DOI:
10.1039/D3NH00401E
(Communication)
Nanoscale Horiz., 2024,
9, 295-304
Hydrogel-mediated tumor T cell infiltration and immune evasion to reinforce cancer immunotherapy†
Received
15th September 2023
, Accepted 4th December 2023
First published on 5th December 2023
Abstract
Cancer immunotherapy has received increasing attention in tumor therapy. However, insufficient infiltration of T cells and over-expressed PD-L1 checkpoint in tumor cells severely impede cancer immunotherapy. Here, an injectable hydrogel was designed to reinforce T cell infiltration and inactivate PD-L1 for powerful cancer immunotherapy. The hydrogel was created by sodium alginate (SA) as the gelator, where linagliptin particles and BMS-202 particles were present in hydrogel micropores. After gelation in the tumor site, the linagliptin powerfully suppressed chemokine CXCL10 degradation, enabling the introduced CXCL10 to realize sustainable chemotaxis towards strong T cell infiltration. Meanwhile, the BMS-202 inactivated PD-L1 of tumor cells, thereby eliminating the PD-L1-governed immune evasion. Therefore, the hydrogel in combination with CXCL10 demonstrated powerful cancer immunotherapy against primary and distant tumors, along with efficient inhibition of lung metastasis. Our study not only offers a potent platform against tumors, but also provides a conceptually new approach to reinforce cancer immunotherapy.
New concepts
Cancer immunotherapy has become a promising approach to combat tumors by activating T cells. However, the inadequate infiltration of T cells into tumors remains a challenge to the efficacy of cancer immunotherapy. In this study, an injectable hydrogel approach was proposed to address the issues of insufficient T cell infiltration and PD-L1-dominated immune evasion. This hydrogel demonstrated strong tumor T cell infiltration by regulating dipeptidyl peptidases 4 (DPP4)-mediated CXCL10 degradation to achieve a sustainable CXCL10 chemotaxis, a new example of in situ application of a degradation inhibitor to protect chemokines. Meanwhile, the PD-L1 was greatly blocked by the BMS-202 of the hydrogel, effectively disrupting the PD-L1-dominated immune evasion. This work offers a valuable therapeutic platform and approach to reinforce cancer immunotherapy, demonstrating great potential for clinical application.
|
Introduction
Cancer immunotherapy by activating T cells to eliminate tumor cells has become a promising approach for tumor treatment in recent years.1–4 Although cytotoxic T cells (CTLs) have powerful lethality to tumor cells, the clinic therapeutic efficacy of cancer immunotherapy is still low owing to the inadequate infiltration of T cells into tumors.5,6 To address the insufficient infiltration of T cells caused by the tumor microenvironment, strategies such as inducing immunogenic cell death of tumor cells or destroying the cell matrix within tumor tissue have been largely studied.7–10 However, these approaches remain unsatisfactory to stimulate adequate infiltration of T cells against tumors. Hence, it is of great interest to develop a new approach to enable powerful infiltration of T cells in cancer immunotherapy.
CXCL10 is a chemokine capable of recruiting T cells to promote their infiltration in tumors.11,12 Due to its strong recruitment ability, CXCL10 has exhibited great potential for application clinically. However, the short half-life of CXCL10 is a major drawback when using CXCL10 to promote infiltration of T cells.13 Researchers have modified the DNA of CXCL10 to construct new CXCL10 chimeric proteins to prolong circulation time.13 Examples of the use of plasmids for in situ expression of CXCL10 in vivo are also available.14,15 However, these approaches are complex and have high requirements for carriers. The short half-life of CXCL10 is discovered to be caused by dipeptidyl peptidases 4 (DPP4)-catalyzed degradation, resulting in a loss of function in vivo.16–18 Therefore, we suppose that regulation of DPP4 activity is a promising approach to address the issue of the short half-life of CXCL10 for realizing its sustainable recruitment of T cells to amplify the cancer immunotherapy.
Besides the insufficient infiltration of T cells, the over-expressed programmed cell death-ligand 1 (PD-L1) checkpoint on tumor cells significantly impairs the immunotherapeutic effect via the immune evasion caused by the programmed cell death receptor-1 (PD-1)/PD-L1 pathway.19,20 Recently, a small molecule immune checkpoint blocker such as BMS-202 has demonstrated promising application in immune checkpoint PD-1/PD-L1 blockade therapy via inducing PD-L1 dimerization outside tumor cells, owing to its advantage of being inexpensive and having low immunological side effects over PD-L1 antibody.21,22 Such PD-1/PD-L1 blockade therapy offers a valuable approach in addressing the immune evasion caused by the PD-L1 checkpoint in tumor cells.
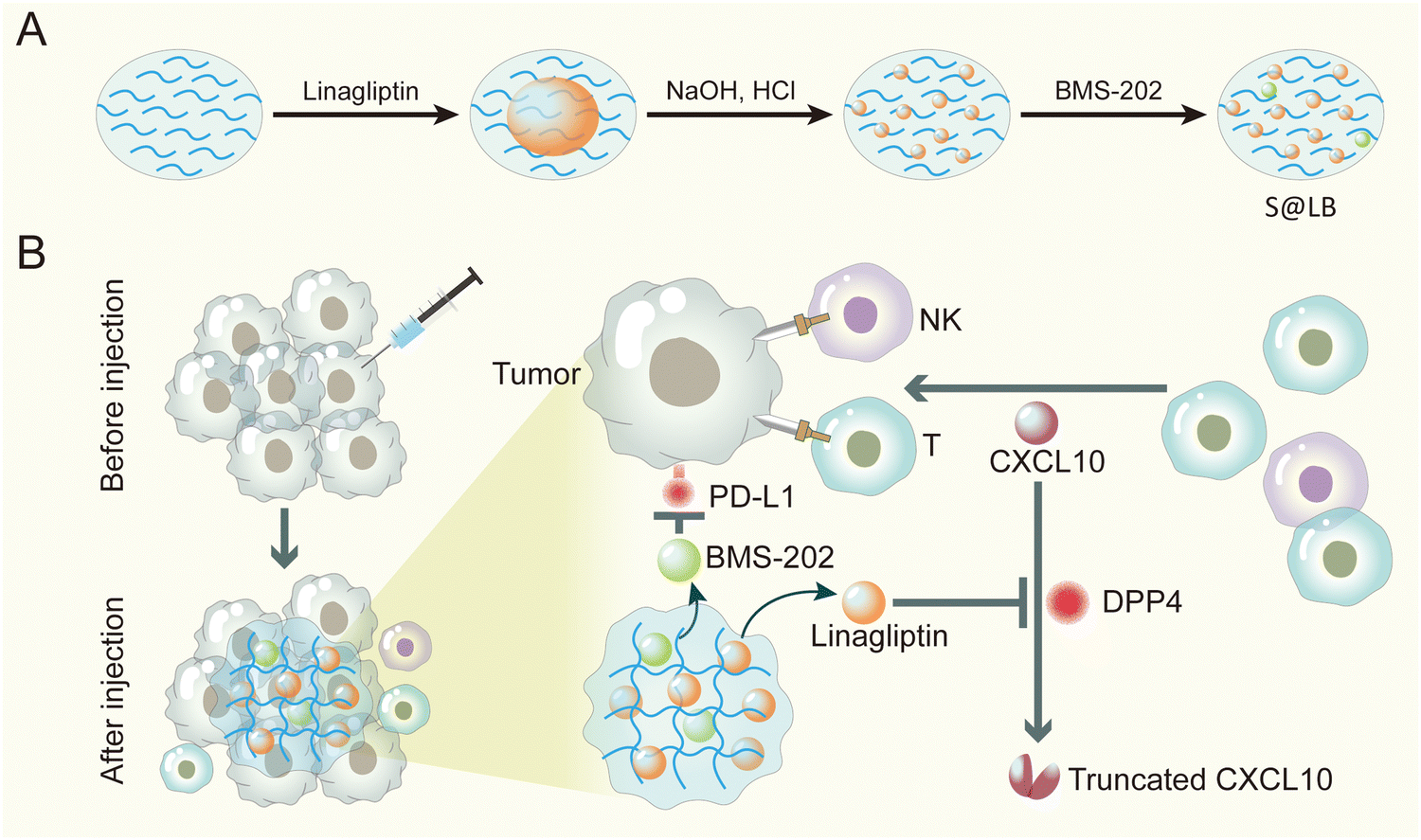
Taken together, here an innovative injectable hydrogel approach was proposed to address the insufficient infiltration of T cells and the immune evasion by over-expressed PD-L1 on tumor cells in cancer immunotherapy. Considering the poor solubility of linagliptin and BMS-202, we formulated them into particles. Furthermore, in order to precisely enhance CXCL10 expression in the tumor tissue without causing side effects of systemic administration, we chose intratumoral injection and gel-based delivery. Sodium alginate (SA) was chosen as the gelator for constructing the hydrogel, owing to its advantages of low cost, biocompatibility and tumor environmental Ca2+-responsiveness.23 Linagliptin was utilized as an inhibitor of DPP4 because it enables inactivity of DPP4 through non-covalent interaction with residues in the catalytic pocket.24 Moreover, BMS-202 was applied as the small molecule immune checkpoint blocker to regulate PD-L1-induced immune evasion. The hydrogel was established by SA in the tumor site, where the linagliptin and BMS-202 particles were encapsulated in the micropores of the hydrogel (Scheme 1). When the constructed SA@linagliptin/BMS-202 (S@LB) hydrogel was combined with CXCL10, the linagliptin enabled powerful suppression of CXCL10 degradation, resulting in its sustainable chemotaxis for achieving strong tumor T cell infiltration. Meanwhile, the BMS-202 disrupted the PD-L1-dominated immune evasion. As a result, the S@LB hydrogel in combination with CXCL10 exhibited potent cancer immunotherapy against tumors. This study offers an innovative strategy to reinforce cancer immunotherapy, holding great promise in clinical application.
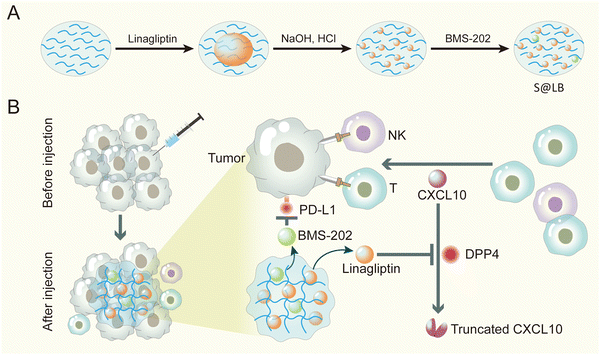 |
| | Scheme 1 Preparation and mechanism scheme of S@LB. (A) The preparation process of the S@LB solution. (B) Schematic illustration of an injectable hydrogel to reinforce cancer immunotherapy by promoting infiltration of T cells and regulating immune evasion. | |
Results and discussion
Preparation and characterization of S@LB hydrogel
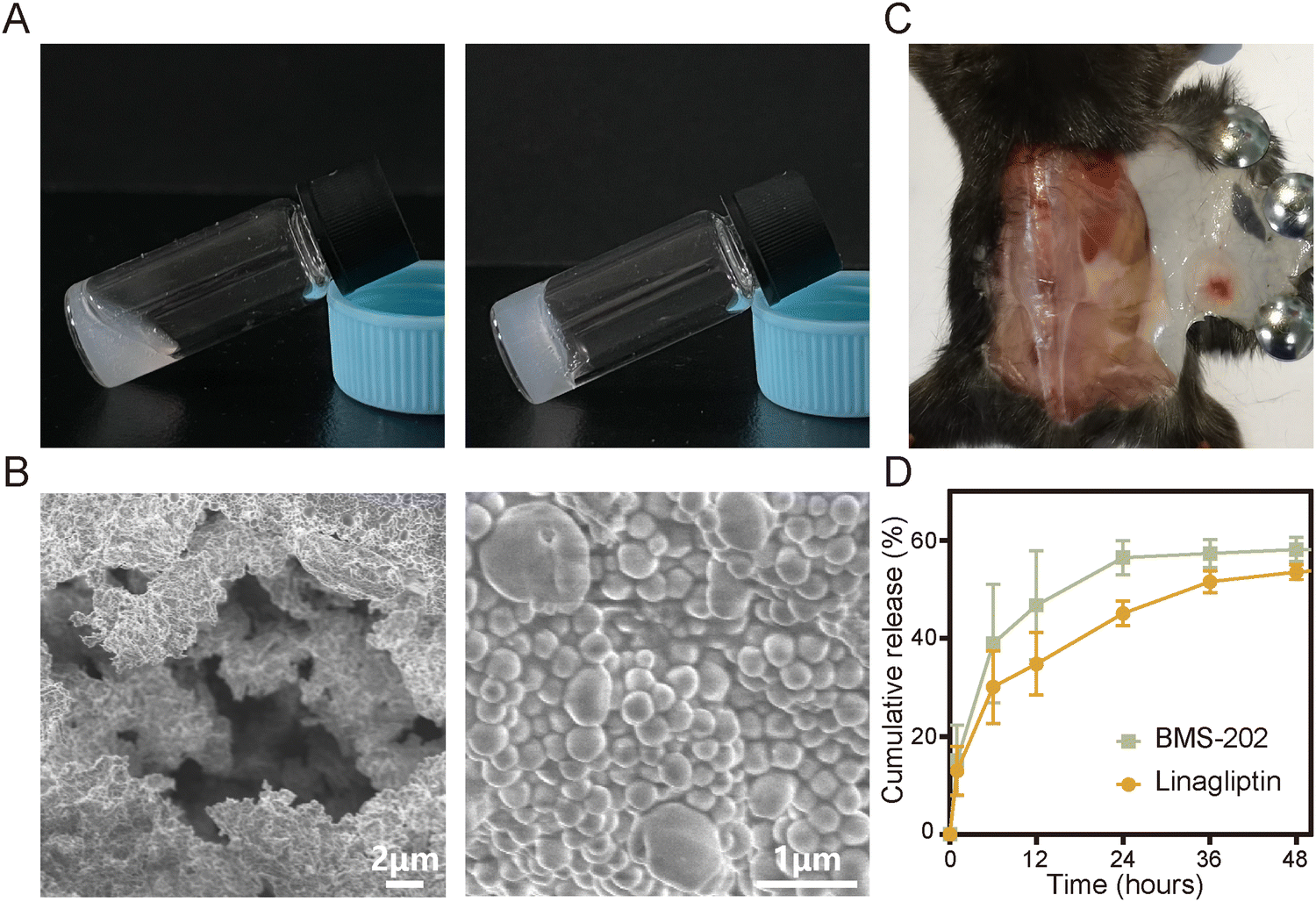
S@LB solution containing linagliptin and BMS-202 particles was prepared by adjusting the pH of linagliptin hydrochloride in SA aqueous solution with subsequently introducing BMS-202. Besides S@LB solution, SA@Linagliptin (S@Linagliptin) and SA@BMS-202 (S@BMS-202) were also prepared as a control. Since SA has –COO− groups with the ability to chelate Ca2+, the SA-based solution can convert into a hydrogel in the presence of Ca2+.23 The in vitro gelation performance was studied by introducing physiological-concentration Ca2+.25 After adding the Ca2+ solution, the S@LB changed from the liquid state to the hydrogel state, verifying its Ca2+-responsive formation of a hydrogel (Fig. 1A). Scanning electron microscopy (SEM) was utilized to study the microstructure of the formed S@LB hydrogel with S@Linagliptin and S@BMS-202 hydrogels as the control. The SEM results in Fig. 1B and Fig. S1 (ESI†) demonstrated the porous microstructure of the formed hydrogels. Moreover, particles were shown in the SEM images, which were attributed to the linagliptin particles (≈400 nm) and BMS-202 particles (≈1000 nm) in the hydrogel.
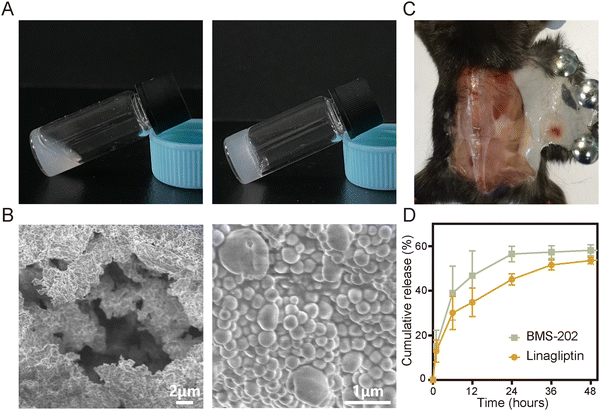 |
| | Fig. 1 Characterization of the physical and chemical properties of S@LB hydrogel. (A) Photographs of S@LB taken before and after the addition of Ca2+ solution. (B) SEM images of S@LB hydrogel. (C) Photograph of S@LB after subcutaneous injection. (D) Release behavior of S@LB in PBS (phosphate buffer solution, 0.5% Tween-80) at pH 6.5 (n = 3). | |
Furthermore, the S@LB solution could easily pass through the needle of a syringe, and rapidly converted into a hydrogel state in the bottle containing Ca2+, indicating its favorable needle-passing property to enable in vivo injection (Fig. S2A, ESI†). In addition, the rheological property assay results showed that the viscous modulus (G′′) was greater than the elastic modulus (G′) before gelation, and the G′ was greater than the G′′ after gelation, which further indicated that S@LB had a satisfactory injectable ability (Fig. S2B, ESI†). The gelation behavior in vivo was further studied by injecting S@LB solution subcutaneously into mice. As shown in Fig. 1C, the S@LB could change to a hydrogel state in the subcutaneous tissue of mice, thanks to the inherent Ca2+ in mice. The cytotoxicity assay showed that SA was safe and nontoxic as a carrier (Fig. S2C, ESI†). We further investigated the in vitro release behavior of the S@LB hydrogel. Fig. 1D shows that the S@LB hydrogel enabled a sustained release of linagliptin and BMS-202. The above results thus indicated that the S@LB solution was feasible as an injectable hydrogel in cancer therapy.
In vivo regulation of DPP4 to inhibit CXCL10 degradation via S@LB hydrogel
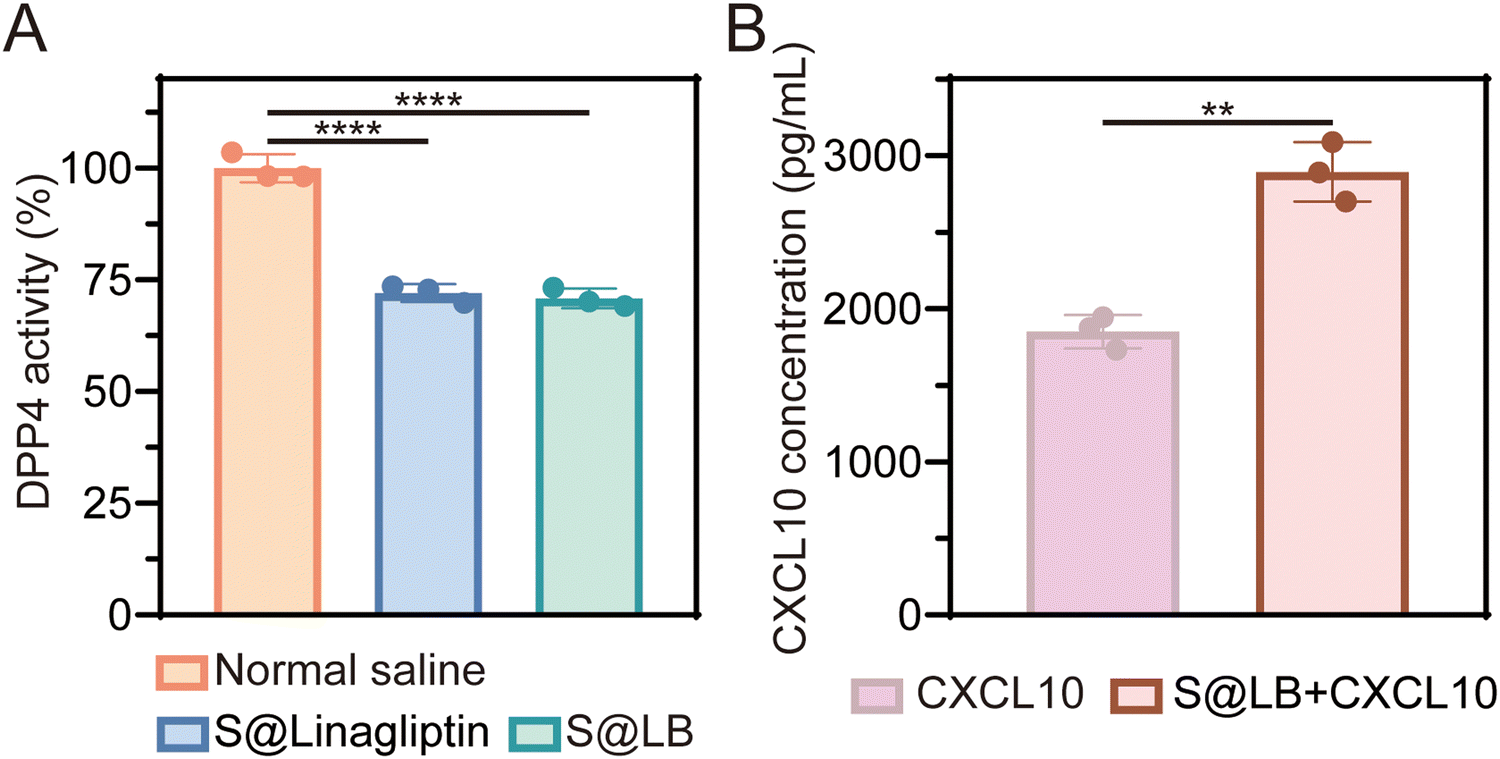
DPP4 can degrade CXCL10 at tumor sites while linagliptin, as a DPP4 inhibitor, can inhibit the activity of DPP4 to protect CXCL10 from degradation.16 The DPP4 substrate glycine-proline-7-amido-4-methylcoumarin (Gly-Pro-AMC) was used to study the inhibitory effect of S@LB hydrogel on DPP4 since Gly-Pro-AMC can be decomposed by DPP4 to produce fluorescent 7-amido-4-methylcoumarin (AMC). In particular, the fluorescence intensity of AMC is positively correlated with the activity of DPP4.26Fig. 2A shows that S@LB hydrogel could significantly inhibit the DPP4 activity in tumors in vivo, thanks to the linagliptin in the hydrogel. The hydrogel-enabled upregulation of CXCL10 in the tumor by suppressing DPP4 activity was further studied. As shown by the ELISA results in Fig. 2B, the CXCL10 content in the S@LB hydrogel + CXCL10 group was greatly enhanced in comparison with the free CXCL10 group, demonstrating the efficient inhibition of CXCL10 degradation by the hydrogel. This result indicated that the S@LB hydrogel could be an effective platform to enable sufficient CXCL10 to induce strong infiltration of T cells.
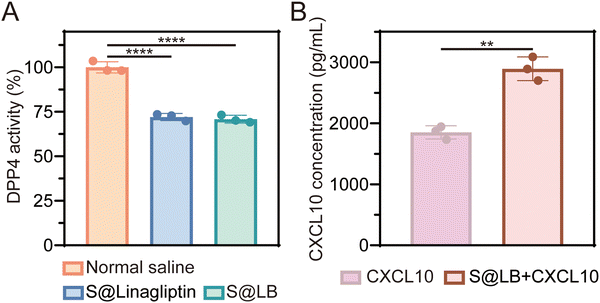 |
| | Fig. 2 The effect of S@LB on DPP4 and CXCL10. (A) DPP4 activity in tumor tissues after different treatments (n = 3). (B) Concentration of CXCL10 at primary tumors (n = 3). | |
In vitro PD-L1 blocking effect via the S@LB hydrogel
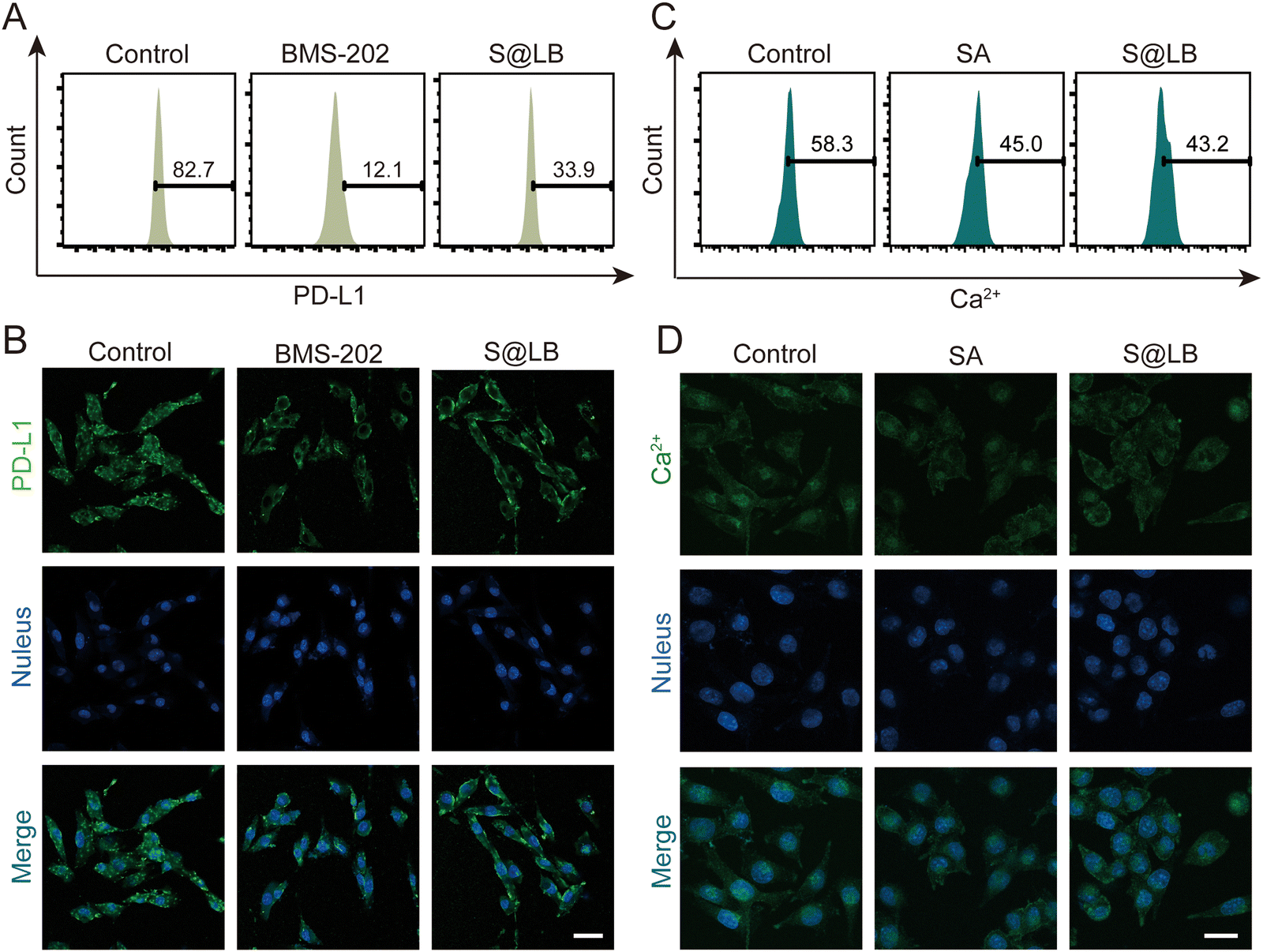
BMS-202 is capable of restoring the tumor-killing function of T cells by blocking PD-L1 on the surface of tumor cells.27 The efficacy of blocking PD-L1 by S@LB hydrogel was further studied. To this end, the fluorescent PE-aPD-L1 was utilized, in which the fluorescence was correlated with the amount of PD-L1. From the flow cytometry result (Fig. 3A and Fig. S3A, ESI†), the proportion of surface PD-L1-positive tumor cells significantly decreased to 33.9% after administration of S@LB in comparison with 82.7% in the control group, thanks to the BMS-202 component in the hydrogel. The blocking effect of PD-L1 was further demonstrated by laser scanning confocal microscopy (LSCM), where the fluorescence intensity demonstrated that the S@LB hydrogel group greatly blocked PD-L1 (Fig. 3B). The results indicated that the effect of the hydrogel on PD-L1 blockade is satisfactory.
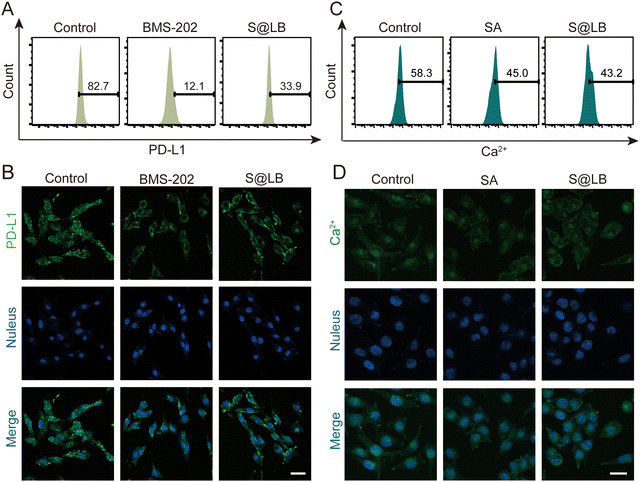 |
| | Fig. 3 The effect of S@LB on PD-L1. The blocking effect of different treatments on PD-L1 of B16F10 cells characterized by (A) flow analysis and (B) fluorescence imaging. The scale bar is 40 μm. Intracellular Ca2+ concentration in B16F10 cells after different treatments characterized by (C) flow analysis and (D) fluorescence imaging. The scale bar is 20 μm. | |
Studies have shown that the decrease of intracellular Ca2+ can increase the autophagy of PD-L1 and downregulate the expression of PD-L1 in tumor cells.28 The SA in S@LB can chelate Ca2+ in the tumor environment, thus resulting in the reduction of intracellular Ca2+ to further downregulate PD-L1. To demonstrate the ability of intracellular Ca2+ reduction enabled by the S@LB hydrogel, the Ca2+ fluorescent probe Fluo-3 AM was used for determination. As seen from the result of flow cytometry (Fig. 3C and Fig. S3B, ESI†) and LSCM (Fig. 3D), the intracellular Ca2+ concentration decreased from 58.3% to 43.2% after introducing S@LB. Therefore, SA and BMS-202 in the S@LB hydrogel enabled synergistic downregulation of PD-L1.
Intratumoral retention of the drug in vivo
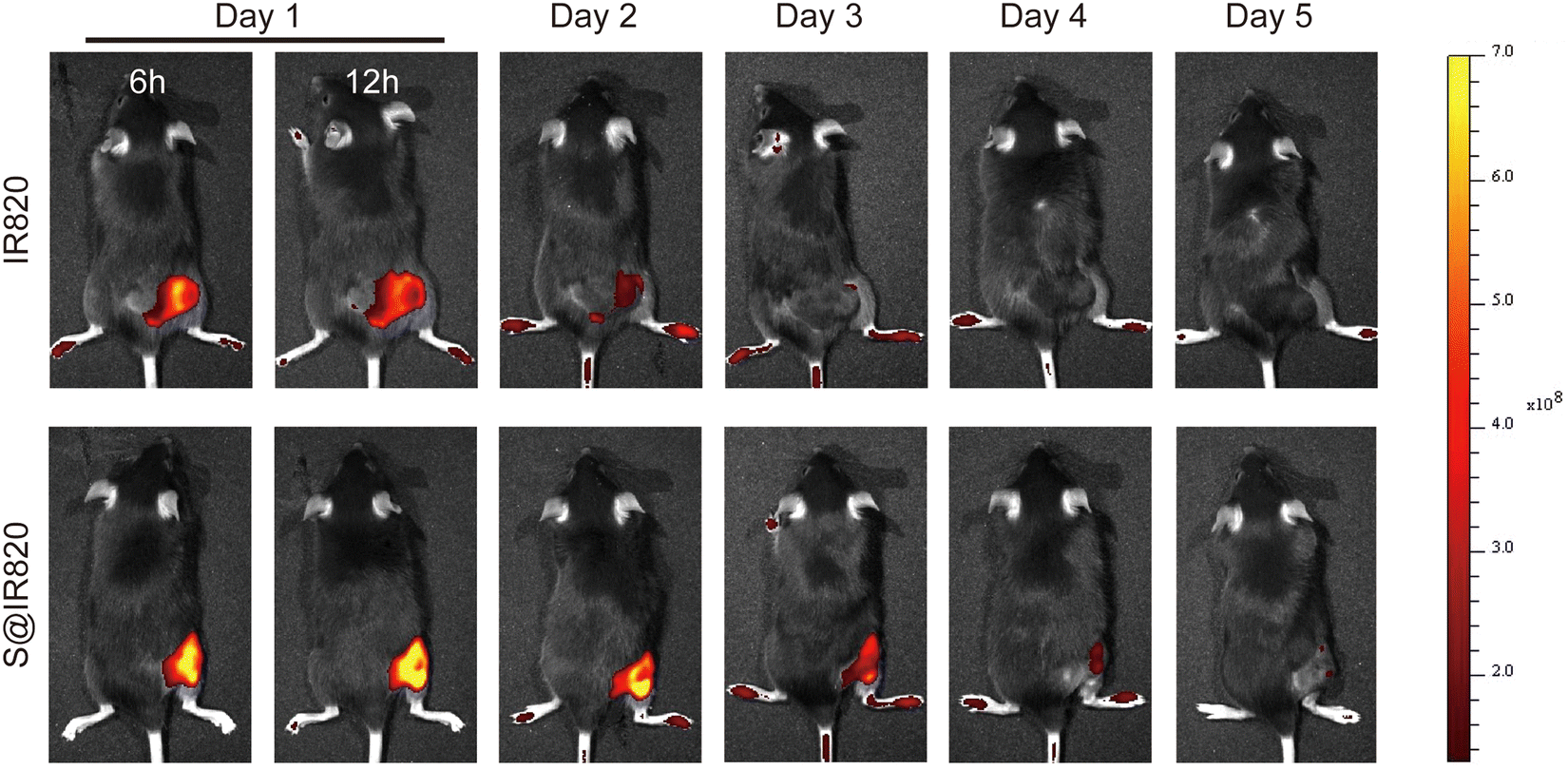
The intratumoral drug retention property of the hydrogel was further studied. In this case, the dye IR820 was used as the probe to determine the drug retention ability in the tumor. The S@IR820 was prepared by mixing the SA solution with the dye IR820, which enabled the drug retention property of the S@LB hydrogel to be determined as IR820 has fluorescence properties. The tumor-bearing mice were injected intratumorally with free IR820 and S@IR820, respectively. The fluorescence intensity of IR820 in the tumor was characterized by in vivo imaging at the predetermined time. From Fig. 4 and Fig. S4 (ESI†), with the extension of time, the free IR820 group demonstrated quick IR820 spread and the fluorescence signal in the tumor vanished after 3 days. However, the fluorescence signal in the S@IR820 hydrogel group was still strong in the tumor site after 4 days. This result thus indicated that the intratumoral retention of the drug was greatly enhanced by the hydrogel.
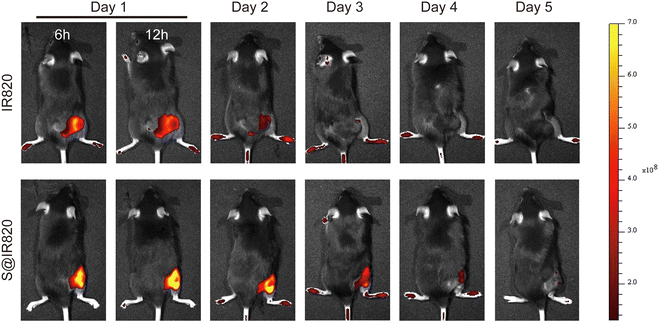 |
| | Fig. 4 Fluorescence images of mice after intratumoral injection of IR820 and S@IR820. | |
In vivo anti-tumor effect
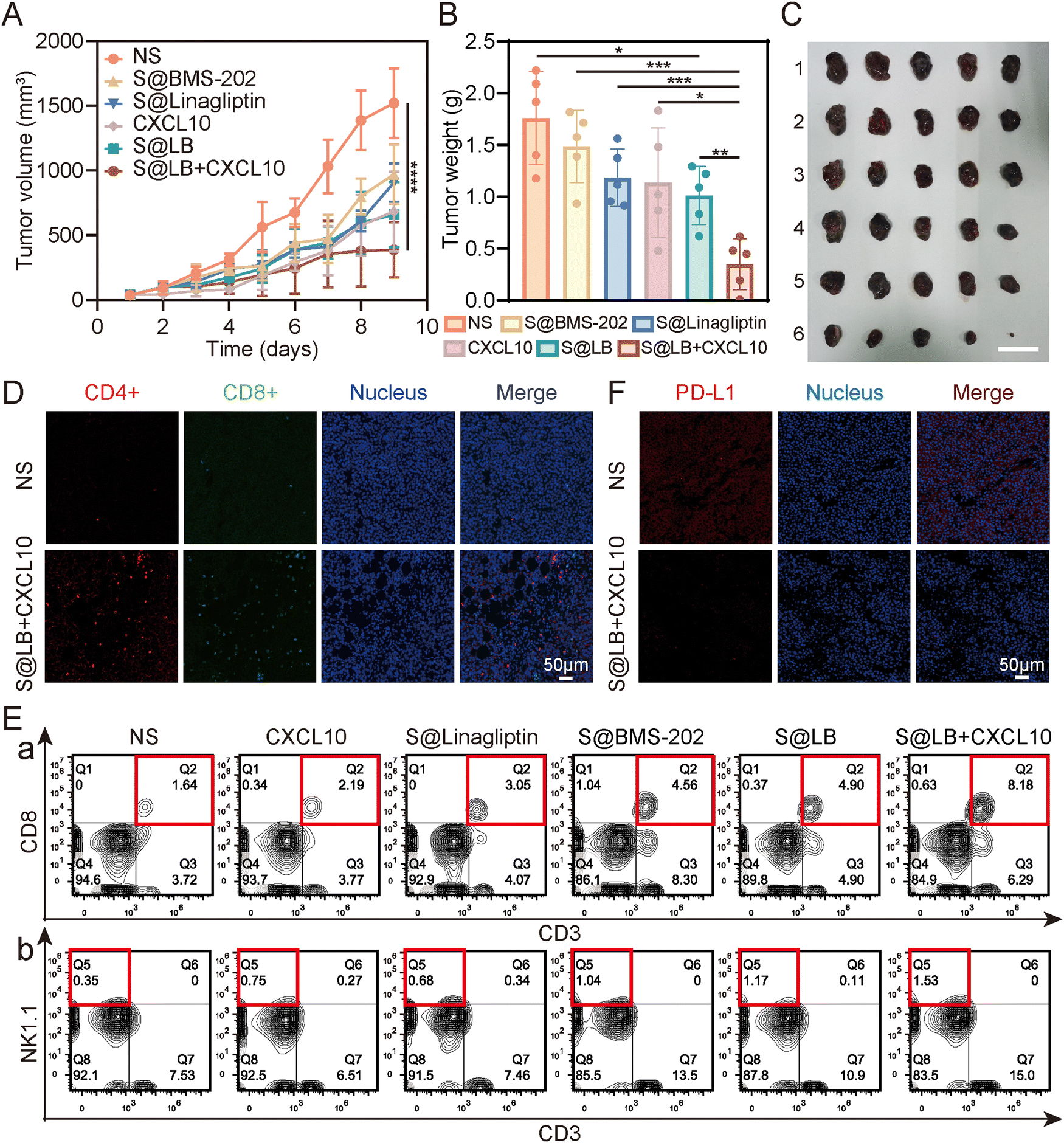
To understand the therapeutic efficacy of the S@LB hydrogel in combination with CXCL10, a mouse bilateral tumor model was established (Fig. S5A, ESI†). The primary tumor was treated by intratumoral injection. Based on the result of the drug release assay of S@LB hydrogel in vitro, the release of linagliptin needs a certain time to realize the inhibition of the CXCL10 degradation. In addition, given the characteristics of easy deactivation of CXCL10, we injected CXCL10 intratumorally the day after administration of S@LB. The tumor volume of the mice in the bilateral tumor model was monitored during the period of treatment. It showed that the tumor volume and weight in the normal saline (NS) group increased rapidly either in primary or distant tumors (Fig. 5A–C, Fig. S5B–D, ESI†). However, the S@LB + CXCL10 group significantly suppressed the tumor growth in comparison with the groups of S@LB, free CXCL10, S@Linagliptin, S@BMS-202 and NS. Moreover, there was no significant difference in the body weight between different treatment groups, indicating good safety of the hydrogel in cancer therapy (Fig. S5E, ESI†).
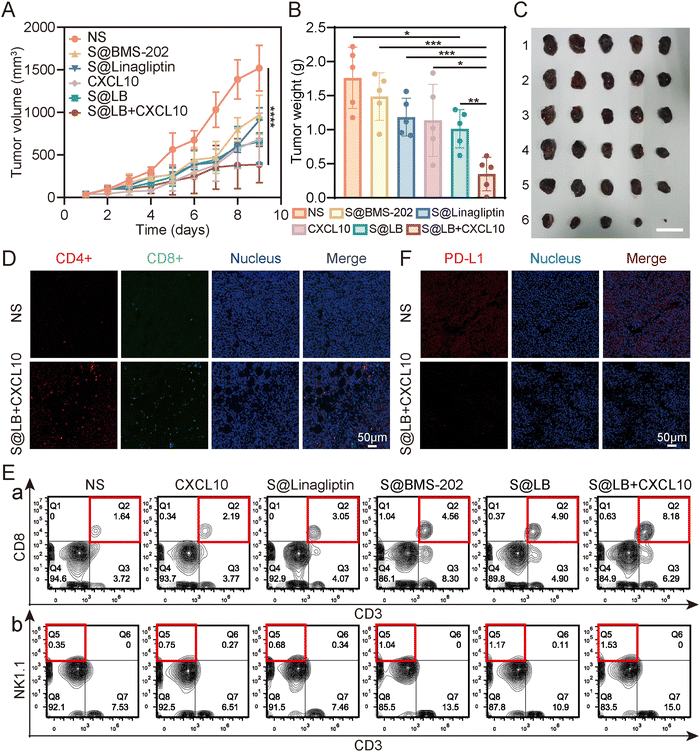 |
| | Fig. 5
In vivo anti-tumor effect of S@LB + CXCL10. (A) Changes in the tumor volume of primary tumors of mice (n = 5). (B) Primary tumor weights of mice on day 10 of treatment (n = 5). (C) Photograph of primary tumors on day 10 of treatment. 1: NS, 2: S@BMS-202, 3: S@Linagliptin, 4: CXCL10, 5: S@LB, 6: S@LB + CXCL10. The scale bar is 2 cm. (D) Fluorescence imaging of Ths (CD4+) and CTLs (CD8+) at primary tumors after NS and S@LB + CXCL10 treatments. (E) Flow analysis of CTLs (CD3+ CD8+, a) and NK cells (CD3-NK1.1+, b) at primary tumors. (F) Fluorescence images of PD-L1 at primary tumors after NS and S@LB + CXCL10 treatment. | |
The high anti-tumor ability of the S@LB + CXCL10 group against tumors could be attributed to the effective inhibition of CXCL10 degradation to enhance T cell infiltration and the effective blockage of the PD-L1 checkpoint by the hydrogel. To demonstrate this, flow analysis and fluorescence imaging of cells in the tissue of primary tumors were further performed. Fig. 5D and E and Fig. S6A–C (ESI†) show that the infiltration of cytotoxic T cells (CTLs), natural killer (NK) cells, and helper T cells (Ths) in the primary tumors was greatly increased in the S@LB + CXCL10 group in contrast to the NS, free CXCL10, S@Linagliptin, S@BMS-202 and S@LB groups. For example, the percentage of CTLs in the S@LB + CXCL10 group is 8.18% in the primary tumor, while the NS and free CXCL10 groups are 1.64% and 2.19%. The CTLs in the distant tumors demonstrated similar trends (Fig. S7A and B, ESI†). These results clearly demonstrated the hydrogel enabled suppression of CXCL10 degradation to realize its sustainable chemotaxis. The change of PD-L1 was further studied. From Fig. 5F, the S@LB hydrogel exhibited powerful ability to block PD-L1 at the tumor site. As a result, the present hydrogel enabled effective regulation of T cell infiltration and PD-L1 expression to realize a satisfactory therapeutic effect in cancer therapy.
In vivo immune response
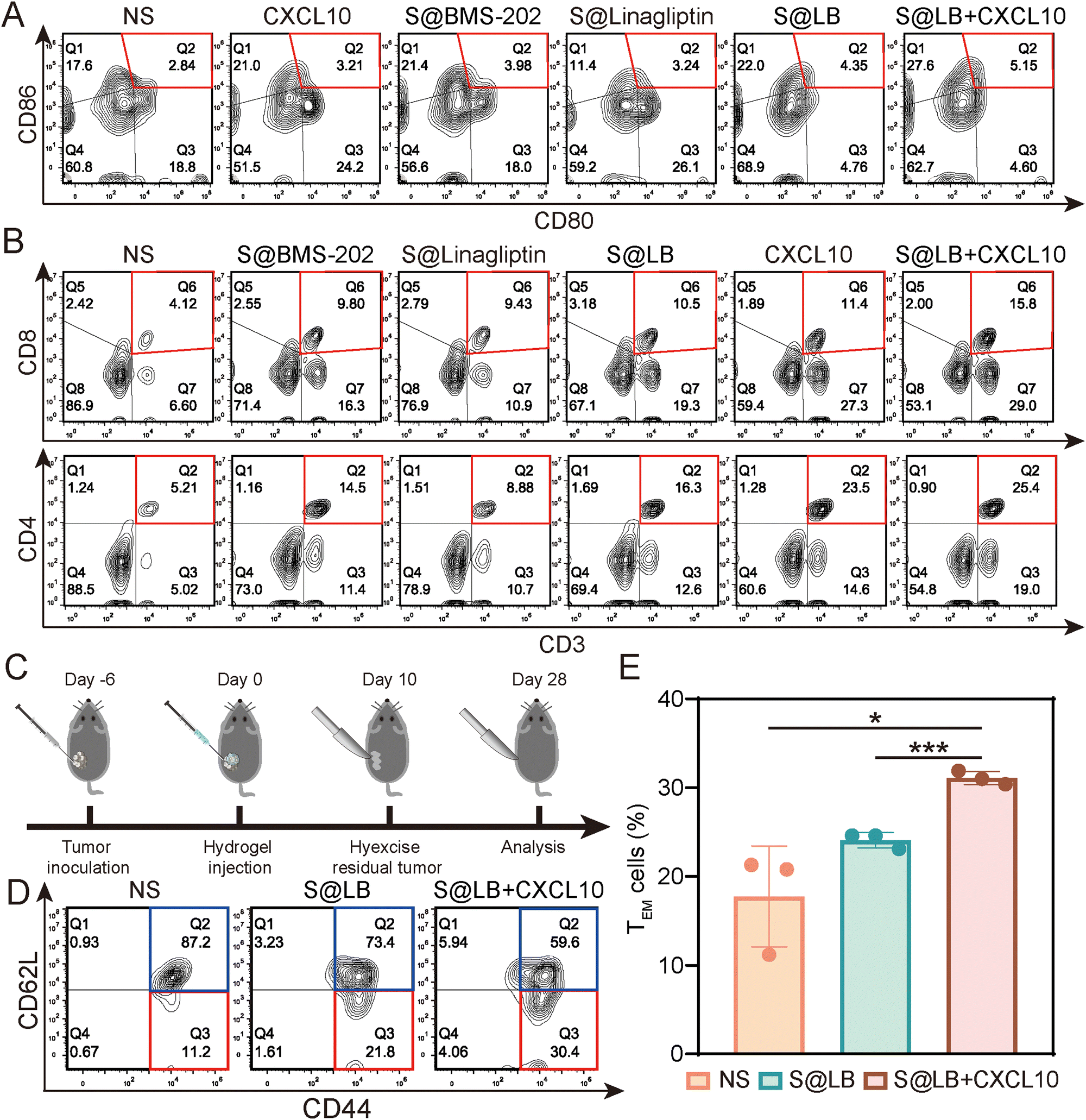
To understand the in vivo immune response, the tumor-draining lymph nodes and spleens of the bilateral-tumor mice after treatment were removed for characterization. Using CD80+ CD86+ as a marker of dendritic cell (DC) maturation, the flow cytometry result of mouse lymph node cells indicated that S@LB + CXCL10 could promote DC maturation, where the mature proportion increased to 5.15%, compared with 2.84% in the NS group (Fig. 6A and Fig. S7C, ESI†). The systemic immune response was further demonstrated by measuring the ratio of cytotoxic T cells (CTLs) and helper T cells (Ths) in the spleens. Flow cytometry analysis showed that S@LB hydrogel could increase the proportion of CTLs and Ths (Fig. 6B, Fig. S7D and E, ESI†). For example, the proportion of CTLs was 10.5%, 2.5 times that of the NS group, while the proportion of Ths was 16.3%, 3.1 times that of the NS group. In particular, when S@LB hydrogel was combined with CXCL10, CTLs and Ths became more abundant, 3.8 and 4.9 times that of the NS group. From these results, it was demonstrated that S@LB in combination with CXCL10 could induce the strong immune response of the body. We suggested that PD-L1 blockade preserved the tumor-killing function of T cells after they were attracted to the tumor site by CXCL10, leading to the death of tumor cells and the exposure of a lot of antigens. After antigen presentation by DCs, the immune cycle was initiated, which in turn triggered a systemic immune response.29
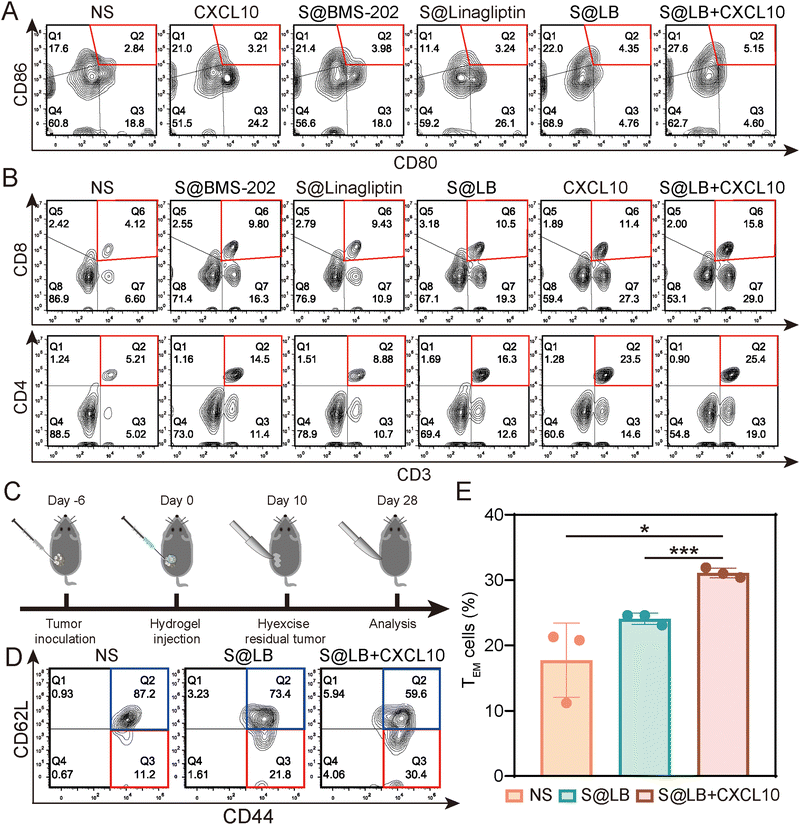 |
| | Fig. 6
In vivo immune response of S@LB + CXCL10. (A) Flow cytometric analysis of DC maturation in the lymph nodes after different treatments. (B) Flow analysis of CTLs (CD3+CD8+) and Ths (CD3+CD4+) in the spleens. (C) Scheme illustrating immunological memory assessment. (D) The proportion of TEM (CD62L−CD44+) and TCM (CD62L+CD44+) in the spleens by flow cytometry after different treatments. (E) The percent of TEM in the spleens (n = 3). | |
To test whether S@LB + CXCL10 could induce an immune memory effect, we built the memory model and tumor tissues were surgically removed at the end of the treatment period. 18 days after tumor removal, effector memory T cells (TEM) and central memory T cells (TCM) in the spleens were detected by flow cytometry (Fig. 6C–E). It can be seen that the percentage of TCM decreased and TEM increased in the treatment group. In contrast to 11.2% in the NS group, the proportion of TEM of the S@LB group increased to 21.8%, and that of the S@BL + CXCL10 group increased to 30.4%. As a result, it was verified that the S@BL hydrogel in combination with CXCL10 could effectively induce an immune memory effect.
In vivo anti-metastatic effect
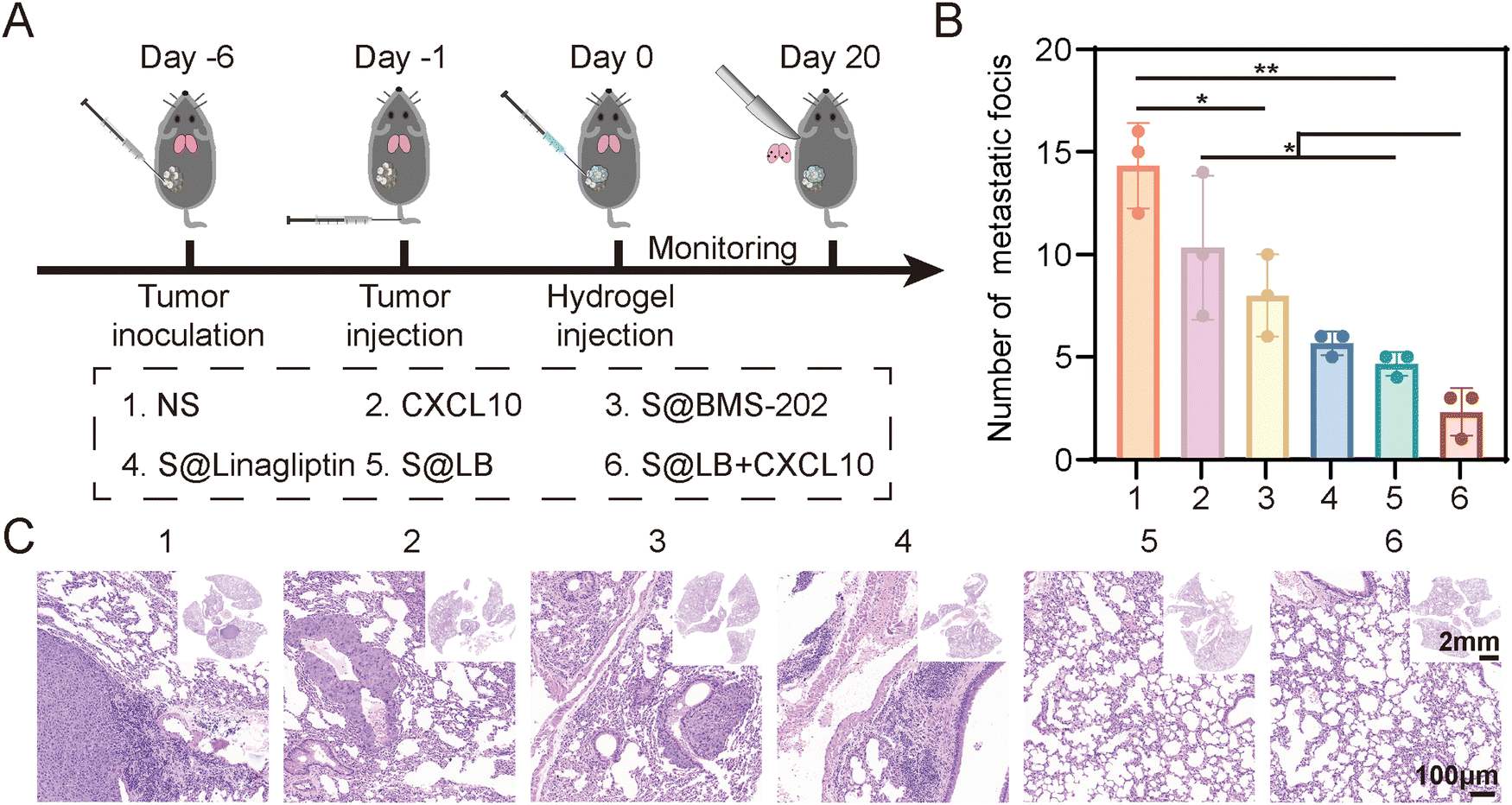
As shown in Fig. 7A, a lung metastasis model was constructed. Although free CXCL10 had no significant inhibitory effect on lung metastasis, the S@LB hydrogel exhibited an obvious inhibitory effect (Fig. 7B and C, and Fig. S8, ESI†). In particular, the combination of S@LB hydrogel with CXCL10 demonstrated the best inhibitory effect on lung metastasis, with a significant reduction in the number of metastatic foci on the lung surface and less metastatic parts in the sections. For instance, the number of foci decreased from 14 in the NS group to 2 in the S@LB + CXCL10 group. Taken together, due to the increased lethality of T cells and the mobilization of systemic immune responses, S@LB + CXCL10 not only enabled a powerful therapeutic effect on primary and distant tumors, but also demonstrated a strong ability against lung metastasis.
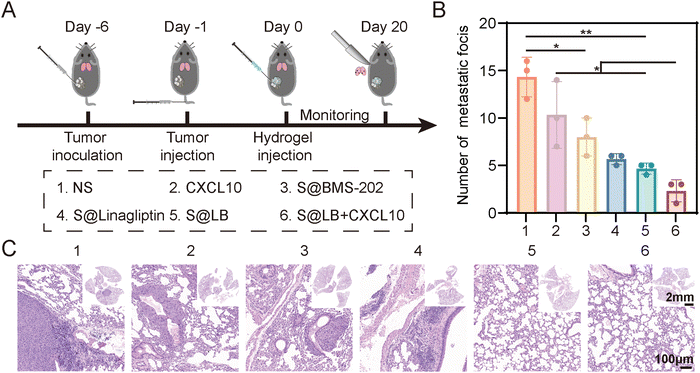 |
| | Fig. 7
In vivo anti-metastatic effect of S@LB + CXCL10. (A) Scheme illustrating anti-metastasis assessment. (B) The number of lung metastatic foci after different treatments (n = 3). (C) Hematoxylin–eosin (H&E) staining images of lungs after different treatments. | |
Conclusions
In summary, an injectable hydrogel approach was reported to solve the issues of insufficient tumor T cell infiltration and PD-L1-governed immune evasion in cancer immunotherapy. The S@LB hydrogel was established in the tumor site by the Ca2+-responsive SA gelator, with encapsulated linagliptin and BMS-202 particles. When the S@LB hydrogel was combined with chemotactic CXCL10, the hydrogel enabled strong tumor T cell infiltration by regulating DPP4-mediated CXCL10 degradation to realize sustainable CXCL10 chemotaxis. Furthermore, the PD-L1 was greatly blocked by the hydrogel, effectively disrupting the PD-L1-dominated immune evasion. As a result, the S@LB hydrogel in combination with CXCL10 exhibited remarkable cancer immunotherapy against tumor growth as well as lung metastasis. This work offers an innovative therapeutic platform and strategy to reinforce cancer immunotherapy, holding great promise in clinical application.
Materials and methods
Materials
BMS-202 was purchased from Dalian Meilun Biotech Co., Ltd. Linagliptin and SA were provided from Shanghai Aladdin Bio-Chem Technology Co., Ltd. Murine CXCL10 was obtained from PeproTech Inc. Alexa Fluor 488-labeled goat anti-rabbit IgG, PD-L1 rabbit polyclonal antibody and Fluo-3 AM were purchased from Beyotime Biotech Inc. Hoechst 33342 was provided by Beijing Solarbio Science & Technology Co., Ltd. IFN-γ and anti-mouse antibodies (PE-labeled PD-L1, APC-labeled CD3, FITC-labeled CD8a, PE-labeled CD8a, PE-labeled CD4, FITC-labeled NK-1.1, PE-labeled CD80, APC-labeled CD86, FITC-labeled CD11c, APC-labeled CD62L, and PerCP-labeled CD44) were obtained from BioLegend. Gly-Pro-AMC was supplied by ATT Bioquest Inc. ELISA kits for CXCL10 were provided by Multisciences Biotechnology Co., Ltd.
Preparation and characterization of S@LB hydrogel
Linagliptin hydrochloride was obtained by adding linagliptin to HCl solution, which was further lyophilized. Typically, 7.8 mg linagliptin hydrochloride was dissolved into 376 μL sodium alginate (SA) aqueous solution, in which the amount of SA is 20 mg. The pH of the above solution was adjusted to be 7.0 by NaOH and HCl, so that linagliptin particles were formed with uniform dispersion in the SA solution to result in 1 mL of the SA@Linagliptin (S@Linagliptin) solution. Then, 100 μL BMS-202 (1 mg) ethanol solution was added by drops to the S@Linagliptin solution under high-speed stirring, enabling the quick formation of dispersed BMS-202 particles in the S@Linagliptin solution for achieving S@Linagliptin/BMS-202 (S@LB). The achieved solution was further stirred for 2 hours to volatilize the ethanol. For the preparation of SA@BMS-202 (S@BMS-202), the fabrication procedure is similar to the above. The gelation behavior of S@LB was studied by introducing calcium chloride solution for the formation of S@LB hydrogel, where the Ca2+ concentration was 1.8 mmol L−1 in the solution. To determine the rheological properties of S@LB, we use the Thermo HAAKE rotation rheometer under the condition of 37 °C, with a parallel plate. Scanning electron microscopy (SEM) was used to study the morphology of the hydrogel.
In vitro drug-releasing characteristics of S@LB hydrogel
The release behavior of linagliptin and BMS-202 in S@LB hydrogel was studied using phosphate buffer solution (PBS, 0.5% Tween-80, pH = 6.5) as the release medium at 37 °C with slow shaking. Samples were taken at specified time points and quantified by high performance liquid chromatography (HPLC).
Cells
B16F10 cells (murine melanoma cell line) were cultured in RPMI 1640 with fetal bovine serum (FBS, 10%) and penicillin/streptomycin (1%) in the incubator containing 5% CO2 at 37 °C.
Animals
Female C57BL/6 mice (6–8 weeks) were supplied by Jinan Pengyue Laboratory Animal Co., Ltd. The whole animal studies were approved by Shandong University Animal Experiment Ethics Review and the Health Guide for the Care and Use of Laboratory Animals of National Institutes.
In vitro cytotoxicity assay
After incubating in plates (96-well) overnight (6 × 103 cells per well), B16F10 cells were co-cultured with SA for 24 hours, and the untreated cells were used as the control. Then, the cells were incubated with thiazolyl blue tetrazolium bromide (MTT) for 4 hours. After that, dimethyl sulfoxide (DMSO) was added and the absorbance at 570 nm was detected by a microplate reader to characterize cell viability.
In vitro PD-L1 blocking assay
The PD-L1 blocking ability of the S@LB hydrogel was studied by flow cytometry and laser scanning confocal microscopy (LSCM). For quantitative analysis, B16F10 cells were seeded at 2 × 105 cells per well in 6-well plates, which were further added with IFN-γ. One day later, the cells were incubated with free BMS-202 and S@LB to give a BMS-202 concentration of 22.5 μg mL−1 in the medium, and cells with no drug treatment were applied as the control group. The cells were stained with the PE-labeled PD-L1 antibody to quantitatively determine the PD-L1 on cells by flow cytometry. For qualitative analysis, cells were stained using the PD-L1 rabbit polyclonal antibody, the Alexa Fluor 488-labeled goat anti-rabbit IgG, and Hoechst 33342 sequentially and then imaged by LSCM.
Intracellular Ca2+ detection
Fluo-3 AM was used as a probe to detect intracellular Ca2+. Cells were seeded in 6-well plates (3 × 105 cells per well) overnight, and treated by 10 μL SA and S@LB, and the untreated cells were used as the control. Cells were stained with Fluo-3 AM, and intracellular calcium was measured by flow cytometry and LSCM for quantitative and qualitative analysis, respectively.
DPP4 activity inhibition assay
A tumor-bearing mouse model was built via subcutaneous injection of 1 million B16F10 cells into the right back of the mice. After the tumor volume of the mice became about 50 mm3, the mice were intratumorally injected with S@Linagliptin and S@LB at an equal linagliptin dose of 36.15 mg kg−1, accompanied by normal saline (NS) as a control. Four days later, tumors from the mice were dissected and homogenized, and the homogenate supernatants were incubated with glycine-proline-7-amido-4-methylcoumarin (Gly-Pro-AMC), a sensitive fluorogenic substrate for detecting the activity of DPP4, which can be cleaved by DPP4 to produce fluorescence (excitation: 380 nm, emission: 500 nm). The intensity of the fluorescence was then tested by a microplate reader to evaluate the DPP4 activity.
In vivo drug retention study
Mice were subcutaneously injected with B16F10 cells (1 × 106 per mouse) on the right back. After the tumor size reached ≈50 mm3, the mice were then divided into two groups and intratumorally injected with the free IR820 or S@IR820 containing IR820 at a dose of 4 mg kg−1. The S@IR820 was prepared by just mixing the SA solution with the dye IR820, which enabled the drug retention property of the S@LB hydrogel to be determined as the IR820 possessed fluorescence properties. The property of hydrogel-enabled drug retention was observed utilizing an in vivo imaging system (IVIS) kinetic imaging system, characterized by the fluorescence intensity of the IR820 at the tumor site.
In vivo tumor therapeutic effect
The in vivo tumor therapeutic effect was evaluated by establishing a bilateral tumor model. Mice were subcutaneously injected with B16F10 cells (1 × 106 per mouse) on the right back to build primary tumors, and 2 days later, the same amount of B16F10 cells was injected on the left side to establish distant tumors. After the primary tumor volume became about 50 mm3, mice were then divided into five groups and intratumorally injected into the primary tumors with NS, S@BMS-202, S@Linagliptin, CXCL10 and S@LB, respectively, at an equal linagliptin dose of 36.15 mg kg−1, and BMS-202 dose of 5 mg kg−1. And a portion of the S@LB group of mice was injected with CXCL10 one day later. The body weight and tumor volume of the mice were monitored daily, and after 10 days, the mice were sacrificed. Quantitative analysis of lymphocytes in the tumor, spleen and lymph nodes was performed using flow cytometry. Intratumoral T cell infiltration was qualitatively evaluated using immunofluorescence sections. After treatment, the primary tumors of the treated mice were removed and homogenized. The CXCL10 content in the tumors was detected by an ELISA kit.
In vivo long-term immune-memory effect
B16F10 cells (1 × 106 per mouse) were subcutaneously injected into the right back of mice to build tumors, and 6 days later, the mice were treated with NS, S@LB and S@LB + CXCL10. At the end of treatment (10 days later), the tumors were surgically removed. After 18 days, the spleens of the mice were collected to detect memory T cells, including central memory T cells (TCM) and effector memory T cells (TEM), by flow cytometry to evaluate the memory effect.
In vivo anti-metastasis effect
B16F10 cells (1 × 106 per mouse) were subcutaneously injected into the right back of mice to build tumors, and 5 days later, B16F10 cells (2 × 105 per mouse) were injected into the tail vein to establish a lung metastasis model. The next day, various treatments were performed (NS, S@BMS-202, S@Linagliptin, CXCL10, S@LB, S@LB + CXCL10). After 20 days’ treatment, the lungs were dissected out for counting the number of metastatic foci and hematoxylin–eosin (H&E) staining to understand the anti-metastasis effect.
Statistical analysis
All data are shown as mean ± SD. Student's t-tests or one-way analysis followed by Turkey's multiple comparisons tests were used in the statistical analysis, where *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001. The statistical software to evaluate differential significance was GraphPad Prism 8.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (NSFC, No. 82061148009, 22272091), the Israel Science Foundation (ISF, No. 3304/20), the Fundamental Research Funds for the Central Universities (No. 2022JC010), and the Natural Science Foundation General Project of Shandong Province (ZR2021MH061). We also acknowledged the Pharmaceutical Biology Sharing Platform and the Advanced Medical Research Institute of Shandong University.
References
- J. M. Hu, Q. Yang, W. D. Zhang, H. W. Du, Y. H. Chen, Q. N. Zhao, L. Dao, X. Q. Xia, F. N. Wall, Z. T. Zhang, K. Mahadeo, R. Gorlick, S. Kopetz, G. Dotti and S. L. Li, J. Immunother. Cancer, 2022, 10, e003633 CrossRef PubMed.
- Y. S. Tian, K. Wang, C. M. Xu, J. Feng and Z. L. Zhang, Chem. Eng. J., 2022, 439, 135839 CrossRef CAS.
- D. Zhang, Q. Li, X. W. Chen, X. X. Nie, F. M. Xue, W. Xu and Y. X. Luan, Small, 2022, 18, 2202663 CrossRef CAS PubMed.
- L. T. Zheng, S. S. Qin, W. Si, A. Q. Wang, B. C. Xing, R. R. Gao, X. W. Ren, L. Wang, X. J. Wu, J. Zhang, N. Wu, N. Zhang, H. Zheng, H. Q. Ouyang, K. Y. Chen, Z. D. Bu, X. D. Hu, J. F. Ji and Z. M. Zhang, Science, 2021, 374, 1462 CrossRef PubMed.
- S. T. Paijens, A. Vledder, M. de Bruyn and H. W. Nijman, Cell. Mol. Immunol., 2021, 18, 842 CrossRef CAS PubMed.
- Y. Zhang and Z. Zhang, Cell. Mol. Immunol., 2020, 17, 807 CrossRef CAS.
- Y. W. Chen, S. C. Hu, Y. H. Shu, Z. B. Qi, B. Zhang, Y. T. Kuang, J. H. Ma and P. Cheng, Hum. Gene Ther., 2022, 33, 237 CrossRef CAS PubMed.
- X. J. Sun, B. G. Wu, H. C. Chiang, H. Deng, X. W. Zhang, W. Xiong, J. Q. Liu, A. M. Rozeboom, B. T. Harris, E. Blommaert, A. Gomez, R. E. Garcia, Y. F. Zhou, P. Mitra, M. Prevost, D. Y. Zhang, D. Banik, C. Isaacs, D. Berry, C. E. Lai, K. Chaldekas, P. S. Latham, C. A. Brantner, A. Popratiloff, V. Jin, N. Y. Zhang, Y. F. Hu, M. A. Pujana, T. J. Curiel, Z. Q. An and R. Li, Nature, 2021, 599, 673 CrossRef CAS PubMed.
- Y. X. Wang, Y. W. Ding, D. Yao, H. Dong, C. W. Ji, J. H. Wu, Y. Q. Hu and A. Yuan, Small, 2021, 17, 2006231 CrossRef CAS.
- S. W. Zhang, J. Wang, Z. Q. Kong, X. X. Sun, Z. G. He, B. J. Sun, C. Luo and J. Sun, Biomaterials, 2022, 282, 121433 CrossRef CAS PubMed.
- T. H. Harris, E. J. Banigan, D. A. Christian, C. Konradt, E. D. Tait Wojno, K. Norose, E. H. Wilson, B. John, W. Weninger, A. D. Luster, A. J. Liu and C. A. Hunter, Nature, 2012, 486, 545 CrossRef CAS PubMed.
- N. Karin and H. Razon, Cytokine, 2018, 109, 24 CrossRef CAS PubMed.
- U. Barash, Y. Zohar, G. Wildbaum, K. Beider, A. Nagler, N. Karin, N. Ilan and I. Vlodavsky, Leukemia, 2014, 28, 2178 CrossRef CAS PubMed.
- J. He, S. L. Duan, X. Yu, Z. Y. Qian, S. F. Zhou, Z. Y. Zhang, X. N. Huang, Y. Huang, J. Su, C. H. Lai, J. Y. Meng, N. Zhou, X. L. Lu and Y. X. Zhao, Theranostics, 2016, 6, 752 CrossRef CAS PubMed.
- E. Regulier, S. Paul, M. Marigliano, J. Kintz, Y. Poitevin, C. Ledoux, D. Roecklin, G. Cauet, V. Calenda and H. E. Homann, Cancer Gene Ther., 2001, 8, 45 CrossRef CAS PubMed.
- D. S. R. Barreira, M. E. Laird, N. Yatim, L. Fiette, M. A. Ingersoll and M. L. Albert, Nat. Immunol., 2015, 16, 850 CrossRef PubMed.
- S. Nishina, A. Yamauchi, T. Kawaguchi, K. Kaku, M. Goto, K. Sasaki, Y. Hara, Y. Tomiyama, F. Kuribayashi, T. Torimura and K. Hino, Cell. Mol. Gastroenterol. Hepatol., 2019, 7, 115 CrossRef PubMed.
- A. L. Wilson, L. R. Moffitt, K. L. Wilson, M. Bilandzic, M. D. Wright, M. D. Gorrell, M. K. Oehler, M. Plebanski and A. N. Stephens, Cancers, 2021, 13, 487 CrossRef CAS PubMed.
- B. Li, T. Fang, Y. Li, T. Xue, Z. Zhang, L. Li, F. Meng, J. Wang, L. Hou, X. Liang, X. Zhang and Z. Gu, Nano Today, 2022, 46, 101606 CrossRef CAS.
- W. Fang, L. Li, Z. Lin, Y. Zhang, Z. Jing, Y. Li, Z. Zhang, L. Hou, X. Liang, X. Zhang and X. Zhang, Extracell. Vesicles, 2023, 2, 100021 CrossRef.
- B. R. Huck, L. Kotzner and K. Urbahns, Angew. Chem., Int. Ed., 2018, 57, 4412 CrossRef CAS PubMed.
- K. M. Zak, P. Grudnik, K. Magiera, A. Domling, G. Dubin and T. A. Holak, Structure, 2017, 25, 1163 CrossRef CAS PubMed.
- Z. P. Zhao, Q. Li, X. H. Qin, M. Z. Zhang, Q. Du and Y. X. Luan, Adv. Funct. Mater., 2022, 32, 2200801 CrossRef CAS.
- C. F. Deacon, Nat. Rev. Endocrinol., 2020, 16, 642 CrossRef CAS PubMed.
- Y. Chao, L. G. Xu, C. Liang, L. Z. Feng, J. Xu, Z. L. Dong, L. L. Tian, X. Yi, K. Yang and Z. Liu, Nat. Biomed. Eng., 2018, 2, 611 CrossRef CAS PubMed.
- Y. Jiang, B. W. Brandt, M. J. Buijs, L. Cheng, R. Exterkate, W. Crielaard and D. M. Deng, Appl. Environ. Microbiol., 2021, 87, e02371 CAS.
- R. Zhang, Z. Zhu, H. Lv, F. Li, S. Sun, J. Li and C. S. Lee, Small, 2019, 15, 1903881 CrossRef CAS PubMed.
- C. Li, H. Yao, H. Wang, J. Y. Fang and J. Xu, Oncogene, 2021, 40, 1128 CrossRef CAS PubMed.
- D. S. Chen and I. Mellman, Immunity, 2013, 39, 1–10 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *a
*a