
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUnveiling the catalytic potency of a novel hydrazone-linked covalent organic framework for the highly efficient one-pot synthesis of 1,2,4-triazolidine-3-thiones†
Pankaj
Teli
,
Shivani
Soni
,
Sunita
Teli
and
Shikha
Agarwal
 *
*
Synthetic Organic Chemistry Laboratory, Department of Chemistry, Mohanlal Sukhadia University, Udaipur, Rajasthan, India. E-mail: shikhaagarwal@mlsu.ac.in; pankajteli802@gmail.com
First published on 21st October 2024
Abstract
A novel hydrazone-linked covalent organic framework (TRIPOD-DHTH COF) was synthesized through the ultrasonic treatment of 2,5-dihydroxyterephthalohydrazide (DHTH) and 4,4′,4′′-[1,3,5-triazine-2,4,6-triyltris(oxy)]tris-benzaldehyde (TRIPOD). The COF was extensively analyzed using FT-IR, PXRD, SEM, TEM, BET, XPS, TGA, and DTA techniques. The characterization studies revealed the presence of mesoporous properties and high thermal stability, with a surface area measuring 2.78 m2 g−1 and an average pore size of 8.88 nm. The developed COF demonstrated exceptional catalytic activity in synthesizing 1,2,4-triazolidine-3-thiones from thiosemicarbazide and various ketones and aldehydes using a water![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :ethanol (1:2) medium at room temperature. A significant yield (80–98%) of 1,2,4-triazolidine-3-thiones was obtained in a low reaction time (4–20 min). The role of TRIPOD as a precursor in the synthesis of the COF and as a reactant in the synthesis of 1,2,4-triazolidine-3-thione (3l) was found to be fascinating. The synthesized COF maintained its catalytic activity over eight runs, underscoring its efficiency and reusability, highlighting its potential for sustainable chemical syntheses.
:ethanol (1:2) medium at room temperature. A significant yield (80–98%) of 1,2,4-triazolidine-3-thiones was obtained in a low reaction time (4–20 min). The role of TRIPOD as a precursor in the synthesis of the COF and as a reactant in the synthesis of 1,2,4-triazolidine-3-thione (3l) was found to be fascinating. The synthesized COF maintained its catalytic activity over eight runs, underscoring its efficiency and reusability, highlighting its potential for sustainable chemical syntheses.
Introduction
Covalent Organic Frameworks (COFs) have emerged as a revolutionary category of crystalline porous polymers, constructed from organic molecules linked by strong covalent bonds.1 These substances are acclaimed for their structural diversity, high surface area, and tunable porosity, making them highly versatile for various applications.2 COFs are formed through the reticulation of organic units into ordered two-dimensional (2D) or three-dimensional (3D) frameworks, enabling the design of materials with specific functionalities.3The synthesis of COFs involves the condensation of organic monomers with complementary functional groups, such as aldehydes, amines, and boronic acids, resulting in a variety of linkage types including imine, boronate ester, and triazine bonds. These linkages contribute to the robustness and stability of the COFs, allowing them to maintain their structure under various conditions.4,5 COFs have demonstrated tremendous potential in various sectors due to their exceptional properties. They are used in catalysis,6–9 gas storage and separation,10,11 sensing,12,13 drug delivery,14,15 energy storage,16 and environmental remediation.17 Their unique structural properties and functional capabilities continue to drive research and innovation, promising exciting future developments in various scientific and industrial fields.
In recent years, the potential of COFs as heterogeneous catalysts in organic transformations has attracted considerable attention.18,19 Due to their ordered porous structure, they provide a unique environment that allows for the encapsulation of reactants and promotes catalytic activity through enhanced mass transfer and stabilization of reaction intermediates.20 Several studies have demonstrated the catalytic efficiency of COFs in C–C coupling reactions,21 cycloadditions, and other organic transformations.22 The modular nature of COF synthesis allows for the design of materials with specific functional groups or active sites tailored for catalytic applications.23 However, despite these advances, the COFs in heterocyclic synthesis, particularly for the preparation of biologically active compounds such as triazolidine derivatives, remain underexplored.24
Synthesis of heterocyclic moieties is a pivotal aspect of organic chemistry.25–30 Triazolidines are an important class of heterocyclic compounds, particularly due to their pharmacological relevance. These five-membered heterocycles, containing nitrogen and sulfur atoms, exhibit a wide range of biological activities, including acetylcholinesterase inhibitory activity,31 and antimicrobial,32,33 antioxidant,34 antifungal,35 antitubercular,36 and anticancer37 properties. Their structural versatility and ability to interact with biological targets make them attractive scaffolds in drug discovery. As a result, the development of efficient and green methods for the synthesis of triazolidines has become a key area of research in synthetic organic chemistry.
The preparation of triazolidines traditionally involves the condensation of thiosemicarbazides with aldehydes or ketones under acidic or basic conditions. Several classical methods rely on heating the reactants in organic solvents, which often leads to low yields and harsh reaction conditions. Recently, green synthetic approaches have gained attention, emphasizing the need for environmentally friendly solvents, lower temperatures, and the use of heterogeneous catalysts to promote the reaction. The development of green, efficient, and sustainable methodologies for the synthesis of these heterocycles has been a major focus in organic chemistry. COFs, with their tunable functionalization and porous structure, present an ideal platform for promoting such reactions under mild and environmentally benign conditions. Specifically, hydrazone-linked COFs, due to the presence of reactive C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bonds, can provide catalytic sites for facilitating nucleophilic additions and cyclization reactions, making them suitable candidates for the synthesis of triazolidine derivatives.
N bonds, can provide catalytic sites for facilitating nucleophilic additions and cyclization reactions, making them suitable candidates for the synthesis of triazolidine derivatives.
COFs have emerged as efficient catalysts for such transformations, as their tunable porosity, stability, and surface functionalities make them ideal platforms for facilitating cyclization reactions and enhancing reaction efficiency under mild conditions. In this study, we have explored the green synthesis of 1,2,4-triazolidine-3-thiones using a novel hydrazone-linked COF, offering an eco-friendly and efficient pathway for the preparation of these biologically significant molecules.
In this research, the TRIPOD-DHTH COF was synthesized through the ultrasonic treatment of 2,5-dihydroxyterephthalohydrazide (DHTH) and 4,4′,4′′-[1,3,5-triazine-2,4,6-triyltris(oxy)]tris-benzaldehyde (TRIPOD). Furthermore, the catalytic activity of the synthesized novel hydrazone-linked COF was explored for the green synthesis of 1,2,4-triazolidine-3-thiones (T-3-Ts) through the reaction of thiosemicarbazide (TSC) with various aldehydes and ketones. Moreover, the role of TRIPOD as a precursor in the synthesis of COFs and as a reactant in the synthesis of 1,2,4-triazolidine-3-thione (3l) was found to be fascinating.
Experimental
Preparation of TRIPOD
TRIPOD was synthesized by a previously reported method.38 4.17 mmol (510 mg) of p-hydroxybenzaldehyde, 1.35 mmol (250 mg) of cyanuric chloride, 4.72 mmol (500 mg) of Na2CO3, and p-dioxane (20 mL) were introduced to a flask. Then, the mixture was stirred and maintained at reflux under nitrogen for 18 h. After refluxing, the mixture was subjected to vacuum concentration, and the resulting residue was added to chilled water (10 mL). The precipitate was isolated through filtration, rinsed with 5 mL of cold water, and further dried to yield the pure product (441 mg, 74%).Synthesis of diethyl-2,5-dihydroxyterephthalate (DHTP)
DHTP was synthesized using previous literature.39 In a flask, 2,5-dihydroxyterephthalic acid (750 mg, 3.78 mmol) and dry ethanol (30 mL) was added. To this mixture, 3 mL of concentrated H2SO4 was introduced dropwise and further refluxed for 18 h until the solid fully dissolved. After cooling to RT, 60 mL of DI was added. The resulting residue was extracted using filtration, thoroughly washed with DI water, and then the compound was subjected to drying in an oven (800 mg, 83.06%).Synthesis of DHTH
DHTH was synthesized using previous literature.39 1.96 mmol (500 mg) of DHTP and ethanol (20 mL) were mixed in a round-bottomed flask (RBF). Then, 51 mmol of hydrazine hydrate (2.5 mL) was introduced to the flask. This solution was heated for 15 h at 90 °C. A pale-yellow precipitate formed after cooling the mixture to RT. The resulting crude product was separated by filtration, rinsed thrice with ethanol, and dried under reduced pressure to obtain the pure compound (330 mg, 74.5%).Preparation of the TRIPOD-DHTH COF
The method for preparation of a novel COF was adopted from the reported method.17 A solution of 1,4-dioxane (12 mL) and 6.0 M aqueous acetic acid (2 mL) was added to a beaker containing 0.72 mmol of synthesized DHTH (162 mg) and 0.48 mmol of TRIPOD (112 mg). This mixture was sonicated for 8 hours using an ice water bath. The resulting fluffy yellow solid, which swelled in the solvent system, was isolated through filtration. The formed solid was sequentially rinsed thrice with anhydrous dichloromethane, anhydrous acetone, and tetrahydrofuran, and then activated under vacuum at 120 °C for 8 h to produce the COF (280 mg).General synthetic approach for triazolidine-3-thione derivatives
In 100 mL RBF, 10 mL of a water:ethanol mixture (1:2) was added to a mixture of aldehyde (1.0 mmol), TSC (1.0 mmol), and COF (20 mg) at RT. The progress of the reaction was tracked with TLC. After completion, extra EtOH was used to separate the compound and catalyst. The catalyst was filtered out to achieve separation. The desired compound was obtained by evaporating ethanol under vacuum. The products of the reaction were analyzed and verified with FT-IR, mass, 1H, and 13C NMR spectroscopy.
Results and discussion
We synthesized a novel COF by condensing TRIPOD and DHTH as organic building blocks to create novel hydrazone-linked structures. The synthesis process of COFs was straightforward and involved ultrasonic treatment of a mixture containing the monomers DHTH and TRIPOD in a blend of aqueous CH3COOH and 1,4-dioxane (Scheme 1). The resulting COF was obtained as a pale-yellow powder, showing insolubility in typical organic solvents, including acetone, alcohols, chloroform, acetonitrile, ethers, dimethyl sulfoxide and N,N-dimethylformamide.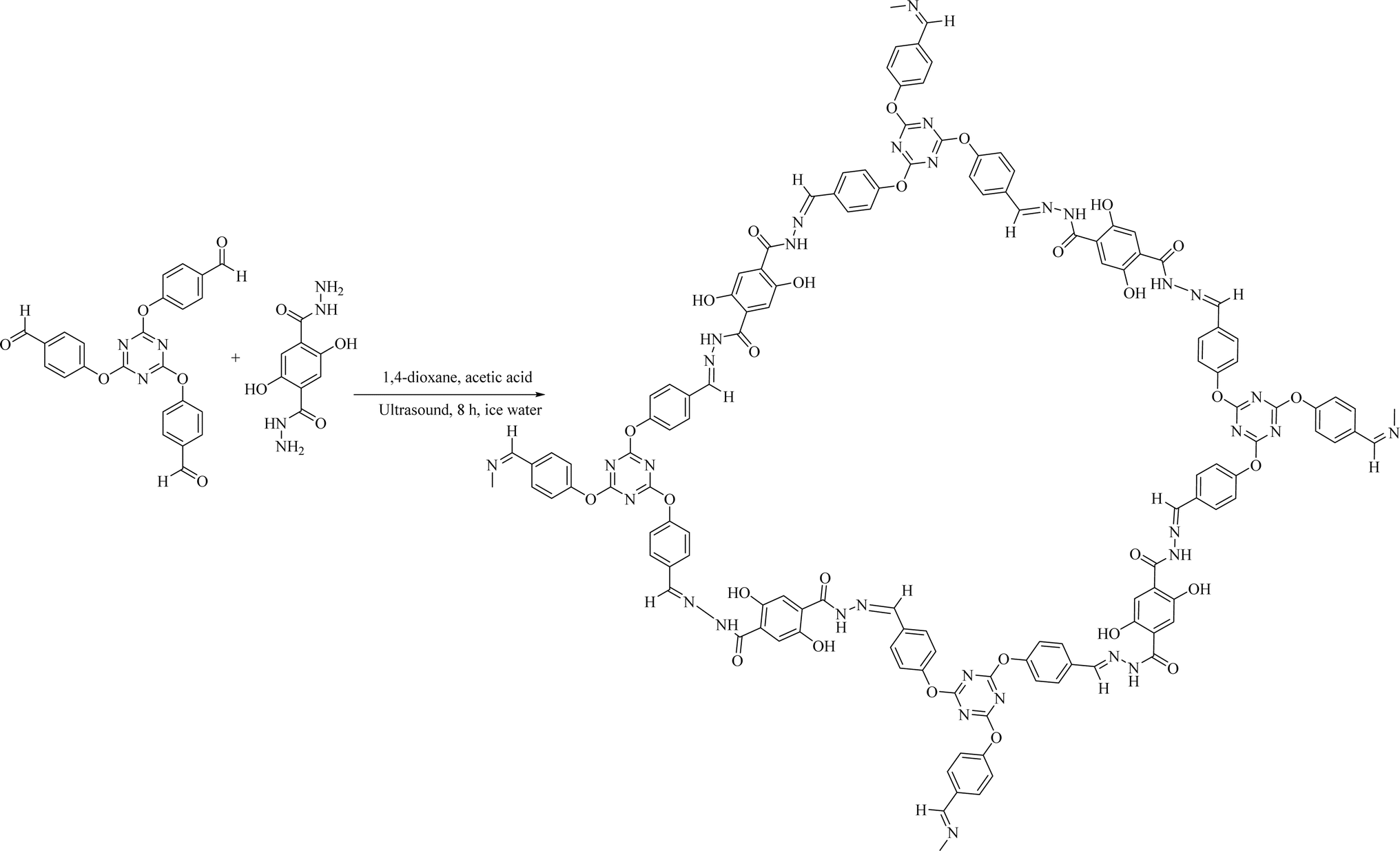 | ||
| Scheme 1 Preparation of the COF. | ||
The synthesized COF was subjected to a series of characterization techniques to evaluate its structural, morphological, surface, and thermal properties. The data obtained from FTIR, XRD, SEM, TEM, BET, XPS, TGA, and DTA analyses are discussed below.
FTIR analysis
Fourier Transform Infrared (FTIR) spectroscopy was employed to confirm the successful formation of the COF and to identify its functional groups. The FTIR spectrum (Fig. 1a) exhibited characteristic absorption bands at 1611 cm−1, corresponding to the CN stretching vibration in the hydrazone linkage, and at 1234 cm−1, indicative of the CN stretching in the triazole ring.40 These peaks confirmed the successful formation of the desired linkages within the COF structure. The absence of significant shifts in these peaks suggests that the COF's framework is chemically stable, a crucial factor for its potential applications in catalysis and adsorption.
 | ||
| Fig. 1 (a) FT-IR spectrum of the synthesized COF; (b) PXRD pattern of the prepared COF. | ||
XRD analysis
The crystalline structure of the COF was investigated using Powder X-ray Diffraction (PXRD). The PXRD pattern (Fig. 1b) revealed a sharp, intense peak at 28.88° and several weaker peaks at 8.3°, 16.74°, 18.07°, 23.39°, 24.43°, 26.96°, 30.42°, and 30.95°, indicating a well-ordered crystalline framework.41 The crystallite size, calculated using the Debye–Scherrer equation, was found to be 23.24 nm. This degree of crystallinity is essential for the COF's functionality, as it impacts the material's stability and its effectiveness in applications requiring a high degree of structural order, such as catalysis.SEM and TEM analyses
Scanning Electron Microscopy (SEM) and Transmission Electron Microscopy (TEM) were used to analyze the morphology and size distribution of the COF particles. The SEM images (Fig. 2a and b) showed that the COF has a rough surface texture, which is beneficial for adsorption processes, as it increases the available surface area for interactions. The TEM images (Fig. 2c and d) revealed a consistent spherical morphology with an average particle size of approximately 135.90 nm. This uniformity in size and shape indicates that the synthesis process was well-controlled, which is vital for ensuring reproducibility in large-scale applications.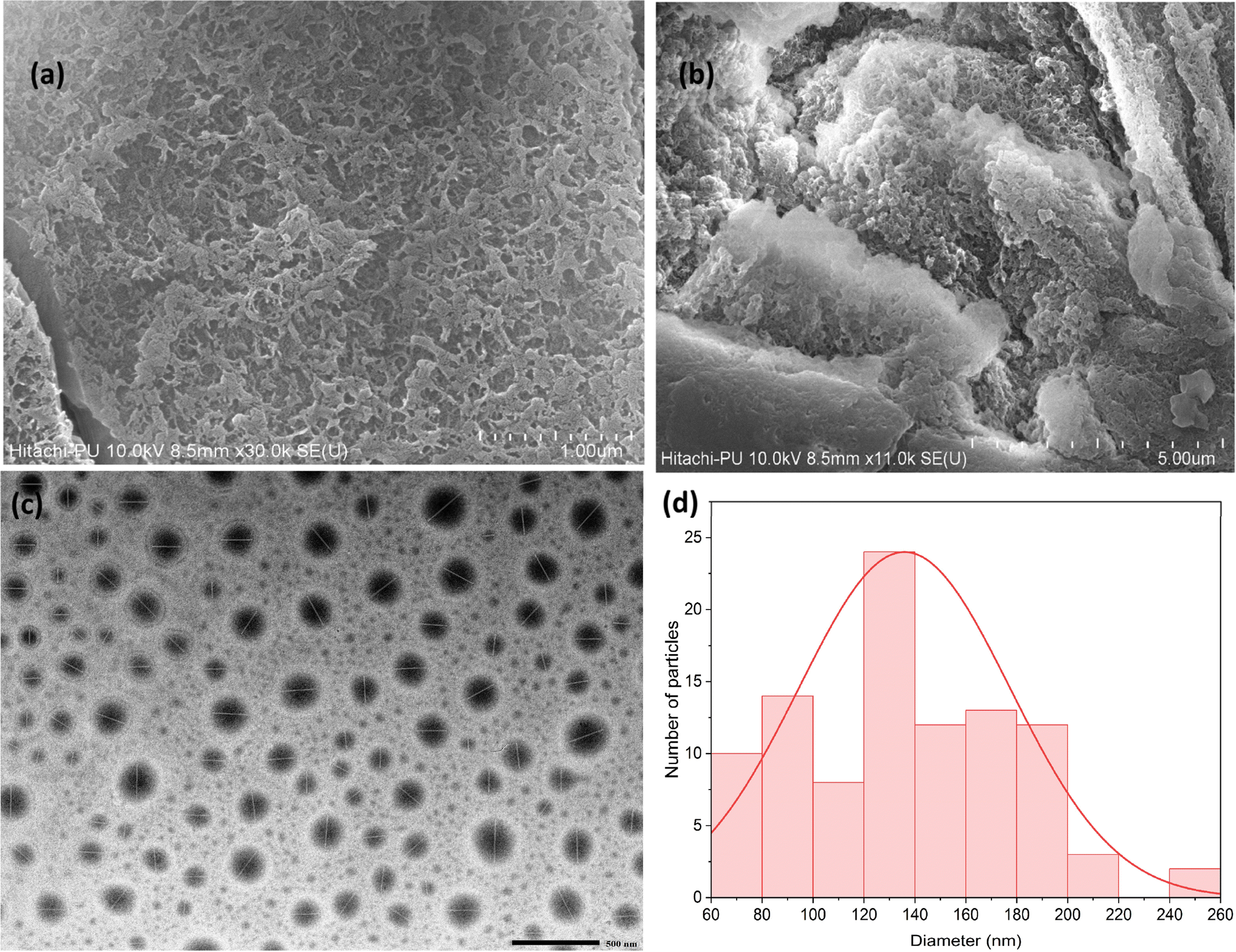 | ||
| Fig. 2 Surface morphology of the synthesized COF: (a) & (b) the SEM images of the COF; (c) the TEM image of the COF; (d) histogram showing the particle size distribution of the COF. | ||
BET surface area and porosity analysis
The surface area and porosity of the COF were characterized using Brunauer–Emmett–Teller (BET) analysis. The nitrogen adsorption–desorption isotherms (Fig. 3) were a type IV isotherm, indicative of a mesoporous structure. The BET surface area was measured to be 2.78 m2 g−1, and moderate, consistent with the COF's mesoporous nature. The Barrett–Joyner–Halenda (BJH) method was used to calculate an average pore size of 8.88 nm and a total pore volume of 0.014 cm3 g−1. These characteristics are crucial for catalytic applications, where pore size and surface area directly influence the material's efficiency in facilitating chemical reactions.42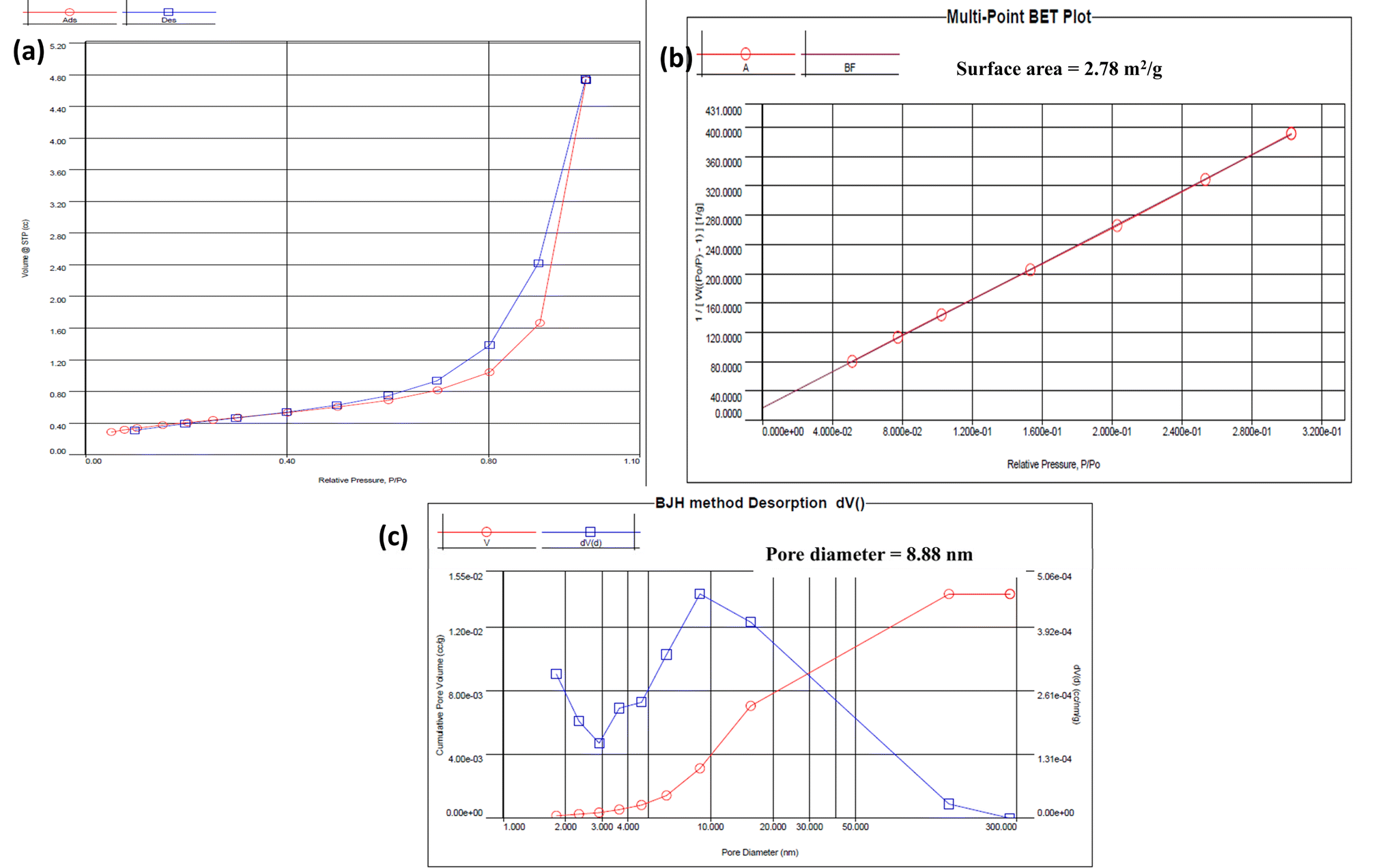 | ||
| Fig. 3 BET analysis of the COF; (a) N2 adsorption–desorption isotherms of the COF; (b) BET surface area plot of the COF calculated from the adsorption data; (c) average pore diameter calculation through the BJH desorption method. | ||
XPS analysis
X-ray Photoelectron Spectroscopy (XPS) was utilized to investigate the COF's surface elemental composition and chemical states. The survey XPS spectrum (Fig. 4) detected carbon (C 1s), nitrogen (N 1s), and oxygen (O 1s) as the main elements on the surface. High-resolution C 1s spectra showed peaks at 284.8 eV (C–C/C–H), 286.2 eV (C–N), and 288.5 eV (CO), confirming the presence of these functional groups. The N 1s peak at 400.2 eV was attributed to sp2-hybridized nitrogen in triazole rings, assuring the integrity of the COF's nitrogenous components. The O 1s peak at around ∼531 eV reflects oxygen atoms, likely originating from hydroxyl or ether groups, enhancing the COF's chemical functionality and adsorption properties. The XPS data suggest that the COF has a surface composition of 64.97% carbon, 14.57% nitrogen, and 19.3% oxygen, consistent with the designed molecular structure.
 | ||
| Fig. 4 XPS study of the COF. | ||
TGA
Thermogravimetric Analysis (TGA) was conducted to assess the thermal stability of the COF. The TGA curve (Fig. 5a) shows an initial minor weight loss at around 100 °C, attributed to the desorption of adsorbed moisture. The major decomposition event occurred between 300 °C and 400 °C, with a weight loss of approximately 48.4%, corresponding to the breakdown of the organic framework. Beyond 400 °C, the TGA curve stabilized, indicating the formation of a thermally stable carbonaceous residue. These results suggest that the COF is thermally stable up to 300 °C, after which significant degradation occurs, limiting its application in high-temperature environments.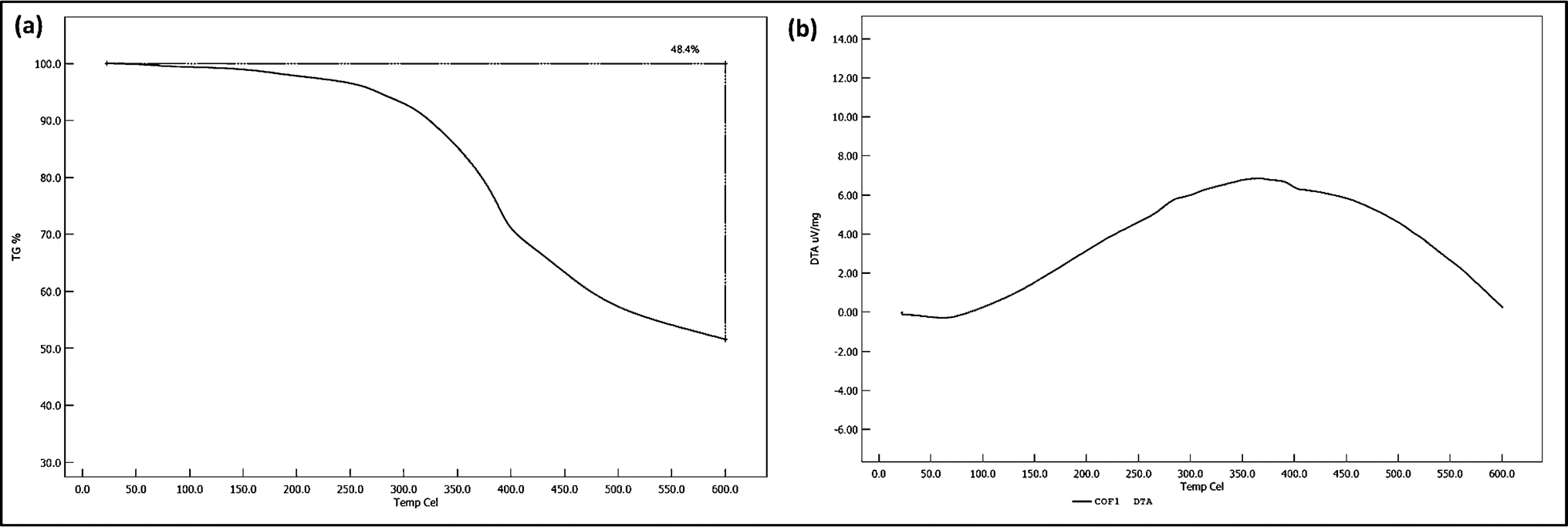 | ||
| Fig. 5 Study about thermal stability of the COF: (a) TGA curve of the COF and (b) DTA curve of the COF. | ||
DTA
Differential Thermal Analysis (DTA) was performed alongside TGA to identify the thermal events associated with the COF's decomposition. The DTA curve (Fig. 5b) displayed a slight endothermic peak at around 100 °C, corresponding to the removal of adsorbed water. A broad exothermic peak between 300 °C and 400 °C was observed, associated with the exothermic decomposition of the organic framework, consistent with the major weight loss seen in TGA. The stabilization of the DTA curve beyond 400 °C aligns with the TGA data, confirming that the remaining residue is thermally stable and does not undergo further significant thermal events.The comprehensive characterization of the synthesized COF reveals a material with promising structural, morphological, and thermal properties. The FTIR and XPS analyses confirmed the successful formation of the desired chemical linkages, while XRD showed a well-ordered crystalline structure. SEM and TEM analyses highlighted the uniformity in particle size and morphology, which is essential for reproducibility. BET analysis indicated moderate surface area and mesoporosity, which are advantageous for catalytic applications. Thermal analysis via TGA and DTA demonstrated that the COF is stable up to 300 °C, after which it decomposes, limiting its use in higher-temperature environments. These findings suggest that the synthesized COF could be a suitable candidate for applications requiring moderate thermal stability and well-defined structural properties.
After the characterization of the prepared COF, their catalytic application was explored in organic synthesis for the synthesis of T-3-Ts through the reaction of TSC with various ketones and aldehydes. A set of experiments were systematically conducted on the reaction of 4-nitrobenzaldehyde and TSC to establish the best reaction conditions, including catalyst loading, solvent choice, and temperature (Scheme 2). The findings are outlined in Table 1.
 | ||
| Scheme 2 The model reaction involving thiosemicarbazide and 4-nitrobenzaldehyde to optimize conditions. | ||
| S. No. | Catalyst loading | Solvent | Temperature | Yield | Time (min) |
|---|---|---|---|---|---|
| 1 | No catalyst | H2O | RT | 42% | 120 |
| 2 | 5 mg | H2O | RT | 60% | 30 |
| 3 | 10 mg | H2O | RT | 68% | 30 |
| 4 | 15 mg | H2O | RT | 80% | 20 |
| 5 | 20 mg | H2O | RT | 90% | 10 |
| 6 | 25 mg | H2O | RT | 89% | 10 |
| 7 | 20 mg | EtOH | RT | 92% | 10 |
| 8 | 20 mg | MeOH | RT | 90% | 20 |
| 9 | 20 mg | Dioxane | RT | 82% | 25 |
| 10 | 20 mg | THF | RT | 85% | 25 |
| 11 | 20 mg | H2O:EtOH (1:1) |
RT | 98% | 6 |
| 12 | 20 mg | H2O:EtOH (1:2) |
RT | 98% | 4 |
| 13 | 20 mg | H2O:EtOH (1:2) |
50 °C | 96% | 5 |
| 14 | 20 mg | H2O:EtOH (1:2) |
80 °C | 96% | 5 |
This study explores the optimization of catalyst loading and solvent choice to enhance yield and efficiency in a chemical reaction. The results revealed a strong correlation between catalyst loading and yield, with an increase from 0 mg to 25 mg elevating the yield from 42% to 89% and reducing the reaction time from 120 minutes to 10 minutes (Table 1, entries 1–6). Notably, the most significant improvement occurred between 5 mg and 15 mg, where yield increased from 60% to 80%. When evaluating solvent effects, a 1:2 mixture of water and ethanol outperformed other solvents, achieving a yield of 98% in just 4 minutes, compared to 90% in 10 minutes with water alone. Other solvents, such as methanol, dioxane, and THF, yielded between 82% and 92% but require longer reaction times (Table 1, entries 7–12). The temperature variations from room temperature to 50 °C or 80 °C show minimal impact on yield or reaction time with the optimal solvent and catalyst loading, maintaining a yield of 96% and a reaction time of 5 minutes (Table 1, entries 13 and 14). Ultimately, the optimal conditions identified are 20 mg of catalyst, a 1:2 water: ethanol solvent ratio, and room temperature, resulting in a yield of 98% in 4 minutes, indicating a well-balanced approach to maximize the reaction efficiency.
To evaluate the protocol's effectiveness and explore the range of the two-component cyclocondensation reaction involving aryl and heteroaryl aldehydes with TCS, a variety of structurally different aldehydes including both aromatic and hetero aromatic species were tested (Scheme 3). These aldehydes reacted efficiently with TCS, yielding the corresponding T-3-Ts in very high to excellent yields. Notably, the newly synthesized TRIPOD smoothly converted with TCS, producing the corresponding 3l (novel) in excellent yields (92%). Further investigation into the scope involved use of acetophenones and cyclic ketones with TCS under optimized experimental parameters, which also resulted in the desired T-3-Ts in remarkable yields Fig. 6.
 | ||
| Scheme 3 General reaction for the preparation of T-3-Ts. | ||
 | ||
| Fig. 6 Synthesized derivatives of T-3-Ts. | ||
Based on the review of existing literature,43 a plausible mechanism has been proposed (Scheme 4). The reactants were adsorbed onto the surface of the COF and the mechanism proceeds through the following steps.
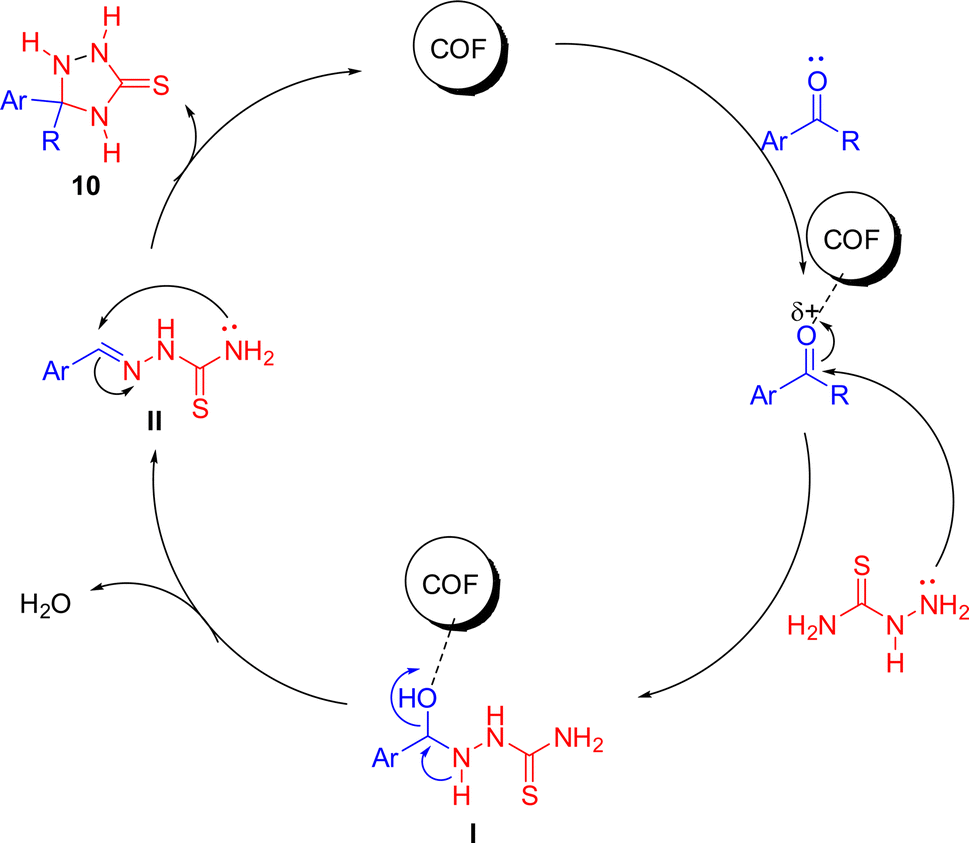 | ||
| Scheme 4 Proposed reaction mechanism for synthesis of T-3-Ts derivatives. | ||
O) of aldehydes or ketones, leading to its activation by polarization. This step weakened the carbonyl bond, making the carbonyl carbon more electrophilic.
• This reaction led to the formation of a CN bond between the TSC and the carbonyl compound, which aligned with the hydrazone linkages present in the COF structure. The porous and moderate surface area of the COF, as suggested by BET analysis, facilitated this adsorption and reaction.
N bond, leading to the formation of the 1,2,4-triazolidine ring.
• During this cyclization process, a proton transfer helped in stabilizing the ring structure, forming a five-membered heterocyclic system.
• The COF served as a heterogeneous catalyst throughout the reaction, facilitating the adsorption, activation, and cyclization steps.
Role of the COF as a catalyst
• The mesoporous structure of the COF, as indicated by BET surface area and pore size distribution, provided a high surface area for reactant molecules to adsorb, allowing the efficient interaction between the carbonyl compounds and thiosemicarbazide.• The nitrogen- and oxygen-rich sites and hydrazone linkages in the COF framework, confirmed by FTIR and XPS, contributed to the activation of carbonyl groups, enhancing the electrophilicity of the substrate and promoting nucleophilic attack.
• The heterogeneous nature of the COF catalyst allowed for the easy separation of the catalyst from the reaction mixture, with no contamination of the final product.
Furthermore, the reusability study of the catalyst was performed on the model reaction. After the completion of the reaction, more ethanol was mixed with the reaction mixture to separate the product, and the COF was separated through simple filtration. Then the catalyst was again washed with ethanol and reused for the next run. The yield was calculated in 4 minutes of reaction time. In six reusability tests, no appreciable change in the yield was seen; afterwards the yield was significantly decreased but the COF was reused up to eight runs with significant catalytic activity (Fig. 7). Furthermore, a comparative study of the present method for the synthesis of T-3-Ts compounds was performed with previous methods as summarized in Table 2. The results illustrated the effectiveness of the developed protocol for the synthesis of T-3-Ts scaffolds and efficient catalytic activity of the prepared COF as shown in Fig. 7.
| S. No. | Catalyst | Reaction conditions | Yield | Time | Reference |
|---|---|---|---|---|---|
| 1 | — | 25% EtOH; 80 °C | 86–95% | 7–15 min | 44 |
| 2 | 3% Sm/FAp | H2O; R.T. | 90–97% | 15–30 min | 45 |
| 3 | VB1 | H2O; R.T. | 65–94% | 12–180 min | 43 |
| 4 | CoCr2O4 NPs | H2O:EtOH (1:1); R.T. |
96–99% | 2.5–5 min | 46 |
| 5 | Meglumine | H2O; R.T. | 85–92% | 5–8 min | 47 |
| 6 | Hydrazone-based COF | H2O:EtOH (1:2); R.T. |
80–98% | 4–20 min | Present work |
 | ||
| Fig. 7 Recyclability study of the COF as a catalyst for synthesis of T-3-Ts scaffolds. | ||
Conclusion
In this study, we have successfully synthesized a novel hydrazone-linked covalent organic framework (COF) using synthesized TRIPOD and DHTH. The synthesis process, which involved the ultrasonic treatment of TRIPOD and DHTH in a blend of 1,4-dioxane and aqueous acetic acid, yielded pale-yellow COF powders, insoluble in common organic solvents. The characterization through FT-IR, PXRD, SEM, TEM, XPS, BET, TGA, and DTA confirmed the successful formation and crystalline structure of the stable COF, with a consistent spherical morphology and a crystallite size of 23.24 nm. The COF showed a surface area of 2.78 m2 g−1 and mesoporous characteristics, essential for catalytic activities. Moreover, the synthesized COF showed thermal stability up to 300 °C. This stability and porosity played a key role in its catalytic performance in the production of 1,2,4-triazolidine-3-thiones from thiosemicarbazide and various ketones and aldehydes. The optimization of reaction conditions revealed that a catalyst loading of 20 mg, a 1:2 water:ethanol solvent ratio, and room temperature were ideal, achieving high to excellent yields (80–98%) of products in a very low reaction time (4–20 min). The reusability tests demonstrated that the COF exhibited consistent catalytic activity across eight cycles of reuse without substantial degradation. Overall, this research highlights the potential of hydrazone-linked COFs in catalysis, providing a new avenue for their application in synthetic chemistry. The straightforward synthesis, combined with excellent catalytic performance and reusability, underscores the viability of these COFs for future catalytic processes.
Data availability
The data related to the manuscript are available in the ESI file.†Conflicts of interest
The authors confirmed that this article has no conflict of interest.Acknowledgements
The authors are thankful to the Department of Chemistry, MLSU, Udaipur, for providing research facilities. They are also thankful to SAIF, Chandigarh, for SEM, TEM, BET, and HRMS, PDEU for XRD, SAIF, IIT Mumbai for TGA and DTA, and IIT Jammu for XPS studies. P. Teli wishes to acknowledge CSIR, India (09/172(0099)2019-EMR-I) for a senior research fellowship as financial support. S. Agarwal also sincerely acknowledges the Ministry of Education, SPD-RUSA Rajasthan for providing the NMR facility under the RUSA 2.0, Research and Innovation project (File no./RUSA/GEN/MLSU/2020/6394). S. Agarwal and S. Teli also acknowledge DST-SERB, SURE (no. SUR/2022/001312) for financial support.References
- S.-Y. Ding and W. Wang, Chem. Soc. Rev., 2013, 42, 548–568 RSC.
- N. Huang, P. Wang and D. Jiang, Nat. Rev. Mater., 2016, 1, 16068 CrossRef CAS.
- M. S. Lohse and T. Bein, Adv. Funct. Mater., 2018, 28, 1705553 CrossRef.
- J. Xiao, J. Chen, J. Liu, H. Ihara and H. Qiu, Green Energy Environ., 2023, 8, 1596–1618 CrossRef CAS.
- J. Hu, Z. Huang and Y. Liu, Angew. Chem., Int. Ed., 2023, 62, e202306999 CrossRef CAS PubMed.
- P. T. Parvatkar, S. Kandambeth, A. C. Shaikh, I. Nadinov, J. Yin, V. S. Kale, G. Healing, A.-H. Emwas, O. Shekhah, H. N. Alshareef, O. F. Mohammed and M. Eddaoudi, J. Am. Chem. Soc., 2023, 145, 5074–5082 CrossRef CAS PubMed.
- S. Liu, Q. Su, W. Qi, K. Luo, X. Sun, H. Ren and Q. Wu, Catal. Sci. Technol., 2022, 12, 2837–2845 RSC.
- M. Tian, S. Liu, X. Bu, J. Yu and X. Yang, Chem.–Eur. J., 2020, 26, 369–373 CrossRef CAS.
- A. Jiménez-Almarza, A. López-Magano, L. Marzo, S. Cabrera, R. Mas-Ballesté and J. Alemán, ChemCatChem, 2019, 11, 4916–4922 CrossRef.
- P. Pachfule, A. Acharjya, J. Roeser, T. Langenhahn, M. Schwarze, R. Schomäcker, A. Thomas and J. Schmidt, J. Am. Chem. Soc., 2018, 140, 1423–1427 CrossRef CAS PubMed.
- K. Asokan, M. K. Patil, S. P. Mukherjee, S. B. Sukumaran and T. Nandakumar, Chem.–Asian J., 2022, 17, e202201012 CrossRef CAS.
- Q. N. Tran, H. J. Lee and N. Tran, Polymers, 2023, 15, 1279 CrossRef CAS PubMed.
- X. Zhang, G. Li, D. Wu, B. Zhang, N. Hu, H. Wang, J. Liu and Y. Wu, Biosens. Bioelectron., 2019, 145, 111699 CrossRef CAS.
- R. Davis, R. A. Urbanowski and A. K. Gaharwar, Curr. Opin. Biomed. Eng., 2021, 20, 100319 CrossRef CAS.
- A. Murali, G. Lokhande, K. A. Deo, A. Brokesh and A. K. Gaharwar, Mater. Today, 2021, 50, 276–302 CrossRef CAS.
- H. H. Hegazy, S. S. Sana, T. Ramachandran, Y. A. Kumar, D. K. Kulurumotlakatla, H. S. M. Abd-Rabboh and S. C. Kim, J. Energy Storage, 2023, 74, 109405 CrossRef.
- S.-X. Gan, C. Jia, Q.-Y. Qi and X. Zhao, Chem. Sci., 2022, 13, 1009–1015 RSC.
- H. Hu, Q. Yan, R. Ge and Y. Gao, Chin. J. Catal., 2018, 39, 1167–1179 CrossRef CAS.
- Z. Alsudairy, N. Brown, A. Campbell, A. Ambus, B. Brown, K. Smith-Petty and X. Li, Mater. Chem. Front., 2023, 7, 3298–3331 RSC.
- D. Ma, Y. Wang, A. Liu, S. Li, C. Lu and C. Chen, Adv. Energy Mater., 2018, 8, 1702170 CrossRef.
- S. Pasricha, A. Chaudhary and A. Srivastava, ChemistrySelect, 2022, 7, e202200576 CrossRef CAS.
- Y. Zhi, Z. Wang, H.-L. Zhang and Q. Zhang, Small, 2020, 16, 2001070 CrossRef CAS PubMed.
- S. S. A. Shah, M. S. Javed, T. Najam, M. A. Nazir, A. ur Rehman, A. Rauf, M. Sohail, F. Verpoort and S.-J. Bao, Mater. Today, 2023, 67, 229–255 CrossRef CAS.
- J. Guo and D. Jiang, ACS Cent. Sci., 2020, 6, 869–879 CrossRef CAS PubMed.
- N. Khaleghi, M. Esmkhani, M. Noori, N. Dastyafteh, M. K. Ghomi, M. Mahdavi, M. H. Sayahi and S. Javanshir, Nanoscale Adv., 2024, 6, 2337–2349 RSC.
- R. Bakhshali-Dehkordi, M. A. Ghasemzadeh and J. Safaei-Ghomi, J. Mol. Struct., 2020, 1206, 127698 CrossRef CAS.
- M. A. Ghasemzadeh, B. Mirhosseini-Eshkevari and M. H. Abdollahi-Basir, Comb. Chem. High Throughput Screening, 2016, 19, 592–601 CrossRef CAS.
- R. H. Althomali, E. A. Musad Saleh, R. H. Mohammed Ali, I. I. Mamadoliev, M. F. Ramadan, A. T. Kareem, S. Aggarwal and S. K. Hadrawi, Nanoscale Adv., 2023, 5, 6177–6193 RSC.
- B. Mirhosseini-Eshkevari, M. A. Ghasemzadeh and M. Esnaashari, Appl. Organomet. Chem., 2019, 33, e5027 CrossRef.
- J. Safaei-Ghomi, M. A. Ghasemzadeh and A. Kakavand-Qalenoei, J. Saudi Chem. Soc., 2016, 20, 502–509 CrossRef CAS.
- P. G. Mahajan, N. C. Dige, B. D. Vanjare, H. Raza, M. Hassan, S.-Y. Seo, C.-H. Kim and K. H. Lee, Mol. Diversity, 2020, 24, 1185–1203 CrossRef CAS PubMed.
- H. Adibi, R. Abiri, S. Mallakpour, M. A. Zolfigol and M. B. Majnooni, J. Rep. Pharm. Sci., 2012, 1 Search PubMed.
- W. M. Huggins, B. M. Minrovic, B. W. Corey, A. C. Jacobs, R. J. Melander, R. D. Sommer, D. V. Zurawski and C. Melander, ACS Med. Chem. Lett., 2017, 8, 27–31 CrossRef CAS PubMed.
- N. U. Hebbar, P. S. Hebbar, P. P. Rathode, B. Negalura, A. Hiremath, P. Gudimani, L. A. Shastri, S. K. Praveen Kumar, A. J. Kadapure and S. Joshi, J. Mol. Struct., 2023, 1280, 135003 CrossRef CAS.
- V. Kanagarajan, J. Thanusu and M. Gopalakrishnan, Med. Chem. Res., 2012, 21, 3965–3972 CrossRef CAS.
- F. Shaikh, S. L. Shastri, N. S. Naik, R. Kulkarni, J. M. Madar, L. A. Shastri, S. D. Joshi and V. Sunagar, ChemistrySelect, 2019, 4, 105–115 CrossRef CAS.
- H. Adibi, L. Hosseinzadeh, M. Mahdian, A. Foroumadi, M. A. Zolfigol and S. Mallakpour, J. Rep. Pharm. Sci., 2013, 2 Search PubMed.
- J. A. Mikroyannidis, S. S. Sharma, Y. K. Vijay and G. D. Sharma, ACS Appl. Mater. Interfaces, 2010, 2, 270–278 CrossRef CAS.
- L. R. Ahmed, A. F. M. El-Mahdy, C.-T. Pan and S.-W. Kuo, Mater. Adv., 2021, 2, 4617–4629 RSC.
- S. Haldar, K. Roy, S. Nandi, D. Chakraborty, D. Puthusseri, Y. Gawli, S. Ogale and R. Vaidhyanathan, Adv. Energy Mater., 2018, 8, 1702170 CrossRef.
- H. M. El-Kaderi, J. R. Hunt, J. L. Mendoza-Cortés, A. P. Côté, R. E. Taylor, M. O'Keeffe and O. M. Yaghi, Science, 2007, 316, 268–272 CrossRef CAS PubMed.
- S. Zeghdi, S. E. Laouini, H. A. Mohammed, A. Bouafia, M. L. Tedjani, M. M. S. Abdullah and T. Trzepieciński, Materials, 2024, 17, 2358 CrossRef CAS.
- P. J. Patil, G. D. Salunke, M. B. Deshmukh, S. P. Hangirgekar, D. R. Chandam and S. A. Sankpal, ChemistrySelect, 2019, 4, 13071–13078 CrossRef CAS.
- R. Ramesh and A. Lalitha, ChemistrySelect, 2016, 1, 2085–2089 CrossRef CAS.
- S. N. Maddila, S. Maddila, K. K. Gangu, W. E. van Zyl and S. B. Jonnalagadda, J. Fluorine Chem., 2017, 195, 79–84 CrossRef CAS.
- S. Ghotekar, D. Sanap, K.-Y. A. Lin, H. Louis, D. Pore and R. Oza, Res. Chem. Intermed., 2024, 50, 49–68 CrossRef CAS.
- L. B. Masram, S. S. Salim, A. B. Barkule, Y. U. Gadkari and V. N. Telvekar, J. Chem. Sci., 2022, 134, 94 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: FTIR, PXRD, SEM, TEM, BET, XPS, TGA, and DTA studies of the synthesized catalyst and 1H, 13C NMR, and mass spectra of COF precursors and synthesized heterocyclic compounds. See DOI: https://doi.org/10.1039/d4na00650j |
| This journal is © The Royal Society of Chemistry 2024 |