
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceViral capsid structural assembly governs the reovirus binding interface to NgR1†
Rita
dos Santos Natividade‡
a,
Andra C.
Dumitru‡
 a,
Alessandro
Nicoli
bc,
Michael
Strebl
d,
Danica M.
Sutherland
ef,
Olivia L.
Welsh
ef,
Mustafa
Ghulam
b,
Thilo
Stehle
d,
Terence S.
Dermody
efg,
Antonella
Di Pizio
*bc,
Melanie
Koehler
*ab and
David
Alsteens
*ah
a,
Alessandro
Nicoli
bc,
Michael
Strebl
d,
Danica M.
Sutherland
ef,
Olivia L.
Welsh
ef,
Mustafa
Ghulam
b,
Thilo
Stehle
d,
Terence S.
Dermody
efg,
Antonella
Di Pizio
*bc,
Melanie
Koehler
*ab and
David
Alsteens
*ah
aLouvain Institute of Biomolecular Science and Technology, Université catholique de Louvain, Louvain-la-Neuve, Belgium. E-mail: david.alsteens@uclouvain.be
bLeibniz Institute for Food Systems Biology, Technical University of Munich, Freising, Germany. E-mail: a.dipizio.leibniz-lsb@tum.de; m.koehler.leibniz-lsb@tum.de
cChemoinformatics and Protein Modelling, Department of Molecular Life Sciences, School of Life Sciences, Technical University of Munich, 85354 Freising, Germany
dInterfaculty Institute of Biochemistry, University of Tübingen, Tübingen, Germany
eDepartment of Pediatrics, University of Pittsburgh School of Medicine, Pittsburgh, Pennsylvania, USA
fInstitute of Infection, Inflammation, and Immunity, UPMC Children's Hospital of Pittsburgh, Pittsburgh, Pennsylvania, USA
gDepartment of Microbiology and Molecular Genetics, University of Pittsburgh School of Medicine, Pittsburgh, Pennsylvania, USA
hWELBIO department, WEL Research Institute, 1300 Wavre, Belgium
First published on 30th September 2024
Abstract
Understanding the mechanisms underlying viral entry is crucial for controlling viral diseases. In this study, we investigated the interactions between reovirus and Nogo-receptor 1 (NgR1), a key mediator of reovirus entry into the host central nervous system. NgR1 exhibits a unique bivalent interaction with the reovirus capsid, specifically binding at the interface between adjacent heterohexamers arranged in a precise structural pattern on the curved virus surface. Using single-molecule techniques, we explored for the first time how the capsid molecular architecture and receptor polymorphism influence virus binding. We compared the binding affinities of human and mouse NgR1 to reovirus μ1/σ3 proteins in their isolated form, self-assembled in 2D capsid patches, and within the native 3D viral topology. Our results underscore the essential role of the concave side of NgR1 and emphasize that the spatial organization and curvature of the virus are critical determinants of the stability of the reovirus–NgR1 complex. This study highlights the importance of characterizing interactions in physiologically relevant spatial configurations, providing precise insights into virus–host interactions and opening new avenues for therapeutic interventions against viral infections.
New conceptsThis study demonstrates a novel concept in viral entry mechanisms by elucidating the unique bivalent interaction between reovirus and Nogo-receptor 1 (NgR1). Unlike previous research that primarily focused on isolated viral components or lacked precise spatial configurations, our work reveals how NgR1 specifically binds at the interface between adjacent heterohexamers on the reovirus capsid, arranged in a well-defined structural pattern. This concept is differentiated by its integration of single-molecule techniques and molecular dynamics simulations to compare binding affinities of human and mouse NgR1 to reovirus proteins in various assemblies—isolated, 2D patches, and the native 3D viral topology. Our findings provide new insights into the critical role of the capsid's molecular architecture and spatial organization in stabilizing the virus-receptor complex. This research advances nanoscience by highlighting the importance of characterizing interactions in physiologically relevant configurations, thereby offering a deeper understanding of virus–host dynamics. Furthermore, it opens new pathways for developing targeted therapeutic interventions by leveraging the structural specificity of these interactions. |
Introduction
Viruses must cross host cell membranes to initiate infection and rely on viral attachment proteins and host cell receptors to mediate this crucial step in viral replication. Virus-receptor interactions often involve diverse cell–surface components, such as glycans and proteins, and influence tissue tropism and pathogenesis.1,2 However, much remains to be understood about the mechanisms by which host–cell receptors facilitate viral dissemination in vivo and infection of discrete tissues to produce organ-specific disease.Mammalian orthoreovirus (reovirus) is a non-enveloped, double-stranded RNA virus that can infect most mammalian species.3 There are three main reovirus serotypes exemplified by prototype strains type 1 Lang (T1L), type 2 Jones (T2J), and type 3 Dearing (T3D).4 Reovirus infection is linked to rare instances of neonatal meningitis, and the virus has been isolated from cerebrospinal fluid samples, suggesting that it infects cells in the human central nervous system (CNS).5 Since reovirus infection in humans is generally asymptomatic, little is known about neuronal spread or specific strains that cause human CNS disease. Conversely, reovirus infection in mice is highly pathogenic and can lead to neuropathology.5,6 Reovirus tropism in the CNS is serotype-specific. Serotype 1 reovirus causes non-lethal hydrocephalus by infecting ependymal cells that line cerebral ventricles, while serotype 3 reovirus causes lethal encephalitis by infecting neurons in the CNS.7–10
Reovirus forms icosahedral particles that contain two protein shells. The outer capsid contains the σ1 protein, which is an elongated receptor-binding fiber protruding from the center of pentameric core-turret λ2 (yellow; Fig. 1A).11,12 The capsid also contains the σ3 and μ1 proteins, which form μ13σ33 heterohexamers (called heterohexamers hereafter), composed of three σ3 monomers bound to a central μ1 trimer base.13 There are 200 heterohexamers that form the bulk of the reovirus outer capsid (white and grey; Fig. 1A) arranged in an icosahedral lattice. These proteins function in viral attachment14,15 and disassembly.16–18
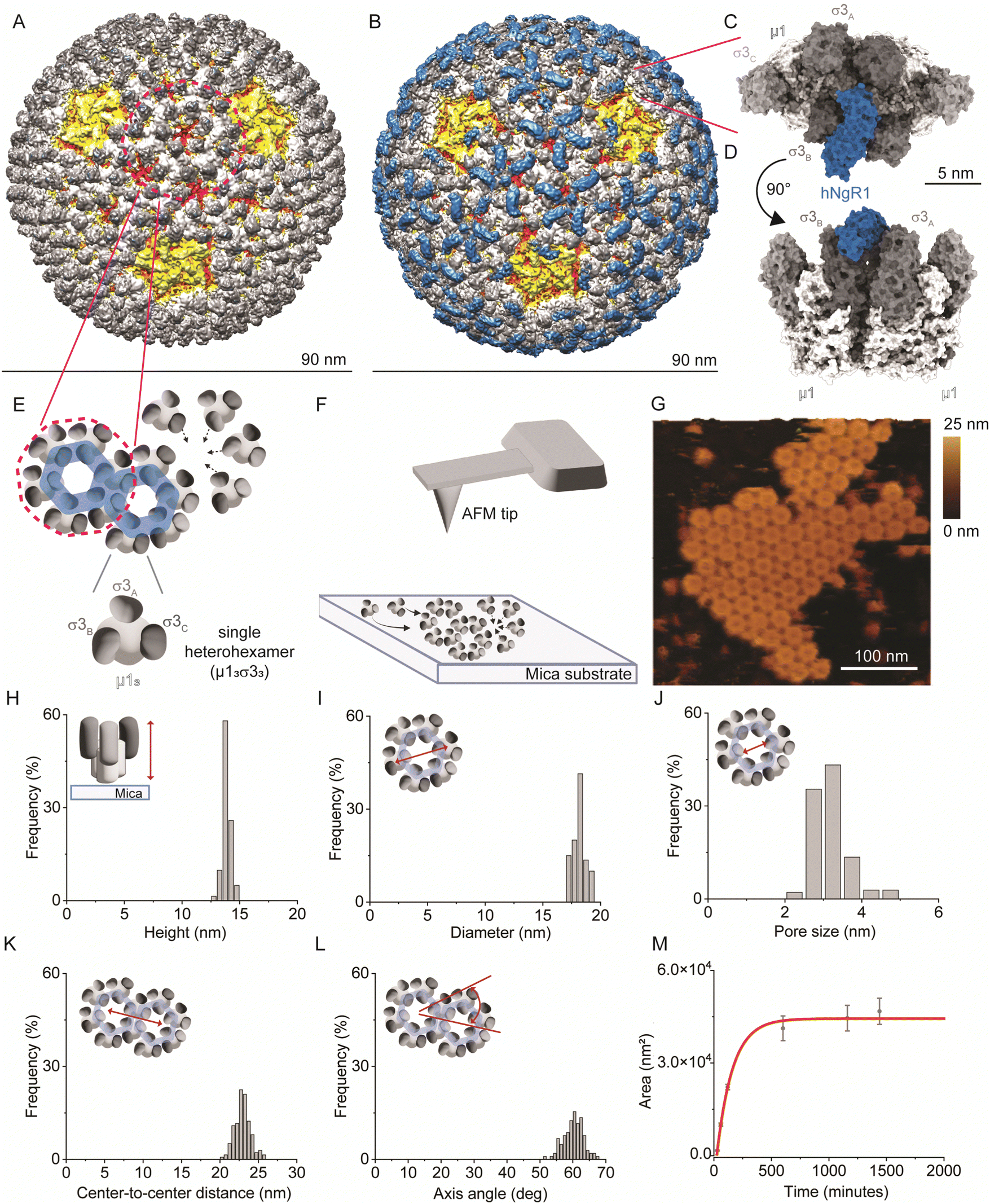 | ||
| Fig. 1 Imaging the self-assembly of reovirus μ1σ3 heterohexamers. (A) and (B) Density maps of reovirus alone (A) or in complex with hNgR1 (B). Viral capsid proteins λ2, λ1/σ2, μ1, and σ3 are depicted in yellow, red, white, and shades of grey, respectively. The receptor hNgR1 is depicted in blue. (C) and (D) Surface representation of two σ3μ1 heterohexamers in complex with hNgR1 receptor, from the top view (C) and side view (D). (E) Schematics of the assembly of μ1σ3 heterohexamers to form on hexagonal structures, as present on the viral capsid. (F) Schematics of the imaging setup, with a bare AFM tip and self-assembled μ1σ3 heterohexamers on a mica substrate. (G) AFM topography image of a patch of self-assembled heterohexamers. (H)–(L) Histograms of the measured parameters from self-assembled heterohexamers structures: height of single heterohexamers (H), diameter of the hexagonal structure formed (I) and its pore size (J), center-to-center distance between hexagonal structures (K), and their axis angle (L). (M) Area of the imaged self-assembled patches is plotted in function of time and fitted with an exponential fit (red line). | ||
Several cell–surface receptors have been identified for reovirus that appear to mediate a multistep infection process. JAM-A, the best characterized reovirus receptor, is required for reovirus hematogenous dissemination but dispensable for reovirus CNS tropism.7,19 Nogo-66 receptor 1 (NgR1) is a neural receptor for reovirus in some mammalian species.20 NgR1 is a leucine-rich repeat protein expressed by neurons and plays a role in axonal growth.21,22 While human NgR1 (hNgR1) functions as a receptor for T1L and T3D reovirus strains, the efficiency of murine NgR1 (mNgR1) engagement by reovirus is uncertain. Mice lacking NgR1 remain susceptible to reovirus encephalitis,23 suggesting that this receptor is dispensable for reovirus neurovirulence in mice. The key residues responsible for differences in reovirus binding to hNgR1 and mNgR1 are unknown. Reovirus T1L exhibits a slightly higher affinity for hNgR1 compared to T3D. The unusual binding interface between reovirus outer-capsid protein σ3 and NgR1 involves two binding interfaces: a smaller surface on the convex face of NgR1 and a larger one on the more conserved concave face.15 Additionally, the convex and concave faces of NgR1 appear to bind simultaneously to two different σ3 monomers (σ3A and σ3B) belonging to adjacent heterohexamers (Fig. 1B–D),15 suggesting a key role of viral capsid protein organization in receptor binding. Polymorphisms at NgR1 binding sites in σ3 proteins or their spatial accessibility may account for the disparities in binding for T1L and T3D strains, although the implicated residues are still unknown.
In this study, we used a novel approach integrating atomic force microscopy (AFM) with molecular dynamics (MD) simulations to delve deeper into the assembly of the viral capsid and unravel molecular mechanisms underlying reovirus–NgR1 interactions using strain T1L. Using high-resolution AFM imaging, we resolved the assembly dynamics of heterohexamers under native conditions at sub-nanometer resolution. With the high-force sensitivity of the AFM technique, we employed hNgR1-functionalized AFM tips to investigate the role of reovirus capsid protein architecture in receptor binding. Specifically, we compared the binding affinities of hNgR1 to force-probed heterohexamers in their isolated form, assembled into 2D patches, and within the native 3D viral topology. Although previous studies have been conducted using 2D assemblies of viral capsid proteins,24 most research is focused on nanomechanical probing of the virus capsid surface, as summarized in ref. 25. Here, we used an innovative approach with AFM to probe adhesion of 2D assemblies of viral proteins. This strategy enabled us to gather comprehensive data highlighting the crucial roles of spatial organization and curvature in stabilizing the T1L–NgR1 virus-receptor interaction. A comparative analysis of binding to mNgR1 was conducted, offering valuable insights into reovirus host-range governed by NgR1. Complementing these experimental findings, MD simulations provide a molecular view of the residues involved in stabilizing the reovirus–NgR1 complex and emphasize a crucial role of the NgR1 concave face and the conservation of key interaction interfaces across human and murine NgR1. This work pioneers a nanotechnology-based method to dissect the thermodynamics of a virus binding interface, paving the way for a deeper understanding of viral tropism and pathogenesis.
Results
Experimental design to study heterohexamer-receptor interactions
To study NgR1-heterohexamer interactions at the single-molecule level, we first reconstituted σ33μ13 heterohexamers assemblies, mimicking the organization of these proteins on native reovirus virions. Next, reovirus heterohexamers self-assembled on mica surfaces were imaged by force–distance (FD) curve-based AFM (FD-based AFM) (Fig. 1E and F) using native conditions.26 The negatively charged mica substrate acts as an assembly-promoting template, allowing the acquisition of high-resolution AFM topography maps showing that a bidimensional (2D) lattice of heterohexamers is formed at physiological pH, temperature, and ionic strength, in the absence of other chaperone molecules (see Methods) (Fig. 1G). These structures, despite not representing the full viral capsid, constitute a relevant 2D model of specific capsid proteins that can be probed at the single-molecule level in an assembly closer to physiological conditions than isolated viral proteins. Single heterohexamers probed by AFM are 13.8 ± 0.2 nm (mean ± standard deviation (S.D.)) in height (Fig. 1H). The closely-packed hexagonal structures are 18.0 ± 0.3 nm in diameter, with a pore opening of 3.0 ± 0.3 nm in size (Fig. 1I and J). This lattice is formed of monomers with a center-to-center distance of 22.9 ± 0.7 nm oriented at an axis angle of 60° (Fig. 1K and L). We also studied the dynamics of lattice formation and found that heterohexamers assembled on mica display an exponential growth phase up to ∼500 minutes, followed by a stationary phase (Fig. 1M). The maximum area covered by heterohexamers lattices is 46.7 × 103 nm2, corresponding to 175 heterohexamers, which approximates the maximum number on the viral capsid surface.Probing hNgR1 interaction with self-assembled heterohexamers
Since σ33μ13 heterohexamers are capable of binding to hNgR1, we probed a surface of assembled heterohexamers with an hNgR1-functionalized AFM tip (Fig. 2A), reproducing in vitro the spatial configuration found in physiological environments. The hNgR1-functionalized tip was scanned across the self-assembled heterohexamers using the FD-based AFM mode, allowing us to collect both topography and adhesion images (Fig. 2B and C). Although tip functionalization slightly reduces the resolution of the topographical images, the heterohexamers remained clearly visible, and the corresponding adhesion image shows some sparsely distributed adhesive pixels. We simultaneously collected FD curves (Fig. 2D and E) and force–time curves (Fig. S1A, ESI†) on the self-assembled patches of heterohexamers. These data mostly depicted specific adhesion events, i.e., curves with a single adhesive peak were located at a separation distance of >5 nm, which accounts for the extension of the polyethylene glycol (PEG) tip-to-virion spacer. Thus, these data can be fitted with the worm-like chain model describing the extension of the polymer chain under external load,27 while FD curves on the underlying substrate show nonspecific binding (curve #3 in Fig. 2D and E). For each specific binding event, the rupture forces were plotted against the loading rate (LR) in a so-called dynamic force spectroscopy (DFS) plot using a semi-logarithmic scale (Fig. 2F). The Bell–Evans model predicts a linear dependence of the most probable rupture force on the DFS plot.28,29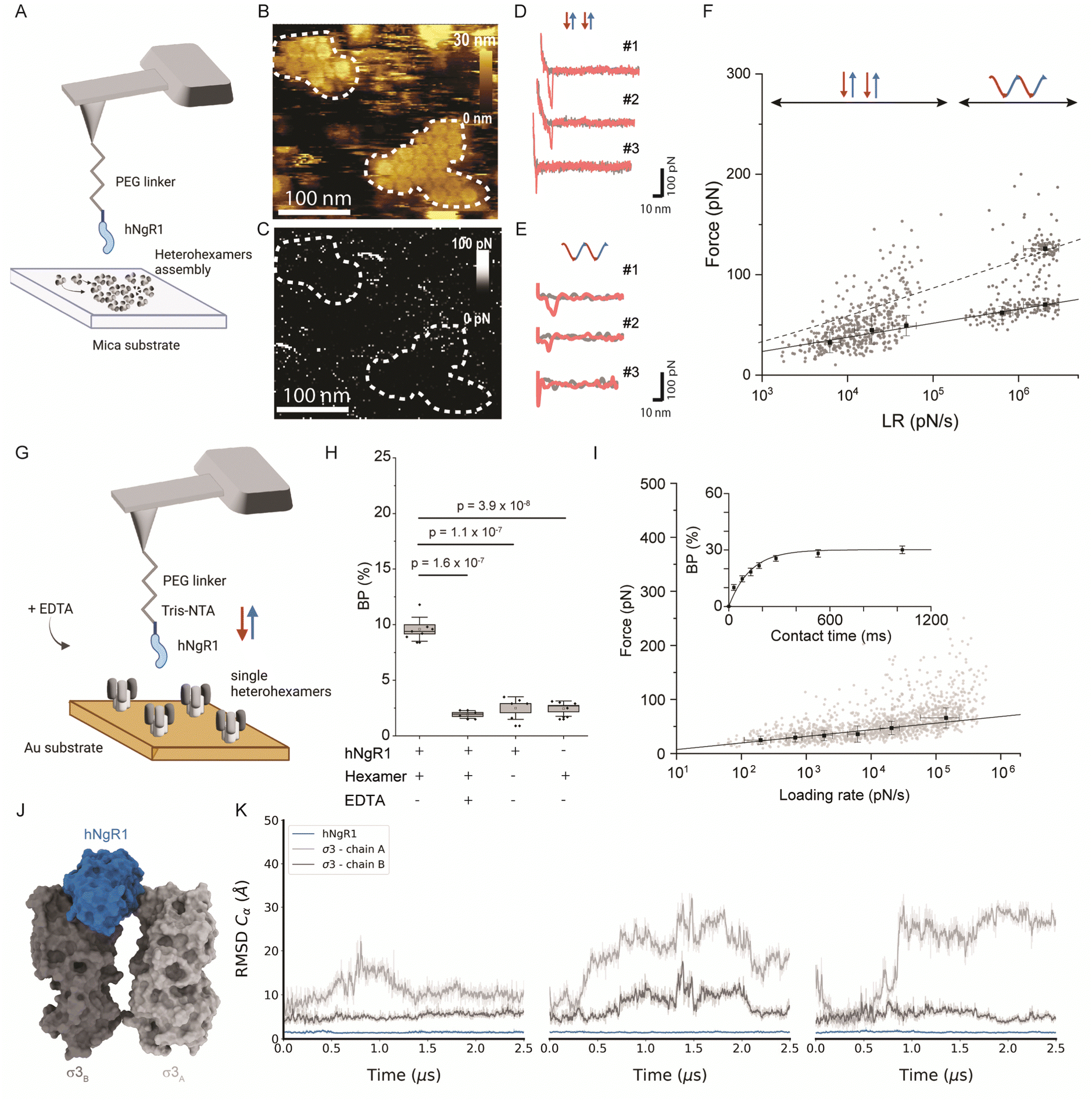 | ||
| Fig. 2 Probing the hNgR1 interaction with μ1σ3 heterohexamers and predicting the hNgR1–σ3 binding interface. (A) Representation of the experimental setup, with an hNgR1-functionalized AFM tip and self-assembled σ3μ1 heterohexamer patches on a mica substrate. FD and force–time curves are collected, from which forces and LRs are extracted. (B) AFM topography image of the scanned σ3μ1 heterohexamers and (C) the corresponding adhesion map of the same area. Colored scales indicate height and force, respectively. (D) Examples of FD curves recorded in FFV mode (using a linear movement of the tip) that display either specific (#1 and #2) or nonspecific (#3) adhesion events. (E) Examples of FD curves recorded in PFT mode (using a sinusoidal movement of the tip) that display either specific (#1 and #2) or nonspecific (#3) adhesion events. (F) Distribution of rupture forces as a function of their LR measured between hNgR1 and self-assembled σ33μ13 heterohexamers, from AFM experiments conducted with rectangular (data points on the left; lower LRs) or sinusoidal (data points on the right; higher LRs) tip movement. The solid line represents the Bell–Evans fit (for simple ligand–receptor bond) and the dashed line represents the Williams–Evans model prediction (for multiple simultaneous uncorrelated bonds). Error bars indicate SD. (G) Representation of the experimental AFM setup, operated in FV mode with an hNgR1-functionalized tip and single σ33μ13 heterohexamers grafted onto a gold-coated surface. (H) Box plot of the BP calculated for the hNgR1-single heterohexamers interaction and control experiments, including this same interaction with the addition of EDTA (5 mM), the interaction between hNgR1 and a surface coated with NHS/EDC (without heterohexamers), and an AFM tip with tris-NTA (without hNgR1) and a single-heterohexamers-coated surface. (I) DFS plot showing the distribution of rupture forces as a function of their LR, measured between hNgR1 and single heterohexamers. The solid line represents the Bell–Evans fit (for simple ligand–receptor bond). For contact time plots, data points represent mean BP calculated for each contact time and were fitted using a least-squares fit of a monoexponential decay. For all DFS and contact time plots, the error bars indicate SD. For all BP plots, one data point represents the BP obtained for one map of 1024 FD curves. The horizontal line within the box indicates the median, boundaries of the box indicate the SEM, and the whiskers indicate the SD. All data are representative of at least n = 3 independent experiments. (J) Structural representation of the complex between hNgR1 (in blue surface) and the two σ3A (in light-grey surface) and σ3B (in dark-grey surface) subunits. (K) Root-mean-square deviation (RMSD) of indicated chains computed using hNgR1 as the reference for the MD trajectory alignment. Plots are colored in blue, light grey, and dark grey for hNgR1, σ3A, and σ3B, three MD replicates are shown. | ||
For weak rupture forces, it is not straightforward to discriminate between multiple bonds at the lowest LRs. Therefore, we explored a wider range of LRs by combining the FD curve-based mode with rectangular or sinusoidal modulation of the distance (Fig. S1B, ESI†) with oscillation frequencies in the range of 0.25 to 0.5 kHz. This approach enabled us to more accurately evaluate the linearity of Bell–Evans fit and determine whether multiple bonds are established. We analyzed a total of 1088 curves, which show specific breaking forces ranging from 15 to 300 pN (Fig. S2, ESI†). From the Bell–Evans fit (Fig. 2F, solid line), we extracted a dissociation rate constant (koff) of 3.51 ± 2.02 s−1 and a distance to the transition state (xu) of 0.67 ± 0.04 nm, which describes the binding free-energy landscape of hNgR1 interacting with self-assembled heterohexamers. At the lower LRs, a single population of data points is observed, while at higher LRs, a second population becomes more evident. This second population correlates well with the Williams–Evans prediction30 for a double bond (Fig. 2F, dashed line), which can originate from either from parallel interactions involving two separate hNgR1 molecules bound to the AFM tip or a single hNgR1 molecule bound to the tip interacting with multiple sites on the heterohexamer. In the latter case, hNgR1 could bind using two opposing interfaces with adjacent heterohexamers, as suggested by the cryo-EM data. However, the exact nature of this second force population requires further investigation.
Human NgR1 engages single heterohexamers with high-affinity
The kinetics extracted for hNgR1 binding to assembled heterohexamers show a significantly higher koff (∼7-fold) relative to hNgR1 binding to T1L virions,15 which we did not expect. This kinetic difference is likely due to the three-dimensional arrangement of the heterohexamers, which adopt a denser conformation when reconstituted in vitro on a planar support, potentially hindering NgR1 access to adjacent heterohexamers.To ensure that the surface-assembled geometry of the heterohexamers was not responsible for the observed differences in binding kinetics, and to assess the requirement for the two interfaces in the formation of the binding complex, we examined the interaction using unassembled, or isolated heterohexamers, referred to as single heterohexamers (Fig. 2G). Our results show that hNgR1 can bind to single heterohexamers, with a BP of 9.6 ± 1.1% (mean ± SD). The specificity was further validated by the addition of EDTA or the absence of one of the binding partners (Fig. 2H). We did not observe a significant difference in the extracted rupture force (distribution of forces in Fig. S3, ESI†), which was again in the range of 15–300 pN (Fig. 2I). Analysis of rupture forces at the single-heterohexamer level enables calculation of koff of 0.32 ± 0.21 s−1 and xu of 0.86 ± 0.08 nm. With a lower dissociation rate constant than that of assembled heterohexamers, single heterohexamer results show a more stable interaction compared to the assembled heterohexamers, indicating that accessibility of NgR1 to the heterohexamer binding interface is critical for complex stability. Conversely, the association rate of the receptor to single heterohexamers is somewhat less favorable than that of virions, with a kon of 2.17 ± 0.20 μM−1 s−1, suggesting that complex initiation is facilitated not only by the multimeric arrangement of heterohexamers, but also by the curvature of viral particles.
It is conceivable that initial interactions with the convex face of hNgR1 facilitate formation of the complex. The affinity of NgR1 for single heterohexamers lies in the high-affinity range, also observed for virion binding15 (Fig. S4, ESI†). Thus, the stability of the binding complex appears to be largely dependent on a single interface established with the concave face of hNgR1. However, by comparing the various koff values extracted for the three systems used, we observed that the stability of the binding interactions is as follows: single heterohexamers > single virus ≫ assembled heterohexamers. We conclude that the accessibility of the NgR1-binding groove between σ3 subunits from adjacent heterohexamers plays a key role in the stability of the complex.
MD simulations of the hNgR1 in complex with σ3A and σ3B
To validate the predominant interaction interface between the concave face of hNgR1 and σ3 protein, we conducted three independent MD simulations of 2.5 μs with explicit solvent (Fig. 2J). The root-mean-square deviation (RMSD) analysis of the backbone alpha carbon (Cα) of each subunit shows that the proteins are stable during the simulations (Fig. S5, ESI†). The RMSF analysis highlights the most flexible loops in the σ3 subunits (Fig. S5, ESI†). To monitor flexibility of the two σ3 monomers with respect to hNgR1, we calculated the RMSD using hNgR1 as the reference for the MD trajectory alignments (Fig. 2K). Lower RMSD values were observed for the σ3B subunit than for σ3A, suggesting greater stability for the concave interface of the σ3–hNgR1 binding interaction. Taken together, the in vitro and in silico experiments confirm that hNgR1 stably interacts with σ3 protein between adjacent heterohexamers and that the binding interface is predominantly located between the external surface of the σ3B subunit and the concave surface of hNgR1.Influence of NgR1 polymorphism on σ3 binding
While hNgR1 and mNgR1 share structural and functional homology,31 subtle polymorphisms within the virus-bound interfaces may wield substantial influence, potentially heralding a paradigm shift in our understanding of reovirus engagement dynamics. These polymorphisms may contribute to the differences observed in experiments using cultured cells and mice.20,23 Therefore, to determine whether the few differences in hNgR1 and mNgR1 amino acid sequences alter reovirus–NgR1 engagement, we investigated interactions between mNgR1 and σ3 using T1L virions (fully functional viral particle) (Fig. 3A–C) or single heterohexamers (Fig. 3D–F).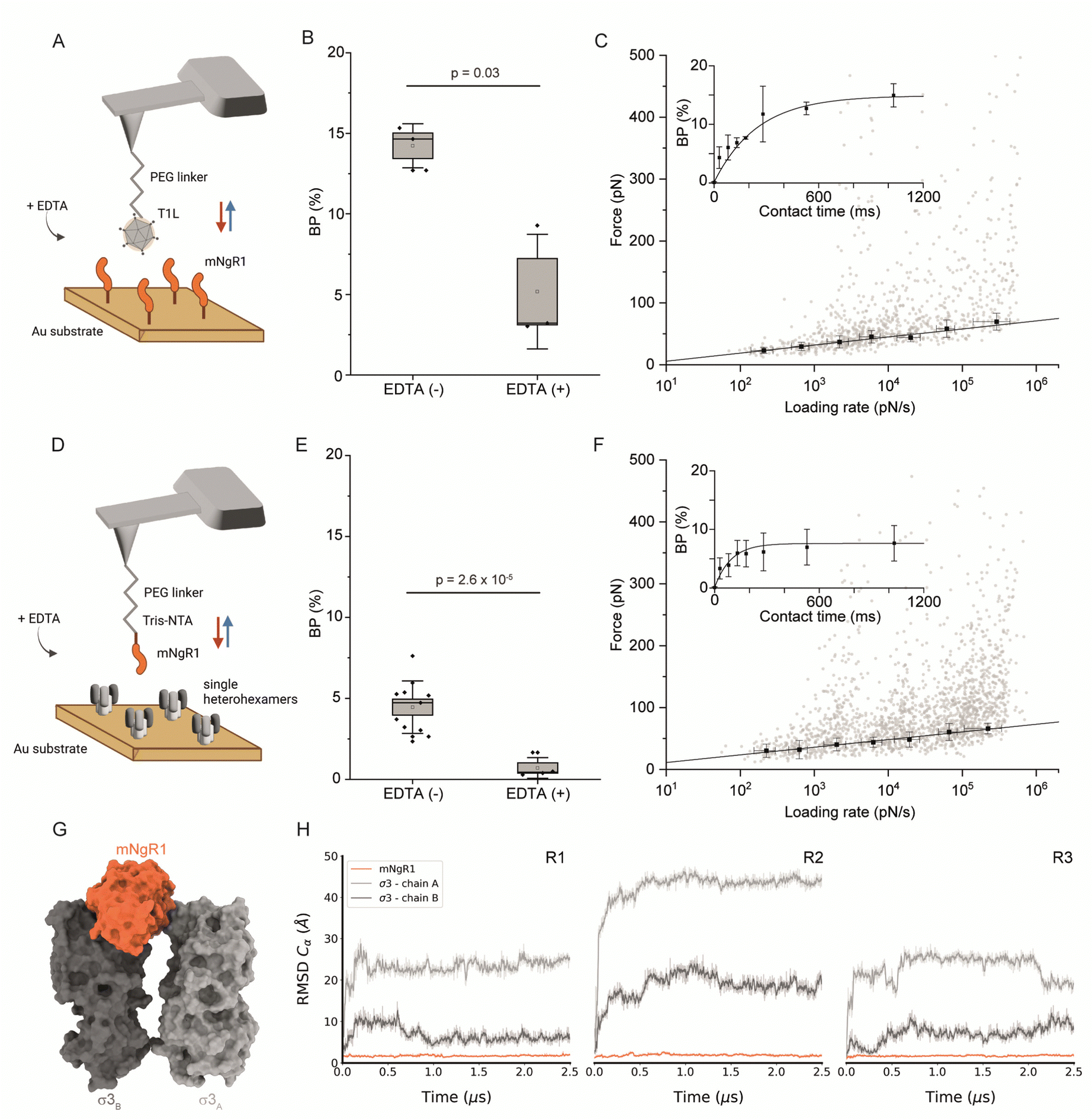 | ||
| Fig. 3 Thermodynamical characterization of the interactions between mNgR1 and either reovirus T1L or single μ1σ3 heterohexamers, and predicting the mNgR1–σ3 binding interface. (A) Representation of the experimental setup, with an AFM tip functionalized with a reovirus T1L virion and mNgR1 proteins grafted on a gold-coated surface, operated in FV mode. (B) Box plot of the BP calculated for the T1L–mNgR1 interaction, with (control) and without addition of EDTA (5 mM). (C) DFS plot showing the distribution of rupture forces as a function of their LR, measured between T1L virion and mNgR1. Binding probability is plotted (as inset) as a function of the contact time. (D) Representation of the experimental setup, with an AFM tip functionalized with mNgR1 and single σ3μ1 heterohexamers grafted on a gold-coated surface, operated in FV mode. (E) Box plot of the BP calculated for the mNgR1-single heterohexamers, with (control) and without addition of EDTA (5 mM). (F) DFS plot showing the distribution of rupture forces as a function of their LR, measured between mNgR1 and single heterohexamers. For all DFS plots, the solid line represents the fit of the data with the Bell–Evans fit (for simple ligand–receptor bond). For contact time plots, data points represent mean BP calculated for each contact time and were fitted using a least-squares fit of a monoexponential decay. For all DFS and contact time plots, the error bars indicate SD. For all BP plots, one data point represents the BP obtained for one map of 1024 FD curves. The horizontal line within the box indicates the median, boundaries of the box indicate the SEM, and the whiskers indicate the SD. All data are representative of at least n = 3 independent experiments. (G) Structural prediction of the complex between mNgR1 (in orange surface) and the two σ3A (in light-grey surface) and σ3B (in dark-grey surface) subunits. (H) RMSD of indicated chains computed using mNgR1 as the reference for the MD trajectory alignment. The plots are colored in orange, light grey, and dark grey for mNgR1, σ3A and σ3B, respectively. R1, R2, and R3 denote MD replicates. | ||
We first validated the specificity of the interaction by analyzing the BPs before and after addition of EDTA (Fig. 3B and E). Next, we conducted DFS analyses (Fig. 3C, F and Fig. S6, ESI†), which enabled extraction of the kinetic parameters of the binding interactions. Similar to hNgR1 binding, a higher kon of 6.94 ± 0.23 μM−1 s−1 was observed for mNgR1 binding to virions relative to single heterohexamers (2.99 ± 0.46 μM−1 s−1), suggesting that the display of heterohexamers on the viral capsid facilitates receptor binding. Similarly, like hNgR1 binding interactions, mNgR1 binding to virions occurred with lower stability (koff of 0.65 ± 0.35 s−1, xu of 0.73 ± 0.07 nm) relative to single heterohexamers (koff of 0.23 ± 0.12 s−1, xu of 0.76 ± 0.05 nm), highlighting that access to the σ3 interface influences stability of the complex. These analyses revealed high-affinity interactions for virions and heterohexamers (KD = 93.5 ± 53.6 nM or 76.8 ± 51.8 nM, respectively). Thus, both hNgR1 and mNgR1 establish stable complexes with reovirus T1L, despite their subtle polymorphisms.
Depicting the σ3–mNgR1 interface using MD
Since AFM experiments suggest that hNgR1 and mNgR1 bind reovirus comparably, we sought to determine whether MD simulations would offer an explanation for the differences in the capacity of these NgR1 homologs to function as reovirus receptors. As before, MD simulations were conducted using a geometry mimicking the assembly observed on the particle surface with mNgR1 binding between two σ3 subunits (σ3A and σ3B) (Fig. 3G). For simulations with mNgR1, the initial complex was obtained using the coordinates of σ3A and σ3B from the hNgR1 complex and superimposing coordinates of mNgR1 (PDB: 5O0L) in to the hNgR1-bound site.32 The RMSD values for both hNgR1 and mNgR1 fall within a similar range, and the RMSF shows a similar flexibility profile, indicating comparable stability of the binding complex (Fig. 3H and Fig. S7, ESI†). This finding reinforces the results obtained from AFM experiments and underscores the consistency of the binding behavior of the NgR1 orthologues. Furthermore, the observed higher flexibility at the convex interface in both cases suggests a lesser role of this interface in stabilizing the complex, aligning with our previous observations of reovirus–hNgR1 interactions. Collectively, these results suggest that the interface stability and dynamics of reovirus binding to mNgR1 and hNgR1 are comparable, which is consistent with our AFM findings.Comparison of key NgR1 residues involved in the interaction with σ3
Using MD simulations for both hNgR1 and mNgR1, we also conducted an in-depth analysis of the frequency of interactions with σ3. Our simulations reveal that σ3B consistently maintains interactions observed in the structures of both hNgR1 and mNgR1 at high frequency (Fig. 4). The majority of the frequent interactions with σ3B are shared by the two receptors, with only minor variations in the observed interaction patterns (Fig. 4A and B, all interactions depicted in Fig. S8, ESI†). Consistent with experimental mutagenesis data,15 key interactions mediated by residues Y160, Q162, and R206 of hNgR1 are also observed for mNgR1 (Y160 and Q162 form hydrogen bonds with σ3B residues D231 and E227, respectively, while R206 interacts with H230 via cation–π or hydrogen bond interactions, Fig. 4C and D). Additionally, our analysis identifies H230 of σ3B as potentially functioning in the binding interaction, given its high contact frequency with a network of residues from both hNgR1 and mNgR1, including NgR1 residue Y232 (Fig. 4C and D).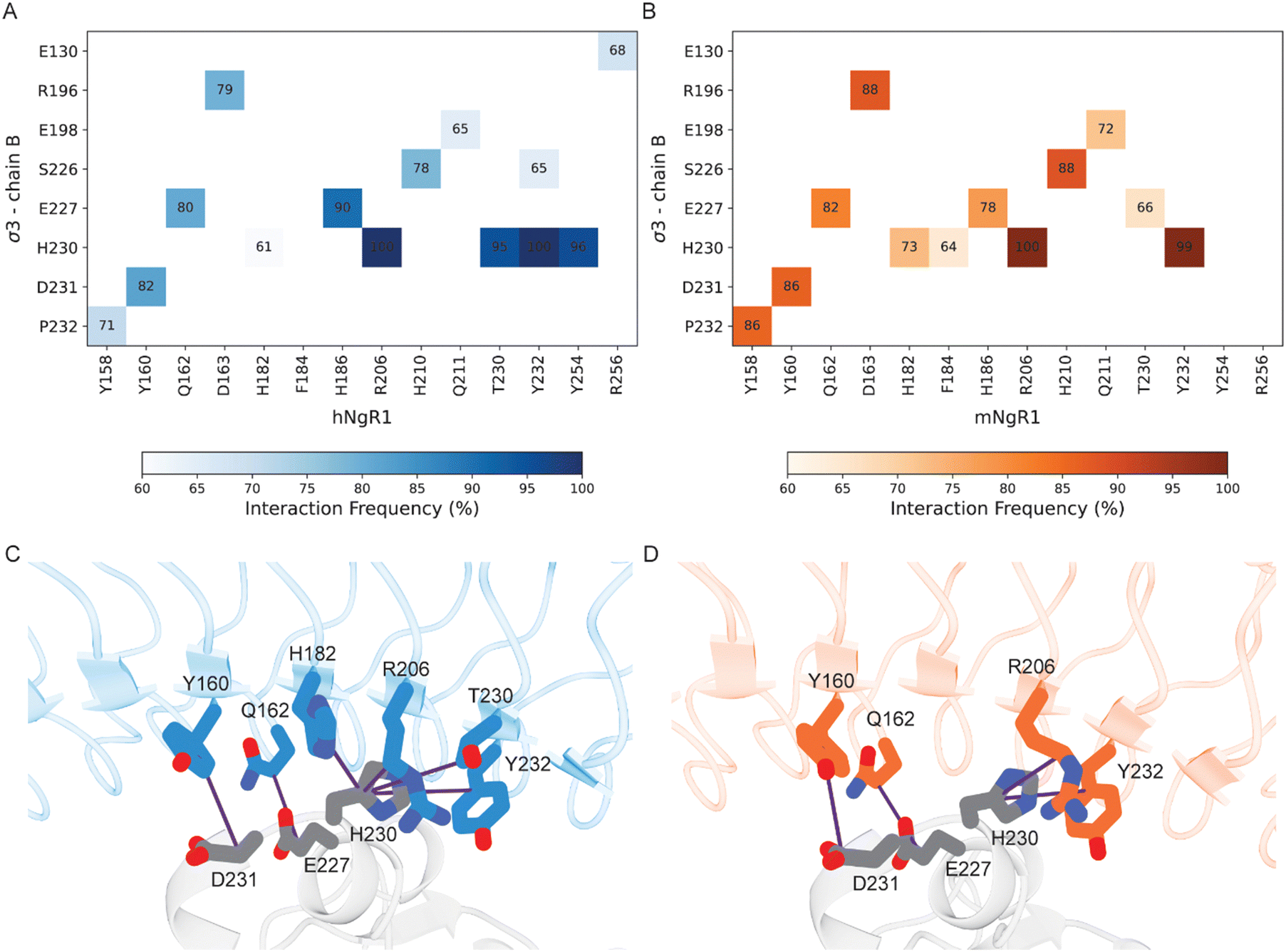 | ||
| Fig. 4 Residues in common that are involved in the interactions between human and murine NgR1 with σ3B. (A) and (B) Heat maps of interacting residues in the initial complexes that occur frequently in the simulations. Cells of the heat maps are colored according to the increased number of interactions, from white to dark blue for the hNgR1 complex (A) and white to dark orange for the mNgR1 complex (B). (C) and (D) 3D representation of the key interactions established by residues Y160, Q162, and R206 of hNgR1 and mNgR1, and H230 of σ3B. The σ3, hNgR1 (C), and mNgR1 (D) structures are shown in grey, blue, and orange cartoons, respectively. Violet lines indicate the residue connections that are found during the simulations (for the type of interactions, see Table S1, for the complete overview of all contacts see Fig. S8, ESI†). | ||
Conversely, residues within σ3A do not have high-frequency interactions with either hNgR1 or mNgR1 during the simulations, and none of the interactions observed in the initial structures are maintained (Table S2, ESI†). This discrepancy suggests that the high flexibility of the interface with σ3A leads to changes in interaction patterns and binding modes throughout the simulations. The majority of the residues that differ in sequence between hNgR1 and mNgR1 (Fig. S9, ESI†) are located in this convex interface. These differences suggest that the interface contributing most importantly to binding also is the most similar in both receptors.
Discussion
In this study, we investigated molecular interactions between reovirus strain T1L and NgR1, a host cell receptor that mediates reovirus entry into some neuronal cells. Using a combination of experimental and computational methods, we aimed to elucidate the assembly dynamics and interaction kinetics between reovirus σ33μ13 heterohexamers and NgR1.Our high-resolution AFM imaging experiments provided unprecedented insights into the assembly dynamics of reovirus heterohexamers at nanoscale (Fig. 1). We observed the formation of a two-dimensional lattice of heterohexamers under native conditions, closely mirroring the assembly of viral proteins on the surface of a reovirus particle that typically encapsidates the dsRNA genome. This lattice structure on the surface was strikingly similar to that observed on the actual virus particle. As this assembly mimics physiological conditions, we subsequently force-probed these patches with an hNgR1-functionalized AFM tip and determined hNgR1 binding strength and stability of the complex (Fig. 2A–F). Unexpectedly, we found that the complex has a lower stability compared with observations made previously using whole virions.15
Further evidence of the influence of viral assembly on binding strength was obtained by studying the interactions of NgR1 with isolated heterohexamers (Fig. 2G–I). These findings underscore the limitations of reconstituting molecular assemblies on flat substrates. The geometry of the capsid subunits in this setup differs significantly from their native arrangement on the virus particle, potentially altering the binding configuration of the complex. Our results suggest that these deviations from the native curvature as well as the lateral organization of the virus particle alter the kinetics and possibly the thermodynamics of the virus-receptor interaction (Fig. 5).
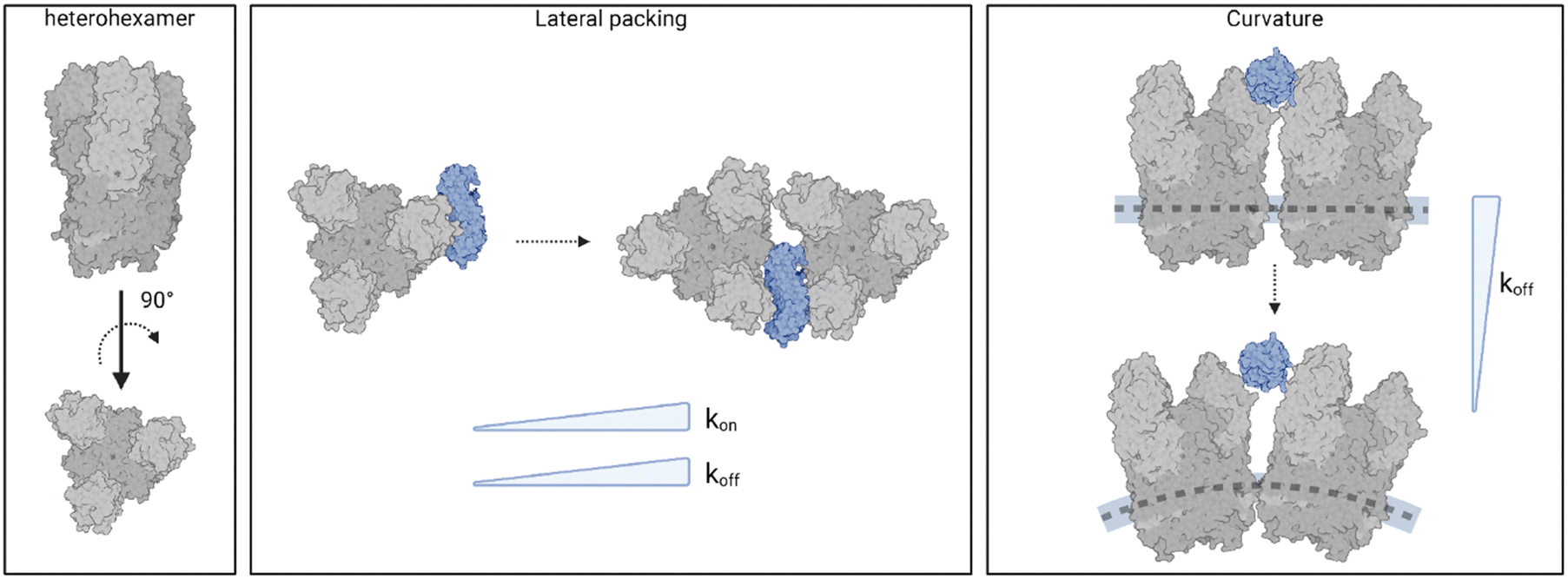 | ||
| Fig. 5 Spatial organization and curvature are critical determinants of NgR1–reovirus complex stability. Models of NgR1 binding interfaces with reovirus heterohexamers reconstituted on a planar support or within the curved virus capsid. The concave NgR1 interface binds more strongly to individual heterohexamers compared to densely packed self-assembled structures (lower koff), however the multimeric heterohexamer arrangement promotes a higher association rate (higher kon). The local curvature of the viral capsid in reovirus native topology allows for a greater accessibility of the NgR1-binding groove between σ3 subunits from adjacent heterohexamers, which plays a key role in the stability of the complex (lower koff). Created with BioRender.com. | ||
Complementing our AFM experiments, MD simulations allowed an examination of the protein–protein interfaces in the reovirus–NgR1 complex. Both techniques consistently indicate that the binding complex is predominantly stabilized by the concave interface of NgR1. Examination of the effect of NgR1 polymorphisms on σ3 binding reveals only subtle differences in the interaction patterns between hNgR1 and mNgR1. Despite these differences, both NgR1 orthologues formed stable complexes with σ3, underscoring the conservation of key interaction interfaces across species. The binding association as depicted by the AFM experiments suggests that the establishment of the binding complex is influenced by the convex NgR1 surface with the σ3B subunit, as reflected by the higher kon extracted in conditions where the assembly geometry mimics viral assembly. This interface could serve as a template to facilitate proper orientation of the final binding pose.
Overall, our study provides valuable insights into the intricate molecular choreography orchestrating reovirus–host interactions, particularly within the central nervous system. The identification of subtle polymorphisms within virus-bound interfaces challenges conventional wisdom about NgR1–reovirus binding and unveils a novel dimension in understanding viral tropism and pathogenesis. By elucidating these dynamics at the molecular level, our research advances our comprehension of viral infections and sets the stage for targeted therapeutic interventions against reovirus infections. This breakthrough underscores the critical importance of exploring virus–host interactions at a subcellular level and offers promising avenues for combating viral diseases with precision and efficacy.
Materials and methods
Virus production
The T1L strain of reovirus was recovered from BHK-T7 cells using plasmid-based reverse genetics.33 Virus was plaque purified from BHK-T7 cell lysates, propagated in L929 cells, and purified from infected L929 cell lysates by cesium chloride gradient centrifugation as described.34 Infected cells were lysed by freeze thaw and sonication. Virions were extracted from lysates using Vertrel-XF (TMC Industries), layered onto step gradients of cesium chloride (1.2–1.4 g cm−3), and centrifuged at 87![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 × g at 4 °C for 18 h. The visible band corresponding to the density of reovirus particles (∼1.36 g cm−3) was collected and exhaustively dialyzed against virion-storage buffer (150 mM NaCl, 15 mM MgCl2, and 10 mM Tris [pH 7.4]). Particle number of purified virions was estimated by spectral absorbance at 260 nm (1 OD260 = 2.1 × 1012 particles per mL).
000 × g at 4 °C for 18 h. The visible band corresponding to the density of reovirus particles (∼1.36 g cm−3) was collected and exhaustively dialyzed against virion-storage buffer (150 mM NaCl, 15 mM MgCl2, and 10 mM Tris [pH 7.4]). Particle number of purified virions was estimated by spectral absorbance at 260 nm (1 OD260 = 2.1 × 1012 particles per mL).
Purified human and mouse NgR1-His protein
His-tagged hNgR1 protein were expressed and purified as described.15 FreeStyle™ 293-F cells (Thermo Fisher Scientific) were transiently transfected at a cell density of 106 cells mL−1 using a 1:3 ratio of plasmid DNA (encoding amino acids 1–310 of hNgR1 appended to a 7X-His tag) to polyethyleneimine (PEI 25K™, Polysciences). Cells were incubated at 37 °C for 7 days and pelleted. The supernatant was collected, supplemented with cOmplete™ protease inhibitor EDTA-free (Roche), and loaded onto a 5-mL HisTrap FF crude column (Cytiva) preequilibrated with running buffer (10 mM HEPES [pH 7.4], 150 mM NaCl) supplemented to contain 20 mM imidazole. A 20-to-500 mM imidazole gradient was applied to the column to first wash and then elute hNgR1-His. hNgR1-His was further purified by size-exclusion chromatography using a HiLoad 16/60 Superdex 75 column (Cytiva) equilibrated with running buffer. Fractions containing hNgR1-His were collected, concentrated to 8 mg mL−1 and frozen at −80 °C until further use.
His-tagged mNgR1 (amino acids 1–447) was acquired through Bio-Connect (CAT# 50106-M08H).
Expression and purification of the σ3μ1 heterohexamer
High Five™ insect cells were infected with a P3 recombinant baculovirus stock expressing μ1 and σ3 of reovirus strain T1L at an MOI of five. Cells were supplemented with 0.1% FBS, incubated at 27 °C for 96 h and the supernatant was harvested at 3500 × g for 10 min. The supernatant was loaded onto a HiTrap Q FF 5-mL ion-exchange column (Cytiva) and washed with buffer A (20 mM Tris [pH 8.5], 2 mM MgCl2, 2 mM BME) supplemented to contain 100 mM NaCl. Heterohexamer protein complexes (σ33μ13) were eluted with a linear gradient of buffer A from 100 to 550 mM NaCl. Fractions containing μ1 and σ3 were pooled and concentrated to 10 mL with a Amicon® Ultra-15 Centrifugal Filter Unit with a 100-kDa molecular weight cut-off (MWCO). Protein was diluted dropwise with ammonium sulfate buffer (buffer A supplemented to contain 4 M ammonium sulfate) to a final concentration of 0.7 M ammonium sulfate. Protein was loaded onto a HiTrap Phenyl HP 5-mL column (Cytiva) and eluted with a linear gradient of buffer A from 0.7 to 0 M ammonium sulfate. Fractions containing heterohexamer were pooled, concentrated, and the protein was further purified using a HiLoad 16/600 SD200 size-exclusion column equilibrated with buffer B (20 mM Bicine [pH 9], 150 mM NaCl, 2 mM MgCl2, 2 mM BME). Heterohexamer fractions were pooled and stored at −80 °C until further use.Preparation of assembled σ33μ13 heterohexamer patches on mica
Heterohexamer fractions previously stored in buffer B were used to prepare assembled μ1σ3 heterohexamer patches on mica. First, the heterohexamer sample (3 mg mL−1) was buffer exchanged by dialysis at 4 °C overnight with HEPES/NaCl buffer (20 mM HEPES, 300 mM NaCl, pH 7.4). Protein was stored at −20 °C until further use. On the day before the experiment, the heterohexamer sample (diluted to 0.015 mg mL−1 in incubation buffer [20 mM HEPES, 300 mM NaCl, 5 mM MgCl2]) was adsorbed at room temperature overnight to freshly cleaved mica using conditions to ensure the sample did not dry out. Divalent cations present in the incubation buffer allow electrostatic coupling of protein to the cleaved, negatively charged mica surface. Mica was subsequently washed with incubation buffer, immediately prior to imaging.Functionalization of AFM tips
AFM tips were coated with T1L virions, hNgR1, or mNgR1, as follows. AFM tips (MSCT probes, Bruker; AC-40, Olympus; PeakForce HIRS-B, Bruker) were functionalized with primary amino groups with aminopropyl-triethoxysilane (APTES; Sigma-Aldrich) from gas phase, as described.35 Tips were cleaned with chloroform (Sigma-Aldrich) for 5 min and treated with UV radiation and ozone (UV-O; Jetlight) for 15 min. Then, two plastic trays were placed inside a desiccator, which was flooded with argon gas for 15 min. AFM tips were placed inside the desiccator, followed by adding 30 μL of APTES and 10 μL of triethylamine to their respective trays and the desiccator was closed. After a reaction time of 2 h, both trays were removed and the desiccator was flooded with argon gas for 10 min. The desiccator was closed and tips were incubated for at least two days to “cure” the APTES coating. In a second step, AFM tips were coupled with Ald-Ph-PEG24-NHS ester linkers, as described previously. In a second step, AFM tips were coupled with Ald-Ph-PEG24-NHS ester linkers, as described previously.36 The tips were immersed in a solution of 1 portion of Ald-Ph-PEG24-NHS (3.3 mg) in chloroform (0.5 mL) and triethylamine (30 μL) for 2 h. Tips were rinsed three times in chloroform, allowed to dry, and placed onto parafilm in a polystyrene Petri dish.For AFM tips functionalized with reovirus T1L virions, the Petri dish was stored at 4 °C and 100 μL of virus solution (109 particles mL−1) were pipetted onto the tips. Then, 2 μL of freshly prepared sodium cyanoborohydride solution (∼6% [wt/vol] in 0.1 M NaOH) were added to the virus droplet and tips were incubated in this solution at 4 °C for 1 h. Ethanolamine (5 μL; 1 M [pH 8.0]) was carefully mixed with the droplet. Tips were incubated at 4 °C for 10 min, rinsed three times in ice-cold virus buffer (150 mM NaCl, 15 mM MgCl2, 10 mM Tris [pH 7.4]), placed in individual wells of a multiwell-plate containing virus buffer, and stored at 4 °C until further use.
For AFM tips functionalized with NgR1 protein, following coupling to PEG linkers, 100 μL of a 100 μM Tris nitrilotriacetic amine 540 trifluoroacetate (tris-NTA; Toronto Research Chemicals, Canada) solution was added to the tips. Freshly prepared sodium cyanoborohydride solution (2 μL; ∼6% [wt/vol] in 0.1 M NaOH) was mixed in and tips were incubated in this solution for 1 h on ice. Next, 5 μL of ethanolamine (1 M [pH 8.0]) was added to the droplet, mixed carefully, and tips were incubated for 10 min on ice. A solution of 50 μL of hNgR1-His (1 μM) or mNgR1-His (1 μM) and 2.5 μL of 5 mM NiCl2 was pipetted onto tips and incubated for 2 h on ice. Finally, tips were rinsed three times in phosphate-buffered saline (PBS), placed in individual wells of a multiwell-plate containing PBS, and stored at 4 °C until further use.
Preparation of model surfaces using surface chemistry
Gold-coated surfaces were coupled with either hNgR1, mNgR1, or σ3μ1 heterohexamers. Surfaces were rinsed with ethanol, dried with low nitrogen flow, and treated with UV radiation and ozone (UV-O; Jetlight) for 15 min. Surfaces were immersed in a solution containing 1% of 16-mercaptohexadecanoic acid and 99% of 11-mercapto-1-undecanol [V/V] and incubated overnight. The next day, surfaces were rinsed with ethanol, dried with low nitrogen flow, and incubated in a solution of NHS and EDC (10 mg mL−1 NHS and 25 mg mL−1 1-ethyl-3-[3-dimethylaminopropyl]-carbodiimide [EDC]) for 30 min. Then, surfaces were rinsed three times with milliQ water, without drying.For surfaces coated with σ3μ1 heterohexamers, a 50-μL droplet of 100 μg mL−1 solution of heterohexamer was added to surfaces. Surfaces were incubated for 1 h, rinsed three times with wash buffer (PBS containing 0.05% Tween20), and stored in PBS at 4 °C until further use.
Fc-tagged hNgR1 (R&D systems) or mNgR1 (Bio-Techne) proteins were immobilized on surfaces via protein A chemistry, as previously described.37 Following NHS and EDC incubation, surfaces were incubated in 100 μL of protein A (100 μg mL−1) in PBS at RT for 1 h. Surfaces were rinsed three times with wash buffer and incubated for 1 h in 300 μL of blocking buffer (PBS supplemented to contain 0.05% Tween20 and 1% BSA). Then, 50 μL of 100 μg mL−1 hNgR1-Fc or mNgR1-Fc protein was incubated on surfaces for 1 h. Finally, surfaces were rinsed three times with wash buffer and stored in PBS at 4 °C until further use.
FD-based AFM on assembled σ33μ13 heterohexamers patches
AFM (Bioscope, Resolve, Bruker) operated in PeakForce tapping (PFT) mode was used to image assembled heterohexamers. PeakForce HIRS-B (Bruker) cantilevers were used to first scan a large area, between 5 to 10 μm2, at a 128 × 128-pixel resolution. Then, smaller areas with specific patches of assembled heterohexamers were imaged with a PFT frequency of 0.25 or 0.5 kHz. The force setpoint was set to 150 pN, with an amplitude of 50 nm, and a 1–3 Hz scan rate.To probe interacting forces between hNgR1 and assembled heterohexamers, AC-40 (BioLever, Olympus) or PeakForce HIRS-B (Bruker) cantilevers were used. The AFM was operated in either FASTForce volume (FFV) or PFT mode. For FFV experiments, a scan rate of 0.125–0.25 Hz and a setpoint of 100–150 pN was used, whit a ramp size distance of 100 nm. Areas of 128 × 128- or 256 × 256-pixel resolution were scanned. When scanning a smaller area of the surface of a heterohexamer patch, a 64 × 64-pixel resolution was used, with 3 or 5 Hz scan rate. For PFT experiments, the setpoint was 150 pN, with a PFT frequency of 0.25 or 0.5 kHz, and a 0.25 Hz scan rate. The amplitude was set to 50 nm.
FD-based AFM on model surfaces
ForceRobot 300 (Bruker, Germany) and NanoScope Multimode 8 (Bruker, NanoScope software v9.1) were operated in force–volume (contact) mode to conduct force distance (FD) curve-based AFM experiments on hNgR1-, mNgR1- or single σ3μ1 heterohexamers-coated surfaces. Virus-functionalized or NgR1-functionalized MSCT-D probes (spring constants calculated using thermal tune38) were used to scan areas of 5 × 5 μm, with a ramp size set to 500 nm and a resolution of 32 × 32 pixels (corresponding to 1024 FD curves). Surfaces were mounted on a piezoelectric scanner using a magnetic carrier and all experiments were conducted in PBS at RT. Approach velocity was maintained at 1 μm s−1 and the maximum force was set to 500 pN.To probe interactions between NgR1 and reovirus or heterohexamers over a wide range of loading rates, dynamic force spectroscopy (DFS) experiments were conducted. In these experiments, retract velocities were varied between 0.1, 0.2, 1, 2, 5, 10, and 20 μm s−1 without surface delay. Kinetic on-rate (kon) experiments were conducted by extracting the binding probability when surface delay is added, allowing the tip to stay in contact with the surface for different periods of time. Hold times were set to 0, 50, 100, 150, 250, 500 and 1000 ms.
Additional independent control experiments were conducted in order to ensure the specificity of the interactions. Blocking was conducted by adding 5 mM EDTA to the buffer, resulting in the chelation of Ni2+ and subsequent loss of ligation between NgR1 and NTA. Measurements were taken before and after addition of the blocking agents, at a retraction velocity of 1 μm s−1. The same sample area was probed several times, using the same tip and without surface delay.
AFM data analysis
Depending on the instrument used for data acquisition, JPK data processing (version 6.1.149) or NanoScope analysis software (v2.0) were used for F–D curve data analyses. FD curves that displayed adhesion events, with peaks corresponding to PEG-linker extension, were selected and the retraction curve was fitted with the worm-like chain model for polymer extension.27 For DFS data, rupture forces were extracted and loading rates were determined using the slope of adhesion peaks in force–time curves. Histograms of rupture force distributions for distinct LR ranges were fitted with various force spectroscopy models, allowing the estimation of koff and xu.39,40 For kinetic on-rate analysis, the binding probability for each hold time (the time the tip is in contact with the surface) was determined from the fraction of curves that displayed a specific binding event. Those data were fitted and KD calculated as described previously.41 Origin software (OriginLab) was used to graph the DFS and kon results, as well as fitting of the data. Statistical significance of BP values was assessed by conducting two-sample t tests. P-Values were obtained using https://www.statskingdom.com/. Image processing of assembled heterohexamers was performed using Gwyddion (v2.5) and NanoScope analysis software (v2.0). Assembly dynamics of heterohexamers was analyzed with origin software (OriginLab) and the area of lattice formation was fitted with an exponential fit, according to the equation: y = A1 × exp(−x/t1) + y0.MD simulations
The structure of hNgR1 (chain N) bound to two σ3 subunits (chains s and m) was retrieved from the structure EMD-13149 in the electron microscopy data bank (https://www.ebi.ac.uk/emdb/). The structure of mNgR1 was retrieved from the structure 5O0L in the RCSB protein data bank (PDB ID: 5O0L) and superimposed to hNgR1 (chain N) with the protein structure alignment tool available in Maestro (Schrödinger Release 2022-1, Maestro, Schrödinger, LLC, New York, NY, 2022).Missing hydrogen atoms were added to the proteins, and side chains were optimized using the protein preparation Wizard tool at physiological pH (Schrödinger Release 2022-1, Maestro, Schrödinger, LLC, New York, NY, 2022). Protonation states of histidine, aspartate and glutamate residues are reported in Table S3 (ESI†). The prepared protein was solvated in a cubic box with a padding of 15 Å, using the TIP3P water model,42 and neutralized with a proper number of sodium and chloride ions to reach a salt concentration of 0.154 M. The system was built with the leap software implemented in AMBER (The Amber Molecular Dynamics Package, at https://ambermd.org/),43,44 using the ff14SB force field.43–45 The initial system (169730 atoms for the human complex and 190906 atoms for the complex with murine NgR1) was minimized with AMBER. The restraints force on the water and ions were gradually reduced starting from 1000 kcal mol−1 Å−2. The steepest descent algorithm was used for 1000 steps, followed by 800 steps of the conjugate gradient algorithm in each stage.
The equilibration stage was then performed in three different passages under canonical NVT (N: number of particles, V: volume, T: temperature) ensemble in AMBER. Firstly, the minimized systems were gradually heated to 300 K and equilibrated using a timestep of 1 fs for 200 ps in which positional restraints applied to heavy atoms (excluding hydrogen, sodium, and chlorine) were gradually decreased from 100 to 50 kcal mol−1 Å−2. The second equilibration stage was 200 ps long with a timestep of 1 fs with positional restraints of 10 kcal mol−1 Å−2. In the last step of 10 ns, the systems were equilibrated without positional restraints. The temperature was maintained at 310 K using a Langevin thermostat46 with a low damping constant of 1 ps−1. The M-SHAKE algorithm47 was used to constrain the bond lengths involving hydrogen atoms. Long-range coulombic interactions were handled using the particle mesh Ewald summation method (PME).48
Three independent replicates of 2.5 μs unrestrained MD simulations for both the human and mouse complexes using the isotropic ensemble (NPT) with an integration time step of 2 fs using the GROMACS software (version 2022.3).49,50 In each replicate, new velocities were computed according to a Maxwell distribution at temperature 300 K using gen-vel flag in GROMACS. The temperature was set at 300 K, by setting the damping constant at 0.1 ps−1. The pressure was set to 1 bar using a Parrinello–Rahman barostat51 with a coupling constant of 2 ps and a compressibility of 4.5 × 10−05 bar−1. All bonds involving hydrogen atoms were constrained using LINear constraint solver (LINCS) algorithm. The cutoff distance for long-range and van der Waals interactions was set at 10.0 Å for both equilibration and production stage.
MD frames (one per ns) were extracted for the analyses. Root mean square deviation (RMSD) values were computed with an in-house Python script based on MDAnalysis (v2.2.0).52,53 We conducted the RMSD and RMSF analyses for analyzing the dynamic behavior of the individual monomers (each chain aligned to itself, Fig. S5 and S7, ESI†) and for evaluating the complex stability (using the coordinates of NgR1 as a reference for the structural alignment, Fig. 2 and 3). GetContacts was used to analyze interactions of hNgR1 and mNgR1 with σ3 protomers (using geometric criteria with default settings https://getcontacts.github.io/, except for distance cutoff of 3.5 Å and angle range of 70–120° for hydrogen bonds). Replica 2 of the mNgR1 simulations was not used for interaction analysis because of the higher RMSD of the σ3 subunits compared to other trajectories (Fig. 3H). Data were visualized using the Matplotlib Python library.54
Author contributions
M. K. and D. A. conceived the project. R. D. S. N., A. C. D., and M. K. conducted all AFM experiments. A. N. and M. G. performed MD simulations, A. N. and A. D. P. analyzed them. D. M. S., O. L. W., and T. S. D. designed experiments, contributed materials, and analyzed data. All authors wrote the manuscript.Data availability
The source data underlying Fig. 1G–L, 2D–F, H, I and 3B, C, E, F are provided as a source data file. MD trajectories and related files (topology, parameter, and coordinates) are available at https://zenodo.org/records/13241238. All other relevant data are available from the corresponding authors upon reasonable request.Conflicts of interest
The authors declare no competing interests.Acknowledgements
This work was supported by the Université catholique de Louvain, the Fonds National de la Recherche Scientifique (F. R. S.-FNRS) and the FNRS-Welbio (Grant # CR-2019S-01). M. K. was a postdoctoral researcher at the F. R. S-FNRS, and her research is now supported by the Leibniz Program for Junior Research groups (grant: J112/2021). A. C. D. and D. A. are research associate and senior research associate at the F. R. S.-FNRS, respectively. This work was also funded by the European Union (ERC, SingViroPhys, 101088316). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. A. D. P. research is supported by the Leibniz Program for Women Professors (grant: P116/2020). Additional support was provided by U.S. Public Health Service award R01 AI174526 (D. M. S., O. L. W., and T. S. D.) and the Heinz Endowments (T. S. D.). The authors gratefully acknowledge the computational resources provided by the Leibniz Supercomputing Centre (https://www.lrz.de).References
- J. Grove and M. Marsh, The cell biology of receptor-mediated virus entry, J. Cell Biol., 2011, 195, 1071–1082 CrossRef CAS PubMed.
- Y. Yamauchi and A. Helenius, Virus entry at a glance, J. Cell Sci., 2013, 126, 1289–1295 CrossRef CAS PubMed.
- K. Guglielmi, E. Johnson, T. Stehle and T. Dermody, Attachment and cell entry of mammalian orthoreovirus, Reoviruses: Entry, Assembly and Morphogenesis, 2006, pp. 1–38 Search PubMed.
- A. B. Sabin, Reoviruses: a new group of respiratory and enteric viruses formerly classified as ECHO type 10 is described, Science, 1959, 130, 1387–1389 CrossRef CAS PubMed.
- K. Tyler, Pathogenesis of reovirus infections of the central nervous system, Reoviruses II: Cytopathogenicity and Pathogenesis, 1998, pp. 93–124 Search PubMed.
- C. S. Raine and B. N. Fields, Reovirus type III encephalitis—a virologic and ultrastructural study, J. Neuropathol. Exp. Neurol., 1973, 32, 19–33 CrossRef CAS PubMed.
- A. A. Antar, et al., Junctional adhesion molecule-A is required for hematogenous dissemination of reovirus, Cell Host Microbe, 2009, 5, 59–71 CrossRef CAS PubMed.
- D. Bass, J. Trier, R. Dambrauskas and J. Wolf, Reovirus type I infection of small intestinal epithelium in suckling mice and its effect on M cells, Lab. Invest., 1988, 58, 226–235 CAS.
- H. L. Weiner, D. Drayna, D. R. Averill Jr and B. N. Fields, Molecular basis of reovirus virulence: role of the S1 gene, Proc. Natl. Acad. Sci. U. S. A., 1977, 74, 5744–5748 CrossRef CAS PubMed.
- J. L. Wolf, et al., Intestinal M cells: a pathway for entry of reovirus into the host, Science, 1981, 212, 471–472 CrossRef CAS PubMed.
- J. D. Chappell, A. E. Prota, T. S. Dermody and T. Stehle, Crystal structure of reovirus attachment protein σ1 reveals evolutionary relationship to adenovirus fiber, EMBO J., 2002, 21, 1–11 CrossRef CAS PubMed.
- D. M. Reiter, et al., Crystal structure of reovirus attachment protein σ1 in complex with sialylated oligosaccharides, PLoS Pathog., 2011, 7, e1002166 CrossRef CAS PubMed.
- X. Zhang, et al., Features of reovirus outer capsid protein μ1 revealed by electron cryomicroscopy and image reconstruction of the virion at 7.0 Å resolution, Structure, 2005, 13, 1545–1557 CrossRef CAS PubMed.
- P. Shang, et al., Paired immunoglobulin-like receptor B is an entry receptor for mammalian orthoreovirus, Nat. Commun., 2023, 14, 2615 CrossRef CAS PubMed.
- D. M. Sutherland, et al., NgR1 binding to reovirus reveals an unusual bivalent interaction and a new viral attachment protein, Proc. Natl. Acad. Sci. U. S. A., 2023, 120, e2219404120 CrossRef CAS PubMed.
- G. J. Wilson, et al., A single mutation in the carboxy terminus of reovirus outer-capsid protein σ3 confers enhanced kinetics of σ3 proteolysis, resistance to inhibitors of viral disassembly, and alterations in σ3 structure, J. Virol., 2002, 76, 9832–9843 CrossRef CAS PubMed.
- K. A. Dryden, et al., Early steps in reovirus infection are associated with dramatic changes in supramolecular structure and protein conformation: analysis of virions and subviral particles by cryoelectron microscopy and image reconstruction, J. Cell Biol., 1993, 122, 1023–1041 CrossRef CAS PubMed.
- J. D. Doyle, et al., Molecular determinants of proteolytic disassembly of the reovirus outer capsid, J. Biol. Chem., 2012, 287, 8029–8038 CrossRef CAS PubMed.
- E. S. Barton, et al., Utilization of sialic acid as a coreceptor is required for reovirus-induced biliary disease, J. Clin. Invest., 2003, 111, 1823–1833 CrossRef CAS PubMed.
- J. L. Konopka-Anstadt, et al., The Nogo receptor NgR1 mediates infection by mammalian reovirus, Cell Host Microbe, 2014, 15, 681–691 CrossRef CAS PubMed.
- A. W. McGee and S. M. Strittmatter, The Nogo-66 receptor: focusing myelin inhibition of axon regeneration, Trends Neurosci., 2003, 26, 193–198 CrossRef CAS PubMed.
- X. Wang, et al., Localization of Nogo-A and Nogo-66 receptor proteins at sites of axon–myelin and synaptic contact, J. Neurosci., 2002, 22, 5505–5515 CrossRef CAS PubMed.
- P. Aravamudhan, et al., The Murine Neuronal Receptor NgR1 Is Dispensable for Reovirus Pathogenesis, J. Virol., 2022, 96, e00055 CrossRef PubMed.
- A. Valbuena and M. G. Mateu, Quantification and modification of the equilibrium dynamics and mechanics of a viral capsid lattice self-assembled as a protein nanocoating, Nanoscale, 2015, 7, 14953–14964 RSC.
- P. J. de Pablo, Seminars in cell & developmental biology, Elsevier, 2018, vol. 73, pp. 199–208 Search PubMed.
- D. J. Müller, et al., Atomic force microscopy-based force spectroscopy and multiparametric imaging of biomolecular and cellular systems, Chem. Rev., 2020, 121, 11701–11725 CrossRef PubMed.
- C. Bustamante, J. F. Marko, E. D. Siggia and S. Smith, Entropic elasticity of λ-phage DNA, Science, 1994, 265, 1599–1600 CrossRef CAS PubMed.
- E. Evans and K. Ritchie, Dynamic strength of molecular adhesion bonds, Biophys. J., 1997, 72, 1541–1555 CrossRef CAS PubMed.
- E. A. Evans and D. A. Calderwood, Forces and bond dynamics in cell adhesion, Science, 2007, 316, 1148–1153 CrossRef CAS PubMed.
- P. M. Williams, Analytical descriptions of dynamic force spectroscopy: behaviour of multiple connections, Anal. Chim. Acta, 2003, 479, 107–115 CrossRef CAS.
- J. Lauré, et al., Characterization of myelin ligand complexes with neuronal Nogo-66 receptor family members, J. Biol. Chem., 2007, 282, 5715–5725 CrossRef PubMed.
- M. F. Pronker, R. P. Tas, H. C. Vlieg and B. J. Janssen, Nogo Receptor crystal structures with a native disulfide pattern suggest a novel mode of self-interaction, Acta Crystallogr., Sect. D: Struct. Biol., 2017, 73, 860–876 CrossRef CAS PubMed.
- T. Kobayashi, L. S. Ooms, M. Ikizler, J. D. Chappell and T. S. Dermody, An improved reverse genetics system for mammalian orthoreoviruses, Virology, 2010, 398, 194–200 CrossRef CAS PubMed.
- K. M. Coombs, Mammalian Reoviruses: Propagation, Quantification, and Storage, Curr. Protocols, 2023, 3, e716 CrossRef CAS PubMed.
- A. Ebner, P. Hinterdorfer and H. J. Gruber, Comparison of different aminofunctionalization strategies for attachment of single antibodies to AFM cantilevers, Ultramicroscopy, 2007, 107, 922–927 CrossRef CAS PubMed.
- L. Wildling, et al., Linking of sensor molecules with amino groups to amino-functionalized AFM tips, Bioconjugate Chem., 2011, 22, 1239–1248 CrossRef CAS PubMed.
- J. M. Lee, et al., Direct immobilization of protein G variants with various numbers of cysteine residues on a gold surface, Anal. Chem., 2007, 79, 2680–2687 CrossRef CAS PubMed.
- H.-J. Butt and M. Jaschke, Calculation of thermal noise in atomic force microscopy, Nanotechnology, 1995, 6, 1 CrossRef.
- D. Alsteens, et al., Nanomechanical mapping of first binding steps of a virus to animal cells, Nat. Nanotechnol., 2017, 12, 177–183 CrossRef CAS PubMed.
- R. Newton, et al., Combining confocal and atomic force microscopy to quantify single-virus binding to mammalian cell surfaces, Nat. Protoc., 2017, 12, 2275–2292 CrossRef CAS PubMed.
- C. Rankl, et al., Determination of the kinetic on-and off-rate of single virus–cell interactions, At. Force Microsc. Biomed. Res., 2011, 197–210 CAS.
- W. L. Jorgensen and J. Tirado-Rives, The OPLS [optimized potentials for liquid simulations] potential functions for proteins, energy minimizations for crystals of cyclic peptides and crambin, J. Am. Chem. Soc., 1988, 110, 1657–1666 CrossRef CAS PubMed.
- D. A. Case, et al., The Amber biomolecular simulation programs, J. Comput. Chem., 2005, 26, 1668–1688 CrossRef CAS PubMed.
- R. Salomon-Ferrer, D. A. Case and R. C. Walker, An overview of the Amber biomolecular simulation package, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2013, 3, 198–210 CAS.
- J. A. Maier, et al., ff14SB: improving the accuracy of protein side chain and backbone parameters from ff99SB, J. Chem. Theory Comput., 2015, 11, 3696–3713 CrossRef CAS PubMed.
- R. J. Loncharich, B. R. Brooks and R. W. Pastor, Langevin dynamics of peptides: the frictional dependence of isomerization rates of N-acetylalanyl-N′-methylamide, Biopolymers, 1992, 32, 523–535 CrossRef CAS PubMed.
- V. Kräutler, W. F. Van Gunsteren and P. H. Hünenberger, A fast SHAKE algorithm to solve distance constraint equations for small molecules in molecular dynamics simulations, J. Comput. Chem., 2001, 22, 501–508 CrossRef.
- U. Essmann, et al., A smooth particle mesh Ewald method, J. Chem. Phys., 1995, 103, 8577–8593 CrossRef CAS.
- M. J. Abraham, et al., GROMACS: high performance molecular simulations through multi-level parallelism from laptops to supercomputers, SoftwareX, 2015, 1, 19–25 CrossRef.
- H. Bekker, et al., 4th international conference on computational physics (PC 92), World Scientific Publishing, 1993, pp. 252–256 Search PubMed.
- R. Martoňák, A. Laio and M. Parrinello, Predicting crystal structures: the Parrinello-Rahman method revisited, Phys. Rev. Lett., 2003, 90, 075503 CrossRef PubMed.
- R. J. Gowers, et al., Proceedings of the 15th python in science conference, SciPy Austin, TX, 2016, vol. 98, pp. 105 Search PubMed.
- N. Michaud-Agrawal, E. J. Denning, T. B. Woolf and O. Beckstein, MDAnalysis: a toolkit for the analysis of molecular dynamics simulations, J. Comput. Chem., 2011, 32, 2319–2327 CrossRef CAS PubMed.
- J. D. Hunter, Matplotlib: a 2D graphics environment, Comput. Sci. Eng., 2007, 9, 90–95 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Fig. S1–S9 and Tables S1–S3. See DOI: https://doi.org/10.1039/d4nh00315b |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |