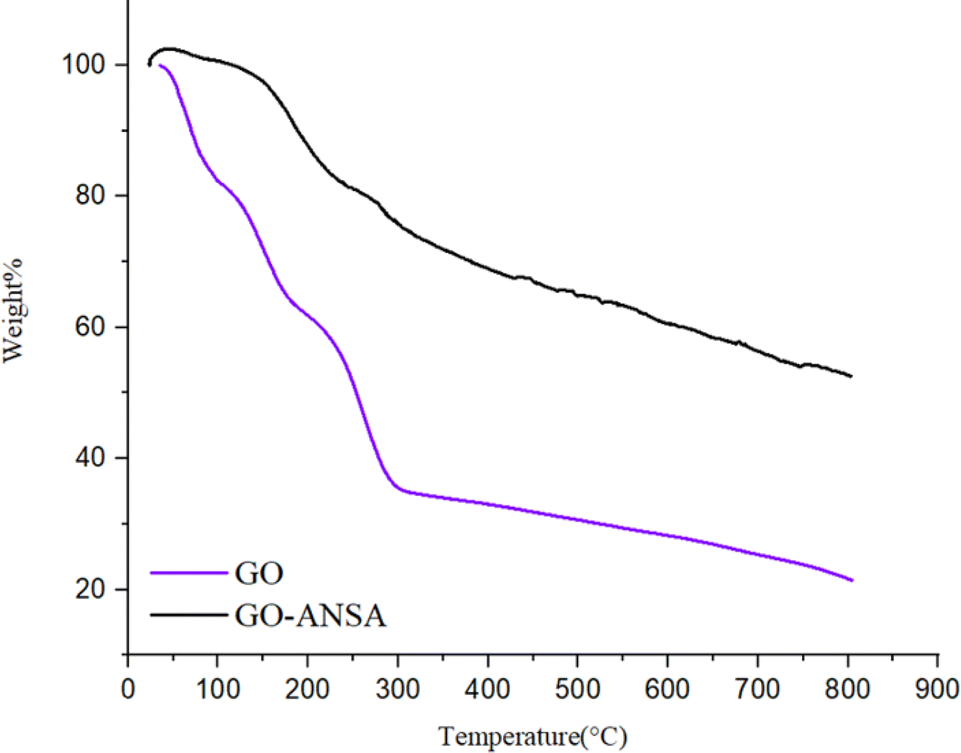
Thermogravimetric analysis (TGA) and differential thermogravimetric analysis (DTA) were carried out in the inert atmosphere of Ar at the range of 25–800 °C for both GO and GO-ANSA (1) (Fig. 6). The TGA diagram of GO showed three sharp decreases in the initial weight. The decrease of sample's weight before 100 °C and between 100–200 °C was related to the removal of absorbed water and pyrolysis of groups containing oxygen in GO (as CO and CO2), respectively.104 The third sharp drop occurred at 250–300 °C and can be attributed to the loss of carbon framework connections.105 In the TGA of GO-ANSA, the increase in weight shown before 50 °C is due to the Buoyancy effects as well as absorption of water and CO2. Also, the drop in weight at 150–200 °C is related to the removal of absorbed water. After that, the weight of the sample decreased with a gentle slope, which is due to the decomposition of functional groups in GO-ANSA and the desulfonation of SO3H groups. After reaching 800 °C, GO lost more than 80% of its initial weight. Meanwhile, up to 50% of the weight of the GO-ANSA sample remained at the same thermal conditions. Therefore, the thermal stability of GO-ANSA is greatly increased by covalent bonding.
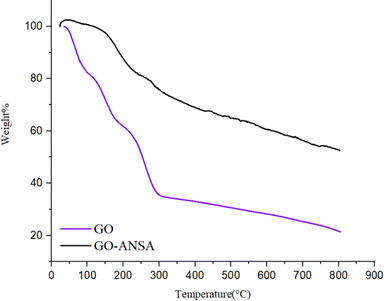 |
| | Fig. 6 The TGA analysis of GO and GO-ANSA (1). | |
3.2. Catalytic activity assessment of the GO-ANSA nanomaterial (1)
To show the efficiency of GO-ANSA (1) and determine the optimized conditions, the three-component reaction of 4-chlorobenzaldehyde (2a, 1.0 mmol) and dimedone (3b, 2.0 mmol) for synthesis of the corresponding tetraketone (2,2′-((4-chlorophenyl)methylene)bis(3-hydroxy-5,5-dimethylcyclohex-2-en-1-one), 4a) was studied as the model reaction. The results are summarized in Table 1. Therefore, various parameters including the type of solvent, amount of the catalyst loading, and temperature of the reaction were evaluated on the model reaction. Initially, the reaction was carried out without a catalyst and only a trace of the desired product 4a was formed after 3 h (Table 1, entry 1). Then, GO was used in EtOH as a solvent, which resulted in a moderate isolated yield of 58% (entry 2). Furthermore, the reaction was carried out in the presence of GO-ANSA nanomaterial (1) in both water and EtOH under reflux conditions (entries 3 and 4) with higher efficiencies than the reaction in other solvents including THF, CH2Cl2, MeCN, MeOH and EtOAc (entries 6–10). In conclusion, EtOH as an effective and green solvent under reflux conditions is the best medium for the reaction. Therefore, all subsequent experiments to investigate the catalytic activity of GO-ANSA nanomaterial (1) were performed in EtOH. By increasing the amount of catalyst loadings in the model reaction, due to more concentration of acidic sites, the reaction efficiency reached the maximum efficiency (97%) in the minimum possible time (15 min). Indeed, increasing the catalyst loading to 15.0 mg resulted in higher production of the desired product 4a in a shorter reaction time under the same conditions (entry 14). However, a further increase in the amount of loaded catalyst 1 did not significantly affect the yield and reaction time (entry 15).
Table 1 Optimization of the three-component reaction for the synthesis of 4a catalyzed by the GO-ANSAa
| Entry |
Catalyst 1 loading (mg) |
Solvent |
Temp. (°C) |
Time (min) |
Yieldb (%) |
|
Reaction conditions: 4-chlorobenzaldehyde (2, 1.0 mmol) and dimedone (3b, 2.0 mmol) catalyzed by GO-ANSA (1).
Yields refer to the isolated products.
GO was used instead of GO-ANSA.
|
| 1 |
— |
EtOH |
100 |
180 |
Trace |
| 2 |
10c |
EtOH |
Reflux |
60 |
58 |
| 3 |
10 |
H2O |
Reflux |
150 |
88 |
| 4 |
10 |
EtOH |
Reflux |
35 |
93 |
| 5 |
10 |
EtOH/H2O (1:1) |
Reflux |
140 |
91 |
| 6 |
10 |
THF |
Reflux |
330 |
70 |
| 7 |
10 |
CH2Cl2 |
Reflux |
360 |
42 |
| 8 |
10 |
MeCN |
Reflux |
270 |
82 |
| 9 |
10 |
MeOH |
Reflux |
180 |
85 |
| 10 |
10 |
EtOAc |
Reflux |
120 |
78 |
| 11 |
10 |
EtOH |
R.T. |
180 |
43 |
| 12 |
10 |
EtOH |
50 |
120 |
55 |
| 13 |
5 |
EtOH |
Reflux |
45 |
89 |
| 14 |
15 |
EtOH |
Reflux |
20 |
97 |
| 15 |
20 |
EtOH |
Reflux |
20 |
96 |
Different derivatives of tetraketone 4a–p were obtained under the optimized reaction conditions (4-amino-3-hydroxy-1-naphthalene sulfonic acid-functionalized GO (GO-ANSA) loading of 15.0 mg in EtOH under reflux conditions) using various aldehydes and enolizable compounds with very high efficiency (Table 2). The results showed that the desired products 4 were prepared in excellent yields and short reaction times. Furthermore, the presence of electron-withdrawing groups on the aromatic ring of aldehyde has a favourable effect on the reaction time compared to electron-donating groups. In addition, the presence of halogen atoms on the aromatic ring of the aldehyde acted as electron-withdrawing groups due to predominance of inductive effects on resonance ones. Therefore, the presence of the halogen atoms on the aromatic ring increases the yield and reduces the required reaction time. Furthermore, sensitive aldehydes having CC bonds such as cinnamaldehyde survived very well under the optimized conditions and afforded corresponding tetraketone (4j) in high yield without polymerization.106
Table 2 Synthesis of tetraketone derivatives 4a–p from different aldehydes 2 and enolizable compounds 3a–b catalyzed by the GO-ANSA (1), as a heterogeneous nanocatalyst, under the optimized conditionsa
| Entry |
R–CHO 2 |
R
2
|
Time (min) |
Product 4 |
Yieldb (%) |
Mp (°C) found |
Mp (°C) reported |
|
Condition of reaction: different aldehydes (2, 1.0 mmol) and enolizable compounds (3a–b, 2.0 mmol) catalyzed by the GO-ANSA (1) loading of 15.0 mg in EtOH under the reflux condition.
Yields refer to isolated products.
|
| 1 |
4-Chlorobenzaldehyde |
3b
|
20 |
4a
|
97 |
145–146 |
140–142 (ref. 107) |
| 2 |
2-Chlorobenzaldehyde |
3b
|
40 |
4b
|
90 |
204–206 |
205 (ref. 108) |
| 3 |
4-Bromobenzaldehyde |
3b
|
25 |
4c
|
94 |
157 |
158–159 (ref. 109) |
| 4 |
4-Formylbenzonitrile |
3b
|
20 |
4d
|
96 |
165–168 |
165–167 (ref. 109) |
| 5 |
4-Hydroxybenzaldehyde |
3b
|
40 |
4e
|
88 |
187–188 |
188–190 (ref. 109) |
| 6 |
4-Methoxybenzaldehyde |
3b
|
35 |
4f
|
88 |
141 |
142–143 (ref. 83) |
| 7 |
2-Methoxybenzaldehyde |
3b
|
45 |
4g
|
89 |
179–181 |
187–188 (ref. 108) |
| 8 |
4-Nitrobenzaldehyde |
3b
|
20 |
4h
|
96 |
188–190 |
188–190 (ref. 110) |
| 9 |
3-Nitrobenzaldehyde |
3b
|
30 |
4i
|
91 |
195–198 |
197–198 (ref. 71) |
| 10 |
(Z)-3-Phenylacrylaldehyde (cinnamaldehyde) |
3b
|
45 |
4j
|
86 |
215–217 |
215–217 (ref. 108) |
| 11 |
4-Chlorobenzaldehyde |
3a
|
20 |
4k
|
96 |
205 |
204–206 (ref. 111) |
| 12 |
2-Chlorobenzaldehyde |
3a
|
45 |
4l
|
87 |
230–232 |
229–231 (ref. 111) |
| 13 |
4-Bromobenzaldehyde |
3a
|
25 |
4m
|
92 |
239–242 |
240–241 (ref. 83) |
| 14 |
4-Hydroxybenzaldehyde |
3a
|
45 |
4n
|
88 |
192–195 |
192–194 (ref. 111) |
| 15 |
4-Methoxybenzaldehyde |
3a
|
35 |
4o
|
86 |
196 |
193–195 (ref. 111) |
| 16 |
3-Nitrobenzaldehyde |
3a
|
35 |
4p
|
90 |
207–211 |
209–210 (ref. 112) |
To demonstrate the superior catalytic activity of GO-ANSA (1) for the synthesis of tetraketone derivatives under the optimized conditions, the obtained results have been compared with the previous methods reported in the literature (Table 3).
Table 3 Comparison of the catalytic efficiency of GO-ANSA with some previously reported heterogenous or homogeneous catalysts
| Entry |
Catalyst |
Catalyst loading |
Conditions |
Time |
Yield (%) |
| 1 |
NaOH113 |
5 mg |
H2O/sonic./R.T. |
60–90 min |
65–84 |
| 2 |
HClO4–SiO2 (ref. 107) |
50 mg |
MeCN/reflux |
6 h |
54 |
| 3 |
TiO2/SO42− (ref. 114) |
100 mg |
Solvent free/R.T. |
48 h |
38–90 |
| 4 |
L-Histidine/ionic liquid69 |
30 mg |
[bmim]BF4/60 °C |
40 min |
60–75 |
| 5 |
Nano SiO2Cl70 |
10 mg |
CHCl3/40 °C |
1–4 h |
70–90 |
| 6 |
Amino-appended β-cyclodextrin71 |
64 mg |
H2O/R.T. |
1–5 h |
58–96 |
| 7 |
Nano Cu–SiO2 (ref. 115) |
100 mg |
EtOH/80 °C |
30–75 min |
90–97 |
| 8 |
Cu2(NH2-BDC)2(DABCO)72 |
40 mg |
H2O/reflux |
30 min |
98 |
| 9 |
Fe-zeolite73 |
30 mg |
Toluene/160 °C |
8 h |
84 |
| 10 |
2-Aminopyrazine74 |
285 mg |
ACN/80 °C |
2 h |
76–86 |
| 11 |
GO-ANSA |
15 mg |
EtOH/reflux |
20–45 min |
86–97 |
Additionally, to evaluate the effectiveness of the optimized conditions and catalytic performance of GO-ANSA (1), another model reaction was chosen involving multi-component heteroannulation of 4-chlorobenzaldehyde (2), dimedone (3b) and malononitrile (5) in a 1:1:1 molar ratio for the synthesis of tetrahydrobenzo[b]pyrans. The results are summarized in Table 4. Although the reaction can be performed in the presence of GO, as a catalyst rather than a support, it afforded a low yield and required a longer reaction time (entry 1). Interestingly, in the synthesis of tetrahydrobenzo[b]pyran derivatives, the best catalytic performance was observed in EtOH under reflux conditions similar to tetraketones (entries 2–8). The effect of temperature on the yield of model reaction was investigated (entries 9 and 10). The optimal quantity of catalyst 1 loading was also studied in the next experiments and the obtained data showed that the yield of the reaction was improved by increasing the amount of GO-ANSA (1) loading in the reaction mixture (entries 11–13). However, increasing the amount of loaded catalyst further than 10.0 mg did not significantly impact the yield of the reaction or have an adverse effect on the reaction time (entry 13).
Table 4 Optimization of the three-component reaction for synthesis of 6b catalyzed by the GO-ANSAa
| Entry |
Catalyst loading (mg) |
Solvent |
Temp. (°C) |
Time (min) |
Yieldb (%) |
|
Reaction conditions: 4-chlorobenzaldehyde (2, 1.0 mmol), dimedone (3b, 1.0 mmol) and malononitrile (5, 1.0 mmol) catalyzed by the GO-ANSA (1).
Yields refer to the isolated products.
GO was used instead of GO-ANSA.
|
| 1 |
10c |
EtOH |
Reflux |
180 |
39 |
| 2 |
10 |
H2O |
Reflux |
60 |
71 |
| 3 |
10 |
EtOH |
Reflux |
25 |
94 |
| 4 |
10 |
EtOH/H2O (1:1) |
Reflux |
50 |
82 |
| 5 |
10 |
THF |
Reflux |
220 |
40 |
| 6 |
10 |
MeCN |
Reflux |
150 |
54 |
| 7 |
10 |
MeOH |
Reflux |
210 |
50 |
| 8 |
10 |
EtOAc |
Reflux |
240 |
38 |
| 9 |
10 |
EtOH |
R.T. |
260 |
44 |
| 10 |
10 |
EtOH |
50 |
140 |
67 |
| 11 |
2.5 |
EtOH |
Reflux |
70 |
86 |
| 12 |
5 |
EtOH |
Reflux |
50 |
89 |
| 13 |
15 |
EtOH |
Reflux |
30 |
95 |
Different tetrahydrobenzo[b]pyran derivatives 6a–k were obtained under optimized reaction conditions (GO-ANSA loading of 10.0 mg in EtOH under reflux conditions) using various aldehydes, dimedone or 1,3-cyclohexanedione (3a–b), and malononitrile with very high efficiency (Table 5). After completion of the reaction, the nanocatalyst 1 was easily separated from the reaction mixture by filtration. All tetrahydrobenzo[b]pyran derivatives were prepared in high to excellent yields and very short times. The rate of reactions and efficiency were significantly affected by the nature of the functional groups on the aromatic ring of aldehydes. Consequently, the preparation of tetrahydrobenzo[b]pyrans by using aldehydes with electron-withdrawing groups requires shorter times than aldehydes with electron-donating groups.
Table 5 Synthesis of tetrahydrobenzo[b]pyran derivatives 6a–k from different aldehydes 2, enolizable compounds 3a–b and malononitrile (5) catalyzed by the GO-ANSA (1), as heterogeneous nanocatalyst, under optimized conditionsa
| Entry |
R–CHO 2 |
R
2
|
Product 6 |
Time (min) |
Yieldb (%) |
Mp (°C) found |
Mp (°C) reported |
|
Reaction conditions: different aldehydes (2, 1.0 mmol), enolizable compounds (3a–b, 1.0 mmol) and malononitrile (5, 1.0 mmol) catalyzed by the GO-ANSA (1) loading of 10.0 mg in EtOH under reflux conditions.
Yields refer to the isolated products.
|
| 1 |
Benzaldehyde |
3b
|
6a
|
30 |
89 |
230–232 |
231–233 (ref. 76) |
| 2 |
4-Chlorobenzaldehyde |
3b
|
6b
|
25 |
94 |
210–212 |
209–211 (ref. 79) |
| 3 |
4-Formylbenzonitrile |
3b
|
6c
|
25 |
92 |
211 |
212–213 (ref. 76) |
| 4 |
3-Nitrobenzaldehyde |
3b
|
6d
|
15 |
96 |
214–216 |
214–216 (ref. 80) |
| 5 |
4-Methoxybenzaldehyde |
3b
|
6e
|
35 |
91 |
187–189 |
186–188 (ref. 76) |
| 6 |
Benzaldehyde |
3a
|
6f
|
45 |
85 |
267–239 |
239–241 (ref. 82) |
| 7 |
4-Chlorobenzaldehyde |
3a
|
6g
|
35 |
91 |
226–229 |
224–226 (ref. 77) |
| 8 |
2-Chlorobenzaldehyde |
3a
|
6h
|
20 |
92 |
210–213 |
212–214 (ref. 77) |
| 9 |
4-Hydroxybenzaldehyde |
3a
|
6i
|
35 |
82 |
235–339 |
234–236 (ref. 82) |
| 10 |
4-Nitrobenzaldehyde |
3a
|
6j
|
30 |
90 |
234–237 |
234–236 (ref. 60) |
| 11 |
4-Methylbenzaldehyde |
3a
|
6k
|
50 |
84 |
224.5 |
223–225 (ref. 82) |
3.3. Reusability of the catalyst
To test the reusability of GO-ANSA (1) as a heterogeneous nanocatalyst, it was separated from the reaction mixture (4a and 6b) after completion of the reaction and washed with hot EtOH, then dried in an oven at 60 °C. The separated catalyst 1 was added to the model reaction. It was repeated five times under the same conditions and no significant change in the reaction yields was observed in both studied model reactions. The results in the synthesis of both tetraketone 4a and tetrahydrobenzo[b]pyran 6a derivatives are shown in Fig. 7. The recyclability of GO-ANSA catalyst (1) demonstrates that GO-ANSA could be used at least five times without meaningful loss in its catalytic performance.
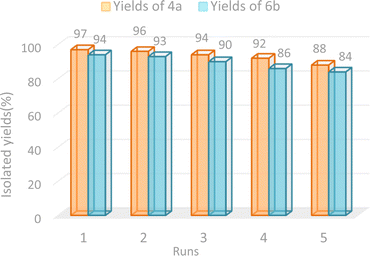 |
| | Fig. 7 Recycling results of GO-ANSA for the synthesis of 4a and 6b. | |
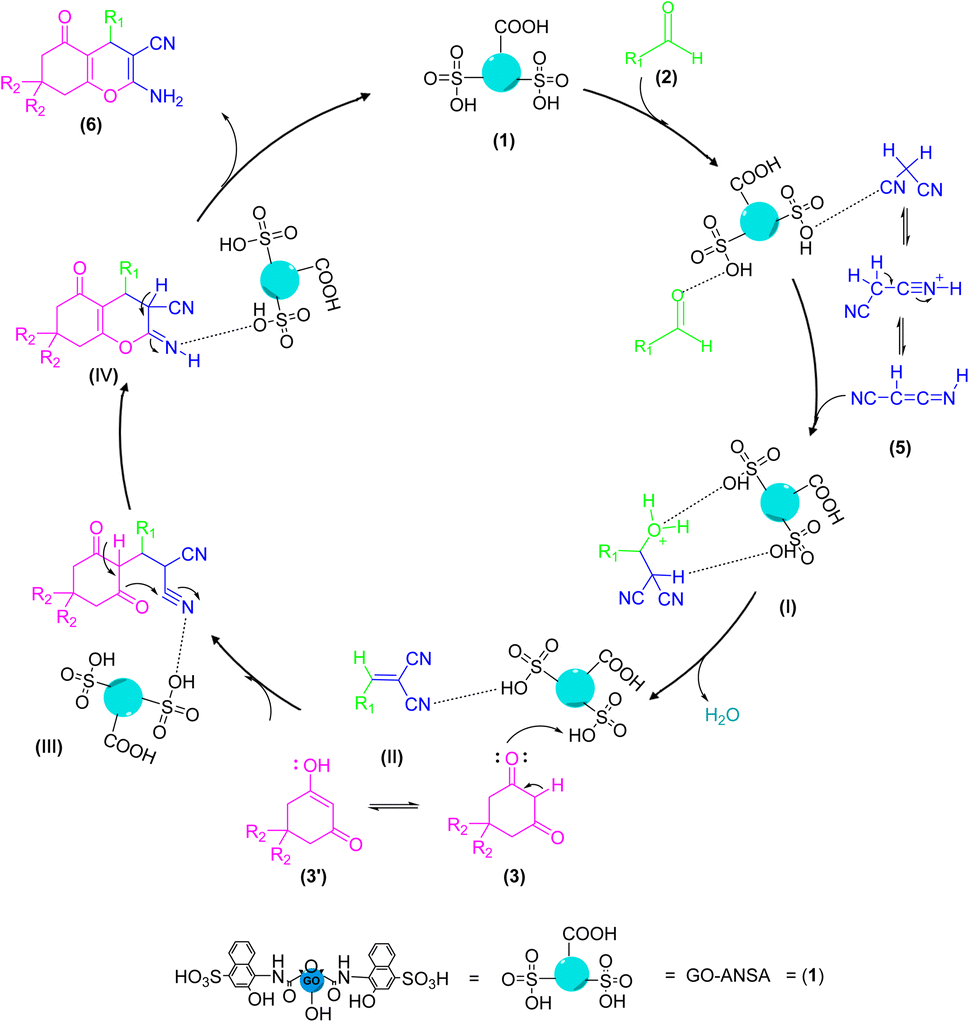
3.4. Proposed mechanism
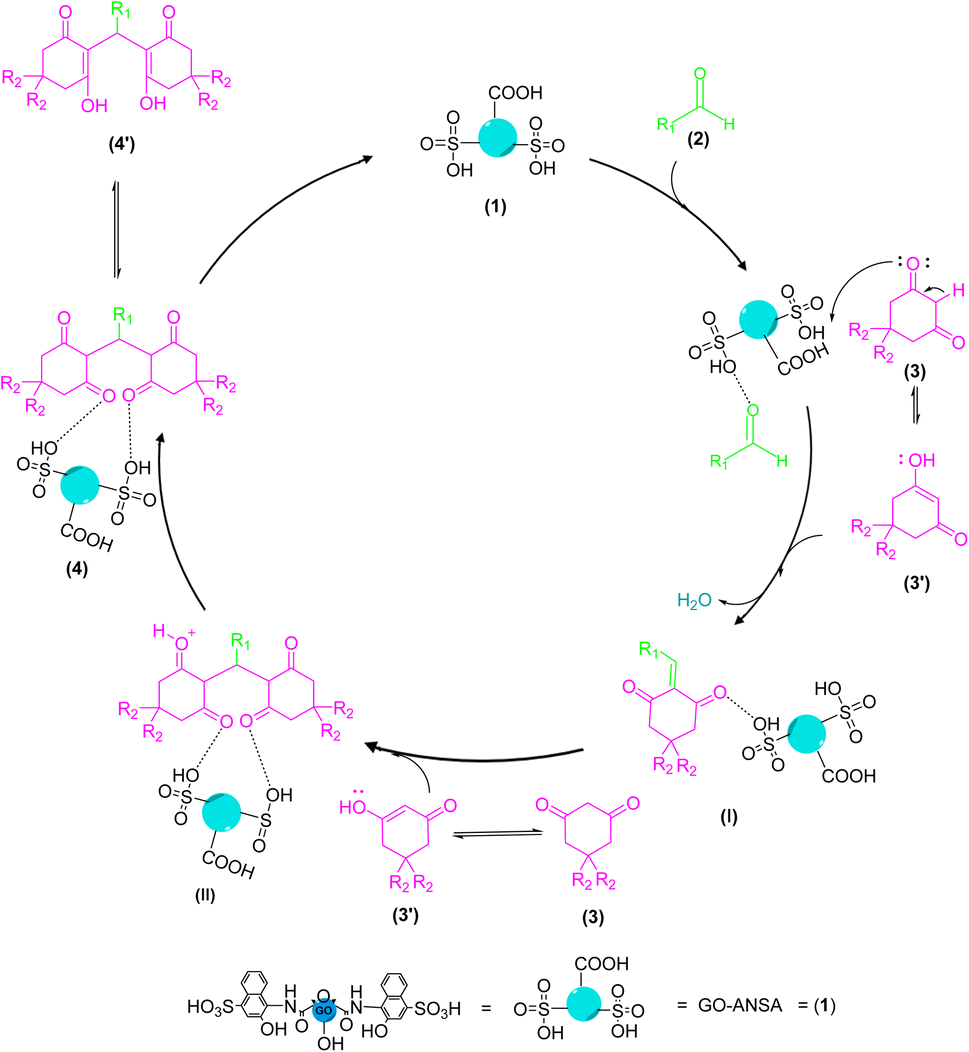
The proposed mechanism for the GO-ANSA (1)-catalyzed reaction of aldehydes and two equivalent of enolizable compounds is shown in Scheme 3. GO-ANSA contains SO3H and carboxylic acid functional groups in its structure and leading to protonation of the carbonyl group of aldehydes 2, thus increasing its nucleophilicity.88 On the other hand, enolizable compounds are in equilibrium with their enolic form and the acidic catalyst accelerates this process. Then, by removing a water molecule, the Knoevenagel intermediate (I) is formed. Another equivalent of the enolizable compound 3, after taking proton from the catalyst 1 and subsequent transformation into its enolic form), reacts with the intermediate I to form the intermediate IIvia Michael addition. After formation of the tetraketone compounds 4, it can be in equilibration with its enol tautomer 4′ (Scheme 3).83
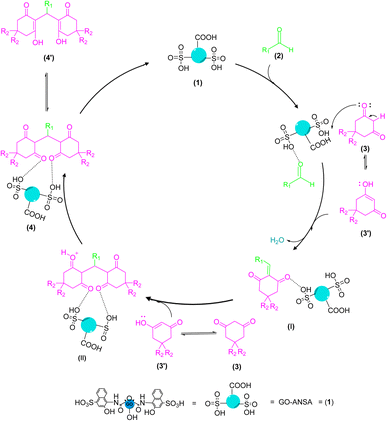 |
| | Scheme 3 A proposed mechanism for the synthesis of tetraketone derivatives 4 catalyzed by the GO-ANSA (1). | |
The proposed mechanism for the GO-ANSA (1)-catalyzed one-pot reaction of aldehydes 2, enolizable compounds 3, and malononitrile (5) is shown in Scheme 4. Protonation of all three compounds, i.e., carbonyl groups of aldehydes, as well as enolizable compounds, and nitrile of malononitrile, occurred through the SO3H groups present in the structure of the nanocatalyst 1. This initial activation accelerates the Knoevenagel condensation of malononitrile (its imine–ketene resonance form) and aldehydes to afford intermediate I. Then, by removing a water molecule, intermediate II was formed through Knoevenagel condensation. The addition of the activated enolizable compound with this intermediate forms the intermediate III through Michael addition. Eventually, the final product 6 is obtained by through heteroannulation of the intermediate III and subsequent tautomerization of the intermediate IV.78
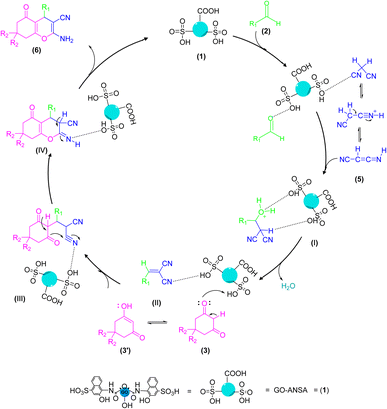 |
| | Scheme 4 A proposed mechanism for the synthesis of tetrahydrobenzo[b]pyran derivatives 6 catalyzed by the GO-ANSA (1). | |
4. Conclusions
In this research, GO-ANSA was prepared as a new reusable carbocatalyst. The efficiency of this new nano-ordered catalyst was evaluated for the green synthesis of tetraketone as well as tetrahydrobenzo[b]pyran derivatives through one-pot MCRs in EtOH as a green solvent. The GO-ANSA catalyst showed high efficiency in both reactions and all of the derivatives were obtained in high to excellent yields and short reaction times. In this work, we used a novel heterogeneous nanocatalyst, which showed very beneficial activity compared to the previously reported methods in the literature. In addition, the catalyst was easily recovered and reused at least five times without a significant decrease in its activity. Among the advantages of the presented method, other features including high yield of the products without the formation of by-products, simple process, and mild and eco-friendly reaction conditions can be mentioned.
Author contributions
Sara Gharghish: writing – original draft, software, methodology, investigation, formal analysis, data curation. Mohammad G. Dekamin: writing – review & editing, visualization, validation, supervision, resources, project administration, funding acquisition, conceptualization. Sepideh Hasanzadeh Banakar: methodology.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We are grateful for the partial financial support from The Research Council of Iran University of Science and Technology (IUST), Tehran, Iran (grant no 160/23372). We would also like to acknowledge the support of The Iran Nanotechnology Initiative Council (INIC), Iran.
Notes and references
- R. Yang, Y. Wang, D. Wu, Y. Deng, Y. Luo, X. Cui, X. Wang, Z. Shu and C. Yang, ACS Nano, 2017, 11, 7710–7718 CrossRef CAS
 .
.
- E. Alonso, F. R. Field and R. E. Kirchain, Environ. Sci. Technol., 2012, 46, 12986–12993 CrossRef CAS .
- A. H. C. Hart, R. Koizumi, J. Hamel, P. S. Owuor, Y. Ito, S. Ozden, S. Bhowmick, S. A. S. Amanulla, T. Tsafack, K. Keyshar, R. Mital, J. Hurst, R. Vajtai, C. S. Tiwary and P. M. Ajayan, ACS Appl. Mater. Interfaces, 2017, 9, 13742–13750 CrossRef CAS .
- S. Hao, B. Ouyang, C. Li, B. Zhang, J. Feng, J. Wu, M. Srinivasan and Y. Huang, J. Phys. Chem. C, 2019, 123, 8599–8606 CrossRef CAS .
- N. Zulfiqar, R. Nadeem and O. A. Musaimi, ACS Omega, 2024, 9, 7986–8004 CAS .
- R. K. Sharma, S. Gulati and S. Mehta, J. Chem. Educ., 2012, 89, 1316–1318 CrossRef CAS .
- R. P. Sourkouhi, M. G. Dekamin, E. Valiey and M. Dohendou, Carbohydr. Polym. Technol. Appl., 2024, 7, 100420 CAS .
- S. J. Jeon, H. Li and P. J. Walsh, J. Am. Chem. Soc., 2005, 127, 16416–16425 CrossRef CAS .
- M. Toorbaf, L. Moradi and A. Dehghani, J. Mol. Struct., 2023, 1294, 136335 CrossRef CAS .
- M. G. Dekamin, M. Azimoshan and L. Ramezani, Green Chem., 2013, 15, 811–820 RSC .
- J. W. J. Ang, J. Chem. Educ., 2021, 98, 203–207 CrossRef CAS .
- A. Mishra, P. Yadav and S. K. Awasthi, ACS Org. Inorg. Au, 2023, 3, 254–265 CrossRef CAS .
- N. Nikooei, M. G. Dekamin and E. Valiey, Res. Chem. Intermed., 2020, 46, 3891–3909 CrossRef CAS .
- C. Wang, R. Ciganda, L. Yate, J. Tuninetti, V. Shalabaeva, L. Salmon, S. Moya, J. Ruiz and D. Astruc, J. Mater. Chem. A, 2017, 5, 21947–21954 RSC .
- M. S. Islam, S. M. Sarkar, M. L. Rahman, K. Hasan and E. J. O'Reilly, Chem. Eng. J., 2024, 483, 149271 CrossRef CAS .
- N. Ghanbari and H. Ghafuri, Heliyon, 2023, 9, e20978 CrossRef CAS .
- B. Zheng, L. Chen, L. He, H. Wang, H. Li, H. Zhang and S. Yang, Ind. Crops Prod., 2024, 210, 118058 CrossRef CAS .
- H. M. A. Sharif, A. Mahmood, H. Y. Cheng, R. Djellabi, J. Ali, W. L. Jiang, S. S. Wang, M. R. Haider, N. Mahmood and A. J. Wang, ACS Appl. Nano Mater., 2019, 2, 5310–5319 CrossRef CAS .
- Z. Alirezvani, M. G. Dekamin, F. Davoodi and E. Valiey, ChemistrySelect, 2018, 3, 10450–10463 CrossRef CAS .
- Y. H. Song, Q. T. Xu, T. He, Z. Y. Wang and L. Yu, ACS Omega, 2020, 5, 21137–21144 CrossRef CAS .
- C. L. Weaver, J. M. Larosa, X. Luo and X. T. Cui, ACS Nano, 2014, 8, 1834–1843 CrossRef CAS .
- Z. Zhang, M. Wang, D. Gao, D. Luo, Q. Liu, J. Yang and Y. Li, Langmuir, 2016, 32, 10253–10258 CrossRef CAS .
- G. P. Kotchey, B. L. Allen, H. Vedala, N. Yanamala, A. A. Kapralov, Y. Y. Tyurina, J. Klein-Seetharaman, V. E. Kagan and A. Star, ACS Nano, 2011, 5, 2098–2108 CrossRef CAS .
- Y. Zhou, K. Maleski, B. Anasori, J. O. Thostenson, Y. Pang, Y. Feng, K. Zeng, C. B. Parker, S. Zauscher, Y. Gogotsi, J. T. Glass and C. Cao, ACS Nano, 2020, 14, 3576–3586 CrossRef CAS .
- M. Cobos, I. De-La-pinta, G. Quindós, M. D. Fernández and M. J. Fernández, Nanomaterials, 2020, 10, 376 CrossRef CAS .
- W. Dai, Y. Liu, M. Wang, M. Lin, X. Lian, Y. Luo, J. Yang and W. Chen, ACS Appl. Mater. Interfaces, 2021, 13, 19915–19926 CrossRef CAS .
- D. D. Kulkarni, I. Choi, S. S. Singamaneni and V. V. Tsukruk, ACS Nano, 2010, 4, 4667–4676 CrossRef CAS .
- C. H. Rodríguez, J. d. J. P. Bueno, A. X. M. Pérez, M. R. Flores and G. Oza, RSC Adv., 2023, 13, 10621–10635 RSC .
- S. Rana, G. B. B. Varadwaj and S. B. Jonnalagadda, Nanoscale Adv., 2019, 1, 1527–1530 RSC .
- M. Eslami, M. G. Dekamin and E. Mahdavi, Surf. Interfaces, 2024, 104363 CrossRef .
- D. Sah, J. Shabir, S. Surabhi, P. Gupta and S. Mozumdar, Dalton Trans., 2021, 50, 5644–5658 RSC .
- Z. Zhang, X. Xiao, Y. Zhou, L. Huang, Y. Wang, Q. Rong, Z. Han, H. Qu, Z. Zhu, S. Xu, J. Tang and J. Chen, ACS Nano, 2021, 15, 13178–13187 CrossRef CAS .
- D. Bradley, S. Sarpaki, V. Mirabello, S. G. Giuffrida, G. I. Kociok-Köhn, D. G. Calatayud and S. I. Pascu, Nanoscale Adv., 2024, 6, 2287–2305 RSC .
- S. Caputo, A. Kovtun, F. Bruno, E. Ravera, C. Lambruschini, M. Melucci and L. Moni, RSC Adv., 2022, 12, 15834–15847 RSC .
- K. Taheri, D. Elhamifar, S. Kargar and A. Zarnegaryan, RSC Adv., 2023, 13, 16067–16077 RSC .
- J. Soni, N. Sahiba, A. Sethiya, P. Teli, D. K. Agarwal, A. Manhas, P. C. Jha, D. Joshi and S. Agarwal, Polycyclic Aromat. Compd., 2022, 42, 2970–2990 CrossRef CAS .
- Z. Ghadamyari, A. Khojastehnezhad, S. M. Seyedi and A. Shiri, ChemistrySelect, 2019, 4, 10920–10927 CrossRef CAS .
- Z. Ghadamyari, A. Khojastehnezhad, S. M. Seyedi, F. Taghavi and A. Shiri, ChemistrySelect, 2020, 5, 10233–10242 CrossRef CAS .
- A. Khojastehnezhad, M. Bakavoli, A. Javid, M. M. K. Siuki and M. Shahidzadeh, Res. Chem. Intermed., 2019, 45, 4473–4485 CrossRef CAS .
- Z. Ghadamyari, A. Shiri, A. Khojastehnezhad and S. M. Seyedi, Appl. Organomet. Chem., 2019, 33, 1–10 CrossRef .
- S. Paul, S. Das, B. Mitra, G. C. Pariyar and P. Ghosh, RSC Adv., 2023, 13, 5457–5466 RSC .
- M. G. Dekamin and M. Eslami, Green Chem., 2014, 16, 4914–4921 RSC .
- S. D. Pasuparthy and B. Maiti, ACS Omega, 2022, 7, 39147–39158 CrossRef CAS .
- S. Allameh, A. Davoodnia and A. Khojastehnezhad, Chin. Chem. Lett., 2012, 23, 17–20 CrossRef CAS .
- A. Yaghoubi, M. G. Dekamin and B. Karimi, Catal. Lett., 2017, 147, 2656–2663 CrossRef CAS .
- A. Malihishoja, M. G. Dekamin and M. Eslami, RSC Adv., 2023, 13, 16584–16601 RSC .
- M. G. Dekamin, K. Varmira, M. Farahmand, S. Sagheb-Asl and Z. Karimi, Catal. Commun., 2010, 12, 226–230 CrossRef CAS .
- S. Kamalifar and H. Kiyani, Polycyclic Aromat. Compd., 2022, 42, 3675–3693 CrossRef CAS .
- H. Ostadzadeh and H. Kiyani, Polycyclic Aromat. Compd., 2023, 43, 9318–9337 CrossRef CAS .
- B. Borah, S. Swain, M. Patat, B. Kumar, K. K. Prajapat, R. Biswas, R. Vasantha and L. R. Chowhan, Sci. Rep., 2023, 13, 1–17 CrossRef .
- P. Teli, A. Sethiya and S. Agarwal, Res. Chem. Intermed., 2022, 48, 731–750 CrossRef CAS .
- S. Singh, A. Kumar, L. Nebhani and C. K. Hazra, JACS Au, 2023, 3, 3400–3411 CrossRef CAS .
- S. Siddiqui and Z. N. Siddiqui, Nanoscale Adv., 2020, 2, 4639–4651 RSC .
- B. Maleki, M. Raei, E. Akbarzadeh, H. Ghasemnejad-Bosra, A. Sedrpoushan, S. S. Ashrafi and M. N. Dehdashti, Org. Prep. Proced. Int., 2016, 48, 62–71 CrossRef CAS .
- F. M. Moghaddam, S. Aghili, M. Daneshfar, H. Moghimi and Z. Daneshfar, Res. Chem. Intermed., 2023, 49, 1507–1543 CrossRef CAS .
- G. M. Maharvi, S. Ali, N. Riaz, N. Afza, A. Malik, M. Ashraf, L. Iqbal and M. Lateef, J. Enzyme Inhib. Med. Chem., 2008, 23, 62–69 CrossRef CAS .
- K. M. Khan, G. M. Maharvi, M. T. H. Khan, A. J. Shaikh, S. Perveen, S. Begum and M. I. Choudhary, Bioorg. Med. Chem., 2006, 14, 344–351 CrossRef CAS .
- J. M. Khurana and K. Vij, J. Chem. Sci., 2012, 124, 907–912 CrossRef CAS .
- T. Josephrajan and V. T. Ramakrishnan, Can. J. Chem., 2007, 85, 572–575 CrossRef CAS .
- A. R. Shendabadi, H. Tebyanian, R. Zare, M. G. Dekamin, H. Kooshki and J. Rashidiani, Biointerface Res. Appl. Chem., 2020, 10, 6706–6717 CAS .
- Z. Akhlaghi, M. R. Naimi-Jamal, L. Panahi, M. G. Dekamin and B. F. Far, Heliyon, 2023, 9, e13522 CrossRef CAS .
- M. A. Zolfigol, A. Khazaei, A. R. Moosavi-Zare, J. Afsar, V. Khakyzadeh and O. Khaledian, J. Chin. Chem. Soc., 2015, 62, 398–403 CrossRef CAS .
- K. K. Gangu, S. Maddila, S. B. Mukkamala and S. B. Jonnalagadda, Ind. Eng. Chem. Res., 2017, 56, 2917–2924 CrossRef CAS .
- F. Kalantari, H. Esmailipour, H. Ahankar, A. Ramazani, H. Aghahosseini, O. Kaszubowski and K. Ślepokura, ACS Omega, 2023, 8, 25780–25798 CrossRef CAS .
- I. A. Azath, P. Puthiaraj and K. Pitchumani, ACS Sustain. Chem. Eng., 2013, 1, 174–179 CrossRef CAS .
- S. Agarwal, A. Sethiya, J. Soni, N. Sahiba and P. Teli, Appl. Organomet. Chem., 2022, 36, e6604 CrossRef CAS .
- P. Teli, N. Sahiba, A. Sethiya, J. Soni and S. Agarwal, J. Heterocycl. Chem., 2021, 58, 1393–1407 CrossRef CAS .
- M. Zabihzadeh, F. Shirini, H. Tajik and N. Daneshvar, Polycyclic Aromat. Compd., 2021, 41, 1972–1987 CrossRef CAS .
- Z. Yan and S. Zhicai, Chin. J. Chem., 2010, 28, 1184–1188 CrossRef .
- R. Karimian, F. Piri, B. Karimi and A. Moghimic, Croat. Chem. Acta, 2011, 84, 111–115 CrossRef CAS .
- Y. Ren, B. Yang and X. Liao, RSC Adv., 2016, 6, 22034–22042 RSC .
- A. Džambić, S. Muratović, E. Veljović, A. Softić, E. Dautović, M. Š. Husejnović, E. Horozić and A. Smajlović, Eur. Chem. Bull., 2020, 9, 285–290 CrossRef .
- S. Mondal, A. M. Pandey and B. Gnanaprakasam, React. Chem. Eng., 2022, 8, 855–862 RSC .
- D. Thakur, M. Kaur, D. S. Malhi, S. Garg, A. Sharma and H. S. Sohal, Monatsh. Chem., 2021, 152, 537–543 CrossRef CAS .
- M. G. Dekamin, S. Z. Peyman, Z. Karimi, S. Javanshir, M. R. Naimi-Jamal and M. Barikani, Int. J. Biol. Macromol., 2016, 87, 172–179 CrossRef CAS .
- A. Khazaee, R. Jahanshahi, S. Sobhani, J. Skibsted and J. M. Sansano, Green Chem., 2020, 22, 4604–4616 RSC .
- H. Alinezhad, M. Tarahomi, B. Maleki and A. Amiri, Appl. Organomet. Chem., 2019, 33, 1–14 Search PubMed .
- N. Rostami, M. G. Dekamin, E. Valiey and H. Fanimoghadam, Sci. Rep., 2022, 12, 1–12 CrossRef .
- B. Maleki and S. S. Ashrafi, RSC Adv., 2014, 4, 42873–42891 RSC .
- M. Kazemzad, A. A. Yuzbashi, S. Balalaie and M. Bararjanian, Synth. React. Inorg., Met.-Org., Nano-Met. Chem., 2011, 41, 1182–1187 CrossRef CAS .
- A. Yaghoubi and M. G. Dekamin, ChemistrySelect, 2017, 2, 9236–9243 CrossRef CAS .
- M. A. Nasseri and S. M. Sadeghzadeh, J. Iran. Chem. Soc., 2013, 10, 1047–1056 CrossRef CAS .
- S. H. Banakar, M. G. Dekamin and A. Yaghoubi, New J. Chem., 2018, 42, 14246–14262 RSC .
- M. G. Dekamin, F. Mehdipoor and A. Yaghoubi, New J. Chem., 2017, 41, 6893–6901 RSC .
- M. Ishani, M. G. Dekamin and Z. Alirezvani, J. Colloid Interface Sci., 2018, 521, 232–241 CrossRef CAS .
- F. Davoodi, M. G. Dekamin and Z. Alirezvani, Appl. Organomet. Chem., 2019, 33, e4735 CrossRef .
- A. Alinasab Amiri, S. Javanshir, Z. Dolatkhah and M. G. Dekamin, New J. Chem., 2015, 39, 9665–9671 RSC .
- M. G. Dekamin and Z. Mokhtari, Tetrahedron, 2012, 68, 922–930 CrossRef CAS .
- D. R. Joya-Cárdenas, J. P. Rodríguez-Caicedo, A. Gallegos-Muñoz, G. A. Zanor, M. S. Caycedo-García, C. E. Damian-Ascencio and A. Saldaña-Robles, Nanomaterials, 2022, 12, 1–18 CrossRef .
- S. S. Mortazavi, A. Abbasi and M. Masteri-Farahani, Appl. Organomet. Chem., 2020, 34, 1–12 CrossRef .
- G. Surekha, K. V. Krishnaiah, N. Ravi and R. P. Suvarna, J. Phys.: Conf. Ser., 2020, 1495, 12 CrossRef .
- A. A. Olorunkosebi, M. A. Eleruja, A. V. Adedeji, B. Olofinjana, O. Fasakin, E. Omotoso, K. O. Oyedotun, E. O. B. Ajayi and N. Manyala, Diamond Relat. Mater., 2021, 117, 108456 CrossRef CAS .
- G. Santamaria, E. G. B. Arojas, E. Q. Gonzalez, E. S. Mora, M. Q. Ruiz and J. D. S. Juarez, Mater. Res. Express, 2019, 6, 31 Search PubMed .
- Y. Ji, X. Yang, Z. Ji, L. Zhu, N. Ma, D. Chen, X. Jia, J. Tang and Y. Cao, ACS Omega, 2020, 5, 8572–8578 CrossRef CAS .
- A. Amoozadeh, S. Rahmani, M. Bitaraf, F. B. Abadi and E. Tabrizian, New J. Chem., 2016, 40, 770–780 RSC .
- I. Sengupta, S. S. S. S. Kumar, S. K. Pal and S. Chakraborty, J. Mater. Res., 2020, 35, 1197–1204 CrossRef CAS .
- L. Irannejad, S. J. Ahmadi, S. Sadjadi and M. Shamsipur, J. Iran. Chem. Soc., 2018, 15, 111–119 CrossRef CAS .
- P. Li, S. Sun, A. Dong, Y. Hao, S. Shi, Z. Sun, G. Gao and Y. Chen, Appl. Surf. Sci., 2015, 355, 446–452 CrossRef CAS .
- M. Mirza-Aghayan, M. Mohammadi and R. Boukherroub, J. Organomet. Chem., 2022, 957, 122160 CrossRef CAS .
- T. Kuanyshbekov, Z. Sagdollin, E. Zhasasynov, K. Akatan and B. Kurbanova, J. Compos. Sci., 2023, 1–16 Search PubMed .
- Y. Esmaeili, E. Bidram, A. Zarrabi, A. Amini and C. Cheng, Sci. Rep., 2020, 1–13 Search PubMed .
- M. D. Pravin, C. F. S and A. Gnanamani, RSC Adv., 2018, 38416–38424 CAS .
- N. Huang, H. Lim, C. Chia, M. Yarmo and M. Muhamad, Int. J. Nanomed., 2011, 3443–3448 CrossRef CAS .
- T. Wu, X. Wang, H. Qiu, J. Gao, W. Wang and Y. Liu, J. Mater. Chem., 2012, 22, 4772–4779 RSC .
- Y. Feng, N. Feng, Y. Wei and G. Zhang, RSC Adv., 2014, 4, 7933–7943 RSC .
- M. G. Dekamin, J. Mokhtari and M. R. Naimi-Jamal, Catal. Commun., 2009, 10, 582–585 CrossRef CAS .
- S. Kantevari, R. Bantu and L. Nagarapu, J. Mol. Catal. A: Chem., 2007, 269, 53–57 CrossRef CAS .
- E. C. Horning and M. G. Horning, J. Org. Chem., 1946, 95–99 CrossRef CAS .
- F. Darviche, S. Balalaie, F. Chadegani and P. Salehi, Synth. Commun., 2007, 37, 1059–1066 CrossRef CAS .
- N. P. Chandia, J. C. Canales, I. Azocar, V. G. Pawar, W. M. D. Borggraeve and W. Dehaen, Synth. Commun., 2000, 927–939 Search PubMed .
- J. K. Rajput and G. Kaur, Catal. Sci. Technol., 2014, 4, 142–151 RSC .
- F. E. King and D. G. I. Felton, J. Chem. Soc., 1946, 1371–1372 Search PubMed .
- G. Cravotto, A. Demetri, G. M. Nano, G. Palmisano, A. Penoni and S. Tagliapietra, Eur. J. Org. Chem., 2003, 4438–4444 CrossRef CAS .
- T. S. Jin, J. S. Zhang and A. Q. Wang, Synth. Commun., 2005, 2339–2345 CrossRef CAS .
- M. Gupta and M. Gupta, J. Chem. Sci., 2016, 128, 849–854 CrossRef CAS .
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ,
Mohammad G.
Dekamin
,
Mohammad G.
Dekamin