
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhotocatalytic activity of a 2D copper porphyrin metal–organic framework for visible light overall water splitting†
Andrés
Uscategui-Linares
,
Horatiu
Szalad
,
Josep
Albero
* and
Hermenegildo
García
 *
*
Instituto Universitario de Tecnología Química (CSIC/UPV), Universitat Politécnica de València, Avda. de los Naranjos s/n, 46022 Valencia, Spain. E-mail: hgarcia@qim.upv.es
First published on 18th September 2024
Abstract
A 2D copper tetrakis(4-carboxyphenyl)porphyrin metal–organic framework has been prepared and used as a photocatalyst for overall water splitting, measuring under visible light irradiation (λ > 450 nm) under one sun power conditions a H2 production rate of 120 μmolH2 gcatalyst−1 h−1 that is among the highest ever reported. While the 2D Cu porphyrin MOF undergoes substantial degradation in 3 h upon UV irradiation (320–380 nm) in the presence of air, it appears to be photostable under the conditions of the overall water splitting and visible light exposure, exhibiting similar temporal profiles for H2 and O2 evolution. Photocurrent experiments and band energy measurements indicate that the photocatalytic efficiency derives from an efficient charge separation in the visible region (apparent electron charge extraction efficiency at 540 nm of 0.1%) and adequate alignment of the redox potential of the conduction (−0.59 V vs. NHE) and valence (+1.48 V vs. NHE) bands for water splitting.
Introduction
Metal–Organic Frameworks (MOFs) have become among the preferred photocatalysts due to their flexibility in the design, wide variety in the metal elements that can be used in the nodes and diversity in the structure and connectivity of organic linkers.1,2 In some cases, MOFs have reached activity comparable to that of the most efficient photocatalysts with the advantage that MOFs still offer promise of further activity enhancement by optimization of their multiple structural parameters, including defects, particle size and morphology.3 In comparison with metal oxide photocatalysts and due to the organic linker, MOFs have shown considerable flexibility for the design of visible light photocatalysts.4 Visible light photoactivity is important for solar energy conversion into chemicals and fuels, since the solar UV radiation energy reaching the Earth surface is very low, about 4%, in comparison to the visible light energy that is over 40% of the total solar energy. In this way, MOFs having dyes as organic linkers can exhibit photoresponse in the whole wavelength range of the visible region.5Among visible light responsive MOFs, those containing porphyrin linkers have been well studied.6–8 In a previous study, we reported that porphyrin PCN-222 MOFs exhibit photocatalytic activity for overall water splitting,9 the performance depending on the nature of the metal complexing the porphyrin ring. Other reports have also confirmed the high performance of porphyrin MOFs as photocatalysts.10,11 In one of the possible photocatalytic mechanisms, the porphyrin acts as a light harvesting unit and, upon excitation, transfers one electron to the metal node, resulting in a ligand-to-metal charge separation state that has a lifetime long enough on the microsecond time scale to promote photocatalytic reactions.12,13
Going one step further, one current trend in MOF photocatalysis is to explore the photocatalytic activity of thin, nanometer-thick 2D MOFs. Compared to 3D bulk particles, 2D MOFs offer a higher exposure of the activity sites and an easier accessibility of substrates to them, considering the shorter diffusion path length required in these nanoplatelets to access all sites.14,15 Short diffusion paths can be important in photocatalysis, since charge carrier mobility in MOFs is low and, somehow, MOFs have to be considered an ordered array of molecular complexes placed in a porous lattice. Since charge mobility in MOFs is low and the lifetime of the charge separation state is a few microseconds,16,17 the substrates have to have fast access to these sites in order to observe an efficient photocatalytic process.
Herein we report the notable photocatalytic activity of a 2D Cu porphyrin MOF denoted as Cu2(CuTCPP) having tetrakis-meso-(carboxyphenyl)porphyrin (TCPP) ligands with Cu2+ ions both in nodal lattice positions of the MOF and complexed within the porphyrin macroring. The use of this 2D MOF has been previously investigated in energy harvesting and conversion18–20 and sensing,21 among other fields.22 However, its use in photocatalysis has been limited to environmental remediation23 to the best of our knowledge. In this study, Cu2(CuTCPP) has reached a H2 production rate of 120 μmol gphotocatalyst−1 h−1 that is among the highest reported for a noble metal-free MOF in photocatalytic overall water splitting under visible light irradiation (λ > 450 nm) in the absence of noble metal co-catalysts.
Results and discussion
The synthesis of 2D Cu2(CuTCPP) was achieved by a modified synthetic procedure previously reported18 and it is described in detail in the ESI† and Scheme 1. In brief, 2D Cu2(CuTCPP) MOF was synthesized by solvothermal synthesis in DMF at 85 °C using Cu(NO3)2 and H2TCPP as precursors. During the process, simultaneous H2TCPP complexation by Cu2+ and 2D MOF formation takes place as illustrated in Scheme 1. | ||
| Scheme 1 Preparation procedure of Cu2(CuTCPP). | ||
The success of the 2D MOF synthesis is supported by combining the information provided by different characterization techniques. ICP elemental analysis of Cu and combustion chemical analysis agree with the formula Cu1.8(CuTCPP)·2H2O (TCPP: C48H30N4O8). Thus, as can be seen in Fig. 1a, XRD shows a pattern corresponding to the 2D MOF with peaks at 2θ values of 7.5, 11.9, 19.3 and 30.4 corresponding to the diffraction along the 110, 210, 002 and 440 planes, respectively.24 IR spectroscopy shows the changes from free carboxylic groups of the H2TCPP precursor characterized by a broad OH vibration band from 3500 to 2000 cm−1 and a C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibration at 1686 cm−1 to the corresponding carboxylate group appearing at 1607 cm−1. Fig. 1b also shows a comparison of the IR spectrum of free H2TCPP in comparison to that of Cu2(CuTCPP). Complexation of the H2TCPP macro-ring was firmly confirmed by UV-vis spectroscopy by observation of the change of the characteristic four degenerate Q bands of the free TCPP that are replaced by two more intense Q bands with relative λmax at 453 and 540 nm,25 as seen in Fig. 1c. The position of the Soret band also redshifts upon formation of the 2D Cu2(CuTCPP) MOF, appearing at 371 nm. A Tauc plot considering Cu2(CuTCPP) as an indirect semiconductor allows an optical bandgap of 2.07 eV to be estimated (Fig. 1d).
O stretching vibration at 1686 cm−1 to the corresponding carboxylate group appearing at 1607 cm−1. Fig. 1b also shows a comparison of the IR spectrum of free H2TCPP in comparison to that of Cu2(CuTCPP). Complexation of the H2TCPP macro-ring was firmly confirmed by UV-vis spectroscopy by observation of the change of the characteristic four degenerate Q bands of the free TCPP that are replaced by two more intense Q bands with relative λmax at 453 and 540 nm,25 as seen in Fig. 1c. The position of the Soret band also redshifts upon formation of the 2D Cu2(CuTCPP) MOF, appearing at 371 nm. A Tauc plot considering Cu2(CuTCPP) as an indirect semiconductor allows an optical bandgap of 2.07 eV to be estimated (Fig. 1d).
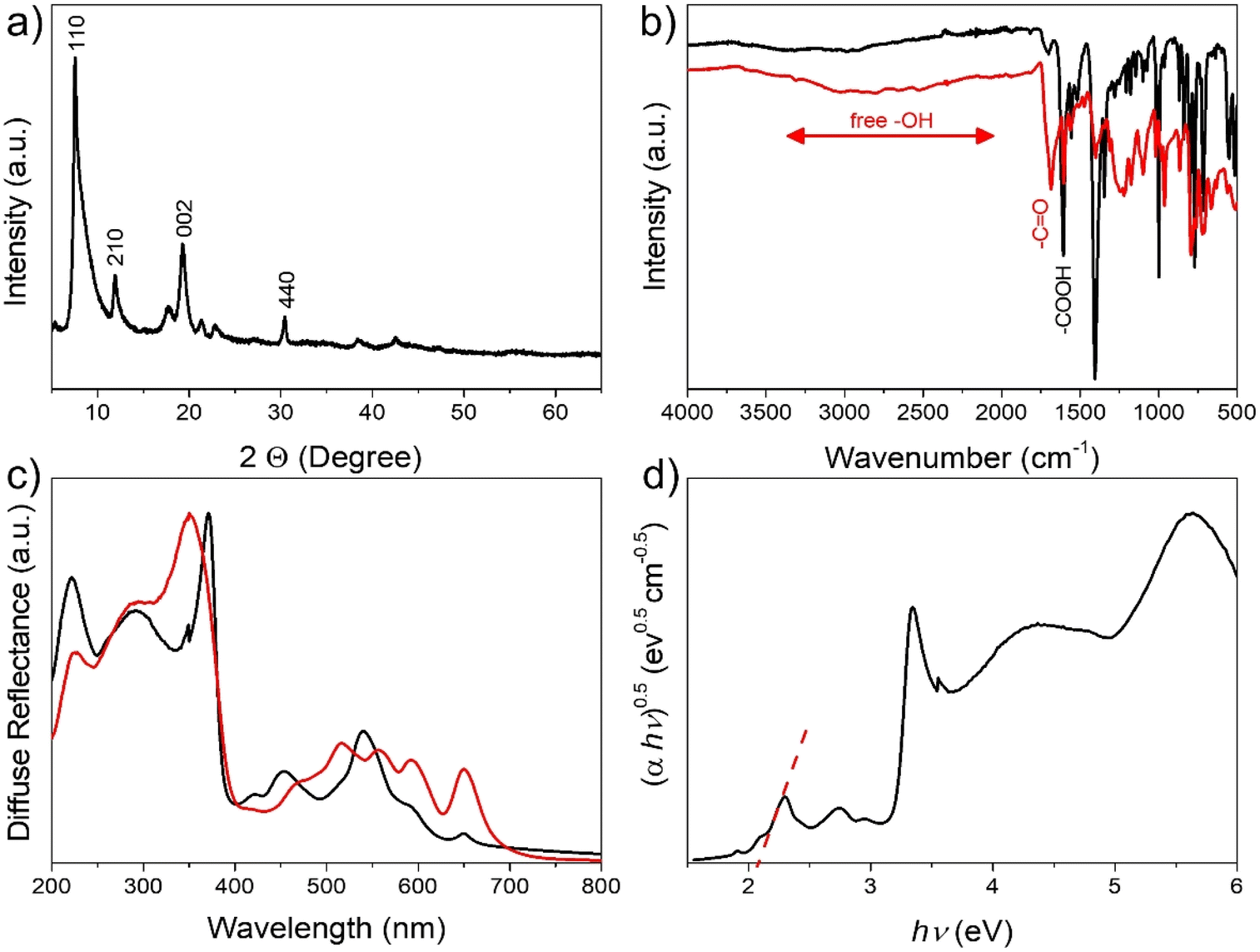 | ||
| Fig. 1 (a) XRD pattern of Cu2(CuTCPP), (b) FTIR spectra of H2TCPP (red) and Cu2(CuTCPP) (black), (c) diffuse reflectance UV-vis spectra of H2TCPP (red) and Cu2(CuTCPP) (black) and (d) Tauc plot obtained from the Cu2(CuTCPP)diffuse reflectance spectrum. The dashed line indicates the intersection used to calculate the optical band gap. | ||
In order to further investigate the chemical composition of the 2D Cu2(CuTCPP)MOF, X-ray Photoelectron (XPS) spectroscopy was employed. Fig. S1 in the ESI† shows the high resolution XPS spectra of C 1s, O 1s, N 1s and Cu 2p3/2, and the best deconvolution to their individual components. As can be seen, the C 1s spectrum shows three main components assigned to CC, C–O and O–CO and centred at 284.50, 286.18 and 288.48 eV, respectively. The O 1s spectrum also presents three components attributed to Cu–O, O–C, and OC–O, located at 528.56, 531.47 and 532.70 eV, respectively. The N 1s spectrum show two components ascribed to pyridinic and pyrrolic N at 398.40 and 399.82 eV, respectively. Finally, the Cu 2p3/2 spectrum shows a main peak at 934.5 eV constituted by two components centred at 934.3 and 935.5 eV and attributed to Cu–O and N-coordinated Cu ions, respectively, and their corresponding satellites.26–28 These results confirm the presence of Cu2+ oxo-clusters as nodes and Cu2+ ions coordinated in the porphyrin linkers.
The 2D morphology of Cu2(CuTCPP) nanoplatelets was clearly visualized in FESEM, TEM and AFM images (Fig. 2). The FESEM image of as-prepared Cu2(CuTCPP) showing aggregates of sub-nanometric nanoplatelets is presented as Fig. 2a. TEM images of the 2D Cu2(CuTCPP) MOF have also been acquired, and they are presented in Fig. 2b. These images show ultrathin Cu2(CuTCPP) sheets of 400–800 nm of lateral size, approximately. HRTEM images of the Cu2(CuTCPP)OF have also been acquired, and they are presented in Fig. S2 in the ESI.† The 2D morphology has been further confirmed with AFM. Fig. 2d shows an image of a Cu2(CuTCPP) sheet. Cross section measurements presented in Fig. 2e show a thickness value of approximately 3.7 nm for the Cu2(CuTCPP) sheet in Fig. 2d. The photocatalytic activity of Cu2(CuTCPP) for H2 evolution under visible light irradiation was evaluated using a 450 nm cut-off filter at 1 mgphotocatalyst ml−1 in the presence of CH3OH as a sacrificial agent. The H2 evolution temporal profile using Cu2(CuTCPP) as a photocatalyst in a 3 h irradiation experiment is presented in Fig. 3a. The decrease in the H2 evolution rate after the first 60 min observed in the temporal profile could be due to the competition between H2 and CH3OH for photogenerated holes, when the H2 concentration is high, and/or the blocking of active sites by the CH3OH oxidation products, among other possible reasons.
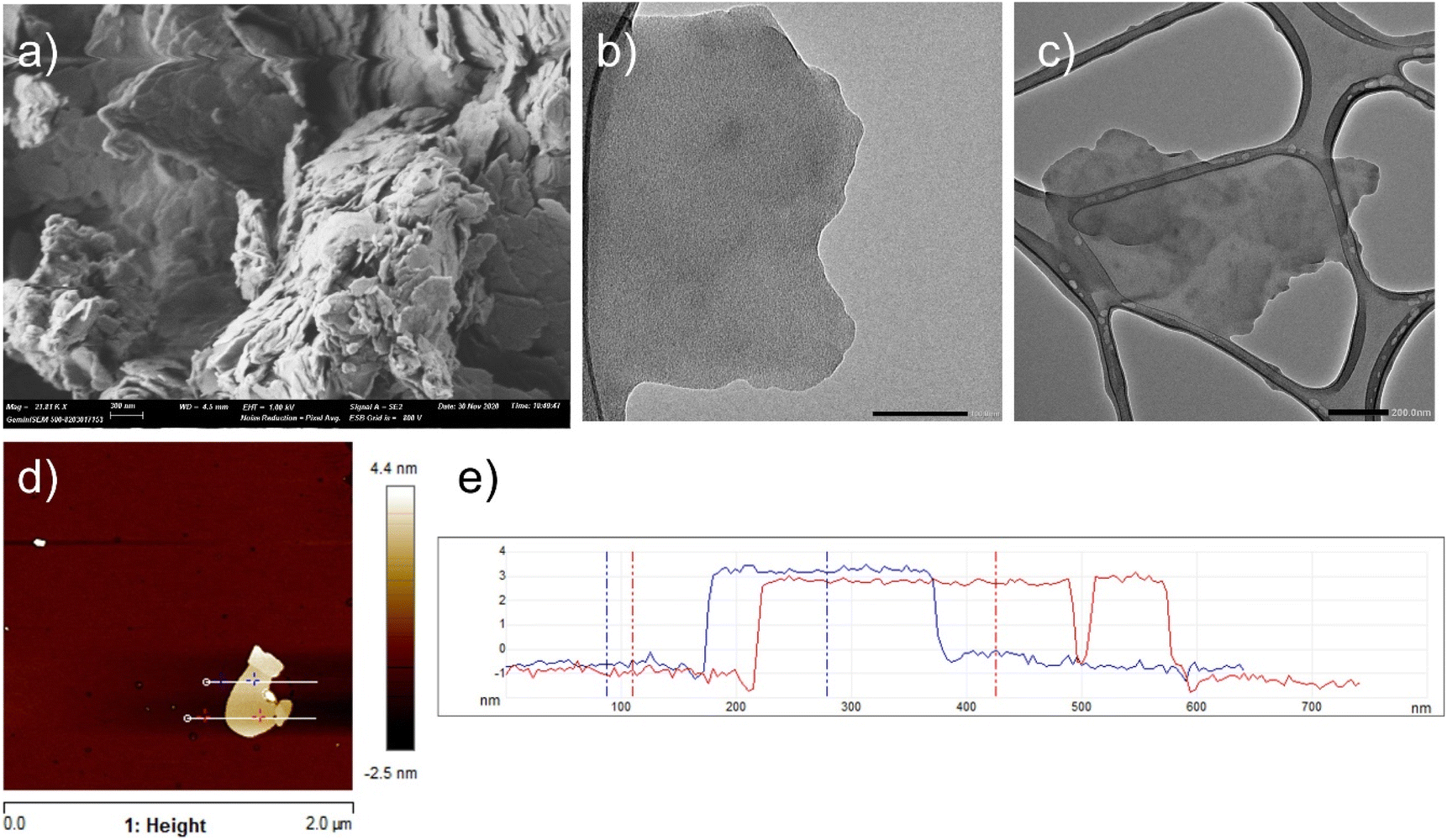 | ||
| Fig. 2 FESEM image of Cu2(CuTCPP) (scale bar 300 nm) (a). TEM images of Cu2(CuTCPP) (b) and (c). Scale bars are 100 nm and 200 nm, in (b) and (c), respectively. The AFM image of Cu2(CuTCPP) (d). Cross section measurements (e) of the Cu2(CuTCPP) sheet presented in panel (d). | ||
 | ||
Fig. 3 (a) Temporal profile of H2 evolution using Cu2(CuTCPP) (black), CuTCPP molecular complex (red) and Cu2(FeTCPP) (blue) dispersions at 1 mg mL−1 in H2O![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :MeOH mixtures (70:30, v:v) under 1080 mW cm−2 illumination from a Xe lamp using a 450 nm cut-off filter. (b) H2 evolution from five consecutive uses of the Cu2(CuTCPP) photocatalyst for 2 h. :MeOH mixtures (70:30, v:v) under 1080 mW cm−2 illumination from a Xe lamp using a 450 nm cut-off filter. (b) H2 evolution from five consecutive uses of the Cu2(CuTCPP) photocatalyst for 2 h. | ||
TON and TOF values have been estimated based on the number of Cu2(CuTCPP) molecules (formula weight 980.65 g mol−1) used in each experiment (20.39 μmol), obtaining 2.45 and 2.27 × 10−4 s−1, respectively. These values are remarkable for a solid photocatalyst using visible light.
Two controls were used to determine the relative photocatalytic efficiency of Cu2(CuTCPP) in comparison to its related CuTCPP molecular complex and other 2D metal porphyrin MOFs. Fig. 3a also presents the temporal H2 evolution profiles for these two controls. Comparison of the photocatalytic activity of 2D Cu2(CuTCPP) MOF with that of the CuTCPP complex showed that the latter exhibits about five-fold lower H2 evolution activity. This comparison shows the influence of having the CuTCPP complex assembled in a porous 2D structure in which the carboxylate groups are interacting with Cu2+ ions. This interaction would allow a more efficient charge separation in the MOF by fast quenching of the excited state of the CuTCPP by nodal Cu2+ in the lattice as electron acceptors. In addition, these reduced nodal Cu+ ions are catalytically more active to interact with H2O or H+, therefore, being more efficient in H2 generation.
Cu2(CuTCPP) as a photocatalyst was also more efficient than Cu2(FeTCPP). Fig. S3 and Table S1 in the ESI† provide analytical data, the XRD pattern and diffuse reflectance UV-vis absorption spectra of the Cu2(FeTCPP) sample used in this study. The higher activity of Cu2(CuTCPP) in comparison to Cu2(FeTCPP) reflects the influence of the electronic configuration of the metal ion complexing the porphyrin ligand. Prior precedents on 3D porphyrin MOFs have also observed the influence of the metal ion in the porphyrin complex on the photocatalytic activity that should be related to the operation of deactivation pathways of the excited state competing with charge separation.9 Photostability of Cu2(CuTCPP) was addressed by performing a series of consecutive runs with the same sample. Reuses were carried out by recovering the photocatalyst from the aqueous suspension and exhaustively washing the solid before a subsequent reuse under the same conditions. Fig. 3b presents the time-H2 evolution plots of these reuse experiments. The accumulated TON value of the five uses of the same Cu2(CuTCPP) sample was 5.0, thus reconfirming that the 2D MOF is acting as a catalyst in the H2 evolution reaction.
Regarding Fig. 3b, we observed that while no decrease in the photocatalytic activity was observed in the four uses, the lower photocatalytic activity in the fifth use from 1050 to 800 μmolH2 g−1 in 2 h was investigated in detail. Structural stability of the 2D MOF was confirmed by characterization of the Cu2(CuTCPP) sample after the photocatalytic experiments by XRD and TEM, whereby no changes were observed in comparison with the properties of the fresh sample (Fig. S4†). Also, XPS analysis of Cu2(CuTCPP) after the five uses shows negligible changes in the high resolution XPS spectra of C 1s, O 1s and N 1s (Fig. S5†). However, the Cu 2p3/2 spectrum shows differences with respect to the peak of the as-prepared sample (see Fig. S1†) and those related to N-coordinated Cu ions could not be observed. This XPS Cu 2p3/2 change reports variation in the mean oxidation state and coordination sphere of Cu, with the formation of Cu–O bonds, as a consequence of the prolonged operation of the photocatalytic overall H2O splitting.
Similarly to H2 generation in the presence of methanol, the ability of Cu2(CuTCPP) to generate O2 under visible light irradiation (λ > 450 nm) in the presence of Ce(NH4)2(NO3)6 as a sacrificial electron acceptor agent was also tested. After 3 h of irradiation time, the evolution of 200 mmolO2 gphotocatalyst−1 was observed. The results are presented in Fig. S6.†
After having shown the activity for H2 generation in the presence of CH3OH as a sacrificial agent and O2 using Ce(NH4)2(NO3)6 as an electron acceptor, the photocatalytic activity of Cu2(CuTCPP) for overall water splitting upon visible light irradiation (λ > 450 nm) was studied. Under these conditions, generation of O2 accompanying H2 evolution was observed. The temporal production of H2 and O2 in overall water splitting in three consecutive uses is presented in Fig. 4. It was noted that the TON value under these conditions was much lower than the value shown in Fig. 3a and b, being less than 1. This lower TON value should be a consequence of the operation during the H2O splitting of the backward reaction, in which photogenerated H2 and O2 react to form H2O, either thermally or photocatalytically. In this way, photogenerated electrons could be captured by O2, resulting in H2O formation, while photogenerated holes could oxidize H2 leading to H+ required in H2O formation. However, even though the TON value is below 1, Cu2(CuTCPP) still works as a photocatalyst under these conditions, as confirmed by the fact that H2 and O2 are simultaneously formed and the photocatalyst is reusable. Otherwise, the photocatalyst should become decomposed in the process and form either H2 by reduction or O2 by oxidation, but not both.
 | ||
| Fig. 4 Temporal profile of H2 (black lines) and O2 (red lines) evolution of three consecutive uses of a Cu2(CuTCPP) dispersion at 1 mg mL−1 in pure water under visible light at 1080 mW cm−2 illumination from a Xe lamp using a 450 nm cut-off filter. | ||
We also noted that the amount of O2 was somewhat about 20% below the stoichiometric amount of H2, hinting the occurrence of spurious consumption of h+. There are precedents in the literature reporting similar lower-than-expected O2 evolution and attributed this fact to the strong oxidizing potential required for O2 evolution that can determine the competitive oxidation of other species, including parts of the photocatalyst. In the present case, the photocatalytic stability of Cu2(CuTCPP) after three consecutive overall H2O splitting experiments was confirmed by XRD, as shown in Fig. S7.†
Since porphyrins in general were considered photochemically labile, we were intrigued by the stability observed in the previous photocatalytic reactions. We considered that this photostability derives from the use of visible light and the subambient concentration of O2. To test this hypothesis and determine the role that UV light would have in the 2D Cu2(CuTCPP) MOF, a photocatalytic study irradiating Cu2(CuTCPP) with UV light using a 320–380 nm band-pass filter exposed to the ambient air for 3 h was carried out. At the end of the irradiation, the sample was analyzed by XRD and IR spectroscopy. The results are presented in Fig. S8.† As can be seen there, XRD indicates a considerable decrease in the intensity of the diffraction peaks, meaning that the sample has become largely amorphous. Accordingly, IR spectroscopy presents significant changes in the fresh and UV-irradiated samples, the most important ones being the presence of a broad –OH vibration band spanning from 3750 to 2600 cm−1 and a decrease in the intensity of the characteristic carboxyl group vibrations at cm−1, all being compatible with the generation of structural defects by decarboxylation. In a previous study on MOFs having polycarboxylate linkers, evolution of CO2 by photodecarboxylation was observed,29 this being compatible with the present results. Therefore, it can be proposed that high-energy UV radiation and O2 can degrade in part Cu2(CuTCPP), something that was not observed in the visible light photocatalytic overall water splitting experiments.
To determine the reaction mechanism, transient photocurrent measurements of Cu2(CuTCPP) on a FTO electrode using a 0.1 M KCl electrolyte were carried out. It was observed that the transient photocurrent decreased as the voltage increased from −0.78 to +0.22 V. As an example, Fig. S9a† shows the transient photocurrent measured at −0.28 V vs. NHE, while Fig. S8b† presents the influence of the bias voltage on the photocurrent. As can be seen there, upon illumination a significant anodic current was observed, meaning that light induces charge separation on Cu2(CuTCPP). For comparison purposes, transient photocurrent measurements of Cu2(CuTCPP) and CuTCPP-based electrodes have been carried out (Fig. S10†). It can be clearly observed that the photogenerated current from the CuTCPP molecular complex is up to 100 times lower than that of the 2D Cu2(CuTCPP) MOF. Hence, the coordination of Cu ions with the CuTCPP molecular complex, forming the MOF, promotes the improvement in charge separation.
Photoresponse as a function of the excitation wavelength and the corresponding apparent external quantum efficiency for charge abstraction at each excitation wavelength was calculated at −0.56 V in 0.1 M KCl electrolyte for the Cu2(CuTCPP) electrode. The results are presented in Fig. 5a. Under the present conditions, a maximum of 0.1% of electron abstraction efficiency was measured at 540 nm. Photoresponse was observed in the visible region range with efficiency following the absorbance intensity of the porphyrin Q bands.
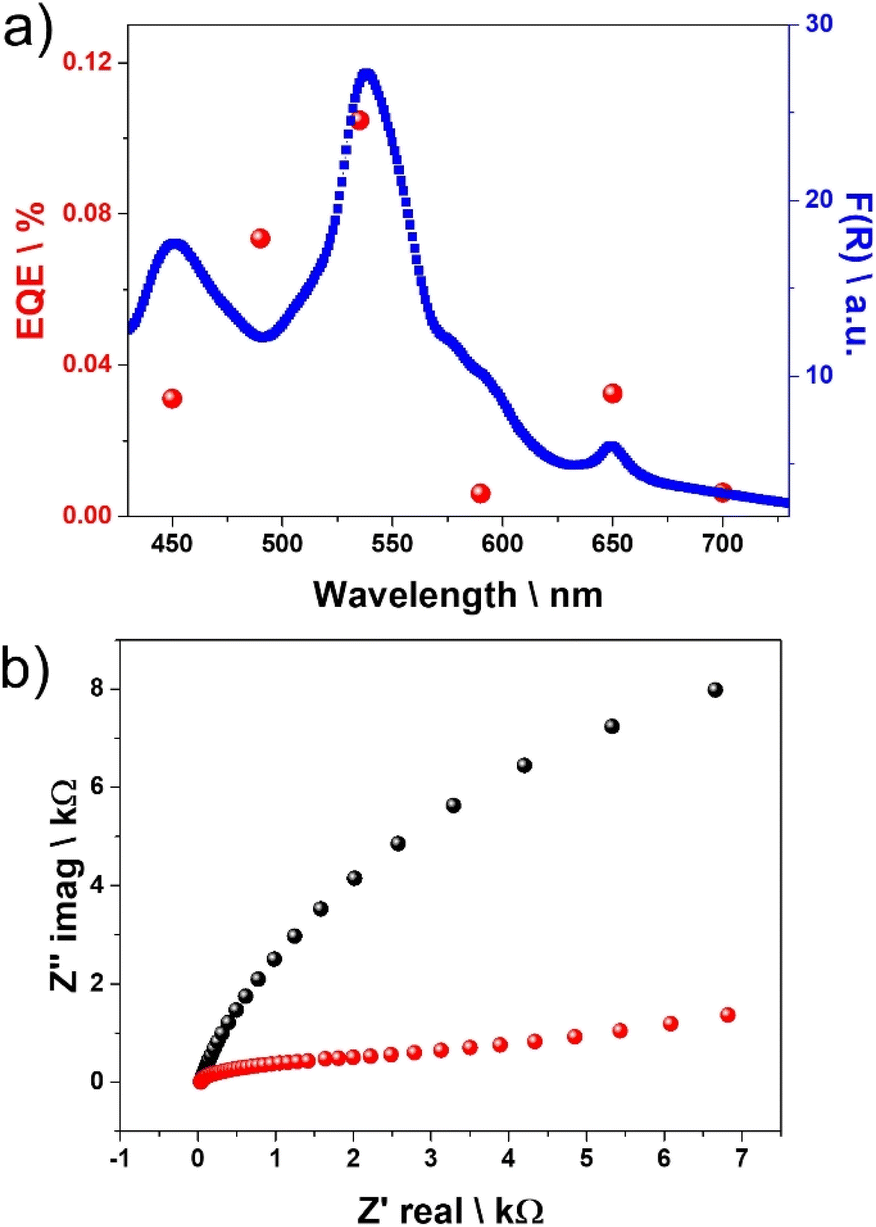 | ||
| Fig. 5 (a) Comparison of the apparent external quantum efficiency for charge abstraction (red dots) at different excitation wavelengths calculated at −0.56 V in 0.1 M KCl electrolyte for the Cu2(CuTCPP) electrode and the diffuse reflectance UV-vis spectrum (blue dots) of Cu2(CuTCPP). (b) Nyquist plots in the range of frequencies from 0.1 Hz to 106 Hz were also measured for Cu2(CuTCPP) films on FTO electrodes using 0.1 M KCl electrolyte in the dark (black dots) and under illumination (red dots). | ||
Nyquist plots in the range of frequencies from Hz to MHz were also measured for Cu2(CuTCPP) films on FTO electrodes using 0.1 M KCl electrolyte in the dark and under illumination. The results are presented in Fig. 5b. It was observed a significant arc radius decrease of the plot under illumination, indicating that light markedly decreases the charge transfer resistance.
Band alignment for Cu2(CuTCPP) was determined from the optical bandgap and the valence band maximum. The Tauc plot of the Kubelka–Munk function of the reflectivity, assuming that Cu2(CuTCPP) corresponds to an indirect semiconductor, is presented in Fig. 1d, resulting in a value of 2.07 eV. The valence band maximum was estimated assuming that it is about 0.1 eV higher than the work function of the Cu2(CuTCPP) electrode measured from the Mott–Schottky plot of the inverse of the capacitance square vs. the applied potential at four different frequencies. Fig. S11 in the ESI† presents the Mott–Schottky plot and the interception value. From these measurements, the energy diagram for the highest occupied crystal orbital (HOCO) and the lowest unoccupied crystal orbital (LUCO) for Cu2(CuTCPP) presented in Scheme 2 was determined. These values are similar, but not coincident, with those previously reported in the literature. Specifically, a LUCO reduction potential of −0.96 V has been reported, while the HOCO oxidation potential in our measurements is lower by about 0.34 V.30
 | ||
| Scheme 2 Structures and band alignment of the 2D Cu2(CuTCPP) MOF (left) and the Cu TCPP molecular complex (right) referred to the NHE at pH 0 and vs. vacuum. The blue line indicates the H+/H2 reduction potential and red line indicates O2/H2O oxidation potential. | ||
For the sake of comparison, Scheme 2 also includes the band energy diagram of molecular CuTCPP obtained from previous reports in the literature31 that has a significantly lower oxidation potential. It seems that coordination of the CuTCPP complex in the 2D MOF lattice with coordination of the four carboxylic groups with Cu2+ ions significantly increases the oxidation potential which is beneficial from the photocatalytic point of view to be able to promote water oxidation.
Moreover, in situ ATR-FTIR measurements (see Fig. S12 in ESI†) of an aqueous Cu2(CuTCPP) dispersion in the dark and upon visible light irradiation point out electron transfer from the metalloporphyrin to the Cu nodes through the carboxylic moieties. This is evidenced by a red shift in the carbonyl vibrational band together with a red shift in the –OH stretching vibration upon light irradiation. These shits support a strength of CO in the carboxylic groups and less free H2O molecules in the paddle-wheel Cu nodes of this MOF.
Conclusions
In summary, the present study has shown the ability of 2D Cu2(CuTCPP) MOF to promote photocatalytic overall water splitting under visible light irradiation (λ > 450 nm) with a H2 evolution rate of 120 μmolH2 gcatalyst−1 h−1 under one sun power conditions. These values compare favorably among those reported in the literature under the same conditions (see Table S2†) and rank Cu2(CuTCPP) among the most efficient photocatalysts for this reaction. Furthermore, although Cu2(CuTCPP) undergoes considerable photoinduced degradation upon irradiation with UV light in the presence of air, Cu2(CuTCPP) is considerably more stable upon visible light irradiation under the conditions of overall water splitting. Photocurrent measurements show that this photocatalytic activity derives from an efficient charge separation and an adequate band alignment to promote both semi-reactions, water reduction and oxidation.Data availability
Data are available from the corresponding authors upon reasonable request.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the Spanish Ministry of Science and Innovation (CEX-2021-001230-S and PDI2021-0126071-OB-CO21 funded by MCIN/AEI/10.13039/501100011033), Generalitat Valenciana (Prometeo 2021/038 and Advanced Materials programme Graphica MFA/2022/023 with funding from European Union NextGenerationEU PRTR-C17.I1), and the European Commission (MOF2H2; Grant Agreement 101084131) is gratefully acknowledged. J. A. thanks the Spanish Ministry of Science and Innovation for a Ramon y Cajal research associate contract (RYC2021-031006-I funded by MCIN/AEI/10.13039/501100011033 and by European Union NextGenerationEU/PRTR) and the financial support (PID2022-141099OA-I00 funded by MCIN/AEI/10.13039/501100011033 and by European Union NextGenerationEU/PRTR).References
- S. Navalón, A. Dhakshinamoorthy, M. Álvaro, B. Ferrer and H. García, Chem. Rev., 2023, 123, 445–490 CrossRef PubMed.
- K. Sun, Y. Qian and H.-L. Jiang, Angew. Chem., Int. Ed., 2023, 62, e202217565 CrossRef CAS PubMed.
- Z. Qian, R. Zhang, Y. Xiao, H. Huang, Y. Sun, Y. Chen, T. Ma and X. Sun, Adv. Energy Mater., 2023, 13, 2300086 CrossRef CAS.
- Z. Ma, B. Guan, J. Guo, X. Wu, Y. Chen, J. Zhang, X. Jiang, S. Bao, L. Chen, K. Shu, H. Dang, Z. Guo, Z. Li, S. Yao and Z. Huang, Catal. Sci. Technol., 2023, 13, 4285–4347 RSC.
- Y. Jiao, Y. Zuo, H. Yang, X. Gao and C. Duan, Coord. Chem. Rev., 2021, 430, 213648 CrossRef CAS.
- X. Zhang, M. C. Wasson, M. Shayan, E. K. Berdichevsky, J. Ricardo-Noordberg, Z. Singh, E. K. Papazyan, A. J. Castro, P. Marino, Z. Ajoyan, Z. Chen, T. Islamoglu, A. J. Howarth, Y. Liu, M. B. Majewski, M. J. Katz, J. E. Mondloch and O. K. Farha, Coord. Chem. Rev., 2021, 429, 213615 CrossRef CAS PubMed.
- C. Lin, C. Han, H. Zhang, L. Gong, Y. Gao, H. Wang, Y. Bian, R. Li and J. Jiang, Inorg. Chem., 2021, 60, 3988–3995 CrossRef CAS PubMed.
- A. Aziz, A. R. Ruiz-Salvador, N. C. Hernández, S. Calero, S. Hamad and R. Grau-Crespo, J. Mater. Chem. A, 2017, 5, 11894–11904 RSC.
- T. G. Semerci, A. Melillo, Y. Ç. Mutlu and H. Garcia, Catal. Today, 2023, 423, 113931 CrossRef CAS.
- X. Wang, X. Zhang, W. Zhou, L. Liu, J. Ye and D. Wang, Nano Energy, 2019, 62, 250–258 CrossRef CAS.
- B. Gikonyo, E. Montero-Lanzuela, H. G. Baldovi, S. De, C. Journet, T. Devic, N. Guillou, D. Tiana, S. Navalon and A. Fateeva, J. Mater. Chem. A, 2022, 10, 24938–24950 RSC.
- M. A. Nasalevich, C. H. Hendon, J. G. Santaclara, K. Svane, B. van der Linden, S. L. Veber, M. V. Fedin, A. J. Houtepen, M. A. van der Veen, F. Kapteijn, A. Walsh and J. Gascon, Sci. Rep., 2016, 6, 23676 CrossRef CAS PubMed.
- M. Alvaro, E. Carbonell, B. Ferrer, F. X. Llabrés i Xamena and H. Garcia, Chem.–Eur. J., 2007, 13, 5106–5112 CrossRef CAS PubMed.
- Y. Xue, G. Zhao, R. Yang, F. Chu, J. Chen, L. Wang and X. Huang, Nanoscale, 2021, 13, 3911–3936 RSC.
- K. A. Adegoke, O. R. Adegoke, R. A. Adigun, N. W. Maxakato and O. S. Bello, Coord. Chem. Rev., 2022, 473, 214817 CrossRef CAS.
- C. G. Silva, A. Corma and H. García, J. Mater. Chem., 2010, 20, 3141–3156 RSC.
- D. Wang, J. Albero, H. García and Z. Li, J. Catal., 2017, 349, 156–162 CrossRef CAS.
- J.-X. Wu, S.-Z. Hou, X.-D. Zhang, M. Xu, H.-F. Yang, P.-S. Cao and Z.-Y. Gu, Chem. Sci., 2019, 10, 2199–2205 RSC.
- Z. Weng, Y. Wu, M. Wang, J. Jiang, K. Yang, S. Huo, X.-F. Wang, Q. Ma, G. W. Brudvig, V. S. Batista, Y. Liang, Z. Feng and H. Wang, Nat. Commun., 2018, 9, 415 CrossRef PubMed.
- J. Wang, Z. Song, M. He, Y. Qian, D. Wang, Z. Cui, Y. Feng, S. Li, B. Huang, X. Kong, J. Han and L. Wang, Nat. Commun., 2024, 15, 2125 CrossRef CAS PubMed.
- M. Deng, W. Jin, W. Yang, L. Tian, X. Gao, X. Wang, L. Feng, M. Janyasupab, B. Zhou and Y. Zhang, ACS Appl. Nano Mater., 2023, 6, 22979–22988 CrossRef CAS.
- L. Gao, Y. Song, J. Zhong, X. Lin, S.-F. Zhou and G. Zhan, ACS Biomater. Sci. Eng., 2022, 8, 1074–1086 CrossRef CAS PubMed.
- L. Qin, S. Zhao, C. Fan and Q. Ye, RSC Adv., 2021, 11, 18565–18575 RSC.
- J. M. Taylor, K. W. Dawson and G. K. H. Shimizu, J. Am. Chem. Soc., 2013, 135, 1193–1196 CrossRef CAS PubMed.
- W. Zheng, N. Shan, L. Yu and X. Wang, Dyes Pigm., 2008, 77, 153–157 CrossRef CAS.
- S. Carniato, G. Dufour, F. Rochet, H. Roulet, P. Chaquin and C. Giessner-Prettre, J. Electron Spectrosc. Relat. Phenom., 1994, 67, 189–209 CrossRef CAS.
- L. Li, S. Zhang, Y. Lu, J. Zhang, X. Zhang, R. Wang and J. Huang, Adv. Mater., 2021, 33, 2104120 CrossRef CAS PubMed.
- T. M. Ivanova, K. I. Maslakov, A. A. Sidorov, M. A. Kiskin, R. V. Linko, S. V. Savilov, V. V. Lunin and I. L. Eremenko, J. Electron Spectrosc. Relat. Phenom., 2020, 238, 146878 CrossRef CAS.
- D. Mateo, A. Santiago-Portillo, J. Albero, S. Navalón, M. Alvaro and H. García, Angew. Chem., Int. Ed., 2019, 58, 17843–17848 CrossRef CAS PubMed.
- Z. Zhang, H. Liu, J. Xu and H. Zeng, J. Photochem. Photobiol., A, 2017, 336, 25–31 CrossRef CAS.
- J. R. Darwent, P. Douglas, A. Harriman, G. Porter and M.-C. Richoux, Coord. Chem. Rev., 1982, 44, 83–126 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4na00594e |
| This journal is © The Royal Society of Chemistry 2024 |